Данное изобретение касается лечения амилоидоза, новых соединений для такого лечения, способов их получений и содержащих их фармацевтических композиций.
Связь между амилоидозом, смертью клеток и потерей функции ткани, по-видимому, имеет отношение к различным типам заболеваний, в том числе нейродегенеративных заболеваний. Поэтому предотвращение образования амилоида и/или индуцирование деградации амилоида могут быть важными терапевтическими инструментами для всех патологических нарушений, связанных с амилоидозом, в том числе AL амилоидозом и нейродегенеративными заболеваниями типа болезни Альцгеймера.
Более конкретно настоящее изобретение относится к использованию при изготовлении лекарственного средства для применения в лечении амилоидоза антрациклина формулы А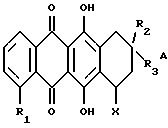
где R1 обозначает водород или гидрокси; группу формулы OR4, в которой R4 представляет собой C1-C6-алкил, C5-C6-циклоалкил или CH2Ph с фенильным (Ph) кольцом, необязательно замещенным 1, 2 или 3 заместителями, выбранными из F, Cl, Br, C1-C6-алкила, C1-C6-алкокси и CF3; или группу формулы OSO2R5, в которой R5 обозначает C1-C6-алкил или Ph, необязательно замещенный 1,2 или 3 заместителями, выбранными из галогена, такого как F, Cl или Br, и C1-C6-алкила;
R2 обозначает водород, гидрокси или OR4, где R4 имеет указанное выше значение;
R3 обозначает водород, метил или группу формулы YCH2R6, в которой Y обозначает CO, CH2, CHOH или группу формулы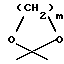
в которой m обозначает 2 или 3, а R6 обозначает водород или гидрокси; группу формулы NR7R8, в которой R7 и R8, каждый независимо, выбраны из:
а) водорода,
b) C1-C6-алкила или C2-C6-алкенила, необязательно замещенного гидрокси, CN, COR9, COOR9, CONR9R10, O(CH2)nNR9R10 (n-обозначает 2-4) или NR9R10, в которой R9R10, каждый независимо, выбран из водорода, C1-C12-алкила, C2-C12-алкенила или фенила, необязательно замещенного одним или несколькими, например 1,2 или 3, заместителями, выбранными из C1-C6-алкила, C1-C6-алкокси, F, Br, Cl, CF3, OH, NH2 или CN;
c) C3-C6 -циклоалкила, необязательно замещенного COR9, COOR9 или OH, где R9 имеет указанное выше значение;
d) фенил C1-C4-алкила или фенил-C2-C4-алкенила, необязательно замещенных в фенильном кольце одним или более, например, 1,2 или 3, заместителями, выбранными из C1-C6-алкила, C1-C6-алкокси, F, Br, Cl, CF3, OH, NH2 или CN1 и
e) COR9, COOR9, CONR9R10, COCH2NR9R10, CONR9COOR10 или SO2R9, где R9 и R10 имеют указанные выше значения, или -R7 и R8 вместе с азотом образуют:
f) морфолиновое кольцо, необязательно замещенное C1-C4-алкилом или C1-C4-алкокси;
g) пиперазиновое кольцо, необязательно замещенное C1-C6-алкилом, C2-C6-алкенилом или фенилом, необязательно замещенным одним или более, например, 1, 2 или 3, заместителями, выбранными из C1-C6-алкила, C1-C6-алкокси, F, Br, Cl, CF3, OH, NH2 или CN, и
h) пирролидиновое, пиперидиновое или тетрагидропиридиновое кольцо, необязательно замещенное OH, NH2, COOH, COOR9 или CONR9R10, где R9 и R10 имеют указанные выше значения, C1-C6-алкилом, C2-C6-алкенилом или фенилом, необязательно замещенным одним или более, например, 1,2 или 3, заместителями, выбранными их C1-C6-алкила, C1-C6-алкокси, F, Br, Cl, CF3 OH, NH2 или CN;
- группу формулы OR4 или SR4, в которых R4 имеет указанное выше значение;
- группу формулы O-Ph, где фенильное кольцо необязательно замещено нитро, амино или NR7R8, как указано выше; или
- группу формулы B или C:
где D обозначает группу формулы COOR9 или CONR7R8, в которых R7, R8 и R9 имеют указанные выше значения; и
X обозначает сахарный остаток формулы X1 или X2: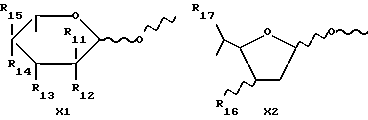
где R11 и R12 оба представляют собой водород или один из R11 и R12 является водородом, а другой является F, Cl, Br или J;
R13 обозначает водород, гидрокси, C1-C4-алкокси, амино, NHCOCF3, N= C(C6H5)2, NHCOR9, NHCONR7R8 или группу формулы E или F: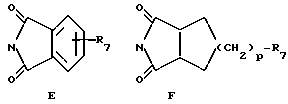
в которых R7, R8 и R9 имеют значения, указанные выше, и p обозначает 0 или 1;
R14 и R15 обозначает водород или один из R14 и R15 является водородом, а другой является OH, F, Cl, Br, I или группой формулы OSO2R5, где R5 имеет указанное выше значение;
R16 обозначает CH2OH или R13, как указано выше;
R17 обозначает F, Cl, Br, I или группу формулы OSO2R5, где R5 имеет указанное выше значение;
и их фармацевтически приемлемых солей; при условии, что соединение формулы A не является 4'-иод-4'-дезоксидоксорубицином (R1 =OCH3, R2=OH, R3= COCH2OH, X=X1, R11=R12=R15=H, R13=NH2 и R14=I).
По следующему аспекту настоящего изобретения предложены новые антрациклины формулы A, как указано выше, где:
- X1 не является остатком, в котором оба R14 и R15 являются атомами водорода или один из R14 или R15 является гидрокси и R13 является аминогруппой, когда R3 является группой формулы YCH3, COCH2NR'7R'8, COСH2R'4 или YCH2OH, где Y имеет указанное выше значение, R'4 обозначает фенил, бензил, C1-C6-алкил или C5-C6-циклоалкил, R'7 и R'8, каждый независимо, обозначают водород, C1-C12-алканоил или, взятые вместе, образуют морфолиновый, пиперазиновый или пиперидиновый остаток;
- X1 не является остатком, в котором R11 и R12 оба являются атомами водорода, R13 обозначает аминогруппу и R14 обозначает иод, когда R1 представляет собой метокси, R2 представляет собой гидрокси и R3 представляет собой COCH2OH.
Каждая алкильная, алкокси или алкенильная группа может быть группой с прямой или разветвленной цепью.
C1C12-алкильная группа, предпочтительно, является C1-C6-алкильной группой. C1-C6-алкильная группа представляет собой, предпочтительно, C1-C4-алкильную группу. C1-C6-алкильная группа, предпочтительно, представляет собой метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, втор-бутил или н-пентил. C1C4-алкильная группа представляет собой, предпочтительно, метил, этил, н-пропил, изопропил, н-бутил, трет-бутил или втор-бутил.
C3-C6-циклоалкильная группа представляет собой, предпочтительно, C5-C6-циклоалкильную группу. C5-C6-циклоалкильная группа представляет собой, предпочтительно, циклопентил или циклогексил.
Пептидильный остаток может содержать до 6, например от 1 до 4, аминокислотных остатков. Подходящими остатками являются
Gly, Ala, Phe, Leu, Gey-Phe, Leu-Gly, Val-Ala, Phe-Ala, Leu-Phe, Phe-Leu-Gly, Phe-Phe-Leu, Leu-Leu-Gly, Phe-Tyr-Ala, Phe-Gly-Phe, Phe-Leu-Gly-Phe, Gly-Phe-Leu-Gly, Glu-Phe-Leu-Gly.
В настоящем изобретении R1 предпочтительно представляет собой водород или метокси. R2 предпочтительно является гидрокси. R3 предпочтительно является группой формулы YCH2R6, в которой Y обозначает CO или группу формулы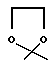
и R6 обозначает водород или гидрокси, группу формулы NR7R8, в которой R7 и R8 независимо выбраны из:
a') водорода,
b') C1-C4-алкила,
c') фенил-C1-C2-алкила, необязательно замещенного в фенильном кольце одной или двумя метоксигруппами, и
d') COCH2NR9R10, в которой R9 и R10 представляют собой метильные группы;
или образуют R7 и R8 вместе образуют:
e') морфолиновое кольцо или
f') пиперазиновое кольцо;
g') тетрагидропиридиновое кольцо,
группу формулы O-Ph, в которой фенильное (Ph) кольцо необязательно замещено NR7R8, как описано выше;
или группу формулы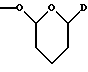
где D обозначает группу формулы CONR7R8, где NR7R8 имеет указанное выше значение; и
x обозначает сахарный остаток формулы XI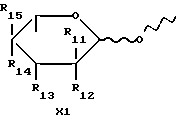
где R11 и R12 обозначают водород, или один из R11 или R12 является водородом, а другой I;
R13 обозначает амино, NHCOCF3, N=C(C6H5)2 или группу формулы
где R14 и R15 обозначают водород, или один из R14 или R15 является водородом, а другой является OH, J или OSO2CH3.
Настоящее изобретение относится к солям тех соединений формулы A, которые имеют образующие соль группы, в частности соли соединений, имеющих карбоксильную группу, основную группу (например, аминогруппу).
Эти соли являются, в частности, физиологически приемлемыми солями, например солями щелочных металлов или щелочноземельных металлов (например солями натрия, калия, лития кальция и магния), солями аммония, солями с подходящими органическим амином или аминокислотой (например, солями, аргинина, прокаина) и солями добавления кислот, образованными с подходящими органическими или неорганическими кислотами, например соляной кислотой, серной кислотой, карбоновой кислотой и сульфоорганическими кислотами (например, уксусной, трифторуксусной, п-толуолсульфоновой кислотой).
Настоящее изобретение включает в себя все возможные стереоизомеры, а также их рацемические или оптически активные смеси.
Конкретные примеры предпочтительных соединений по настоящему изобретению перечислены ниже:
A1: 14-N-(морфолино)-3'-N-трифторацетил-4'-иоддауномицин
R1=OCH3, R2=OH, 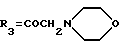 R11=R12=R16=H, R13=NHCOCF3, R14=I
R11=R12=R16=H, R13=NHCOCF3, R14=I
A2: 14-N-(3,4-диметоксибензиламино)-3'-N-трифторацетил-4'- иоддауномицин
R1=OCH3, R2=OH, R3=COCH2 NHCH2[C6H3(OCH3)2], R11=R12=R15=H, R13=NHCOCF3, R14=I
A3: 14-O-[2-(1-пиперазинил)карбонилтетрагидропиран-6-ил] -3'-N- трифторацетил-4'-иоддауномицин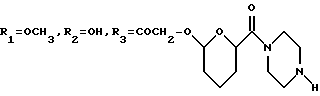
R11=R12=R15=H, R13=NHCOCF3, R14=I
A4: 14-[n-(диметиламинокарбониламино)фенилокси] -3'-N- трифторацетил-4'-иоддауномицин
R1=OCH3, R2=OH, R3=COCH2O- C6H4[n NHCOCH2N (CH3)2],
R11=R12=R15=H, R13=HCOCF3, R14=I
A5: 13-деоксо-13-этилендиокси-14-N-(морфолино)-3'-N- трифторацетил-4'-иоддауномицин
R1= OCH3, R2= OH, R3=C(OCH2CH2O)CH2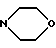 R11=R12=R15=H, R13=NHCOCF3, R14=I
R11=R12=R15=H, R13=NHCOCF3, R14=I
A6: 13-деоксо-13-этилендиокси-14-[n-диметиламинокарбониламино)- фенилокси]-3'-N-трифторацетил-4'-иоддауномицин
R1= OCH3, R2= OH, R3=C(OCH2CH2O)CH2O-C6H4 [n NHCOCH2N(CH3)2], R11=R12= R15=H, R13=HCOCF3, R14=I
A7: 13-дигидро-14-N-(морфолино)-3'-N-трифторацетил-4'- иоддауномицин
R1=OCH3, R2=OH, R3=CHOHCH2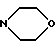 , R11=R12=R15=H, R13=NHCOCF3, R14=I
, R11=R12=R15=H, R13=NHCOCF3, R14=I
A8: 14-N-(морфолино)-3'-N-фталоил-4'-иоддауномицин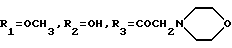
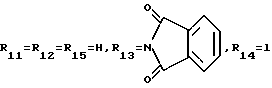
A9: 14-N-(3,4-диметоксибензиламино)-3'-N-фталоил-4'- иоддауномицин
R1= OCH3, R2=OH, R3=COCH2N HCH2[C6H3(OCH2)2], R1=OCH3, R2=OH, R3=COCH2N HCH2[C6H3(OCH3)2]
A10: 14-O-[2-(1-пиперазинил)-карбонилтетрагидропиран-6-ил] - 3'-N-фталоил-4'-иоддауномицин

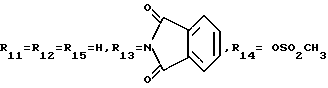
A11: 14-N-(морфолино)-3'-N-трифторацетил-4'-метансульфонат- дауномицин
R1= OCH3, R2= OH, R3= COCH2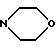 , R11=R12=R15=H, R13=HCOCF3, R14= OSO2CH3
, R11=R12=R15=H, R13=HCOCF3, R14= OSO2CH3
A12: 14-O-[2-(1-пиперазинил)-карбонилтетрагидропиран-6-ил] - 3'-N-трифторацетил-4'-метансульфонатдауномицин
R11=R12=R15=H, R13=NHCOCF3, R14=OSO2CH3
A13: 14-[n-(диметиламинокарбониламино)фенилокси] -3'-N-трифторацетил-4'- метансульфонатдауномицин
R1=OCH3, R2=OH, R3=COCH2O-C6H4 [nNHCOCH2N(CH3)2],
R11=R12=R15=H, R13=HCOCF3, R14=OSO2CH3
A14:14-N-(морфолино)-3'-N-фталоил-4'-метансульфонатдауномицин
A15: 3'-N-дифенилметилен-4'-эпидаунорубицин
R1=OCH3, R2=OH, R3=COCH3, R11=R12=R14=H, R13=NC(C6H5)2,
R15=OH
A16: 3'-N-дифенилметилен-4'-иодоксорубицин
R1=OCH3, R2=OH, R3=COCH2OH, R11=R12=R15=H, R13=C(C6H5)2,
R14=I
A17: 14-N-(морфолино)-3'-N-дифенилметилен-4'-иоддауномицин
R1=OCH3, R2=OH, R3=COCH2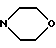 , R11=R12=R15=H,
, R11=R12=R15=H,
R13=NC(C6H5)2, R14=I
A18: 4-деметокси-2'-иоддаунорубицин
R1=H, R2=OH, R3=COCH3, R11=R15=H, R12=1, R13=NH2, R14=OH
Соединения формулы A могут быть приготовлены, в зависимости от природы заместителей, исходя из известных антрациклинов, подходящими химическими модификациями агликоновой или сахарной части молекулы или обеих частей молекулы или соединением антрациклинонов с сахарами.
Способами получения соединений формулы A и их фармацевтически приемлемых солей являются следующие способы:
(i) Предпочтительный способ получения соединений формулы A, где R3 представляет собой группу формулы COCH2NR7R8, где R7 и R8 имеют указанные выше значения, при условии, что R7 и R8 не являются группами COR9, CONR9R10, CONR9COOR10 или SO2R9, в которых R9 и R10 имеют указанные выше значения, заключается в следующем:
1) превращение соединения формулы G
где R6 обозначает водород,
R1, R2, X имеют указанные выше значения,
при условии, что в G не присутствуют остатки алкенила, и в сахарном остатке X R13 не является гидрокси, когда один из остальных заместителей X является гидрокси, в соответствующее 14-бромпроизводное, затем
2) взаимодействие полученного бромпроизводного формулы H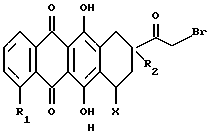
в которой R1, R2 и X имеют данные выше значения, с подходящим амином формулы NHP7R8, где R7 и R8 имеют данные выше значения, при условии, что R7 и R8 не являются группами COR9, CONR9R10, CONR9COOR10 или SO2R9, описанными выше, и, если желательно, превращение полученного соединения формулы A в его фармацевтически приемлемую соль.
(ii) В другом примере соединения A, описанные в (i), могут далее быть преобразованы в другие антрациклины формулы A, в которых один или оба R7 и R8 обозначают группу формулы COR9 или SO2R9, где R9 имеет данное выше значение, путем взаимодействия 14-аминопроизводного формулы A, описанного в (i), при условии, что один или оба R7 и R8 обозначают атом водорода, с ацильным производным формулы HalCOR9 или HalSO2R9, где Hal представляет собой галоген, а R9 имеет указанное выше значение, и, если желательно, превращения полученного указанного соединения формулы A и его фармацевтически приемлемую соль.
(iii) В другом примере предпочтительный способ получения соединений формулы A, где R3 представляет собой группу формулы B или C, указанные выше, при условии, что R1 и заместители сахарного остатка X не являются первичными гидроксигруппами, заключается в следующем:
1) взаимодействие соединения, представленного формулой G, в которой R6 обозначает гидрокси, а R1 и X указаны выше, с соединением формулы B1 или C1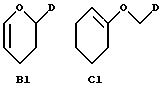
где D имеет указанное выше значение, и, если желательно, удаление защиты у защищенных гидроксигрупп, и, если, желательно, превращение полученного указанного соединения формулы A в его фармацевтически приемлемую соль.
(iv) В другом примере предпочтительный способ получения соединений формулы A, где R3 представляет собой CHOHCH2R6, заключается в восстановлении соединения формулы A, в которой R3 обозначает COCH2R6, где R6 имеет указанное выше значение, при условии, что в A не присутствуют дополнительные кетогруппы, и, если желательно, превращение полученного указанного соединения формулы A в его фармацевтически приемлемую соль.
(v) В другом примере предпочтительный способ получения соединений формулы A, где R3 является группой формулы CH2CH2R6, включает:
1) преобразование соединения формулы A, в котором R3 является COCH2R6, при условии, что в A не присутствуют дополнительные кетогруппы, в 13-(замещенный)-бензолсульфонилгидразон, предпочтительно, 13-(n-фтор)бензолсульфонилгидразон, затем
2) восстановление его в условиях, способных сохранять гликозидную связь, и, если желательно, превращение полученного указанного соединения формулы A в его фармацевтически приемлемую соль.
(vi) В другом примере способ получения соединений формулы A, в которой R11 и R12 являются атомами водорода, включает:
1) конденсацию агликона формулы K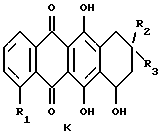
где R1, R2 и R3 имеют указанные выше значения, при условии,
что R1, R2 и R3 не являются группами, несущими первичные или вторичные гидроксигруппы,
с производным сахара формулы L1 или L2
где R18 обозначает подходящую отщепляемую группу, такую как атом галогена, например атом хлора, или активированный эфирный остаток, такой как OCOCF3 или OCO (n-NO2C6H5),
R19 и R20 обозначают атомы водорода,
R21 обозначает водород, C1-C4-алкокси, остаток эфира,
такой как OCOCF3 или OCO (n-NO2C6H5) или группу NHCOCF3,
R22 и R23 оба обозначают водород или один из R22 или R23 является водородом, а другой является эфирным остатком, таким как OCOCF3 или OCO(n-NO2C6H5) или группой NHCOCF3.
R24 обозначает CH2OCOCF3, или имеет то же значение, что и R21 и выше, и
R25 обозначает OCOCF3 или OCO(n-NO2C6H5), затем
(vi) удаление защиты амино- и гидроксигрупп, и, если желательно, превращение полученного соединения формулы A в его фармацевтически приемлемую соль.
(vii) В другом примере предпочтительный способ получения соединений формулы A, в которой R13 представляет собой E или F, предусматривает:
1) взаимодействие антрациклина формулы A, как указано выше, который имеет только первичную аминогруппу, с галогенацилпроизводным формулы E1 или F1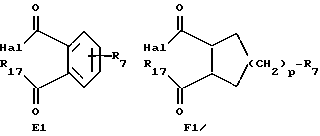
где R7 имеет указанное выше значение,
Hal обозначает атом галогена, и
R17 является алкоксиостатком, предпочтительно этокси, и, если желательно,
2) обработку полученного моноаминоацилпроизводного основанием с образованием групп формул E или F, и, если желательно, превращение полученного указанного соединения формулы A в его фармацевтически приемлемую соль.
Соединения формулы A, как указано в (i), могут быть получены, как описано в DE-A-2557537, например взаимодействием соединения формулы H, как указано выше, полученного из соединения G согласно DE-A-1197874, с 1-1,2 эквивалентными подходящего амина формулы NHR7 R8, где R7 и R8 указаны выше, при условии, что R7 и R8 не являются группой формул COP9, CONR9R10, CONR9COOR10 или SO2R9, как указано выше, в сухом полярном растворителе, таком как ацетон или диметилформамид, при температуре приблизительно 0-30oC, предпочтительно при комнатной температуре, в течение 4 - 24 часов, и, если желательно, превращением полученного указанного соединения формулы A в его фармацевтически приемлемую соль, предпочтительно с безводным хлористым водородом в метаноле.
Соединения формулы A, описанные в (ii), могут быть получены путем взаимодействия соединения формулы A, указанного в (i), с ацилпроизводным формулы HalCOR9 или HalSO2R9, где Hal обозначает галоген, а R9 имеет указанное выше значение, в сухом полярном растворителе, таком как ацетон или диметилформамид, при температуре приблизительно 0 - 30oC, предпочтительно при комнатной температуре, в течение 4 - 24 часов.
Соединения формулы A, как указано в (iii), могут быть получены, как описано в WO 92/10212 и WO 92/02255, например взаимодействием антрациклина, описанного в (iii) (1), с производными формулы B1 или C1 в апротонном растворителе, таком как метиленхлорид, в присутствии кислотного катализатора, такого как п-толуолсульфонат пиридиния, при температуре 10 - 30oC, предпочтительно при комнатной температуре, в течение 3 - 24 часов, и, если желательно, превращением полученного указанного соединения формулы A в фармацевтически приемлемую соль, предпочтительно с безводным хлористым водородом в метаноле; или гидролизом эфирного производного разбавленным водным гидроксидом натрия.
Соединения формулы A, описанные в (iv), могут быть получены восстановлением антрациклинов формулы A, указанных в (iv), в воде или в одном или более органических растворителях, в зависимости от природы соединения, или при помощи микробного восстановления. Например, водорастворимые антрациклины восстанавливают в воде при pH 8-9, предпочтительно при pH 8,5, в присутствии восстановителя, такого как боргидрид натрия, при температуре от 0oC до комнатной температуры, в течение 1 - 10 минут, как описано в Gaz. Chim. Ital., 114, 185 (1984). Нерастворимые в воде антрациклины предпочтительно растворяют в безводном апротонном органическом растворителе, таком как сухой тетрагидрофуран, охлаждают до -50oC, обрабатывают 1,5 эквивалентами эфирата бромида магния и 1,5 эквивалентами боргидрида натрия в течение 5 - 30 минут, затем добавляют каплями метанол. 13-Дигидропроизводное формулы A, описанное выше, извлекают из реакционной смеси экстракцией метиленхлоридом и промыванием водой. Микробное восстановление антрациклинов, описанных в (iv), можно проводить, например, с применением блокированного мутанта Streptomyces peucetius, как описано в Gaz. Chim. Ital. 114, 185, (1984).
Соединения формулы A, описанные в (v), могут быть получены, как описано с GB-A-2238540, например восстановлением производного 13-[(4-фтор)бензолсульфонил)гидразона антрациклина формулы A, описанного в (v) (1), цианоборгидридом натрия в органическом растворителе, таком для толуол или диметилформамид, при температуре 25 - 80oC, в течение 6 - 24 часов.
Соединения формулы A, описанные в (iv), могут быть получены конденсацией антрациклинона формулы K, описанного выше, при помощи реакции Кенинга-Кнорра с производным галогенированного сахара формулы L1, или L2, описанных выше, в сухом аполярном растворителе, таком как оксид ртути, в присутствии конденсирующего агента, такого как оксид ртути, бромид ртути, и молекулярных сит, описанных в DE-A-2525633. Альтернативный способ предусматривает конденсацию антрациклинона формулы K с производным галогенированного сахара формулы L1 или L2, описанным выше, в сухом аполярном растворителе, таком как метиленхлорид, с трифторметансульфонатом серебра, растворенным в этиловом эфире, при температуре 0 - 25oC в течение 1 - 6 часов, как описано в BE-A-842930.
Соединения формулы A, описанные в (vii), могут быть получены взаимодействием антрациклина, имеющего только первичную аминогруппу, с галогенпроизводным формулы E1 или F1, описанным выше, в органическом растворителе, таком как тетрагидрофуран или диметилформамид, при 0oC - комнатной температуре в течение 1 - 24 часов. Моноацилпроизводное обрабатывают затем конденсирующим агентом, таким как тетрабутиламмонийфторид, с получением (циклический ацил)антрациклина.
Другие антрациклины формулы A можно получить аналогичным образом, исходя из известных соединений при помощи известных процедур.
Например, следующие соединения известны и могут быть представлены той же формулой A, в которой X обозначает сахар формулы X1:
даунорубицин (A19: R1= OCH3, R2=OH, R3=COCH3, R11=R12=R15=H, R13=NH2, R14=OH);
доксорубицин (A20: R1=OCH3, R2=OH, R3=COCH2OH, R11=R12=R15=H, R13=NH2, R14=OH);
4-деметоксидаунорубицин (A21: R1=H, R2=OH, R3=COCH3, R11=R12=R15=H, R13= NH2, R14=OH);
4-деметоксидоксорубицин (A22: R1= H, R2=OH, R3=COCH2OH, R11=R12=R15=H, R13=NH2, R14=OH);
4'-эпидаунорубицин (A23: R1=OCH3, R2OH, R3COCH3, R11=R12=R14=H, R13=NH2, R15=OH);
4'-эпидоксорубицин (A24: R1=OCH3, R2=OH, R3=COCH2OH, R11R12=R14=H, R13= NH2, R15=OH);
4'-деоксидаунорубицин (A25: R1=OCH3, R2=OH, R3=COCH3, R11R12=R14=R15=H, R13=NH2);
4'-деоксидоксорубицин (A26: R1=OCH3, R2=OH, R3=COCH2OH, R11=R12=R14=R15= H, R13=NH2);
4'-иоддаунорубицин (A27: R1=OCH3, R2=OH, R3=COCH3, R11=R12=R15=H, R13= NH2, R14=I);
4'-иоддоксорубицин (A28: R1=OCH3, R2=OH, R3=COCH2OH, R11=R12=R15=H, R13= NH2, R14=I);
9-деоксидаунорубицин (A29: R1=OCH3, R2=H, R3=COCH3, R11=R12 R15=H, R13= NH2, R14=OH);
9-деоксидоксорубицин (A30: R1= OCH3, R2=H, R3=COCH2OH, R11=R12=R15=H, R13=NH2, R14=OH);
9-деацетилдаунорубицин (A31: R1=OCH3, R2=OH, R3=H, R11=R12=R15=H, R13= NH2, R14=OH);
9-деацетил-9-деоксидаунорубицин (A32: R1=OCH3, R2=R3=H, R11= R12= R15=H, R13= NH2, R14=OH);
9-деацетил-9-гидроксиметилендаунорубицин (A33: R1=OCH3, R2=CH2OH,R3=H, R11=R12=R15=H, R13=NH2, R14=OH);
13-дигидродаунорубицин (A34: R1=OCH3, R2=OH, R3=CHOHCH3, R11=R12=R15=H, R13=NH2, R14=OH);
13-дигидродоксорубицин (A35: R1=OCH3, R2=OH, R3=CHOHCH2OH, R11=R12R15=H, R13=NH2, R14=OH);
13-дигидро-4-деметоксидаунорубицин (A36: R1=H, R2=OH, R3=CHOHCH3, R11= R12=R15=H, R13=NH2, R14=OH);
13-дигидро-4'-эпидаунорубин (A37: R1=OCH3, R2=OH, R3=CHOHCH3, R11=R12= R14=H, R13=NH2, R15=OH);
13-дигидро-4'-эпидоксорубицин (A38: R1=OCH3, R2=OH, R3=CHOHCH2OH, R11= R12=R14=H, R13=NH2, R15=OH);
13-дигидро-4-иоддоксорубицин (A39: R1= OCH3, R2=OH, R3=CHOHCH2OH, R11= R12=R15=H, R13=NH2, R14=I);
13-деоксодаунорубицин (A40: R1= OCH3, R2=OH, R3=CH2CH3, R11=R12=R15=H, R13=NH2, R14=OH);
N-трифторацетилдаунорубицин (A41: R1=OCH3, R2=OH, R3=COCH3, R11=R12=R15= H, R13=HCOCF3, R14=OH);
N-трифторацетилдоксорубицин (A42: R1=OCH3, R2=OH, R3=COCH2OH, R11=R12= R15=H, R13=NHCOCF3, R14=OH);
N-трифторацетил-4-деметоксидаунорубицин (A43: R1= H, R2=OH, R3=COCH3, R11=R12=R15=H, R13=NHCOCF3, R14=OH);
N-трифторацетил-4'-эпидаунорубицин (A44: R1=OCH3, R2=OH, R3=COCH3, R11= R12=R14=H, R13=NHCOCF3, R15=OH);
N-трифторацетил-4'-иоддаунорубицин (A45: R1=OCH3, R2=OH, R3=COCH3, R11= R12=R15=H, R13=NHCOCF3, R14I);
(см. F. Arcamone in "Doxorubicn" Medicinal Chemisty, vol. 17, Academic Press. 1981) или
4'-деокси-4'-метансульфонатдаунорубицин (A46: R1=OCH3, R2=OH, R3=COCH3, R11=R12=R15=H, R13-NH2, R14=OSO2CH3);
4'-деокси-4'-метансульфонатдоксорубицин (A47: R1= OCH3, R2= OH, R3= COCH2OH, R11=R12=R15=H, R13=NH2, R14=OSO2=CH3) (см. WO 95/16693).
Некоторые из вышеупомянутых антрациклинов, в частности 4'-эпидоксорубицин, являются также предпочтительными соединениями в объеме данного изобретения.
Некоторые агликоны, применяемые для получения антрациклинов формулы A, описанных в (vi), также известны и могут быть представлены формулой K, описанной выше, например;
дауномицинон (K1: R1=OCH3, R2=OH, R3=COCH3;
адриамицинон (K2: R1=OCH3, R2=OH, R3=COCH2OH);
4-деметоксидауномицион (K2: R1=H, R2=OH, R3=COCH3).
Известны также некоторые сахара, применяемые для получения антрациклинов формулы A, описанных в (vi), и могут быть представлены формулой M: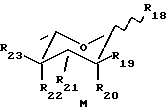
такие как аминосахара дауносамин, 3-амино-2,3,6-тридеокси-L-ликсо-гексопираноза (M1: R18= OH, R19=R20=R23=H, R21=NH2, R22=OH) (см. J. Am. Chem. Soc. , 86, 5334, 1964) или акозамин, 3-амино-2,3,6-тридеокси-L-арабино-гексопираноза (M2: R18=OH, R19=R20=R22=H, R21=NH2, R23=OH) (см. J. Med. Chem, 18, 703, 1975) или соответствующие 1-хлор-3,4-дитрифторацетилдаунозаминилпроизводные (M3: R18=Cl, R19=R20R23=H, R21=NHCOCF3, R22=OCOCF3) или 1-хлор-3,4-дитрифторацетилакозаминилпроизводные (M3: R18= Cl, R19=R20=R22=H, R21= NHCOCF3, R23=OCOCF3) или диаминосахара, такие как L-фруктоза (M4: R18= R19=R21=R22=OH, R20=R23=H) и L=рамноза (M5: R18=R20=R21=R23=OH, R19=R22=H).
Соединения по данному изобретению характеризуются высокой активностью ингибирования амилоидоза.
Термин амилоидоз обозначает многочисленные заболевания, общей чертой которых является склонность к полимеризации и осаждению определенных белков в виде нерастворимых фибрилл во внеклоточном пространстве, что вызывает структурное и функциональное повреждение органа и тканей. Классификация амилоида и амилоидоза недавно пересмотрена в Bulletin of the World Health Organization, 71 (1):105 (1993).
Все разные типы амилоида имеют общую ультраструктурную организацию в виде антипараллельных β-сложенных слоев, несмотря на тот факт, что они содержат множество сильно отличающихся белковых субъединиц [см.: Glenner G.G., New England J,. Med., 302 (23): 1283-8(1980)]. AL амилоидоз обусловлен необычными легкими цепями многоклонального иммуноглобулина, которые образуют амилоидные фибриллы. Эти моноклональные легкие цепи продуцируются моноклональными клетками плазмы с низким митотическим индексом, что объясняет их хорошо известную нечувствительность к химиотерапии. Злокачественность этих клеток состоит в их белоксинтезирующей активности.
Клиническое течение этого заболевания зависит от селективности поражения органов; прогноз может быть крайне неблагоприятным в случае инфильтрации сердца (среднее выживание менее 12 месяцев) или более обнадеживающим в случае поражения почки (среднее выживание приблизительно 5 лет).
Принимая во внимание относительную нечувствительность амилоидогенных отложений к протеолитическому разрушению, можно полагать, что только разработка молекулы, которая может блокировать или замедлять образование амилоида и увеличивать растворимость уже существующих отложений амилоида, дает, по-видимому, вероятную надежду в случае больных с AL амилоидозом.
Кроме того, поскольку надмолекулярная организация амилоидных фибрилл одинакова для всех типов амилоида, наличие лекарственного средства, которое препятствует образованию амилоида и увеличивает растворимость существующих отложений, делая возможным очищение при помощи нормальных механизмов, могло бы привести к большому успеху в случаях всех типов амилоидоза и, в частности, при лечении болезни Альцгеймера.
Действительно, основной патологической особенностью болезни Альцгеймера (AL), синдрома Дауна, старческого слабоумия (Dementia pugilistica) и церебральной амилоидной ангиопатии является отложение амилоида в паренхиме мозга и в стенках сосудов. Эти маркеры ассоциированы с потерей нервных клеток в коре головного мозга, лимбических районах и подкорковых ядрах. Некоторые исследования показали, что селективное повреждение различных нейронных систем и потеря синапса в лобной части коры головного мозга коррелировали с прогрессированием ухудшения познавательной способности. Патогенез и молекулярная основа нейродегенеративных процессов в АД не известны, по роль β -амилоида, отлагающегося в паренхиме мозга и стенках сосудов, была выдвинута на первый план недавним сообщением о его нейротоксической активности in vitro и in vivo (Janker et al, Science, 245: 417, 1990. Kowall et al., PNAS, 88: 7247, 1991). Кроме того, сегрегация хорошо знакомой болезни Альцгеймера (АД) с мутацией гена предшественника белка амилоида (APP) вызвала интерес к потенциальной патогенетической функции β-амилоида в болезни Альцгеймера [Mullan M. et al., TINS, 16 (10): 392 (1993)].
Нейротоксичность β -амилоида была ассоциирована с фибриллогенными свойствами белка. Исследования с гомологичными синтетическими пептидами показывают, что клетки гиппокампа были нечувствительны к обработке свежим раствором β 1-42 в течение 24 часов, тогда как их жизнеспособностью снижалась при экспозиции нервных клеток с β 1-42, предварительно хранившимся в солевом растворе в течение 2-4 дней при 37oC для способствования агрегации пептидов. Связь между фибриллами и нейротоксичностью подтверждается далее недавним результатом, показывающим, что растворимая форма β -амилоида продуцируется in vivo и in vitro во время нормального клеточного метаболизма (Hass et al, Nature, 359, 322, 1993), и только при агрегации амилоида в конгофильное образование он ассоциировался с дистрофическим невритом. С другой стороны, не-конгофильное "преамилоидное" образование, состоящее из единственной молекулы β -амилоида, не было связано с изменением нейронов (Tagliavini et al, Neurosci. Lett. 93: 191, 1988).
Нейротоксичность β -амилоида также была подтверждена с применением пептидного гомолога β -амилоидного фрагмента 25-35 ( β 25-35), сохраняющего свойства самоагрегации полного β -амилоидного фрагмента β 142.
Хроническое, но неострое воздействие нейронов гиппокампа на микромолярную концентрацию β 25-35 индуцировало смерть нервных клеток путем активации механизма запрограммированной смерти клеток, известной как апоптоз (Forloni et al. Neuro-Report, 4:523, 1993). В этом случае нейротоксичность также ассоциировалась со свойством самоагрегации β 25-35.
Другие нейродегенеративные нарушения, такие как губкообразная энцефалопатия (SE), характеризуются смертью нервных клеток и внеклеточным отложением амилоида, в этом случае происходящего из белка Приона (PrP). Параллельно с наблюдением, что β -амилоид является нейротоксичным, исследовали действие синтетических пептидов, гомологичных разным сегментам PrP, на жизнеспособность первичных нейронов гиппокампа крыс.
Длительное применение пептида, соответствующего PrP 106-126, вызвало смерть нейронов в результате апоптоза, тогда как при тех же условиях все другие испытанные пептиды и беспорядочно собранная последовательность PrP 106-126 не снижали жизнеспособность клеток (Forloni et al, Nature, 363: 543). PrP 106-126 был высокофибриллогенным in vitro и при окрашивании Конго красным пептидный агрегат обнаружил зеленое окрашивание, свидетельствующее о конформации в виде β -слоев (пластов), характерной для амилоида.
Соединения по данному изобретению можно применять для приготовления лекарственных средств, применимых для предотвращения или остановки прогрессирования заболеваний, вызываемых амилоидными белками, таких как AL амилоидоз, синдром Альцгеймера или синдром Дауна и т.п.
Данное изобретение также включает в свой объем фармацевтические композиции, содержащие одно или более соединений формулы A или их фармацевтически приемлемые соли в качестве активных ингредиентов в сочетании с фармацевтически приемлемыми носителями, наполнителями и другими добавками, если необходимо.
Фармацевтические композиции, содержащие соединение формулы A или его соли, могут быть приготовлены обычным способом с использованием обычных нетоксичных фармацевтических носителей или разбавителей в виде различных лекарственных форм для различных путей введения.
В частности, соединения формулы A могут быть введены:
A) перорально, например, в виде таблеток, пастилок, лепешек водной или масляной суспензии, диспергируемых порошков или гранул, эмульсий, твердых или мягких капсул или сиропов или эликсиров. Композиции, предназначенные для перорального введения, могут быть приготовлены согласно любому способу, известному в этой области для производства фармацевтических композиций, и такие композиции могут содержать один или несколько агентов из группы, содержащей подслащивающие средства, ароматизаторы и консервирующие средства, для обеспечения приятных на вид и съедобных фармацевтических препаратов.
Таблетки содержат активный ингредиент в смеси с нетоксичными фармацевтическими приемлемыми наполнителями, которые пригодны для производства таблеток. Эти наполнители могут представлять собой, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие и разрыхляющие агенты, например кукурузный крахмал или альгиновую кислоту; связующие средства, например кукурузный крахмал и аравийскую камедь, и смачивающие агенты, например стеарат магния или стериновую кислоту или тальк. Таблетки могут не иметь покрытия или они могут быть покрыты известными способами для замедления разрушения и поглощения в желудочно-кишечном тракте и, следовательно, обеспечения пролонгированного действия в течение более длительного периода. Например, можно использовать такой материал для пролонгирования действия, как глицерилмоностеарат или глицерилдистеарат. Готовая лекарственная форма для перорального применения может быть также представлена в виде жесткой желатиновой капсулы, в которой активный ингредиент смешан с инертным твердым разбавителем, например карбонатом кальция, фосфатом кальция или каолином, или мягкими желатиновыми капсулами, в которых активный ингредиент смешан с водой или масляной средой, например арахисовыми маслом, жидким парафином или оливковым маслом. Водные суспензии содержат активные материалы в смеси с наполнителями, пригодными для изготовления водных суспензий.
Такие наполнители представляют собой суспендирующие агенты, например такие как карбоксиметилцеллюлоза натрия, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, смола трагаканта и аравийская камедь; диспергирующие или смачивающие агенты могут представлять собой встречающиеся в природе фосфатиды, например лецитин, или продукты конденсации алкиленоксида с жирными кислотами, например полиэтиленстеарат, или продукты конденсации этиленоксида с длинноцепочечными алифатическими спиртами, например гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с неполными сложными эфирами, производными жирных кислот и гексита, например полиоксиэтиленсорбитмоноолеат, или продукты конденсации этиленоксида с неполными сложными эфирами, производными жирных кислот и ангидридов гексита, например полиоксиэтиленсорбитанмоноолеат. Водные суспензии могут также содержать один или несколько консервантов, например н-пропил-п-гидроксибензоат, один или более красителей, один или более ароматизаторов, или один или более подслащивающих средств, таких как сахароза или сахарин. Масляная суспензия может быть получена суспендированием активного ингредиента в растительном масле, например в арахисовом масле, оливковом масле, кунжутном масле или кокосовом масле, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загуститель, например пчелиный воск, твердый парафин или цетиловый спирт. Подслащивающие вещества такие, которые указаны выше, и ароматизаторы могут быть добавлены для придания пероральному препарату вкуса. Эти композиции могут содержать консерванты, такие как автооксиданты, например аскорбиновую кислоту. Диспергируемые порошки и гранулы, пригодные для приготовления водной суспензии путем добавления воды, обеспечивают активный ингредиент в смеси с диспергирующим для смачивающим агентом, суспендирующим агентом, и одним или нескольким консервантами. Примеры пригодных диспергирующих и смачивающих и суспендирующих агентов приведены выше. Могут также присутствовать дополнительные наполнители, например подслащивающие, ароматизирующие агенты.
Фармацевтические композиции по изобретению могут быть также в форме эмульсий типа масло в воде. Масляной фазой может быть растительное масло, например оливковое масло или арахисовое масло, или минеральное масло, например жидкий парафин, или их смеси.
Подходящие эмульгаторы могут представлять собой природные камеди, например, такие как аравийская камедь или трагакантовая камедь, встречающиеся в природе фосфатиды, например лецитин сои, и эфиры или неполные эфиры, производные жирных кислот и ангидридов гексита, например сорбитанмоноолеат, и продукты конденсации неполных эфиров с этиленоксидом, например полиоксиэтиленсорбитанмоноолеат. Эмульсия может также содержать подслащивающие и ароматизирующие вещества. Сиропы и эликсиры могут быть приготовлены с подслащивающими веществами, например глицерином, сорбитом или сахарозой. Такие формы могут также содержать средство, уменьшающее раздражение, консервант, ароматизаторы и красители.
B) парентерально, либо подкожно, либо внутривенно, либо внутримышечно, либо внутригрудинно или инфузионным способом, в форме стерильной инъецируемой водной или маслянистой суспензии. Фармацевтические композиции могут быть в форме стерильных инъецируемых водных или масляных суспензий.
Эти суспензии могут быть приготовлены известным способом с применением указанных выше диспергирующих или смачивающих агентов и суспендирующих агентов. Стерильный инъецируемый препарат может также представлять собой стерильные инъецируемые раствор или суспензии в нетоксичном парентерально приемлемом разбавителе или растворителе, например в виде раствора в 1,3-бутандиоле. Среди приемлемых носителей и растворителей, которые можно использовать, можно указать воду, раствор Рингера и изотонический раствор хлористого натрия. Кроме того, стерильные жирные масла (с большим содержанием жира) являются обычно используемыми в качестве растворителя или суспендирующей среды.
Для этой цели могут использоваться любые мягкие жирные масла, в том числе синтетические моно- или диглицериды. Кроме того, такие жирные кислоты, как олеиновая кислота, находит применение в приготовлении инъецируемых препаратов.
Еще одним объектом изобретения является способ борьбы со связанными с амилоидозом заболеваниями путем введения терапевтически эффективного количества одного или более активных соединений формулы A людям, нуждающимся в подобном лечении.
Суточные дозы находятся в диапазоне приблизительно 0,1-50 мг на кг веса пациента, в зависимости от активности конкретного соединения, возраста, веса и состояния подвергаемого лечению субъекта, типа и тяжести заболевания и частоты и пути введения; предпочтительно уровни суточных доз находятся в диапазоне 5 мг - 2 г. Количество активного ингредиента, которое может быть вместе с материалами-носителями для получения формы разовой дозы, будет изменяться в зависимости от подвергаемого лечению субъекта и конкретного способа введения. Например, состав, предназначенный для перорального введения, может содержать 5 мг - 2 г активного агента вместе с приемлемым и удобным количеством материала-носителя, которое может изменяться от около 5 до около 95% от всей композиции. Единичные дозированные формы обычно будут содержать от около 5 мг до около 500 мг активного ингредиента.
Следующие примеры иллюстрируют изобретение, но не ограничивают его.
Пример 1. Получение 14-N-(морфолино)-3'-N-трифторацетил-4'-иоддауномицина (A1)
Гидрохлорид 4'-иоддаунорубицина (A27, 1,34 г, 2 ммоль) растворяли в смеси диоксана (30 мл) и метанола (20 мл) и обрабатывали раствором 10% брома в хлороформе (2 мл) при 0oC, как описано в US-A-4438105. Спустя 1 час, в реакционной смеси добавляли этиловый эфир (200 мл) и осадок собирали, промывали этиловым эфиром (100 мл), вновь растворяли в безводном тетрагидрофуране (80 мл) и обрабатывали морфолином (0,25 мл) в течение ночи при комнатной температуре. После этого реакционную смесь концентрировали до малого объема при пониженном давлении, разбавляли метиленхлоридом, охлаждали до 0oC и обрабатывали раствором трифторуксусного ангидрида (2 мл) в метиленхлориде (10 мл). Через 30 минут реакционную смесь промывали водным 5% гидрокарбонатом калия и дважды водой. Органическую фазу концентрировали при пониженном давлении и подвергали флаш-хроматографии на силикагеле с применением смеси метиленхлорид/метанол (90:10 об/об) в качестве элюирующей системы, получая указанное в заголовке соединение А1, которое превращали в соответствующий гидрохлорид путем добавления стехиометрического количества хлористого водорода с последующим осаждением этиловым эфиром. Выход: 0,9 г. Тонкослойная хроматография на Kieselgel F254 (Merck), элюирующая система метиленхлорид/метанол (90:10 об/об) Rf= 0,5. FD - MS: m/e 816 [M]+
Пример 2. Получение 13-дигидро-14-N-(морфолино)-3'-N-трифторацетил-4'-иоддауномицина (А7)
14-N-(морфолино)-3'-N-трифторацетил-4'-иоддауномицин (А1, 0,4 г, 0,05 ммоль), полученный, как описано в примере 1, в виде свободного основания растворяли в безводном тетрагидрофуране (20 мл) и к раствору добавляли эфират бромида магния (0,7 г). Через 10 минут при комнатной температуре, в атмосфере азота, смесь охлаждали до -50oC, добавляли боргидрид натрия (50 мг) и затем обрабатывали безводным метанолом (1х2 мл) в течение 5 минут. После этого добавляли ацетон (5 мл). Реакционную смесь доводили до температуры 0oC и экстрагировали 0,1 н. водной соляной кислотой. Водную фазу доводили до pH 8,5 0,1 н. гидроксидом натрия и экстрагировали метиленхлоридом, получая указанное в заголовке соединение А7, которое превращали в соответствующий гидрохлорид добавлением стехиометрического количества хлористого водорода с последующим осаждением этиловым эфиром. Выход: 0,3 г.
TLC на Kieselgel F254 (Merck), система, элюирующая метиленхлорид и метанол (90:10 об./об.) Rf = 0,3. FD - MS: m/e 818 [M]=.
Пример 3. Получение 14-N-(3,4-диметоксибензиламино)-3'-N-трифторацетил-4'-иоддауномицина (А2)
Указанное в заголовке соединение А2 получали взаимодействием 14-бром-4'-иоддауномицина (0,65 г, 1 ммоль) с 3,4-ди метоксибензиламином (0,3 г, 1 ммоль) в безводном тетрагидрофуране (50 мл), как описано в примере 1. Выход: 0,3 г. TLC на Kieselgel F254 (Merck), элюирующая система - метиленхлорид/метанол (90:10 об/об) Rf = 0,45. FD - MS: m/e 797 [M]+.
Пример 4. Получение 14-N-(морфолино)-3'-N-фталоил-4'-иоддауномицина (А8)
14-N-(морфолино)-4'-иоддауномицин (0,7 г, 1 ммоль), полученный, как описано в примере 1, подвергали взаимодействию в фталоилхлоридом (0,4 г, 2 ммоль) в безводном тетрагидрофуране (50 мл) при 0oC в течение 4 часов. Смесь разбавляли метиленхлоридом (100 мл) и промывали водным гидрокарбонатом натрия и водой, затем сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении и сырой продукт подвергали флаш-хроматографии на силикагеле с применением смеси метиленхлорид/ацетон (95:5 об/об) в качестве элюирующей системы, получая указанное в заглавии соединения А8 (0,5 г). TLC на Kieselgel F254 (Merck), элюирующая система метиленхлорид/метанол (90:10 об./об) Rf = 0,25. FD-MS: m/e 851 [M]+.
Пример 5. Получение 3'-N-дифенилметилен-4'-иоддоксорубицина (А16).
Гидрохлорид иоддоксорубицина (А27, 0,65 г, 1 ммоль) растворяли в тетрагидрофуране (50 мл) и обрабатывали бензофенонимином (0,36 г, 2 ммоля). Смесь выдерживали при комнатной температуре в течение ночи, затем растворитель удаляли при пониженном давлении и сырой продукт подвергали флаш-хроматографии на силикагеле, получая указанное в заголовке соединения А16 (0,6 г).
TLC на Kieselgel F254 (Merck), элюирующая система метиленхлорид/ацетон (95:5 об/об) Rf = 0,55. FD-MS: m/e 816 [M]+.
Пример 6. Получение 14-N-(морфолино)-3'-N-дифенилметилен-4'-иоддаунорубицина (А17)
14-N-(морфолино)-4'-иоддауномицин (0,7 г, 1 ммоль), полученный, как описано в примере 1, подвергали взаимодействию с бензофенонимином (0,36 г, 2 ммоля), как описано в примере 5, получая указанное в заголовке соединения А17 (0,65 г). TLC на Kieselgel F254 (Merck), элюирующая система - метиленхлорид/ацетон (95:5 об/об) Rf = 0,40. FD-MS:m/e 885 [M]+.
Биологическое исследование
Производные антрациклина формулы А препятствуют самоагрегирующей активности фрагмента 25-35 β -аминоида и PrP фрагмента 106-126 при испытании анализом светорассеяния.
β 25-35 (GSNKGAIIGLH) и PrP 106-126 (KTNMKHMAGAAAAGA-UUGGLG) синтезировали при помощи твердофазной химии с применением 430А Applied Biosystems Instruments и очищали при помощи HPLC (ЖХВР) с обращенной фазой (Beckman Inst. mod. 243) согласно Forloni et al., Nature 362: 543, 1993.
Светорассеяние пептидных растворов оценивали при помощи спектрофлуорометрии (Perkin Elmer LS50В), возбуждение и эмиссию наблюдали при 600 нм. β -Амилоидный фрагмент 25-35 и PrP 106-126 растворяли при концентрации 0,5-1 мг/мл (0,4-0,8 мМ и 0,2-0,4 мМ соответственно), в растворе фосфатного буфера pH 5, 10 мМ самопроизвольно агрегировали в пределах часа.
13-дигидро-4'-иодоксорубицин (А39), растворенный при нескольких концентрациях (0,2-2 мМ) в Трис буфере 5 мМ pH 7,4, добавляли к пептидным растворам в момент их приготовления для оценки процесса фибриллогенеза.
Соединение А29, добавленное при эквимолярной концентрации с β -амилоидным фрагментом 25-35 и PrP 106-126, показало полное предотвращение агрегации.
Соединения, описанные в данном изобретении, препятствуют агрегации мономерного пептида Aβ1-40, стимулируемой посевом амилоидных фибрилл Aβ1-40. Активность соединений была оценена согласно процедуре, изложенной ниже.
Исходный раствор мономера пептида Aβ1-40 получают путем растворения пептида в диметилсульфоксиде при концентрации 33,33 мг/мл. Исходный раствор далее разводят в 11,5 раз диметилсульфоксидом. Затем этот раствор разводят 10 м фосфатного буфера pH 7,4, содержащего 150 мМ хлорида натрия, чтобы получить испытуемый раствор. В пробирку Эппендорфа, содержащую 47 мкл раствора мономера пептида Aβ1-40, добавляют 3 мкл 830 мкМ водного раствора исследуемого соединения, содержащего 66,4 мкМ, в расчете на содержание мономера Aβ1-40, предварительно сформированных обработанных ультразвуком фибрилл Aβ1-40: полученный раствор является 20 мкМ по мономеру Aβ1-40, 50 мкМ по испытуемому соединению и содержит 4 мкМ, в расчете на содержание мономера Aβ1-40, предварительно сформированных обработанных ультразвуком фибрилл Aβ1-40. Агрегации позволяют происходить в течение двух часов при 37oC. Затем суспензию центрифугируют при 15000 об/мин в течение 15 минут при +4oC, супернатант собирают и производят количественный анализ мономера путем ЖХВР.
Активность некоторых представительных соединений представлена в таблице 1. Активность выражена как процент ингибирования агрегации раствора 20 мкМ мономера Aβ1-40, стимулированной 4 мкМ, в расчете на содержание мономера Aβ1-40, предварительно сформированных обработанных ультразвуком фибрилл Aβ1-40.
Характеристики продуктов, полученных по примерам 1-6, представлены в табл. 2.
Пример 7
Получение 14-(1,2,3,6-тетрагидропиридин-1-ил)-4'-дезокси-4'-иод-даунорубицина (FCE29767A)
4'-Дезокси-4'-иододаунорубицин (А27: 500 мг, 0,742 ммоль) преобразуют в соответствующее 14-бромпроизводное, следуя стандартным процедурам (описаны в патенте США 3803124, 9 апреля 1974). Соединение растворяют в ацетоне (10 мл), добавляют 1,2,3,6-тетрагидропиридин (0,5 мл, 5 ммоль) и реакционную смесь перемешивают при 35oC в течение одного часа. Реакционную смесь обрабатывают дихлорэтаном и водой, органическую фазу отделяют, сушат над безводным сульфатом натрия и добавляют гексан, чтобы осадить сырой продукт. Осажденный продукт очищают путем флэш-хроматографии на силикагеле, используя смесь дихлорметан/метанол/уксусную кислоту.
Пример 8
Получение дигидрохлорида 14-N-(морфолино)-4'-дезокси-4'-иододаунорубицина (FCE29648A)
Действуя, как описано в примере 7, но используя морфолин вместо 1,2,3,6-тетрагидропиридина, получают указанное в заголовке соединение с выходом 68%.
Брутто-формула: C31H35IN2O10•2HCl, молекулярная масса 795,46.
Пример 9
Таблетки, содержащие следующие ингредиенты, могут быть получены обычным способом:
Ингредиент - На таблетку
Соединение А1 - 25,0 мг
Лактоза - 125,0 мг
Маисовый крахмал - 75,0 мг
Тальк - 4,0 мг
Стеарат магния - 1,0 мг
Общая масса - 230,0 мг
Пример 10
Капсуы, содержащие следующие ингредиенты, могут быть получены обычным способом:
Ингредиент - На капсулу
Соединение А39 - 50,0 мг
Лактоза - 165,0 мг
Маисовый крахмал - 20,0 мг
Тальк - 5,0 мг
Масса капсулы - 240,0 мгь
Изобретение относится к области медицины и касается применения производных антрациклина и новых производных антрациклина для лечения амилоидоза, а также фармацевтических композиций, содержащих указанные соединения. Изобретение позволяет повысить эффективность ингибирования амилоидоза, что в свою очередь позволяет увеличить эффективность лечения нейродегеративных заболеваний. 3 с. и 5 з.п. ф-лы, 2 табл.

где R1 обозначает водород или гидрокси, группу формулы OR4, где R4 представляет собой C1 - C6-алкил, C5 - C6-циклоалкил или CH2Ph, с фенильным (Ph) кольцом, необязательно замещенным 1, 2 или 3 заместителями, выбранными из F, Cl, Br, C1 - C6-алкила, C1 - C6-алкокси и CF3, или группу формулы OSO2R5, в которой R5 представляет собой C1 - C6-алкил или Ph, необязательно замещенные 1, 2 или 3 заместителями, выбранными из галогена, такого как F, Cl, Br, и C1 - C6-алкила;
R2 обозначает водород, гидрокси или OR4, как указано выше;
R3 обозначает водород, метил или группу формулы YCH2R6, в которой Y обозначает CO, CH2, CHOH или группу формулы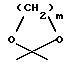
в которой m обозначает 2 или 3, R6 обозначает водород или гидрокси, группу формулы NR7R8, в которой R7 и R8, каждый, независимо, выбраны из: а) водорода, b) C1 - C6-алкильной или C2 - C6-алкенильной группы, необязательно замещенных гидрокси, CN, COR9, COOR9, CONR9R10, O(CH2)nNR9R10 (n равно 2 - 4) или NR9R10, в которых R9 и R10, каждый, независимо, выбраны из водорода, C1 - C12-алкила, C2 - C12-алкенила или фенила, необязательно замещенных одним или несколькими заместителями, выбранными из C1 - C6-алкила, C1 - C6-алкокси, F, Br, Cl, CF3, OH, NH2 или CN, с) C3 - C6-циклоалкила, необязательно замещенного COR9, COOR9 или OH, где R9 имеет указанное выше значение, d) фенил-C1 - C4-алкила или фенил-C2 - C4-алкенила, необязательно замещенных в фенильном кольце одним или несколькими заместителями, выбранными из C1 - C6-алкила, C1 - C6-алкокси, F, Br, Cl, CF3, OH, NH2 или CN, е) COR9, COOR9, CONR9R10, COCH2NR9R10, CONR9COOR10, SO2R9, в которых R9 и R10 имеют указанные выше значения, или R7 и R8 вместе с азотом образуют: f) морфолиновое кольцо, необязательно замещенное C1 - C4-алкилом или C1 - C4-алкокси, g) пиперазиновое кольцо, необязательно замещенное C1 - C6-алкилом, C2 - C6-алкенилом или фенилом, необязательно замещенными одним или несколькими заместителями, выбранными из C1 - C6-алкила, C1 - C6-алкокси, F, Br, Cl, CF3, OH, NH2 или CN, и h) пирролидиновое или пиперидиновое, или тетрагидропиридиновое кольцо, необязательно замещенное OH, NH2, COOH, COOR9 или CONR9R10, где R9 и R10 имеют указанные выше значения, C1 - C6-алкилом, C2 - C6-алкенилом или фенилом, необязательно замещенными одним или несколькими заместителями, выбранными из C1 - C6-алкила, C1 - C6-алкокси, F, Br, Cl, CF3, OH, NH2 или CN, группу формулы OR4 или SR4, в которых R4 имеет указанное выше значение, группу формулы O-Ph, где фенильное (Ph) кольцо необязательно замещено нитро, амино или NR7R8, как указано выше, или группу формулы В или С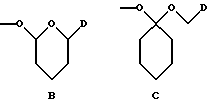
где D представляет собой группу формулы COOR9 или CONR7R8, в которых R7, R8 и R9 имеют указанные выше значения, X представляет собой остаток сахара формулы X1 или X2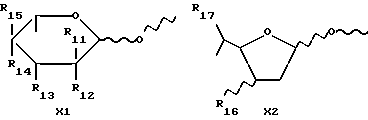
где R11 и R12, оба, представляют собой водород, или один из R11 и R12 является водородом, а другой представляет собой F, Cl, Br или I;
R13 представляет собой водород, гидрокси, C1 - C4-алкокси, амино, NHCOCF3, N=C(C6H5)2, NHCOR9, NHCONR7R8 или группу формулы E или F
в которых R7, R8 и R9 имеют указанные выше значения, и p равно 0 или 1;
R14 и R15 обозначают водород, или один из R14 или R15 обозначает водород, а другой обозначает OH, F, Cl, Br, I или группу формулы OSO2R5, где R5 имеет указанное выше значение;
R16 обозначает CH2OH или R13, как указано выше;
R17 обозначает F, Cl, Br, I или группу формулы OSO2R5, в которой R5 имеет указанное выше значение,
и его фармацевтически приемлемых солей, при условии, что соединение формулы А не является 4'-йод-4'-деоксидоксорубицином, и при условии, что X1 является остатком, в котором оба, R14 и R15, являются атомами водорода, или один из R14 и R15 представляет собой гидрокси, и R13 является амино, когда R3 является группой формулы YCH3, COCH2NR'7R'8, COCH2OR'4 или YCH2OH, где Y указан выше, R'4 обозначает фенил, бензил, C1 - C6-алкил или C5 - C6-циклоалкил, R'7 и R'8, каждый, независимо, обозначают водород, C1 - C12-алканоил, или, взятые вместе, образуют морфолиновый, пиперазиновый или пиперидиновый остаток, или X1 является остатком, в котором оба R11 и R12 являются атомами водорода, R13 обозначает амино и R14 обозначает йод, когда R1 представляет собой метокси, R2 представляет собой гидрокси и R3 представляет собой COCH2OH, при лечении амилоидоза или для изготовления лекарственного средства для использования при лечении амилоидоза.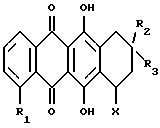
где R1 обозначает водород или гидрокси, группу формулы OR4, где R4 представляет собой C1 - C6-алкил, C5 - C6-циклоалкил или CH2Ph, с фенильным (Ph) кольцом, необязательно замещенным 1, 2 или 3 заместителями, выбранными из F, Cl, Br, C1 - C6-алкила, C1 - C6-алкокси и CF3, или группу формулы OSO2R5, в которой R5 представляет собой C1 - C6-алкил или Ph, необязательно замещенные 1, 2 или 3 заместителями, выбранными из галогена, такого как F, Cl, Br, и C1 - C6-алкила;
R2 обозначает водород, гидрокси или OR4, как указано выше;
R3 обозначает водород, метил или группу формулы YCH2R6, в которой Y обозначает CO, CH2, CHOH или группу формулы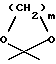
в которой m обозначает 2 или 3, R6 обозначает водород или гидрокси, группу формулы NR7R8, в которой R7 и R8, каждый, независимо, выбраны из: а) водорода, b) C1 - C6-алкильной или C2 - C6-алкенильной группы, необязательно замещенных гидрокси, CN, COR9, COOR9, CONR9R10, O(CH2)nNR9R10 (n равно 2 - 4) или NR9R10, в которых R9 и R10, каждый, независимо, выбраны из водорода, C1 - C12-алкила, C2 - C12-алкенила или фенила, необязательно замещенных одним или несколькими заместителями, выбранными из C1 - C6-алкила, C1 - C6-алкокси, F, Br, Cl, CF3, OH, NH2 или CN, с) C3 - C6-циклоалкила, необязательно замещенного COR9, COOR9 или OH, где R9 имеет указанное выше значение, d) фенил-C1 - C4-алкила или фенил-C2 - C4-алкенила, необязательно замещенных в фенильном кольце одним или несколькими заместителями, выбранными из C1 - C6-алкила, C1 - C6-алкокси, F, Br, Cl, CF3, OH, NH2 или CN, е) COR9, COOR9, CONR9R10, COCH2NR9R10, CONR9COOR10, SO2R9, в которых R9 и R10 имеют указанные выше значения, или R7 и R8 вместе с азотом образуют: f) морфолиновое кольцо, необязательно замещенное C1 - C4-алкилом или C1 - C4-алкокси, g) пиперазиновое кольцо, необязательно замещенное C1 - C6-алкилом, C2 - C6-алкенилом или фенилом, необязательно замещенными одним или несколькими заместителями, выбранными из C1 - C6-алкила, C1 - C6-алкокси, F, Br, Cl, CF3, OH, NH2 или CN, и h) пирролидиновое, или пиперидиновое, или тетрагидропиридиновое кольцо, необязательно замещенные OH, NH2, COOH, COOR9 или CONR9R10, где R9 и R10 имеют указанные выше значения, C1 - C6-алкилом, C2 - C6-алкенилом или фенилом, необязательно замещенными одним или несколькими заместителями, выбранными из C1 - C6-алкила, C1 - C6-алкокси, F, Br, Cl, CF3, OH, NH2 или CN, группу формулы OR4 или SR4, в которых R4 имеет указанное выше значение, группу формулы O-Ph, где фенильное (Ph) кольцо необязательно замещено нитро, амино или NR7R8, как указано выше, или группу формулы В или С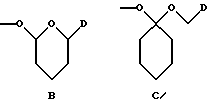
где D представляет собой группу формулы COOR9 или CONR7R8, в которых R7, R8 и R9 имеют указанные выше значения, и X представляет собой остаток сахара формулы X1 или X2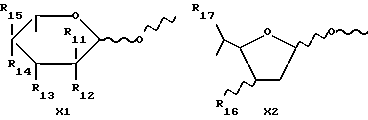
где R11 и R12, оба, представляют собой водород, или один из R11 и R12 является водородом, а другой представляет собой F, Cl, Br или I;
R13 представляет собой водород, гидрокси, C1 - C4-алкокси, амино, NHCOCF3, N=C(C6H5)2, NHCOR9, NHCONR7R8 или группу формулы E или F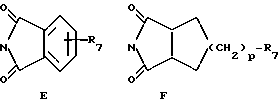
в которых R7, R8 и R9 имеют указанные выше значения, и p равно 0 или 1;
R14 и R15 обозначают водород, или один из R14 или R15 обозначает водород, а другой обозначает OH, F, Cl, Br, I или группу формулы OSO2R5, где R5 имеет указанное выше значение;
R16 обозначает CH2OH или R13, как указано выше;
R17 обозначает F, Cl, Br, I или группу формулы OSO2R5, в которой R5 имеет указанное выше значение,
и его фармацевтически приемлемых солей, при условии, что соединение формулы А не является 4'-йод-4'-деоксидоксорубицином, и при условии, что X1 не является остатком, в котором оба, R14 и R15, являются атомами водорода, или один из R14 и R15 представляет собой гидрокси, и R13 является амино, когда R3 является группой формулы YCH3, COCH2NR'7R'8, COCH2OR'4 или YCH2OH, где Y указан выше, R'4 обозначает фенил, бензил, C1 - C6-алкил или C5 - C6-циклоалкил, R'7 и R'8, каждый, независимо, обозначают водород, C1 - C12-алканоил, или, взятые вместе, образуют морфолиновый, пиперазиновый или пиперидиновый остаток, или X1 не является остатком, в котором оба R11 и R12 являются атомами водорода, R13 обозначает амино и R14 обозначает йод, когда R1 представляет собой метокси, R2 представляет собой гидрокси и R3 представляет собой COCH2OH.
где R1 обозначает водород или гидрокси, группу формулы OR4, где R4 представляет собой C1 - C6-алкил, C5 - C6-циклоалкил или CH2Ph, с фенильным (Ph) кольцом, необязательно замещенным 1, 2 или 3 заместителями, выбранными из F, Cl, Br, C1 - C6-алкила, C1 - C6-алкокси и CF3, или группу формулы OSO2R5, в которой R5 представляет собой C1 - C6-алкил или Ph, необязательно замещенные 1, 2 или 3 заместителями, выбранными из галогена, такого как F, Cl, Br, и C1 - C6-алкила;
R2 обозначает водород, гидрокси или OR4, как указано выше;
R3 обозначает водород, метил или группу формулы YCH2R6, в которой Y обозначает CO, CH2, CHOH или группу формулы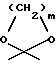
в которой m обозначает 2 или 3, и R6 обозначает водород или гидрокси, группу формулы NR7R8, в которой R7 и R8, каждый, независимо, выбраны из: а) водорода, b) C1 - C6-алкильной или C2 - C6-алкенильной группы, необязательно замещенных гидрокси, CN, COR9, COOR9, CONR9R10, O(CH2)nNR9R10 (n равно 2 - 4) или NR9R10, в которых R9 и R10, каждый, независимо, выбран из водорода, C1 - C12-алкила, C2 - C12-алкенила или фенила, необязательно замещенных одним или несколькими заместителями, выбранными из C1 - C6-алкила, C1 - C6-алкокси, F, Br, Cl, CF3, OH, NH2 или CN, с) C3 - C6-циклоалкила, необязательно замещенного COR9, COOR9 или OH, где R9 имеет указанное выше значение, d) фенил-C1 - C4-алкила или фенил-C2 - C4-алкенила, необязательно замещенных в фенильном кольце одним или несколькими заместителями, выбранными из C1 - C6-алкила, C1 - C6-алкокси, F, Br, Cl, CF3, OH, NH2 или CN, е) COR9, COOR9, CONR9R10, COCH2NR9R10, CONR9COOR10, SO2R9, в которых R9 и R10 имеют указанные выше значения, или R7 и R8 вместе с азотом образуют: f) морфолиновое кольцо, необязательно замещенное C1 - C4-алкилом или C1 - C4-алкокси, g) пиперазиновое кольцо, необязательно замещенное C1 - C6-алкилом, C2 - C6-алкенилом или фенилом, необязательно замещенными одним или несколькими заместителями, выбранными из C1 - C6-алкила, C1 - C6-алкокси, F, Br, Cl, CF3, OH, NH2 или CN, и h) пирролидиновое, или пиперидиновое, или тетрагидропиридиновое кольцо, необязательно замещенные OH, NH2, COOH, COOR9 или CONR9R10, где R9 и R10 имеют указанные выше значения, C1 - C6-алкилом, C2 - C6-алкенилом или фенилом, необязательно замещенными одним или несколькими заместителями, выбранными из C1 - C6-алкила, C1 - C6-алкокси, F, Br, Cl, CF3, OH, NH2 или CN, группу формулы OR4 или SR4, в которых R4 имеет указанное выше значение, группу формулы O-Ph, где фенильное (Ph) кольцо необязательно замещено нитро, амино или NR7R8, как указано выше, или группу формулы В или С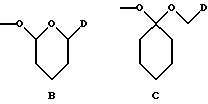
где D представляет собой группу формулы COOR9 или CONR7R8, в которых R7, R8 и R9 имеют указанные выше значения, и X представляет собой остаток сахара формулы X1 или X2
где R11 и R12, оба, представляют собой водород, или один из R11 и R12 является водородом, а другой представляет собой F, Cl, Br или I;
R13 представляет собой водород, гидрокси, C1 - C4-алкокси, амино, NHCOCF3, N=C(C6H5)2, NHCOR9, NHCONR7R8 или группу формулы E или F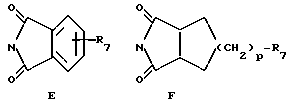
в которых R7, R8 и R9 имеют указанные выше значения, и p равно 0 или 1;
R14 и R15 обозначают водород, или один из R14 или R15 обозначает водород, а другой обозначает OH, F, Cl, Br, I или группу формулы OSO2R5, где R5 имеет указанное выше значение;
R16 обозначает CH2OH или R13, как указано выше;
R17 обозначает F, Cl, Br, I или группу формулы OSO2R5, в которой R5 имеет указанное выше значение,
при условии, что соединение формулы А не является 4'-йод-4'-деоксидоксорубицином, или его фармацевтически приемлемой соли вместе с фармацевтически приемлемым носителем или разбавителем.
| ПРЕДОХРАНИТЕЛЬНЫЙ МЕХАНИЗМ ОТ НЕСАНКЦИОНИРОВАННОГО СРАБАТЫВАНИЯ КУРКА В ОРУЖИИ С НАРУЖНЫМ ВЗВЕДЕНИЕМ КУРКА | 1999 |
|
RU2164335C2 |
| ЭКСТРУДИРОВАННЫЙ СОТОВЫЙ ЭЛЕМЕНТ, В ЧАСТНОСТИ КОРПУС-НОСИТЕЛЬ КАТАЛИЗАТОРА, С УСИЛЕННОЙ СТРУКТУРОЙ СТЕНОК | 1998 |
|
RU2195998C2 |
| ЛИГАНДНОЕ СОЕДИНЕНИЕ α7-НИКОТИНОВОГО АЦЕТИЛХОЛИНОВОГО РЕЦЕПТОРА И ЕГО ПРИМЕНЕНИЕ | 2017 |
|
RU2756604C2 |
| ПРОИЗВОДНЫЕ 7, -8, -9(ЗАМЕЩЕННЫХ)-6-ДЕМЕТИЛ-6-ДЕГИДРОКСИТЕТРАЦИКЛИНОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2127255C1 |

