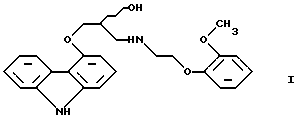
Настоящее изобретение относится к новому способу получения соединения формулы
его оптически активным R или S энантиомерам, смесям этих энантиомеров и их фармацевтически приемлемым солям присоединения кислот.
Соединение формулы I известно под названием карведилол, который используется в качестве лекарства, обладающего антигипертензивным, бета-адреноблокирующим и сосудорасширяющим действием. Его химическое название 1-(9Н-карбазол-4-илокси)-3-[/2-(2-метоксифенокси)этил/амин]-2-пропанол.
Способ получения карведилола известен из патентной публикации DE-OS No. 2815926, а способ получения R и S энантиомеров описан в патентной публикации DE-OS No. 3319027. Согласно известному способу 4-(оксиранилметокси)-9Н-карбазол формулы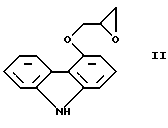
или его R или S энантиомеры реагируют с 2-(2-метоксифенокси)этиламином формулы
с получением соединения формулы I, выход которого составляет от 39 до 42%. Недостаток известного способа состоит в том, что одновременно с получением карведилола также образуется бис-соединение формулы
в результате реакции двух молярных эквивалентов 4-(оксиранилметокси)-9Н-карбазола формулы II и одного молярного эквивалента 2-(2-метоксифенокси)этиламина формулы III. Избежать этой побочной реакции нельзя, и бис-соединение формулы IV образуется в количестве, сравнимом с количеством карведилола. Следовательно, данный способ является не экономичным.
Другие возможности получения карведилола описаны в патентной публикации DE-OS No. 2815926. Согласно этим способам 4-(3-аминогидроксипропокси)-9Н-карбазол может реагировать с
a) 2-(2-метоксифенокси)этилгалидом или сульфонатом;
b) 2-(2-метоксифенокси)ацетальдегидом с последующим каталитическим гидрированием;
c) 2-(2-метоксифенокси)ацетилхлоридом с последующим восстановлением полученного кислотного амида комплексным гидридом металла.
Ни один из способов от а) до с) не подходит для экономичного получения карведилола. Для каждого способа необходима стадия очистки продукта с помощью колоночной хроматографии, что делает получение исходного соединения не экономичным. В реакции по способу а) также образуется бис-соединение формулы IV. В реакции по способу b) выход составляет 41%, в то время как по способу с) - максимум 24%. В реакции по способу с) использование комплексного гидрида металла, практически гидрида лития и алюминия, также является недостатком, поскольку эта реакция сопровождается повышенной опасностью воспламенения. Данная реакция требует особых условий - даже следы влаги должны быть удалены.
В принципе можно избежать образования бис-соединения формулы IV, если использовать вместо первичного амина формулы III вторичный амин, который является производным первичного амина формулы III, содержащего защитную группу. Таким вторичным амином является, например, N-/2-(2-метоксифенокси)этил/бензиламин формулы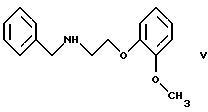
В примере 5 патентной публикации DE OS No. 2815926 4-оксиранилметокси-9Н-карбазол формулы II реагирует со вторичным амином формулы V с получением бензил-карведилола формулы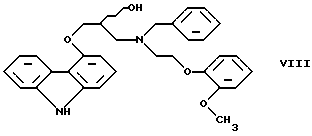
который можно выделить только благодаря очистке с помощью колоночной хроматографии. Такая процедура является не экономичной для производства в промышленном объеме. Реакцию соединений формул I и V проводили в диметиловом эфире этиленгликоля.
Пример 5 из DE OS 2815926 был воспроизведен четыре раза. Как показано в примере сравнения 1 выход продукта был 60%, причем температура плавления загрязненного продукта была 92oС, что значительно ниже значения, указанного в примере 5 (97-99oС). Было невозможно получить продукт в кристаллическом виде без очистки с помощью колоночной хроматографии, таким образом, нельзя исключить эту стадию очистки. Это является существенным недостатком для применения в промышленности. Так как увеличение объема производства продукта по данному известному способу сопровождается снижением как выхода, так и качества продукта, указанный способ не пригоден для применения в промышленности.
Способ, описанный в примере 5 DE OS 2815926 был модифицирован путем использования вместо диметилового эфира этиленгликоля этилацетата или диоксана, как показано в примерах сравнения 2 и 3. Даже после 28 часов реакции около половины исходного соединения осталось не прореагировавшим в диоксане, а в этилацетате реакция практически не прошла совсем.
Целью настоящего изобретения было разработать экономичный способ получения карведилола формулы I.
Указанная цель была достигнута в результате создания способа по настоящему изобретению, в котором 4-оксиранилметокси-9Н-карбазол формулы II либо его S или R энантиомеры реагируют со вторичным амином формулы V в протонном органическом растворителе, затем полученный бензил-карведилол формулы VIII дебензилируют путем каталитического гидрирования и полученный бензил-карведилол формулы VIII дебензилируют путем каталитического гидрирования; при необходимости полученный продукт реагирует с неорганической или органической кислотой для получения его фармацевтически приемлемой соли присоединения кислоты.
Согласно способу настоящего изобретения в качестве протонного растворителя используется предпочтительно С1-C4- алканол, в первую очередь этанол или изопропанол, и реакцию проводят при температуре от 0 до 120oС, предпочтительно при температуре кипения реакционной смеси.
Вторичный амин формулы V используется в виде масла, то есть в виде соединения, в котором отсутствует кристаллизационная вода, либо предпочтительно в виде соединения, в котором присутствует кристаллизационная вода.
Полученный бензил-карведилол формулы VIII либо сначала выделяют, а затем дебензилируют, либо, что предпочтительно, не выделяют из реакционной смеси, в которой он был получен, перед реакцией дебензилирования. Дебензилирование проводят путем каталитического гидрирования способом, известным для удаления бензильной группы.
Предпочтительней в качестве катализатора используют палладий на угле. Для дебензилирования бензил-карведилола катализатор можно использовать несколько раз без регенерации. Предпочтительно гидрирование проводят с использованием гидрата гидразина.
Оптически активные энантиомеры бензил-карведилола формулы VIII получают способом по настоящему изобретению, то есть R-(+)-1-/N-бензил-2-(2-метоксифенокси)этиламин/-3-(9Н-карбазол-4-илокси)-2-пропанол и S-(-)-1-/N-бензил-2-(2-метоксифенокси)этиламин/-3-(9Н-карбазол-4-илокси)-2-пропанол являются новыми соединениями. Изобретение включает эти соединения и их фармацевтически приемлемые соли присоединения кислот.
R и S энантиомеры 4-оксиранилметокси-9Н-карбазола формулы II получают способом, известным из патентной публикации DE OS 3319027. Вторичный амин формулы V получают методом Аугстейна (Augstein, J. Med.Chem., 8, 356-367, 1965) либо способом, данным в настоящем описании.
Карведилол очень высокой чистоты с выходом около 80% получают способом по настоящему изобретению. В данном способе не образуется бис-соединения формулы IV.
Бензил-карведилол формулы VIII получается очень высокой чистоты, следовательно, не требуется его очистки, и реакцию дебензилирования можно проводить в той реакционной смеси, в которой он был получен.
Приведенные выше данные свидетельствуют о том, что способ по настоящему изобретению является экономичным и простым в исполнении.
Специалисты вряд ли могли ожидать, что по способу настоящего изобретения при использовании протонного органического растворителя в результате реакции 4-оксиранилметокси-9Н-карбазола формулы II и вторичного амина формулы V будет получен бензил-карведилол формулы VIII в хорошо кристаллизующемся чистом виде с выходом более 90% без специальной процедуры очистки, так как исходя из примеров сравнения можно предположить обратное.
Далее настоящее изобретение поясняется следующими примерами.
Получение исходных соединений.
Получение соединения формулы V.
Дигидрат N-/2-(2-метоксифенокси)этил/бензиламина.
К 131 см3 (128,64 г, 1,2 моль) бензиламина, нагретого до 80oС, добавляют 69,33 г (0,3 моль) 1-(2-бромэтокси)-2-метоксибензола в течение 25 минут с такой скоростью, чтобы температура реакционной смеси была от 95 до 105oС. Затем реакционную смесь перемешивают при температуре от 95 до 105oС в течение 2 часов, полученную суспензию охлаждают ледяной водой до 25oС и добавляют в течение 5 минут к 1000 см3 10%-ной соляной кислоты, охлажденной ледяной водой, обращая внимание на то, чтобы температура смеси не превысила 50oС.
Полученный раствор охлаждали ледяной водой. Через 5-10 минут гидрохлорид продукта осаждался в виде кристаллов. Суспензию кристаллов перемешивали при 5-10oС в течение 0,5 часа, фильтровали и промывали водой. Неочищенный гидрохлорид перекристаллизовывали из 400 см3 воды, фильтровали и промывали водой.
Таким образом было получено 71,9 г (81,6%) гидрохлорида названного соединения с температурой плавления 148-150oС.
Гидрохлорид суспендировали, суспензию нагревали до растворения кристаллов и затем к теплому раствору прибавляли по каплям 100 см3 10%-ного водного раствора гидроокиси натрия. Раствор охлаждали до температуры 5-10oС ледяной водой, кристаллы отфильтровывали, промывали 100 см3 ледяной воды и сушили на воздухе при комнатной температуре.
Таким образом было получено 43,1 г (48,9%) названного соединения с температурой плавления 53-55oС. Продукт содержал кристаллизационную воду (C16H19NO2• 2Н2O).
Пример 1.
1-/N-Бензил-2-(2-метоксифенокси)этиламин/-3-(9Н-карбазол-4-илокси)-2-пропанол.
К 20 см3 этанола добавляли 2,7 г (10,5 ммоль) N-/2-(2-метоксифенокси)этил/бензиламина и 1,92 г (8,0 ммоль) 4-оксиранилметокси-9Н-карбазола, реакционную смесь кипятили 5 часов при перемешивании, охлаждали, и еще перемешивали 16 часов при комнатной температуре. Кристаллы отфильтровывали, промывали один раз 10 см3 этанола, один раз 15 см3 диизопропилового эфира и сушили под лампой инфракрасного излучения.
Таким образом было получено 3,44 г (86,3%) названного соединения в виде белых кристаллов с температурой плавления 93-95oС.
Пример 2.
1-/N-Бензил-2-(2-метоксифенокси)этиламин/-3-(9Н-карбазол-4-илокси)-2-пропанол.
К 25 см3 изопропанола добавляли 3,85 г (13,1 ммоль) N-/2-(2-метоксифенокси)этил/бензиламина и 2,4 г (10,0 ммоль) 4-оксиранилметокси-9Н-карбазола, реакционную смесь кипятили 5 часов при перемешивании, охлаждали и еще перемешивали 15 минут при комнатной температуре. Кристаллы отфильтровывали, промывали один раз 4 см3 пропанола, два раза по 2 см3 диизопропилового эфира и сушили под лампой инфракрасного излучения.
Таким образом было получено 4,8 г (96,3%) названного соединения в виде белых кристаллов с температурой плавления 94-96oС.
Пример 3.
S-(-)-1-/N-Бензил-2-(2-метоксифенокси)этиламин/-3-(9Н-карбазол-4-илокси)-2-пропанол.
К 25 см3 изопропанола добавляли 3,85 г (13,1 ммоль) N-/2-(2-метоксифенокси)этил/бензиламина и 2,4 г (10,0 ммоль) S-(+)-4-оксиранилметокси-9Н-карбазола, реакционную смесь кипятили 7 часов при перемешивании и упаривали при пониженном давлении. При стоянии осадок кристаллизовался, кристаллы перемешивали с 12 см3 диэтилового эфира в течение 0,5 часа, отфильтровывали и промывали дважды по 10 см3 диэтилового эфира. Неочищенный продукт перекристаллизовывали из 40 см3 диизопропилового эфира.
Таким образом было получено 3,52 г (70,6%) названного соединения с температурой плавления 102,5-103,5oС.
[α]D = -9,6° (20oС, с=1, уксусная кислота).
[α]435нм = -14,5° (20oС, с=1, уксусная кислота).
Пример 4
R-(+)-1-/N-бензил-2-(2-метоксифенокси)этиламин/-3-(9Н-карбазол-4-илокси)-2-пропанол
К 25 см3 изопропанола добавляли 3,85 г (13,1 ммоль) N-/2-(2-метоксифенокси)этил/бензиламина и 2,4 г (10,0 ммоль) R-(-)-4-оксиранилметокси-9Н-карбазола, реакционную смесь кипятили 7 часов при перемешивании и упаривали при пониженном давлении. Кубовый осадок кристаллизовался из диизопропилового эфира, кристаллы отфильтровывали. Неочищенный продукт суспендировали в 50 см3 диизопропилового эфира, суспензию нагревали до кипения, охлаждали и отфильтровывали.
Таким образом было получено 4,09 г (82,1%) названного соединения с температурой плавления 103-104oС.
[α]D = +8,2° (20oС, с=1, уксусная кислота).
[α]435нм = +12,4° (20oС, с=1, уксусная кислота).
Пример 5.
1-(9Н-Карбазол-4-илокси)-3-/2-(2-метоксифенокси)этиламин-2-пропанол.
К 100 см3 этанола добавляли 15,4 г (52,5 ммоль) N-/2-(2-метоксифенокси)этил/бензиламина, содержащего 2 молекулы кристаллизационной воды и 9,6 г (40,0 ммоль) 4-оксиранилметокси-9Н-карбазола, суспензию нагревали до кипения. Полученный раствор кипятили 5 часов при перемешивании, охлаждали до комнатной температуры и добавляли 10,4 г катализатора - увлажненного палладия на угле. Содержание влаги в катализаторе 50,6%, содержание палладия 16,2% в расчете на сухой катализатор. К смеси, охлажденной холодной водой, добавляли по каплям 40 см3 (0,806 моль) 98%-ного гидрата гидразина в течение 15 минут, затем реакционную смесь перемешивали при температуре 25oС в течение 18 часов. После этого катализатор отфильтровывали, промывали трижды по 100 см3 этанола, фильтрат упаривали при пониженном давлении. К кубовому остатку прибавляли 200 см3 воды и 200 см3 1,2-дихлорэтана, смесь перемешивали до растворения осадка, фазы разделяли, водную фазу экстрагировали еще 2 раза по 150 см3 1,2-дихлорэтана. Органическую фазу сушили и упаривали при пониженном давлении. Кубовый остаток нагревали с 40 см3 этилацетата до кипения, горячую смесь фильтровали через бумажный складчатый фильтр, фильтрат охлаждали при перемешивании. После выпадения кристаллов смесь перемешивали еще один час при 25oС, кристаллы отфильтровывали и промывали три раза по 10 см3 холодного этилацетата. Полученный продукт сушили под лампой инфракрасного излучения.
Таким образом было получено 13,05 г (80,0%) неочищенного продукта в виде белых кристаллов с температурой плавления 113-116oС. Неочищенный продукт перекристаллизовывали из 75 см3 этилацетата и получали 12,1 г (74,2%) названного соединения с температурой плавления 114-116oС.
Пример 6.
1-(9Н-Карбазол-4-илокси)-3-/2-(2-метоксифенокси)этиламин/-2-пропанол.
В колбу на 100 см3 помещали 1,99 г (4 ммоль) 1-/N-бензил-2-(2-метоксифенокси)этиламин/-3-(9Н-карбазол-4-илокси)-2-пропанола, 40 см3 этанола, 4 см3 воды и 1,0 г катализатора - увлажненного палладия на угле с содержанием влаги 50,6% и содержанием палладия 16,2% в расчете на сухой катализатор. К смеси добавляли по каплям 4 см3 (80 ммоль) 98%-ного гидрата гидразина при температуре от 5 до 10oС в течение 10 минут, затем реакционную смесь перемешивали при температуре 25oС в течение 2 часов. После этого катализатор отфильтровывали, промывали дважды по 20 см3 этанола, фильтрат упаривали при пониженном давлении. К кубовому остатку прибавляли 50 см3 воды и 50 см3 1,2-дихлорэтана, смесь перемешивали до растворения осадка, фазы разделяли, водную фазу экстрагировали еще 2 раза по 50 см3 1,2-дихлорэтана. Органическую фазу сушили и упаривали при пониженном давлении. Кубовый остаток нагревали с 9 см3 этилацетата до кипения, горячую смесь фильтровали через бумажный складчатый фильтр, фильтрат охлаждали при перемешивании. После выпадения кристаллов смесь перемешивали еще один час при 25oС, кристаллы отфильтровывали и дважды промывали по 1 см3 холодного этилацетата. Полученный продукт сушили под лампой инфракрасного излучения.
Таким образом было получено 1,33 г (81,6%) названного соединения в виде белых кристаллов с температурой плавления 114-116oС.
Пример 7.
1-(9Н-Карбазол-4-илокси)-3-/2-(2-метоксифенокси)этиламин/-2-пропанол.
К 60 см3 этанола прибавляли 1,1 г (2,2 ммоль) 1-/N-бензил-2-(2-метоксифенокси)этиламин/-3-(9Н-карбазол-4-илокси)-2-пропанола и 0,5 г катализатора - увлажненного палладия на угле с содержанием влаги 50,6% и содержанием палладия 16,2% в расчете на сухой катализатор. Смесь тщательно перемешивали в атмосфере водорода с давлением 5 бар при комнатной температуре. После этого катализатор отфильтровывали, промывали дважды по 20 см3 этанола, фильтрат упаривали при пониженном давлении. К кубовому остатку прибавляли 5 см3 этилацетата, смесь перемешивали 2 часа при 25oС, полученные кристаллы отфильтровывали и дважды промывали по 1 см3 холодного этилацетата. Полученный продукт сушили под лампой инфракрасного излучения.
Таким образом было получено 0,69 г (76,6%) названного соединения в виде белых кристаллов с температурой плавления 114-116oС.
Пример 8.
R-(+)-1-(9Н-Карбазол-4-илокси)-3-/2-(2-метоксифенокси)этиламин/-2-пропанол.
К 40 см3 этанола добавляли 1,99 г (4,0 ммоль) R-(+)-1-/N-бензил-2-(2-метоксифенокси)этиламин/-3-(9Н-карбазол-4-илокси)-2-пропанола и 1,0 г катализатора - увлажненного палладия на угле с содержанием влаги 50,6% и содержанием палладия 16,2% в расчете на сухой катализатор. К смеси добавляли по каплям 4 см3 (80 ммоль) 98%-ного гидрата гидразина при температуре от 20 до 30oС в течение 20 минут, затем реакционную смесь перемешивали при температуре 25oС в течение 1,5 часов. После этого катализатор отфильтровывали, промывали дважды по 30 см3 этанола, фильтрат упаривали при пониженном давлении. К кубовому остатку прибавляли 80 см3 1,2-дихлорметана, смесь перемешивали, фазы разделяли, водную фазу экстрагировали еще 2 раза по 80 см3 1,2-дихлорметана. Органическую фазу сушили и упаривали при пониженном давлении. Кубовый остаток нагревали с 5 см3 этилацетата до кипения, горячую смесь фильтровали через бумажный складчатый фильтр, фильтрат охлаждали при перемешивании. После выпадения кристаллов смесь перемешивали еще 8 часов при 25oС, кристаллы отфильтровывали и дважды промывали по 3 см3 холодного этилацетата. Полученный неочищенный продукт перекристаллизовывали из 8 см3 этилацетата.
Таким образом было получено 1,16 г (71,2%) названного соединения в виде белых кристаллов с температурой плавления 112-113oС.
[α]D = +17,2° (20oС, с=1, уксусная кислота).
Пример 9.
S-(-)-1-(9Н-Карбазол-4-илокси)-3-/2-(2-метоксифенокси)этиламин/-2-пропанол.
К 35 см3 этанола добавляли 1,74 г (3,5 ммоль) S-(-)-1-/N-бензил-2-(2-метоксифенокси)этиламин/-3-(9Н-карбазол-4-илокси)-2-пропанола и 0,9 г катализатора - увлажненного палладия на угле с содержанием влаги 50,6% и содержанием палладия 16,2% в расчете на сухой катализатор. К смеси добавляли по каплям 3,5 см3 (70 ммоль) 98%-ного гидрата гидразина при температуре от 20 до 30oС в течение 10 минут, затем реакционную смесь перемешивали при температуре 25oС в течение 1,5 часов. После этого катализатор отфильтровывали, промывали дважды по 30 см3 этанола, фильтрат упаривали при пониженном давлении. К кубовому остатку прибавляли 70 см3 воды и 70 см3 1,2-дихлорметана, смесь перемешивали, фазы разделяли, водную фазу экстрагировали еще 2 раза по 70 см3 1,2-дихлорметана. Органическую фазу сушили и упаривали при пониженном давлении. Кубовый остаток нагревали с 5 см3 этилацетата до кипения, горячую смесь фильтровали через бумажный складчатый фильтр, фильтрат охлаждали при перемешивании. После выпадения кристаллов смесь перемешивали еще 2 часа при 25oС, кристаллы отфильтровывали и дважды промывали по 2 см3 холодного этилацетата. Полученный неочищенный продукт перекристаллизовывали из 7,5 см3 этилацетата.
Таким образом было получено 1,15 г (80,7%) названного соединения в виде белых кристаллов с температурой плавления 110-111,5oС.
[α]D = -18,1° (20oС, с=1, уксусная кислота).
Пример 10.
1-/N-Бензил-2-(2-метоксифенокси)этиламин/-3-(9Н-карбазол-4-илокси)-2-пропанол.
К 2450 см3 изопропанола добавляли 385 г дигидрата N-/2-(2-метоксифенокси)этил/бензиламина и 290 г 4-оксиранилметокси-9Н-карбазола, смесь кипятили при перемешивании в течение 6 часов, охлаждали и перемешивали при комнатной температуре еще 16 часов. Кристаллический продукт отфильтровывали, промывали 400 см3 пропанола, затем дважды по 200 см3 диизопропилового эфира и сушили.
Таким образом было получено 469 г (94%) названного соединения в виде белых кристаллов с температурой плавления 94-96oС.
Пример сравнения 1
(воспроизведение примера 5 патентной публикации DE OS 2815926).
Смесь 60,4 г 4-оксиранилметокси-9Н-карбазола, 64,8 г N-/2-(2-метоксифенокси)этил/бензиламина и 200 мл диметилового эфира этиленгликоля кипятили в течение 24 часов. При охлаждении смеси продукт не выпал в осадок. Смесь упаривали при пониженном давлении, кубовый остаток не могли выделить в виде кристаллов из этанола, изопропанола и ацетона, следовательно, его перенесли на колонку, заполненную силикагелем. При хроматографии на колонке в качестве растворителя использовали 1,2-дихлорметан; смесь, содержащую 9 объемов 1,2-дихлорметана и 1 объем этилацетата; смесь, содержащую 7 объемов 1,2-дихлорметана и 3 объема этилацетата; этилацетат. Содержащие продукт фракции собирали и упаривали. В результате получали 75 г (60%) 1-(9Н-карбазол-4-илокси)-3-/2-(2-метоксифенокси)этиламин/-2-пропанола с температурой плавления 92oС.
Пример сравнения 2.
Смесь 1,92 г (8 ммоль) 4-оксиранилметокси-9Н-карбазола, 3,08 г (10,5 ммоль) N-/2-(2-метоксифенокси)этил/бензиламина и 20 см3 этилацетата кипятили. После этого продукт выделяли с помощью ВЭЖХ. После 28 часов реакции реакционная смесь содержала только 3% бензил-карведилола, практически весь исходный 4-оксиранилметокси-9Н-карбазол остался не прореагировавшим.
Характеристики ВЭЖХ:
Колонка: LiChrospher 100RP-18 (5 um), λ=254 нм,
Ацетонитрил : буферный раствор=1:1,
Скорость потока 0,5 см3 /мин,
Буферный раствор: 7,74 г ацетата аммония и 10,5 см3 уксусной кислоты на 1000 см3 водного раствора,
Время удерживания, мин:
N-/2-(2-метоксифенокси)этил/бензиламин - 2,0
4-Oксиранилметокси-9Н-карбазол - 4,6
Бензил-карведилол - 14,1
Пример сравнения 3.
Повторяли процедуру, описанную в примере сравнения 2, которая отличалась тем, что в качестве растворителя использовали диоксан. Через 28 часов реакции в реакционной смеси присутствовало около 50% исходного 4-оксиранилметокси-9Н-карбазола, что было определено с помощью ВЭЖХ.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКОГО ПРОДУКТА | 1998 |
|
RU2245875C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-ГИДРОКСИ-9H-КАРБАЗОЛА | 1998 |
|
RU2204551C2 |
| СПОСОБ ПОЛУЧЕНИЯ {2-[4-(АЛЬФА-ФЕНИЛ-П-ХЛОРБЕНЗИЛ)ПИПЕРАЗИН-1-ИЛ]-ЭТОКСИ}-УКСУСНОЙ КИСЛОТЫ И НОВЫЕ ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2000 |
|
RU2248974C2 |
| СПОСОБ ПОЛУЧЕНИЯ 5-ХЛОР-4-АМИНОЗАМЕЩЕННОГО ПРОИЗВОДНОГО 3(2Н)-ПИРИДАЗИНОНА | 1998 |
|
RU2211217C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФЛУОКСЕТИНА | 1997 |
|
RU2173679C2 |
| СПОСОБ ПОЛУЧЕНИЯ АМОРФНОГО АТОРВАСТАТИНА КАЛЬЦИЯ | 2000 |
|
RU2255932C2 |
| СПОСОБ ПОЛУЧЕНИЯ КВЕТИАПИНА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2001 |
|
RU2258067C2 |
| СПОСОБ ПОЛУЧЕНИЯ 6-МЕТИЛ-2-(4-МЕТИЛФЕНИЛ)-ИМИДАЗОЛО[1,2-А]-ПИРИДИН-3-(N, N-ДИМЕТИЛАЦЕТАМИДА), ЭФИРЫ, КРИСТАЛЛИЧЕСКИЕ ЭФИРЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ | 2000 |
|
RU2254334C2 |
| СПОСОБ ПОЛУЧЕНИЯ СОЛИ 3-ЭТИЛ-5-МЕТИЛ-2-(2-АМИНОЭТОКСИМЕТИЛ)-4-(2-ХЛОРФЕНИЛ)-6-МЕТИЛ-1,4-ДИГИДРО-3,5-ПИРИДИНДИКАРБОКСИЛАТА И БЕНЗОЛСУЛЬФОКИСЛОТЫ (БЕЗИЛАТА АМЛОДИПИНА) | 1998 |
|
RU2163597C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ 2,3-БЕНЗОДИАЗЕПИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 2000 |
|
RU2243228C2 |
Настоящее изобретение относится к способу получения карведилола формулы I, его оптически активных R или S энантиомеров, смесей этих энантиомеров и их фармацевтически приемлемых солей присоединения кислот. Согласно настоящему изобретению 4-оксиранилметокси-9Н-карбазол или его R или S энантиомеры подвергают взаимодействию с N-[2-(2-метоксифенокси)этил]бензиламином в протонном органическом растворителе и полученный бензил-карведилол дебензилируют путем каталитического гидрирования. Настоящий способ позволяет повысить экономические показатели при выходе 80%. 3 с. и 1 з.п.ф-лы.

его оптически активных R или S энантиомеров, смесей этих энантиомеров и их фармацевтически приемлемых солей присоединения кислот, отличающийся тем, что 4-оксиранилметокси-9Н-карбазол формулы
или его R или S энантиомеры реагируют со вторичным амином формулы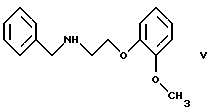
в протонном органическом растворителе и полученный бензил-карведилол формулы
дебензилируют путем каталитического гидрирования и, при необходимости, полученный продукт подвергают реакции с неорганической или органической кислотой для получения его фармацевтически приемлемой соли присоединения кислоты.
| Способ получения 1-( @ -карбазолил)-пропандиола-2,3 | 1981 |
|
SU1068429A1 |
| АНТИТЕЛА ПРОТИВ КЛАУДИНА И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2815926C2 |
| DE 3319027 А, 29.11.1984. | |||

