Изобретение относится к области химии гетероциклических соединений, а также к области фармацевтической промышленности, а именно к новому способу получения амлодипинбензолсульфоната общей формулы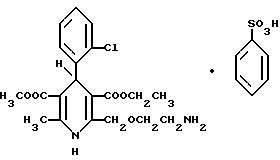
Амлодипинбензолсульфонат является потенциальным блокировщиком кальциевого канала и представляет собой ценный антиишемический и антигипертонический агент.
Существует постоянная необходимость получения амлодипинбензолсульфоната простым и легко осуществимым путем, который дает желаемое вещество с высоким выходом и высокой чистоты, без необходимости предварительного получения и выделения амлодипина в виде его основания.
Амлодипин (родовое название для 3-этил-5-метил-(±)-2- [(2-аминоэтокси)-метил] -4-(2-хлорфенил)-1,4-дигидро-6-метил-3,5- пиридинкарбоксилата), описан в EP-BI-0089167 в качестве нового вещества и пригодного антиишемического и антигипертонического агента. Известны также фармацевтически приемлемые кислотно-аддитивные соли амлодипина.
EP-BIN-0244944 рассматривает новую бензолсульфонатную (безилатную) соль амлодипина и содержащие ее галеновые формы. Благодаря своей необычайной растворимости, высокой стабильности и хорошей перерабатываемости, эта соль является особенно пригодной для получения галеновых форм амлодипина. В соответствии с указанным патентом амлодипинбензолсульфонат получают взаимодействием амлодипина в виде его свободного основания с бензолсульфокислотой или бензолсульфонатом аммония в инертном растворителе, таком как технический метанол при 5oC.
Амлодипин в виде его свободного основания может быть получен различными путями (как описано в вышеуказанном EP-BI N 0089167).
В соответствии с этими способами амлодипин получают удалением аминозащитной группы из предшественника амлодипина, т.е. 1,4-дигидропиридинпроизводного, где аминогруппа в положении 2 защищена выбранными защитными группами.
В случае, когда аминогруппа защищена бензилгруппой, последняя удаляется каталитическим гидрированием на палладиевом катализаторе в растворителе, таком как метанол, при комнатной температуре. Когда защитной группой является 2,2,2-трихлорэтоксикарбонильная (-COOCH2CCl3) группа, она удаляется восстановлением цинком либо в муравьиной, либо в уксусной кислоте. В случае, когда защитной группой является фталимидогруппа, фталоил-остаток этой группы удаляется реакцией с первичным амином, таким как метиламин, или гидразингидратом в растворителе, таком как этанол, или с гидроокисью щелочного металла, такой как гидроокись калия, с последующим взаимодействием с хлористоводородной или серной кислотой в смеси тетрагидрофурана с водой в температурном ряду от комнатной температуры до температуры дефлегмации. При желании, получаемый амлодипин преводится в его фармацевтически приемлемую кислотно-аддитивную соль.
В EP-BI-0089167 также описывается способ получения амлодипина из его предшественника, имеющего азидогруппу во втором положении, которая превращается в аминогруппу восстановлением, например, трифенилфосфином или цинком и хлористоводородной кислотой, или гидрированием на палладиевом катализаторе.
Недостатком указанных способов является относительно низкий выход указанного производного амлодипина, который получается синтезом Ганча из асимметричных 1,4-дигидропиридиновых эфиров. То же самое справедливо для восстановления азидосоединения до амлодипина, кроме того, обращение с азидосоединениями является менее пригодным благодаря хорошо известной взрывчатости азидных структур.
Амлодипин, получаемый в соответствии со способами, описанными в опубликованной литературе, окончательно выделяется, очищается и переводится при желании в кислотно-аддитивные соли.
Целью настоящего изобретения является получение амлодипинбензолсульфоната простым и легко осуществимым способом, который дает желаемое вещество с высоким общим выходом и высокой чистотой без какой-либо необходимости в предварительном получении и выделении амлодипина в виде его свободного основания и выделения его предшественника из сырой реакционной массы, получаемой синтезом Ганча.
Цель достигается синтезом Ганча несимметричных 1,4-дигидропиридиновых эфиров конденсацией этил-4-[2-(N- трифенилметиламино)этокси]ацетоацетата, метил-(E)-3- аминокротоната и 2-хлорбензальдегида и метанольной среде при температуре кипения реакционной смеси с получением 3-этил-5-метил (±)-2-[2-(N-трифенилметиламино)-этоксиметил] -4- (2-хлорофенил)-1,4-дигидро-6-метил-3,5-пиридиндикарбоксилата, который является новым соединением, не описанным в литературе, и предшественником амлодипина, аминогруппа которого защищена трифенилметилгруппой. Способ согласно настоящему изобретению основан на одновременном отделении защитной трифенилметилгруппы от получаемого предшественника амлодипина, который в процессе не выделяется из реакционной массы, а сразу взаимодействует с бензолсульфокислотой, и амлодипинбензолсульфонат непосредственно выделяют без какого-либо образования в виде его основания. Реакцию проводят при температуре от 20oC до температуры кипения. Однако, для того, чтобы удалить отделенный трифенилметилметиловый эфир (возможно с примесью некоторого количества трифенилметанола) из реакционной смеси, необходимо перемешивание при температуре ниже 0oC, предпочтительно около -10oC.
Из сырой смолообразной реакционной массы амлодипинбензолсульфонат выделяют с помощью экстрактора непрерывного действия из растворителя, имеющего низкую удельную плотность. По этому способу полярные и неполярные примеси удаляют из трехфазной системы смола-вода-органический растворитель с последующим вывариванием желаемого соединения в подходящем органическом растворителе, таком как этилацетат, при пониженной температуре (около 0oC), чтобы удалить примеси и сделать возможным выделение кристаллов амлодипинбензолсульфоната, который, для того, чтобы получить его высокую чистоту, окончательно затем очищают перекристаллизацией из соответствующего органического растворителя, такого как метанол, с последующим вывариванием в том же растворителе при низкой температуре (около 0oC).
Преимуществом настоящего изобретения является упрощение процесса за счет его одновременного отделения защитной трифенилметилгруппы от предшественника амлодипина и прямого отделения амлодипинбензолсульфоната без необходимости промежуточного получения и выделения амлодипина в виде его свободного основания и взаимодействия этого основания с солью бензолсульфоната, как описано в известном способе. Бензолсульфокислота является одновременно и средством отделения защитной трифенилметилгруппы от предшественника амлодипина, а также средством прямого получения желаемой соли.
В способе согласно изобретению не выделяют предшественник амлодипина из сырой реакционной массы, а добавляют бензолсульфокислоту для получения сырой метанольной реакционной массы, и после окончания реакции выделяют целевой продукт. Таким образом одна стадия процесса исключается, желаемое соединение получается с хорошим выходом, и восстановления, которое являлось прежде совершенно необходимым, удается избежать. 2-Хлорбензальдегид и метил(E)-3-аминокротонат, которые являются необходимыми для синтеза Ганча асимметричных 1,4-дигидропиридиновых диэфиров, являются коммерчески доступными, тогда как этил-4-[2-(N-трифенилметиламино)- этокси] -ацетоацетат получают путем, который описан в примере 3 или 4. N-Трифенилметил-2-аминоэтанол, необходимый для получения ацетоацетата, получают согласно P.F. Buckus, P.J. Saboniene and D.B. Lemetiene, Zh. Obsch. Khim 6, 1984 (1970), но предпочтительно, улучшенным способом, приведенным в примере 1 или 2, где реакция выполняется не в зловонном пиридине и при температуре кипения, а в среде изопропилового спирта и при комнатной температуре, получая при этом с высоким выходом и высоким содержанием желаемый N-трифенилметил-2-аминоэтанола, что представляет вклад в развитие технологии.
Получение исходных материалов.
Пример 1. N-трифенилметил-2-аминоэтанол. Смесь изопропилового спирта (750 мл), диэтиламина (75 мл), этаноламина (98% 62,3 г, 1 моль) и трифенилхлорметана (97% 143,7 г, 0,5 моля) перемешивают при комнатной температуре 3 ч. Полученный раствор выливают в охлажденную льдом воду (4 л) при перемешивании, а полученный осадок отфильтровывают и высушивают. Неочищенный продукт выдерживают в толуоле (340 мл), затем фильтруют и высушивают под вакуумом при 60oC. Получают 130 г (85%) чистого желаемого соединения.
Пример 2. N-трифенилметил-2-аминоэтанол. Смесь этаноламина (2-аминоэтанола) (98% 1,750 кг, 28,0 молей) в изопропиловом спирте (7,0 л) перемешивают до получения гомогенного раствора. Затем трифенилхлорметан (97% 2,020 кг, 7,03 моля) добавляют медленно в течение 1 ч при комнатной температуре с перемешиванием, температура не превышает 30oC. После полного растворения трифенилхлорметана (при этом этаноламингидрохлорид выделяют в виде белого осадка) реакционную смесь перемешивают еще 1 ч и охлаждают до комнатной температуры. Выпавший осадок фильтруют и высушивают под вакуумом при температуре, не превышающей 50oC. Получают 2,120 кг (98%) желаемого соединения.
Пример 3. Этил-4[2-(N-трифенилметиламино)этокси]ацетоацетат. Гидрид натрия (60% 45,7 г, 1,14 моля) в жидком парафине и безводный тетрагидрофуран (300 мл) загружают в реактор, который продувают азотом. Чистый N-трифенилметил-2-аминоэтанол (100% чистоты, 130 г, 0,42 моля), растворенный в тетрагидрофуране (380 мл), добавляют по каплям в течение 1 ч при комнатной температуре с перемешиванием, после чего реактор продувают азотом, и температура после этого не превышает 40oC. После завершения добавления по каплям реакционную смесь перемешивают еще 1 ч при комнатной температуре и затем охлаждают до 0oC. Затем в течение 1 ч с перемешиванием к реакционной смеси добавляют раствор этил-4-хлорацетацетата (98% 70,9 г, 0,42 моля) в безводном тетрагидрофуране (80 мл), с продувкой реактора азотом и охлаждением таким образом, чтобы температура не превышала 20oC. После окончания добавления реагентов реакционную смесь перемешивают при комнатной температуре в течение дополнительных 20 ч. Затем к реакционной смеси добавляют этиловый спирт (55 мл) и полученную смесь разбавляют водой (550 мл) и нейтрализуют хлористоводородной кислотой (около 60 мл) до значения pH 7. Органическую фазу отделяют, испаряют под вакуумом, а полученный сырой продукт растворяют в метаноле (500 мл), а жидкий парафин выделяют и удаляют. Оставшийся раствор загружают во вращающийся вакуумный испаритель, после чего добавляют воду (170 мл) при высокой скорости вращения для получения гомогенной эмульсии, из которой воду и метанол испаряют с получением 160,5 г (85%) продукта, содержащего 96% указанного в заголовке соединения (определено методом жидкостной хроматографии высокого разрешения), который может быть использован на следующей стадии реакции без дальнейшей очистки. При желании, чистый целевой продукт получают очисткой хроматографией на силикагеле (разбавитель: диэтиловый эфир: н-пентан 1:1 (объем/объем)).
Анализ: ЯМР (ТМС, CDCl3), 60 Гц):
δ 1,20 (т, 3H, J 7 Гц), 2,37 (т, 2H, J 5,5 Гц), 3,47 (е, 2H); 3,57 (т, 2H, J 5,5 Гц), 4,00 (е, 2H), 4,10 (ч, 2H, I 7, Гц); 7,1 7,6 (м, 15H).
Пример 4. Этил-4-[2-(N-трифенилметиламино)этокси]ацетацетат. Гидрид натрия (60% 0,680 кг, 17,0 молей) в жидком парафине и безводный тетрагидрофуран (3,20 л) загружают в реактор, продуваемый азотом. N-трифенилметил-2-аминоэтанол (2,120 кг, 6,94 моля), полученный в примере 2, растворенный в безводном тетрагидрофуране (5,45 л), добавляют в реактор при комнатной температуре с перемешиванием и продувкой реактора азотом, при этом температура не превышала 30oC. После окончания добавления реакционную смесь перемешивают еще в течение получаса при комнатной температуре и затем охлаждают до температуры между -5 и -10oC. К реакционной смеси добавляют раствор этил-4-хлорацетацетата (98% 1,160 кг, 6,90 моля) в безводном тетрагидрофуране (1,35 л) с перемешиванием, продувкой реактора азотом и охлаждением. После окончания добавления реагентов реакционную смесь перемешивают в течение 5 ч, при этом температура не превышала 5oC, а затем перемешивают дополнительно при комнатной температуре в течение 15 ч. Затем в реактор добавляют абсолютный этиловый спирт (0,5 л) с продувкой азотом и интенсивным перемешиванием. Продувку азотом и перемешивание затем прекращают, реакционную смесь разбавляют водой (30 л) и жидкий парафин, который выделяется, удаляют. Полученный раствор затем нейтрализуют хлористоводородной кислотой (0,8 кг концентрированной кислоты) до pH 7. Органическую фазу отделяют и концентрируют во вращающемся вакуумном испарителе сначала при температуре 40oC. Температура постепенно достигает 70oC с получением 2,520 кг (75%) продукта, содержащего 87,5% указанного в заголовке соединения (определено методом жидкостной хроматографии высокого разрешения), который используют на следующей реакционной стадии без дальнейшей очистки.
Способ согласно изобретению
Пример 5. 3-этил-5-метил(±)2- [(2-аминоэтокси)-метил]-4-(2-хлорфенил)-1,4-дигидро-6-метил-3,5- пиридиндикарбоксилатмонобензолсульфонат.
Смесь 2-хлорбензальдегида (95% 50,2 г, 0,35 моля), метил(E)-3-аминокротоната (97% 41,5 г, 0,35 моля) и метил-4[2-(N- трифенилметиламино)этокси]ацетацетата (160,5 г, 0,35 моля содержание 96% полученного в примере 3) растворяют в метаноле (350 мл). Реакционную смесь нагревают с обратным холодильником в течение 10 ч, а затем охлаждают до комнатной температуры. К сырой смеси полученного 3-этил-5-метил(±)2- [2(N-трифенилметиламино)-этоксиметил] -4-(2-хлорфенил)-1,4-дигидро-6- метил-3,5-пиридиндикарбоксилата добавляют водный раствор бензолсульфокислоты (105,0 г, 0,53 моля, 80%). Реакционную смесь повторно нагревают с обратным холодильником в течение 3 ч, затем охлаждают до -10oC и перемешивают при этой температуре в течение 10 ч. Выпавший осадок фильтруют. Фильтрат концентрируют в вакууме до получения вязкой смолы. Теплую смолу перегружают в двухлитровый экстрактор непрерывного действия для экстракции растворителем, имеющим низкую удельную плотность. Горячую воду (1500 мл) и толуол (1500 мл) добавляют, соответственно к смоле и в сборник дистиллата, и неполярные примеси непрерывно экстрагируют в течение 24 ч из трехфазной системы смолы-вода-толуол (смола не растворялась ни в воде, ни в толуоле). Масло, полученное из смолы, отделяют от воды и толуола и растворяют в хлороформе (1500 мл). Хлороформный раствор загружают в двухлитровый экстрактор непрерывного действия для экстракции растворителями с низкой удельной плотностью и воду (1500 мл) и снова подают в приемник дистиллата. Полярные примеси в течение 24 ч непрерывно экстрагируют водой, хлороформный слой отделяют, растворитель испаряют под вакуум. Получена бурая пена (37,2 г), которая затвердевала в аморфное твердое вещество, к которому добавляют этилацетат (90 мл), и полученную смесь перемешивают в течение 1 ч при 0oC. В результате из аморфного материала получают кристаллы неочищенного амлодипинбензолсульфоната, которые отфильтровывают и сушат под вакуумом при 60oC. Сухой неочищенный продукт (24,1 г) перекристаллизовывают из метанола (50 мл). Еще влажный продукт вываривают 2 ч при 0oC в метаноле (25 мл), затем фильтруют и высушивают под вакуумом при 60oC. Получают 13,8 г белых кристаллов амлодипинбензолсульфоната высокой чистоты (>99% чистоты, как определено методом жидкостной хроматографии высокого разрешения) с т. пл. 201,0oC.
Пример 6. 3-Этил-5-метил(±)2-[(2-аминоэтокси)-метил]-4-(2- хлорфенил)-1,4-дигидро-6-метил-3,5-пиридиндикарбоксилатмонобензолсульфонат.
Смесь этил-4-[(2-(N-трифенилметиламино)-этокси)] ацетацетата (2,520 кг, 5,12 моля, содержание 87,5%), полученного в примере 4, метил(E)-3-аминокротоната (97% 0,608 кг, 5,12 моля) и 2-хлорбензальдегида (98% 0,734 кг, 5,12 моля) растворяют в метаноле (6,40 л), реакционную смесь нагревают с обратным холодильником в течение 15 ч и затем охлаждают до комнатной температуре. К охлажденному сырому раствору полученного 3-этил-5-метил(±)2- [2-(N-трифенилметиламино)-этоксиметил] -4-(2-хлорфенил)-1,4 -дигидро-6-метил-3,5-пиридиндикарбоксилата в метаноле медленно добавляют с перемешиванием раствор технической бензолсульфокислоты (0,980 кг, 92%) в метаноле (1,95 л). Реакционную смесь снова нагревают 3 ч с обратным холодильником с перемешиванием, медленно охлаждают до температуры около 0oC и перемешивают при этой температуре дополнительно 5 ч. Осадок трифенилметилметилэфира, который осаждается при этом, фильтруют концентрируют под вакуумом сначала при 40oC, которая постепенно выросла до 70oC, при этом получают бурую вязкую смолу. Эту смолу перегружают вместе с водой (4,2 л) в экстрактор непрерывного действия для экстракции растворителем с низкой удельной плотностью, и неполярные примеси непрерывно экстрагируют в течение 48 ч смесью толуола (1,38 л) и н-пентана 1: 1 (объем/объем). Содержимое реактора перегружают в делительную воронку, остатки в реакторе растворяют в хлороформе (0,47 л) и добавляют в делительную воронку. Масло, которое получают из смолы, отдельно как от воды, так и от органической фазы и затем выпаривают в вакуумном вращающемся испарителе с получением аморфного твердого вещества, к которому добавляют или перемешивают этилацетат (0,75 л) при температуре 0oC. В результате этого аморфное твердое вещество превращается в кристаллической неочищенный амлодипинбензолсульфонат. Кристаллы фильтруют и высушивают под вакуумом при 70oC. Сухой продукт (0,640 кг) кристаллизуют из метанола (1,15 л), и еще влажный продукт вываривают в течение 1 ч при 0oC в метаноле (0,290 л), затем отфильтровывают и сушат под вакуумом при температуре, не превышающей 70oC. Так получают 0,300 кг кристаллов высокой чистоты амлодипинбензолсульфоната (содержание > 98% ). Продукт перекристаллизовывают из метанола (0,540 л), и еще влажный продукт вываривают в метаноле (0,200 л) в течение 1 ч при 0oC и затем отфильтровывают и высушивают под вакуумом при температуре, не превышающей 70oC. получают 0,200 кг белых кристаллов амлодипинбензолсульфоната высокой чистоты (> 99% чистоты, как было определено методом жидкостной хроматографии высокого разрешения) с т. пл. 201,0oC.
Пример сравнения.
Для характеристики предшественника амлодипинбензолсульфоната из примера 5 неочищенный 3-этил-5-метил(±)-2- [2-(N-трифенилметиламино)-этоксиметил]-4-(2-хлорфенил-1,4-дигидро -6-метил-3,5-пиридиндикарбоксилат очищают колоночной хроматографией на силикагеле6 (разбавитель: диэтиловый эфир: петролейный эфир 1:3 (объем/объем)).Очищенный продукт получают в виде желтой пены, имеющей следующмй спектр:
ЯМР (CDCl3, ТМС, 60 Гц).
d 1,19 (т, 3H, I 7 Гц); 1,45 (с, 3H); 2,46 (т2, 2H, I 5 Гц); 3,64 (е, 2H); 3,7 (т, нечетко 2H); 4,10 (ч, 2H, I 7 Гц); 4,75 (е, 2H); 5,50 (е, 1H); 7,0 7,8 (м. 19H).
Использование: в медицине в качестве антиишемического и антигипертонического средства. Продукт: 3-этил-5-метил-(±) 2 / (2-аминоэтокси)-метил/ -4-(2-хлорфенил)-1,4 дигидро-6-метил-3,5 пиридиндикарбоксилатмонобензол сульфоната, т. пл. 201oС. Реагент 1: 3-этил-5-метил (±)2- / 2-(N-трифенилметиламино)-этоксиметил / - 4-(2-хлорфенил) 1,4-дигидро-6-метил-3,5-пиридинкарбоксилат. Реагент 2: бензолсульфокислота. Условия реакции: в метанольной или водно-метанольной среде при температуре от 20oС до температуры кипения. 2 с.п. ф-лы.
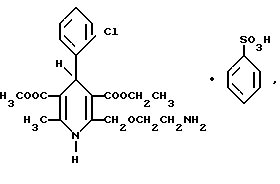
отличающийся тем, что 3-этил-5-метил (±)2-[2-(N-трифенилметиламино)-этоксиметил] -4-(2-хлорфенил)-1,4-дигидро-6-метил-3,5-пиридиндикарбоксилат взаимодействует с бензолсульфокиcлотой в метанольной или водно-метанольной среде при температуре от 20oС до температуры кипения с последующим отделением и очисткой продукта.
| EP, заявка, 0089167 A2, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, заявка, 0244944 А2, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

