Настоящее изобретение относится к производным 5-гидроксибензо[b]тиофен-3-карбоновой кислоты, которые являются основными исходными материалами для получения соединений, полезных в области лекарственных средств.
Предпосылки создания изобретения
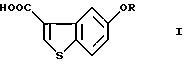
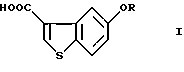
Производные 5-гидроксибензо[b]тиофен-3-карбоновой кислоты общей формулы (I)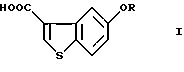
где R представляет водород или гидрокси защитную группу,
являются важными исходными материалами в синтезе фармакологически активных соединений. Например, соединение формулы (I) играет важную роль в синтезе производных бензотиофенкарбоксамида, имеющих общую формулу (VI)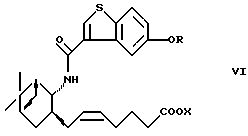
где R определен, как указано выше, и Х представляет водород или алкил. Производные бензотиофенкарбоксамида являются специфическими антагонистами PGD2 и, как известно, полезны в качестве лекарственного вещества для лечения различных заболеваний, связанных с дисфункцией тучных клеток, вызванной избыточной продукцией PGD2, например системного мастоцитоза, нарушения системной активации тучных клеток, трахеального спазма, астмы, аллергического ринита, аллергического конъюнктивита, крапивницы, повреждения в результате ишемической реперфузии, воспаления и атонического дерматита (W097/00853, PCT/JP97/04527 (W098/25919)). Из соединений формулы (VI) соединение, в котором OR представляет 5-гидрокси и Х представляет водород (далее называемое "соединение А"), оказывает особенно сильное антагонистическое действие на PGD2, обладает превосходной активностью против носовой окклюзии и, как ожидается, будет многообещающим лекарственным средством для лечения носовой окклюзии.
Сущность изобретения
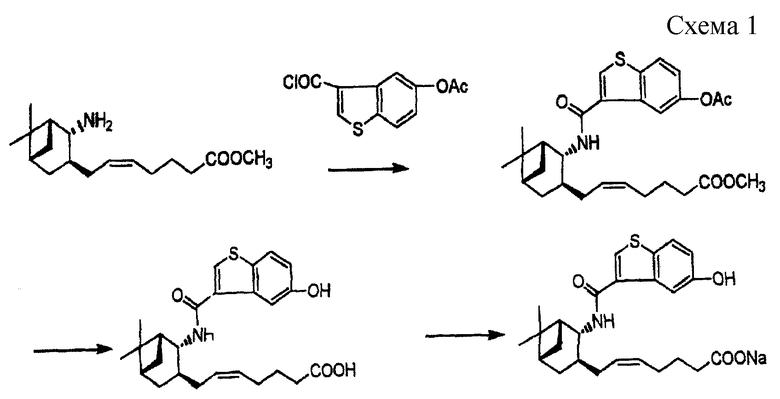
Способ получения вышеуказанного соединения показан на реакционной схеме 1 (W098/25919):
Для широкого клинического применения соединения А важно создать способ получения исходного материала - соединения (I), который (способ) был бы безопасным, эффективным и промышленно применимым.
Однако синтезировать производные бензотиофена, имеющие, подобно соединению (I), 5-гидроксильную группу, трудно и потому до сих пор не было промышленных способов синтеза. Существующие способы включают различные сложные процессы, являются непроизводительными и дают низкий выход. Например, уже есть способы, в которых осуществляют бромирование 5-ацетоксибензо[b]тиофена с получением 3-бром-5-ацетоксибензо[b]тиофена, который в свою очередь повторно защищают по 5-ацетоксигруппе бензильной группой с получением 3-бром-5-бензилоксибензо[b] тиофена, который затем подвергают металлизации магнием, введению диоксида углерода и удалению бензильной группы (J. Chem. Soc. (С), 1967, 1899-1905), или 5-бромбензо[b] тиофен подвергают реакции Фриделя-Крафтса с получением 3-ацетил-5-бромбензо[b]тиофена, который затем окисляют гипохлоритом натрия с получением 5-бромбензо[b]тиофен-3-карбоновой кислоты (Nippon-Kagaku Zasshi, т.86, 10, 1067-1072 (1965), J. Chem. Soc. (С), 1967, 2084-2089). Затем из указанных выше продуктов реакций синтезируют 5-гидроксибензо[b] тиофен-3-карбоновую кислоту или 5-ацетоксибензо[b]тиофен-3-карбоновую кислоту. Однако исходный материал, такой как 5-гидроксибензо[b]тиофен или 5-бромбензо[b] тиофен, не является коммерчески доступным и во всех случаях должен быть синтезирован из подходящего реагента (например, J. Chem. Soc., 57, 1611 (1935); J. Heterocyclic Chem., 25, 1271 (1988)), что удлиняет и усложняет процесс синтеза.
Настоящее изобретение решает проблемы существующих способов и дает способ получения соединений формулы (I), промышленно применимый, производительный и безопасный.
Таким образом, в соответствии с настоящим изобретением предлагается способ получения соединения формулы (I)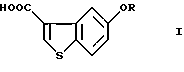
где R представляет водород или гидроксизащитную группу,
или его реакционноспособного производного, включающий введение 4-меркаптофенола в реакции, обеспечивающие введение пропаргильной группы и защиту гидроксильной группы, с получением соединения формулы (II)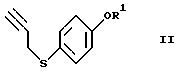
где R1 представляет гидроксизащитную группу,
окисление соединения (II) с получением соединения формулы (III)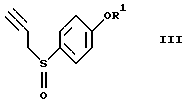
где R1 представляет гидроксизащитную группу,
введение соединения (III) в реакцию термической перегруппировки с получением соединения формулы (IV)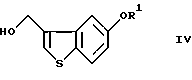
где R1 определен, как указано выше,
и введение соединения (IV) в реакцию ступенчатого окисления гидроксиметильной группы и, необязательно, удаления защитной группы.
В соответствии с настоящим изобретением предлагается также способ получения соединения формулы (I)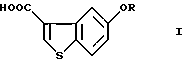
где R представляет водород или гидроксизащитную группу,
или его реакционноспособного производного, включающий введение 5-гидроксибензо[b] тиофена в реакцию введения защитной группы с получением соединения формулы (VII)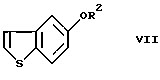
где R2 представляет гидроксизащитную группу,
осуществление взаимодействия соединения (VII) с ацетилгалогенидом в условиях реакции Фриделя-Крафтса с получением соединения формулы (VIII)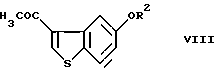
где R2 представляет гидроксизащитную группу,
и введение соединения (VIII) в реакцию окисления ацетильной группы и, необязательно, удаление защитной группы.
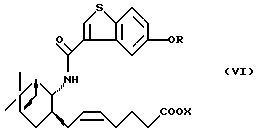
Кроме того, в соответствии с настоящим изобретением предлагается способ получения вышеуказанного производного 5-гидроксибензо[b]тиофен-3-карбоновой кислоты, имеющего общую формулу (VI), с использованием соединения формулы (I). Так, в соответствии с настоящим изобретением предлагается способ получения соединения формулы (VI)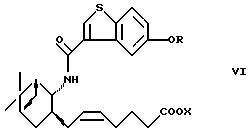
где R определен, как указано выше, и Х представляет водород или алкил, а двойная связь представляет Е- или Z-конфигурацию,
или его фармацевтически приемлемой соли или гидрата, который (способ) включает введение соединения формулы (I) или его реакционноспособного производного в следующие реакции:
(1) взаимодействие с соединением формулы (V)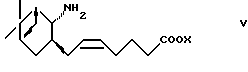
где X представляет водород или алкил, или
(2) взаимодействие с соединением формулы (V')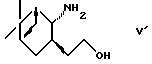
или его солью с последующим окислением и взаимодействием с илидом в условиях реакции Виттига и
(3) необязательное удаление защитной группы.
Наилучший способ осуществления изобретения
Ниже дано определение терминов, использованных в данном описании.
Термин "гидроксизащитная группа" означает алкил, алкоксиалкил, ацил, аралкил, алкилсульфонил, арилсульфонил, алкилзамещенный силил, алкоксикарбонил, арилоксикарбонил, аралкилоксикарбонил или тетрагидропиранил.
Термин "алкил" означает C1-C20 неразветвленный или разветвленный алкил, в частности метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, неопентил, трет-пентил, гексил, гептил, октил, нонил, децил, ундецил, додецил, тридецил, тетрадецил, пентадецил, гексадецил, гептадецил, октадецил, нонадецил, икозил и тому подобное, причем предпочтительным является C1-С6 алкил.
Термин "алкокси" означает C1-С6 неразветвленный или разветвленный алкокси, в частности метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси, н-пентилокси, изопентилокси, неопентилокси, втор-пентилокси, трет-пентилокси, н-гексилокси, неогексилокси, изогексилокси, втор-гексилокси, трет-гексилокси и тому подобное, причем предпочтительным является C1-С3 алкокси.
Термин "алкоксиалкил" означает алкильную группу, замещенную алкоксигруппой, и включает метоксиметил, этоксиметил, метоксиэтоксиметил, этоксиэтил, метоксипропил и тому подобное.
Термин "ацил" означает C1-С11 ацил, произведенный от алифатической или ароматической карбоновой кислоты. Примеры ацила алифатической кислоты включают ацетил, хлорацетил, трихлорацетил, пропионил, бутирил, валерил и тому подобное, а примеры ацила ароматической кислоты включают бензоил, п-нитробензоил, п-метоксибензоил, п-бромбензоил, толуоил, нафтоил и тому подобное.
Термин "арил" означает фенил, нафтил или полициклическую ароматическую углеводородную группу и тому подобное. Кроме того, арил может быть замещенным следующими заместителями.
Примеры заместителей включают алкил, такой как метил, этил, н-пропил, изопропил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, неопентил или трет-пентил, низший алкокси, такой как метокси или этокси, галоген, такой как фтор, хлор, бром или иод, нитро, гидрокси, карбокси, циано, сульфонил, амино, низший алкиламино, такой как метиламино, диметиламино, этиламино или диэтиламино, и тому подобное. Арильная группа может иметь один или несколько заместителей в любых возможных положениях. Конкретные примеры арила включают 2-метилфенил, 3-метилфенил, 4-метилфенил, 2-этилфенил, 3-этилфенил, 4-этилфенил, 4-пентилфенил, 4-карбоксифенил, 4-ацетилфенил, 4-(N, N-диметиламино)фенил, 4-нитрофенил, 4-гидроксифенил, 4-метоксифенил, 4-фторфенил, 4-хлорфенил, 4-иодфенил и тому подобное.
Арильная группа в описанном ниже "аралкиле", "арилсульфониле", "арилоксикарбониле" или "аралкилоксикарбониле" может иметь заместители, аналогичные определенным выше.
Термин "аралкил" означает алкильную группу, замещенную арильной группой, и включает бензил, 4-метилбензил, 4-метоксибензил, 3,4-диметоксибензил, нафтилметил, фенэтил и тому подобное.
Термин "алкилсульфонил" означает сульфонильную группу, замешенную алкильной группой, и включает метансульфонил, этансульфонил и тому подобное.
Термин "арилсульфонил" означает сульфонильную группу, замещенную арильной группой, и включает бензолсульфонил, п-толуолсульфонил и тому подобное.
Термин "алкилзамещенный силил" означает моно-, ди- или триалкилзамещенный силил, например метилсилил, диметилсилил, триметилсилил, трет-бутилдиметилсилил и тому подобное.
Термин "алкоксикарбонил" означает метоксикарбонил, изопропоксикарбонил, трет-бутоксикарбонил и тому подобное.
Термин "арилоксикарбонил" означает феноксикарбонил и тому подобное.
Термин "аралкилоксикарбонил" означает бензилоксикарбонил и тому подобное.
Все указанные гидроксизащитные группы являются предпочтительными в качестве гидроксизащитной группы, показанной символами R1, R2 или R в соответственных формулах, приведенных выше, но из них более предпочтительным является арилсульфонил, а особенно предпочтительным - бензолсульфонил.
Примеры солей соединения общей формулы (VI) включают соли щелочных металлов, такие как литиевая соль, натриевая соль или калиевая соль и тому подобное, соли щелочноземельных металлов, такие как кальциевая соль и тому подобное, аммониевая соль, соли с органическими основаниями, такими как триметамин, триметиламин, триэтиламин; 2-аминобутан, трет-бутиламин, диизопропилэтиламин, н-бутилметиламин, н-бутилдиметиламин, три-н-бутиламин, циклогексиламин, дициклогексиламин, N-изопропилциклогексиламин, фурфуриламин, бензиламин, метилбензиламин, дибензиламин, N,N-диметилбензиламин, 2-хлорбензиламин, 4-метоксибензиламин, 1-нафталинметиламин, дифенилбензиламин, трифениламин, 1-нафтиламин, 1-аминоантрацен, 2-аминоантрацен, дегидроабиетиламин, N-метилморфолин или пиридин, или аминокислотные соли, такие как лизиновая соль или аргининовая соль.
Соли аминоспирта формулы (V') включают соли с органической кислотой, такой как бензойная кислота и т.д., и минеральной кислотой, такой как хлороводородная кислота, серная кислота и т.д.
Конечное соединение по настоящему изобретению представлено приведенной выше формулой (VI), в которой двойная связь алкениленовой боковой цепи (5-гептениленовой цепи) может быть в Е- или Z-конфигурации.
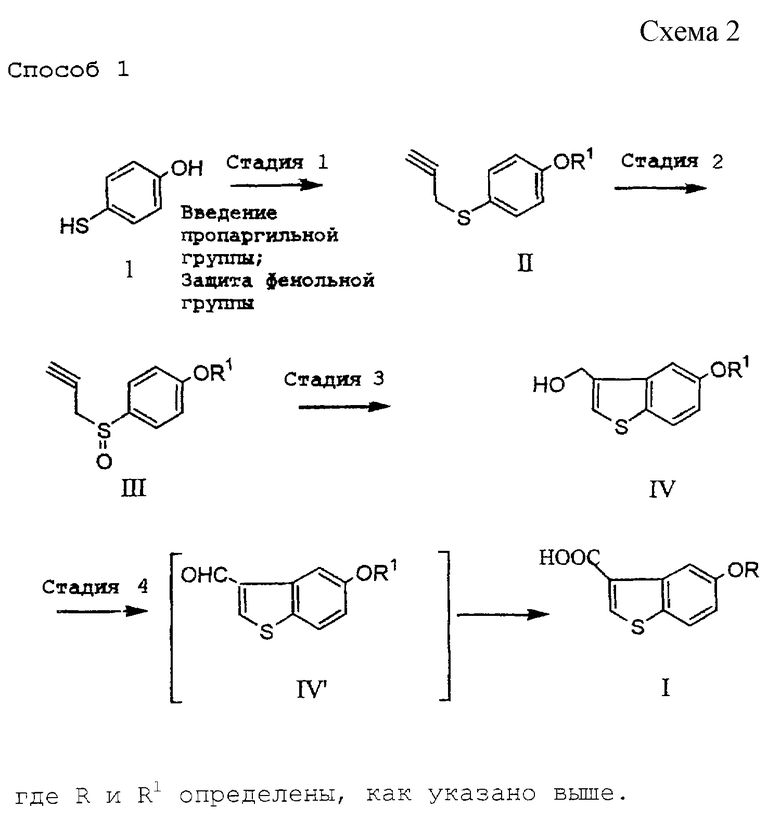
Способ по настоящему изобретению описан ниже более подробно. При наличии заместителя(ей), возможно мешающего(их) реакции, его (их) можно подходящим образом защитить, а затем на требуемой стадии освободить от защиты. Такие защиту или освобождение от защиты можно осуществить по методике, известной в данной области техники. Далее см. схему 2.
[Стадия 1]
Эта стадия связана с введением пропаргильной группы в меркаптогруппу 4-меркаптофенола (1) и защитой гидроксильной группы.
Введение пропаргильной группы осуществляют, используя пропаргилгалогенид, такой как пропаргилбромид, пропаргилхлорид и тому подобное, в присутствии основных агентов. Реакция может быть осуществлена в течение времени в пределах от нескольких десятков минут до нескольких часов при комнатной температуре с использованием в качестве основного агента неорганического основания, такого как карбонат калия, карбонат натрия или тому подобное, или органического основания, такого как триэтиламин, пиридин, 4-диметиламинопиридин или тому подобное, в растворителе, таком как ацетон, этилацетат, тетрагидрофуран, ацетонитрил или тому подобное.
При использовании сильного основания, такого как гидроксид калия или гидроксид натрия, реакцию можно также осуществлять в двухслойной растворительной системе, такой как толуол-вода или ксилол-вода.
Защита гидроксильной группы может быть проведена с использованием обычной гидроксизащитной группы обычным образом. Предпочтительными защитными группами, используемыми в предлагаемом способе, являются те, которые не подвержены изменениям в ходе окислительных реакций на 2-ой и 4-ой стадиях данного процесса и 2-ой стадии описанного ниже процесса IV получения соединения формулы (VI), а также в ходе реакции Виттига на 3-ей стадии указанного процесса, и могут быть легко удалены на 4-й стадии с образованием уходящих групп, легко отделяемых, например, от соединения А при его очистке, которое соответствует соединению формулы (VI), где OR представляет 5-гидрокси, Х представляет водород и двойная связь находится в Z-конфигурации. Примеры такой гидроксизащитной группы включают алкил, алкоксиалкил, ацил, аралкил, алкилсульфонил, арилсульфонил, алкилзамещенный силил, алкоксикарбонил, арилоксикарбонил, аралкилоксикарбонил или тетрагидропиранил.
С учетом требований, состоящих в том, что защитная группа должна уцелеть в ходе реакции Виттига, проводимой в сильно основных условиях, быть легко удаляемой, например, на 4-ой стадии при получении соединения А и быть отделяемой от соединения А, более предпочтительным является арилсульфонил и особенно предпочтительным - бензолсульфонил. Бензолсульфонильная группа является относительно стойкой к основанию в безводных растворителях и при удалении для снятия защиты дает бензолсульфоновую кислоту, которая растворима в воде и легко отделяется от целевого продукта формулы (VI). Защиту и снятие защиты можно осуществить методом, известным в данной области. Например, в случае использования бензолсульфонильной группы ее введение осуществляют способом, аналогичным введению пропаргильной группы, с использованием бензолсульфонилхлорида.
[Стадия 2]
Эта стадия относится к окислению соединения (II). Известны методы окисления, в которых используют, например, смесь безводный пероксид водорода-уксусная кислота (J. Am. Chem. Soc., 87, 1109-1114 (1965)), смесь водный пероксид водорода-хлорид титана (III) (Synthesis 1981, 204-206), м-хлорпербензойную кислоту (Org. Synth., 64, 157-163 (1985)) или метапериодат натрия (J. Org. Chem., 27, 282-284 (1962)). На этой стадии является предпочтительным использовать слегка избыточное количество 30%-ного водного пероксида водорода в спиртовом растворителе, как, например, раствор в этаноле, метаноле, изопропаноле или трет-бутаноле, содержащий муравьиную кислоту. Реакцию осуществляют в течение времени в пределах от нескольких десятков минут до нескольких часов при охлаждении или при комнатной температуре.
[Стадия 3]
Эта стадия относится к превращению соединения (III) в гидроксиметильное соединение (IV) путем термической перегруппировки. Реакцию термической перегруппировки на этой стадии осуществляют в соответствии со способом, описанным в J. С. S. Chem. Соmm., 1974, 848-849. Примеры предпочтительных растворителей для этой реакции включают диоксан, 1,2-диметоксиэтан, пропилацетат и 3-пентанон. Реакцию осуществляют кипячением с обратным холодильником в растворителе в течение нескольких часов с последующим добавлением к полученному промежуточному продукту кислоты (п-толуолсульфоновой кислоты, метансульфоновой кислоты, серной кислоты и т.д.).
[Стадия 4]
Эта стадия относится к окислению соединения (IV) с получением карбоновой кислоты (I). Окисление может быть осуществлено непосредственно или ступенчато. Примеры окислителя для превращения ароматического первичного спирта непосредственно в соответствующую карбоновую кислоту включают хромовые кислоты (Synthesis, 1986, 285-288), перманганат калия (J. Org. Chem., 18, 806-809 (1,953)) и оксиды рутения (J. С. S. Chem. Comm., 1979, 58-59)). Однако эти способы имеют недостатки не только с точки зрения выхода, но и по следующим причинам. Например, велико время реакции, необходимо обезвреживание окислителя после реакции, неустойчивы реагенты и/или они включают сложные операции.
Наоборот, в некоторых случаях может быть выгодным, с точки зрения выхода, ступенчатое окисление, при котором первичный спирт окисляют до альдегида и затем до карбоновой кислоты. Обычно окисление спирта до альдегида осуществляли с использованием окислителя из семейства хромовых кислот, например реактивов Джонса (J. Org. Chem., 40, 1664-1665 (1975)), реактивов Коллинза (J. С. S. Chem. Соmm., 1972, 1126), пиридинийхлорхромата (Tetrahedron Lett. , 2647-2650 (1975)). Известен также метод с использованием диоксида марганца (Helv. Chem. Acta. , 39, 858-862 (1956)) или диметилсульфоксида (Swern oxidation, J. Org. Chem., 43, 2480-2482 (1978)). Однако эти существующие методы имеют недостатки. Например, хромовые кислоты ядовиты для организма человека и должны быть обезврежены после применения. Кроме того, окисление по Суорну (Swern) с использованием смеси диметилсульфоксид-оксалилхлорид не годится для крупномасштабного производства, потому что оно сопровождается выделением моноксида углерода, опасного для работников, и запахом серы, и к тому же его нужно осуществлять при низкой температуре, например, между -50oС и -78oС.
Спирт (IV) может быть преобразован в альдегид (IV') почти количественно способом, при котором спирт (IV) окисляют с помощью окислителя, такого как галогеноксокислота, в присутствии 2,2,6,6-тетраметилпиперидин-1-оксила или подобных ему соединений (обозначаемых как "ТЕМПО"), согласно описанию в литературе (например, J. Org. Chem., 52, 2559-2562 (1987)), что решает проблемы существующих методов. Примеры используемых TEMPO включают 2,2,6,6-тетраметилпиперидин-1-оксил, 4-метокси-2,2,6,6-тетраметилпиперидин-1-оксил, 4-ацетиламино-2,2,6,6-тетраметилпиперидин-1-оксил, 4-бензоилокси-2,2,6,6-тетраметилпиперидин-1-оксил, 4-циано-2,2,6,6-тетраметилпиперидин-1-оксил. Примеры используемых галогеноксокислот включают гипохлорит натрия, гипобромит натрия, бромит натрия и высшую хлорную известь. Раствор окислителя может быть доведен, например, до значения рН 8,5-9,5 добавлением минеральной кислоты, такой как соляная или серная кислота. В соответствии с другим вариантом раствор окислителя может быть добавлен в присутствии гидрокарбоната натрия. Реакция может быть осуществлена за время от нескольких минут до нескольких десятков минут при температуре от температуры охлаждения льдом до комнатной температуры в растворителе, таком как этилацетат, ацетонитрил или дихлорметан.
Когда реакционный раствор, содержащий полученный альдегид (IV'), подкисляют и добавляют к нему хлорит натрия и водный пероксид водорода, альдегид превращают в карбоновую кислоту при охлаждении льдом в течение времени от нескольких десятков минут до нескольких часов.
При необходимости продукт может быть дополнительно обработан для удаления 5-гидроксизащитной группы и/или преобразования в реакционноспособные производные по 3-карбоксильной группе. Такое "реакционноспособное производное" включает соответствующие галогенангидриды кислоты (например, хлорид, бромид, иодид), ангидриды кислоты (например, смешанный ангидрид кислоты с муравьиной или уксусной кислотой), активированные сложные эфиры (например, сукцинимидоэфир) и тому подобное и включает ацилирующие агенты, обычно используемые для ацилирования аминогруппы. Например, чтобы получить галогенангидриды кислоты, карбоновую кислоту подвергают взаимодействию с тионилгалогенидом (например, тионилхлоридом), фосфористым галогенидом (например, трихлоридом фосфора, пентахлоридом фосфора), оксалилгалогенидом (например, оксалилхлоридом) или тому подобным в соответствии с известным методом (например, Shin-jikken Kagaku Koza, т.14, стр.1787 (1978); Synthesis, 852-854 (1986); Shin-jikken Kagaku Koza, т.22, стр.115 (1992)), (см. схему 3).
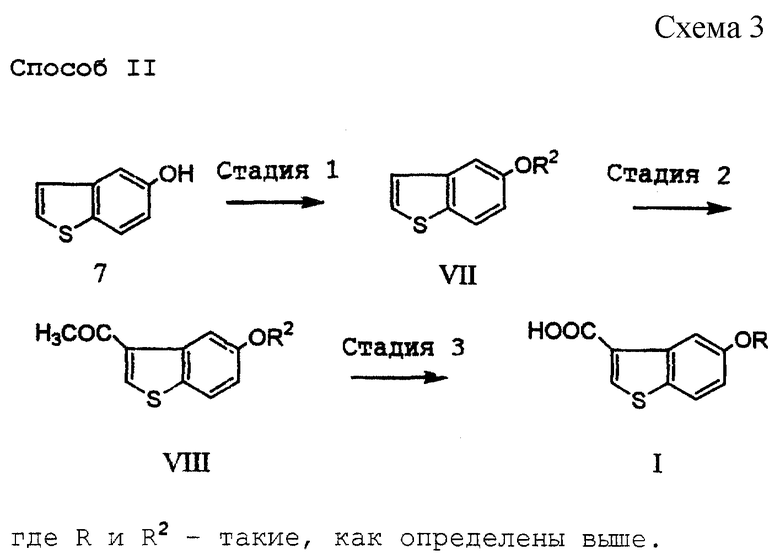
[Стадия 1]
Эта стадия связана с защитой 5-гидроксигруппы соединения (7).
Соединение (7) как исходный материал данной стадии известно в литературе (J. Am. Chem. Soc., 57, 1611-1616 (1935), Ann. Chem., 527, 83-114 (1938), J. Am. Chem. Soc., 78, 5351-5357 (1956), J. Org. Chem., 41, 1118-1124 (1976)). Гидроксильную группу этого соединения соответствующим образом защищают подобно тому, как описано для 1-ой стадии процесса I выше. Например, при использовании бензолсульфонильной группы соединение добавляют к бензолсульфонилхлориду в присутствии неорганического основания, такого как карбонат натрия или карбонат калия, или органического основания, такого как триэтиламин или трипропиламин. Примеры предпочтительного растворителя включают ацетон, этилацетат и тетрагидрофуран. Реакцию осуществляют в течение времени в пределах от нескольких минут до нескольких часов при температуре от комнатной до температуры кипения растворителя. Соединение (VII) может быть также синтезировано с помощью широко применяемого метода, известного как "реакция Шоттена-Баумана".
[Стадия 2]
Эта стадия связана с введением ацетильной группы в положение 3 соединения (VII) путем проведения реакции Фриделя-Крафтса. Введение ацетильной группы осуществляют, например, используя ацетилхлорид или ацетилбромид в присутствии катализатора, например кислоты Льюиса, такой как хлорид алюминия, хлорид железа (III), хлорид цинка, хлорид олова и трифторид бора. Примеры используемого растворителя включают дисульфид углерода, нитробензол или галогенированные углеводороды, такие как дихлорметан или дихлорэтан. Реакции обычно осуществляют в течение нескольких часов при температуре в пределах от температуры охлаждения льдом до комнатной температуры. Полученное в небольшом количестве в качестве побочного продукта 2-ацетильное соединение может быть легко отделено путем перекристаллизации.
[Стадия 3]
Эта стадия относится к превращению соединения (VIII) в карбоновую кислоту (I) или ее реакционноспособное производное путем окисления ацетильной группы в присутствии соли галогенноватистой кислоты. Примеры предпочтительного гипогалогенита включают соли галогенноватистых кислот со щелочными или щелочноземельными металлами, причем особенно предпочтительной является калиевая, натриевая или кальциевая соль хлорноватистой или бромноватистой кислоты.
В водном растворе такой соли окисление проходит при относительно низкой температуре. Однако, чтобы повысить растворимость окисляемого соединения, можно в качестве растворителя использовать диоксан или 1,2-диметоксиэтан. Реакцию осуществляют в течение времени от нескольких часов до нескольких десятков часов при комнатной температуре или при нагревании, (см. схему 4).
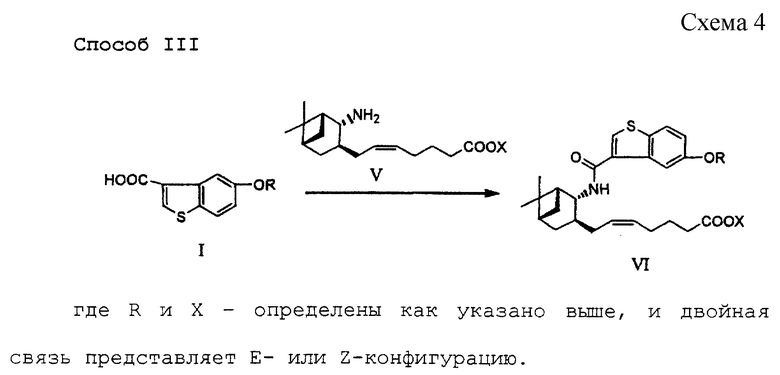
Этот способ связан с синтезом соединения формулы (VI) путем осуществления взаимодействия соединения формулы (I) или его реакционноспособного производного, полученного в описанном выше способе I или II, с соединением формулы (V).
Соединение (V), используемое в данном процессе, может быть получено методом, описанным в публикации патента Японии (KOKOKU) 6-23170 (23170/1994).
Реакция может быть осуществлена в обычных условиях ацилирования аминогруппы. Например, при использовании галогенида карбоновой кислоты реакцию осуществляют согласно способу, общеизвестному как "реакция Шоттена-Баумана". Обычно галогенид карбоновой кислоты добавляют по каплям к водному щелочному раствору амина при перемешивании и охлаждении с удалением образующейся кислоты щелочью. В соответствии с другим вариантом, когда карбоновую кислоту используют в виде свободной кислоты, а не ее реакционноспособного производного, реакцию можно проводить стандартным образом в присутствии сочетающего агента, обычно используемого в реакции сочетания между амином и карбоновой кислотой, такого как дициклогексилкарбодиимид (DCC), 1-этил-3-(3-диметиламинопропил)карбодиимид или N,N'-карбонилдиимидазол, (см. схему 5).
[Стадия 1]
Эта стадия относится к получению соединения формулы (IX) путем осуществления взаимодействия соединения формулы (I) иди его реакционноспособного производного с соединением формулы (V') или его солью подобно тому, как описано выше для способа III. Получение некоторых из соединений формулы (V') описано в Chem. Pharm. Bull., т.37, 6, 1524-1533 (1989).
[Стадия 2]
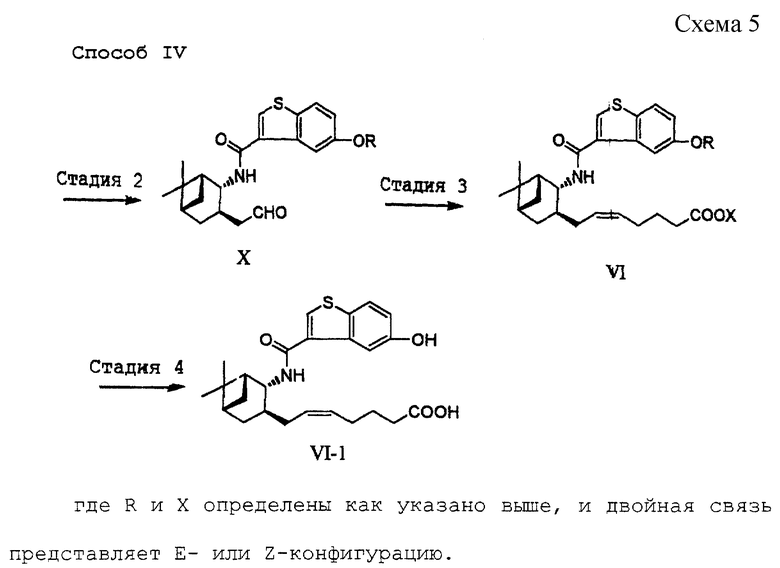
Эта стадия относится к получению альдегида формулы (X) путем окисления соединения формулы (IX). Реакция может быть осуществлена в течение нескольких часов при охлаждении или при комнатной температуре с использованием окислителя, выбранного из семейства хромовых кислот, такого как реактивы Джонса, реактивы Коллинза, пиридинийхлорхромат, пиридинийдихромат, или смеси диметилсульфоксид-оксалилхлорид, в растворителе, таком как хлорированные углеводороды (хлороформ, дихлорметан и т. д.), простые эфиры (диэтиловый эфир, тетрагидрофуран и т.д.), ацетон или бензол.
[Стадия 3]
Эта стадия связана с образованием двойной связи путем осуществления взаимодействия соединения формулы (X) с илидом (Рh3P=CH(CH2)3COOH). Реакция образования двойной связи может быть осуществлена традиционным для реакции Виттига образом. Используемые в реакции илиды могут быть синтезированы в присутствии основания путем обработки фосфониевой соли, синтезированной из трифенилфосфина, и алкилгалогенида, имеющего требуемую для конденсации алкильную группу, например, 5-бромпентановой кислоты. Примеры основания включают диметилсилилнатрий, диметилсилилкалий, гидрид натрия, н-бутиллитий, трет-бутилат калия и диизопропиламид лития. Реакцию осуществляют в течение нескольких часов при комнатной температуре в растворителе, таком как диэтиловый эфир, тетрагидрофуран, н-гексан, 1,2-диметоксиэтан или диметилсульфоксид.
[Стадия 4]
На этой стадии производят необязательное снятие защиты у соединения (VI), в котором R представляет гидроксизащитную группу, с получением соединения (VI-1). Реакция может быть осуществлена традиционным образом с использованием катализатора, такого как хлороводородная кислота, серная кислота, гидроксид натрия, гидроксид калия или гидроксид бария или тому подобное. Реакцию осуществляют в течение времени от нескольких десятков минут до нескольких часов при нагревании в растворителе, таком как метанол-вода, этанол-вода, ацетон-вода, ацетонитрил-вода или тому подобное.
Для более подробной иллюстрации настоящего изобретения далее даны примеры, которые не должны рассматриваться как ограничивающие каким-либо образом объем изобретения. Используемые в примерах сокращения имеют следующие значения:
Ph - фенил,
Ас - ацетил,
TEMPO - 2,2,6,6-тетраметилпиперидин-1-оксид.
Пример 1
(1) Стадия 1:
4-(2-Пропин-1-илтио)фенилбензолсульфонат (2)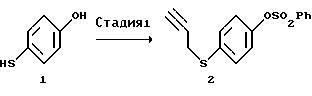
4-Меркаптофенол (1) (37,85 г, 300 ммоль) и пропаргилбромид (42,82 г, 360 ммоль) растворяли в этилацетате (757 мл). К раствору добавляли по каплям триэтиламин (42,5 г, 420 ммоль) в течение 25 минут при перемешивании и охлаждении льдом. После перемешивания в течение еще 1,5 часов при той же температуре добавляли триэтиламин (42,5 г, 420 ммоль) одной порцией и в течение 30 минут добавляли по каплям бензолсульфонилхлорид (63,58 г, 360 ммоль). Выдержав 1 час при той же температуре, убирали охлаждающую ванну и смесь перемешивали 30 минут при комнатной температуре и разделяли на два слоя добавлением ледяной воды (500 мл) и 2 н. хлороводородной кислоты (110 мл). Водный слой экстрагировали этилацетатом (200 мл). Объединенный органический слой промывали водой, сушили над безводным сульфатом магния и затем отгоняли растворитель при пониженном давлении с получением 100,04 г указанного в заголовке соединения (2) в виде масла. Выход неочищенного продукта 109%.
ИК (СНСl3): 3306, 3071, 3031, 3019, 3009, 1585, 1486, 1449, 1378 см-1.
1H ЯМР δ (CDCl3), 300 МГц: 2,23 (1Н, т, J=2,7 Гц), 3,56 (2Н, д, J=2,7 Гц), 6,94 и 7,34 (каждый 2Н, каждый д, J=8,7 Гц), 7,51-7,56 (2Н, м), 7,68 (1Н, м), 7,82-7,85 (2Н, м).
(2) Стадия 2:
4-(2-Пропин-1-илтио)фенилбензолсульфонат (3)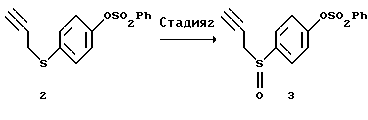
Соединение (2) (60,8 г, 183 ммоль), полученное на описанной выше стадии (1), растворяли в муравьиной кислоте (30,4 мл) и метаноле (122 мл) и затем в раствор добавляли 31%-ный водный пероксид водорода (26,29 г, 240 ммоль). Через 3,5 часа добавляли ледяную воду (240 мл) и смесь экстрагировали этилацетатом (2 х 300 мл). Объединенный органический слой промывали 5%-ным водным раствором карбоната натрия и водой, сушили над безводным сульфатом магния и затем отгоняли растворитель при пониженном давлении с получением 65,47 г указанного в заголовке соединения (3) в виде масла. Выход неочищенного продукта 117%.
ИК (CHCl3): 3305, 3066, 3032, 3012, 1586, 1486, 1449, 1382 см-1.
1H ЯМР δ (CDCl3), 300 МГц: 2,34 (1Н, т, J=3,9 Гц), 3,58 и 3,68 (каждый 1Н, каждый дд, J= 3,9 и 23,7 Гц), 7,18 и 7,67 (каждый 2Н, каждый д, J=9,9 Гц), 7,51-7,59 (2Н, м), 7,66 (1Н, м), 7,82-7,87 (2Н, м).
(3) Стадия 3:
5-Бензолсульфонилокси-3-гидроксиметилбензо[b]тиофен (4)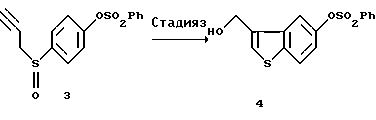
Соединение (3) (65,47 г, 183 ммоль), полученное на описанной выше стадии 2, растворяли в 1,2-диметоксиэтане (1,6 л) и раствор нагревали с обратным холодильником в течение 4 часов. К раствору добавляли воду (64 мл) и моногидрат п-толуолсульфоновой кислоты (19,2 г, 100 ммоль) и продолжали нагревание с обратным холодильником в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении. После добавления к полученному маслу воды (200 мл) смесь экстрагировали этилацетатом (300 мл). Органический слой промывали водным раствором гидрокарбоната натрия и водой, сушили над безводным сульфатом магния и затем отгоняли при пониженном давлении растворитель с получением 60,18 г указанного в заголовке соединения (4) в виде масла. Выход неочищенного продукта 103%.
ИК (СНС13): 3609, 3067, 3033, 3013, 2935, 2878, 1589, 1566, 1449, 1435, 1376 см-1.
1H ЯМР  (CDCl3), 300 МГц: 4,78 (2Н, д, J=0,9 Гц), 6,98 (1H, дд, J=2,4 и 8,7 Гц), 7,26 (1Н, с), 7,43-7,45 (2Н, м), 7,50-7,55 (2Н, м), 7,66 (1Н, м), 7,73 (1Н, д, J=8,7 Гц), 7,83-7,86 (2Н, м).
(CDCl3), 300 МГц: 4,78 (2Н, д, J=0,9 Гц), 6,98 (1H, дд, J=2,4 и 8,7 Гц), 7,26 (1Н, с), 7,43-7,45 (2Н, м), 7,50-7,55 (2Н, м), 7,66 (1Н, м), 7,73 (1Н, д, J=8,7 Гц), 7,83-7,86 (2Н, м).
(4) Стадия 4:
5-Бензолсульфонилоксибензо[b]тиофен-3-карбоновая кислота (6)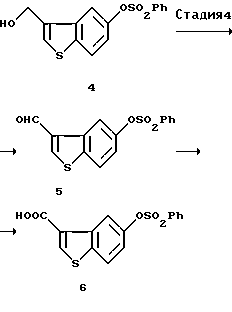
Соединение (4) (51,26 г, 155 ммоль), полученное на описанной выше стадии 3, растворяли в ацетонитриле (1,54 л) и к этому раствору добавляли TEMPO (2,2,6,6-тетраметилпиперидин-1-оксил, 250 мг, 0,01 экв.). К смеси добавляли по каплям 0,81 н. водный раствор гипохлорита натрия, который был получен путем разбавления 1,63 н. водного раствора гипохлорита натрия (150 мл) водой (75 мл), доведения до рН 8,6 добавлением 1 н. серной кислоты и доведения общего объема до 300 мл, в течение 15 минут с поддержанием при этом внутренней температуры в пределах между -1 и 8oС. После перемешивания в течение 15 минут при указанной температуре добавляли 1 н. водный раствор сульфита натрия (32 мл). Затем добавляли при охлаждении льдом 79%-ный хлорит натрия (27,48 г, 240 ммоль) и 31%-ный йодный раствор пероксида водорода (23,26 г, 212 ммоль). Убирали охлаждающую ванну и смесь перемешивали 2 часа. Реакционную смесь разбавляли водой (1,5 л), доводили до рН 3 добавлением 1 н. хлороводородной кислоты и выпавшие в осадок кристаллы отфильтровывали и промывали два раза водой (200 мл), ацетонитрилом (50 мл) с получением 32,4 г неочищенных кристаллов. Неочищенные кристаллы (32,4 г) суспендировали в ацетонитриле (224 мл), нагревали с обратным холодильником в течение 15 минут и охлаждали на льду. Кристаллы отфильтровывали и промывали ацетонитрилом (65 мл) с получением 26,79 г указанного в заголовке соединения (6).
Выход 51,7%, т. пл. 202-203oС.
ИК (нуйлон): 3102, 2925, 2854, 2744, 2640, 2577, 1672, 1599, 1558, 1500, 1460, 1451 см-1.
ЯМР δ (CDC13), 300 МГц: 7,16 (1Н, дд, J=2,7 и 9, 0 Гц), 7,55-7,61 (2Н, м), 7,73 (1Н, м), 7,81 (1H, д, J=9,0 Гц), 7,90-7,94 (2Н, м), 8,16 (1H, д, J= 2,7 Гц), 8,60 (1H, с).
Элементарный анализ для С15Н10O5S2.
Вычислено, %: С 53,88; Н 3,01; S 19,18.
Найдено, %: С 53,73; Н 3,24; S 19,09.
Пример 2
(l) Стадия 1:
5-Бензолсульфонилоксибензо[b]тиофен (8)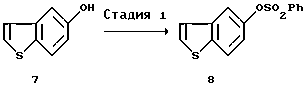
Соединение (7) [J. Am. Chem. Soc., 57, 1611-1616 (1935); Ann. Chem., 52, 83-114 (1938); J. Am. Chem. Soc., 78, 5351-5357 (1956); J. Org. Chem., 41, 1118-1124 (1976)] (1,36 г, 9,05 ммоль) и триэтиламин (1,89 мл, 13,6 ммоль) растворяли в тетрагидрофуране (10 мл). К раствору добавляли по каплям раствор бензолсульфонилхлорида (1/92 г, 10,9 ммоль) в тетрагидрофуране (3 мл). После перемешивания реакционной смеси в течение 2 часов ее разбавляли водой и экстрагировали толуолом. Органический слой промывали водой, сушили над безводным сульфатом магния и затем отгоняли при пониженном давлении растворитель. Остаток хроматографировали на силикагеле (гексан:этилацетат = 5: 1) и затем перекристаллизовывали из гексана, содержавшего небольшое количество этилацетата, с получением 2,28 г указанного в заголовке соединения (8).
Выход 86,8%, т. пл. 80-81oС.
ИК (нуйлон): 1599, 1579, 1564, 1497, 1448, 1440, 1415, 1352 см-1.
1H ЯМР δ (СDСl3), 300 МГц: 6,92 (1Н, дд, J=2,4 и 8,7 Гц), 7,26 (1H, дд, J= 0,9 и 5,4 Гц), 7,47 (1Н, д, J=2,4 Гц), 7,51 (1H, д, J=5,4 Гц), 7,52-7,55 (2Н, м), 7,67 (1H, м), 7,74 (1H, д, J=8,7 Гц), 7,83-7,87 (2Н, м).
Элементарный анализ для С14Н10О3S2.
Вычислено, %: С 57,91; Н 3,47; S 22,09.
Найдено, %: С 57,72; Н 3,45; S 21,98.
(2) Стадия 2:
3-Ацетил-5-бензолсульфонилоксибензо[b]тиофен (9)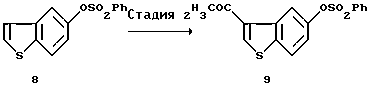
Порошкообразный хлорид алюминия (1,34 г, 10 ммоль) суспендировали в дихлорметане (10 мл). К суспензии добавляли по каплям ацетилхлорид (1,02 мл, 14,3 ммоль) в течение 5 минут при перемешивании и охлаждении на льду. Затем добавляли по каплям в течение 15 минут раствор соединения (8) (2,075 г, 7,2 ммоль), полученного так, как описано выше, в дихлорметане (6 мл). После перемешивания в течение 2 часов при той же температуре, а затем в течение 2,5 часов при комнатной температуре, раствор выливали в ледяную воду и экстрагировали дихлорметаном. Органический слой промывали водой, сушили над безводным сульфатом магния и затем отгоняли при пониженном давлении растворитель. Полученный остаток перекристаллизовывали из этилацетата (3 мл) и гексана (3 мл) с получением 2,01 г указанного в заголовке соединения (9).
Выход 84,4%, т. пл. 129-130oС.
ИК (нуйлон): 3094, 1672, 1619, 1596, 1556, 1494, 1450, 1437, 1428, 1369 см-1.
1H ЯМР δ (СDСl3), 300 МГц: 2,58 (3Н, с), 7,22 (1H, ддд, J=0,6, 2,4 и 9,0 Гц), 7,52-7,58 (2Н, м), 7,69 (1H, м), 7,79 (1H, д, J=9,0 Гц), 7,87-7,91 (2Н, м), 8,27 (1H, дд, J=0, 6 и 2,4 Гц), 8,31 (1H, с).
Элементарный анализ для C16H12O4S2.
Вычислено, %: С 57,82; Н 3,64; S 19,29.
Найдено, %: С 57,62; Н 3,71; S 19,23.
(3) Стадия 3:
5-Бензолсульфонилоксибензо[b]тиофен-3-карбоновая кислота (6)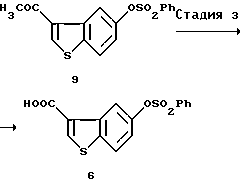
Соединение (9) (6,65 г, 20 ммоль), полученное на описанной выше стадии 2, растворяли в диоксане (50 мл) и добавляли 10%-ный гипохлорит натрия (46,2 мл) в течение 20 минут при перемешивании с поддержанием температуры на уровне 10-12oС.
Через 7 часов реакционную смесь разбавляли ледяной водой (80 мл) и подкисляли концентрированной хлороводородной кислотой (5,2 мл). Выпавшие в осадок кристаллы отфильтровывали, промывали водой, высушивали с получением 5,84 г неочищенных кристаллов. Неочищенные кристаллы (5,84 г) перекристаллизовывали из метанола (66 мл) и воды (16 мл) с получением 5,51 г указанного в заголовке соединения (6).
Выход 82,4%, т.пл. 203-204oС.
Это соединение идентично соединению (6), полученному в примере 1.
Ссылочный пример 1
5-Бензолсульфонилоксибензо[b]тиофен-3-карбонилхлорид (10)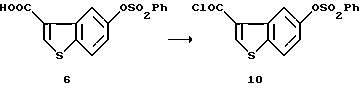
5-Бензолсульфонилоксибензо[b] тиофен-3-карбоновую кислоту (6) (5,582 г, 16,7 ммоль), полученную в примерах, описанных выше, нагревали с обратным холодильником в течение 1,5 часов с диметилформамидом (1 капля), тионилхлоридом (3,57 мл, 50 ммоль) и толуолом (22 мл) и затем удаляли при пониженном давлении растворитель с получением 5,89 г указанного в заголовке соединения (10).
Ссылочный пример 2
(1) Стадия 1:
5-Гидроксибензо[b]тиофен-3-карбоновая кислота (11)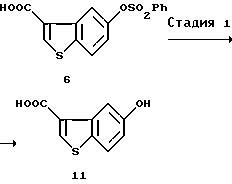
5-Бензолсульфонилоксибензо[b] тиофен-3-карбоновую кислоту (6) (100 мг, 0,3 ммоль), полученную в примерах, описанных выше, растворяли в 1 н. гидроксиде натрия (1,2 мл) и нагревали при 40oС в течение 8 часов при перемешивании. К реакционному раствору добавляли 1 н. хлороводородную кислоту (1,2 мл) и выпавшие в осадок кристаллы отфильтровывали, промывали водой и высушивали с получением 58 г указанного в заголовке соединения (11).
Выход 96,6%, т.пл. 262-263oС.
Это соединение (11) идентично 5-гидроксибензо[b]тиофен-3-карбоновой кислоте, описанной у М. Martin-Smith et al., J. Chem. Soc. (C), 1899-1905 (1967).
(2) Стадия 2:
5-Ацетоксибензо[b]тиофен-3-карбоновая кислота (12)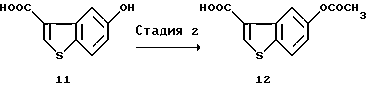
5-Гидроксибензо[b]тиофен-3-карбоновую кислоту (11) (1140 мг), полученную на описанной выше стадии (1), растворяли в уксусном ангидриде (2 мл) и пиридине (4 мл). Через 3 часа добавляли воду и смесь непрерывно перемешивали при охлаждении на льду в течение 1,5 часов. Выпавшие в осадок кристаллы отфильтровывали, промывали водой и высушивали с получением 1,349 мг указанного в заголовке соединения (12).
Выход 47,3%, т.пл. 239-240oС.
1H ЯМР  (СDСl3), 300 МГц: 2,37 (Н, с), 7,20 (1Н, дд, J=2,4 и 8,7 Гц), 7,87 (1Н, д, J=8,7 Гц), 8,34 (1Н, д, J=2,4 Гц), 8,57 (1Н, с).
(СDСl3), 300 МГц: 2,37 (Н, с), 7,20 (1Н, дд, J=2,4 и 8,7 Гц), 7,87 (1Н, д, J=8,7 Гц), 8,34 (1Н, д, J=2,4 Гц), 8,57 (1Н, с).
(3) Стадия 3:
5-Ацетоксибензо[b]тиофен-3-карбонилхлорид (13)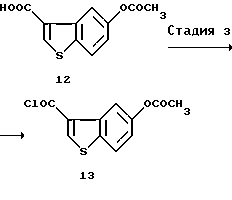
5-Ацетоксибензо[b]тиофен-3-карбоновую кислоту (12) (1349 мг), полученную так, как описано выше, нагревали с обратным холодильником в течение 1,5 часов с диметилформамидом (1 капля), тионилхлоридом (1,22 мл) и толуолом (25 мл). Удаляли при пониженном давлении растворитель с получением 1,454 мг указанного в заголовке соединения (13).
Пример 3
(5Z)-7-[(1R, 2R, 3S,5S)-2-(5-Гидроксибензо[b]тиофен-3-илкарбониламино)-10-норпинан-3-ил]-5-гептеновая кислота (17)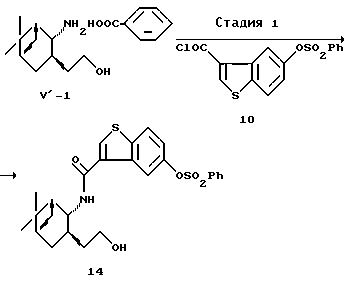
(1) Стадия 1:
Получение [3-[(1R, 2R,3R,5S)-3-(2-гидроксиэтил)-10-норпинан-2-ил]карбамоилбензо[b]тиофен-5-ил]бензолсульфоната (14)
Соль бензойной кислоты и (+)-2-[(1R,2R,3R,53)-2-амино-10-норпинан-3-ил] этанола (описана в Chem. Pharm. Dull., т.37, 6, 1524-1533 (1989) (V'-1)) (5,1 г, 16,7 ммоль) суспендировали в воде (10 мл). К суспензии добавляли 1 н. хлороводородную кислоту (17 мл) и осажденную бензойную кислоту удаляли путем экстрагирования этилацетатом. Органический слой промывали водой (10 мл). К объединенному водному слою добавляли 4 н. раствор гидроксида натрия (9,2 мл, 36,8 ммоль) при охлаждении льдом. Затем добавляли по каплям в течение 15 минут при перемешивании раствор 5-бензолсульфонилоксибензо[b]тиофен-3-карбонилхлорида (10) (5,89 г, 16,7 ммоль) в тетрагидрофуране (16 мл). После перемешивания в течение 1 часа при той же температуре добавляли 1 н. хлороводородную кислоту (4 мл) и смесь экстрагировали этилацетатом. Органический слой промывали водой, сушили над безводным сульфатом магния и затем отгоняли при пониженном давлении растворитель с получением 8,00 г (95,6%) указанного в заголовке соединения (14) в виде бесцветного аморфного вещества.
1H ЯМР δ (CDCl3), 300 МГц: 0,96 (1H, д, J=9,9 Гц), 1,12 и 1,26 (каждый 3Н, каждый с), 1,50-2,42 (9Н, м), 3,69-3,82 (2Н, м), 4,30 (1H, м), 6,21 (1H, д, J= 8,1 Гц), 7,06 (1H, дд, J=2,4 и 8,7 Гц), 7,51-7,56 (2Н, м), 7,67 (1H, м), 7,73 (1H, д, J=8,7 Гц), 7,85-7,88 (2Н, м), 7,88 (1H, с), 8,06 (1H, д, J= 2,4 Гц).
[α]
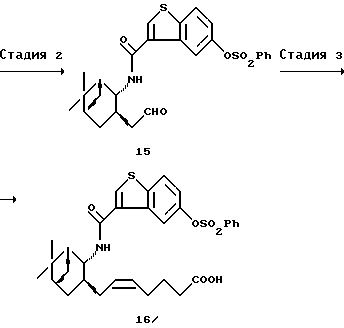
(2) Стадия 2:
Получение [3-[(1R, 2R, 3R,5S)-3-формилметил-10-норпинан-2-ил]карбамоилбензо[b]тиофен-5-ил]бензолсульфоната (15)
К диметилсульфоксиду (3,16 мл, 44,5 ммоль), растворенному в диметоксиэтане (50 мл), добавляли оксалилхдорид (1,91 мл, 21,9 ммоль) при охлаждении до температуры в пределах от -60 до -65oС. Добавляли по каплям при той же температуре раствор соединения (14) (7,352 г, 14,7 ммоль) в 1,2-диметоксиэтане (58 мл). После перемешивания смеси при температуре в пределах от -55 до -60oС в течение 30 минут добавляли триэтиламин (6,1 мл) и, спустя 30 минут, удаляли охлаждающую ванну, давая смеси возможность нагреться до комнатной температуры. Реакционную смесь разбавляли водой (100 мл) и экстрагировали толуолом. Органический слой промывали водой, сушили над безводным сульфатом магния и затем отгоняли при пониженном давлении растворитель. Полученный остаток очищали путем хроматографии на силикагеле (гексан:этилацетат = 5:5-4:6) с получением 7,32 г (100%) указанного в заголовке соединения (15) в виде бесцветного аморфного вещества.
ИК (СНС13): 3443, 3093, 3066, 3030, 3016, 2925, 2871, 2828, 2729, 1720, 1655, 1599, 1558, 1513, 1377 см-1.
1Н ЯМР δ (СDСl3), 300 МГц: 0,97 (1Н, д, J=10,2 Гц), 1,17 и 1,28 (каждый 3Н, каждый с), 2,69 (1Н, ддд, J=1,2, 8,7 и 17,4 Гц), 3,14 (1Н, дд, J=4,5 и 17,4 Гц), 4,28 (1Н, м), 6,18 (1Н, д, J=8,1 Гц), 7,09 (1Н, дд, J=2,4 и 8,7 Гц), 7,50-7,55 (2Н, м), 7,67 (1Н, м), 7,75 (1Н, д, J=8,7 Гц), 7,85-7,89 (2Н, м), 7,89 (1Н, с), 8,03 (1Н, д, J=2,4 Гц), 9,80 (1Н, д, J=1,2 Гц).
[α]
(3) Стадия 3:
Получение (5Z)-7-[(1R,2R,3S,5S]-2-(5-бензолсульфони-локсибензо[b]тиофен-3-илкарбониламино)-10-норпинан-3-ил]-5-гептеновой кислоты (16)
4-Карбоксибутилтрифенилфосфонийбромид (12,17 г, 27,5 ммоль) и трет-бутоксид калия (7,19 г, 64,1 ммоль) суспендировали в тетрагидрофуране (64 мл) и перемешивали в течение 1 часа при охлаждении на льду. К реакционной смеси добавляли в течение 15 минут раствор соединения (15) (9,11 г, 18,3 ммоль), полученного на описанной выше стадии 2, в тетрагидрофуране (27 мл) и смесь непрерывно перемешивали в течение 2 часов при той же температуре. Реакционную смесь разбавляли водой и (80 мл) и промывали толуолом (2 х 105 мл). После доведения рН водного слоя до 8,1 добавлением 5 н. хлороводородной кислоты (4,8 мл) добавляли безводный хлорид кальция (8,1 г, 73 ммоль), растворенный в воде (16 мл), и смесь экстрагировали этилацетатом (2 х 100 мл). В органический слой добавляли воду (100 мл) и рН водного слоя доводили до значения менее 2 добавлением 5 н. хлороводородной кислоты и экстрагировали этилацетатом. Органический слой промывали водой, сушили над безводным сульфатом магния и затем отгоняли при пониженном давлении растворитель с получением 11,06 г соединения (16). Соединение (16) использовали в следующей реакции без дополнительной очистки.
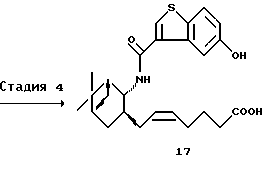
(4) Стадия 4:
Получение (5Z)-7-[(1R, 2R,3S,5S)-2-(5-гидроксибензо[b]тиофен-3-илкарбониламино)-10-норпинан-3-ил]-5-гептеновой кислоты (17) (соединение А)
Соединение (16) (11,06 г, 18,3 ммоль), полученное на описанной выше стадии 3, растворяли в диметилсульфоксиде (22 мл). После добавления 4 н. гидроксида натрия (27,5 мл) смесь нагревали при 55oС в течение 2 часов при перемешивании. Реакционную смесь разбавляли водой (130 мл) и промывали толуолом (2 х 65 мл). Водный слой подкисляли 5 н. хлороводородной кислотой и экстрагировали этилацетатом. Органический слой промывали водой, сушили над безводным сульфатом магния и отгоняли при пониженном давлении растворитель с получением 8,26 г неочищенного целевого соединения, которое затем растворяли в метаноле (40 мл) и воде (16 мл). Раствор затравляли и постепенно охлаждали при перемешивании. Выпавшие в осадок кристаллы отфильтровывали и промывали смесью вода-метанол (2: 5) с получением 6,35 г целевого соединения. Выход 78,6%. Кристаллы растворяли в метаноле (40 мл). К раствору добавляли воду (12 мл) в течение 7 минут при перемешивании. Смесь затравляли и непрерывно перемешивали 1 час при 25oС. После добавления воды (7 мл) в течение 40 минут смесь перемешивали 1,5 часа при 25oС. Выпавшие в осадок кристаллы отфильтровывали и промывали смесью вода-метанол (3:5) (8 мл) с получением 6,14 г целевого соединения (17), которое было почти бесцветным.
Выход 76,0%, т.пл. 145-146oС.
ИК (нуйлон): 3313, 3096, 3059, 3001, 1717, 1672, 1603, 1548, 1469, 1440 см-1.
1Н ЯМР δ (CDCl3), 300 МГц: 1,02 (1Н, д, J=10,2 Гц), 1,12 и 1,24 (каждый 3Н, каждый с), 1,56-2,55 (14Н, м), 4,29 (1Н, м), 5,32-5,51 (2Н, м), 6,20 (1Н, д, J=9,3), 7,01 (1Н, дд, J=2,4 и 9,0 Гц), 7,66 (1H, д, J=9,0 Гц), 7,69 (1H, с), 8,03 (1H, д, J=2,4 Гц).
[α]
Элементарный анализ для C25H31NO4S.
Вычислено, %: С 68,00; Н 7,08; N 317; S 7,26.
Найдено, %: С 67,84; Н 7,08; N 3,24; S 7,31.
Изобретение относится к производным бензо[b]тиофеновой кислоты формулы I, к способу их получения, которые полезны как исходные материалы для получения лекарственных средств, и к способу получения производных 5-гидроксибензо[b] тиофен-3-карбоновой кислоты формулы VI, являющихся специфическими антагонистами PGD2, с использованием указанных выше производных формулы I. Соединения VI могут быть полезны в качестве лекарственного вещества для лечения системного мастоцитоза, астмы, крапивницы. 5 с. и 5 з.п.ф-лы.
где R представляет водород или гидроксизащитную группу,
или его реакционноспособного производного, включающий введение 4-меркаптофенола в реакции введения пропаргильной группы и защиты гидроксильной группы с получением соединения формулы II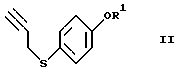
где R1 представляет гидроксизащитную группу,
окисление соединения II с получением соединения формулы III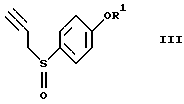
где R1 представляет гидроксизащитную группу,
введение соединения III в реакцию термической перегруппировки с получением соединения формулы IV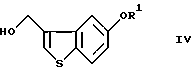
где R1 определен, как указано выше,
и введение соединения IV в реакцию ступенчатого окисления гидроксиметильной группы и, необязательно, удаления защитной группы.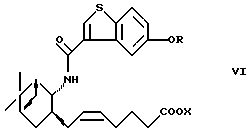
где R представляет водород или гидроксизащитную группу;
Х представляет водород или алкил;
двойная связь обозначает либо Е-, либо Z-конфигурацию,
или его фармацевтически приемлемой соли, или гидрата, который включает введение 4-меркаптофенола в реакции введения пропаргильной группы и защиты гидроксильной группы с получением соединения формулы II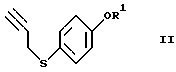
где R1 представляет гидроксизащитную группу,
окисление соединения II с получением соединения формулы III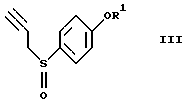
где R1 представляет гидроксизащитную группу,
введение соединения III в реакцию термической перегруппировки с получением соединения формулы IV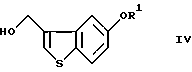
где R1 определен, как указано выше,
и введение соединения IV в реакцию ступенчатого окисления гидроксиметильной группы и, необязательно, удаления защитной группы с получением соединения формулы I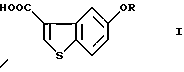
где R определен, как указано выше,
или его реакционноспособного производного, введение соединения формулы I или его реакционноспособного производного в следующие реакции: (1) взаимодействие с соединением формулы V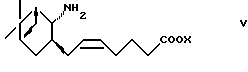
где Х представляет водород или алкил,
или (2) взаимодействие с соединением формулы V'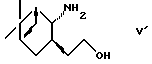
или его солью с последующим окислением и взаимодействием с илидом в условиях реакции Виттига и (3) необязательное удаление защитной группы.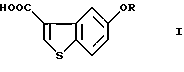
где R представляет водород или гидроксизащитную группу,
или его реакционноспособного производного, включающий введение 5-гидроксибензо[b] тиофена в реакцию введения защитной группы с получением соединения формулы VII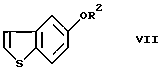
где R2 представляет гидроксизащитную группу,
введение соединения VII во взаимодействие с ацетилгалогенидом в условиях реакции Фриделя-Крафтса с получением соединения формулы VIII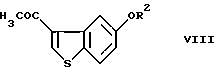
где R2 представляет гидроксизащитную группу,
и введение соединения VIII в реакцию окисления ацетильной группы и, необязательно, удаления защитной группы.
где R представляет водород или гидроксизащитную группу;
Х представляет водород или алкил;
двойная связь представляет либо Е-, либо Z-конфигурацию,
или его фармацевтически приемлемой соли или гидрата, который включает введение 5-гидроксибензо[b] тиофена в реакцию введения защитной группы с получением соединения формулы VII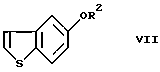
где R2 представляет гидроксизащитную группу,
введение соединения VII во взаимодействие с ацетилгалогенидом в условиях реакции Фриделя-Крафтса с получением соединения формулы VIII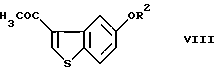
где R2 представляет гидроксизащитную группу,
и введение соединения VIII в реакцию окисления ацетильной группы и, необязательно, удалению защитной группы с получением соединения формулы I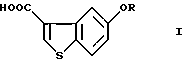
где R определен, как указано выше,
или его реакционноспособного производного, введение соединения формулы I или его реакционноспособного производного в следующие реакции: (1) взаимодействие с соединением формулы V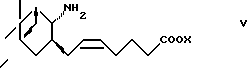
где Х определен, как указано выше,
или (2) взаимодействие с соединением формула V'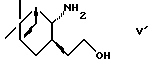
или его солью с последующим окислением и взаимодействием с илидом в условиях реакции Виттига и (3) необязательно удаление защитной группы.
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| RU 94045150, A1, 10.08.1996. | |||

