Настоящее изобретение относится к лечению амилоидоза, к новым соединениям, пригодным для такого лечения, к способам их получения, а также к содержащим их фармацевтическим композициям.
Связь между амилоидозом, смертью клеток и потерей функции тканей, по-видимому, имеет место при заболеваниях самого различного типа, включая нейродегенеративные заболевания. В этой связи предупреждение образования амилоида и/или индукция амилоидного разрушения может рассматриваться как важный терапевтический инструмент для всех патологических нарушений, связанных с амилоидозом, включая амилоидоз с L-цепями амилоида (AL амилоидоз) и нейродегенеративные заболевания типа болезни Альцгеймера.
Более конкретно, настоящее изобретение относится к применению антрациклинона формулы А при получении лекарственного препарата для лечения амилоидоза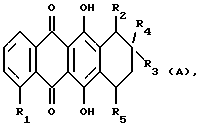
где R1 обозначает:
водород или гидрокси;
группу формулы OR6, в которой R6 представляет собой C1-C6 алкил, C5-C6 циклоалкил или CH2Ph, с фенильным (Ph) кольцом, необязательно замещенным 1, 2 или 3 заместителями, выбранными из F, Cl, Br, C1-C6 алкила, C1-C6-алкокси и CF3; или
- группу формулы OSO2R7, в которой R7 представляет собой C1-C6 алкил или Ph, необязательно замещенный 1, 2 или 3 заместителями, выбранными из галогена, такого, как
F, Cl или Br, или C1-C6 алкила;
R2 обозначает водород, гидрокси, OR6, COOH или COOR6, где R6 указан выше;
R3 обозначает водород, гидрокси или OR6, определенный выше;
R4 обозначает водород, метил или группу формулы XCH2R8, где X обозначает CO, CH2, CHOH, или группу формулы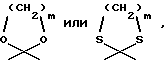
в которой m обозначает 2 или 3, а R8 представляет собой:
- водород или гидрокси;
- группу формулы NR9R10, в которой:
- R9 и R10, каждый, независимо выбран из:
(а) водорода,
(б) C1-C6 алкильный или C2-C6 алкенильной группы, необязательно замещенных гидрокси, CN, COR11, COOR11, CONR11R12, O(CH2)nNR11R12 (n равно от 2 до 4) или NR11R12, где R11 и R12 каждый, независимо, выбран из водорода, C1-C12 алкильной или C1-C12 алкенильной группы или фенила, необязательно замещенного одним или более, например 1, 2 или 3, заместителями, выбранными из C1-C6 алкила, C1-C6 алкокси, F, Br, Cl, CF3, OH, NH2 или CN,
(в) C3-C6 циклоалкила, необязательно замещенного COR11, COOR11 или OH, где R11 указан выше,
(г) фенил (C1-C4 алкила или C2-C4 алкенила), необязательно замещенного в фенильном кольце одним или более, например 1, 2 или 3, заместителями, выбранными из C1-C6 алкила, C1-C6 алкокси, F, Br, Cl, CF3, OH, NH2 или CN или
(д) COR11, COOR11, CONR11R12, COCH2NR11R12, CONR11COOR12 или SO2R12, где R11 и R12 указаны выше, или
-R9 и R10 вместе с атомом азота, к которому они присоединены, образуют:
(е) морфолиновое кольцо, необязательно замещенное C1-C4 алкилом или C1-C4 алкокси,
(ж) пиперазиновое кольцо, необязательно замещенное C1-C6 алкилом, C2-C6 алкенилом или фенилом, необязательно замещенным одним или более, например 1, 2 или 3, заместителями, выбранными из C1-C6 алкокси, F, Br, Cl, CF3, OH, NH2 или CN, или
(з) пирролидиновое или пиперидиновое, или тетрагидропиридиновое кольцо, необязательно замещенное OH, NH2, COOH, COOR11 или CONR11R12, где R11 и R12 указаны выше, C1-C6 алкилом, C2-C6 алкенилом или фенилом, необязательно замещенным одним или более, например 1, 2 или 3, заместителями, выбранными из C1-C6 алкила, C1-C6 алкокси, F, Br, Cl, CF3, OH, NH2 или CN;
- группу формулы OR6 или SR6, в которой R6 указан выше;
- группу формулы O-Ph, где фенильное кольцо (Ph) необязательно замещено нитро, амино или NR9R10, как указано выше;
- группу формулы В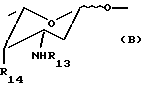
где R13 обозначает водород, COR11, где R11 указан выше, или пептидильный остаток, и R14 обозначает галоген или группу формулы OSO2R7, где R7 указан выше; или
- группу формулы C или D: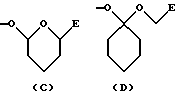
где Е обозначает группу формулы COOR11 или CONR9R10, в которых R9, R10 и R11 указаны выше; и
R5 обозначает водород, гидрокси, группу формулы OR6 или NR9R10, где R6, R9 и R10 указаны выше, или группу формулы F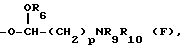
где R6, R9 и R10 указаны выше, и p равно от 1 до 6; и его фармацевтически приемлемых солей.
Другим аспектом настоящего изобретения являются новые антрациклиноны формулы А, указанной выше, при следующих условиях:
- R5 не обозначает NR9R10, где R9 и R10 указаны в пунктах от а) до в) или от д) до з), когда R1 является H, OH или OCH3, R2 представляет H, R3 представляет собой OH, а R4 является группой формулы XCH2OH или XCH3, где X указан выше;
- R5 не является H или OH, когда R1 представляет H, OH или OCH3, R2 представляет H, OH, COOCH3, а R4 обозначает группу формулы XCH3 или XCH2OH, где X указан выше;
- R4 не является COCH2OR'6, где R'6 обозначает фенил, бензил, C1-C6 алкил или C5-C6 циклоалкил, когда R1 представляет H или OH, R5 и R4 представляют OH, и R2 обозначает H;
- соединение формулы А не является ни одним из приведенных ниже производных:
14-(N-морфолино)-дауномицинон;
14-(N-пиперидино)-дауномицинон;
14-ацетамидодауномицинон;
14-ацетамидо-4-деметоксидауномицинон;
14-(N-морфолино)-карминомицинон;
14-(N-метил-N-пиперазино)-дауномицинон;
14-(N-морфолино)карминомицинон;
14-(N-метил-N-пиперазин) карминомицинон.
Каждая алкильная, алкокси или алкенильная группа могут быть как прямоцепочечной, так и разветвленной группой.
C1-C12 алкильная группа представляет собой предпочтительно C1-C6 алкильную, более предпочтительно C1-C4 алкильную группу. C1-C6 алкильная группа представляет собой предпочтительно C1-C4 алкильную группу. C1-C6 алкильная группа представляет собой предпочтительно метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, втор-бутил или н-пентил. C1-C4 алкильная группа представляет собой предпочтительно метил, этил, н-пропил, изопропил, н-бутил, трет-бутил или втор-бутил.
C3-C6 алкильная группа представляет собой предпочтительно C5-6 циклоалкильную группу. C5-6 циклоалкильная группа представляет собой предпочтительно циклопентил или циклогексил.
C2-C12 алкенильная группа представляет собой предпочтительно C2-C6 алкенильную группу и более предпочтительно C2-C4 алкенильную группу. C2-C6 алкенильная группа представляет собой предпочтительно C2-C4 алкенильную группу. Предпочтительными алкенильными группами являются этенил и пропенил.
Пептидный остаток может включать до 6, например от 1 до 4, аминокислотных остатков. Подходящие пептидные остатки выбраны из Gly, Ala, Phe, Leu, Gly-Phe, Leu-Gly, Val- Ala, Phe-Ala, Leu-Phe, Phe-Leu-Gly, Phe-Phe-Leu, Leu-Leu-Gly, Phe- Tyr-Ala, Phe-Gly-Phe, Phe-Leu-Gly-Phe, Gly-Phe-Leu-Gly, Gly-Phe-Leu-Gly.
В настоящем изобретении R1 обозначает предпочтительно водород или метокси. R2 обозначает предпочтительно водород. R3 относится предпочтительно к гидроксигруппе. R4 является предпочтительно группой формулы XCH2R8, в которой X обозначает CO, CH2 или группу формулы: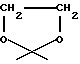
и R8 обозначает водород, группу формулы NR9R10, группу формулы O-Ph, где Ph кольцо необязательно замещено NR9R10, группу формулы В или группу формулы C, где R9 и R10, каждый, независимо, выбран из:
(а') водорода;
(б') C1-C4 алкила, необязательно замещенного O(CH2)n, NR11R12 или NR11R12, где n, R11 и R12 указаны выше,
(г') бензила, необязательно замещенного по фенильному кольцу одним или более, например 1, 2 или 3? заместителями, выбранными из C1-C4 алкила, C1-C4 алкокси, F, Br, Cl, CF3, OH, NH2 или CN, или
(д') COCF3 или COCH2NR11R12, где R11 и R12 указаны выше,
или R9 и R10 вместе с атомом азота, к которому они прикреплены, образуют:
(е') морфолиновое кольцо,
(ж') пиперазиновое кольцо, необязательно замещенное C1-C4 алкилом, или
(з') пирролидиновое, или пиперидиновое кольцо, или тетрагидропиридиновое кольцо,
R13 в группе формулы В обозначает водород, R14 в группе формулы В представляет собой I или OSO2(C1-C4 алкил) и Е в группе формулы C обозначает группу формулы CONR'9R'10, где R'9 и R'10 вместе с атомом азота, к которому они присоединены, образуют пиперазиновое кольцо, необязательно замещенное C1-C4 алкилом. Более предпочтительно R4 обозначает группу формулы: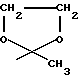
или группу формулы XCH2R8, где X обозначает CO или CH2, и R8 обозначает водород, группу формулы NR9R10, группу формулы O-Ph, где Ph кольцо необязательно замещено NH2 или NHCOCH2(C1-C4 алкилом)2, группа формулы В или группа формулы C, где R9 и R10, каждый, независимо выбран из:
(а'') водорода,
(б'') метильной или этильной групп, необязательно замещенных O(CH2)nNH2 или NH2, где n указано выше,
(г'') бензила, необязательно замещенного в фенильном кольце 1, 2 или 3 заместителями, выбранными из C1-C4 алкила и C1-C4 алкокси, или
(д'') COCF3 или COCH2N(C1-C4 алкила)2,
или R9 и R10 вместе с атомом азота, к которому они присоединены, образуют:
(е'') морфолиновое кольцо,
(ж'') пиперазиновое кольцо, необязательно замещенное C1-C4 алкилом, или
(з'') пирролидиновое, пиперидиновое или 1, 2, 3, 6-тетрагидропиридиновое кольцо,
R13 в группе формулы В обозначает водород, R14 в группе формулы В обозначает Т или OSO2(C1-C4 алкил), и Е в группе формулы C обозначает группу формулы CONR'9R'10, где R'9 и R'10 вместе с атомом азота, к которому они присоединены, образуют пиперазиновое кольцо, необязательно замещенное C1-C4 алкилом.
R5 представляет собой предпочтительно водород, гидрокси или группу формулы NR9R10, как указано выше.
Нaстоящее изобретение относится к солям тех соединений формулы А, которые имеют солеобразующие группы, особенно к солям соединений, имеющих карбоновую группу, основную группу (например, аминогруппу); соли являются особенно физиологически переносимыми солями, например соли щелочных и щелочноземельных металлов, например соли натрия, калия, лития, кальция и магния, соли аммония, соли с соответствующим органическим амином или аминокислотой, (например, соли аргенина, прокаина), и солями прибавления, образованными с соответствующими органическими или неорганическими кислотами, например соляной кислоты, серной кислоты, карбоновой кислоты и органических сульфоновых кислот (например, уксусной, трифторуксусной, п-толуолсульфоновой кислотой).
Нaстоящее изобретение охватывает все возможные стереоизомеры, а также рацемические или оптические активные смеси.
Предпочтительно R3 находится в альфа-конфигурации, т.е. расположен ниже плоскости кольца.
Конкретные примеры предпочтительных соединений для использования по настоящему изобретению перечислены ниже:
А1: 14-(N-морфолино)дауномицинон
R1=OCH3, R2=H, R3=R5=OH, 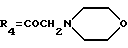
A2: 14-(N-пиперидино)дауномицинон
R1=OCH3, R2=H, R3=R5=OH, 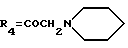
А3: 14-(N-пирролидино)дауномицинон
R1=OCH3, R2=H, R3=R5=OH, 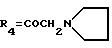
А4: 14-[N-(N'-метил)пиперазино]дауномицинон
R1=OCH3, R2=H, R3=R5=OH, 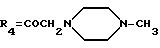
A5: 14-(3',4'-диметоксибензиламино)]дауномицинон
R1=OCH3, R2=H, R3=R5=OH, R4= COCH2NHCH2[C6H3(OCH3)2]
A6: 14-аминоэтилоксиэтиламинодауномицинон
R1=OCH3, R2=H, R3=R5=OH, R4=COCH2NH(CH2)2O(CH2)2NH2
A7: 14-аминоэтиламинодауномицинон
R1=OCH3, R2=H, R3=R5=OH, R4=COCH2NH(CH2)2NH2
A8: 14-(N-аминоэтил-N-трифторацетиламино)дауномицинон
R1=OCH3, R2=H, R3=R5=OH, R4=COCH2N(COCF3)(CH2)2NH2
A9: 14-(N-аминоэтилоксиэтил-N-трифторацетиламино)дауномицинон
R1=OCH3, R2=H, R4=COCH2N(COCF3)(CH2)2O(CH2)2NH2, R3=R5=OH
А10: 4-деметокси-14-(N-морфолино)дауномицинон
R1=R2=H, R3=R5=OH, 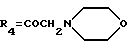
A11: 4-деметокси-14-(N-пиперидино)дауномицинон
R1=R2=H, R3=R5=OH, 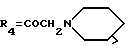
А12: 4-деметокси-14-(N-пирролидино)дауномицинон
R1=R2=H, R3=R5=OH, 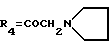
А13: 4-деметокси-14-N-[(N'-метил)пиперазино]-дауномицинон
R1=R2=H, R3=R5=OH, 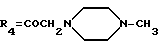
А14: 4-деметокси-14-(3',4'-диметоксибензиламино)дауномицинон
R1=R2=H, R3=R5-OH, R4=COCH2NHCH2[C6H3(OCH3)2]
А15: 4-деметокси-14-аминоэтилоксиэтиламинодауномицинон
R1=R2=H, R3=R5-OH, R4=COCH2NH(CH2)2O(CH2)2NH2
A16: 4-деметокси-14-(N-аминоэтилоксиэтил-N-трифторацетиламино) дауномицинон
R1=R2=H, R3=R5-OH, R4=COCH2N(COCF3)(CH2)2O(CH2)2NH2
А17: 7-дезокси-14-(N-морфолино)дауномицинон
R1=OCH3, R2=R5=H, R3=OH, 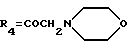
А18: 7-дезокси-14-(N-пиперидино)дауномицинон
R1=OCH3, R2=R5=H, R3=OH, 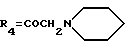
А19: 7-дезокси-14-(N-пирролидино)дауномицинон
R1=OCH3, R2=R5=H, R3=OH, 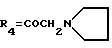
А20: 7-дезокси-14-[N-(N'-метил)-пиперазино]-дауномицинон
R1=OCH3, R2=R5=H, R3=OH, 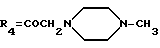
А21: 7-дезокси-14-(3',4'-диметоксибензиламино)дауномицинон
R1=OCH3, R2=R5=H, R3=OH, R4=COCH2NHCH2[C6H3(OCH3)2]
А22: 7-дезокси-14-аминоэтилоксиэтиламино)дауномицинон
R1=OCH3, R2=R5=H, R3=OH, R4=COCH2NH(CH2)2O(CH2)2NH2
А23: 7-дезокси-14-(N-аминоэтилоксиэтил-N-трифторацетиламино) дауномицинон
R1=OCH3, R2=R5=H, R3=OH, R4=COCH2N(COCF3)(CH2)2 O(CH2)2NH2
А24: 4-деметокси-7-дезокси-14-(N-морфолино)-дауномицинон
R1=R2=R5=H, R3=OH, 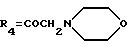
А25: 4-деметокси-7-дезокси-14-(N-пиперидино)-дауномицинон
R1=R2=R5=H, R3=OH, 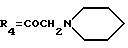
А26: 4-деметокси-7-дезокси-14-(N-пирролидино)-дауномицинон
R1=R2=R5=H, R3=OH, 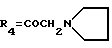
А27: 4-деметокси-7-дезокси-14-[N-(N'-метил)-пиперазино]дауномицинон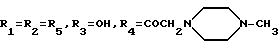
А28: 4-деметокси-7-дезокси-14-(3',4'-диметоксибензиламино) дауномицинон
R1=R2=R5=H, R3=OH, R4=COCH2NHCH2[C6H3(OCH3)2]
А29: 4-деметокси-7-дезокси-14-аминоэтилоксиэтиламино)дауномицинон
R1=R2=R5=H, R3=OH, R4=COCH2NH(CH2)2O(CH2)2NH2
А30: 7-дезокси-7-(N-морфолино)дауномицинон
R1=OCH3, R2=H, R3=OH, R4=COCH3, 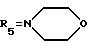
А31: 7-дезокси-7-[бис(2'-гидроксиэтил)]аминодауномицинон
R1=OCH3, R2=H, R3=OH, R4=COCH3, R5=N(CH2CH2OH)2
A32: 7-дезокси-7-(3',4'-диметоксибензиламино)]-13-дезоксо-13-этилендиоксидауномицинон
R1=OCH3, R2=H, R3=OH, R4=C(OCH2CH2O)CH3, R5=NHCH2C6H3(OCH3)2
А33: 7-дезокси-7-бензиламино-13-дезоксо-13-этилендиокси-дауномицинон
R1=OCH3, R2=H, R3=OH, R4=C(OCH2CH2O)CH3, R5=NHCH2C6H5
А34: 7-дезокси-7-(2'-гидроксиэтиламино)-13-дезоксо-13- этилендиоксидауномицинон
R1=OCH3, R2=H, R3=OH, R4=C(OCH2CH2O)CH3, R5=NHCH2CH2OH
А35: 4-деметокси-7-дезокси-7-(3',4'-диметоксибензиламино)-13-дезоксо-13-этилендиоксидауномицинон
R1=R2=H, R3=OH, R4=C(OCH2CH2O)CH3, R5=NHCH2C6H3(OCH3)2
А36: 7-дезокси-7-(3',4'-диметоксибензиламино)дауномицинон
R1=OCH3, R2=H, R3=OH, R4=COCH3, R5=NHCH2C6H3 (OCH3)2
А37: 7-дезокси-7-бензиламинодауномицинон
R1=OCH3, R2=H, R3=OH, R4=COCH3, R5=NHCH2C6H5
А38: 7-дезокси-7-(2'-гидроксиэтиламино)дауномицинон
R1=OCH3, R2=H, R3=OH, R4=COCH3, R5=NHCH2CH2OH
А39: 4-деметокси-7-дезокси-7-(3',4'-диметоксибензиламино)дауномицинон
R1=R2=H, R3=OH, R4=COCH3, R5=NHCH2C6H3(OCH3)2
A40: 7-дезокси-7-амино-13-дезоксо-13-этилендиоксидауномицинон
R1=OCH3, R2=H, R3=OH, R4=C(OCH2CH2O)CH3, R5=NH2
А41: 4-деметокси-7-дезокси-7-амино-13-дезоксо-13-этилен-диоксидауномицинон
R1=R2=H, R3=OH, R4=C(OCH2CH2O)CH3, R5=NH2
A42: 7-дезокси-7-аминодауномицинон
R1=OCH3, R2=H, R3=OH, R4=COCH3, R5=NH2
А43: 4-деметокси-7-дезокси-7-аминодауномицинон
R1=R2=H, R3=OH, R4=COCH3, R5=NH2
А44: 13-дезоксо-14-(N-морфолино)дауномицинон
R1=OCH3, R2=H, R3=R5=OH, 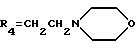 R4=COCH3, R5=NH2
R4=COCH3, R5=NH2
А45: 4-деметокси-13-дезоксо-14-(N-морфолино)-дауномицинон
R1=R2=H, R3=R5=OH, 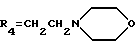
А46: 13-дезоксо-14-аминоэтилоксиэтиламинодауномицинон
R1=OCH3, R2=H, R3=R5=OH, R4=CH2CH2NH(CH2)2O(CH2)2 NH2
А47: 7-дезокси-14-O-(3'-амино-4'-метансульфонил)-2', 3', 4',6'-тетрадезокси-L-ликсогексопиранозил)дауномицинон
R1= OCH3, R2= R5=H, R3=OH, R4=COCH2R8, где R8 представляет собой группу формулы В, в которой R13=H, а R14=OSO2CH3
А48: 7-дезокси-14-O-(3'-амино-4'йод-2', 3', 4',6'-тетра-дезокси-L- ликсогексопиранозил)дауномицинон
R1= OCH3, R2= R5=H, R3=OH, R4=COCH2R8, где R8 представляет собой группу формулы В, в которой R13=H, а R14=I
А49: 7-дезокси-14-O-[2'-(1''-пиперазинил)-карбонилтетрагидропиран- 6'-ил]дауномицинон
R1=OCH3, R2=R5=H, R3=OH, 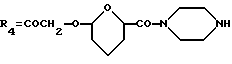
А50: 14-(п-аминофенилокси)дауномицинон
R1=OCH3, R2=H, R3=R5=OH, R4=COCH2O-C6H4(pNH2)
А51: 14-[п-(диметиламинометилкарбониламино)фенилокси] дауномицинон
R1=OCH3, R2=H3, R3=R5=OH, R4=COCH2O-C6H4[pNHCOCH2N (CH3)2]
A52: 4-деметокси-14-(п-аминофенилокси)дауномицинон
R1=R2=H, R3=R5=OH, R4=COCH2O-C6H4(p-NH2)
А53: 4-деметокси-14-[п-(диметиламинометилкарбониламино)фенилокси] дауномицинон
R1=R2=H, R3=R5=OH, R4= COCH2O-C6H4[p-NHCOCH2N(CH3)2]
А54: 7-дезокси-14-(п-аминофенилокси)дауномицинон
R1=OCH3, R2=R5=H, R3=R5=OH, R4=COCH2O-C6H4(p-NH2)
А55: 7-дезокси-14-[п-(диметиламинометилкарбониламино)фенилокси] дауномицинон
R1=OCH3, R2=R5=H, R3=OH, R4= COCH2O-C6H4[p-NHCOCH2N(CH3)2]
А56: 7-дезокси-4-деметокси-14-(п-аминофенилокси)дауномицинон
R1=R2=R5=H, R3=OH, R4=COCH2O-C6H4(p-NH2)
А57: 7-дезокси-4-деметокси-14[п-(диметиламинометилкарбониламинометил) фенилокси]дауномицинон
R1=R2=R5=H, R3=OH, R4= COCH2O-C6H4[p-NHCOCH2N(CH3)2]
А58: 14-[N-диэтиламино]дауномицинон
R1=OCH3, R2=H, R3=R5=OH, R4=COCH2N(C2H5)2
А59: 13-дигидро-14-(N-морфолино)дауномицинон
R1=OCH3, R2=H, R3=R5=OH, R4=CHOHCH2N(CH2)2O
А60: 7-дезокси-13-дигидро-14-(N-морфолино)дауномицинон
R1=OCH3, R2=R5=H, R3=OH, R4=CHOHCH2N(CH2)2O
А61: 4-деметокси-7-дезокси-10-гидрокси-14-(N-морфолино)- дауномицинон
R1=R5=H, R2=OH, R4=COCH2N(CH2)2O
А62: 4-деметокси-4-гидрокси-7-дезокси-7-N-морфолино) дауномицинон
R1=OH, R2=H, R3=OH, R4=COCH3, R5=N(CH2)2O
А63: 4-деметокси-7,9-дидезокси-14-(N-морфолино)дауномицинон
R1=R2=R3=R5=H, R4=COCH2N(CH2)2O
А64: 4-деметокси, 4-гидрокси, 14-(N-морфолино)дауномицинон
R1=R3=R5=OH, R2=H, R4=COCH2N(CH2)2O
Дополнительные примеры приведены в конце текста описания.
Соединения формулы А могут быть получены, в зависимости от природы заместителей, исходя из известных антрациклинонов, путем соответствующих химических модификаций.
Способы получения соединений формулы А и их фармацевтически приемлемых солей следующие:
(i) Предпочтительный способ получения соединения формулы А, где R1 обозначает OR6, где R6 указан выше, R2 обозначает водород или COOCH3, R3 обозначает OH, R4 обозначает C1 или C2 алкил или COCH3, и R5 обозначает водород, OH или OCOOC2H5, или его фармацевтически приемлемой соли включает:
(1) защиту 6-, 11- и, если имеется, 7-гидрокси групп соединения формулы G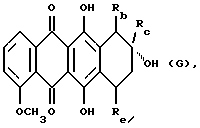
где Rb обозначает водород или COOCH3, Rc обозначает C1 или C2 алкил или COCH3 и Re обозначает водород или гидрокси, с получением производного формулы G1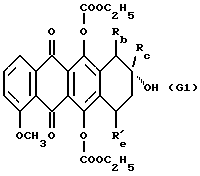
где Rb и Rc определены выше и R'е обозначает водород или группу OCOOC2H5;
(2) деметилирование такого производного формулы G1 и взаимодействие полученного 4-гидрокси соединения формулы G2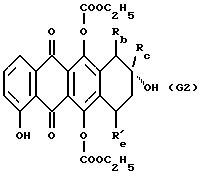
где Rb, Rc и R'е указаны выше, с соответствующим галогенпроизводным формулы R6Hal, в котором R6 указан выше, и Hal обозначает галоген, предпочтительно йод;
(3) снятие защиты 6- и 11-фенольных гидроксигрупп полученного 4-O-алкильного производного, что дает соединение формулы G3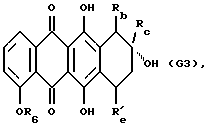
где R6, Rb, Rc и R'е указаны выше, и, если желательно, когда R'е обозначает OCOOC2H5, снятие защиты у 7-гидрокси группы соединения G3; и
(4) при желании, преобразование полученного указанного соединения формулы А в его фармацевтически приемлемую соль.
(ii) В другом примере предпочтительный способ получения соединения формулы А, где R1 обозначает группу OSO2R7, указанную выше, R2 представляет собой гидрокси или COOCH3, R3, обозначает OH, R4 является C1 или C2 алкилом или COCH3, и R5 обозначает водород или гидроксигруппу, или его фармацевтически приемлемой соли включает обработку антрациклинона формулы H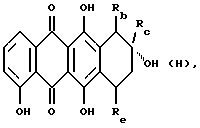
где Rb обозначает гидроксигруппу или COOCH3, Rc является C1 или C2 алкилом или COCH3 и Rе обозначает водород или гидрокси, соответствующие галогенпроизводным формулы HalSO2R7 (Hal обозначает галоген, предпочтительно атом хлора); и, при желании, преобразование полученного указанного соединения формулы А в его фармацевтически приемлемую соль.
(iii) В другом примере предпочтительный способ получения соединений формулы А, где R3 является OH, R4 обозначает COCH3 и R5 представляет собой группу формулы NR9R10, где R9 и R10 указаны выше, при условии, что R9 и R10 не могут представлять собой водород или группу формулы COR11 или COOR11, как указано выше, или его фармацевтически приемлемой соли включает взаимодействие агликона формулы К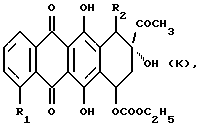
где R1 и R2 указаны выше, с соответствующим производным амина формулы NHR9R10, R9 и R10 указаны выше; и, при желании, преобразование полученного указанного соединения формулы А в его фармацевтически приемлемую соль.
(iv) В другом примере предпочтительный способ получения соединений формулы А, где R3 обозначает OH, R4 является COCH3 и R5 представляет собой группу формулы NR9R10, где один из R9 и R10 представляет собой атом водорода, а другой не представляет водород или группу формулы COR11 или COOR11, как указано выше, или его фармацевтически приемлемой соли, включает:
(1) защиту агликона формулы К, указанной выше, с получением 13-этилендиокси-производного формулы К1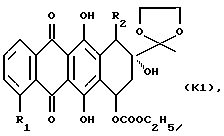
где R1 и R2 указаны выше;
(2) взаимодействие указанного производного формулы К1 с соответствующим соединением формулы NHR9R10, R9 и R10 указаны выше;
(3) снятие защиты у 13-карбонильной группы полученного 7-амино-замещенного производного формулы К2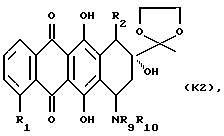
где R1, R2, R9 и R10 указаны выше; и, при желании, преобразования указанного соединения формулы А в его фармацевтически приемлемую соль, например, подкислением соединения А с получением соли добавления кислоты.
(v) В другом примере предпочтительный способ получения соединений формулы А, где R3 является OH, R4 обозначает COCH3 и R5 представляет собой NH2, или его фармацевтически приемлемой соли, включает:
(1) обработку производного формулы К2, указанного выше, в которой NR9R10 обозначает 3',4'-диметоксибензиламино, окислительным агентом;
(2) снятие защиты у 13-карбонильной группы полученного 7-аминозамещенного соединения формулы К3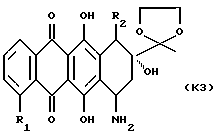
где R1 и R2 указаны выше; и
(3) при желании, преобразование полученного указанного соединения формулы А в его фармацевтически приемлемую соль, например, подкислением соединения А с получением соли добавления кислоты.
(vi) В другом примере предпочтительный способ получения соединений формулы А, где R3 обозначает OH или H, R4 обозначает COCH2NR9NR10, где R9 и R10 указаны выше, при условии, что они не обозначают группу формулы COR11 или COOR11, и R5 обозначает водород или OH, или его фармацевтически приемлемую соль, включает:
(1) преобразование соединение формулы L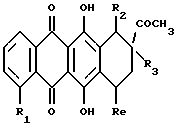
где R1, R2, R3 и Rе указаны выше, в соответствующее 14-бром-производное формулы L1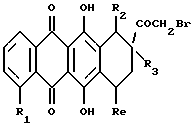
где R1, R2, R3 и Rе указаны выше;
(2) взаимодействие 14-бромпроизводного формулы L1 с соответствующим амином формулы NHR9R10, где R9 и R10 были определены ранее, при условии, что они не представляют собой группу формулы COR11 или COOR11; и
(3) при желании, превращение полученного указанного соединения формулы А в его фармацевтически приемлемую соль, например, подкислением соединения А с получением соли добавления кислоты.
(vii) В другом примере предпочтительный способ получения соединений формулы А, где R4 обозначает группу формулы COCH2O-Ph, где фенильное кольцо (Ph) необязательно замещено нитро, амино или NR9R10, указанным выше, и R5 обозначает водород или гидрокси, или его фармацевтически приемлемой соли включает:
(1) взаимодействие соединения формулы L1, указанного выше, с фенолом, необязательно замещенным, как указано выше, предпочтительно нитрофенолом, в виде соли; и
(2) при желании, преобразование полученного указанного соединения формулы А в его фармацевтически приемлемую соль.
(viii) В другом примере предпочтительный способ получения соединений формулы А, где R4 обозначает группу формулы XCH2R8, где R8 обозначает группу формулы C и D, указанные выше, или их фармацевтически приемлемых солей, включает взаимодействие антрациклинона, несущего гидроксилированную боковую цепь по 9-му положению, такую как COCH2OH или CH2CH2OH, с производным формулы C' или D'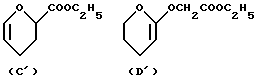
(2) при желании, гидролиз полученного эфирного производного с получением антрациклинонов формулы А, несущих карбоксигруппу в ацетальной части молекулы; и
(3) при желании, преобразование полученного указанного соединения формулы А в его фармацевтически приемлемую соль.
(ix) В другом примере предпочтительный способ получения соединений формулы А, где R4 обозначает группу CH2CH2R8, включает:
(1) преобразование соединения формулы А, указанного выше, где R4 обозначает группу формулы COCH2R8, в соответствующее 13-гидразоновое производное, предпочтительно в 13-[(4-фтор)бензолсульфонил]гидразон;
(2) восстановление вышеуказанного производного гидразона с помощью восстановителя в условиях, способствующих сохранению природы хиноновой системы соединения формулы А; и
(3) при желании, преобразование полученного указанного соединения формулы А, где R4 обозначает указанную группу CH2CH2R8, в ее фармацевтически приемлемую соль.
Следует отметить, что, при желании, производные формулы А, полученные в соответствии со способами (i), (ii), (iii), (iv), (v), (vi), (vil), (viii) и (ix), могут быть далее модифицированы в различных частях молекулы путем сочетания вышеописанных способов или синтетическими методами, описанными для антрациклинов или антрациклинонов (см.: F.Arcamone in "Doxorubicin", Medicinal Chemistry, Vol. 17, Academic Press, 1981) или обычными синтетическими методами (см.: J.March, "Advanced Organic Reaction", Fourth., J. Wiley & Sons, 1992). Например, соединения формулы А, где X является CO группой, могут быть преобразованы в соединение формулы А, где X обозначает CHOH восстановлением, например боргидридом натрия. Соединение формулы А, где R5 обозначает OH, может быть преобразовано в соответствующее соединение, имеющее R5= H, путем обработки дитионитом натрия.
Соединение формулы А, указанное в (i), может быть получено по способу, описанному в DE-A-2 750 812, например, путем взаимодействия соединения формулы G, указанного выше, с избытком этоксикарбонилхлорида в пиридине при комнатной температуре в течение периода времени от 1 до 2 часов; затем обработкой защищенного производного G1 бромидом алюминия в сухом апротонном растворителе, предпочтительно в метиленхлориде, при комнатной температуре в течение периода времени от 3 до 6 часов; алкилированием полученного 4-гидроксипроизводного формулы G2, предпочтительно йод-производным формулы R6I в апротонном растворителе, таком как метиленхлорид или хлороформ, в присутствии конденсирующего агента, предпочтительно оксида серебра, при температуре от 40 до 60oC в течение периода времени от 6 до 24 часов; с последующим снятием защиты у гидроксигрупп соединения формулы G3 вначале за счет удаления групп, защищающих фенол обработкой морфолином в полярном протонном растворителе, таком как метанол, при комнатной температуре в течение периода времени от 1 до 3 часов и затем гидролизом 7-O-этокси-карбонильной группы, если она имеется, очень разбавленным водным раствором гидроксида натрия.
Соединения формулы А, указанные в (ii), могут быть получены, как описано в US-A-4 965 351, например, путем растворения соединения формулы H, указанного выше, в сухом аполярном растворителе, таком как метиленхлорид, и последующей обработкой соединением формулы HalSO2R7, указанного выше, (Hal обозначает галоген), предпочтительно хлор, в присутствии органического основания, такого как N,N-диизопропилэтиламин, и каталитического количества 4-диметиламинопиридина при температуре от 0 до 30oC предпочтительно при комнатной температуре в течение периода времени от нескольких минут до нескольких часов.
Соединения формулы A, указанные в (iii), могут быть получены путем взаимодействия соединения формулы К, указанного выше, с производным амина формулы NHR9R10, указанным выше, в сухом апротонном растворителе, таком как безводный метиленхлорид, при температуре от 10 до 30oC в течение периода времени от нескольких часов до нескольких дней; и, при желании, подкислением полученного продукта предпочтительно безводным хлористым водородом в метаноле с получением соли присоединения кислоты.
Соединения формулы А, указанные в (iv), могут быть получены путем взаимодействия соединения формулы К, указанного выше, с этиленгликолем в толуоле, в присутствии кислотного катализатора, предпочтительно п-толуолсульфоновой кислоты, при температуре кипения с обратным холодильником в течение периода времени от 3 до 6 часов; с последующим взаимодействием защищенного производного К1 с соединением формулы NHR9R10, как указано выше, в полярном растворителе, таком как пиридин или тетрагирофуран, предпочтительно при комнатной температуре в течение периода времени от одного до нескольких дней; и далее снятием защиты у защищенной карбонильной группы путем обработки производного формулы К2 трифторуксусной кислотой с добавлением нескольких капель анизола, предпочтительно при комнатной температуре в течение периода времени от 30 минут до двух часов; и, при желании, преобразование полученного соединения в соль добавления кислоты предпочтительно с использованием безводного хлористого водорода в метаноле.
Соединения формулы A, указанные в (v), могут быть получены, например, взаимодействием соединения формулы К2, указанного выше, где NR9R10 обозначает 3,4-диметоксибензиламино, с 2,3-дихлор-5,6-дициан-1,4-бензохиноном [ДДХ (DDG)] в смеси воды и метиленхлорида в течение одного дня; с последующим снятием зашиты у полученного производного формулы К3, указанного выше, с помощью трифторуксусной кислоты и анизола при комнатной температуре в течение одного часа; и, при желании, с последующим превращением полученного соединения А в соль добавления кислоты предпочтительно с помощью безводного хлористого водорода в метаноле.
Соединения формулы А, указанные в (vi), могут быть получены, как описано в DE-A-2 557 537, например, путем взаимодействия соединения формулы L1, полученного из соединения, описанного в DE-A-1 917 874, с соответствующим амином формулы NНR9R10, где R9 и R10 соответствуют данным ранее определениям, при условии, что R9 и R10 не могут представлять собой группы формулы COR11 или COOR11, указанные выше, в сухом полярном растворителе, таком как ацетон или диметилформамид, при температуре от 20 до 60oC в течение периода времени от 4 до 24 часов и, при необходимости, преобразуя полученное соединение в соль присоединения кислоты предпочтительно с безводным хлористым водородом в метаноле.
Соединения формулы А, указанные в (vii), могут быть получены, как описано в DE-A-1 917 874, например, взаимодействием соединения формулы L1, указанного выше, с фенольным производным, описанным ранее, в апротонном органическом растворителе, таком как ацетон, в присутствии основания, предпочтительно карбонате калия или натрия при температуре от 20 до 60oC.
Соединения формулы А, указанные в (viii), могут быть получены, как описано в WO 92/10212 и WO 92/02255, например, путем взаимодействия антрациклинона, указанного выше, с производными формулы C' или D' в присутствии кислотного катализатора, например п-толуолсульфоната пиридиния в апротонном растворителе, таком как метиленхлорид, при температуре от 10 до 30oC, предпочтительно при комнатной температуре, в течение периода времени от 3 до 24 часов; и, при желании, гидролизом эфирного производного разбавленным водным раствором гидроксида натрия.
Соединения формулы А, указанные в (ix), могут быть получены, как описано, в GB-A-2238540, например, восстановлением 13-[(4-фтор)бензолсульфонил] гидразонового производного антрациклинона формулы А, указанной выше, с помощью цианборгидрида натрия в органическом растворителе, таком как толуол или диметилформамид, при температуре от 25 до 80oC в течение периода от 6 до 24 часов.
Некоторые из исходных веществ для получения соединений формулы А известны. Другие могут быть получены аналогично, исходя из известных соединений с помощью известных методов.
Например, приведенные ниже соединения известны и могут быть представлены той же формулой А:
дауномицинон (R1=OCH3, R2=H, R3=R5=OH, R4=COCH3),
адриамицинон (R1=OCH3, R2=H, R3=R4=OH, R5=COCH2OH),
4-деметоксидауномицинон (R1=R2=H, R3=R5=OH, R4=COCH3),
карминомицинон (Ra=OH, R2=H, R3=R5=OH, R4=COCH3),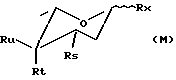 -родомицинон (R1=R2=R3=R5=OH, R4=CH2CH3),
-родомицинон (R1=R2=R3=R5=OH, R4=CH2CH3),
β-родомицинон (R1=R3=R5=OH, R2=COOCH3, R4=CH2CH3),
соответствующие 7-дезокси-производные (R5= H) (см. : F. Acramone in "Doxorubicin", Medicinal Chemistry, vol. 17, Academic Press, 1981)
или производные сахаров формулы М
β
такие как аминосахара даунозамин, 3-амино-2,3,6-тридезокси- L-ликсо-гексопираноза, (M1: R8= NH2, Rt=Rx=OH, Ru=H) (см.: J. Am. Chem. Soc., 86, 5334, 1964) или акозамин, 3-амино-2,3,6-тридезокси-1-арабино-геусопираноза (М2: R8= NH2, Ru=Rx=OH, Rt=H) (см.: J. Med. Chem., 18, 703, 1975) или соответствующие 1-хлор-3,4-ди-трифторуксусные производные даунозамина (Rx=Cl и Rs= NHCOCF3, Rt=OCOCF3) или 1-хлор-3,4-дитрифторацетильные производные акозамина (Rх =Cl и R8=NHCOCF3), Rи = OCOCR3).
Соединения по настоящему изобретению отличаются высокой ингибирующей активностью в отношении амилоидоза.
Термин амилоидоз указывает на наличие общей характерной тенденции некоторых белков к полимеризации и осаждению во внеклеточном пространстве в виде нерастворимых фибрилл, вызывая структурные и функциональные повреждения органов и тканей. Недавно в Бюллетене Всемирной Организации Здравоохранения (Bulletin of the World Health Organisation 71(1): 105, 1993) была приведена пересмотренная классификация амилоида и амилоидоза.
Все самые различные типы амилоида обладают общей ультраструктурной организацией антипараллельных β-складчатых полос, несмотря на то, что они содержат множество очень различающихся белковых субъединиц (см.: Glenner G.G., New England J. Med., 302(23): 1283, 1980). AL амилоидоз вызывает особый моноклональный иммуноглобулин, его легкие цепи, которые образуют амилоидные фибриллы. Такие легкие моноклональные фибриллы продуцируются моноклональными плазматическими клетками, которые имеют низкий митотический индекс и известны по их нечувствительности к химиотерапии. Злокачественное перерождение этих клеток связано с их белоксинтезирующей активностью.
Клинический курс лечения болезни зависит от того, какой орган вовлечен в патологический процесс; при этом прогноз может быть крайне неблагоприятен в случае сердечного инфильтрата (средний срок выживания >12 месяцев), несколько лучше он в случае почечного инфильтрата (средний срок выживания примерно 5 лет).
При этом достижение относительной нечувствительности амилоидогенных молекул, которые могут блокировать или замедлять образование амилоида и увеличивать растворимость имеющихся амилоидных отложений, может рассматриваться, по-видимому, как единственная обоснованная надежда для пациентов с AL амилоидозом. Кроме того, поскольку супермолекулярная организация амилоидных фибрилл одинакова для всех типов амилоида, доступность препарата, который препятствует образованию амилоида и повышает растворимость имеющихся отложений, что приводит к возможности осуществлять клиренс за счет функционирования нормальных механизмов, может иметь большое значение при использовании в случае всех типов амилоидоза и в особенности для лечения болезни Альцгеймера.
И в действительности основным патологическим признаком, общим для болезни Альцгеймера (БА, АD), синдрома Дауна, dementia pugilistica и церебральной амилоидной ангиопатии является образование амилоидных отложений в паренхиме головного мозга и на стенках сосудов. Такого рода маркеры связаны с потерей нейрональных клеток в коре головного мозга, лимбических регионах и в подкорковых ядрах. Ряд исследований показал наличие корреляции селективного повреждения различных нейрональных систем и утраты синапса в лобной доле коры головного мозга со снижением познавательной способности. Патогенез и молекулярные основы нейродегенеративных процессов, происходящих при БА, до сих пор не известны, однако в последних сообщениях четко вырисовывается роль β-амилоида, откладывающегося в паренхиме головного мозга и на стенках сосудов в связи с показанной in vitro и in vivo нейротоксичностью (Yanker et al. Science, 245: 417, 1990; Kowall et al., PNAS, 88: 7247, 1991). Кроме того, выделение сходных AD с мутацией в гене белкового предшественника амилоида (БПА, АРР) вызвало интерес к возможной патогенетической роли β-амилоида в развитии БA [Mullan M. et al., TINS, 16(10): 393, 1993].
Нейротоксичность β-амилоида связана с фибрилогенными свойствами белка. Исследования гомологичных синтетических пептидов указывают на то, что клетки гиппокампа нечувствительны к 24-часовому воздействию свежеприготовленного раствора β 1-42, в то время как их жизнеспособность снижалась в том случае, если нейроны подвергали воздействию β 1-42, который перед этим хранился в течение 2-4 дней при температуре 37oC в солевом растворе, что в результате способствовало агрегации пептидов. Наличие связи между фибриллами и нейротоксичностью подтверждается также недавно полученными данными о том, что in vivo и in vitro в ходе нормального клеточного метаболизма продуцируется растворимая форма β-амилоида (Hass et al. Nature, 359, 322, 1993) и только в том случае, когда агрегация носит конгофильный характер, она связана с дистрофическими невритами. С другой стороны, неконгофильное "предамилоидное" образование, содержащее единичные молекулы β-амилоида, не связано с изменениями нейронов (Tagliavini et al. Neurosci. Let., 93, 193, 1998).
Нейротоксичность β-амилоида была также подтверждена с использованием пептидного гомолога фрагмента β-амилоида 25- 35 (β25-35), сохраняющего самоагрегирующие свойства полного β-амилоидного фрагмента β 142.
Хроническое, но не одноразовое воздействие микромолярных концентраций β 25-35 на нейроны гиппокампа индуцировало гибель нейронов за счет активации механизма программированной гибели клеток, известной как апоптоз (Forloni et al. , NeuroReport, 4, 523, 1993). Это также указывает на связь нейротоксичности со способностью β 25-35 к самоагрегации.
Другое нейодегенеративное заболевание, такое как губковидная энцефалопатия (spongiform encephalopathy) (СЭ, SE), характеризуется гибелью нейронов и отложением во внеклеточном пространстве амилоида, который в этом случае происходит из Прион белка (Prion, PrP). Основываясь на данных о нейротоксичности β-амилоида, аналогично было исследовано воздействие синтетических пептидов, гомологичных к различным сегментам PrP, на жизнеспособность первичных нейронов гиппокампа крысы. Хроническое применение пептида, соответствующего PrP 106-126, индуцировало гибель нейронов посредством апоптоза, тогда как в тех же самых условиях при исследовании всех других пептидов и восходящей последовательности PrP 106-126 не было выявлено снижения жизнеспособности клеток (Forioni et al. Nature, 362, 543). PrP 106-126 обладает высокой фибриллогенной активностью in vitro, а также при окраске красителем конго ред, при этом пептидные агрегаты демонстрируют двойное лучепреломление в зеленом свете (green biifrangence), указывающее на наличие β-складчатой конформации, характерной для амилоида.
Соединения по настоящему изобретению могут использоваться для изготовления медикаментов, используемых с целью профилактики или остановки прогрессирования заболеваний, вызванных амилоидными белками, такими как AL амилоидоз, болезнь Альцгеймера, синдром Дауна и др.
Настоящее изобретение включает также фармацевтические композиции, содержащие в качестве активных ингредиентов одно или более из соединений А в сочетании, при необходимости, с фармацевтически приемлемыми носителями, наполнителями или другими добавками.
Фармацевтические композиции, содержащие соединение формулы А или его соли, могут быть получены обычным способом при использовании обычных нетоксичных фармацевтических носителей или разбавителей в широком диапазоне дозирования форм и способом их введения.
В частности, соединения формулы А могут вводиться:
А) перорально, например, в виде таблеток, пастилок, лепешек, водных или масляных суспензий, диспергированных порошков или гранул, эмульсий, твердых или мягких капсул, а также сиропов или эликсиров. Композиции, предназначенные для перорального использования, могут быть приготовлены по любому известному в технике методу, применяемому для производства фармацевтических композиций, при этом такие композиции могут содержать один или более агентов, выбранных из группы, состоящей из подсластителей, ароматизаторов, красителей и консервантов, вводимых для создания фармацевтических препаратов, обладающих хорошими эстетическими и вкусовыми показателями.
Таблетки содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми наполнителями, которые применяются для изготовления таблеток. Упомянутые наполнители могут представлять собой, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактозу, фосфат кальция или фосфат натрия; гранулирующие и дезинтегрирующие агенты, например кукурузный крахмал или альгиновую кислоту; связующие вещества, в частности кукурузный крахмал, желатин или аравийскую камедь, и замасливатели, такие как стеарат магния, стеариновую кислоту или тальк. Таблетки могут или вовсе не содержать покрытия или с помощью известных методов могут быть покрыты оболочкой, способствующей задержке процесса их разрушения и всасывания в желудочно-кишечном тракте, оказывая, таким образом, пролонгированное воздействие в течение более длительного периода времени. В частности, для этой цели могут применяться глицерил моностеарат или глицерил дистеарат как вещества, замедляющие процесс разрушения. Композиции для перорального использования могут также представлять собой твердые желатиновые капсулы, в которых активный ингредиент смешивается с инертным твердым разбавителем, таким, например, как карбонат кальция, фосфат кальция или каолин, а также мягкие желатиновые капсулы, в которых активный ингредиент смешивается с водной или масляной средой, например с арахисовым маслом, жидким парафином или оливковым маслом. Водные суспензии содержат активные материалы в смеси с наполнителями, пригодными для производства водных суспензий.
Упомянутые наполнители могут представлять собой суспендирующие агенты, например натрий-карбоксиметилцеллюлозу, метилцеллюлозу, гидроксипропилметилцеллюлозу, альгинат натрия, поливинилпирролидон, трагакантовую камедь и аравийскую камедь; диспергирующие или смачивающие вещества, которые могут представлять собой либо натуральные фосфатиды, например лецитин, либо продукты конденсации алкиленоксида с жирными кислотами, например полиоксиэтилен стеарат, или продукты конденсации этиленоксида с длинноцепочечными алифатическими спиртами, например гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с неполными эфирами, полученными из жирных кислот и гексита, такого как полиоксиэтилен сорбит моноолеат, или продукты конденсации этиленоксида с неполными эфирами, полученными из жирных кислот и ангидридов гексита, например полиоксиэтилен сорбитан моноолеат. Указанные водные суспензии могут также содержать один или более консервантов, например этил- или н-пропилоксибензоат, один или более красителей, один или более ароматизаторов, один или более подсластителей, таких как сахароза или сахарин. Масляные суспензии могут быть получены при суспендировании активного ингредиента в растительном масле, например в арахисовом масле, оливковом масле, кунжутном масле или кокосовом масле, или в минеральном масле, таком как жидкий парафин. Водные суспензии могут содержать загустители, например пчелиный воск, твердый парафин или цетиловый спирт. Указанные выше подсластители, а также ароматизаторы могут добавляться в препарат для придания ему приятного вкуса. Сохранность таких композиций может быть улучшена за счет добавления антиоксиданта, такого как аскорбиновая кислота. Диспергированные порошки и гранулы, пригодные для приготовления водной суспензии при добавлении воды, представляют смесь активного ингредиента с диспергирующим или смачивающим веществом, с добавлением суспендирующего агента, а также одного или более консервантов. Выше уже были приведены примеры материалов, которые могут использоваться в качестве диспергирующих или смачивающих веществ, а также суспендирующих агентов. В композициях могут быть представлены также другие наполнители, например подсластители, красители и другие агенты.
Фармацевтические композиции настоящего изобретения могут также иметь вид эмульсии масла в воде. Масляная фаза может быть представлена растительным маслом, например оливковым маслом или арахисовым маслом, или минеральным маслом, в частности жидким парафином, а также их смесями.
Подходящие эмульгаторы могут включать натуральные камеди, например аравийскую камедь или трагакантовую камедь, натуральные фосфатиды, например из соевых бобов, лецитин, а также эфиры или неполные эфиры, полученные из жирных кислот и ангидридов гексита, такие, например, как сореитан моноолеат, а также продукты конденсации упомянутых неполных эфиров с этиленоксидом, например полиоксиэтилен сорбитан моноолеат. Эмульсия может также содержать подсластители и ароматизаторы. Сиропы и эликсиры могут содержать подсластители, такие как глицерин, сорбит или сахароза. Описанные композиции могут также содержать средство, уменьшающее раздражение, консервант, ароматизатор и краситель.
Б) Парентерально, т.е. либо подкожно, либо внутривенно, либо интерстернально, либо с применением техники инфузий в виде стерильных водных или маслянистых инъецируемых суспензий. Фармацевтические композиции могут представлять собой инъецируемые водные или маслянистые суспензии.
Такие суспензии могут быть получены известными методами с использованием указанных выше подходящих диспергирующих и увлажняющих веществ, а также суспендирующих агентов. Стерильный препарат для инъекций может представлять стерильный раствор или суспензию для инъекций в нетоксичном подходящем для парентерального введения разбавителе или растворителе, например, может быть в виде раствора в 1,3-бутандиоле. Среди подходящих для использования по настоящему изобретению носителей и растворителей могут найти применение такие, как вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно используются стерильные жирные масла.
Для описываемых целей может найти применение любая смесь жирных масел, включая синтетические моно- и диглицериды. Кроме того, для изготовления инъецируемых форм могут использоваться жирные кислоты, такие как олеиновая кислота. Еще один объект настоящего изобретения касается способа лечения амилоидозных заболеваний путем введения людям, нуждающимся в таком лечении, терапевтически эффективного количества одного или более активных соединений, охватываемых формулой А.
Дневные дозы варьируют в диапазоне примерно от 0,1 до 50 мг на кг веса тела, в зависимости от активности конкретного соединения, возраста, веса и состояния здоровья пациента, типа и тяжести заболевания, а также от частоты и способа введения; предпочтительно дневные дозы находятся в диапазоне от 5 мг до 2 г. Количество активного ингредиента, которое может быть объединено с материальными наполнителями для получения единичной дозированной формы, может изменяться в зависимости от состояния пациента и применяемого способа введения. Так, например, композиция, предназначенная для перорального введения, может содержать от 5 мг до 2 г активного вещества, объединенного с подходящим количеством соответствующего материала наполнителя, содержание которого может изменяться от 5 до 95% от всей композиции. Дозированные формы содержат обычно от примерно 5 мг до примерно 500 мг активного ингредиента.
Приведенные ниже примеры иллюстрируют настоящее изобретение, не ограничивая его никоим образом.
Пример 1: Получение 14-(N-морфолино)дауномицинона (А1)
14-Бромдауномицинон (L1': R1=OCH3, R2=H, R3=OH) (0,95 г, 2 ммоль), полученный, как описано в J. Org. Chem., 42, 3653, 1977, растворяют в сухом метиленхлориде (50 мл), обрабатывают морфолином (0,34 г, 4 ммоль) и выдерживают при комнатной температуре в течение 24 часов. После этого удаляют при пониженном давлении растворитель, и полученный сырой продукт подвергают флэш хроматографии на силикагеле с использованием смеси метиленхлорида и ацетона (90: 10 по объему) в качестве элюента с получением указанного в заголовке соединения А1, которое затем преобразуют в соответствующий гидрохлорид (0,8 г, выход 77%), путем добавления стехиометрического количества хлористого водорода в метаноле с последующим высаживанием диэтиловым эфиром.
ТСХ проводят на Кизельгеле F254 (Мерк), элюируя системой метиленхлорид/ацетон (9:1 по объему), Rf=0,5.
FD-MS:m/e 483 [M]+
1H ЯМР (200 МГц, DMSO-O6) δ:
2,03 (дд. , J = 4,5, 14,2 Гц, 1H, H-8ax); 2,32 (д., J=14,2 Гц, 1H, H-8eq); 2,95 (д. , J=18,5 Гц, 1H, H-10ax); 3,17 (д., J=18,5 Гц, 1H, H-10eg); 3,2, 3,5 (м. , 4H, -CH2-N-CH2-); 3,7, 4,0 (м., 4H, -CH2-O-CH2-); 4,02 (с., 3H, OCH3); 4,87 (м., 2H, CH2-14); 5,16 (м., 1H, H-7); 5,70 (широкий сигнал, 1H, OH-7); 6,36 (с., 1H, OH-9); 7,68 (м., 1H, H-3); 7,93 (м., 2H, H-1 + H-2); 10,40 (широкий сигнал, 1H, NH+); 13,29 (с., 1H, OH-11); 14,01 (с., 1H, OH-6).
Пример 2: Получение 7-дезокси-7-(N-морфолино)дауномицинона (А30)
7-Этоксикарбонилдауномицинон (К': R1= OCH3, R2= H) (0,94 г, 2 ммоль), приготовленный, как описано в DE-A-2 750 812, растворяют в смеси метиленхлорида (50 мл) и метанола (5 мл), добавляемой вместе с морфолином (3 мл), после чего смесь выдерживают при комнатной температуре в течение 20 часов. После этого при пониженном давлении удаляют растворитель и сырой продукт подвергают флэш-хроматографии на силикагеле с использованием в качестве элюирующей системы смеси метиленхлорида и ацетона (95:5 по объему), получая при этом указанное в заголовке соединение А30, которое затем преобразуют в соответствующий гидрохлорид (0,5 г, выход 53%) путем добавления стехиометрического количества хлористого водорода в метаноле с последующим высаживанием с помощью диэтилового эфира.
ТСХ проводят на Кизельгеле F254 (Мерк), элюируя системой метиленхлорид/ацетон (9:1 по объему), Rf =0,58.
FD-MS: m/e 467 [M]+
1H ЯМР (200 МГц, CDCl3) δ :
1,77 (дд. , J =3,3, 14,5 Гц, 1H, H-8ax); 2,32 (дд., J=2,0, 14,5 Гц, 1H, H-8eg); 2,40 (с., 3H, COCH3); 2,50, 3,00 (м., 4H, -CH2-N-CH2-); 3,10, 3,20 (Abg, J =18,7 Гц, 2H, CH2-10); 3,64 (м., 4H, -CH2-O-CH2-); 4,08 (с., 3H, OCH3); 4,35 (дд., J=2,0, 3,3 Гц, 1H, H-7); 7,38 (д., J=8,3 Гц, 1H, H-3); 7,78 (дд. , J =7,7, 8,3 Гц, 1H, H-2); 8,02 (д., J =7,7 Гц, 1H, H-1); 13,29 (с., 1H, OH-11); 14,11 (с., 1H, OH-6).
Пример 3: Получение 7-дезокси-7-(3',4'-диметокси- бензиламино)-13-дезоксо-13-этилендиоксидауномицинона (А32)
7-Этоксикарбонилдауномицинон (K': 1,88 г, 4 ммоль), приготовленный по вышеописанному методу, растворяют в толуоле (100 мл), который добавляют вместе с этиленгликолем (3 мл) и п-толуолсульфонатом пиридиния (0,1 г). Смесь нагревают при температуре кипячения с обратным холодильником в течение 6 часов для удаления воды с использованием аппарата Дина Старка (Dean Stark). Затем реакционную смесь охлаждают при комнатной температуре, промывают 1% водным раствором однозамещенного карбоната натрия и водой. Органическую фазу высушивают над безводным сульфатом натрия, и органический растворитель удаляют при пониженном давлении с получением 13-этилендиоксо производного K' (0,92 г).
ТСХ проводят на Кизельгеле F254 (Мерк), элюируя системой метиленхлорид/ацетон (9:1 по объему), Rf =0,25.
FD-MS: m/e 518 [M]+
13-Этилендиоксопроизводное K' (0,85 г, 1,64 ммоль) растворяют в смеси пиридина (20 мл) и сухого тетрагидрофурана (20 мл), добавляемой вместе с 3,4-диметоксибензиламином (1 мл), и выдерживают при комнатной температуре в течение двух дней. После этого реакционную смесь разбавляют метиленхлоридом (100 мл) и промывают 1H водным раствором хлористого водорода, водой и 1% водным раствором однозамещенного карбоната натрия. Отделяют органическую фазу, и растворитель удаляют при пониженном давлении. Сырой продукт подвергают флеш-хроматографии на силикагеле, элюируя системой смесь метиленхлорида и ацетона (95:5 по объему) и получая указанное в заголовке соединение А32, который затем преобразуют в соответствующий гидрохлорид путем добавления стехиометрического количества хлористого водорода в метаноле с последующим осаждением диэтиловым эфиром.
ТСХ проводят на Кизельгеле F254 (Мерк), элюируя системой метиленхлорид/ацетон (9:1 по объему), Rf =0,8.
FD-MS: m/e 607 [M]+
1H ЯМР (200 МГц, CDCl3) δ :
1,49 (с., 3H, CH3); 1,62 (дд., J=4,0, 14,2 Гц, 1H, H-8ах); 2,45 (ддд., J =1,8, 1,8, 14,2 Гц, 1H, H-8eg); 2,86 (д., J=19,1 Гц, 1H, H-10ах); 3,21 (дд., J=1,8, 19,1 Гц, 1H, H-10ag);
3.85, 3,88 (2хс, 6H, OCH3); 4,06 (с., 7H, 4-OCH3 + -O-CH2-CH2-O-); 4,44 (дд. , J =1,8, 4,0 Гц, 1H, H-7); 6,8-6,9 (м., 3H, H-2'- + H-5' + H-6'); 7,34 (дд. , J= 1,0, 8,5 Гц, 1H, H-3); 7,75 (дд., J =7,8, 8,5 Гц, 1H, H-2); 8,01 (дд., J=10, 7,8 Гц, 1H, H-1).
Пример 4: Получение 7-дезокси-7-(3',4'-диметокси- бензиламино)дауномицинона (А36)
Соединение А32 (0,3 г), полученное, как описано в примере 3, растворяют в трифторуксусной кислоте (3 мл) и добавляют одну каплю анизола. Через один час реакционную смесь разбавляют метиленхлоридом, промывают 1% водным раствором однозамещенного карбоната натрия, сушат над безводным сульфатом натрия. При пониженном давлении удаляют органический растворитель, а сырой продукт подвергают флэш-хроматографии на силикагеле с использованием в качестве элюирующей системы смеси метиленхлорида и метанола (95:5 по объему) с получением 7-дезокси-7-(3',4'- диметоксибензиламино)дауномицинона (А36), который преобразуют в соответствующий гидрохлорид путем добавления стехиометрического количества хлористого водорода в метаноле с последующим высаживанием диэтиловым эфиром.
ТСХ проводят на Кизельгеле F254 (Мерк), элюируя системой метиленхлорид/метанол (8:2 по объему), Rf=0,5.
FD-MS: m/e 547 [M]+
1H ЯМР (200 МГц, DMSO-d6) δ :
2,02 (дд. , J=5,0, 14,4 Гц, 1H, H-8ax); 2,34 (с., 3H, COCH3); 2,48 (м., 1H, H-8eq); 2,98, 3,08 (Abg, J=19,0 Гц, 2H, CH1-10); 3,74, 3,78 (2xc., 6H, OCH3); 4,00 (с., 3H, 4-OCH3); 4,35 (м., 2H, NH-CH2-арил); 4,59 (м., 1H, H-7); 6,80 (широкий сигнал, 1H, OH-9); 6,99 (д., J=8,1 Гц, 1H, H-5'); 7,15 (д. , J= 8,1 Гц, 1H, H-6'); 7,31 (с., 1H, H-2'); 7,69 (м., 1H, H-3); 7,92 (м., 2H, H-1 + H-2); 8,5, 9,9 (широкий сигнал, 2H, NH2+); 12,99 (широкий сигнал, 1H, OH-11); 13,94 (широкий сигнал, 1H, OH-6).
Пример 5: Получение 7-дезокси-7-бензиламино-13-дезоксо-13- этилендиоксидауномицинона (А33)
13-Этилендиоксопроизводное К1' (0,85 г, 1,64 ммоль), полученное, как описано в Примере 3, растворяют в смеси метиленхлорида (40 мл) и метанола (4 мл), обрабатывают бензиламином (0,5 мл) и выдерживают при комнатной температуре в течение 18 часов. После этого растворитель удаляют при пониженном давлении, и сырой продукт подвергают флэш-хроматографии на силикагеле с использованием в качестве элюирующей системы смеси метиленхлорида и ацетона (9: 1 по объему) с получением указанного в заголовке соединения А33 (0,55 г), который преобразуют в соответствующий гидрохлорид путем добавления стехиометрического количества хлористого водорода в метаноле с последующим высаживанием диэтиловым эфиром.
ТСХ проводят на Кизильгеле F254 (Мерк), элюируя системой метиленхлорид/ацетон (9:1 по объему), Rf=0,20.
Пример 6: Получение 7-дезокси-7-бензиламинодауномицинона (А37)
Соединение А33, полученное, как описано в Примере 5, обрабатывают трифторуксусной кислотой, как описано в Примере 4, с получением 7-дезокси-7-бензиламинодауномицинона (А37), который преобразуют в соответствующий гидрохлорид путем добавления стехиометрического количества хлористого водорода в метаноле с последующим высаживанием диэтиловым эфиром.
ТСХ проводят на Кизельгеле F254 (Мерк), элюируя системой метиленхлорид/ацетон (9:1 по объему), Rf= 0,60.
FD-MS: m/e 487 [M]+
1H ЯМР (200 МГц, CDCl3) δ :
1,76 (дд., J=4,1, 14,4 Гц, 1H, H-8ax); 2,33 (ддд., J=1,7, 1,9, 14,4 Гц, 1H, H-eg); 2,42 (с. , 3H, COCH3); 2,88 (д., J =18,8 Гц, 1H, H-10ax); 3,14 (дд. , J=1,9, 18,8 Гц, 1H, H-10eq); 3,91, 4,08 (Abg, J=12,4 Гц, 2H, NH-CH2- арил); 4,04 (с. , 3H, 4-OCH3); 4,41 (дд., J=1,7, 4,1 Гц, 1H, H-7); 7,3-7,4 (м. , 6H, C6H5-+H-3); 7,72) дд., J=7,8, 8,5 Гц, 1H, H-2); 7,94 (дд., J =1,1, 7,8 Гц, 1H, H-1); 13,20 (широкий сигнал, 1H, OH-11), 13,40 (широкий сигнал, 1H, OH-6).
Пример 7: Получение 7-дезокси-7-(2'-гидроксиэтиламино) - 13-дезоксо-13-этилендиоксидауномицинона (А34)
Указанное в заголовке соединение А34 получают в соответствии со способом Примера 3, используя при этом этаноламин.
ТСХ проводят на Кизельгеле F254 (Мерк), элюируя системой метиленхлорид/метанол (9:1 по объему), Rf=0,2.
1H ЯМР (200 МГц, CDCl3) δ :
1,45 (с. , 3H, CH3); 1,57 (дд., J=4,1, 14,1 Гц, 1H, H-8ax); 2,37 (ддд., J=1,4, 1,4, 14,1 Гц, 1H, H-8eq); 2,80 (д., J=19,0 Гц, 1H, H-10ax); 3,03 (м., 2H, NH-CH2-CH2-OH); 3,11 (дд., J=1,4, 19,0 Гц, 1H, H-10eq); 3,6-4,0 (м., 2H, NH-CH2-CH2-OH); 3,99 (с., 3H, OCH3); 4,04 (с., 4H, -O-CH2-O-); 4,34 (м., 1H, H-7); 7,29 (д., J=7,7 Гц, 1H, H-3); 7,69 (дд., J=7,7, 7,7 Гц, 1H, H-2); 7,88 (д., J=7,7 Гц, 1H, H-1).
Пример 8: Получение 7-дезокси-7-(2'-гидроксиэтиламино)дауномицинона (А38)
Указанное в заголовке соединение А38 получают из соединения А34 в соответствии со способом, описанным в Примере 4.
ТСХ проводят на Кизельгеле F254 (Мерк), элюируя системой метиленхлорид/метанол (9:1 по объему), Rf = 0,5.
FD-MS: m/e 441 [M]+
1H ЯМР (200 МГц, CDCl3) δ :
1,80 (дл., J=4,2, 14,4 Гц, 1H, H-8ах); 2,28 (ддд., J=1,8, 2,0, 14,4 Гц, 1H, H-8eq); 2,41 (с., 3H, COCH3); 2,96 (д., J=18,5 Гц, 1H, H-10ax); 3,03 (м. , 2H, NH-CH2-CH2-OH); 3,20 (дд., J=1,8, 18,5 Гц, 1H, H-10eq); 3,5-4,0 (м., 2H, NH-CH2-CH2-OH); 4,08 (с., 3H, OCH3); 4,45 (дд., J=2,0, 4,2 Гц, 1H, H-7); 7,39 (J=1,0, 8,5 Гц, 1H, H-3); 7,78 (J=7,7, 8,5 Гц, 1H, H-2); 8,03 (дд., J= 1,0, 7,7, 1H, H-1).
Пример 9: Получение 7-дезокси-13-этилендиокси-7-амино- дауномицинона (А40)
7-Дезокси-7-(3', 4'-диметоксибензиламино)-13-дезоксо-13- этилендиоксидауномицинон (А32, 0,5 г), полученный, как описано в Примере 3, растворяют в смеси метиленхлорида (80 мл) и воды (4 мл), добавляемой вместе с 2,3-дихлор-5,6-дициано-1,4- бензохиноном (ДДХ, DDQ), и выдерживают при комнатной температуре в течение 24 часов. После этого реакционную смесь промывают 1% водным раствором однозамещенного карбоната натрия. Органическую фазу отделяют, и растворитель удаляют при пониженном давлении с получением указанного в заголовке соединения А40 (0,3 г).
ТСХ проводят на Кизельгеле F254 (Мерк), элюируя системой метиленхлорид/метанол (6:1 по объему), Rf=0,25.
Пример 10: Получение 7-дезокси-7-аминодауномицинона (А42)
Соединение A40 (0,2 г), полученное, как описано в Примере 9, обрабатывают трифторуксусной кислотой, как описано в Примере 4, с получением, после проведения флэш-хроматографии на силикагеле с использованием смеси метиленхлорида и метанола (95: 5 по объему), 7-дезокси-7-аминодауномицинона (А42, 0,14 г), который преобразуют в соответствующий гидрохлорид путем добавления стехиометрического количества хлористого водорода в метаноле с последующим высаживанием диэтиловым эфиром.
ТСХ проводят на Кизельгеле F254 (Мерк), элюируя системой метиленхлорид/метанол (6:1 по объему), Rf= 0,33.
FD-MS: m/e 397 [M]+
1H ЯМР (200 МГц, DMSO-d6) δ :
1,79 (дл., J=5,3, 14,9 Гц, 1H, H-8ax); 2,02 (д., J=14,9 Гц, 1H, H-8eq); 2,29 (с., 3H, COCH3); 2,76 (д., J=18,6 Гц, 1H, H-10ax); 2,89 (д., J=18,6 Гц, 1H, H-10eq); 3,96 (с., 3H, 4-OCH3); 4,31 (д., J=5,3 Гц, 1H, H-7); 7,60 (м., 1H, H-1 + H-2); 8,00 (широкий сигнал, 2H, NH2).
Пример 11: Получение 14-(N-пиперидино)-дауномицинона (А2)
14-Бромдауномицинон (L1': R1=OCH3, R2=H, R3=OH) (0,95 г, 2 ммоль), полученный, как описано в J. Org. Chem., 42, 3653, 1977, растворяют в сухом метиленхлориде (50 мл), обрабатывают пиперидином (0,34 г, 4 ммоль) и выдерживают при комнатной температуре в течение 16 часов. Затем при пониженном давлении удаляют растворитель, и сырой продукт подвергают флэш-хроматографии на селикагеле с использованием в качестве элюирующей системы смеси этиленхлорида и метанола (96:4 по объему) с получением указанного в заголовке соединения, которое переводят в соответствующий гидрохлорид (0,55 г, выход 53%) путем добавления стехиометрического количества хлористого водорода в метаноле с последующим высаживанием диэтиловым эфиром.
ТСХ проводят на Кизельгеле F254 (Мерк), элюируя системой метиленхлорид/метанол (9:1 по объему) Rf=0,5.
FAB-MS: m/e 482 [M]+
1H ЯМР (200 МГц, DMSO-d6) δ :
1,2-1,9 (м. , 6H, пиперидин CH2-3 + CH2-4 + CH2-5); 1,97 (дд., J=4,6, 14,1 Гц, 1H, H-8ax); 2,30 (д., J=14,1 Гц, 1H, H-8eq); 2,89 (д., J=18,4 Гц, 1H, H-10ax); 3,0, 3,4 (м., 4H, пиперидин CH2-1 + CH2-6); 3,13 (д., J=18,4 Гц, 1H, H-10eq); 3,97 (с., 3H, OCH3-4); 4,76 (м., 2H, CH2-14); 5,10 (м., 1H, H-7); 5,60 (д., J=6,6 Гц, 1H, OH-7); 6,39 (с., 1H, OH-9); 7,64 (м., 1H, H-3); 7,90 (м., 2H, H-1 + H-2); 9,7 (широкий сигнал, 1H, NH+); 13,23 (с., 1H, OH-11); 13,95 (с., 1H, OH-6).
Пример 12: Получение 14-[N-(N'-метил)пиперазино]дауномицина (А4)
Способ получения указанного в заголовке соединения аналогичен описанным в примерах 1 и 2.
ТСХ проводят на Кизельгеле F254 (Мерк), элюируя системой метилекнхлорид/метанол (8:2 по объему) Rf=0,36.
FAВ-MS: m/e = 497[M+H]+
1H ЯМР (200 МГц, CDCl3) δ :
2,14 (дд., J=4,8, 14,5 Гц, 1H, H-8ax); 2,32 (с., 3H, NCH3); 2,36 (ддд., J=2,0, 2,0, 14,5 Гц, 1H, H-8eq); 2,4-2,7 (м., 8H, водороды пиперазина); 2,98 (д. , J =18,5 Гц, 1H, H-10ax); 3,17 (дд., J=2,0, 18,5 Гц, 1H, H-10eq); 3,60, 3,72 (два дуплета, J=16,7 Гц, 2H, CH2-14); 4,0 (широкий сигнал, 1H, OH-7); 4,09 (с. , 3H, OCH3-4); 5,27 (дд., J=2,0, 4,8 Гц, 1H, H-7); 6,1 (широкий сигнал, 1H, OH-9); 7,39 (дд., J=1,1, 8,6 Гц, 1H, H-2); 7,78 (дд., J=7,7, 8,6 Гц, 1H, H-2); 8,02 (дд., J=1,1,7,7 Гц, 1H, H-1); 13,31 (широкий сигнал, 1H, OH-11); 13,97 (с., 1H, OH-6).
Пример 13: Получение 14-[N-диэтиламино]дауномицинона (А58)
Способ получения указанного в заголовке соединения аналогичен описанным в примерах 1 и 2.
ТСХ проводят на Кизельгеле F254 (Мерк), элюируя системой метиленхлорид/метиловый спирт (9:1 по объему), Rf=0,45.
FAВ-MS: m/e = 470 [M+H]+
1H ЯМР (200 МГц, CDCl3) δ :
1,15 (т., J=7,2 Гц, 6H, N(CH2CH3)2); 2,14 (дд., J=4,8, 14,5 Гц, 1H, H-8ax); 2,37 (ддд., J=2,0, 2,0, 14,5 Гц, 1H, H-8eq); 2,69 (м., 4H, N(CH2CH3)2); 2,97 (д., J =18,5 Гц, 1H, H-10ax); 3,21 (дд., J=2,0, 18,5 Гц, 1H, H-10eq); 3,58, 3,73 (два дуплета, J=15,4 Гц, 2H, CH2-14); 4,08 (с., 3H, OCH3-4); 5,23 (дд., J=2,0, 4,8 Гц, 1H, H-7); 7,38 (дд., J=1,0, 8,3 Гц, 1H, H-3); 7,76 (дд. , J= 7,7, 8,3 Гц, 1H, H-2); 8,02 (дд., J =1,0, 7,7 Гц, 1H, H-1); 13,3 (широкий сигнал, 1H, OH-11); 14,0 (широкий сигнал, 1H, OH-6).
Пример 14: Получение 4-деметокси-14-(N-морфолино)- дауномицинона (А10)
Указанное в заголовке соединение получают, как описано в примере 1, используя в качестве исходного соединения 4-деметокси- 14-бромдауномицинон, который получен путем бромирования 4- деметоксидауномицинона по способу, описанному в J.Org. Chem., 42, 3653, 1977, для дауномицинона.
ТСХ проводят на Кизельгеле F254 (Мерк), элюируя системой метиленхлорид/метиловый спирт (96:4 по объему), Rf=0,21.
FAВ-MS: m/e 454 [M+H]+
1H ЯМР (200 МГц DMSO-d6) δ :
1,96 (дд. , J=4,6, 14,3 Гц, 1H, H-8ax); 2,16 (дд., J=2,0, 14,3 Гц, 1H, H-8eq); 2,44 (м., 4H, N(CH2CH2)2O); 2,92, 3,00 (два дуплета, J=18,7 Гц, 2H, CH2-10); 3,57 (м., 4H, N(CH2CH2)2O); 3,67, 3,72 (два дуплета, J=18,9 Гц, 2H, CH2-14); 5,03 (м., 1H, H-7); 5,4 (широкий сигнал, 1H, OH-7); 6,05 (с., 1H, OH-9); 7,96 (м., 2H, H-2 + H-3); 8,26 (м., 2H, H-1 + H-4); 13,3 (широкий сигнал, 2H, OH-6 + OH-11).
Пример 15: Получение 4-деметокси-7-дезокси-14-(N-морфолино) дауномицинона (А24)
Указанное в заголовке соединение получают, как описано в примере 1, используя в качестве исходного соединения 4-деметокси-7-дезокси- 14-бромдауномицинон, полученный путем бромирования 4-деметокси-7-дезокси-лауномицинона по способу, описанному в J. Org. Chem., 42, 3653, 1977, для дауномицина. ТСХ проводят на Кизельгеле F254 (Мерк), элюируя системой метиленхлорид/метиловый спирт (96:4 по объему), Rf=0,33.
FAВ-MS: m/e 438 [M+H]+
1H ЯМР (200 МГц, CDCl3) δ :
1,9-2,0 (м. , 2H, CH2-8); 2,63 (м., 4H, N(CH2CH2)2O); 1,8-3,2 (м., 4H, CH2-7 + CH2-10); 3,46, 3,60 (два дуплета, J=15,0 Гц, 2H, CH2-14); 3,78 (м., 4H, N(CH2CH2)2O); 7,82 (м., 2H, H-2 + H-3); 8,32 (м., 2H, H-1 + H-4); 13,44, 13,46 (два синглета, OH-6 + OH-11).
Пример 16: Получение 7-дезокси-14-(N-морфолино)-дауномицинона (А17)
Смесь 14-(N-морфолино)дауномицинона (А1) (1,5 г, 3,3 ммоль) и 5% палладия на активированном угле (300 мг) в 250 мл диоксана перемешивают в атмосфере водорода под давлением 2 атм в течение одного часа. Затем катализатор отфильтровывают, растворитель выпаривают при пониженном давлении, а остаток чистят с помощью колоночной хроматографии на силикагеле (элюент: метиленхлорид/метиловый спирт 96: 4 по объему). Указанное в заголовке соединение (0,4 г, 28%) выделяют в виде красного порошка, затем превращают его в соответствующий гидрохлорид путем добавления стехиометрического количества хлористого водорода в метаноле с последующим высаживанием диэтиловым эфиром.
ТСХ проводят на Кизельгеле F254 (Мерк), элюируя системой метиленхлорид/метиловый спирт (96:4 по объему), Rf = 0,20.
FAВ-MS: m/e 468 [M+H]+
1H ЯМР (200 MГц, DMSO-d6) δ :
1,7-2,2 (м. , 2H, CH2-8), 2,83 (м., 2H, CH2-7), 2,91 (м., 3H, CH2-10); 3,3-3,8 (м. , 8H, водороды морфолина); 3,97 (с., 3H, OCH3-4); 4,8 (м., 2H, CH2-14); 6,14 (широкий сигнал, OH-9); 7,65 (м., 1H, H-3); 7,91 (м., 2H,H-1 + H-2); 10,4 (широкий сигнал, 1H, HN+); 13,34 (с., 1H, OH-11); 13,85 (с., 1H, OH-6).
Пример 17: Получение 13-дигидро-14-(N-морфолино)-дауномицинона (А59)
Эфират бромида магния (2,24 г, 8,68 ммоль) добавляют по каплям к перемешиваемой суспензии 14-(N-морфолино)дауномициноиа (А1) (2,10 г, 4,34 ммоль) в тетрагидрофуране (80 мл) в атмосфере аргона. К полученной смеси, которую охлаждают до температуры -40oC, добавляют порциями боргидрид натрия (0,164 г, 4,34 ммоль). После перемешивания в течение 1,5 часа при температуре -40oC реакцию гасят, добавляя по каплям метиловый спирт (25 мл). Летучие продукты выпаривают при пониженном давлении, и остаток чистят на колонке с силикагелем (элюент: хлороформ/метиловый спирт 94:6 по объему. Указанное в заголовке соединение (1,39 г, 66%) выделяют в виде красного порошка и затем переводят его в соответствующий гидрохлорид путем добавления стехиометрического количества хлористого водорода в метаноле с последующим высаживанием диэтиловым эфиром.
ТСХ проводят на Кизельгеле F254 (Мерк), элюируя системой хлороформ/метиловый спирт (94:6 по объему), Rf= 0,30.
FAВ-MS: m/e 486 [M+H]+
1H ЯМР (200 МГц, DMCO-d6) δ :
1,7-2,2 (м., 2H, CH2-8); 2,3-2,8 (м., 6H, CH2-14 + N(CH2CH2)2O; 2,86 (м. , 2H, CH2-10); 3,57 (м., 5H, N(CH2CH2)2O + CH-13); 3,97 (с., 3H, OCH3-4); 4,81 (д.,J=5,5 Гц, 1H, OH-13); 5,03 (м., 1H, H-7); 5,20 (м., 1H, OH-7); 5,6, 5,8 (широкий сигнал, 1H, OH-9); 7,5-8,0 (м., 3H, H-1 + H-2 + H-3); 13,3 (широкий сигнал, 1H, OH-6).
Пример 18: Получение 7-дезокси-13-дигидро-14-(N-морфолино) дауномицинона (А60)
К раствору 13-дигидро-14-(N-морфолино)дауномицинона (0,240 г, 0,494 ммоль), полученного, как описано в предыдущем примере, в диметилформамиде (16 мл) при перемешивании добавляют при комнатной температуре в атмосфере аргона по каплям раствор дитионита натрия (2,15 г, 1,23 ммоль) в воде (8 мл). После перемешивания в течение 1 часа при комнатной температуре реакционную смесь вливают в воду (250 мл) и экстрагируют с помощью этилацетата (6 х 25 мл).
Слои разделяют, органический слой промывают водой, сушат над сульфатом натрия и выпаривают с получением 300 мг сырого продукта, который далее чистят с применением колоночной хроматографии на силикагеле (элюент: хлороформ/метиловый спирт 96:4 по объему). Указанное в заголовке соединение (116 мг, 50%) выделяют в виде красного порошка и затем переводят его в соответствующий гидрохлорид путем добавления стехиометрического количества хлористого водорода в метаноле с последующим высаживанием диэтиловым эфиром.
ТСХ проводят на Кизельгеле F254 (Мерк), элюируя системой хлороформ/метиловый спирт (96:4 по объему), Rf= 0,20.
FAВ-MS: m/e 470 [M+H]+
1H ЯМР (400 МГц, DMSO-d6) δ :
1,56 (м. , 1H, H-8ax); 1,82 (м., 1H, H-8eq); 2,38 (дд., J=7,7, 12,8 Гц, 1H, CH(H)-14); 2,63 (дд. , J= 3,7, 12,8 Гц, 1H, CH(H)-14); 2,45 (м., 4H, N(CH2CH2)2O); 2,6-2,9 (м., 4H, CH2-10 + CH2-7); 3,53 (м., 4H, N(CH2CH2)2O); 3,53 (м., 1H, CH-13); 3,94 (с., 3H, OCH3-4); 4,65 (д.,J=5,1 Гц, 1H, OH-13); 4,76 (широкий сигнал, 1H, OH-9); 7,58 (м., 1H, H-3); 7,86 (м., 2H, H-1 + H-2); 13,36 (с., 1H, OH-11); 13,88 (с., 1H, OH-6).
Пример 19: Получение 4-деметокси-7-дезокси-10-гидрокси-14- (N-морфолино) дауномицинона (А61)
К суспензии 4-деметокси-7-дезокси-9,10-ангидро-дауномицинона, полученного, как описано в J. Org. Chem., 48, 2820, 1983, (0,5 г, 1,5 ммоль) в этилацетате/ацетонитриле (1:1) (20 мл) добавляют при перемешивании по каплям при температуре 0oC в атмосфере аргона раствор гидрата трихлорида рутения (27 мг, 0,1 ммоль) и периодата натрия (0,48 г, 2,2 ммоль в воде (3 мл). После перемешивания в течение 0,5 часа при температуре 0oC реакционную смесь выливают в водный раствор тиосульфата натрия (20 мл) и затем экстрагируют метиленхлоридом. Далее слои разделяют, органический слой промывают водой, высушивают над сульфатом натрия и выпаривают с получением 300 мг сырого продукта, который затем подвергают чистке с применением колоночной хроматографии на силикагеле (элюент: хлороформ/метиловый спирт 50:0,2 по объему), 4-деметокси-7- дезокси-10-гидрокси-дауномицинон выделяют (194 мг, 35%) в виде красного порошка, Т.пл. 241-242oC (разлож.).
4-Деметокси-7-дезокси-10-гидрокси-14-бромдауномицинон получают из 4-деметокси-7-дезокси-10-гидрокси-дауномицинона, который подвергают бромированию, как описано в J. Org. Chem., 42, 3653, 1977, для дауномицинона. Красный порошок; Т.пл. 223-225oC (разлож.).
В соответствии со способом, указанным в примере 1, 4-деметокси-7- дезокси-10-гидрокси-14-бромдауномицинон преобразуют в указанное в заголовке соединение, которое выделяют в виде красного порошка и затем переводят в соответствующий гидрохлорид путем добавления стехиометрического количества хлористого водорода в метаноле с последующим высаживанием диэтиловым эфиром.
ТСХ проводят на Кизельгеле F254 (Мерк), элюируя системой хлороформ/метиловый спирт (96:4 по объему), Rf =0,30.
FAВ-MS: m/e 453 [M+H]+
1H ЯМР (200 MГц, DMSO-d6) δ :
1,8-3,0 (м. , 4H, CH2-8 + CH2-7); 2,36 (м., 4H, N(CH2CH2)2O); 3,47 (м., 4H, N(CH2CH2)2O); 3,59 (с., 2H, CH2-14); 4,90 (с., 1H, H-10); 5,60 (широкие сигналы, 1H, OH-10); 5,67 (с., 1H, H-9); 7,94 (м., 2H, H-2 + H-2 + H-3); 8,27 (м., 2H, H-1 + H-4); 13,30 (широкие сигналы, 2H, OH-6 + OH-11).
Пример 20: получение 4-дeмeтoкcи-4-гидpoкcи-7-дeзoкcи 7- (N-морфолино) дауномицинона (А62)
4-Деметокси-4-гидрокси-O6, O7- диэтоксикарбонилдауномицинон (1,3 г, 2,5 ммоль), полученный из 6,7,11-триэтоксикарбонилдауномицинона (G1: Rb=H, Rc= COCH3, R'e =OCOOC2H5), как описано IО Farmaco, Ed. Sc., 35, 347 1980, растворяют в смеси метиленхлорида (40 мл) и метанола (40 мл) и добавляют 1 мл морфолина. Смесь выдерживают при комнатной температуре в течение 20 часов. При пониженном давлении удаляют растворитель и сырой продукт подвергают флэш-хроматографии на силикагеле с использованием в качестве элюирующей системы смеси метиленхлорида и ацетона (9:1 по объему) с получением указанного в заголовке соединения, которое переводят в соответствующий гидрохлорид (0,3 г, выход 25%) путем добавления стехиометрического количества хлористого водорода в метаноле с последующим высаживанием его диэтиловым эфиром.
ТСХ проводят на Кизельгеле F254 (Мерк), элюируя системой метиленхлорид/метанол (96:4 по объему), Rf =0,8.
FAВ-MS: m/e 502 [M+H]+
1H ЯМР (200 МГц, DMSO-d6) δ :
1,85 (м., 1H, H-8ax); 2,10 (м., 1H, OCH2CH(Hax)N); 2,20 (м., 1H, H-8eq); 2,32 (д., J=10,7 Гц, 1H, CH(H)-14); 2,40 (м., 2H, NCH2CH2OH); 2,68 (м., 1H, OCH2CH(Heg)N); 2,77, 2,86 (д., J=19,2 Гц, 1H, H-10ax); 2,83 (д., J=10,7 Гц, 1H, CH(H)-14); 3,07-3,10 (д. , J= 19,2 Гц, 1H, H-10eq); 3,47 (м., 2H, NCH2CH2OH); 3,57 (м., 1H, NCH(Hax)O); 3,90 (м., 1H, NCH2CH(Heg)O); 3,97 (с., 3H, OCH3-4); 4,40 (м., 1H, CH2OH); 5,32, 5,34, 5,84, 5,93 (с., 2H, OH-9 + OH-13); 7,63 (м., 1H, H-3); 7,87 (м., 2H, H-1 + H-2); 13,31 (широкий сигнал, 1H, OH-11); 13,97 (широкий сигнал, 1H, OH-6).
Пример 21: Получение 4-деметокси-7,9-дидезокси-14-(N- морфолино)дауномицинона (А63)
4-Деметокси-7,9-дидезокси-14-бромдауномицинон получают из 4- деметокси-7,9-дидезокси-10-гидрокси-дауномицинона, полученного, как описано в Synt. Commun. , 15, 1081, 1985, с последующим бромированием, как описано в J.Org. Chem., 42, 3653, 1977, для дауномицинона.
В соответствии со способом, описанным в примере 1, 4-деметокси-7,9-дидезокси-14-бромдауномицинон преобразуют в указанное в заголовке соединение, которое выделяют в виде красного порошка и затем переводят в соответствующий гидрохлорид путем добавления стехиометрического количества хлористого водорода в метаноле с последующим высаживанием диэтиловым эфиром.
ТСХ проводят на Кизельгеле F254 (Мерк), элюируя системой хлороформ/метиловый спирт (98:2 по объему),
Rf=0,23.
FAВ-MS: m/e 422 [M+H]+
1H ЯМР (200 МГц, пиридин-d5) δ :
1,78 (м., 1H, H-8ах); 2,18 (м., 1H, H-8eq); 2,54 (м., 4H, N(CH2CH2)2O); 2,70 (м., 1H, H-9); 2,9-3,3 (м., 4H, CH2-10 + CH2-7); 3,43 (м., 2H, CH2-14); 3,78 (м. , 4H, N(CH2CH2)2O); 7,75 (м., 2H, H-2 + H-3); 8,39 (м., 2H, H-1 + H-4); 13,80 (широкий сигнал, 2H, OH-6 + OH-11).
Пример 22: Получение 4-деметокси, 4-гидрокси, 14-(N-морфолино) дауномицинона (А64)
Суспензию 4-деметокси-4-гидроксидауномицинона (1,4 г, 3,6 ммоль), полученного, как описано в I.Antibiotics, 31, 178, 1978, в 20 мл диоксана, обрабатывают 10,3 мл раствора брома, полученного при разбавлении диоксаном 1 мл брома до конечного объема 50 мл. Реакционную смесь перемешивают при комнатной температуре в течение трех часов, а полученный 4-деметокси-4-гидрокси-14- бромдауномицинон высаживают добавлением 50 мл петролейного эфира. Осадок отфильтровывают, промывают петролейным эфиром и высушивают под вакуумом с получением 1,35 г (80% выход) сырого продукта, который используют как таковой в последующем взаимодействии.
Суспензию 0.75 г (1,6 ммоль) 4-деметокси-4-гидрокси-14- бромдауномицинона в 50 мл метиленхлорида обрабатывают 0,28 г (3,2 ммоль) морфолина, и полученную при этом смесь перемешивают при комнатной температуре в течение 24 часов. Затем при пониженном давлении удаляют растворитель, и сырой продукт подвергают флэш-хроматографии на силикагеле, используя для элюации смесь метиленхлорида в метаноле (95:5 по объему), с получением указанного в заголовке соединения, которое переводят в соответствующий гидрохлорид (0,16 г, выход 20%) путем добавления стехиометрического количества хлористого водорода в метаноле с последующим высаживанием его диэтиловым эфиром.
ТСХ проводят на Кизельгеле F254 (Мерк), элюируя системой метиленхлорид/метанол (9:1 по объему), Rf= 0,6.
FAB-MS: m/e 470 [M+H]+
1H ЯМР (400 МГц, DMSO-d6) δ :
2,01 (дд. , J=4,7, 14,1 Гц, 1H, H-8ax); 2,31 (дд., J=14, 1 Гц, 1H, H-8eq); 2,92, 3,16 (два дуплета, J =18,8 Гц, 2H, CH2-10); 3,1-3,3 (м., 4H, N(CH2CH2)2O); 3,7-4,0 (м., 4H, N(CH2CH2)2O); 4,80, 4,87 (два дуплета, J=18,8 Гц, 2H, CH2-14); 5,11 (м., 1H, H-7); 5,70 (широкие сигналы, 12H, OH-7); 6,34 (с. , 1H, OH-9); 7,41 (м. , 1H, H-3); 7,8-7,9 (м., 2H, H-1 + H-2); 10,40 (широкий сигнал, 1H, HN+); 11,98 (с., 1H, OH-4); 12,80 (с., 1H, OH-6); 13,40 (с., 1H, OH-11).
Биологическое исследование
Способность антрациклиноновых производных формулы А препятствовать проявлению самоагрегационных свойств β-амилоидного фрагмента 25-35 и PrP фрагмента 106-126 может быть продемонстрирована при исследовании светорассеяния.
β 25-35 (GSNKGAIIGLH) и PrP 106-126 (KTNMKHMAGAAAAGAVVGGLG) синтезируют с помощью техники твердофазной химии в аппарате 430А Эпплайд Биосистемз Инструментс (430А Applied Biosystems Instruments) и последующей очистки методом ЖХВД с обращенной фазой [Бэкман Инст. мод. 243 (Beckman Inst. mod. 243)], как описано в Forloni et al. Nature 362, 543, 1993.
Светорассеяние растворов, содержащих пептид, оценивают с помощью спектрофлуориметрии [Перкин Элмер LS 50В (Perkin Elmer LS 50В), в ходе которой процессы возбуждения и эмиссии отслеживают при 600 нм. β-Амилоидный фрагмент 35-35 и PrP 106-126 растворяют в 10 мМ растворе фосфатного буфера, pH 5, до достижения концентрации от 0,5 до 1 мг/мл, (0,4-0,8 мМ и 0,2-0,4 мМ растворы соответственно), при этом в пределах часа отмечается их спонтанная агрегация.
Исследуемые соединения, растворенные в 5 мМ Трис буфере, pH 7,4, до нескольких различных концентраций (0,2-2 мМ), добавляют к раствору пептидов в момент их приготовления для исследования и оценки процесса фибриллогенеза. Показано, что добавление соединений, полученных по примерам 1-22, в эквимолярной концентрации к β-амилоидному фрагменту 25-36 и PrP 106-126, полностью блокирует агрегацию.
Нейротоксичность
Нейрональные клетки получают из коры головного мозга крысиных эмбрионов на 17 день эмбрионального развития и культивируют в присутствии эмбриональной сыворотки теленка (10%), как описано в Forloni et al., Mol. Brain Res., 16, 128, 1992.
Присущую соединениям А цитотоксичность оценивают, проводя повторные экспозиции корковых нейронов с различными концентрациями соединений, изменяющихся в широком диапазоне от наномолярных до микромолярных концентраций.
Смерть нейрональных клеток оценивают количественно с помощью колориметрического метода, описанного Моссманн с соав. (Mossmann et al., J. Immunol. Meth., 65, 55-63, 1983).
Показано, что вплоть до концентрации 10 мкМ все исследованные соединения не оказывают какого-либо нейротоксического воздействия.
Биологическое испытание
Соединения, раскрываемые в настоящей патентной заявке обладают высокой ингибиторной активностью в отношении амилоидоза. Действительно, соединения формулы А препятствуют инициируемой затравкой агрегации А β 1-40 пептида в мономерной форме. Эту активность оценивали в соответствии с описанной ниже процедурой.
Исходный раствор пептидного мономера А β 1-40 готовили путем растворения пептида в диметилсульфоксиде в концентрации 33,33 мг/мл. Исходный раствор затем разбавляли 1 - 11,5 диметилсульфоксидом. Этот раствор затем разбавляли 10 мМ фосфатного буфера, имеющего pH 7,4, содержащего 150 мМ хлорида натрия, для получения раствора для испытания.
В колбу Эппендорфа, содержащую 47 мкл раствора пептидного мономера А β 1-40, добавляли 3 мкл водного раствора испытываемого соединения (830 мкМ), содержащего 66,4 мкМ (в расчете на количество мономера А β 1-40) предварительно образованных обработанных ультразвуком фибрилл А β 1-40; полученный раствор состоял из мономера А β 1-40 (20 мкМ), испытываемого соединения (50 мкМ) и содержал 4 мкМ, в расчете на количество А β 1-40 мономера, предварительно образованных обработанных ультразвуком А β 1-40 фибрилл. Агрегации давали осуществиться в течение двух часов при температуре 37oC. Суспензию затем центрифугировали при 15000 об/мин при температуре +4oC в течение 15 минут, надосадочную жидкость собирали и производили количественную оценку мономера А β 1-40 при помощи ВЭЖХ.
Активность некоторых репрезентативных соединений приведена в Таблице 1. Активность выражена в процентах ингибирования агрегации 20 мкМ раствора мономера А β 1-40, стимулированной 4 мкМ, в расчете на количество мономера А β 1-40, предварительно образованных обработанных ультразвуком А β 1-40 фибрилл.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНТРАЦИКЛИНОВЫЙ ГЛИКОЗИД, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2118328C1 |
| ПРОИЗВОДНЫЕ АЗА-АНТРАЦИКЛИНОНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2159245C2 |
| ПРОИЗВОДНЫЕ 3'-АЗИРИДИНО-АНТРАЦИКЛИНА, СПОСОБЫ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2149163C1 |
| ПРОИЗВОДНЫЕ АНТРАЦИКЛИНА | 1995 |
|
RU2159619C2 |
| БИОЛОГИЧЕСКИ АКТИВНЫЕ ПОЛИМЕРСВЯЗАННЫЕ АНТРАЦИКЛИНЫ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2145965C1 |
| ФТОРИРОВАННЫЕ ПРОИЗВОДНЫЕ 17β-ЗАМЕЩЕННОГО -4-АЗА-5α-АНДРОСТАН-3-ОНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2125061C1 |
| АНТРАЦИКЛИН ГЛИКОЗИД И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1992 |
|
RU2081878C1 |
| КОНЪЮГАТЫ АНТРАЦИКЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 1992 |
|
RU2107690C1 |
| СВЯЗУЮЩИЙ АГЕНТ ДЛЯ БИОАКТИВНЫХ ПРЕПАРАТОВ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2116087C1 |
| 7-ГАЛО- И 7β, 8β-МЕТАНОТАКСОЛЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 1993 |
|
RU2125996C1 |
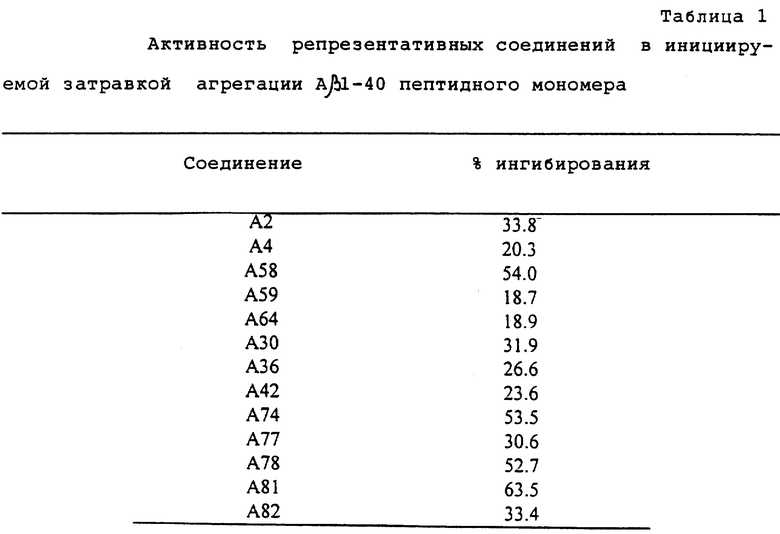
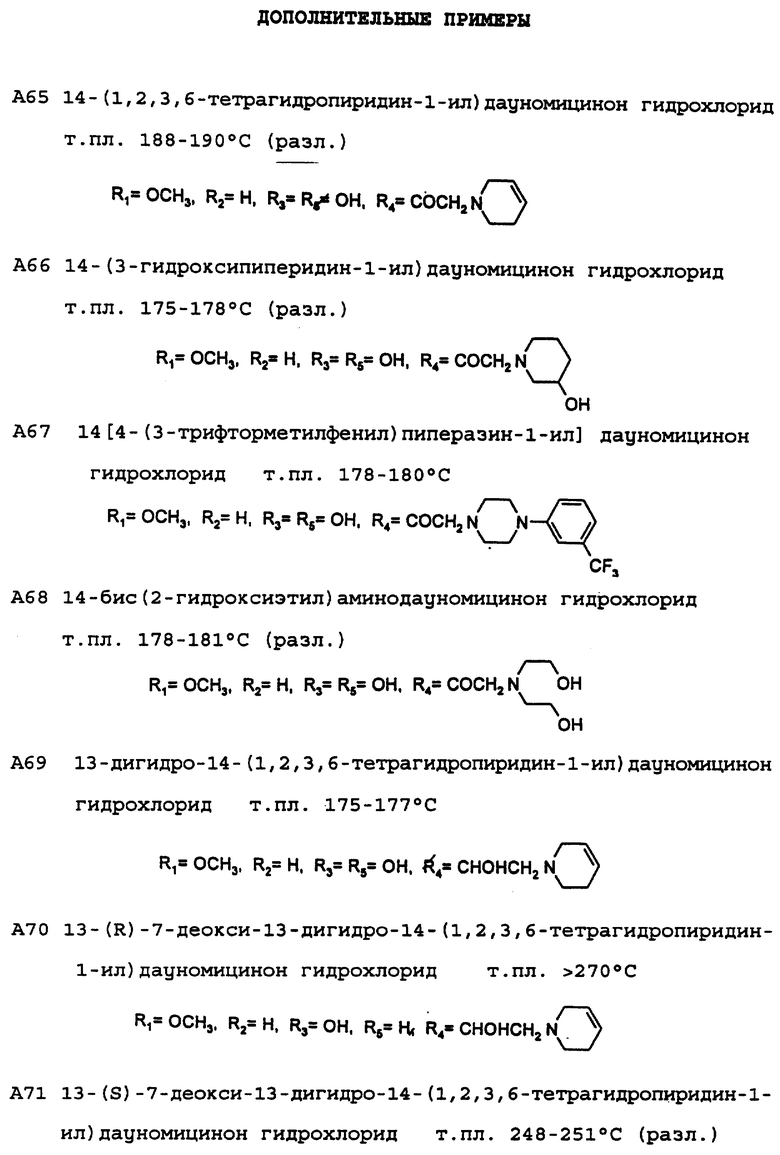
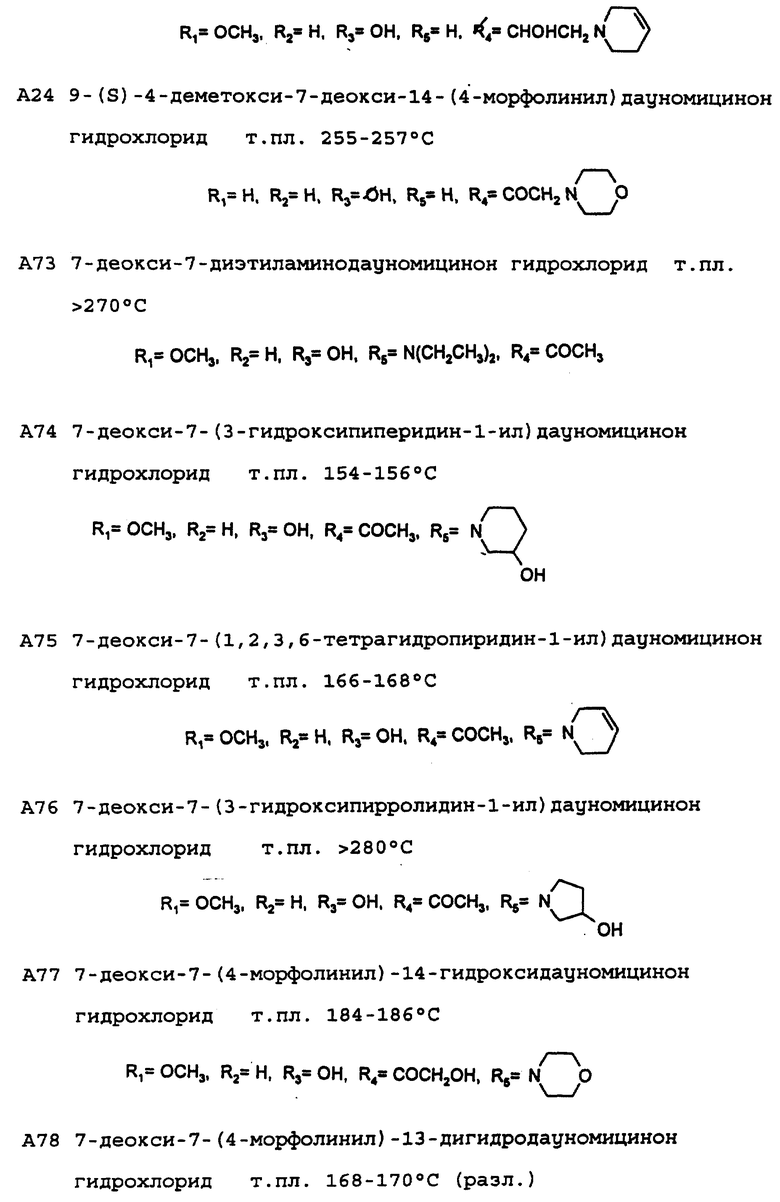
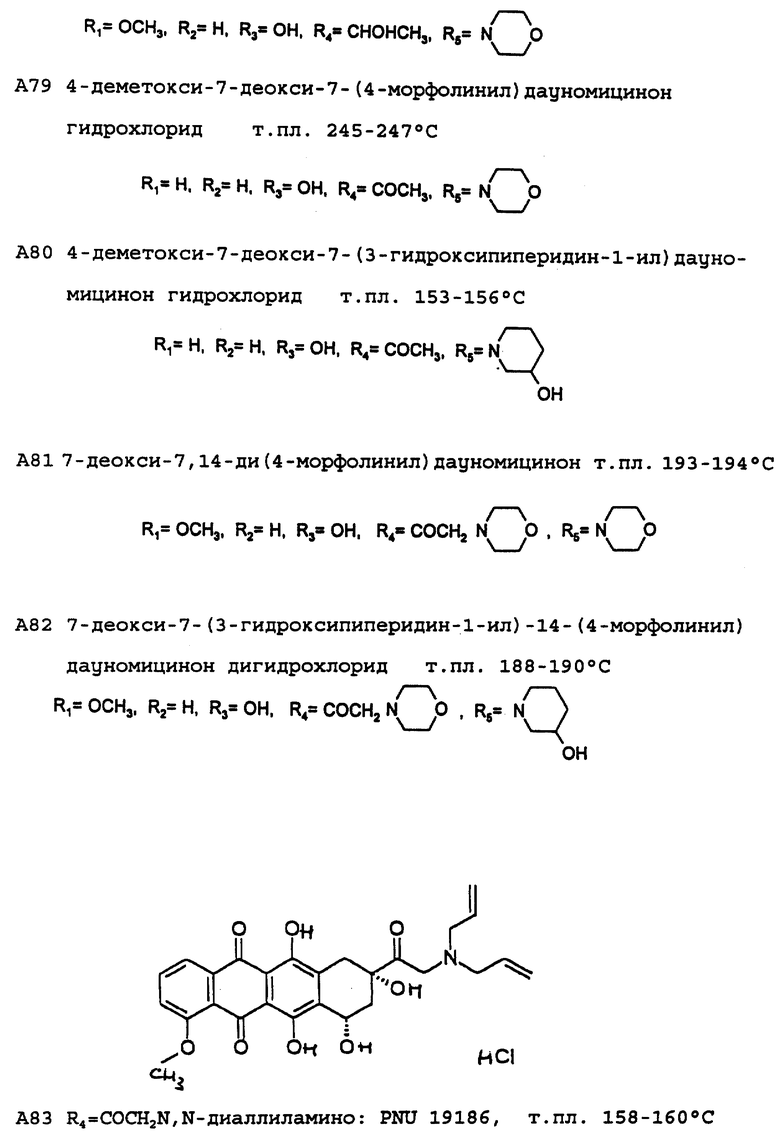
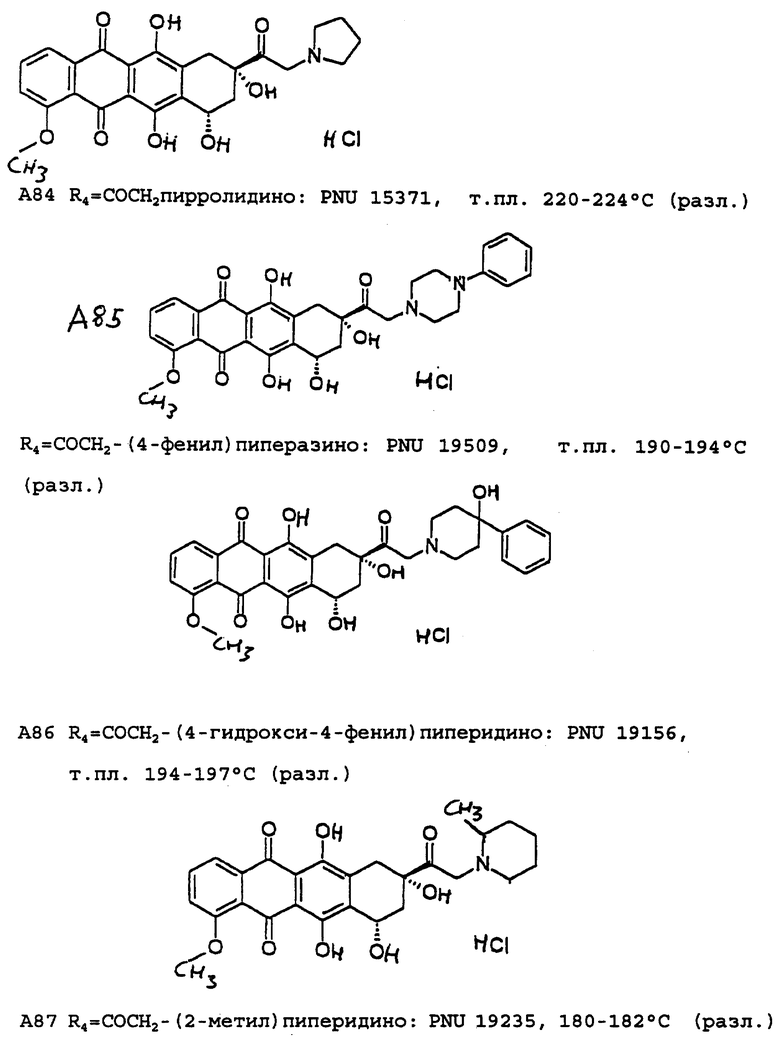
Настоящее изобретение относится к медицине, к новому применению для лечения амилоидоза антрациклинона формулы А, в которой R1, R2, R3, R4 и R5 представляют собой соответствующие заместители. Некоторые соединения формулы А являются новыми. Описаны также способы их получения и содержащие их фармацевтические композиции. Изобретение позволяет повысить эффективность лечения амилоидоза. 3 с. и 14 з.п. ф-лы, 1 табл.
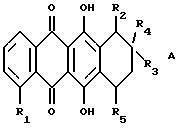
где R1 означает водород, гидрокси или группу OR6, где R6 представляет собой C1-C6 алкил;
R2 означает водород или гидрокси;
R3 означает водород, гидрокси или OR6, где R6 такой, как указано выше;
R4 означает группу формулы XCH2R8, где X означает CO, CHOH, C(OCH2CH2O) и R8 представляет собой водород или OH;
R5 означает водород, гидрокси, группу NR9R10, где R9 означает H, R10 означает водород, фенил(C1-C4 алкил), необязательно замещенный в фенильном кольце одним или более C1-C6 алкокси заместителями, C1-C6 алкил, необязательно замещенный группой гидрокси, или R9 и R10 вместе с атомом азота, к которому они присоединены образуют морфолиновое кольцо, пиперазиновое кольцо, необязательно замещенное C1-C4 алкилом, или пиперидиновое кольцо, или их фармацевтически приемлемых солей для лечения амилоидоза или при получении лекарственного препарата для лечения амилоидоза.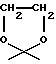
и R8 обозначает водород, группу формулы NR9R10, где R9 и R10, каждый, независимо, выбраны из водорода; C1-C4 алкильной группы; или R9 и R10 вместе с атомом азота, к которому они присоединены, образуют морфолиновое кольцо, пиперазиновое кольцо, необязательно замещенное C1-C4 алкилом, или пиперидиновое кольцо.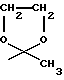
или группу формулы XCH2R8, где X обозначает CO или CH2 и R8 обозначает водород, группу формулы NR9R10, где R9 и R10, каждый, независимо, выбран из водорода, метильной или этильной группы, или R9 и R10, вместе с атомом азота, к которому они присоединены, образуют морфолиновое кольцо, пиперазиновое кольцо, необязательно замещенное C1-C4 алкилом или пиперидиновое кольцо.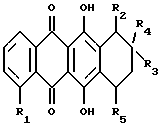
где R1 представляет атом водорода, гидрокси или метокси группу;
R2 представляет водород или гидрокси;
R3 представляет водород или гидрокси;
R4 представляет группу формулы XCH2R8, где X означает CO, CHOH, C(OCH2CH2O) и R8 представляет собой NY, где NY обозначает замещенный атом азота, который может быть частью гетероцикла, выбранного из следующих: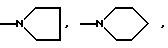

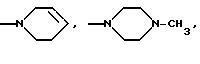
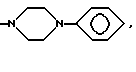
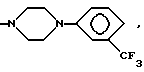
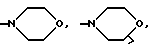
R5 представляет H, OH или содержащую азот группу NX, выбранную из NH2, NHCH2C6H5, NHCH2CH2OH, NHCH2C6H3(OCH3)2,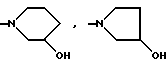
или N(CH2CH3)2, или его фармацевтически приемлемые соли.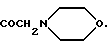
15. Антрациклинон формулы A по п.10 или 11, где R5 является водородом, гидрокси, 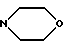 или
или 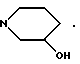
16. Фармацевтическая композиция для лечения амилоидоза, содержащая в качестве активного ингредиента эффективное количество антрациклинона формулы A, указанного в п. 1, или его фармацевтически приемлемой соли в смеси с фармацевтически приемлемым носителем или разбавителем.
| ПРЕДОХРАНИТЕЛЬНЫЙ МЕХАНИЗМ ОТ НЕСАНКЦИОНИРОВАННОГО СРАБАТЫВАНИЯ КУРКА В ОРУЖИИ С НАРУЖНЫМ ВЗВЕДЕНИЕМ КУРКА | 1999 |
|
RU2164335C2 |
| ЭКСТРУДИРОВАННЫЙ СОТОВЫЙ ЭЛЕМЕНТ, В ЧАСТНОСТИ КОРПУС-НОСИТЕЛЬ КАТАЛИЗАТОРА, С УСИЛЕННОЙ СТРУКТУРОЙ СТЕНОК | 1998 |
|
RU2195998C2 |
| ЛИГАНДНОЕ СОЕДИНЕНИЕ α7-НИКОТИНОВОГО АЦЕТИЛХОЛИНОВОГО РЕЦЕПТОРА И ЕГО ПРИМЕНЕНИЕ | 2017 |
|
RU2756604C2 |
| ПРОИЗВОДНЫЕ 7, -8, -9(ЗАМЕЩЕННЫХ)-6-ДЕМЕТИЛ-6-ДЕГИДРОКСИТЕТРАЦИКЛИНОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2127255C1 |
| US 4985548 A, 15.01.91 | |||
| US 4965352 A, 23.10.90. | |||

