Изобретение относится к способу получения нитрофенолов. Более конкретно, изобретение относится к получению п-нитрофенола.
Из пат. США А-3 517075 известен способ получения п-нитрофенола путем окисления п-нитрозофенола. На первой стадии сначала получают п-нитрозофенол путем нитрозирования фенола с помощью азотистой кислоты в присутствии серной кислоты; затем во второй стадии п-нитрозофенол окисляют до п-нитрофенола. Существенным недостатком такого способа является выпадение в осадок п-нитрозофенола, который представляет собой нестабильное соединение, создающее значительную опасность теплового взрыва.
Цель изобретения разработка более безопасного способа получения п-нитрофенола. Эта цель достигается предлагаемым способом получения п-нитрофенола путем нитрозирования фенола, возможно замещенного в присутствии серной кислоты с последующим окислением полученного п-нитрозофенола с помощью азотной кислоты, отличающимся тем, что на первой стадии нитрозирования используют серную кислоту с концентрацией, по крайней мере равной 60% а на второй стадии, по окончании реакции окисления, используют серную кислоту с концентрацией не больше 80% что позволяет осаждать п-нитрофенол, который затем отделяют.
В соответствии с изобретением данный способ применим как к фенолу, так и ко всем другим ароматическим соединениями, имеющим OH-группу, не находящуюся в пара-положении. Среди таких гидроксилированных соединений предпочтительны те из них, которые описываются общей формулой (I):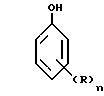
в которой
R атом водорода; алкильный или алкоксильный радикал, включающий от 1 до 4 атомов углерода, преимущественно метильный или этильный радикал; перфторсодержащий алкильный радикал, имеющий от 1 до 4 атомов углерода; атом галогена, преимущественно это хлор, бром или фтор;
n число, равное 0, 1 или 2.
Данное изобретение не исключает присутствия других заместителей в ароматическом кольце при том условии, что они не будут мешать течению реакционного процесса, представленного в изобретении. В частности, это представляется возможным как в отношении функциональных групп или атомов галогена в алкильных цепочках бензольного ядра, так и всей алкильной цепи, поскольку гетероатомы, такие, как кислород, азот или сера могут препятствовать проявлению их активности.
В последующем описании настоящего изобретения термин "фенол" используется в тривиальном смысле, подразумевая при этом и собственно фенол, и все ароматические гидроксисоединения и в особенности те из них, которые удовлетворяют формуле (I).
Способ изобретения благодаря контролированию концентрации серной кислоты на обоих стадиях нитрозирования и окисления позволяет:
с одной стороны, разрешить проблему взрывоопасности п-нитрозофенола, так как, используя концентрацию серной кислоты выше 60% на первой стадии, п-нитрозофенол становится растворимым;
и, с другой стороны, рекуперировать осадившийся п-нитрофенол по окончании стадии окисления, на которой концентрация серной кислоты ниже 80% т.к. п-нитрофенол становится растворимым при более высоких концентрациях.
Согласно способу изобретения на первой стадии осуществляют нитрозирование фенола в присутствии серной кислоты.
Как указано выше, количество используемой серной кислоты является критическим.
Концентрация серной кислоты в реакционной среде, выраженная в весовом соотношении серная кислота /серная кислота + вода, составляет величину, по крайней мере равную 60 мас%
Предпочтительно концентрация серной кислоты составляет 60-90% Предпочтительно она выбирается между 70% и 80%
Присутствие воды на стадии нитрозирования не мешает в той мере, когда ее количество такое, что соблюдается концентрация серной кислоты.
Нитрозирующим агентом является любой источник NO+. Так, можно исходить из диоксида азота NO2, азотистого ангидрида N2O3, диоксида азота N2O4, оксида азота NO, ассоциированного с окислителем, таким, как, например, азотная кислота, диоксид азота или кислород. В случае, когда нитрозирующий агент газообразен в реакционных условиях, его вводят путем барботажа в среду.
Также можно использовать азотистую кислоту, нитрозилсульфат или нитрозосульфат или соль азотистой кислоты, предпочтительно нитрит щелочного металла, или более предпочтительно нитрит натрия.
Количество используемого нитрозирующего агента может изменяться в широких пределах. Когда это количество выражено молярным соотношением фенол/нитрозирующий агент, определенный в виде NO+, то оно по крайней мере равно стехиометрическому количеству, но предпочтительно, чтобы нитрозирующий агент использовался в избытке, причем его количество может достигать 500% от стехиометрического количества, и предпочтительно оно составляет 150-300%
Концентрации фенольного субстрата в реакционной среде составляет предпочтительно 2-20 мас%
Фенол обычно вводят в жидкой форме. Следовательно, он может быть введен либо в расплавленном состоянии, либо в виде смеси с водой. В этом последнем случае хорошо пригодны смеси, содержащие 60-90% фенола. Здесь нужно следить за тем, чтобы содержание воды в реакционной среде было таким, чтобы соблюдалась концентрация серной кислоты.
На следующей стадии осуществляют окисление п-нитрозофенола с помощью азотной кислоты.
Также можно использовать предшественник азотной кислоты, такой, как, например, диоксид азота.
Используют водный раствор азотной кислоты, имеющий концентрацию, которая может изменяться в пределах 30-100% Однако предпочтительна концентрация 60-100%
Количество азотной кислоты, выражаемое молярным соотношением фенол/азотная кислота, обычно изменяется в пределах 0,9-1,2, предпочтительно составляет 0,95-1,05.
Как указано выше, количество используемой серной кислоты на этой стадии должно быть строго контролируемым.
Концентрация серной кислоты ниже или равна 80% Нижний предел не имеет критического характера. Концентрация предпочтительно составляет 50-80% и преимущественно 65-75%
В процессе реакции нитрозирования может выделяться вода. Во время реакции окисления можно при необходимости добавлять воду, чтобы достигать вышеуказанных концентраций серной кислоты. Наиболее часто воду добавляют одновременно с азотной кислотой.
Способ изобретения предпочтительно осуществляют при температуре 0-40oC, преимущественно при 10-30oC.
Способ изобретения обычно осуществляют при атмосферном давлении, но его можно также осуществлять при слегка пониженном давлении, составляющем, например, 500-760 мм рт.ст. (66500 101080 Па), или в условиях повышенного давления, которое может достигать, например, 5 бар.
Согласно одному предпочтительному варианту способа изобретения стадию нитрозитрования проводят в атмосфере инертных газов. Можно использовать атмосферу благородных газов, предпочтительно аргона, но более экономично использовать азот.
С практической точки зрения способ изобретения легко осуществим, так как нет необходимости прибегать к специальной аппаратуре.
Практически способ изобретения может быть осуществлен нижеописанным образом.
Различные составляющие реакционной смеси загружают в выбранную аппаратуру.
Могут быть предусмотрены несколько способов осуществления.
Первый вариант состоит в загрузке сначала раствора серной кислоты, затем в добавлении фенола и нитрозирующего агента.
Согласно другому варианту вводят раствор серной кислоты и нитрозирующий агент, затем добавляют фенол, предпочтительно порциями или путем непрерывного введения.
Следующий вариант заключается во введении параллельно в нижнюю часть резервуара, с одной стороны, фенола и, с другой стороны, серной кислоты и нитрозирующего агента.
После введения реагентов реакционную смесь выдерживают при вышеуказанной температуре. Может быть благоприятным охлаждать реакционную смесь.
На следующей стадии окисления затем в реакционную среду, содержащую п-нитрозофенол, вводят азотную кислоту. Ее можно добавлять за один раз или постепенно порциями или непрерывно, путем приливания. Также можно вводить азотную кислоту с самого начала например, параллельно с добавлением фенола.
Температуру реакционной смеси поддерживают при вышеуказанном температурном интервале.
Согласно другому способу осуществления изобретения можно получать азотную кислоту in situ из диоксида азота, который служит одновременно нитрозирующим агентом на первой стадии и предшественником азотной кислоты во второй стадии. С этой целью диоксид азота вводят в самого начала и во второй стадии нагревают до температуры 20-40oC.
При заданной концентрации серной кислоты в реакционной среде п-нитрофенол осаждается.
Полученный осадок отделяют классическими способами разделения твердое вещество/ жидкость, предпочтительно путем фильтрования.
Полученный осадок промывают предпочтительно раствором серной кислоты такой же концентрации, что и концентрация на стадии окисления.
Для очистки осадка от примесей можно осуществлять промывку водой.
Маточные растворы, получающиеся после каждого отделения, могут быть рециркулированы. Они обогащены нитрозирующим агентом, так как он регенерируется в процессе окисления.
Способ изобретения позволяет получать главным образом п-нитрофенол, так как количество о-нитрофенола обычно ниже 5%
Согласно способу изобретения п-нитрофенол осаждается в преобладающем количестве по сравнению с о-нитрофенолом и динитрофенолами, получаемыми в качестве примесей.
П нитрофенольные соединения, которые могут быть получены по способу данного изобретения, и в особенности сам п-нитрофенол используется как промежуточное соединение при получении N ацетил-п-аминофенола путем с восстановления п-нитрофенола и последующего ацетилирования полученного п-аминофенола. Этот способ хорошо известен и описан, например, KIRKOTHMER (Энциклопедия Химической технологии 2, 3 изд. Encyclopedia of Chemical Technology 2, 3eme edition). Восстановление п-нитрофенола осуществляется традиционным способом, т. е. при взаимодействии с водородом в присутствии катализатора восстановления, предпочтительно на основе благородных металлов, платины или палладия. Указанный металл может быть нанесен на носитель: древесный уголь, ацетиловую сажу, двуокись кремния, оксид алюминия и т.д. Ацетилирование предпочтительно проводить с помощью ангидрида уксусной кислоты.
Для характеристики способа используют следующие обозначения:
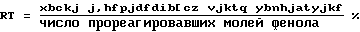
Пример 1. В реактор емкостью 1 л, снабженный двойной рубашкой, механическим перемешиванием и температурным зонтом, загружают 500 мл 80 мас.-ного водного раствора серной кислоты и добавляют туда 97,4 г водного 42,5-ного раствора NaNO2 (или 0,6 моль NaNO2. Охлаждают реакционную смесь до 10oC и затем в течение 40 мин и при поддерживании температуры 10oC приливают 33,7 г водного 80 -го раствора фенола (или 0,286 моль фенола). Затем реакционную смесь выдерживают в течение 5 мин при 10 oC, после чего в течение 10 мин приливают 30,1 г водного 67 мас.-ного раствора азотной кислоты (0,32 моль), все время поддерживая температуру 10oC. Реакционная смесь становится гетерогенной. Реакционную смесь выдерживают при 10 oC, затем отфильтровывают. Осадок на фильтре промывают 2 раза с помощью 50 г водного 70-ного раствора серной кислоты, затем водой и сушат в вакууме, создаваемом водоструйным насосом. Таким образом получают 32,94 г продукта светло-серого цвета с содержанием п-нитрофенола 95,2 (Фильтрат и промывные воды рекуперируют и анализируют путем высокоэффективной жидкостной хроматографии (ВЭЖХ). Результаты, полученные после осуществления реакции, следующие:
TTфенол 100 RTп-нит рофенол 92% - RTо-нит рофенол 0,6%
RTдинит рофенолы 0,93%
Пример 2. В реактор емкостью 250 мл, снабженный механическим перемешиванием и системой регулирования температуры, загружают 100 мл (173 г) водного 80%-ного раствора серной кислоты. Добавляют 13 мл (15,3 г.) водного 27-ного раствора нитрата натрия (0,06 моль NaNO2). Доводят реакционную среду до 10oC. Затем в течение 40 мин приливают 6,7 мл водного 80-ного раствора фенола (0,0605 моль фенола), все время поддерживая температуру 10oC. Спустя 2 мин приливания фенола одновременно и в течение 38 мин приливают водный 40-ный раствор азотной кислоты (0,550 моль азотной кислоты). Реакционная среда становится гетерогенной. Реакционную смесь выдерживают при 10oC в течение 5 мин и отфильтровывают на холоду. Осадок на фильтре промывают 2 раза с помощью 30 мл водного 70-ного раствора серной кислоты, затем 3 раза водой (30 мл), после чего высушивают в вакууме. Получают 5,54 г твердого продукта с содержанием 95 п-нитрофенола. Фильтры и промывные воды объединяют и анализируют. Полученные после реакции результаты следующие: TTфенол 100% RTп-нитр офенол 83,6% RTо-нит рофенол 6,7% RTдинит рофенолы 0,7 RTп-нитр озофенол 6,8
Пример 3. Реакционные условия точно идентичны условиям примера 2, за исключением температуры, которая поддерживается при 26oC в течение всей продолжительности опыта. Полученные результаты следующие: TTфенол 100 RTп-нитр офенол 62,5 RTо-нитр офенол 5,4 RTдинит рофенолы 1,3 RTп-нитро зофенол 12,0
Пример 4. В реактор емкостью 250 мл, снабженный двойной рубашкой и механическим перемешиванием, загружают в нижнюю часть 100 мл водного 80-ного раствора серной кислоты и добавляют 19,5 г водного 42,5-ного раствора нитрата натрия (0,12 моль NaNO2). Затем в течение 40 мин приливают 7,07 г водного 80 -ного раствора фенола (0,0602 моль), все время поддерживая температуру 25oC. Затем реакционную смесь выдерживают при 25oC в течение 5 мин, после чего в течение 15 мин приливают 5,44 г водного 67-ного раствора азотной кислоты (0,058 моль). Смесь выдерживают при 25oC в течение 5 мин. Среда гетерогенная: твердое вещество /жидкость. Анализ с помощью ВЭЖХ различных форм дает следующие результаты: TTфенол 100 RTп-нитр офенол 89,5 RTо-нитр офенол 0,75 RTдини трофенолы 2,05
Пример 5. Условия идентичны таковым примера 4. за исключением температуры, которая составляет 10oC. Загрузку азотной кислоты осуществляют 2 раза. К серной кислоте и нитрату натрия в нижней части реактора перед приливанием фенола добавляют 0,7 г азотной кислоты (0,0074 моль) и после приливания фенола добавляют 5,7 г азотной кислоты (0,0595 моль). Конечная реакционная среда гетерогенная. Среду фильтруют, после чего фазы анализируют. Полученные результаты следующие: TTфенол 100 RTп-нитро фенол 91,1 RTо-нитро фенол 1,5 RTдини трофенолы 0,8
Пример 6. В реактор емкостью 1 л, снабженный двойной рубашкой и механическим перемешиванием, вводят 904 г водного 70-ного раствора серной кислоты и 18,5 г 68-ного раствора азотной кислоты (0,2 моль NHO3). В жидкую смесь при перемешивании пропускают 13 г газообразного оксида азота (0,43 моль). Эту реакционную смесь доводят до 10oC, затем добавляют 35,2 г водного 80-ного раствора фенола в течение 40 мин, все время поддерживая температуру 10oC, затем реакционную смесь выдерживают при 10oC в течение 5 мин и после этого к реакционной смеси приливают 28,2 г (0,3 моль) водного 68-ного раствора азотной кислоты в течение 15 мин и в течение 5 мин выдерживают при 10oC. Затем реакционную среду отфильтровывают. Получают 15,8 г твердого вещества темно-красного цвета. Это твердое вещество также, как оставшиеся маточные растворы, анализируют путем ВЭЖХ. Полученные после реакции результаты следующие: TTфенол 100 RTп-нитро фенол 61 RTо-нитро фенол 4,6% RTдинитр офенолы 8,8
Пример 7. В реактор с двойной рубашкой, снабженный механической мешалкой, загружают в нижнюю часть 440,5 г водного 69-ного раствора серной кислоты, 40,5 г кислого нитрозилсульфата (0,3 моль). Реакционную смесь охлаждают до 10oC и к реакционной смеси в течение 40 мин приливают 17,7 г водного 80 -ного раствора фенола (0,15 моль), поддерживая температуру 10oC. Затем реакционную смесь выдерживают в течение 5 мин при 10oC и после этого в течение 5 мин к реакционной смеси, все время поддерживая температуру 10oC, приливают 13,9 г водного 68-ного раствора азотной кислоты (0,15 моль). Затем реакционную смесь отфильтровывают, осадок на фильтре промывают 2 раза по 30 мл водного 70-ного раствора серной кислоты, затем 3 раза по 30 мл воды, и продукт высушивают в вакууме. Получают 18 г светло-желтого осадка, состоящего главным образом из п-нитрофенола. Анализ различных фаз дает следующий баланс: TTфенол 100 RTп-нитр офенол 88 RTо-нитро фенол 1,1 RTдини трофенолы 1,75
Пример 8. В реактор с двойной рубашкой, снабженный механической мешалкой, загружают в нижнюю часть 440 г водного 70-ного раствора серной кислоты и 40,5 г кислого нитрозилсульфата (0,3 моль). В течение 10 мин все время поддерживая температуру 25oC, приливают 17,7 г водного 80-ного раствора фенола (0,15 моль), после чего реакционную смесь выдерживают в течение 5 мин при 25oC и затем в течение 15 мин, все время поддерживая температуру 25oC, добавляют 14,5 г водного 68-ного раствора азотной кислоты (0,156 моль). Выдерживают в течение 5 мин при 25oC, после чего реакционную смесь отфильтровывают. Осадок на фильтре промывают 2 раза с помощью 30 мл водного 70%-ного раствора серной кислоты, затем 3 раза по 30 мл водой. Получают 12,1 г твердого вещества серого цвета с содержанием 98 п-нитрофенола. Различные фазы анализируют путем ВЭЖХ, и баланс следующий: TTфенол 100 RTп-нитр офенол 90 RTо-нитр офенол 0,2 RTдинитр офенолы 3,38
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПИКРИНОВОЙ КИСЛОТЫ | 1995 |
|
RU2128642C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 3-КАРБОКСИ-4-ГИДРОКСИБЕНЗАЛЬДЕГИДА, СПОСОБ ПОЛУЧЕНИЯ 4-ГИДРОКСИБЕНЗАЛЬДЕГИДА, СПОСОБЫ ПОЛУЧЕНИЯ ВАНИЛИНА И ЭТИЛВАНИЛИНА | 1996 |
|
RU2186055C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВОДОРАСТВОРИМЫХ И/ИЛИ ВОДОДИСПЕРГИРУЕМЫХ СОПОЛИЭФИРОВ | 1992 |
|
RU2102405C1 |
| ОСАЖДЕННЫЙ ДИОКСИД КРЕМНИЯ (ВАРИАНТЫ) И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1994 |
|
RU2087417C1 |
| ОСАЖДЕННЫЙ ДИОКСИД КРЕМНИЯ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1994 |
|
RU2092435C1 |
| СПОСОБ ПОЛУЧЕНИЯ БИСФЕНОЛА-А | 1991 |
|
RU2060984C1 |
| Способ непрерывного синтеза 4-нитрозофенола | 2021 |
|
RU2762969C1 |
| СПОСОБ И УСТАНОВКА ДЛЯ ПОЛУЧЕНИЯ ПАРА-АМИНОФЕНОЛА ИЗ ФЕНОЛА ПУТЁМ ПОСЛЕДОВАТЕЛЬНОГО НИТРОЗИРОВАНИЯ И ВОССТАНОВЛЕНИЯ СУЛЬФИДОМ АММОНИЯ | 2023 |
|
RU2801692C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННОГО 4-ГИДРОКСИБЕНЗАЛЬДЕГИДА | 1996 |
|
RU2164911C2 |
| ДЕТЕРГЕНТНАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2077559C1 |
Предметом изобретения является способ получения нитрофенолов. Более конкретно, изобретение относится к получению п-нитрофенола. Способ получения согласно изобретению заключается в получении п-нитрофенола путем нитрозирования фенола в присутствии серной кислоты, затем окисления п-нитрозофенола с помощью азотной кислоты, и отличается тем, что на первой стадии нитрозирования концентрация серной кислоты по крайней мере равна 60 %, а на второй стадии, по окончании реакции окисления, концентрация серной кислоты самое большее равна 80 %, что позволяет осаждать п-нитрофенол, который затем отделяют. 1 с. и 14 з.п.ф-лы.
3. Способ по п.1 или 2, отличающийся тем, что нитрозирующим агентом является любой источник NO+, предпочтительно оксид азота NO, ассоциированный с окислителем, диоксид азота NO2, азотный ангидрид N2O3, диоксид азота N2O4, азотистая кислота, нитрозилсульфат или соль азотистой кислоты, предпочтительно нитрит щелочного металла, предпочтительно нитрит натрия.
5. Способ по любому из пп.1 4, отличающийся тем, что концентрация фенольного субстракта в реакционной среде составляет 2 20 мас.
7. Способ по любому из пп.1 6, отличающийся тем, что используют водный раствор азотной кислоты или предшественник азотной кислоты, предпочтительно диоксид азота.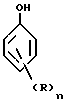
где R атом водорода, алкильный или алкоксильный радикал, содержащий от 1 до 4 атомов углерода, преимущественно метильный или этильный радикал, перфторсодержащий алкильный радикал, имеющий от 1 до 4 атомов углерода, атом галогена, преимущественно это хлор, бром или фтор;
n число, равное 0, 1 или 2.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| US, патент, 3517075, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| US, патент, 3519693, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| US, патент, 4229595, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| DE, патент, 1965384, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

