Изобретение относится к производным камптотецина, к способу их получения, к их применению в качестве активных ингредиентов для приготовления лекарственных средств, пригодных для лечения опухолей, и к фармацевтическим препаратам, их содержащим.
Противоопухолевый агент 20S-камптотецин формулы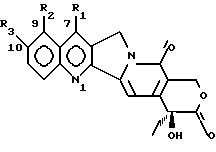
где R1, R2, R3 являются водородом, открытый в 1966 году M.E. Wall et al. (J. Amer. Chem. Soc. 88, 3888-90(1966)), после предварительной клинической оценки был отвергнут как терапевтический агент из-за его токсичности для человека и его низкой растворимости, которая усложняет его введение в приемлемые фармацевтические препараты. Внимание академических и промышленных исследователей было затем посвящено синтезу аналогов камптотецина с улучшенным терапевтическим профилем. Два из многочисленных аналогов, описанных вышеприведенной формулой, а именно топотекан, где R1 является водородом, R2 является -CH2-NH(CH3)2-группой, R3 является OH, и CPT-11, где R1 является этилом, R2 является водородом, R3 является
стали недавно доступны онкологам для лечения некоторых опухолей (J. of Clinical Oncology, 10, 1775-80 (1992); J. of the National Cancer Inst. 85, 271 (1993). Другими производными в клинических испытаниях в настоящее время являются 9-аминокамптотецин и аналог формулы
(Cancer Treatment Reviews 20, 73-96 (1994)).
Наибольшее количество попыток синтеза было посвящено введению подходящих заместителей для решения проблемы, связанной с плохой растворимостью в воде, что характеризует этот класс соединений и что может приводить к сложностям в их технологии приготовления и к непредсказуемым уровням содержания лекарства в плазме. Более того, постоянство лактонового кольца в закрытой форме является важным фактором для противоопухолевой эффективности.
Уместность этого класса соединений возникает также благодаря их особому механизму действия: фактически они проявляют свои противоопухолевые эффекты, ингибируя топоизомеразу I, фермент, который регулирует топологию ДНК и, таким образом, играет критическую роль в существенных клеточных процессах, таких как репликация, транскрипция, рекомбинация и репарация ДНК (C. Capranico and F. Zunino, Current Pharm. Design, 1, 1-14 (1995)). Необходимость в новых лекарствах, эффективных против карциномы толстой кишки, немелкоклеточной карциномы легкого, опухолей яичника и карциномы простаты, все еще слабо поддающихся химиотерапевтическому лечению, делает необходимым поиск новых производных камптотецина с улучшенными фармакологическими свойствами.
Сейчас найдено, что производные камптотецина и 10-гидроксикамптотецина, несущие заместители при углероде C-7, проявляют противоопухолевую активность и проявляют подходящие физико-химические свойства, которые допускают их технологию приготовления в подходящие фармацевтические композиции.
Настоящее изобретение включает в себя соединения формулы (I)
где R1 является -CN, -CH(CN)-R4, -CH=C(CN)-R4, -CH2-CH(CN)-R4, -C(= NOH)-NH2, -C(= NH)-NH2, -CH=C(NO2)-R4, -CH(CN)-R5, -CH(CH2NO2)-R5, 5-тетразолилом, 2-(4,5-дигидрооксазолилом), 1,2,4-оксадиазолин-3-ил-5-оном;
R2 является водородом;
R3 является водородом, OR6;
R4 является водородом, C1-C6 линейным или разветвленным алкилом, CN, COOR7;
R5 является водородом, OR8:
R6 является водородом, C1-C6 линейным или разветвленным алкилом, (C6-C12)арил(C1-C4)алкилом, (C1-C4)алкокси(C1-C4)алкилом, (C1-C4)алкил(C6-C12)арилом, (C6-C12)арил(C2-C4)ацилом, (C2-C4)ацилом, амино(C1-C4)алкилом, амино(C2-C4)ацилом, гликозилом;
R7 является водородом, C1-C6 линейным или разветвленным алкилом, (C6-C12)арил(C1-C4)алкилом, (C1-C4)алкокси(C1-C4)алкилом, (C1-C4)алкил(C6-C12)арилом;
R8 имеет те же значения, что и R6, независимо от последнего;
их N1-окислы, их изомеры, диастереоизомеры, энантиомеры и их смеси, так же как и их метаболиты, в частности активные метаболиты.
Настоящее изобретение включает в себя также фармацевтически приемлемые соли.
Настоящее изобретение включает в себя применение соединений формулы (I) в качестве активных ингредиентов для получения лекарственных средств, в частности лекарственных средств, пригодных для лечения опухолей.
Настоящее изобретение включает в себя фармацевтические композиции, содержащие соединения формулы (I) как активные ингредиенты.
Настоящее изобретение включает в себя способ получения соединений формулы (I).
Настоящее изобретение включает в себя использование соединений формулы (I), где R1 является CN как промежуточных соединений для получения других соединений формулы (I), где R1 является -C=(NOH)-NH2, -C(=NH)-NH2, 5-тетразолилом, 2-(4,5-дигидрооксазолилом).
Примерами C1-C6 алкила являются метил, этил, пропил, изопропил, бутил, втор-бутил, трет-бутил, пентил, 2-метилбутил, изопентил, гексил, 3-метилпентил, 2-этилбутил.
Примером (C6-C12)арил(C1-C4)алкила являются бензил, моно- или полизамещенный C1-C6-алкилбензил, α- или β-фенилэтил, моно- или поли- C1-C4-алкил-замещенный α- или β-фенилэтил, моно- или поли- C1-C4алкил-замещенный α-, β- или γ-фенилпропил, α- или β-нафтилметил, моно- или поли- C1-C2-алкил-замещенный α- или  нафтилметил.
нафтилметил.
Примерами (C1-C2)алкокси(C1-C4)алкила являются метоксиметил, этоксиэтил, этоксиметил, пропоксиэтил, бутоксиэтил.
Примерами (C1-C4)алкил(C6-C12)арила являются толил, ксилил, этилфенил, изопропилфенил, трет-бутилфенил, метилнафтил.
Примерами (C6-C12)арил(C2-C4)ацила являются фенилацетил, нафтилацетил, 2-фенилпропионил, 3-фенилпропионил, 2-, 3- или 4-фенил бутирил, моно-, ди- или три-(C1-C4)алкил-замещенный фенилацетил.
Примерами C2-C4-ацила являются ацетил, пропионил, бутирил и их изомеры.
Примерами амино(C1-C4)алкила и амино(С2-С4)ацила являются C1-C4-алкил и C2-C4-ацил, где амино-заместители могут быть в любом положении углеродной цепи.
Примерами фармацевтически приемлемых солей являются в случае основного атома азота соли фармацевтически приемлемых кислот как неорганических, так и органических, таких как соляная кислота, серная кислота, уксусная кислота, в случае кислой группы, такой как -COOH, соли фармацевтически приемлемых оснований как неорганических, так и органических, таких как гидроксиды щелочных и щелочноземельных металлов, гидроксид аммония, амины.
Соединения формулы (I) могут находиться в форме фармацевтически приемлемых солей и/или N1-оксидов. Список предпочтительных групп соединений приведен ниже.
Первая группа представленных соединений включает в себя соединения формулы (I), где R3 является водородом.
Вторая группа представленных соединений включает в себя соединения формулы (I), где R3 является OR6 и R6 такой, как определено выше.
Третья группа представленных соединений включает в себя соединения формулы (I), где R1 является CN, R3 является водородом или OR6 и R6 такой, как определено выше.
Четвертая группа представленных соединений включает в себя соединения формулы (I), где R1 является CH(CN)-R4, где R4 является предпочтительно CN или COOR7, а R7 такой, как определено выше.
Пятая группа представленных соединений включает в себя соединения формулы (I), где R1 является CH(=NOH)NH2, R3 является OR6, как определено выше.
Шестая группа представленных соединений включает соединения формулы (I), где R1 является CH(=NH)NH2, R3 является OR6, как определено выше.
Седьмая группа представленных соединений включает в себя соединения формулы (I), где R1 является CH=C(CN)R4, где R4 является предпочтительно CN или COOR7, R7 такой, как определено выше, в частности R4 является CN, R2 и R3 являются водородом.
Восьмая группа представленных соединений включает в себя соединения формулы (I), где R1 является CH(CH2NO2)R5, R5 является OR8 в соответствии в данными выше определениями.
Девятая группа представленных соединений включает в себя соединения формулы (I), где R1 является CH=C(NO2)-R4, где R4 является H, R3 является OR6 в соответствии с данными выше определениями.
В частности, представленными соединениями формулы (I) являются те соединения, где R1 является CN или CH=C(CN)2 или -CH2CH(CN)-R4 и R2 и R3 являются водородом.
Соединения формулы (I) могут быть получены, начиная с камптотецин-7-метанола (II, R1 = CH2OH, R2 = H, R3 = H) или с 10-гидроксикамптотецин-7-метанола (II, R1 = CH2OH, R2 = H, R3 = H) или с камптотецин-7-альдегида (II, R1 = CHO, R2 = H, R3 = H), или с камптотецин-N-оксида, все соединения доступны, как описано Sawada et al., Chem. Pharm. Bull. 39, 2572 (1991).

Соединения формулы (I), где R1 = -CN готовят способом, включающим в себя окисление соединений формулы (II, R1 = -CH2OH) до соединений формулы (II, R1 = -CHO) с помощью известных способов окисления спиртов до альдегидов, таких, как окисления Moffatt или Swern, или с помощью иодозобензойной кислоты в диметилсульфоксиде (Frigerio et al., J. Org. Chem. 60, 7272-6 (1995)) или с помощью обработки кислотой, как описано у Sawada et al. (Chem. Pharm. Bull. 39, 2572 (1991)), а затем обработки этих альдегидов гидроксиламином для получения соответствующих оксимов, за чем следует нагревание оксима с муравьиной кислотой и формиатом натрия, или с помощью других известных способов превращения альдегидов в нитрилы.
Соединения формулы (I, R1 = -CN, R1 = -CH(CN)-R4) могут также быть получены путем взаимодействия N-оксидов камптотецина, например тех, что описаны Sawada et al. (Chem. Pharm. Bull. 39, 2572 (1991)), с цианидом калия или триметилсилилцианидом, или с малононитрилом или эфирами цианоуксусной кислоты соответственно (как описано в A. Albini and S. Pietra, Heterocyclic N-Oxides, CRC, 1991, p. 165), или с помощью обработки соединений формулы (II, R1 = -CONH2) при помощи известных способов дегидратации амидов до нитрилов, или с помощью других способов, пригодных для получения квинолин-4-карбонитрилов.
Аминогидроксиимины (I, R1 = -C(=NOH)-NH2) получают взаимодействием соответствующих нитрилов (I, R1 = -CN) с гидроксиламином (F. Eloy and R. Lenaers, Chem. Rev. 61, 157 (1961)). Аминогидроксиимины могут быть восстановлены до соответствующих амидинов (I, R1 = -C(=NH)-NH2) с помощью каталитической гидрогенизации, предпочтительно с катализатором Никель-Ренея (F. Eloy and R. Lenaers, Chem. Rev. 61, 166 (1961)). Те же амидины могут также быть получены из нитрилов (I, R1 = -CN) с помощью известных способов превращения нитрилов в амидины, таких, как взаимодействие с HCl и спиртом, за чем следует обработка аммиаком или солью аммония; или из амидов (II, R = -CONH2) с триэтилоксониумфторборатом (A.I. Meyers et al., Tetrahedron 39, 1991 (1983)).
Соединения формулы (I, R1 = -CH=C(CN)-R4) получают, например, взаимодействием альдегидов (II, R1 = -CHO) с малононитрилом или с эфирами малоновой или цианоуксусной кислоты в присутствии органических или неорганических оснований или без них, или взаимодействием альдегидов или кетонов (II, R1 = -CHO или -CO-алкил) с подходящими илидами или анионами фосфонатов в соответствии с реакциями Wittig или Wadsworth-Emmons. По желанию соединения формулы (I, R1 = -C=C(CN)-R4) могут быть гидрогенизированы в присутствии катализатора, такого как Pd, Pt или Ni с образованием соответствующих соединений формулы I (R1 = CH2CH(CN)R4).
Соединения формулы (I, R1 = -CH(CN)-R4), -CH(CH2NO2)-R5, где R5 является OH могут быть получены взаимодействием альдегидов (II, R1 = -CHO) с цианидом калия или натрия или триметилсилилцианидом и соответственно с нитрометаном в присутствии органического или неорганического основания.
Соединения формулы (I, R1 = -CH=CH(NO2)-R4) получают обработкой кислотой соединений, где R1 является -CH(CH2NO2)-R5.
По желанию соединения формулы (I, R1 = -CN) могут быть превращены с помощью известных подходящих методов в соединения формулы (I), где R1 является гетероциклическим кольцом, предпочтительно 2-(4,5-дигидрооксазолом) (J.F. Bower et al. , J. Chem. Soc. Perkin Trans. 1, 333 (1996)) или 5-тетразолом (Duncia et al., J. Org. Chem. 56, 2395 (1991)).
Соединения формулы (I, R1 = 1,2,4-оксадиазолин-3-ил-5-он) получают из соответствующих амидинов.
N-оксиды соединений формулы (I) получают в соответствии с известными способами окисления гетероароматического азота, предпочтительно с помощью окисления уксусной или трифторуксусной кислотой и перекисью водорода, или с помощью взаимодействия с органическими надкислотами (A. Albini and S. Pietra, Heterocyclic N-oxides, CRC, 1991).
Фармацевтически приемлемые соли соединений формулы (I) могут быть получены в соответствии с описанными в литературе способами.
Соединения, описанные в настоящем изобретении, проявляют сильную антипролиферативную активность и имеют физико-химические свойства, которые делают эти соединения подходящими для включения в фармацевтически приемлемые композиции.
Цитотоксическая активность соединений по настоящему изобретению была протестирована в клеточных системах клеток опухоли человека с использованием антипролиферативного теста как способа для оценки цитотоксического потенциала. Этот способ состоит в определении числа клеток, выживших в течение 72 ч после 1 ч экспозиции с цитотоксическим агентом. Цитотоксическая активность соединений по настоящему изобретению сравнивалась с таковой I) топотекана как контрольного агента среди ингибиторов ДНК топоизомераз I; II) доксорубицина, стандартного противоопухолевого агента, одного из наиболее эффективных среди таковых, используемых при клинической терапии опухолей. Результаты приведенные в табл. 1 показывают, что соединение формулы (I), описанное в примере 1 ниже (I, R1 = -CN, R2 = H, R3 = H), и соединение формулы (I), описанное в примере 4 ниже (I, R1 = CH=C(CN)-R4, R4 = CN, R2 = H, R3 = H), проявляют цитотоксическую активность, большую, чем активность контрольных соединений в системе немелкоклеточной карциномы легкого (не МККЛ) (H-460), которой присуща устойчивость к цитотоксичной терапии и только умеренная чувствительность к ингибиторам топоизомеразы I, несмотря на сверхэкспрессию целевого фермента.
Более того, соединение примера 1 проявляет значительную эффективность при обработке клеточной линии (H460/TPT), отобранной после продолжительной экспозиции с топотеканом и характеризующейся высокой степенью сильной устойчивости к топотекану. Так как линия H460 экспрессирует высокие уровни топоизомеразы I, улучшенная цитотоксичность соединения, приведенного в примере 1, при обработке этой линии опухолевых клеток указывает на улучшенную специфичность этого соединения по отношению к клеточной мишени. Эта интерпретация поддерживается пониженной эффективностью этих соединений на GBM клеточную линию, которая довольно устойчива к этим ингибиторам, благодаря низкой экспрессии топоизомеразы I.
Доклиническое изучение эффективности было разработано для оценки противоопухолевой активности соединений по настоящему изобретению по сравнению с топотеканом (первое поколение камптотецина уже в клинических испытаниях) как контрольным лекарством. Линия опухоли человека NCI-H460, немелкоклеточной карциномы легкого, была выбрана из-за высокой экспрессии топоизомеразы I, известной мишени лекарств на основе камптотецина. Эта модель опухоли относительно устойчива к обработке in vivo общепринятыми цитотоксическими агентами (например, доксорубицином, цисплатином). Клетки опухоли инъецировались внутрибрюшинно "голым" мышам (2,5·106 клеток/мышь) около 10 недель, и через 3 дня лекарства инъецировали в брюшную полость (10 мл/кг по весу) для осуществления прямого контакта лекарств с клетками опухоли. Как топотекан, так и соединение формулы (I) описанное в примере 1, подавалось q4d х 4 раза. Эта схема описана, как оптимальная для лекарств на основе камптотецина в других доклинических исследованиях. Мыши наблюдались ежедневно до момента смерти. Противоопухолевая активность этих лекарств была выражена как T/C%, то есть отношение между средним временем жизни мышей, обработанных лекарством (T), и временем жизни контрольных необработанных мышей (C) · 100. Обработанные мыши, умершие ранее первой контрольной мыши или умершие вскоре после обработки и имеющие пониженный вес тела, были признаны умершими от токсичности лекарства. Мыши, прожившие более 100 дней после инокуляции клеток опухоли, были признаны длительно живущими (ДЖ). (Второй эксперимент еще продолжался и ДЖ признаны прожившими более 70 дней). Результаты двух независимых экспериментов показаны в табл. 2.
Соединение примера 1 по настоящему изобретению, обозначенное как CPT83, было высокоэффективно в увеличении времени жизни мышей, имеющих опухоль внутри брюшины, причем значения T/C% выше 200 при всех исследованных дозах. Что касается токсичности лекарства, то только одна мышь умерла при дозе 14,4 мг/кг х 4 (суммарная накопленная доза: 49,6 мг/кг). Эффективность лекарства CPT83 превосходила эффективность топотекана в эксперименте 1 при условиях, при которых клетки опухоли вызывали замедленную смерть (медленно растущая опухоль). При использовании быстрорастущей опухоли (эксперимент 2) эффективность CPT83 была сравнима с эффективностью топотекана в терминах T/C%. Однако в обоих экспериментах более высокая доля длительно живущих (ДЖ, то есть вылеченных животных) была обнаружена в CPT83-обработанных группах. Это открытие отражает перспективный терапевтический профиль, имеющий отношение к улучшенному терапевтическому индексу. Потенциальное терапевтическое преимущество CPT83 также подчеркивается его хорошей активностью при обработке медленно растущей опухоли, рост которой клинически более показателен. В заключение на ксенографте опухоли NCI-H460 CPT83 имеет сравнимую активность и лучшую толерантность, чем топотекан.
Соединения по настоящему изобретению проявляют особенно выгодные свойства, которые могут быть суммированы в следующих пунктах.
1. Повышенная специфичность по отношению к клеточной мишени и, следовательно, по отношению к клеткам опухоли, экспрессирующим высокие уровни топоизомеразы I. Эта возможность поддерживается повышенной чувствительностью опухолевых клеток H460, которые известны как имеющие высокие уровни топоизомеразы I. Действительно, эта селективность теряется у клеточной линии (GMB), характеризующейся низким уровнем экспрессии мишени.
2. Эта активность, очевидно, меньше зависит от скорости пролиферации опухоли, чем таковая топотекана, как предполагается в экспериментах in vivo и по заметной активности против клеточной линии H460/TPT, характеризующейся очень медленной пролиферацией. Этот профиль активности может иметь клинические аспекты, так как медленный рост типичен для плотных опухолей человека.
3. Эта in vitro цитотоксическая действенность не связана с повышенной токсичностью in vivo, разрешая таким образом использование широкого ряда эффективных доз. Это согласуется с улучшенным терапевтическим индексом.
4. Соединения по настоящему изобретению, в частности CPT83, оказались активными при оральном применении. Интересно, что при применении орально CPT83 более активен, чем топотекан, введенный внутривенно (при применении оптимальной схемы лечения).
Поскольку затрагиваются промышленные аспекты этого изобретения, дополнительным объектом по настоящему изобретению являются фармацевтические композиции, содержащие эффективное количество по меньшей мере соединения формулы I в качестве активного ингредиента в смеси с носителями и эксципиентами.
Фармацевтические составы получают в соответствии с общепринятыми способами, хорошо известными из уровня техники, например, как описано в Remington's Pharmaceutical Sciences Handbook, Mack. Pub., NY, USA.
Примерами фармацевтических композиций являются соединения, инъецируемые композиции, такие как растворы, суспензии, эмульсии в водном или неводном растворителе; энтеральные композиции, такие как капсулы, таблетки, пилюли, сиропы, жидкие препараты для питья. Другие фармацевтические композиции, совместимые с соединениями формулы (I), такие как препараты с регулируемым высвобождением, включены в настоящее изобретение.
Дозировка активного ингредиента в фармацевтической композиции должна определяться квалифицированным специалистом в зависимости от активности и фармакокинетических характеристик активного ингредиента. Дозировка препарата должна решаться врачом на основе типа опухоли, подлежащей лечению, состояний пациента.
Соединения по настоящему изобретению могут быть также использованы в комбинированной терапии с другими противоопухолевыми лекарствами.
Следующие примеры дополнительно иллюстрируют данное изобретение.
Пример 1
20S-камптотецин-7-карбонитрил
1) 400 мг оксима камптотецин-7-альдегида (Sawada et al. Chem. Pharm. Bull. 39, 2572 (1991)), 102 мг формиата натрия и 15 мл 99% муравьиной кислоты кипятили с обратным холодильником в течение 6 ч. К этому раствору было добавлено 150 мл воды и 50 мл CH2Cl2, две фазы разделяли, и водная фаза была снова экстрагирована 4 раза. Органические экстракты были выпарены, и остаток хроматографировали на силикагеле MerckR с CH2Cl2-MeOH в соотношении 96:4. Нитрил (300 мг) был получен как желтое твердое вещество, tплавл. 263o. Масса (М/э %): 374 (16), 373 (98), 344 (36), 329 (48), 314 (55), 301 (53), 300 (53), 273 (100). 1H ЯМР (ДМСО-d6) 0.92 (CH3), 1.92 (CH2), 5.48, 5.51 (CH2-5), 5.56 (CH2-17), 6.62 (OH), 7.13 (CH-14), 8.02 (CH-11), 8.10 (CH-10), 8.30 (CH-9), 8.39 (CH-12).
2) 320 мг камптотецин-7-альдегида, 154 мг NH2OH·HCl, 578 мг формиата натрия и 20 мл муравьиной кислоты кипятили с обратным холодильником 3 ч, добавляли 60 мг NH2OH·HCl, и смесь кипятили с обратным холодильником 2 ч. Вода (90 мл) была добавлена, и смесь экстрагировалась с CH2Cl2. Соединение было выделено и очищено как описано выше.
3) 500 мг камптотецин-N-оксида кипятили с обратным холодильником с 0,86 мл триметилсилилцианида и 0,32 мл перекиси бензоила в 45 мл 1,1,2,2-тетрахлорэтана в течение 12 ч. Смесь была охлаждена и выпарена, а остаток хроматографирован на силикагеле MerckR с гексаном-этилацетатом в соотношении 4:6 как элюентом для получения камптотецин-7-карбонитрила.
Начиная с подходящих 10-замещенных камптотецинов следующие соединения были получены аналогично: 20S-10-гидроксикамптотецин-7-карбонитрил; 20S-10-ацетоксикамптотецин-7-карбонитрил; 20S-10-метоксикамптотецин-7-карбонитрил; 20S-10-метоксиметоксикамптотецин-7-карбонитрил; 20S-10-этоксикамптотецин-7-карбонитрил; 20S-10-бензилоксикамптотецин-7-карбонитрил; 20S-10-β-D-гликозилоксикамптотецин-7-карбонитрил; 20S-10-камптотецин-7-ил-малононитрил; этил 20S-камптотецин-7-ил-цианоацетат.
Пример 2
20S-камптотецин-7-карбамидоксим
Суспензия 60 мг камптотецин-7-карбонитрила, 40 мг гидроксиламин гидрохлорида и 0,2 мл триэтиламина в 5 мл абсолютного этанола кипятили с обратным холодильником в течение 8 ч, с добавлением дополнительных 40 мг NH2OH·HCl и 0,2 мл Et3N после 4 ч. Смесь была выпарена, отобрана водой, отфильтрована, и осадок был хроматографирован на силикагеле MerckR с CH2Cl2-MeOH в соотношении 9:1 для получения камптотецин-7-карбамидоксима.
Следующие соединения были получены аналогично: 20S-10-гидроксикамптотецин-7-карбамидоксим; 20S-10-ацетоксикамптотецин-7-карбамидоксим; 20S-10-метоксикамптотецин-7-карбамидоксим.
Пример 3
20S-7-амидинокамптотецин
100 мг 20S-камптотецин-7-карбамидоксима в 10 мл метанола были гидрогенизированы в присутствии 1 г катализатора Никель-Ренея под давлением 50 атм и при температуре 70oC в течение 5 ч. Фильтрование катализатора и выпаривание дали 20S-7-амидинокамптотецин в виде стеклообразного твердого вещества.
Следующие соединения были получены аналогично: 20S-10-гидрокси-7-амидинокамптотецин; 20S-10-ацетокси-7-амидинокамптотецин; 20S-10-метокси-7-амидинокамптотецин.
Пример 4
20S-7-(2,2-дицианоэтенил)камптотецин
60 мг камптотецин-7-альдегида кипятили с обратным холодильником 4 ч с 3 мл малононитрила в 8 мл 1,1,2,2-тетрахлорэтана и в присутствии 20 мг LiBr. Охлаждение, фильтрование и хроматография на силикагеле с этилацетатом дали 20S-7-(2,2-дицианоэтенил)-камптотецин как стекловидное твердое вещество. Масса (М/э) 424, 380. 1H ЯМР (ДМСО-d6) 0.85 (CH3), 1.88 (CH2), 5.38 (CH2-5), 5.45 (CH2-17), 6.56 (OH), 7.36 (CH-14), 7.82 (CH-11), 7.96 (CH-10), 8.18 (CH-9), 8.26 (CH-12), 9.30 (CH=).
Следующие соединения были получены аналогично: 20S-7-(2,2-дицианоэтенил)-10-гидроксикамптотецин; 20S-7-(2,2-дицианоэтенил)-10-метоксикамптотецин; 20S-7-(2,2-дицианоэтенил)-10-этоксикамптотецин; 20S-7-((2-циано-2-этоксикарбонил)этенил)камптотецин.
Пример 5
20S-7-(2-нитро-1-гидроксиэтил)-камптотецин
150 мг камптотецина, 0,05 мл нитрометана, 0,01 мл триэтиламина в 3 мл изопропанола кипятили с обратным холодильником 10 ч. Выпаривание, обработка разбавл. HCl и CH2Cl2 и хроматография экстракта с 4% метанолом в CH2Cl2 дали 20S-7-(2-нитро-1-гидроксиэтил)-камптотецин.
1H ЯМР (ДМСО-d6) 0.80 (CH3), 1.84 (CH2), 4.90-5.05 (CH2-7), 5.46 (CH2-5), 5.54 (CH2-17), 6.33 (CHOH), 6.56 (OH-16), 6.91 (CHOH), 7.33 (CH-14), 7.70 (CH-11), 7.82 (CH-10), 8.17 (CH-9), 8.20 (CH-12).
Следующие соединения были получены аналогично: 20S-7-(2-нитро-1-гидроксиэтил)-10-метоксикамптотецин; 20S-7-(2-нитро-1-гидроксиэтил)-10-этоксикамптотецин.
Пример 6
20S-7-(2-нитроэтенил)-камптотецин
50 мг 20S-7-(2-нитро-1-гидроксиэтил)-камптотецина в 5 мл тетрагидрофурана кипятили с обратным холодильником 1-2 ч с 20 мг п-толуолсульфокислоты или с 0,03 мл трифтороуксусной кислоты для получения 20S-7-(2-нитроэтенил)-камптотецина как желтого стекловидного твердого вещества.
Следующие соединения были получены аналогично: 20S-7-(2-нитроэтинил)-10-метоксикамптотецин; 20S-7-(2-нитроэтинил)-10-этоксикамптотецин.


Описываются новые производные камптотецина общей формулы (I), где R1 является -CH, -CH(CN)-R4, -CH= C(CN)-R4, -C(=NOH)-NH2, -C(=NH)-NH2, -CH= C(NO2)-R4, -CH(CN)-R5, -CH(CH2NO2)-R5, 5-тетразолилом, 2-(4,5-дигидрооксазолилом), 1, 2, 4-оксадиазолин -3-ил-5-оном; R2 - водород; R3 - водород, OR6; R4 - водород, C1 - C6 линейный или разветвленный алкил, CN, COOR7; R5 - водород, OR8; R6 является водородом, C1 - C6 линейным или разветвленным алкилом, (C6 - C12) арил (C1 - C4) алкилом, (C1 - C4)алкокси(C1 - C4)алкилом, (C1 - C4)алкил (C6 - C12)арилом, (C6 - C12)арил(C2 - C4)ацилом, (C2 - C4)ацилом, амино (C1 - C4)алкилом, амино(C2 - C4)ацилом, гликозилом; R7 является водородом, C1 - C6 линейным или разветвленным алкилом, (C6 - C12) арил (C1 - C4)алкилом, (C1 - C4) алкокси(C1 - C4)алкилом, (C1 - C4)алкил (C6 - C12)арилом; R8 имеет те же значения, что и R6, независимо от последнего, их N1-оксиды, их изомеры, диастереоизомеры, энантиомеры и их смеси, а также их активные метоболиты. Соединения проявляют противоопухолевую активность и подходящие физико-химические свойства, которые допускают их технологию приготовления в подходящие фармацевтические композиции. Описываются также способ их получения и фармацевтическая композиция. 9 с. и 7 з. п. ф-лы, 2 табл.


где R1 является -CN, -CH(CN)-R4, -CH=C(CN)-R4, -CH2-CH(CN)-R4, -C(= NOH)-NH2, -C(= NH)-NH2, -CH=C(NO2)-R4, -CH(CN)-R5, -CH(CH2NO2)-R5, 5-тетразолилом, 2-(4,5-дигидрооксазолилом), 1,2,4-оксадиазолин-3-ил-5-оном;
R2 - водород;
R3 - водород или OR6;
R4 - водород, C1 - C6 линейный или разветвленный алкил, CN, COOR7;
R5 - водород, OR8;
R6 - водород, C1 - C6 линейный или разветвленный алкил, (C6 - C12)арил(C1 - C4)алкил, (C1 - C4)алкокси(C1 - C4)алкил, (C1 - C4)алкил(C6 - C12)арил, (C6 - C12)арил(C2 - C4)ацил, (C2 - C4)ацил, амино(C1 - C4)алкил, амино(C2 - C4)ацилом, гликозил;
R7 - водород, C1 - C6 линейный или разветвленный алкил, (C6 - C12)арил(C1 - C4)алкил, (C1 - C4)алкокси(C1 - C4)алкил, (C1 - C4)алкил(C6 - C12)арил;
R8 имеет те же значения, что и R6, независимо от последнего;
их N1-оксиды, их изомеры, диастереоизомеры, энантиомеры и их смеси, а также их активные метаболиты.
где R1 - CHO;
R3 имеет те же значения, что и в формуле (I),
в соответствующий оксим, затем обрабатывают HCOOH/HCOONa для получения соответствующих соединений формулы (I), где R1 - -CN; и при желании б) обрабатывают соединение формулы (I), полученное на стадии (а), гидроксиламином для получения соответствующих соединений формулы (I), где R1 - -C(=NOH)-NH2; и при желании в) каталитически гидрогенизируют соединение формулы (I), полученное на стадии (б), для получения соответствующего соединения формулы (I), где R1 - -C(=NH)-NH2; или желании трансформируют соединение, полученное на любой из стадий (а), (б) или (в), в соответствующий N1-оксид или фармацевтически приемлемую соль.
где R1 - -CN;
R2 и R3 такие, как определено выше,
как промежуточное соединение на стадии (б) в способе по п.8.
где R1 - -CN;
R2 и R3 такие, как определено выше,
как промежуточное соединение в способе получения соединений по п.1, где R1 выбран из группы, состоящей из -C(=NOH)-NH2, -C(=NH)-NH2, 2-(4,5-дигидроксазол-ила), 5-тетразолила.
где R1 - CHO;
R2 - водород;
R3 имеет те же значения, что в формуле (I),
подвергают взаимодействию с соединением R4-CH2-CN, где R4 такой, как определено выше,
для получения соединения формулы (I), где R1 - -CH=C(CN)-R4, и при желании гидрогенизируют до соединения формулы (I), где R1 - -CH2-CH(CN)-R4, или возможно осуществляют конверсию в фармацевтически приемлемую соль.
где R1 - CHO;
R2 - водород;
R3 имеет те же значения, что и в формуле (I),
подвергают взаимодействию с цианидом калия, натрия или триметилсилила или нитрометаном в присутствии неорганического или органического основания и возможно осуществляют конверсию в фармацевтически приемлемую соль.
| Колосниковая решетка с переменным живым сечением | 1950 |
|
SU88642A1 |
| US 5459269 A, 17.10.1995 | |||
| RU 95112485 A1, 20.04.1997. | |||

