Настоящее изобретение относится к способу получения 4-гидроксибензальдегида, который имеет по меньшей мере один заместитель, находящийся в ортоположении к группе OH.
Изобретение касается, в частности, получения 3-метокси-4-гидроксибензальдегида и 3-этокси-4-гидроксибензальдегида, называемых соответственно "ванилин" и "этилванилин".
Ванилин получают, в основном, из натурального сырья, такого, например, как лигнин, но какую-то часть получают химическим путем.
В литературе описаны многочисленные способы получения ванилина [KIRK-OTHMER-Encyclopedia of Chemical Technology 23, с. 1710 - 3- издание], и многие из них основаны на получении ванилина из гваякола или 2-метоксифенола.
Так можно сослаться на получение ванилина путем взаимодействия гваякола с глиоксиловой кислотой, окисления на воздухе конденсата и последующего выделения ванилина из реакционной среды путем подкисления. Недостаток этого способа заключается в использовании глиоксиловой кислоты, которая является дорогим реактивом.
Другой способ получения ванилина основан на реакции Реймера-Тисмана, и состоит во взаимодействии гваякола и хлороформа в присутствии гидроокиси калия. Образование смолы является недостатком этого метода.
Синтез ванилина согласно реакции Гаттермана заключается в обработке цианистоводородной кислотой гваякола в присутствии соляной кислоты. Кроме того, что в этом способе применяют реактив, требующий осторожного обращения, он имеет и другой недостаток, заключающийся в том, что не является селективным, так как вместе с ванилином получают изованилин и ортованилин.
Наибольшая трудность в синтезе ванилина - это селективное закрепление формильной группы на гваяколе в параположении к гидроксильной группе.
Другая задача, которую необходимо решить, это разработать такой способ, который можно было бы применить в промышленности.
Настоящее изобретение предлагает такой способ, который позволяет устранить указанные выше недостатки, удовлетворяя требованиям, указанным выше.
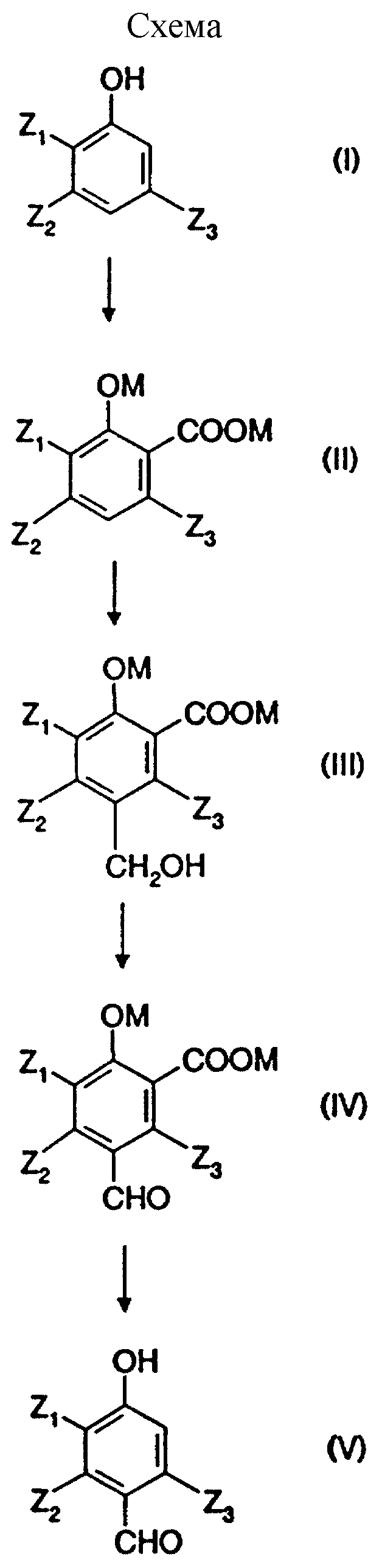
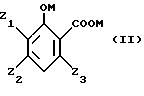
Объектом настоящего изобретения является способ получения 4-гидроксибензальдегида, замещенного, по меньшей мере, в положении 3 алкокси-группой, заключающийся в том, что фенольное соединение, замещенное, по меньшей мере, в положении 2 алкокси-группой, и положения 4 и 6 которого свободны, на первом этапе подвергают карбоксилированию по положению 6, затем подвергают на следующем этапе гидроксиметилированию по положению 4, затем окисляют гидроксиметильную группу до формильной группы и, наконец, подвергают этапу декарбоксилирования.
Способ согласно изобретению основан на получении 2-гидроксибензойных гидроксиметилированных в положении 5 кислот, замещенных по меньшей мере в положении 3 алкокси-группой, которые служат промежуточным продуктом в синтезе 4-гидроксибензальдегидов, замещенных по меньшей мере в положении 3 алкокси-группой.
Другим объектом изобретения является способ окисления 2-гидроксибензойных гидроксиметилированных в положении 5 кислот, замещенных, по меньшей мере, в положении 3 алкокси-группой до соответствующих 2-гидроксибензойных формилсодержащих кислот.
Способ по изобретению очень хорошо подходит для получения ванилина. В самом деле, он позволяет осуществлять селективное формилирование гваякола в параположении, осуществляя последовательно карбоксилирование гваякола в положении 6, гидроксиметилирование с последующим окислением до формильной группы в положении 4, и, наконец, удаление карбоксильной группы, находящейся в положении 6.
Этот способ является не только селективным, но и удовлетворяет с промышленной точки зрения, так как в нем используются недорогостоящие реактивы.
Хотя способ согласно изобретению дает прекрасные результаты с гваяколом и гветолом, он также осуществим и с другими фенольными замещенными соединениями.
Под "замещенным фенольным соединением" понимают любое ароматическое соединение, в котором ароматическое ядро является носителем гидроксильной группы, алкоксильной группы в положении 2 и других возможных заместителей, и положения 4 и 6 которого являются свободными.
В нижеизложенном подразумевают под термином "ароматическое" классическое понятие ароматики, которое определено в литературе, в частности, Jerry March, Advanced Organic Chemistry, 4-е изд, John Wiley and Sons, 1992, с. 40 и следующие.
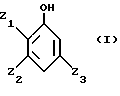
Из замещенных фенольных соединений согласно изобретению используют, в частности те, которые отвечают общей формуле I: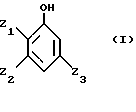
в которой:
- Z1 представляет алкоксильный радикал, линейный или разветвленный, имеющий от 1 до 12 атомов углерода, преимущественно от 1 до 4 атомов углерода, такой как радикалы метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси, трет-бутокси,
- Z2 и Z3, идентичные или разные, представляют атом водорода или одну из следующих групп:
алкильный радикал, линейный или разветвленный, имеющий от 1 до 12 атомов углерода, преимущественно от 1 до 4 атомов углерода, такой как метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил,
алкенильный радикал, линейный или разветвленный, имеющий от 2 до 12 атомов углерода, преимущественно от 2 до 4 атомов углерода, такой как винил, аллил,
алкоксильный радикал, линейный или разветвленный, имеющий от 1 до 12 атомов углерода, преимущественно от 1 до 4 атомов углерода, такой как радикалы метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси, трет-бутокси,
фенильный радикал,
атом галогена, преимущественно атом фтора, хлора или брома.
Согласно изобретению возможно присутствие на ароматическом кольце заместителей разных типов, поскольку они не оказывают влияния на реакции согласно способу изобретения.
В настоящем изобретении предпочтительно используются соединения формулы I, в которой Z1 представляет алкоксильный радикал, линейный или разветвленный, имеющий от 1 до 6 атомов углерода, преимущественно 1-4 атома углерода; Z2 и Z3 означают атом водорода.
В качестве предпочтительных примеров субстратов, применяемых в способе, можно назвать, среди прочих, гваякол и гветол.
Согласно предлагаемому способу применяют в качестве исходных продуктов замещенное фенольное соединение, которым является преимущественно соединение, отвечающее формуле I.
В конце описания дается реакционная схема способа по изобретению, чтобы легче понять изобретение, но она не ограничивает объем изобретения.
Дальше в описании будет даваться ссылка на указанные формулы.
Согласно изобретению на первом этапе осуществляют карбоксилирование замещенного фенольного соединения формулы I взаимодействием названного фенольного соединения в виде соли с двуокисью углерода.
Фенольные замещенные соединения взаимодействуют в способе по изобретению в форме соли. Речь идет преимущественно о солях с металлами группы Ia Периодической системы химических элементов.
Для определения групп металлов и других элементов в данном описании дается ссылка на Периодическую систему элементов, опубликованную в Бюллетене Химического Общества Франции, N 1 (1996).
С точки зрения практической и экономической используют соли натрия и калия.
Можно использовать замещенное фенольное соединение в виде соли, полученное перед самым использованием, но можно также получить соль ин ситу путем взаимодействия замещенного фенольного соединения с основанием.
Используют в способе по изобретению основание, которое может быть минеральным или органическим.
Выбирают преимущественно сильное основание, т.е. основание, имеющее показатель pKb больше 12: значение pKb определяют как сологарифм константы диссоциации измеряемого основания в водной среде при 25oC.
Преимущественно используют минеральные основания, такие как соли щелочных металлов, преимущественно гидроокись щелочного металла, которая может быть гидроокисью натрия или калия; карбонат щелочного металла, преимущественно калия.
Также можно использовать гидроокись четвертичного аммония.
В качестве примеров гидроокиси четвертичного аммония используют преимущественно гидроокиси тетралкиламмония или триалкилбензиламмония, алкиловые радикалы которых, идентичные или разные, представляют алкильную цепь, линейную или разветвленную, имеющую от 1 до 12 атомов углерода, преимущественно от 1 до 6 атомов углерода.
Выбирают преимущественно гидроокись тетраметиламмония, гидроокись тетраэтиламмония или гидроокись тетрабутиламмония.
Согласно изобретению также можно использовать гидроокиси триалкилбензиламмония, и в частности, гидроокиси триметилбензиламмония.
Из экономических соображений из всех оснований выбирают преимущественно гидроокись натрия или карбонат калия.
Концентрация исходного раствора основного характера не является критической. Раствор используемой гидроокиси щелочного металла имеет концентрацию, равную обычно 10-50 вес.%.
Количество основания, вводимого в реакционную среду, зависит от количества, необходимого для превращения в соль гидроксильной функции замещенного фенольного соединения.
Если названное соединение имеет другие солеобразующие функции кроме гидроксильной, то вводят такое количество основания, которое необходимо для превращения в соль всех солеобразующих функций.
Обычно количество основания, выраженное по отношению к замещенному фенольному соединению, составляет от 90 до 120% от стехиометрического количества.
Получают замещенное фенольное соединение в форме соли путем его взаимодействия с основанием при температуре, предпочтительно равной 25-100oC.
Перед тем как ввести двуокись углерода, удаляют воду, образовавшуюся во время реакции солеобразования, путем отгонки под атмосферным давлением, или под уменьшенным давлением, равным от 1 мм рт. ст. до атмосферного давления, или путем сушки. Когда в среде нет больше воды, вводят двуокись углерода.
Другой вариант способа состоит в использовании дополнительно безводного карбоната щелочного металла, преимущественно безводного карбоната калия, что позволяет устранить этап удаления воды (например от 5 до 100 мол.% замещенного фенольного соединения).
Количество используемой двуокиси углерода, выраженное в молярном отношении между двуокисью углерода и замещенным фенольным соединением, изменяется от 1 до 100 и предпочтительнее от 1 до 2.
Способ по изобретению проводят преимущественно при температуре 150-250oC, преимущественно при 160-200oC.
Осуществляют способ по изобретению при атмосферном давлении, пропуская двуокись углерода в виде пузырьков через реакционную среду, поддерживаемую при перемешивании.
Можно также проводить реакцию под давлением двуокиси углерода, которое варьируется между атмосферным давлением и приблизительно 100 барами. Предпочитают давление от 1 до 20.
Предпочтительный метод практического осуществления изобретения состоит в том, что используют замещенное фенольное соединение, основание, удаляют воду в случае необходимости путем дистиляции, а затем вводят двуокись углерода. Речь идет о реакции типа твердое вещество/газ.
В конце реакции выделяют замещенное фенольное соединение, в котором карбоксилатная группа находится в положении 6, и растворяют его в воде до концентрации от 5 до 50 вес.%.
Доводят раствор до pH, равного 6-10, преимущественно близкого к 7, путем добавления кислоты.
Можно использовать любую кислоту, но из-за экономических соображений предпочитают использовать классические минеральные кислоты, преимущественно соляную кислоту или серную кислоту. Концентрация кислоты не имеет критического значения. Она соответствует преимущественно концентрации, выпускаемой в продажу, например, 37 вес.% для соляной кислоты и 92 или 96% для серной кислоты.
Получают, таким образом, двухфазную среду, состоящую из органической фазы, содержащей непрореагировавшее замещенное фенольное соединение, и из водной фазы, содержащей ожидаемый продукт, а именно соль 2-гидроксибензойной кислоты, по меньшей мере, замещенную в положении 3 алкокси-группой, которая представлена формулой I в которой Z1, Z2 и Z3 имеют значения, данные выше, а М представляет атом водорода и/или катион металла группы Ia.
Дальше эта соль будет называться упрощенно, а именно "соль гидроксибензойной кислоты".
Разделяют обе фазы и получают водную фазу.
Следует заметить, что использование в качестве исходного соли гидроксибензойной кислоты формулы II, полученной перед самим употреблением, входит в рамки изобретения.
На следующем этапе осуществляют реакцию гидроксиметилирования в положении пара к группе OH путем реакции соли кислоты, полученной выше, с формальдегидом, в случае необходимости в присутствии основания.
Можно использовать формальдегид или любое соединение, образующее формальдегид, такое как, например, триоксан или параформальдегид, используемый в форме линейных полиформальдегидов с любой степенью полимеризации, имеющей преимущественно число звеньев (CH2O) от 8 до 100.
Используют названный реактив обычно в форме водного раствора, имеющего концентрацию меньше 50 вес.%, преимущественно от 20-50 вес.%.
Количество формальдегида, выраженное в молях формальдегида на моль соли гидроксибензойной кислоты может изменяться в широком диапазоне.
Преимущественно молярное соотношение формальдегид/соль гидроксибензойной кислоты составляет 0,5-3,0.
Можно проводить реакцию в присутствии основания. Основания, перечисленные выше, очень хорошо подходят для этой реакции.
Количество используемого основания, выраженное как соотношение между числом молей основания и числом молей соли гидроксибензойной кислоты, может изменяться от 0 до 2 и преимущественно между 0 и 1,1.
Основание может быть использовано как в твердой форме, так и в форме водного раствора.
Температура реакции составляет диапазон от 50oC до 100oC, и преимущественно от 60oC до 80oC.
Способ проводят преимущественно под автогенным давлением реактивов, чтобы избежать возможные потери параформальдегида, который может быть газообразным при используемых температурах.
Предпочитают проводить реакцию в атмосфере инертных газов, таких как азот или благородные газы, например аргон.
Продолжительность реакции может быть очень разной. Чаще всего она составляет от 30 минут до 24 часов, преимущественно от 4 часов до 8 часов.
С практической точки зрения реакцию легко осуществлять, если в установку подавать соль гидроксибензойной кислоты и формальдегид, в случае необходимости основание, затем при перемешивании довести реакционную смесь до желаемой температуры в течение времени, необходимого для окончания реакции.
Порядок введения реактивов не является критическим и может быть разным.
В конце реакции получают 2-гидроксибензойную гидроксиметилированную в положении 5 кислоту, замещенную, по меньшей мере, в положении 3 алкокси-группой, отвечающей преимущественно формуле III, в которой Z1, Z2 и Z3 имеют значения данные выше, а M представляет атом водорода и/или катион металла группы Ia.
Согласно предпочтительному варианту способа по изобретению не выделяют полученное соединение, а подвергают его прямо окислению.
Предпочтительный метод окисления согласно изобретению, составляющий второй объект настоящего изобретения, состоит в окислении 2-гидроксибензойной гидроксиметилированной в положении 5 кислоты, замещенной, по меньшей мере, в положении 3 алкокси-группой, в жидкой фазе, с помощью молекулярного кислорода или газа, его содержащего. Работают в водной среде, содержащей щелочной агент, в присутствии катализатора на основе платины или палладия, в случае необходимости в присутствии сокатализатора на основе производного висмута.
Что касается благородных металлов, используемых для катализирования реакции, в данном случае платины и палладия, они могут иметь разные формы, как например: платиновая чернь, палладиевая чернь, окись платины, окись палладия или сам благородный металл, осажденный на разные носители, такие как сажа, карбонат кальция, окиси алюминия и активированные двуокиси кремния или эквивалентные вещества. Каталитические массы на основе сажи превосходно подходят для катализирования реакции.
Количество применяемого катализатора в расчете на вес платины или палладия по отношению к весу 2-гидроксибенэойной гидроксиметилированной кислоты может варьироваться от 0,01 до 4% и преимущественно от 0,04 до 2%.
Можно использовать сокатализатор, в частности используют, как правило, минеральное производное или органическое производное висмута, в котором атом висмута имеет степень окисления больше нуля, например, 2, 3, 4 или 5. Остаток, соединенный с висмутом, не является критическим с момента, когда он удовлетворяет этому условию. Сокатализатор может быть растворимым или не растворимым в реакционной среде.
В качестве иллюстрации сокатализаторы, которые могут быть использованы в способе по изобретению, представляют собой: оксиды висмута; гидроокиси висмута; соли минеральных водородных кислот, такие как хлорид, бромид, иодид, сульфид, селенид, теллурид висмута; соли минеральных кислородсодержащих кислот, такие как сульфит, сульфат, нитрит, нитрат, фосфит, фосфат, пирофосфат, карбонат, перхлорат, антимонат, арсенат, селенит, селенат висмута; соли производных кислородсодержащих кислот с переходными металлами, такие как ванадат, ниобат, танталат, хромат, молибдат, вольфрамат, перманганат висмута.
Другими подходящими соединениями являются соли органических алифатических или ароматических кислот, например ацетат, пропионат, бензоат, салицилат, оксалат, тартрат, лактат, цитрат висмута; фенаты, такие как галлат и пирогаллат висмута. Эти соли и фенаты могут быть также солями висмутила.
В качестве других минеральных или органических соединений можно использовать бинарные композиции висмута с такими элементами, как фосфор и мышьяк; гетерополикислоты, содержащие висмут, а также их соли, подходят также алифатические и ароматические висмутины.
В качестве конкретных примеров можно назвать:
- окиси: BiO; Bi2O3; Bi2O4; Bi2O5,
- гидроокиси: Bi(OH)3,
- соли минеральных водородных кислот: хлорид висмута BiCl3, бромид висмута BiBr3, иодид висмута BiI3, сульфид висмута Bi2S3, селенид висмута Bi2Se3, теллурид висмута Bi2Te3,
- соли минеральных кислородсодержащих кислот: основной сульфит висмута Bi2(SO3)3, Bi2O3·5H2O, нейтральный сульфат висмута Bi2(SO4)3, сульфат висмутила (BiO)HSO4, нитрит висмутила (BiO)NO2·0,5H2O, нейтральный нитрат висмута Bi(NO3)3·5H2)O, двойной нитрат висмута и магния 2Bi(NO3)3·3Mg(NO3)2·24H2O; нитрат висмутила (BiO)NO3; фосфит висмута Bi2(PO3H)3·3H2O; нейтральный фосфат висмута BiPO4; пирофосфат висмута Bi4(P2O7)3; карбонат висмутила (BiO)2CO3·0,5H2O; нейтральный перхлорат висмута Bi(ClO4)3·5H2O; перхлорат висмутила (BiO)ClO4; антимонат висмута BiSbO4; нейтральный арсенат висмута Bi(AsO4)3; арсенат висмутила (BiO)AsO4·5H2O; селенит висмута Bi2(SeO3)3.
- соли производных кислородсодержащих кислот переходных металлов: ванадат висмута BiVO4; ниобат висмута BiNbO; танталат висмута BiTaO4; нейтральный хромат висмута Bi2(CrO4); бихромат висмутила ((BiO)2)2Cr2O7, кислый хромат висмутила H(BiO)CrO4; двойной хромат висмутила и калия K(BiO)CrO10; молибдат висмута Bi2(MoO4)3; вольфрамат висмута Bi2(WO4)3; двойной молибдат висмута и натрия NaBi(MoO4)2; основной перманганат висмута Bi2O2(OH)MnO4;
- соли алифатических и ароматических органических кислот: ацетат висмута Bi(C2H3O2)3; пропионат висмутила (BiO)C3H5O2; основной бензоат висмута C6H5CO2Bi(OH)2; салицилат висмутила C6H4CO2(BiO)(OH); оксалат висмута (C2O4)3Bi2; тартрат висмута Bi2(C4H4O6)3·6H2O; лактат висмута (C6H9O5)OBi·7H2O; цитрат висмута C6H5O7Bi;
- фенаты: основной галлат висмута C7H7O7Bi, основной пирогаллат висмута C6H3(OH)2(OBi)(OH).
В качестве других минеральных и органических соединений: фосфид висмута BiP, арсенид висмута Bi3As4, висмутат натрия NaBiO3 висмут-тиоцианистые кислоты H2[Bi(BNS)5] , H3[Bi(CNS)6] и их соли натрия и калия; триметилвисмутин Bi(CH3)3, трифенилвисмутин Bi(C6H5)3.
Производными висмута, которые преимущественно используют для проведения способа по изобретению, являются: окиси висмута, гидроокиси висмута, соли висмута или висмутила минеральных водородных кислот, соли висмута или висмутила минеральных кислородсодержащих кислот; соли висмута или висмутила алифатических или ароматических органических кислот, и фенаты висмута или висмутила.
Группа сокатализаторов, которые особенно подходят для выполнения изобретения, состоит из: окисей висмута Bi2O3 и Bi2O4, гидроокиси висмута Bi(OH)3, нейтрального сульфата висмута Bi2(SO4)3, хлорида висмута BiCl3, бромида висмута BiBr3, иодида висмута BiI3, нейтрального нитрата висмута Bi(NO3)3·5H2O; нитрата висмутила BiO(NO3); карбоната висмутила (BiO)2CO3·0,5H2O; ацетата висмута Bi(C2H3O2)3; салицилата висмутила C6H4CO2(BiO)(OH).
Количество используемых сокатализаторов, выраженное через количество металлического висмута, содержащегося в сокатализаторе, по отношению к весу взятого благородного металла, может варьироваться в широком диапазоне. Например, это количество может быть как небольшим 0,1%, так и может достигать веса используемого благородного металла и даже превысить его без отрицательных последствий.
В частности, это количество выбирают таким образом, чтобы оно вносило в среду окисления от 10 до 900 ppm веса металлического висмута по отношению к 2-гидроксибензойной гидроксиметилированной кислоте. С этой целью могут использоваться количества сокатализатора порядка 900-1500 ppm, однако без значительного дополнительного преимущества.
Согласно способу по изобретению окисление проводят в водной среде, содержащей в растворе щелочной агент. Для этого в качестве щелочного агента используют обычно гидроокись натрия или калия. Количество используемого минерального основания составляет 0,5-3 моля гидроокиси натрия или калия по отношению к 2-гидроксибензойной гидроксиметилированной кислоте.
Концентрация 2-гидроксибензойной гидроксиметилированной кислоты в водном растворе щелочного агента должна быть преимущественно такой, чтобы избежать образования осадка и сохранить однородным раствор.
Весовая концентрация 2-гидроксибензойной гидроксиметилированной кислоты в водной среде обычно равна 1-60%, преимущественно 2-30%.
На практике выполнение способа по изобретению состоит в том, что осуществляют контактирование с молекулярным кислородом или газом, его содержащим, например с воздухом, водного раствора, окисляемой 2-гидроксибензойной гидроксиметилированной кислоты щелочного агента, катализатора на основе платины или палладия и в случае необходимости сокатализатора на основе производного висмута согласно указанным выше соотношениям.
Работают при атмосферном давлении, но в случае необходимости можно также работать при давлении, равном 1-20 бар.
Затем смесь перемешивают при желаемой температуре, пока не будет потреблено то количество кислорода, которое соответствует количеству, необходимому для превращения спиртовой функции в функцию альдегида. Таким образом, ход реакции контролируют путем измерения поглощенного количества кислорода.
Температура реакции варьируется в зависимости от термостойкости получаемых продуктов.
Обычно реакцию проводят в температурном интервале в 50-100oC, преимущественно 60-80oC.
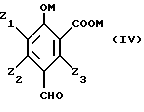
В конце реакции, которая длится преимущественно от 30 минут до 2 часов, получают 2-гидроксибензойную формилированную в положении 5 кислоту, замещенную, по меньшей мере, в положении 3 алкокси-группой и отвечающую преимущественно формуле IV.
Затем, после охлаждения, если таковое имеет место, отделяют каталитическую массу от реакционной среды, например, с помощью фильтрации.
На последнем этапе способа по изобретению осуществляют реакцию декарбоксилирования.
Для этого подкисляют полученную жидкость путем добавления протонной кислоты минерального происхождения, преимущественно соляной кислоты или серной кислоты, до получения pH, меньше или равного 3, преимущественно, равного 0-3.
Нагревают реакционную среду до температуры, например в диапазоне 120-350oC, преимущественно 150-220oC.
Способ проводят преимущественно под автогенным давлением реактивов.
В конце реакции охлаждают реакционную среду до 20-80oC.
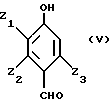
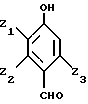
Получают двухфазную среду, состоящую с одной стороны, из органической фазы, включающей 4-гидроксибензальдегид, замещенный, по меньшей мере, в положении 3 алкоксигруппой и отвечающий преимущественно формуле V, и, возможно, исходный субстрат формулы I, а с другой стороны, из водной фазы, содержащей соль.
Разделяют органическую и водную фазы и рекуперируют замещенный 4-гидроксибензальдегид из органической фазы преимущественно дистилляцией.
Как сказано выше, способ по изобретению, в частности, предназначен для получения ванилина и этилванилина.
Ниже даются примеры выполнения изобретения. Эти примеры даются в качестве иллюстрации и не имеют ограничительного характера.
Пример 1
1. Карбоксилирование гваяколята калия под давлением CO2
Загружают в реактор типа Бартон Корбелин емкостью 500 мл из жаропрочного сплава марки B2, снабженный лопастной турбиной, 224 г (1,81 моль) гваякола.
Добавляют 31,5 г (228 моль) карбоната калия.
Продувают реактор потоком CO2. Наблюдается небольшое выделение тепла.
Нагревают до 170o в течение 7 часов, все время поддерживая давление CO2 на уровне 20 бар.
После охлаждения реактора до температуры окружающей среды добавляют 200 мл воды.
Приливают раствор хлористо-водородной кислоты 5н. до получения pH приблизительно 7,0. Происходит расслоение.
Декантируют органическую фазу, состоящую в основном из гваякола.
Водную фазу подкисляют до pH 1 с помощью хлористо-водородной кислоты. Происходит осаждение ортованилиновой кислоты.
Отделяют фильтрацией. Промывают водой и сушат полученный продукт при 40oC под пониженным давлением, равным 20 мм рт. ст.
Получают 38 г ортованилиновой кислоты с концентрацией 96 вес.%.
Выход равен 96% по отношению к карбонату калия.
2. Конденсация ортованилиновая кислота/параформальдегид
К суспензии 16,8 г (0,1 моль) ортованилиновой кислоты в 16,72 г воды добавляют 14,66 г 30%-го водного раствора гидроокиси натрия (0,11 моль) при перемешивании и при нагревании.
Когда среда становится однородной, а температура достигает 70oC, добавляют 3 г параформальдегида (0,1 моль).
После 6-ти часового перемешивания при 70oC осуществляют количественный анализ полученного раствора фиолетового цвета с помощью высокоэффективной жидкостной хроматографии.
Полученные результаты следующие:
- TT (кислота ортованилиновая) = число молей превращенной ортованилиновой кислоты к числу молей введенной ортованилиновой кислоты = 42,5%.
- RR (3-метокси-5-гидроксиметилсалициловая кислота) = число молей образовавшейся 3-метокси-5-гидроксиметилсалициловой кислоты к числу молей введенной ортованилиновой кислоты = 29,95%.
- RT = число молей образовавшейся 3-метокси-5-гидроксиметилсалициловой к числу молей превращенной ортованилиновой кислоты = 70,5%.
3. Окисление
К предыдущему раствору добавляют 1,63 г 2,5%-ной платины на угле (0,2 мол.%), затем 140 мг сульфата висмута (0,2 мол.%).
Температуру доводят до 65oC, а pH до 12, pH поддерживают при такой величине в течение реакции, добавляя 30%-ный гидрат окиси натрия.
При сильном перемешивании в реактор подают кислород со скоростью 1,5 л/час.
После 3 часов добавляют 0,815 г платины на угле, и продолжают реакцию в течение еще трех часов.
Затем осуществляют количественный анализ реакционной массы с помощью высокоэффективной жидкостной хроматографии:
- TT (3-метокси-5-гидроксиметилсалициловая кислота) = число молей превращенной 3-метокси-5-гидроксиметилсалициловой кислоты к числу молей введенной 3-метокси-5-гидроксиметилсалициловой кислоты = 100%.
- RR (5-карбоксиванилин) = число молей образованной 5-карбоксиванилиновой кислоты к числу молей введенной 3-метокси-5-гидроксиметилсалициловой кислоты = 59%.
4. Декарбоксилирование
Реакционную среду, полученную после окисления, разбавляют 100 мл воды, затем ее загружают в реактор типа Бартон Корбелин из жаростойкого сплава B2. Затем вливают 2н. раствор серной кислоты до получения pH приблизительно 1,9.
Продувают реактор потоком азота, затем сушат в течение 30 мин при 200oC.
Быстро охлаждают потоком холодной воды.
Разбавляют реакционную смесь ацетонитрилом, затем анализируют высокоэффективной жидкостной хроматографией.
Полученные результаты следующие:
- TT 5-карбоксиванилин = число молей превращенного 5-карбоксиванилина к числу молей использованного 5-карбоксиванилина = 100%.
- RT ванилин = число молей образованного ванилина к числу молей преобразованного 5-карбоксиванилина = 99,4%.
- RR ванилин/ортованилиновая кислота = число молей образованного ванилина к числу молей использованной ортованилиновой кислоты = 27,4%.
Примеры 2-4
Осуществляют серию испытаний карбоксилирования 3-карбокси-4-гидрокси-5-метоксибензальдегида.
Загружают в реактор Бартон Корбелин емкостью 50 мл из жаростойкого сплава B2, снабженный лопастной турбиной: 0,246 г (1,26 ммоль) 5-карбоксиванилина и 20 мл смеси уксусной кислоты и воды (50/50 в объеме) (пример 2), 20 мл воды (пример 3) и 20 мл раствора серной кислоты (5 ммоль) (пример 4).
Продувают реактор потоком азота.
Греют в течение 20 мин при 160oC (примеры 2 и 3) и при 200oC (пример 4).
Анализируют высокоэффективной жидкостной хроматографией реакционную среду после разбавления ацетонитрилом: колонка Lichro Cart RP-18 - 5 мкм - 250/4 мм, выпускаемая в продажу фирмой Мерк-элюант: 800 мл H2O/200 мл CH3CN/3,5 мл H3PO4 - расход; 1 мл·мин-1 - детекция УФ, 240 мкм - температура окружающей среды.
Результаты помещены в таблицу.

Описывается способ получения замещенного 4-гидроксибензальдегида общей формулы V, в которой Z1 представляет алкокси радикал, линейный или разветвленный, имеющий от 1 до 12 атомов углерода, преимущественно от 1 до 4 атомов углерода, такой как радикалы метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси, трет-бутокси, Z2 и Z3 идентичные или разные, представляют атом водорода или одну из следующих групп: алкил радикал, линейный или разветвленный, имеющий от 1 до 12 атомов углерода, преимущественно от 1 до 4 атомов углерода, такой как метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, алкенил радикал, линейный или разветвленный, имеющий от 2 до 12 атомов углерода, преимущественно от 2 до 4 атомов углерода, такой как винил, аллил; алкокси радикал, линейный или разветвленный, имеющий от 1 до 12 атомов углерода, преимущественно от 1 до 4 атомов углерода, такой как радикалы метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси, трет-бутокси, фенильный радикал, атом галогена, преимущественно атом фтора, хлора или брома. Способ отличается тем, что замещенное фенольное соединение общей формулы I, в которой Z1, Z2, Z3 имеют вышеуказанные значения, в форме соли подвергают карбоксилированию в положении 6 с получением соединения, имеющего формулу II, в которой Z1, Z2, Z3 имеют вышеуказанные значения, М - катион металла группы Ia, или катион четвертичного аммония, затем осуществляют стадию гидроксиметилирования в положении 4 с получением соединения, имеющего формулу III, в которой Z1, Z2, Z3, М имеют вышеуказанные значения, с последующим окислением гидроксиметильной группы до формильной группы, с получением соединения формулы IV, в которой Z1, Z2, Z3, М имеют вышеуказанные значения, и на последней стадии декарбоксилируют соединение формулы IV с получением целевого 4-гидроксибензальдегида формулы V. Технический результат - упрощение технологии процесса. 4 с. и 21 з.п. ф-лы, 1 табл.
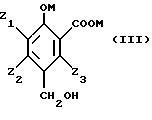
в которой Z1 представляет алкокси радикал, линейный или разветвленный, имеющий от 1 до 12 атомов углерода, преимущественно от 1 до 4 атомов углерода, такой как радикалы метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси, трет-бутокси;
Z2 и Z3 идентичные или разные, представляют атом водорода или одну из следующих групп: алкил радикал, линейный или разветвленный, имеющий от 1 до 12 атомов углерода, преимущественно от 1 до 4 атомов углерода, такой как метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, алкенил радикал, линейный или разветвленный, имеющий от 2 до 12 атомов углерода, преимущественно от 2 до 4 атомов углерода, такой как винил, аллил; алкокси радикал, линейный или разветвленный, имеющий от 1 до 12 атомов углерода, преимущественно от 1 до 4 атомов углерода, такой как радикалы метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси, трет-бутокси, фенильный радикал, атом галогена, преимущественно атом фтора, хлора или брома,
отличающийся тем, что замещенное фенольное соединение общей формулы I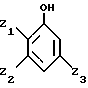
в которой Z1, Z2 и Z3 имеют вышеуказанные значения,
в форме соли подвергают карбоксилированию в положении 6, с получением соединения, имеющего формулу II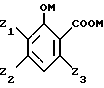
в которой Z1, Z2, Z3 имеют вышеуказанные значения;
M - катион металла группы (Ia) или катион четвертичного аммония,
затем осуществляют стадию гидроксиметилирования в положении 4, с получением соединения, имеющего формулу III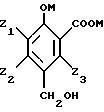
в которой Z1, Z2, Z3, М имеют вышеуказанные значения,
с последующим окислением гидроксиметильной группы до формильной группы, с получением соединения формулы IV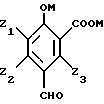
в которой Z1, Z2, Z3, М имеют вышеуказанные значения,
и на последней стадии декарбоксилируют соединение формулы IV с получением целевого 4-гидроксибензальдегида формулы V.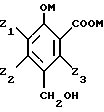
в которой Z1, Z2 и Z3 имеют значения, определенные в п.1;
M означает атом водорода и/или катион металла группы Ia,
отличающийся тем, что осуществляют взаимодействие 2-гидроксибензойной кислоты общей формулы II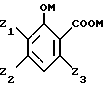
в которой Z1, Z2, Z3 и M имеют значения, указанные выше,
с формальдегидом или источником формальдегида, в случае необходимости, в присутствии основания.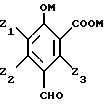
в которой Z1, Z2, Z3, M имеют значения, указанные в п.10,
отличающийся тем, что 2-гидроксибензойную кислоту формулы III по п.10 окисляют в жидкой фазе с помощью молекулярного кислорода или газа, его содержащего, в водной среде, содержащей щелочной агент, в присутствии катализатора на основе платины или палладия, в случае необходимости в присутствии сокатализатора на основе производного висмута.
| OHTA M ET AL: "Microbial degradation of dehydrodiconiferul alcohol, a lignin substructure model", ARCH | |||
| MICROBIOL, 1979, с.26 | |||
| Дуплексное регенеративное устройство | 1941 |
|
SU72600A1 |
| 0 |
|
SU51381A1 | |

