Настоящая заявка является частично продолжением заявки Мерка 185971А, которая в свою очередь является частично продолжением заявки США с серийным номером 07/789,508, поданной 8 ноября 1991 г. Настоящая заявка имеет отношение к следующим патентным документам: заявке США с серийным номером 595913, поданной 11 октября 1990 г. (дело Мерка 18236); заявке США с серийным N 746460, поданной 16 августа 1991 г. (дело Мерка 18466); дело Мерка 18583, поданной 23 октября 1991 г; и дело Мерка 18416.
Настоящее изобретение относится к соединениям, которые ингибируют протеазу, закодированную вирусом иммунодефицита человека (ВИЧ), или фармацевтически приемлемым солям таких соединений, и ценность изобретения состоит в предотвращении ВИЧ-инфицирования, лечении ВИЧ-инфекции и лечении развившегося в результате синдрома приобретенного иммунодефицита (СПИДа). Настоящее изобретение относится также к фармацевтическим композициям, содержащим такие соединения, и к способу применения настоящих соединений и других агентов для лечения СПИДа и вирусного ВИЧ-инфицирования.
Ретровирус, обозначаемый как вирус иммунодефицита человека (ВИЧ), представляет собой этиологический агент комплексного заболевания, включающего прогрессирующее разрушение иммунной системы (синдром приобретенного иммунодефицита, СПИД) и деградацию центральной и периферической нервной системы. Ранее такой вирус был известен как LAV, HTLV-III или ARV. Общим признаком ретровирусной репликации является экстенсивное пост-транляционное развитие полипротеиновых предшественников под действием вирусно- закодированной протеазы, в результате чего возникают зрелые вирусные протеины, требующиеся для сборки вируса и его функционирования. Ингибирование такого процесса предотвращает продуцирование обычного инфицирующего вируса.
Так, например, Н.Е.Кель с сотр. в Proc. Natl. Acad. Sci. 85, 4686 (1988) показали, что генетическая инактивация ВИЧ-закодированной протеазы приводит к образованию незрелых, неинфицирующих вирусных частиц. Этот результат указывает на тот факт, что ингибирование ВИЧ-протеазы является жизнеспособным методом лечения СПИДа и профилактики или лечения ВИЧ-инфицирования.
Установление нуклеотидной последовательности ВИЧ обнаруживает наличие ро1 гена в одном открытом остове считывания генетической информации (Л.Ратнер с сотр. , Nature, 313, 277 (1985)). Гомология аминокислотной последовательности доказывает, что pol- последовательность кодирует обратимую транскриптазу, эндонуклеазу и ВИЧ-протеазу (Г.Тох с сотр., ЕМВО., 1, 1267 (1985); М. Д. Пауэр с сотр., Science, 231, 1567 (198); Л.Г.Пиэл с сотр., Nature 329, 351 (1987)). Доказано, что соединения изобретения являются ингибиторами ВИЧ-протеазы.
Соединения настоящего изобретения используются для ингибирования ВИЧ-протеазы, профилактики ВИЧ-инфицирования, лечения ВИЧ-инфицирования и лечения СПИДа в виде индивидуальных соединений, фармацевтически приемлемых солей, ингредиентов фармацевтической композиции как в индивидуальном порядке, так и в комбинации с другими антивирусными агентами, иммуномодуляторами, антибиотиками или вакцинами. В настоящем изобретении раскрываются также способы лечения СПИДа, способы профилактики ВИЧ-инфицирования и способы лечения ВИЧ-инфицирования.
Далее следуют некоторые сокращения, используемые в тексте заявки.
Обозначение - Сокращения - Защитная группа
BOC (Boc) - Трет.-бутилоксикарбонил
CBZ (Cbz) - Бензилоксикарбонил (карбобензокси)
TBS (TSDMS) - Трет.-бутил-диметилсилил - Активирующая группа
HBT (HOBT или HOBt) - 1-гидроксибензотриазол гидрат - Реагент сочетания
BOP - Реагент бензотриазол-1-ил-окситрис(диметиламино)фосфоний гексафторфосфат
BOP-C1 - Бис(2-оксо-3-оксазолидинил)фосфиний хлорид
EDC - 1-этил-3-(3-диметиламинопропил)-карбодиимид гидрохлорид - Прочее
(BOC)2O (BOC2O) - Ди-трет.-бутилдикарбонат
n-Bu4N+F- - Фтористый тетрабутил аммоний
n-BuLi (n-Buli) - н-бутиллитий
DMF - Диметилформамид
Et3N - Триэтиламин
EtOAc - Этилацетат
TFA - Трифторуксусная кислота
DMAP - Диметиламинопиридин
DME - Диметоксиметан
LDA - Диизопропиламид лития
THF - Тетрагидрофуран - Аминокислоты - Jle L-изолейцин - Val L-валин
Настоящее изобретение относится к соединениям формулы I, их комбинациям или их фармацевтически приемлемым солям при ингибировании ВИЧ-протеазы, профилактике или лечении ВИЧ-инфицирования и лечении возникающего в результате синдрома приобретенного иммунодефицита (СПИД). Соединения формулы I отвечают следующей структуре: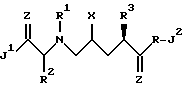
в которой X представляет собой -OH или NH2;
Z представляет собой -O, S, или -NH;
R - водород или C1-4 алкил;
R1 и R2, независимо друг от друга, представляют собой:
1) водород,
2) C1-4 алкил, незамещенный или замещенный одним или более следующими атомами или группами:
а) галоген,
b) гидрокси,
c) C1-4алкокси,
d) арил, незамещенный или замещенный одной или более группами, выбранными из C1-4 алкила, гидрокси или арила,
e) -W-арил или -W-бензил, где W- O-, -S- или -NH-,
f) 5-7-членная циклоалкильная группа, незамещенная или замещенная одним или более из следующих атомов или групп:
I) галоген,
II) гидрокси,
III) C1-3 алкокси, или
IV) арил
g) гетероциклическая группа, незамещенная или замещенная одной или более группами, выбранными из гидрокси, C1-4алкила, необязательно замещенного гидрокси или BOC,
h) 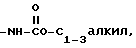
i) 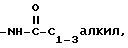
j) -NH-SO2-C1-3 алкил,
k) -NR2,
l) -COOR, или
m) -((CH2)mO)nR, где m равно 2-5, а n - 0, 1, 2 или 3, либо
3) арил, незамещенный или замещенный одним или более атомами или группами, выбранными из следующих значений:
а) галоген,
b) гидрокси,
c) -NO2 или NR2,
d) C1-4алкил,
e) C1-3 алкокси, незамещенная или замещенная одной или более группами, выбранными из -OH или C1-3 алкокси,
f) -COOR,
g) 
h) -CH2NR2,
i) 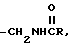
j) -CN,
k) -CF3,
l) 
m) арил C1-3 алкокси,
n) арил,
o) -NRSO2R,
p) -OP(O)(ORx)2, или
q) -R5, значение которого указано ниже, либо R1 и R2 могут быть связаны друг с другом с образованием совместно с атомом азота, к которому присоединен R1, 3-10-членной моноциклической или бициклической насыщенной кольцевой системы, состоящей из атома азота, к которому присоединен R1, и из 2-9 углеродных атомов, и незамещенной или замещенной следующими группами:
1) гидрокси,
2) C1-4 алкилом, незамещенным или замещенным одной или более из следующих групп;
а) галогеном,
b) гидрокси,
c) C1-3 алкокси,
d) арилом,
e) 5-7-членной циклоалкильной группой, незамещенной или замещенной одной или более группами, выбранными из:
I) галогена,
II) гидрокси,
III) C1-3 алкокси, или
IV) арила,
f) гетероцикла, или
g) -NR2,
3) C1-3 алкокси,
4) 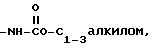
5) 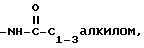
6) -NH-SO2-C1-3 алкилом,
7) гетероциклом,
8) -W-арилом, или
9) 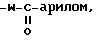 где W имеет указанные выше значения; либо
где W имеет указанные выше значения; либо
R1 и R2 могут быть связаны друг с другом с образованием, совместно с атомом азота, к которому присоединен R1, 3-10-членной моноциклической или бициклической насыщенной кольцевой системы, состоящей из атома азота, к которому присоединен R1, 1-8 углеродных атомов и одного или более незамещенного или замещенного гетероатома, выбранного из:
1) 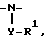 где Y отсутствует или
где Y отсутствует или 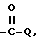 либо -SO2-Q-, R1 имеет указанные выше значения для тех случаев, когда R1 не зависит от R2 и не связан с ним, и где Q отсутствует или -O-, NR-, либо гетероцикл, необязательно замещенный C1-4 алкилом,
либо -SO2-Q-, R1 имеет указанные выше значения для тех случаев, когда R1 не зависит от R2 и не связан с ним, и где Q отсутствует или -O-, NR-, либо гетероцикл, необязательно замещенный C1-4 алкилом,
2) 
3)  незамещенного или замещенного арилом,
незамещенного или замещенного арилом,
4) 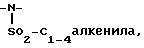 незамещенного или замещенного арилом,
незамещенного или замещенного арилом,
5) -S(O)p-, где p равно 0, 1 или 2, либо
6) -O-; или
R1 и R2 могут быть связаны друг с другом с образованием совместно с атомом азота, к которому присоединен R1, 3-10-членной моноциклической или бициклической насыщенной кольцевой системы, состоящей из атома азота, к которому присоединен R1, и 2-9 углеродных атомов, причем насыщенная кольцевая система сконденсирована* с фенильным кольцом и такое фенильное кольцо незамещено или замещено одной или более группами, выбранными из:
1) галогена,
2) C1-3 алкокси,
3) гидрокси,
4) C1-4 алкила,
5) -NHR1, где R1 имеет указанные выше значения для случая, когда R1 не зависит от R2 и не связан с ним, или
6) -NH-гетероцикла;
R3 представляет собой:
1) -(CH2)r-R4, где r имеет значения от 0 до 5,
* (при.пер. - сшита)
2) C1-4 алкенил-R4, или
3) C1-4 алкинил-R4;
R4 представляет собой:
1) водород,
2) C1-4 алкил,
3) C5-10 циклоалкил, необязательно замещенный гидроксигруппой,
4) C6-10 арил, незамещенный или замещенный одной или более группами, выбранными из:
а) галогена,
b) гидрокси,
c) -NO2 или NR2,
d) C1-4 алкила,
e) C1-3 алкокси, незамещенной или замещенной одной или более группами, выбранными из -OH или C1-3 алкокси,
f) -COOR,
g) 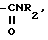
h) -CH2NR2,
i) 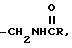
j) -CN,
k) -CF3,
l) 
m) арил-C1-3 алкокси,
n) арила,
o) -NRSO2R,
p) -OP(O)(ORx)2, или
g) -R5, имеющего указанные ниже значения, либо
5) моноциклический или бициклический гетероцикл, содержащий 1-3 гетероатомов, выбранных из группы, состоящей из N, O и S, который может быть незамещенным или замещенным радикалом R5 и, необязательно, одной или более следующими группами:
а) галогеном,
b) C1-4 алкилом, или
c) C1-3 алкокси;
Rx представляет собой H или арил;
R5 представляет собой:
1) -W-(CH2)m-NR6R7, где W имеет указанные выше значения, m равно 2-5, а R6 и R7, независимо друг от друга, представляют собой:
а) водород,
b) C1-6 алкил, незамещенный или замещенный одной или более группами, выбранными из:
I) C1-3 алкокси,
II) -OH, или
III) -NR2,
c) одинаковые или различные группы, соединенные друг с другом с образованием 5-7-членного гетероцикла, такого как морфолино, содержащего до двух дополнительных гетероатомов, выбранных из 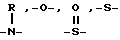 или -SO2-, причем такой гетероцикл может быть необязательно замещен C1-4 алкилом, или
или -SO2-, причем такой гетероцикл может быть необязательно замещен C1-4 алкилом, или
d) ароматический гетероцикл, незамещенный или замещенный одной или более из следующих групп:
I) C1-4 алкилом, или
II) -NR2,
2) -(CH2)q-NR6R7, где q равно 1-5, а R6 и R7 имеют указанные выше значения, за исключением того случая, когда R6 и R7 представляют собой H или незамещенный C1-6 алкил, или
3) бензофурил, индолил, азациклоалкил, азабицикло-C7-11 цциклоалкил, либо бензопиперидинил, незамещенный или замещенный C1-4 алкилом;
В отсутствует или представляет собой группу: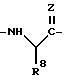
в которой R8 представляет собой:
1) -CH(CH3)2,
2) -CH(CH3)(CH2CH3), или
3) фенил;
J1 и J2, независимо друг от друга, представляют собой:
1) -YR9, в которой Y представляет собой -О- или -NH-, а R9 представляет собой:
а) водород,
b) C1-6 алкил, незамещенный или замещенный одной или более из следующих групп:
I) -NR2,
II) -OR,
III) -NHSO2C1-4 алкилом,
IV) -NHSO2 арилом или -NHSO2 (диалкиламиноарилом)
V) -CH2OR,
VI) -C1-4 алкилом,
VII) 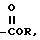
VIII) 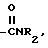
IX) 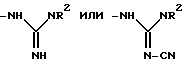
X) 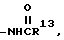 где R13 представляет собой:
где R13 представляет собой:
A) -H,
B) -C1-4 алкил,
C) -арил,
D) -гетероцикл, или
E) -NH-, -O- или -(CH2)n-, где n равно 0, 1, 2 или 3, замещенную:
1) C1-4 алкилом, незамещенным или замещенным одной или более группами, выбранными из арила или гетероцикла, или
II) арилом, незамещенным или замещенным гетероциклом,
XI) -NR3+A-, где A- - противоион,
XII) -NR10R11, где R10 и R11 имеют одинаковые или различные значения и представляют собой C1-5 алкилы, соединенные непосредственно с образованием 5-7-членного гетероцикла, содержащего до одного дополнительного гетероатома, выбранного из -O-, -S- или -NR-.
XIII) арилом,
XIV) -CHO,
XV) -OP(O)(ORx)2,
XVI) 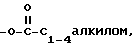 замещенный одним или более амином или четвертичным амином, или -O-((CH2)mO)n-R, либо -OP(O)(ORx),
замещенный одним или более амином или четвертичным амином, или -O-((CH2)mO)n-R, либо -OP(O)(ORx),
XVII)  или
или
XVIII) 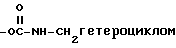 или
или
c) -((CH2)O)m-CH3 или -((CH2)mO)n-H, где m и n имеют указанные выше значения,
-N(R9)2,
3) -NR10R11, где R10 и R11 имеют указанные выше значения, или
4) 
где Y, R9 и n имеют указанные выше значения;
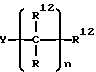
а R12 представляет собой:
1) водород,
2) арил, незамещенный или замещенный одной или более группами, выбранными из:
а) R14 где R14 представляет собой;
I) галоген,
II) -OR,
III) 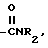
IV) -CH2NR2,
V) -SO2 NR2,
VI) -NR2,
VII) 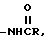
VIII) C1-4 алкил,
IX) фенил,
X) -CF3,
XI) 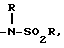
XII) -OP(O)(ORx)2, или
XIII) 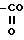
b) -C1-4 алкил-NR2, или
c) 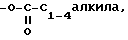 замещенного одним или более амином, либо четвертичным амином, или -OP(O)(ORx)2,
замещенного одним или более амином, либо четвертичным амином, или -OP(O)(ORx)2,
3) такой гетероцикл, как изохроман, хромай, изотиохроман, тиохроман, бензимидазол, бензотиопиран, оксобензотиопиран, бензопиран, бензотиопиранилсульфон, бензотиопиранилсульфоксид, причем указанное кольцо или кольца могут быть незамещенными или замещенными одной или более из следующих групп:
a) R14, имеющей указанные выше значения,
b) -OC1-4 алкенилом,
c) фенил-C1-4 алкилом,
d) 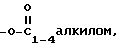 замещенным одной или более группами, выбранными из амина или четвертичного амина, либо - OP(O)(ORx)2, или - O((CH2)mO)n-R, или
замещенным одной или более группами, выбранными из амина или четвертичного амина, либо - OP(O)(ORx)2, или - O((CH2)mO)n-R, или
e) 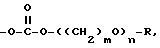
4) 5-7-членное карбоциклическое или 7-10-членное бициклическое кольцо, такое как циклопентан, циклогексан, индан, норборнан, нафталин, тиопиран, изотиопиран, либо бензопиран, причем карбоциклическое кольцо может быть незамещенным или замещенным одной или более из следующих групп:
a) R14, имеющей указанные выше значения,
b) -CH2OR,
c) -(CH2)n-NR2, C5-16 алкилом, пиридином,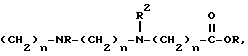
((CH2)mO)n-R, хинуклидинимилом, замещенным R, пиперазин-C1-4 алкил-бензилом, замещенным одним или несколькими R, или морфолино-C1-4 алкилбензилом,
d) 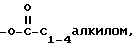 замещенным одним или более амином, четвертичным амином, -OP(O)(ORx)2 или -O-((CH2)mO)n-R,
замещенным одним или более амином, четвертичным амином, -OP(O)(ORx)2 или -O-((CH2)mO)n-R,
e) 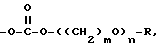 или
или
f) C1-4 алкилфенилом;
или фармацевтически приемлемые соли указанных выше соединений.
Предпочтительными являются соединения, у которых радикалы R1 и R2 связаны друг с другом с образованием совместно с атомом азота, к которому присоединен радикал R1, 3-10-членной моноциклической или бициклической насыщенной кольцевой системы, состоящей из атома азота, к которому присоединен R1 и 2-9 углеродных атомов, которая незамещена или замещена следующими группами:
1) гидрокси,
2) C1-4 алкилом, незамещенным или замещенным одной или более из следующих групп:
a) гидрокси,
b) C1-3 алкокси,
c) арила,
f) 5-7-членной циклорадикальной группы, незамещенной или замещенной одной или более из следующих групп:
I) галогена,
II) гидрокси,
III) C1-3 алкокси, или
IV) арила,
e) гетероцикла, или
f) -NR2,
3) C1-3 алкокси,
4) 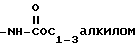
5) 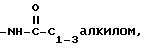
6) -NH-SO2C1-3 алкилом,
7) -W-арилом, или
8) 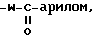 где W представляет собой -O-, -S- или -NH;
где W представляет собой -O-, -S- или -NH;
либо:
R1 и R2 связаны друг с другом с образованием совместно с атомом азота, к которому присоединен R1, 3-10-членной моноциклической или бициклической насыщенной кольцевой системы, состоящей из атома азота, к которому присоединен радикал R10 1-8 углеродных атомов и одного или более замещенных или незамещенных гетероатомов, выбранных из:
1) группы 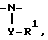 в которой Y отсутствует, либо
в которой Y отсутствует, либо 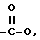 или -SO2-Q-, R1 имеет указанные выше значения для случая, когда R1 не зависит от R2 и не соединен с этим радикалом, либо Q отсутствует, или -O-, -NR-, или гетероцикл, необязательно замещенный C1-4 алкилом,
или -SO2-Q-, R1 имеет указанные выше значения для случая, когда R1 не зависит от R2 и не соединен с этим радикалом, либо Q отсутствует, или -O-, -NR-, или гетероцикл, необязательно замещенный C1-4 алкилом,
2) 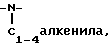 незамещенного или замещенного арилом,
незамещенного или замещенного арилом,
3) -S(O)p- в которой p равно 0, 1 или 2, либо
4) -O-, или
R1 и R2 связаны друг с другом с образованием совместно с атомом азота, к которому присоединен радикал R1, 3-10-членной моноциклической или бициклической насыщенной кольцевой системы, которая состоит из атома азота, к которому присоединен радикал R 1 и 2-9 углеродных атомов, причем в такой системе насыщенное кольцо сконденсировано с фенильным кольцом и указанное фенильное кольцо незамещено или замещено одной или более из следующих групп:
1) C1-3 алкокси,
2) гидрокси,
3) C1-4 алкилом, или
4) -NHR1, в которой R1 имеет указанные выше значения для случая, когда R1 не зависит и не связан с радикалом R2.
Второе, наиболее предпочтительное воплощение настоящего изобретения, дополнительно ограничено соединениями, в которых:
R1 и R2 соединены друг с другом с образованием совместно с атомом азота, к которому присоединен R1, 3-10-членной моноциклической или бициклической насыщенной кольцевой системы, состоящей из атома азота, к которому присоединен R1, и 2-9 углеродных атомов и незамещенной или замещенной следующими группами:
1) гидрокси,
2) C1-4 алкилом, незамещенным или замещенным одной или более из следующих групп:
а) гидрокси,
b) C1-3 алкокси,
c) арилом,
d) 5-7-членной циклоалкильной группой, незамещенной или замещенной одной или более из следующих групп:
I) галогеном,
II) гидрокси,
Ill) C1-3 алкокси, или
IV) арилом,
e) гетероциклом, или
f) -NR2,
3) C1-3 алкокси,
4) 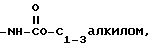
5) 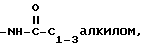
6) -NH-SO2C1-3 алкилом,
7) -W-арилом, или
8) 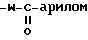
где W представляет собой -O-, -S- или -NH-; либо R1 и R2 связаны друг с другом с образованием совместно с атомом азота, к которому присоединен R1, 3-10-членной моноциклической или бициклической насыщенной кольцевой системы, состоящей из атома азота, к которому присоединен R1, 1-8 углеродных атомов и одного или более незамещенных или замещенных гетероатомов, выбранных из:
1)  где Y отсутствует или
где Y отсутствует или 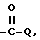 либо -SO2-Q-, R1 имеет указанные выше значения для тех случаев, когда R1 не зависит от R2 и не связан с ним, и где Q отсутствует, или -O-, -NR-, либо гетероцикл необязательно замещенный C1-4-алкилом,
либо -SO2-Q-, R1 имеет указанные выше значения для тех случаев, когда R1 не зависит от R2 и не связан с ним, и где Q отсутствует, или -O-, -NR-, либо гетероцикл необязательно замещенный C1-4-алкилом,
2) -S(O)p-, где p равно 0, 1 или 2, либо
3) -O-;
R3 представляет собой бензил, незамещенный или замещенный одной или более из следующих групп:
а) гидрокси,
b) -NO2 или -NR2,
c) C1-4 алкилом,
d) C1-3 алкокси, незамещенной или замещенной одной или более группами, выбранными из -OH или C1-3 алкокси,
e) 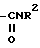
f) -CH2NR2,
g) 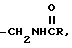
h) -CF3,
i) 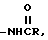
j) -NRSO2R,
k) -OP(O)(ORx)2, или
l) -R5;
а B отсутствует.
Третье, наиболее предпочтительное воплощение настоящего изобретения, дополнительно ограничивается соединениями, в которых:
X представляет собой OH;
Z представляет собой -O;
R1 и R2 соединены друг с другом с образованием совместно с атомом азота, к которому присоединен R1, 3-10-членной моноциклической или бициклической насыщенной кольцевой системы, состоящей из атома азота, к которому присоединен R1, и 2-9 углеродных атомов, которая может быть незамещена или замещена группой -W-арил или 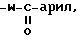 либо
либо
R1 и R2 соединены друг с другом с образованием совместно с атомом азота, к которому присоединен R1, 3-10-членной моноциклической или бициклической насыщенной кольцевой системы, состоящей из атома азота, к которому присоединен R1, 1-8 углеродных атомов и одной из групп 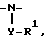 в которой Y отсутствует или
в которой Y отсутствует или 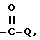 либо -SO2-Q-, R1 имеет выше указанные значения для тех случаев, когда R1 не зависит от R2 и не связан с этим радикалом, а Q отсутствует или -O-, NR- либо гетероцикл, необязательно замещенный C1-4 алкилом;
либо -SO2-Q-, R1 имеет выше указанные значения для тех случаев, когда R1 не зависит от R2 и не связан с этим радикалом, а Q отсутствует или -O-, NR- либо гетероцикл, необязательно замещенный C1-4 алкилом;
R3 представляет собой бензил, незамещенный или замещенный одной или более групп, выбранных из
1) гидрокси,
2) C1-3 алкокси, замещенной одной или более группами -OH, либо,
3) группой 
J1 представляет собой -NH-C1-4 алкил; а
J2 представляет собой группы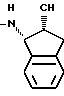
или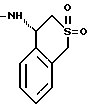
Наиболее предпочтительными соединениями настоящего изобретения являются соединения A-H и J, указанные ниже.
Соединение А:
N-(2(R)-гидрокси-1(S)инданил)-2(R)-фенилметил-4(S)- гидрокси-5-(2-(3(S)-N'-(трет.-бутилкарбамоил)-(4as,8as)- декагидроизохинолин)ил)пентанамид;
Соединение В: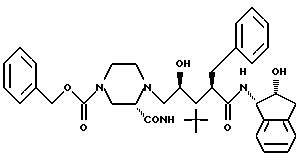
N-(2(R)-гидрокси-1(S)-инданил)-2(R)-фенилметил-4 (S)-гидрокси-5-(1-(4-карбобензилокси-2(S)-N'-(трет.- бутилкарбамоил)-пиперазинил)пентанамид;
Соединение C: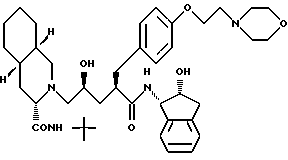
N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4-(2-(4- морфолинил)-этокси)-фенил)метил)-4-(S)-гидрокси-5-(2-(3 (S)-N'-(трет.-бутил-карбамоил)-(4as,8as)- декагидроизохинолин)ил)пентанамид;
Соединение D: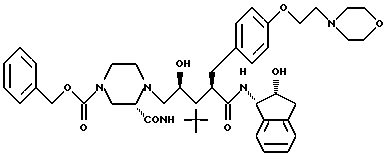
N-(2-(R)-гидрокси-1(S)-инданил)-2(R)-((4-(2-(4- морфолинил)этокси)фенил)метил)-4(S)-гидрокси-5-(1-(4- карбобензилокси-2(S)-N'-(трет.-бутилкарбамоил)пиперазинил)пентанамид;
Соединение E: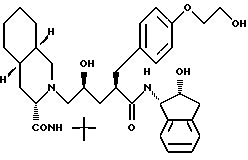
N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4-((2- гидрокси)этокси)-фенил)метил)-4(S)-гидрокси-5-(2-(3-(S)-N'-(трет. - бутилкарбамоил)(4as,8as)-декагидроизохинолин)ил)пентанамид;
Соединение F: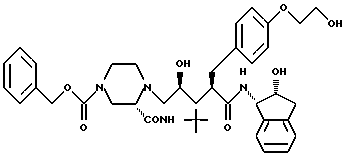
N-(2(R)-гидрокси-1(S)-инданил)-2-(R)-((4-((2- гидрокси)этокси)-фенил)метил)-4(S)-гидрокси-5-(1-(4-карбобензилокси-2 (S)-N'-(трет.-бутилкарбамоил)пиперазинил))-пентанамид;
Соединение G: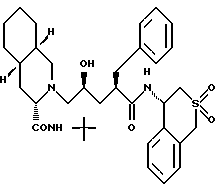
N-4(S)-3,4-дигидро-1H-2,2-диоксобензотиопиранил)-2(R)- фенил-метил-4(S)-гидрокси-5-(2-(3(S)-N'-(трет.-бутилкарбамоил)- (4as, 8as)декагидроизохинолин)ил)пентанамид;
Соединение H: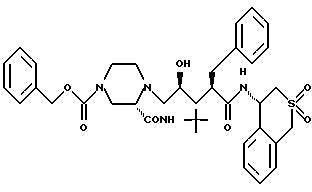
N-(4-(S)-3,4-дигидро-1H-2,2-диоксобензотиопиранил)-2(R) -фенил-метил-4(S)-гидрокси-5-(1-(4-карбобензилокси-2(S)-N'- (трет. -бутилкарбамоил)пиперазинил))пентанамид;
Соединение J: (L - 735,524)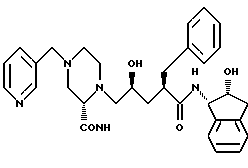
N-(2(R)-гидрокси-1(S)-инданил)-2(R)-фенилметил-4 (S)-гидрокси-5-(1-(4-(3-пиридилметил)-2(S)-N'-(трет.- бутилкарбамоил)пиперазинил))пентанамид.
Новые соединения настоящего изобретения также включают, но не ограничиваются следующими соединениями:
N-(2(R)-гидрокси-1(S)-инданил)-2(R)-фенилметил-4- (S)-гидрокси-5-(1-(N'-(трет.-бутил)-4-(S)феноксипролинамид) ил)пентанамид;
N-(2(R)-гидрокси-1-(S)-инданил)-2(R)-фенилметил-4- (S)-гидрокси-5-(1-(N'-трет.-бутил-4-(S)-2-нафтилокси-пролинамид) ил)-пентанамид;
N-(2-(R)-гидрокси-1(S)-инданил)-2-(R)-фенилметил-4 (S)-гидрокси-5-(1-(N'-трет.-бутил-4-(S)-1-нафтилокси- пролинамид)ил)-пентанамид;
N-(2(R)-гидрокси-1(S)-инданил)-2-(R)-фенилметил-4- (S)амино-5-(2-(3(S)-N'-(трет.-бутилкарбамоил)- (4as,8as)декагидроизохинолин)ил)пентанамид;
N-(2(R)-гидрокси-1-(S)-инданил)-2(R)-фенилметил-4-(S)- гидрокси-5-(1-(4-(3-фенилпропионил)-2(S)-N'-(трет. - бутилкарбамоил)пиперазинил))пентанамид;
N-(2(R)-гидрокси-1(S)-инданил)-2(R)-фенилметил-4- (S)-гидрокси-5-(1-(4-бензоил-2-(S)-N'-(трет. - бутилкарбоксамидо)-пиперазинил))пентанамид
N-(2(R)-гидрокси-1(S)-инданил)-2-(R)-фенилметил-4(S)- гидрокси-5-(1-(4-(3-фенилпропил)-2(S)-N'-(трет.- бутилкарбамоил)-пиперазинил)пентанамид;
N-(2(R)-гидрокси-1(S)-инданил)-2(R)-фенилметил-4- (S)амино-5-(1-(4-карбобензилокси-2(S)-N'-(трет.-бутилкарбамоил) пиперазинил))пентанамид;
N-(2(R)-гидрокси-1(S)-инданил)-2-(R)-((4-(2-(4- морфолинил)-этоксифенил)метил)-4(S)-гидрокси-5-(1-(N'-(трет. -бутил)- 4-(S)-феноксипролинамид)ил)пентанамид;
N-(2(R)-гидрокси-1(S)-инданил)-2-(R)-((4-(2-(4-морфолинил)- этоксифенил)метил-4(S)-гидрокси-5-(1-(-N'-трет. -бутил-4- (S)-2-нафтилоксипролинамид)ил)пентанамид;
N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4-(2-(2-(4- морфолинил)-этокси)фенил)метил)-4(S)-гидрокси-5-(1-(N'-трет. - бутил-4(S)-1-нафтилоксипролинамид)ил)пентанамид;
N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4-(2-(4-морфолинил)-этокси) фенил)метил)-4(S)-амино-5-(2-(3(S)-N-трет. - бутилкарбамоил)-(4as, 8as)-декагидроизохинолин)ил)пентанамид;
N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4-(2-(4- морфолинил)-этокси)фенил)метил-4(S)-гидрокси-5-(1-(4-(3- фенилпропионил)-2(S)-N'-(трет.-бутилкарбамоил)пиперазинил))пентанамид;
N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4-(2-(4- морфолинил)-этокси)фенил)метил)-4(S)-гидрокси-5-(1-(4-бензоил)-2(S)- (трет. -бутилкарбамоил)пиперазинил))пентанамид;
N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4-(2-(4-морфолинил)- этокси)фенил)метил)-4(S)-гидрокси-5-(1-(4-(3-фенилпропил)-2(S)- -N-(трет. -бутилкарбамоил))пиперазинил)пентанамид;
N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4-(2-(2-(4- морфолинил)-этокси)фенил)метил-4(S)-амино-5-(1-(1-(4- карбобензилокси-2(S)-1-N'-(трет.-бутилкарбамоил)пиперазинил)пентанамид;
N-2(R)-гидрокси-1(S)-инданил)-2(R)-((4-(2-гидрокси) этокси)-фенил)метил)-4(S)-гидрокси-5-(1-(N'-(трет. -бутил)-4 (S)-феноксипролинамид)ил)пентанамид;
N-(2(R)-гидрокси-1(S)-инданил)-2-(R)-((4-((2- гидрокси)этокси)-фенил)метил)-4(S)-гидрокси-5-(1-(N'- трет. -бутил-4(S)-2-нафтил-оксипролинамид)ил)пентанамид;
N-(2(R)-гидрокси-1(S)-инданил)-2-(R)-((4-((2- гидрокси)этокси)фенил)метил)-4(S)-гидрокси-5-(1-(N'-трет. - бутил-(S)-1-нафтил-оксипролинамид)ил)пентанамид;
N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4-((2- гидрокси)этокси)-фенил)метил)-4(S)-амино-5-(2-(3(S)-N'-(трет.- бутилкарбамоил)-(4as,8as)-декагидроизохинолин)ил)пентанамид;
N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4-((2- гидрокси)этокси)-фенил)метил)-4(S)-гидрокси-5-(1-(4-(3- фенил-пропионил)-(2(S)-N'-(трет.-бутилкарбамоил)-пиперазинил))пентанамид;
N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4-((2-гидрокси) этокси)-фенил)метил)-4(S)-гидрокси-5-(1-(4-бензоил-2(S)- N'-(трет. -бутил-карбамоил)пиперазинил))пентанамид;
N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4-((2-гидрокси) этокси)-фенил)метил)-4(S)-гидрокси-5-(1-(4-(3-фенилпропил)-2 (S)-N'-(трет.-бутилкарбамоил))пиперазинил)пентанамид;
N-2(R)-гидрокси-1(S)-инданил)-2(R)-((4-((2- гидрокси)этокси)-фенил)метил-4(S)-амино-5-(1-(4-карбобензилокси- 2(S)-N'-(трет. -бутилкарбамоил)пиперазинил))пентанамид;
N-(4(S)-3,4-дигидро-1Н-2,2-диоксобензотиопиранил)-2(R) -фенил-метил-4(S)-гидрокси-5-(1-(N'-(трет.-бутил)-4-(S)- феноксипролинамид)ил)пентанамид;
N-(4(S)-3,4-дигидро-1Н-2,2-диоксобензотиопиранил)-2- (R)-фенил-метил-4(S)-гидрокси-5-(1-(N'-трет.-бутил-4(S)-2- нафтилоксипролинамид)ил)пентанамид;
N-(4(S)-3,4-дигидро-1Н-2,2-диоксобензотиопиранил)-2-(R) -фенил-метил-4(S)-гидрокси-5-(1-(N'-трет.-бутил-4-(S)-1- нафтилокси-пролинамид)ил)пентанамид;
N-(4-(S)-3,4-дигидро-1Н-2,2-диоксобензотиопиранил)-2- (R)-фенил-метил-4(S)-амино-5-(2-(3(S)-N'-(трет. - бутилкарбамоил)-(4as,8as)-декагидроизохинолин)ил)пентанамид;
N-(4(S)-3,4-дигидро-1Н-2,2-диоксобензотиопиранил)-2-(R) -фенил-метил-4(S)-гидрокси-5-(1-(4-(3-фенилпропионил)-2(S)-N'- (трет. -бутилкарбамоил)пиперазинил)пентанамид;
N-(4-(S)-3,4-дигидро-1Н-2,2-диоксобензотиопиранил)-2-(R)- фенил-метил-4(S)-гидрокси-5-(1-(4-бензоил-2(S)-N'-(трет. -бутилкарбамоил) пиперазинил)пентанамид;
N-(4(S)-3,4-дигидро-1Н-2,2-диоксобензотиопиранил)-2- (R)-фенил-метил-4(S)-гидрокси-5-(12(4-(3-фенилпропил)-2(S) -N'-(трет. -бутилкарбамоил)пиперазинил)пентанамид или -(4(S)-3,4-дигидро-1Н-2,2-диоксобензотиопиранил)-2(R)-фенил- метил-4(S)-амино-5-(1-(4-карбобензилокси-2(S)-N'-(трет.- бутил-карбамоил)пиперазинил))пентанамид.
Соединения настоящего изобретения могут иметь асимметрические центры и существовать в виде рацематов, рацемических смесей, а также в виде индивидуальных диастереомеров или энантиомеров, причем все изомерные формы включены в объем настоящего изобретения.
В том случае, когда группы, которые могут иметь различные значения (например, арил, гетероцикл, R, R1, R2 A-, n, z и т.п.), присутствуют более одного раза, как заместители любого сорта или как составляющие формулы I, их конкретные значения в каждом случае не зависят от значений в каком-либо ином случае. Следует отметить, что комбинации заместителей и/или групп, которые могут иметь различные значения, допустимы лишь в том случае, когда такие комбинации обеспечивают существование устойчивых соединений.
Термин "алкил", используемый в настоящем описании, за исключением тех случаев, когда это отмечено особо, охватывает насыщенные алифатические углеводородные группы как нормального, так и изо-строения, содержащие конкретное число углеродных атомов (Me обозначает метил, Et - этил, Pr - пропил, Bu - бутил); термин "алкокси" используется для обозначения алкильной группы с указанным числом углеродных атомов, связанных друг с другом через кислородный мостик; под термином "циклоалкил" подразумеваются такие насыщенные кольцевые группы, как циклопропил, циклобутил, циклопентил, циклогексил (Cyh) и циклогептил. Под термином "алкенил" подразумеваются углеводородные группы нормальной или разветвленной конфигурации, содержащие одну или более двойных углерод-углеродных связей, находящихся в любом устойчивом положении цепи, такие как этенил, пропенил, бутенил, пентенил и т.п. Под термином "алкинил" подразумеваются углеводородные группы нормального или изо-строения, содержащие одну или более тройных углерод-углеродных связей, которые могут быть локализованы в любом стабильном участке цепи, такие как этинил, пропинил, бутинил, пентинил и т.п. Используемый в тексте термин "гало" ("Halo") относится к атомам фтора, хлора, брома и иода; термин "противоион" относится к таким небольшим с зарядом минус 1 анионам, как хлорид, бромид, гидрокси, ацетат, трифторацетат, перхлорат, нитрат, бензоат, малеат, тартрат, полутартрат, бензолсульфонат и т.п.
Используемый в тексте термин "арил", за исключением особо оговоренных случаев, означает фенил (Ph) или нафтил. Термин "карбоциклический" охватывает 5-7-членное углеродное кольцо или 7-10-членное бициклическое углеродное кольцо, причем любое из таких колец может быть как насыщенным, так и ненасыщенным.
Используемый в тексте описания термин "гетероцикл" или "гетероциклический", за исключением особо отмеченных случаев, относится к 5-7-членной моно- или бициклической, либо устойчивой 7-10-членной бициклической гетероциклической кольцевой системе, любое из колец которой может быть насыщенным или ненасыщенным и состоять из углеродных атомов и 1-3 гетероатомов, выбранных из группы, состоящей из N, O и S, причем такие гетероатомы, как азот и сера, могут быть необязательно окисленными, а азотный гетероатом может быть необязательно четвертичным, и такие группы могут быть бициклическими, в которых любое из указанных выше гетероциклических колец может быть сконденсировано с бензольным кольцом. Такое гетероциклическое кольцо может быть присоединено по любому гетероатому или углеродному атому, когда такое присоединение обеспечивает получение устойчивой структуры. Примерами таких гетероциклических элементов могут служить пиперидинил, пиперазинил, 2-оксопиперазинил, 2-оксопиперидинил, 2-оксопирролодинил, 2-оксоазепинил, азепинил, пирролил, 4-пиперидонил, пирролидинил,пиразолил, пиразолидинил, имидазолил, имидазолинил, имидазолидинил, пиридил, пиразинил, пиримидинил, пиридазинил, оксазолил, оксазолидинил, изоксазолил, изоксазолидинил, морфолинил, тиазолил, тиазолидинил, изотиазолил, хинуклидинил, изотиазолидинил, индолил, хинолинил, изохинолинил, бензимидазолил, тиадиазолил, бензопиранил, бензотиазолил, бензоксазолил, фурил, тетрагидрофурил, тетрагидропиранил, тиенил, бензотиенил, тиаморфолинил, тиаморфолинил сульфоксид, тиаморфолинил сульфон и оксадиазолил.
Термин "морфолино" идентичен морфолинилу.
Фармацевтически приемлемые соли соединений формулы I (в виде водо- или маслорастворимых продуктов, либо диспергируемых веществ) включают традиционные нетоксичные соли или соли четвертичного аммония, которые получают из неорганических, либо органических солей или оснований. Примерами таких солей присоединения кислот могут служить ацетат, адипинат, альгинат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, цитрат, камфорат, камфорсульфонат, циклопентанпропионат, диглюконат, додецилсульфонат, этилсульфонат, фумарат, глюкогептаноат, глицерофосфат, полусульфат, гептаноат, гексаноат, гидрохлорид, гидробромид, гидроиодид, 2-гидроксиэтансульфонат, лактат, малеат, метансульфонат, 2-нафталинсульфонат, никотинат, оксалат, памоат, пектинат, персульфат, 3-фенилпропионат, пикрат, пивгалат, пропионат, сукцинат, тартрат, тиоцианат, тозилат и ундеканоат. Основные соли включают соли аммония, такие соли щелочных металлов, как соли натрия и калия, такие соли щелочноземельных металлов, как соли кальция и магния, такие соли с органическими основаниями, как соли дициклогексиламина, N-метил- D-глюкамина, а также такие соли, в которых кислотными компонентами являются такие аминокислоты, как аргинин, лизин и т.д. Кроме того, основные азотсодержащие группы могут быть переведены в форму четвертичных солей с помощью таких агентов, как низшие алкилгалогениды, например метил, этил, пропил и бутил хлориды, бромиды и иодиды; такие диалкилсульфаты, как диметил-, диэтил-, дибутилсульфаты; диамилсульфаты, такие как длинноцепочечные галогениды, как децил, лаурил, миристил и стеарил хлориды, бромиды и иодиды, такие аралкилгалогениды, как бензил и фенетил бромиды и т.п. Другие фармацевтически приемлемые соли включают этоксисульфатные и сульфатные соли.
Ниже представлены схемы I-III, описывающие получение новых соединений настоящего изобретения. В следующих далее таблицах I и II иллюстрируются соединения, которые могут быть получены в рамках схем I-III. Схемы I-III не ограничиваются соединениями, указанными в таблицах, или какими-либо конкретными заместителями, используемыми в схемах для иллюстрации. Примеры конкретно иллюстрируют применение следующих схем (см. в конце описания) на конкретных соединениях.
Реакции амидного сочетания, используемые для получения соединений настоящего изобретения, обычно осуществляют карбодиимидным методом с помощью таких реагентов, как дициклогексилкарбодиимид или 1-этил-3-(3-диметиламинопропил)карбодиимид. Другие методы образования амидной или пептидной связи включают, но не ограничиваются ими, синтетические маршруты, реализуемые через хлорангидрид кислоты, азид, смешанный ангидрид или активированный сложный эфир. Обычно амидное сочетание осуществляют в жидкой фазе, однако может также использоваться твердофазный синтез с применением классического метода Мэррифилда. Присоединение и удаление одной или более защитных групп осуществляют традиционными методами.
Дополнительная информация, касающаяся синтетических аспектов, содержится в EPO 0337714.
Один из методов получения соединений формулы I представлен схемой I. Дигидро-5(S)-(трет. -бутилдиметилсилилоксиметил)-3(2Н)- фуранон (ниже, соединение 1) получают стандартными методами, известными в данной области, из выпускаемого промышленностью дигидро-5(S)-(гидроксиметил)-2(3Н)-фуранона. После алкилирования соединения 1 с образованием соединения 2, защитную группу лактона 1 удаляют с помощью водного раствора HF с получением соединения 3.
Спиртовую группу соединения 3 активируют путем превращения в такую уходящую группу, как мезилат, тозилат или трифилат, в результате обработки спирта хлористым сульфонилом, или таким ангидридом сульфокислоты, как ангидрид трифторметансульфокислоты, в присутствии такого затрудненного аминного основания, как триэтиламин, диэтил-изопропиламин или 2,6-лутидин, в результате чего получают соединение 4. Уходящую группу соединения 4 заменяют на амин 5, например N'-трет.-бутил- (4as,8as)-(декагидроизохинолин)-3(S)-карбоксамид, в среде такого высококипящего растворителя, как ДМФ или ксилол, в результате чего получают соединение 6. Трифторметансульфонил-окси- группа может быть заменена на амин при комнатной температуре в среде такого растворителя, как изопропанол, в результате обработки N,N-диизопропилэтиламином.
Соединение 6 гидролизуют с помощью водного раствора гидроксида лития или натрия и полученную в результате гидроксикислоту 7 превращают в защищенную гидроксикислоту 8. Гидроксильную группу удобнее всего защищать такой стандартной силильной защитной группой, как трет.-бутилдиметилсилил или трет.-бутилдифенилсилил.
Защищенную гидроксикислоту подвергают сочетанию с желаемым амином R12 с образованием соединения 9 и силильную защитную группу удаляют с помощью иона фторида, в результате чего получают соединение 10.
Второй способ получения продуктов общей формулы 1 показан на схеме II. На схеме II алкилирование соединения II включает две стадии, первая из которых представляет собой депротонирование соединения II с помощью н-бутиллития или диизопропиламида лития (IDA), а вторая - присоединение алкенилгалогенида (например, бромистого аллила) с образованием соединения 12.
В результате дигидроксилирования олефина 12 четырехокисью осмия и N-метилморфолин-N-оксидом (NMO) получают диастереомерную смесь диолов 13. В результате селективного мезилирования первичного спирта 13 с помощью хлористого метансульфонила и триэтиламина или пиридина получают мезилат 14.
В результате нагревания мезилата 14 с амином при кипении в таком спиртовом растворителе, как метанол или изопропанол, содержащем избыток карбоната калия, получают такой аминоспирт, как соединение 15. На этой стадии диастереомеры могут быть разделены стандартными методами, хорошо известными специалистам в данной области. С другой стороны, такое разделение может быть осуществлено после удаления кеталя.
Удаление кеталя в соединении 15 осуществляют путем обработки кислотой в присутствии метанола или водным раствором кислоты, либо 1 N раствором HCl в ТГФ, в результате чего получают соединение 16.
Третий способ получения продуктов общей формулы I показан на схеме III. Защиту пирролидиновой -NH-группы соединения 17 осуществляют с помощью BOC-ангидрида и диметиламинопиридина с получением защищенного соединения 18. Алкилирование соединения 18 осуществляют в две стадии, первая из которых заключается в депротонировании 18 таким сильным основанием, как литийгексаметилдисиламид (LHMDS) или литийдиизопропиламид (LDA), после чего осуществляют вторую стадию присоединения галоидного алкила (например, бромистого бензила) с получением соединения 19.
TBS-защитную и BOC-защитную группы соединения 19 удаляют путем обработки водным раствором HF в ацетонитриле с образованием спирта 20. В результате мезилирования первичного спирта 20 с помощью метансульфонилхлорида и триэтиламина, либо пиридина, получают мезилат 21, который нагревают с амином при кипении в таком спиртовом растворителе, как метанол или изопропанол, содержащем избыток карбоната калия, в результате чего получают такой аминопирролидинон, как соединение 22. Пирролидиновую - NH-группу соединения 22 вновь защищают BOC-группой, как описано выше, и полученное в результате соединение 23 гидролизуют с раскрытием цикла с помощью такого основания, как гидроксид лития или натрия, в результате чего получают кислоту 24. Затем соединение 24 подвергают сочетанию с амином NH2R12, используя стандартную методику, и группу BOC удаляют с помощью газообразной HCl или трифторуксусной кислоты с получением желаемого продукта, представленного соединением 25.
Соединение формулы 26: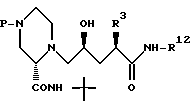
в которой P представляет собой такую азотзащищающую группу,
-BOC или -CBZ,
предпочтительно получают в соответствии со способом, описанным на схеме I, предпочтительно используя 5-трифторметансульфонил- оксиметиловый аналог лактона 4 (см. пример 15, стадия 1).
Соединение формулы 27: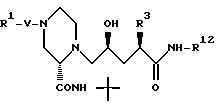
может быть получено различными путями из соединения 28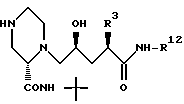
которое получают после удаления азотзащищающей группы в соединении 26 с использованием методов, хорошо известных в данной области, например путем каталитического гидрирования с целью удаления CBZ группы или путем обработки триметилсилилтрифлатом и 2,6-лутидином при примерно 0oC в таком растворителе, как CH2Cl2 с целью удаления BOC группы.
Так, например, атом азота, находящийся в положении 4 пиперазинила в соединении 28, может быть подвергнут алкилированию соединением формулы R1-X в таком растворителе, как ДМФ, в присутствии EtN3 при комнатной температуре, причем X, представляющий собой -Cl, Br или -J либо сульфонамидную группу, может быть получен обработкой соединения 28 сульфонилхлодом формулы R1 SO2Cl в аналогичных условиях. Кроме этого, методы стандартного амидного сочетания могут использоваться для образования амидной группы в положении 4 пиперазинила. Техника таких методов хорошо известна специалистам в данной области. Группа R1 в R1-X или R1 SO2Cl имеет значения, указанные выше в определении соединений формулы I, в которой R1 не зависит от R2 и не связан с ним, за исключением того, что R1 не является водородом или группой со свободным гидрокси- заместителем, например C1-4 алкилом, замещенным гидрокси, и также за исключением того, что R1 может представлять собой арил, замещенный гидрокси-группой.
Соединения настоящего изобретения также проиллюстрированы в таблицах I-IV (см. в конце описания).
Соединение настоящего изобретения используются для подготовки и проведения скрининговых анализов на антивирусные соединения. Так, например, соединения настоящего изобретения используются для выделения энзимных мутантов, представляющих собой отличные инструменты скрининга на более мощные антивирусные соединения. Кроме того, соединения настоящего изобретения используются для установления или определения места присоединения других антивирусных агентов к ВИЧ-протеазе, например, в результате конкурентного ингибирования. Таким образом, соединения настоящего изобретения являются коммерческими продуктами, предназначенными для продажи в указанных выше целях.
Соединения настоящего изобретения используются для ингибирования ВИЧ-протеазы, профилактики или лечения инфицирования вирусом иммунодефицита человека (ВИЧ) и лечения развивающегося в результате патологического состояния, такого как СПИД. Лечение СПИДа или профилактика, либо лечение инфицирования ВИЧ, рассматривается как включающее, но не ограничивающееся лечением большого числа состояний ВИЧ-инфицирования: СПИДа, ARC (родственный СПИДу комплекс), как симптоматический, так и асимптоматический, а также реальной или потенциальной подверженности ВИЧ. Так, например, соединения настоящего изобретения используют для лечения ВИЧ-инфицирования после предполагаемого последующего воздействия ВИЧ, например, в случае переливания крови, трансплантации органов, замене жидкостной среды тела, укусов насекомых, случайного укола иглой или воздействия на кровь человека в ходе хирургического вмешательства.
В этих целях соединение настоящего изобретения применяют орально, парентерально (включая подкожные инъекции, внутривенные, внутримышечные, интрастернальные инъекции или методы вливания), в виде ингаляционного спрэя, либо ректально, в виде единичных дозировочных рецептур, содержащих нетоксичные фармацевтически приемлемые носители, присадки и связующие вещества.
Таким образом, в соответствии с настоящим изобретением дополнительно предусматривается способ лечения и фармацевтическая композиция для лечения ВИЧ-инфицирования и СПИДа. Такое лечение включает применение на пациенте, нуждающемся в таком лечении, фармацевтической композиции, содержащей фармацевтический носитель и терапевтически эффективное количество соединения настоящего изобретения или его фармацевтически приемлемой соли.
Такие фармацевтические композиции могут быть в виде орально применяемых суспензий или таблеток; спрэев для вдыхания через нос; стерильных препаратов для инъекций, например, стерильных водных или маслосодержащих суспензий, или свечей.
При оральном применении в виде суспензий такие композиции готовят в соответствии с методиками, хорошо известными в области создания фармацевтических рецептур, и они могут содержать микрокристаллическую целлюлозу для обеспечения массы, альгиновую кислоту или альгинат натрия в качестве суспендирующего агента, метилцеллюлозу в качестве усилителя вязкости, а также подслащивающие агенты или отдушки, известные в данной области. В качестве таблеток немедленного выделения такие композиции могут содержать микрокристаллическую целлюлозу, дикальций фосфат, крахмал, стеарат магния и лактозу и/или другие эксципиенты, наполнители, расширяющие агенты, дезинтеграторы, разбавители и смазывающие агенты, известные в данной области.
При применении в качестве носового аэрозоля или для ингаляции такие композиции готовят в соответствии с методиками, хорошо известными в области изготовления фармацевтических рецептур, и они могут быть выполнены в виде растворов в физиологическим растворе, с использованием бензилового спирта или других подходящих предохраняющих агентов, промоторов абсорбции для усиления биоприменимости, фторуглеродов и/или других солюбилизирующих или диспергирующих агентов, известных в данной области.
Растворы для инъекций или суспензии формируют в соответствии с известными методами с использованием таких нетоксичных, парентерально применимых разбавителей или растворителей, как маннит, 1,3-бутандиол, вода, раствор Рингера или изотонический раствор хлорида натрия, или таких подходящих диспергирующих или смачивающих и суспендирующих агентов, как стерильные, успокаивающие, устойчивые масла, включающие синтетические моно- или диглицериды, а также жирные кислоты, например олеиновую кислоту.
При ректальном применении в виде свечей такие композиции готовят путем смешивания лекарства с таким подходящим, не вызывающим раздражения эксципиентом, как масло какао, синтетические глицеридные эфиры или полиэтиленгликоли, которые являются твердыми веществами при обычных температурах, но ожижаются и/или растворяются в ректальной полости с выделением лекарства.
Уровни дозировок порядка 0,02-5,0 г или 10,0 г в день являются подходящими для лечения или профилактики указанных выше состояний, причем оральные дозировки в 2-5 раз выше. Так, например, ВИЧ-инфицирование эффективно лечится в результате применения 1,0-50 мг соединения на килограмм веса тела в течение 1-4 раз в день. В соответствии с одним из предпочтительных режимов лечения на каждом пациенте орально применяют 100-400 мг препарата каждые 6 часов. Однако следует иметь в виду, что конкретный уровень дозировок и частота их применения для каждого конкретного пациента может изменяться и будет зависеть от большого числа факторов, включающих активность конкретного используемого соединения, метаболическую стабильность и длительность действия такого соединения, возраст пациента, вес тела, общее состояние здоровья, пол пациента, тип диеты, тип и время применения, скорость экскрекции, комбинацию лекарств, тяжесть конкретного состояния и тип используемой терапии.
Настоящее изобретение также относится к комбинациям соединений, ингибирующих ВИЧ-протеазу с одним или более агентами, используемыми для лечения СПИДа. Так, например, соединения настоящего изобретения могут эффективно применяться в период как до, так и после воздействия, в комбинации с эффективными количествами СПИД-антивирусных агентов, иммуномодуляторов, противоинфекционных агентов или вакцин, известных специалистам в данной области.
Следует иметь в виду, что сфера комбинаций соединений настоящего изобретения со СПИД-антивирусными агентами, иммуномодуляторами, противоинфекционными агентами или вакцинами не ограничивается теми, что перечислены в приведенной таблице С, и в принципе может включать любую комбинацию с любой фармацевтической композицией, используемой для лечения СПИДа.
Некоторые соединения, указанные в таблице С, представляют собой следующие вещества: 1-697,661 или '661' представляет собой 3-(/4,7- дихлор-1,3-бензоксазол-2-ил)метил/амино)-5-этил-6-метилпиридин- 2(1Н)-он; 1-696,229 представляет собой 3-/2-(1,3-бензоксазол-2- ил)этил/-5-этил-6-метил-пиридин-2(1Н)-он. Синтезы 1-697,661 и 1-696,229 описаны в EPO 434071 и EPO 462800 и на них ссылаются в настоящем описании. Синтезы ddC ddI и AZT также описаны в EPO 484071.
Предпочтительными комбинациями являются одновременные или чередующиеся обработки ингибитора ВИЧ-протеазы и ненуклеозидного ингибитора ВИЧ обратной транскриптазы. Необязательным третьим компонентом в комбинации является нуклеозидный ингибитор ВИЧ обратной транскриптазы, такой как AZT, ddC или ddI. Предпочтительным ингибитором ВИЧ протеазы является L-735,524 (соединение 1). Предпочтительные ненуклеозидные ингибиторы ВИЧ обратной транскриптазы включают L-697,661. Такие комбинации могут оказывать синергитические воздействия на ограничение распространения ВИЧ. Предпочтительными комбинациями являются следующие: (1) I-735,524 с I-697,661 и необязательно AZT или ddI, либо ddC; (2) I-735,524 с любым агентом, выбранным из AZT, или ddI, либо ddC.
Анализ на ингибирование микробиально экспрессированной ВИЧ протеазы
Исследование ингибирования реакции экспрессирования протеазы в Escherichia coli в присутствии пептидного субстрата (Val- Ser-Gln-Asn-(бетанафтил)Ala-Pro-Ile-Val, 0,5 мг/мл во время инициирования реакции) проводят в 50 М ацетата натрия, при pH 5.5, температуре 30oC в течение 1 часа. Различные концентрации ингибитора в 1,0 μкл ДМСО добавляют к 25 μкл пептидного раствора в воде. Реакцию инициируют добавлением 15 μкл 0,33 nМ протеазы (0,11 u г) в 0,133 М растворе ацетата натрия при pH 5.5 и 0,1% альбумина бычьей сыворотки. Реакцию прекращают с помощью 160 μкл 5% фосфорной кислоты. Продукты реакции разделяют методом HPLC (обратимая фаза УУДАС С-18 с широкими порами, 5 см, ацетонитрильный градиент, 0,1% фосфорной кислоты). Степень ингибирования реакции определяют по высотам пиков продуктов. Хроматограммы независимо синтезированных продуктов, полученные методом HPLC, служат количественными стандартами и с их помощью подтверждают состав продуктов. С помощью продуктов, синтезированных в примерах 1-7 включительно, установлено, что значения IC50 лежат в интервале 1-100 n М. Значения IC50 соединений А.В и J составляет величины от примерно 0,3 до примерно 6 nМ.
Ингибирование распространения вируса
А.Приготовление суспензии клеток МТ-4, инфицированных ВИЧ
МТ клетки инфицируют в день 0 при их концентрации 250000 на мл 1:1000 разбавлением штамма ВИЧ-1 линии 111в (конечное значение 125 pr р24/мл; достаточно для выхода инфицированных клеток в первый день ≤ 1% и 25-100% на четвертый день). Клетки инфицируют и выращивают в следующей среде: RPMI 1640 (Виттакер Биопродактс), 10% инактивированной сыворотки коровьего плода, 4 мМ глютамина (Гибко Лабс.) и 1:100 пенициллин-стрептомицин (Гибко Лабс.).
Полученную смесь инкубируют в течение ночи при 37oС в атмосфере, содержащей 5% CO2.
В. Обработка ингибиторами
Готовят матрицу наномолярных концентрационных интервалов парных комбинаций (см. таблицу S). В первый день аликвоты в 125 мкл ингибиторов добавляют к равным объемам клеток МТ-4, зараженных ВИЧ (50000 клеток в пробирке), находящихся в 96 пробирках на микротитрической пластине для клеточных культур. Инкубирование продолжают в течение 3 дней при 37oC в атмосфере, содержащей 5% CO2.
C. Измерение распространения вируса
С использованием многоканального пипетора, осажденные клетки ресуспендируют и 125 мкл собирают на отдельную микротитрическую пластину. Верхний слой анализируют на присутствие ВИЧ р24 антигена.
Концентрацию ВИЧ р24 антигена измеряли с помощью энзимного иммуноанализа, описанного ниже. Аликвоты р24 антигена, подлежащие измерению, добавляют в микропробирки, покрытые моноклональным антителом, специфичным на антиген ядра ВИЧ. На этой и других соответствующих стадиях микропробирки подвергают промывке. Добавляют биотинилированное ВИЧ-специфичное антитело и затем добавляют конъюгат стрепавидина и пероксидазы хрена. В результате добавления пероксида водорода и тетраметилбензидинового субстрата происходит цветная реакция. Интенсивность окраски пропорциональна концентрации ВИЧ р24 антигена.
Расчет степени синергизма
Обнаружено, что парные комбинации ингибиторов (см. таблицу S) обеспечивают явно усиленное ингибирование распространения вируса по сравнению с действием каждого ингибитора по отдельности или по сравнению с просто аддитивным ингибированием каждым ингибитором. Так, например, парная комбинация 524 и AZT, как установлено, демонстрирует явное усиление ингибирования распространения вируса по сравнению с действием 524 и AZT по отдельности, или по сравнению с суммарным эффектом ингибирования 524 и AZT.
Полученные данные обрабатывают следующим образом: фракционные ингибиторные концентрационные соотношения (FIC) рассчитывают согласно методу Элиона с сотр., J.Biol. Chem. 208, 477 (1954). Для различных парных комбинаций определяют минимальную сумму FIC, что соответствует максимальному синергизму (см. таблицу S). Полученные результаты указывают на существенный синергизм в ингибировании распространения вируса. Чем меньше число, тем выше синергизм. - Таблица S
Парные комбинации * - Максимальный синергизм
524 + ddJ - 0,7
524 + AZT - 0,7
524 + 661
524 представляет собой L-735,524 (соединение J). Другие соединения также указаны в приведенной выше таблице С.
Пример 1. Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-гидрокси-5-(1-N'-(трет.-бутил)-4(S)- феноксипролинамид)ил)пентанамида.
Стадия 1: Получение N-(2(R)-гидрокси-1(S)-инданил)- -3-фенил-пропанамида.
К холодному (0oC) раствору хлористого метилена (30 мл), содержащему 2(R)-гидрокси-1(S)-аминоиндан (750 мг, 5,0 ммоля) и триэтиламин (606 мг, 6,0 ммоля), добавляют раствор гидроцинамоил хлорида (843 мг, 5,0 ммоля) в 5 мл хлористого метилена. Через 2 часа реакционную смесь переливают в делительную воронку, содержащую 50 мл хлористого метилена, и промывают 10% раствором лимонной кислоты (2х30 мл). Органический слой сушат, фильтруют и концентрируют с получением белого твердого вещества.
Стадия 2: Получение N-(2(R)-гидрокси-1(S)-индан-N,O- изопропилиденил)-3-фенил-пропанамида.
Неочищенное белое твердое вещество со стадии 1 растворяют в 50 мл хлористого метилена и добавляют 5 мл диметоксипропана, после чего добавляют 100 мг п-толуолсульфокислоты. Реакционную смесь в течение 18 часов перемешивают при комнатной температуре и затем переливают в делительную воронку и промывают насыщенным раствором NaHCO3 (2х30 мл). Органический слой сушат, фильтруют и концентрируют с получением масла, которое подвергают хроматографической очистке (SiO2, 40% EtOAc/гексан), в результате чего получают маслянистый продукт, который с течением времени закристаллизовывается.
Стадия 3: Получение N-(2(R)-гидрокси-1(S)-индан-N, O- изопропилиден-ил)-2(S)-фенилметилпент-4-енамида.
К раствору N-(2(R)-гидрокси-1(S)-индан-N,O-изопропилиден- ил)-3-фенил-пропанамида (1,03 г, 2,9 ммоля)в 20 мл ТГФ, охлажденному до -78oC, добавляют н-BuLi (2,5 М, 1,40 мл, 3,5 ммоля). Через 20 минут добавляют бромистый аллил (0,48 г, 3,9 ммоля), реакционную смесь перемешивают в течение 1 часа при -78oC и затем для прекращения реакции добавляют 10 мл насыщенного раствора NH4Cl. Реакционную смесь разбавляют 50 мл воды, экстрагируют этилацетатом (2х50 мл), органическую фазу промывают насыщенным раствором NaCl (50 мл), сушат, фильтруют и концентрируют с получением сырого продукта. Сырой продукт очищают на силикагеле с получением целевого продукта.
Стадия 4: Получение N-(2(R)-гидрокси-1(S)-индан-N, O- изопропилиден-ил)-2(S)-фенилметил-(4(RS),5- дигидроксипентанамида.
К 800 мг (2,2 ммоля) N-(2(R)-гидрокси-1(S)-индан-N,O- изопропилиден-ил)-2(S)-фенилметил-пент-4-енамида, растворенного в 40 мл смеси ацетон/вода в соотношении 9:1, добавляют 0,8 мл 60% раствора N-метилморфолин-N-оксида в воде, после чего добавляют 4 мл 2,5% раствора четырехокиси осмия в трет.-BuOH. Через 18 часов добавляют избыток твердого бисульфата натрия, реакционную смесь перемешивают в течение 2 часов и затем фильтруют через слой целита. Фильтрат концентрируют, разбавляют 50 мл воды, экстрагируют хлористым метиленом (2х50 мл), органическую фазу сушат, фильтруют и концентрируют с получением продукта в виде пены.
Стадия 5: Получение N-(2(R)-гидрокси-1(S)-индан-N, O- изопропилиден-ил)-2(S)-фенилметил-4(RS)-гидрокси-5- метансульфонилокси-пентанамида.
К 200 мг (0,527 ммоля) N-(2(R)-гидрокси-1(S)-индан-N,O- изопропилиденил)-2(S)-фенилметил-(4(RS)-5- дигидрокси)пентанамида, растворенного в 7 мл хлористого метилена, при 0oC добавляют триэтиламин (59 мг, 0,58 ммоля), после чего добавляют метансульфонил хлорид (66 мг, 0,579 ммоля). Через 4 часа реакционную смесь промывают 10% раствором лимонной кислоты (2х50 мл) и органическую фазу сушат, фильтруют и концентрируют с получением мономезилата в виде смеси спиртов.
Стадия 6: Получение N'-трет.-бутил-N-Вос-4(R)-гидрокси-1-пролинамида.
К раствору N-Вос-4(R)-гидроксипролина (2,00 г) в ДМФ (20 мл), охлажденному до 0oC, добавляют ЕДС (1,987 г), HOBt (1,401 г), трет.-бутиламин (1,09 мл) и триэтиламин (2,41 мл). Через 18 часов реакционную смесь разбавляют этилацетатом (150 мл) и промывают 10% раствором HCl, насыщенным раствором NaHCO3, водой и рассолом. Затем полученный раствор сушат над MgSO4 и концентрируют с получением белого твердого вещества.
Стадия 7: Получение N'-трет.-бутил-N-Вос-4(R)-фенокси- -L-пролинамида.
К раствору N'-трет.бутил-N-Вос-4(R)-гидрокси-1-пролинамида (0,6 г) в ТГФ (5 мл) добавляют фенол (0,295 г), трифенилфосфин (0,824 г) и затем прикапывают диэтилазодикарбоксилат (0,495 мл). Реакционную смесь перемешивают в течение 24 часов при окружающей температуре и затем разбавляют этилацетатом (200 мл) и промывают насыщенным раствором NaHCO3 водой, рассолом и сушат над MgSO4. Концентрированием в вакууме получают желтое масло, которое очищают методом хроматографии (элюирование смесью гексан:EtOAc, 1:1, 30 мм колонка).
Стадия 8: Получение N-трет.-бутил-4(S)-фенокси-1- пролинамидтрифторацетата.
К раствору N'-трет.-бутил-N-Вос-4(S)-фенокси-1-пролинамида (0,596 г) в хлористом метилене (4 мл) при 0oC добавляют трифторуксусную кислоту (2 мл). Через 30 минут реакционную смесь нагревают до комнатной температуры и перемешивают в течение 2 часов. Растворитель удаляют в вакууме и получают бледно-желтое масло.
Стадия 9: Получение N-(2(R)-гидрокси-1(S)-индан-N, O- изопропилиден-ил)-2-(R)-фенилметил-4-(S)-гидрокси-5-(1-N'- (трет. бутил)-4(S)-фенокси-пролинамид)ил)пентанамида.
К раствору соли трифторуксусной кислоты N-трет.-бутил- 4(S)-фенокси-1-пролинамида (0,36 г) и N-(2(R)-гидpoкcи-1(S) индан-N,O-изопропилиден-ил)-2(S)-фенилметил-4(R)-гидрокси- 5-метансульфонилокси-пентанамида (0,226 г) в 3 мл изопропанола добавляют карбонат калия (0,441 г) и реакционную смесь нагревают до 80oC. Через 18 часов реакционную смесь охлаждают до комнатной температуры, фильтруют через целит, который промывают дополнительными порциями EtOAc. Фильтрат концентрируют, остаток растворяют в EtOAc (100 мл) и промывают водой, рассолом и сушат над MgSO4. Растворитель удаляют в вакууме и полученное в результате масло очищают методом хроматографии однократного испарения с получением продукта в виде смеси диастереомеров.
Стадия 10: Получение N-(2(R)-гидрокси-1(S)-инданил)-2- (R)-фенилметил-4(S)-гидрокси-5-(1-(N'-трет.бутил-4(S)-фенокси- пролинамид)ил)-пентанамида.
К раствору N-(2(R)-гидрокси-1(S)-индан-N,O-изопропилиден- ил)-2(R)-фенилметил-4-(S)-гидрокси-5-(1-(N'-(трет. бутил) 4-(S)-феноксипролинамид)-ил)пентанамида (0,13 г) в MeOH (5 мл) добавляют камфорсульфокислоту (CSA) (0,070 г) при комнатной температуре. Через 5 часов добавляют еще 0,025 г CSA и реакционную смесь перемешивают в течение 18 часов. Реакцию прекращают с помощью насыщенного раствора NaHCO3 (5 мл) и растворитель удаляют до остаточного объема в 4 мл. Водный раствор тщательно экстрагируют EtOAc и органический слой промывают водой, рассолом и сушат. После удаления растворителя в вакууме полученное в результате масло очищают методом хроматографии однократного испарения с получением целевого соединения в виде белой пены. Такую пену растворяют в смеси EtOAc: гексан и маточную жидкость декантируют с масла. Затем масло сушат в высоковакуумном эксикаторе с получением белой пены.
Пример 2. Получение N-(2(R)-гидрокси-1(S)-инданил)-2 (R)-фенилметил-4(S)-гидрокси-5-(1-(N'-трет.бутил-4(S)-2- нафтилоксипролинамид)ил)пентанамида.
Стадия 1: Получение соли трифторуксусной кислоты N-трет. бутил-4(S)-2-нафтилокси-1-пролинамида.
Следуя той же методике синтеза трифторацетата N-трет. -бутил-4(S)-фенокси-1-пролинамида, что описана в стадиях 6-8 примера 1, но заменяя фенол на 2-нафтол, получают 2-нафтилоксипролинамид.
Стадия 2: Получение N-(2(R)-гидрокси-1(S)-инданил-2 (R)-фенилметил-4(S)-гидрокси-5-(1-(N'-трет. бутил-4-(S)-2- нафтилокси-пролинамид)ил)-пентанамида.
Целевое соединение получают, следуя методике, описанной в стадиях 9 и 10 примера 1, но заменяя используемый в стадии 9 N-трет.бутил-4(S)-фенокси-1-пролинамид трифторацетат на N-трет.бутил-4(S)-2-нафтилокси-1-пролинамид трифторацетат.
Пример 3. Получение N-(2-(R)-гидрокси-1(S)- инданил-2(R)-фенилметил-4(S)-гидрокси-5-(1-(N'-трет. бутил-4(S)-1-нафтилокси-пролинамид(ил)-пентанамид
Стадия 1: Получение соли трифторуксусной кислоты N-трет.бутил-4(S)-1-нафтилокси-1-пролинамида.
Следуя практически той же методике синтеза N-трет.бутил- 4(S)фенокси-1-пролинамида трифторацетата, что описана для стадий 6-8 примера 1, но заменяя фенол на 1-нафтол, получают 1-нафтилоксипролинамид.
Стадия 2: Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R) -фенилметил-4(S)-гидрокси-5-(1-(N-трет. бутил-4(S)-2- нафтилоксипролинамид(ил)-пентанамида.
Целевое соединение получают, следуя практически той же методике, что описана для стадий 9 и 10 примера 1, но заменяя используемый на стадии 9 трифторацетат N-трет.бутил-4(S)- фенокси-1-пролинамида на трифторацетат N-трет.бутил-4(S)-1- нафтилокси-1-пролинамида.
Пример 4. Получение N-2(R)-гидрокси-1(S)-инданил)-2(R) -фенилметил-4(S)-гидрокси-5-(2-(3(S)-N'-(трет. бутил- карбоксамидо)-(4as,8as)-декагидро-изохинолин)ил)-пентанамида.
Стадия 1: Получение дигидро-5(S)-((трет. -бутилдифенилсилил)- оксиметил)-3(R)-фенилметил-3(2Н)-фуранона.
Раствор диизопропиламида лития (LDA)получают путем добавления 1,55 мл n-BuLi (2,5 М в гексане) к 0,55 мл (3,9 ммоля) диизопропиламина в 10 мл ТГФ при -78oC. Через 30 минут добавляют раствор дигидро-5-(S )-((трет.-бутилдифенилсилил)- оксиметил)-3(2Н)-фуранона (1,38 г, 3,89 ммоля) в 5 мл ТГФ. После дополнительного перемешивания в течение 30 минут добавляют бромистый бензил (0,68 г, 3,9 ммоля) и перемешивание продолжают в течение 3 часов, после чего реакцию прекращают путем добавления 10% водного раствора лимонной кислоты. Раствор экстрагируют этилацетатом (2х50 мл), после чего промывают рассолом, сушат, фильтруют и концентрируют с получением масла. Продукт реакции очищают методом хроматографии (SiO2, 20% EtOAc/гексан) с получением целевого соединения.
Стадия 2: Получение дигидро-5(S)-(гидроксиметил)-3(R)- фенилметил-3(2Н)-фуранона.
К 5,26 г дигидро-5(S)-((трет. бутилдифенилсилил)оксиметил)- 3(R)-фенилметил-3(2Н)-фуранона в 40 мл ацетонитрила добавляют 1,34 мл 49% водного раствора HF. Через 18 часов при комнатной температуре реакционную смесь концентрируют досуха и остаток распределяют между водой (50 мл) и этилацетатом (50 мл). Органический слой промывают рассолом, сушат, фильтруют и концентрируют с получением продукта в виде желтовато-коричневого твердого вещества (т.пл. 69-72oC).
Стадия 3: Получение дигидро-5(S)-((метансульфонил)оксиметил- 3(R)-фенилметил-3(2Н)-фуранона.
К раствору 2,93 г (14 ммоля) дигидро-5(S)-(гидроксиметил)-3 (R)-фенилметил-3(2Н)-фуранона в хлористом метилене, охлажденному до 0oC, добавляют триэтиламин (1,98 мл, 15,6 ммоля), после чего добавляют метансульфонил хлорид (1,20 мл, 15,6 ммоля). Через 1 час при 0oC реакционную смесь переливают в 10% водный раствор лимонной кислоты, промывают этилацетатом (2х100 мл), который подвергают обратной промывке водой (100 мл), рассолом (100 мл), сушат, фильтруют и концентрируют с образованием продукта в виде воскообразного коричневого твердого вещества.
Стадия 4: Получение дигидро-5(S)-(2-(3(S)-N-(трет. бутил- карбоксамидо)-(4as, 8as)-(декагидроизохинолин)ил)-метил)- 3(R)-фенилметил-3(2Н)-фуранона.
К 70 мг дигидро-5(S)-((метансульфонил)оксиметил)-3-(R)- фенилметил-3(2Н)-фуранона (0,25 ммоля) в 10 мл ксилола, содержащего 100 мг карбоната калия, добавляют 65 мг (0,27 ммоля) N-трет.- бутил(4as,8as)-декагидроизохинолин)-3(S)-карбоксамида и реакционную смесь нагревают до 140oC. Через 6 часов реакционную смесь охлаждают, переливают в 30 мл воды, которую промывают этилацетатом (2х30 мл). Органическую фазу сушат, фильтруют и концентрируют с получением остатка, который подвергают хроматографической очистке (50/50 EtOAc/гексан) с получением продукта.
Стадия 5: Получение 2(R)-фенилметил-4(S)-(трет.-бутил диметилсилилокси)-5-(2-(3(S)-N-(трет. -бутил карбоксамидо)- (4as, 8as)-декагидроизохинолин)ил)-пентановой кислоты.
К 130 мг (0,305 ммоля)дигидро-5(S)(2-(3(S)-N-(трет. - бутилкарбоксамида)-(4as, 8as)-(декагидроизохинолин)ил)метил) - 3(R)-фенилметил-3-(2Н)-фуранона в 2 мл ДМЕ добавляют 1 мл раствора гидроксида лития. Через 4 часа при комнатной температуре реакционную смесь концентрируют досуха и подвергают азеотропной перегонке с толуолом (3Х) с целью удаления избыточного количества воды. Остаток растворяют в 5 мл ДМФ и добавляют 414 мг (6,10 ммоля) имидазола и 465 мг (3,5 ммоля) трет. бутилдиметилсилил хлорида. Через 2 дня выстаивания при комнатной температуре к реакционной смеси добавляют 1 мл метанола и через 1 час раствор выпаривают досуха. Остаток распределяют между насыщенным раствором NH4Cl (водный раствор) и промывают этилацетатом, который сушат, фильтруют и концентрируют с получением масла, представляющего собой смесь продукта и исходного фуранона. Полученную смесь используют в последующей реакции без дополнительной очистки.
Стадия 6: Получение N-(2(R)-гидрокси-1(S)-инданил-(2(R)- фенилметил-4(S)-(трет. -бутилдиметил-силилокси-5-(2-(3(S)-N'- (трет. -бутилкарбоксамидо)-(4as,8as)-декагидроизохинолин)ил) -пентанамида.
Сырой продукт, полученный на описанной выше стадии 5, растворяют в 3 мл ДМФ, содержащего 47 мг (0,246 ммоля) ЕДС, 33 мг (0,246 ммоля) НОВТ и 37 мг 2(R)-гидрокси-1(S)-аминоиндана. pH раствора устанавливают равным 8.5-9.0 с помощью триэтиламина и через 18 часов проводят обработку, включающую концентрирование досуха, растворение остатка в 10% водном растворе лимонной кислоты и промывку водного слоя этилацетатом. Органический слой сушат, фильтруют и концентрируют, а полученное в результате масло подвергают хроматографической очистке (SiO2, 30% EtOAc/гексан) с получением целевого соединения.
Стадия 7: Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-гидрокси-5-(2-(3(S)-N'-(трет. бутилкарбоксамидо)-(4as, 8as)- декагидроизохинолин)ил)пентанамида.
Продукт, полученный выше на стадии 6, растворяют в 1 мл ТГФ и добавляют 1 мл 1 М раствора фтористого тетрабутиламмония в ТГФ. Через 18 часов при комнатной температуре реакционную смесь разбавляют 20 мл насыщенного водного раствора NaHCO3 и продукт реакции экстрагируют этилацетатом, который сушат, фильтруют и концентрируют с образованием пены. Полученный в результате материал подвергают хроматографической очистке на препаративной пластине (0,5 мм, 5% MeOH/CHCl3) и целевое соединение выделяют обычным способом в виде твердого вещества с т.пл. 105-107oC.
Пример 5. Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-амино-5-(2-(3(S)-N'-(трет. - бутилкарбоксамидо)-(4as, 8as)-декагидроизохинолин)ил)- пентанамида.
Стадия 1: Получение 5(S)-((трет. бутилдиметилсилилокси)- метил)-3(R)-фенилметил-N-BOC-2-пирролидинона.
Раствор 5(S)-((трет.-бутилдиметилсилилокси)метил)-N-BOC-2- пирролидинона (400 мг, 1,26 ммоля) в 2 мл ТГФ добавляют к предварительно охлажденному (-78oC) 1 М раствору гексаметилдисилазида лития (1,3 мл) в 5 мл ТГФ. Через 45 минут добавляют 0,15 мл бромистого бензила (1,3 ммоля) и перемешивание продолжают. Через 5 часов реакционную смесь переливают в делительную воронку, содержащую 30 мл 10% водного раствора лимонной кислоты. Водный слой экстрагируют EtOAc (2х30 мл) и подвергают обратной промывке рассолом (50 мл), сушат, фильтруют и концентрируют до образования масла. Остаток подвергают хроматографической очистке (SiO2, 20% EtOAc/гексан) с получением продукта в виде масла.
Стадия 2: Получение 5(S)-гидроксиметил-3(R)-фенилметил- 2-пирролидинона.
К 130 мг (0,34 ммоля) 5(S)-((трет.бутилдиметилсилилокси) метил)-3(R)-фенилметил-N-BOC-2-пирролидинона в 5 мл ацетонитрила добавляют 0,1 мл 48% водного раствора HF. Через 3 часа при комнатной температуре реакционную смесь концентрируют досуха и разбавляют 30 мл 10% водного раствора NaHCO3. Полученный раствор экстрагируют EtOAc (2х30 мл), сушат, фильтруют и концентрируют с образованием сырого продукта.
Стадия 3: Получение 5(S)-(метансульфонилокси)метил-3(R)- фенилметил-2-пирролидинона.
К раствору сырого продукта со стадии 2 в 5 мл хлористого метилена, охлажденного до 0oC, добавляют триэтиламин (42 мг, 0,41 ммоля) и метансульфонилхлорид (47 мг, 0,41 ммоля).
Реакционной смеси дают медленно нагреваться до комнатной температуры и перемешивают в течение 18 часов, после чего ее разбавляют 30 мл хлористого метилена, промывают 30 мл 10% раствора лимонной кислоты, сушат, фильтруют и концентрируют с получением продукта в виде масла.
Стадия 4: Получение 5(S)-(2-(3(S)-N-(трет. бутил- карбоксамидо)-(4as, 8as)-(декагидроизохинолин)-ил)метил) -3(R)-фенилметил-2-пирролидинона.
К раствору 380 мг (1,34 ммоля) 5(S)-(метансульфонилокси)- метил-3(R)-фенилметил-2-пирролидинона в 20 мл изопропанола добавляют 350 мг карбоната калия и 360 мг N-трет.бутил-(4as,8as) -(декагидроизохинолин)-3(S)-карбоксамида и реакционную смесь нагревают до 85oC. Через 18 часов охлажденную реакционную смесь фильтруют через целит, выпаривают досуха и остаток растворяют в воде, которую экстрагируют EtOAc (2х50 мл). Органический слой сушат, фильтруют и концентрируют, а остаток подвергают хроматографической очистке (SiO2, 50/50 EtOAc/гексан) с получением продукта в виде масла.
Стадия 5: Получение 5(S)-(2-(3(S)-N'-(трет.-бутил- карбоксамидо)-(4as, 8as)-(декагидроизохинолин)-ил)метил-3 (R)-фенилметил-N-BOC-2-пирролидинона.
К раствору продукта со стадии 4 (260 мг, 0,611 ммоля) в 10 мл хлористого метилена добавляют диметиламинопиридин (74 мг, 0,6 ммоля) и 133 мг (0,61 ммоля) BOC-ангидрида. Через 18 часов при комнатной температуре реакционную смесь обрабатывают путем разбавления 30 мл хлористого метилена и органический слой промывают 30 мл 10% раствора лимонной кислоты, рассолом (30 мл), сушат, фильтруют и концентрируют с получением масла. В результате хроматографической очистки (SiO2, 40%, EtOAc/гексан) получают целевое соединение.
Стадия 6: Получение 5-(2-(3(S)-N'-(трет.бутилкарбоксамидо)- (4as,8as)-декагидроизохинолин)ил)-4(S)-(1', 1')- (диметилэтоксикарбонил)-амино-2(R)-фенилметил-пентановой кислоты.
К раствору продукта с описанной выше стадии 5 (260 мг, 0,495 ммоля) с 3 мл диметоксиэтана добавляют 1,5 мл 1 М водного раствора гидроксида лития (1,5 ммоля). Реакционную смесь обрабатывают через 2 часа путем ее концентрирования досуха, растворения остатка в насыщенном водном растворе хлористого аммония и водную фазу промывают этилацетатом (2х50 мл), который сушат, фильтруют и концентрируют с получением сырой кислоты.
Стадия 7: Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-(1,1)-(диметилэтоксикарбонил)- амино-5-(2-(3(S)-N'-(трет. бутилкарбоксамидо)- (4as,8as)-декагидроизохинолин)ил)пентанамида.
К раствору продукта, полученного в описанной выше стадии 6, (260 мг, 0,49 ммоля) в хлористом метилене добавляют ЕДС (94 мг, 0,49 ммоля), НОВТ (66 мг, 0,49 ммоля), 2(R)-гидрокси-1(S)- аминоиндан (73 мг, 0,49 ммоля) и pH реакционной смеси устанавливают в интервале 8.5-9.0 с помощью триэтиламина. Через 5 часов при комнатной температуре реакционную смесь обрабатывают путем разбавления 50 мл хлористого метилена и промывки органического слоя насыщенным водным раствором хлористого аммония. Органическую фазу сушат, фильтруют и концентрируют, а остаток подвергают хроматографической очистке с получением целевого соединения в виде пены.
Стадия 8: Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-гидрокси-5-(2-(3(S)-N'-(трет. бутилкарбоксамидо)-(4as,8as)-декагидроизохинолин)ил)- пентанамида.
К раствору продукта со стадии 7 (180 мг, 0,28 ммоля) в 5 мл хлористого метилена, охлажденного до 0oC, добавляют 1 мл трифторуксусной кислоты. Через 4 часа реакционную смесь концентрируют досуха и остаток растворяют в 50 мл хлористого метилена и промывают 10% водным раствором NaHCO3 Органический слой сушат, фильтруют и концентрируют с получением продукта в виде твердого вещества, которое подвергают хроматографической очистке (SiO2, 7% MeOH/CH2Cl2) с получением целевого соединения, т.пл. 92-95oC.
Пример 6. Получение N-2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-гидрокси-5-(1-(4-карбобензилокси-2-(S)-N'- (трет. бутилкарбоксамидо)пиперазинил))пентанамида.
Используя практически ту же методику, что и в примере 1, но заменяя применяемый на стадии 9 N-трет.-бутил-4(S)-фенокси-1- пролинамид на N-трет.-бутил-4-CBZ-пиперазин-(2-(S)-карбоксамид, получают целевое соединение.
Пример 7. Получение N''-(N-(2-пиридил)-валил)- 2(R)-фенилметил-4(S)-гидрокси-5-(2-(3(S)-(N'-трет. бутилкарбоксамидо)-(4as,8as)-декагидроизохинолин)ил)- пентанамида.
Используя практически ту же методику, что описана в примере 4, но заменяя применяемый на стадии 6 2(R)-гидрокси-1(S) аминоиндан на N-2-пиридил-валин, получают целевое соединение.
Пример 8. Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-гидрокси-5-(2(S)-(N'-трет.бутил-3-фенил- пропионамид)амино)пентанамида.
Используя практически ту же методику, что описана в примере 1, но заменяя применяемый на стадии 9 N'-трет.-бутил- 4(S)-фенокси-N-пролинамид на N-трет-бутил- фенилаланин амид, получают целевое соединение.
Пример 9. Получение N-(4(S)-3,4-дигидро-1Н-2,2- диоксобензотиопиранил)-2(R)-фенилметил-4(S)-гидрокси-5- (2-(3(S)-N'-(трет. бутилкарбоксамидо)-(4as,8as)- декагидроизохинолин)ил)пентанамида.
Стадия 1: Получение N-(4(R)-3,4-дигидро-1Н-бензотио- пиранил)-2(R)-фенилметил-4(S)-гидрокси-5-(2-(3(S)-трет. бутилкарбоксамидо)-(4as,8as)-декагидроизохинолин)ил) пентанамида.
Следуя практически той же методике, что описана в примере 4, но заменяя используемый на стадии 6 этого примера 2(R)-гидрокси- 1(S)-аминоиндан на 4(S)-амино-3,4-дигидро-1Н-бензотиопиран, получают целевое соединение.
Стадия 2: Получение N-(4(S)-3,4-дигидро-1Н-2,2-диоксо- бензотиопиранил)-2(R)-фенилметил-4(S)-гидрокси-5-(2-(3(S) -трет.бутилкарбоксамидо)-(4as, 8as)- декагидроизохинолин)ил)пентанамида.
Соединение, полученное в описанной выше стадии 1, растворяют в смеси метанола с водой в соотношении 1:1. К полученной смеси добавляют 10 эквивалентов OXONE и реакционную смесь перемешивают при комнатной температуре. После завершения реакции реакционную смесь концентрируют досуха, добавляют воду и проводят экстракцию этилацетатом, который сушат, фильтруют и концентрируют с получением целевого соединения.
Пример 10. Получение N-(4(S)-3,4-дигидро-1Н-2,2- диоксобензотиопиранил)-2(R)-фенилметил-4(S)-гидрокси-5-(1- (4-карбобензилокси-2(S)-N'-трет.бутилкарбоксамидо)пиперазинил)) пентанамида.
Стадия 1: Получение дигидро-5(S)-(1-(4-карбобензилокси- 2(S)-N'-(трет. бутилкарбоксамидо)пиперазинил)- метил-3(R)-фенилметил-3(2Н)-фуранона.
Следуя практически той же методике, что описана в примере 4, но заменяя N'-трет. -бутил-(4as, 8as)-(декагидроизохинолин)- 3(S)карбоксамид на 4-карбобензилокси-2(S)-N'-(трет. бутилкарбоксамидо)пиперазин, получают целевое соединение.
Стадия 2: Получение 2(R)-фенилметил-4(S)-(трет.-бутил- диметилсилилокси)-5-(1-(4-карбобензилокси- 2(S)-N-(трет.-бутилкарбоксамидо)пиперазинил))пентановой кислоты.
Следуя практически той же методике, что описана в примере 4, но заменяя дигидро-5(S)-2-(3(S)-N'-(трет.бутилкарбоксамидо)- (4as,8as)-(декагидроизохинолин)ил)метил)-3(R)-фенил-
метил-3(2Н)-фуранон на дигидро-5(S)-(1-(4-карбобензилокси- 2(S)-N'-(трет. бутилкарбоксиамидо)пиперазинил)метил)-3(R)- фенилметил-3(2Н)-фуранон, получают целевое соединение.
Стадия 3: Получение N-(4(S)-3,4-дигидро-1Н-бензо- тиопиранил)2(R)-фенилметил-4(S)-(трет. бутилдиметилсилилокси)- 5-(1-(4-карбобензилокси-2(S)-N'-(трет.-бутилкарбоксамидо) пиперазинил))-пентанамида
Сырую 2(R)-фенилметил-4(S)-(трет.бутилдиметилсилилокси)- 5-(1-(4-карбобензилокси-2(S)-N'-(трет. -бутилкарбоксамидо)- пиперазинил)-пентановую кислоту растворяют в 3 мл ДМФ, содержащих 1 экв. ЕДС, 1 экв. НОВТ и 1 экв. 4(S)-амино-3,4-дигидро-1Н- бензотиопирана. pH раствора устанавливают в интервале 8.5-9.0 с помощью триэтиламина и через 18 часов реакционную смесь концентрируют досуха, остаток растворяют в 10% водном растворе лимонной кислоты и промывают водный слой этилацетатом. Органический слой сушат, фильтруют и концентрируют, а полученный в результате остаток подвергают хроматографической очистке с получением целевого продукта.
Стадия 4: Получение N-(4(R)-3,4-дигидро-1Н-бензотиопиранил) -2(R)-фенилметил-4( )-гидрокси)-5-(1-(4-карбобензилокси-2 (S)-(трет.-бутилкарбоксамидо)-пиперазинил))пентанамида
Продукт, полученный на стадии 3, описанной выше, растворяют в 1 мл ТГФ и добавляют 1 мл 1 М раствора фтористого тетрабутиламмония в ТГФ. Через 18 часов при комнатной температуре реакционную смесь разбавляют 20 мл насыщенного водного раствора NaHCO3 и продукт реакции экстрагируют этилацетатом, который сушат, фильтруют и концентрируют с получением остатка.
Полученный остаток подвергают хроматографической очистке с получением целевого продукта.
Стадия 5: Получение N-(4(S)-3,4-дигидро-1Н-2,2-диоксо- бензотиопиранил)-2(R)-фенилметил-4(S)-гидрокси- -5-(1-(4-карбобензилокси-2(S)- N-(трет. бутил- карбоксамидо)пиперазинил))пентанамида
Соединение, полученное в описанной выше стадии 4, растворяют в смеси метанол/вода в соотношении 1:1. К полученной реакционной смеси добавляют 10 экв. OXONE и реакционную смесь перемешивают при комнатной температуре. После завершения реакции реакционную смесь концентрируют досуха, добавляют воду и проводят экстракцию этилацетатом, после чего экстракт сушат, фильтруют и концентрируют с получением целевого соединения.
Пример 11. Получение N-(2(R)-гидрокси-1(S)-инданил)-2 (R)-((4-((2-гидрокси)-этокси)фенил)метил-4(S)-гидрокси-5- (2-(3-(S)-N'-(трет.бутилкарбоксамидо)- (4as,8as)-декагидроизохинолин)ил)пентанамида
Стадия 1: Получение N-(2(R)-гидрокси-1(S)инданил)-2 (R)-((4-(2-аллилокси)фенил)метил)-4(S)-гидрокси-5-(2-(3(S) -(трет.бутилкарбоксамидо)-(4as, 8as)-декагидроизохинолин)ил) пентанамида
К раствору N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4- гидроксифенил)метил-4(S)-гидрокси-5-(2-(3(S)-трет.бутилкарбоксамидо- (4as,8as)-декагидроизохинолин)ил)пентанамида в диоксане добавляют 6 экв. бромистого аллила и б экв. карбоната цезия. Реакционную смесь нагревают до 90oC. После завершения реакции осадок отфильтровывают, диоксановый экстракт концентрируют досуха и остаток разбавляют водой, которую промывают этилацетатом. Органическую фазу сушат, фильтруют и концентрируют с получением продукта.
Стадия 2: Получение N-(2(R)-гидрокси-1(S)-инданил)-2 (R)-((4-((2-гидрокси)этокси)фенил)метил)-4(S)-гидрокси-5- (2-(3(S)-N'-(трет. -бутилкарбоксамидо)-(4as,8as)- декагидроизохинолин)ил)пентанамида
Продукт, полученный на описанной выше стадии 1, растворяют в метаноле, добавляют 1 экв. п-толуолсульфокислоты и реакционную смесь охлаждают до -78oC. Избыточное количество озона барботируют через реакционную смесь до появления голубой окраски. Колбу продувают азотом с целью удаления озона и добавляют избыток раствора боргидрида натрия. Реакционную смесь нагревают до комнатной температуры и затем добавляют насыщенный раствор NaHCO3. Метанол отгоняют на роторном испарителе и водный остаток промывают этилацетатом, который сушат, фильтруют и концентрируют с получением целевого соединения.
Пример 12. Получение N-(2(R)-гидрокси-1(S)-инданил)-2 (R)-((4-((2-гидрокси-этокси)фенил)метил)-4(S)-гидрокси-5-(1- (4-карбобензилокси-(2(S)-N'-(трет.бутилкарбоксамидо) пиперазинил))пентанамида
Используя практически ту же методику, что описана в примере 11, но заменяя N-(2(R)-гидрокси-1(S)-инданил-2(R)-((4- гидроксифенил)метил)-4(S)-гидрокси-5-(2-(3(S)-трет.бутил- карбоксамидо)-(4as,8as)-декагидроизохинолин)ил- пентанамид на N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4- гидроксифенил)метил-4(S)-гидрокси-5-(1-(4-карбобензилокси-2 (S)-(трет.-бутилкарбоксамидо)-пиперазинил-пентанамид, получают целевое соединение.
Пример 13. Получение N-(2(R)-гидрокси-1(S)-инданил)-2 (R)-((4-(2-(4-морфолинил)-этокси)фенил)метил)-4(S)- гидрокси-5-(2-(3(S)-N'-(трет. бутилкарбоксамидо)-(4as,8as)- декагидроизохинолин)-ил)пентанамида
К раствору N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4- гидроксифенил)метил)-4(S)-гидрокси-5-(2-(3(S)-N'-(трет. бутил- карбоксамидо)-(4as, 8as)-декагидроизохинолин)ил)пентанамида в диоксане добавляют 6 экв. хлорэтилморфолина и б экв. карбоната цезия. Реакционную смесь нагревают до 90oC. После завершения реакции осадок отфильтровывают, диоксановый экстракт концентрируют досуха и остаток разбавляют водой, которую промывают этилацетатом. Органическую фазу сушат, фильтруют и концентрируют с получением целевого соединения.
Пример 14. Получение N-(2(R)-гидрокси-1(S)-инданил)-2 (R)-((4-(2-(4-морфолинил)-этокси)фенил)метил)-4(S)- гидрокси-5-(1-(4-карбобензилокси)-2(S)-N'-(трет. бутилкарбоксамидо)пиперазинил))-пентанамида
К раствору N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4- гидроксифенил)метил)-4(S)-гидрокси-5-(1-(4- карбобензилокси)-2(S)-(трет. бутилкарбоксамидо)- пиперазинил)пентанамида в диоксане добавляют 6 экв. хлорэтилморфолина и 6 экв. карбоната цезия. Реакционную смесь нагревают до 90oC. После завершения реакции осадок отфильтровывают, диоксан отгоняют и смесь концентрируют досуха, а остаток разбавляют водой, которую промывают этилацетатом. Органическую фазу сушат, отфильтровывают и концентрируют с получением целевого соединения.
Пример 15. Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R) -фенилметил-4(S)-гидрокси-5-(1-(4-(3-пиридилметил)-2(R)- N'-(трет. бутилкарбоксамидо)-пиперазинил))пентанамида
Стадия 1: Получение дигидро-5(S)-((трифторметансульфонил)- оксиметил)-3(R)-фенилметил-3(2Н)-фуранона
К раствору 18,4 г(89,4 ммоля)дигидро-5(S)-(гидроксиметил)- 3(R)-фенилметил-3(2Н)-фуранона в 350 мл хлористого метилена, охлажденного до 0oC, добавляют 13,51 мл 2,6-лутидина (115,98 ммоля), после чего прикалывают 16,51 мл ангидрида триметансульфокислоты(98,1 ммоля). Через 1,5 часа при комнатной температуре реакционную смесь переливают в смесь 300 мл льда с рассолом и полученный раствор перемешивают в течение 0,5 часа. Затем водный слой экстрагируют хлористым метиленом (3х150 мл), органические слои промывают 10% HCl (2х75 мл), насыщенным NaHCO3 (100 мл), водой (100 мл), сушат над MgSO4 фильтруют и концентрируют с получением твердого остатка. В результате очистки методом хроматографии однократного испарения (колонка 120х150 мм, градиентное элюирование смесью гексан EtOAc, в диапазоне 4:1 - 3:1) получают целевой продукт, т.пл. 53-54oC.
Стадия 2: Получение 4-(1,1-диметилэтил)-1-(фенилметил)-1,2-(S) -4-пиперазинтрикарбоксилата
Это соединение получают по методике, описанной С.Ф.Биггом С.Дж.Хайсом, П. М. Новаком, Дж. Т.Драммондом, Г.Енсоном, Т.П. Бобовски Tetrahedron Lett, 1989, 30, 5193; исходным веществом служит 2(S)- пиперазинкарбоновая кислота (см. Е.Фелдер, С.Маффи, С.Пьетра, Д.Питри Helv. Chem. Acta 1960, 117, 888.
Стадия 3: Получение N-трет.бутил-4-(1,1-диметилэтокси- карбониламино)-1-(фенилметилкарбониламино)-пиперазин-2(S)- карбоксамида
К 9,90 г (27,16 ммоля) 4-(1,1-диметилэтил)-1-(фенилметил)-1,2 (S)-4-пиперазинтрикарбоксилата, растворенного в 75 мл ДМФ и охлажденного до 0oC, добавляют 5,73 г (29,88 ммоля) ЕДС, 4,03 г (29,88 ммоля) HOBt, 3,14 мл (29,88 ммоля) трет.-бутиламина и, наконец, 4,16 мл (29,88 ммоля) триэтиламина. Реакционную смесь перемешивают в течение 18 часов и реакционный объем концентрируют наполовину. Затем полученную смесь разбавляют 600 мл EtOAc и промывают 100 мл HCl (2х75 мл), насыщенным раствором NaHCO3 (1х75 мл), водой (3х75 мл) и рассолом (1х50 мл), сушат над MgSO4 и концентрируют с получением твердого вещества. Это твердое вещество обрабатывают смесью EtOAc:гексан (1: 2) и фильтруют с получением целевого продукта в виде белого твердого вещества; т.пл. 134-135oC.
Стадия 4: Получение N-трет. бутил-4-(1,1-диметилэтокси- карбониламино)пиперазин-2(S)-карбоксамида
К 1,20 г (2,86 ммоля) N-трет.бутил-4-(1,1-диметилэтоксикарбониламидо)-1-(фенилметилкарбониламино)пиперазин-2(S)- карбоксамида и 1,1 г (0,086 ммоля) 10% Pd/C добавляют 15 мл метанола. Реакционный сосуд заполняют водородом и реакционную смесь перемешивают в течение 2 часов, фильтруют через целит и промывают этанолом. Растворители удаляют в вакууме с получением целевого соединения в виде пены.
Спектр 1H ЯМР (300 МГц, CDCl3) δ : 6.65 (широкий, 1H), 4.10 (мультиплет, 1H), 3.81 (широкий, 1H), 3.21 (двойной дублет, J=18 и 7 Гц, 1H), 3.02-2.70 (мультиплет, 4H), 2.10-2.0 (широкий, 1H), 1.50 (синглет, 9H), 1.41 (синглет, 9Н).
Стадия 5: Получение дигидро-5(S)-(4-(1,1-диметилэтокси- карбониламино))-2(S)-N'-(трет. бутилкарбоксамидо)- пиперазинил)метил-3(R)-фенилметил)-3(2Н)-фуранона
К раствору 22,40 г (0,0662 моля) дигидро-5-(S)-((трифторметансульфонил)оксиметил)-3(R)-фенилметил-3(2Н)-фуранона (полученного на стадии 1) и 18,0 г (0,063 моля н-трет. бутил-4- (1,1-диметилэтоксикарбониламино)пиперазин-2(S)карбоксамида, растворенного в 180 мл изопропанола, добавляют 11,53 мл (0,0662 моля) N,N-диизопропилэтиламина. Через 2,5 часа добавляют еще 1,2 г дигидро-5(S)-((трифторметансульфонил)-оксиметил)-3(R)- фенилметил-3(2Н)-фуранона. Через 3,5 часа реакцию завершают и анализируют реакционную смесь методом тонкослойной хроматографии (ТСХ), после чего реакционную смесь концентрируют до состояния вязкого масла. В результате обработки смесью EtOAc:гексаны (1:2, 200 мл) получают белое твердое вещество, которое удаляют. Масло очищают методом хроматографии однократного испарения (колонка 120х150 мм, элюирование градиентами EtOAc:гексаны 1:1, 2: 1, 3:1 и далее только EtOAc) с получением целевого соединения.
Спектр 1H ЯМР (400 МГц, CDCl3) δ : 7.34-7.17 (мультиплет, 5H), 6.31 (широкий синглет, 1H), 4.38 (широкий мультиплет, 1H), 3.90-3.92 (мультиплет, 1H), 3.79 (широкий мультиплет, 1H), 3.16 (двойной дублет, J=13.6 и 4.4 Гц, 1H), 3.08- 2.99 (мультиплет, 3H), 2.90-2.82 (мультиплет, 1H), 2.80 (двойной дублет, J=13.5 и 8.9 Гц, 1H), 2.78 (мультиплет, 1H), 2.67-2.61 (мультиплет, 1H), 2.58-2.49 (мультиплет, 1H), 2.38- 2.32(мультиплет, 1H), 2.32-2.04 (мультиплет, 1H), 1.99-1.92 (мультиплет, 1H), 1.45 (синглет 9H), 1.29 (синглет, 9Н).
Стадия 6: Получение 2(R)-фенилметил-4(S)-(трет.бутилдиметилсилилокси)-5(1-(4-(1,1-диметилэтокси-карбониламино)- 2(S)-N-(трет.бутилкарбоксамидо)-пиперазинил))-пентанамида
К 25,50 г (52,50 ммоля) дигидро-5(S)-(4-(1,1- диметилэтоксикарбониламино)-2(S)-N-(трет. бутилкарбоксамидо)пиперазинил) метил-3(R)-фенилметил-3(2Н)-фуранона, растворенного в 120 мл ДМФ, охлажденного до 0oC, добавляют раствор, состоящий из 60 мл воды и 1,512 г (63,01 ммоля) гидроксида лития. Через 0,5 часа реакцию прекращают добавлением 10% HCl до pH 6 и полученный раствор концентрируют в вакууме. Остаток растворяют в 50 мл воды и экстрагируют EtOAc (4х75 мл) и органические слои промывают водой (1х20 мл), рассолом (1х20 мл). Водные слои подвергают обратной экстракции EtOAc (2х75 мл) и объединенные органические слои сушат над MgSO4 и концентрируют с получением желтого твердого вещества. Сырой продукт растворяют в 100 мл ДМФ и добавляют 17,87 г (0,262 моля) имидазола, охлажденного до 0oC, после чего добавляют 31,50 г (0,21 моля) трет. бутилдиметилсилилхлорида. Полученную смесь перемешивают в течение 1 часа при 0oC и затем нагревают до комнатной температуры. Через 20 часов реакцию прекращают путем добавления 10 мл метанола и реакционную смесь концентрируют до половины объема. Добавляют 100 мл водного буфера с pH 7 и водный слой экстрагируют EtOAc (4х100 мл), объединенные органические слои промывают 10% HCl (2х50 мл), водой (3х75 мл) и рассолом (1х50 мл), сушат над MgSO4 и концентрируют с получением целевого соединения. Полученный материал используют непосредственно на последующей стадии.
Стадия 7: Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-(трет. бутилдиметилсилилокси)-(5-(1-(4-(1,1- диметилэтоксикарбониламино))-2(S)-N-(трет. бутилкарбоксамидо)-пиперазинил))пентанамида
К 27,0 г (0,0446 моля) сырого материала, полученного на стадии 6, растворенного в 180 мл ДМФ и охлажденного до 0oC, добавляют 8,98 г (0,0468 моля) EDC, 6,32 (0,0468 моля) HOBt и 7,31 г (0,049 моля) аминогидроксииндана. Добавляют триэтиламин (6,52 мл, 0,0468 моля) и реакционную смесь перемешивают в течение 2 часов при 0oC, в течение 16 часов при комнатной температуре и затем реакцию прекращают путем разбавления 500 мл EtOAc. Органический слой промывают 10% HCl (2х100 мл), насыщенным раствором NaHCO3 (1х100 мл), водой (3х150 мл), рассолом (1х75 мл), сушат над MgSO4 и концентрируют с получением целевого соединения в виде белой пены.
Спектр 1H ЯМР (400 МГц, CDCl3): 7.4-7.17 (мультиплет, 9H), 6.51 (широкий синглет, 1H), 5.79 (широкий синглет, 2H), 5.23 (мультиплет, 1H), 4.23 (широкий синглет, 1H), 4.06 (мультиплет, 1H), 3.96-3.84 (мультиплет, 2H), 3.07-2.78 (мультиплет, 8H), 3.65 (двойной дублет, J=9.6 и 4.1 Гц, 1H), 2.56-2.44 (мультиплет, 2H), 2.29 (двойной дублет, J=12.0 и 4,5 Гц, 1H), 2.17-2.09 (мультиплет, 1H), 1.79 (широкий синглет, 1H), 1.44 (синглет, 9H), 1.35 (синглет, 9H), 1.10 (синглет, 1H), 0.84 (синглет, 9H), 0.12 (синглет, 3H), 0.08 (синглет, 3Н).
Стадия 8: Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-(гидрокси)-5-(1-(4-(1,1- диметилэтоксикарбониламино)))-2(S)-N-(трет. бутилкарбоксамидо)пиперазинил))-пентанамида
К 32,20 г (0,0437 моля) N-(2(R)-гидрокси-1-(S)-инданил) 2(R)-фенилметил)-4(S)-(трет. бутилдиметилсилилокси)-5-(1-(4-(1,1- диметилэтоксикарбониламино)))-2(S)-N-(трет. бутилкарбоксамидо) пиперазинил))-пентанамида добавляют 437 мл (0,437 моля) фтористого тетрабутиламмонийфторида (1,0 М раствор в ТГФ, Алдрич). Реакционную смесь перемешивают в течение 18 часов и затем концентрируют до объема в 200 мл и разбавляют 700 мл EtOAc. Полученную смесь промывают водой (2х100 мл), рассолом (1х50 мл) и водные слои подвергают обратной экстракции EtOAc (2х200 мл). Объединенные органические слои сушат над MgSO4 и концентрируют до получения масла. В результате очистки методом хроматографии мгновенного испарения (колонка 120х150 мм, градиентное элюирование смесью CH2Cl2:CHCl3/насыщенной смесью NH3:метанол, с повышающимися концентрациями метанола: 1%, 1.5%, 2%) получают целевое соединение в виде пены.
Спектр 1H ЯМР (400 МГц, CDCl3) δ : 7.31-7.11 (мультиплет,9H), 6.41 (широкий синглет, 1H), 6.24 (дублет, J=8.6 Гц, 1H), 5.25 (двойной дублет, J= 8.6 и 4.7 Гц, 1H), 4.21 (мультиплет, 1H), 3.83-3.82 (мультиплет, 2H), 3.78-3.61 (мультиплет, 2H), 3.22-3.19 (мультиплет, 2H), 3.03-2.78 (мультиплет, 8H), 2.62-2.58 (мультиплет, 1H), 2.41-2.35 (мультиплет, 2H), 2.04-2.02 (мультиплет, 1H), 1.57-1.50 (мультиплет, 1H), 1.45 (синглет, 9H), 1.32 (синглет, 9Н).
Стадия 9: Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-(гидрокси)-5-(1-(2(S)-N-(трет. бутилкарбоксамидо) пиперазинил) -пентанамида
К 21,15 г (0,034 моля) N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-(гидрокси)-5-(1-(4-(1,1-диметилэтокси- карбониламино))))-2(S)-N-(трет. -бутилкарбоксамидо) пиперазинил))пентанамида, растворенного в 350 мл хлористого метилена и охлажденного до 0oC, добавляют 22,43 мл (0,204 моля) 2,6-лутидина и затем в течение 5 минут 32,25 мл (0,170 моля) триметилсилилтрифлата. Через 0,5 часа реакцию прекращают путем добавления 10% HCl (80 мл) и полученную смесь перемешивают в течение 0,5 часа. К полученной смеси добавляют 100 мл насыщенного раствора NaHCO3 затем добавляют твердый NaHCO3 до pH 8. Затем водный слой экстрагируют EtOAc (4х100 мл) и объединенные органические слои промывают водой (4х100 мл), рассолом (1х75 мл), сушат над MgSO4 концентрируют. Остаток очищают методом хроматографии на колонке (120-150 мм, градиентное элюирование смесью CH2Cl2-CHCl3, насыщенной смесью NH3-MeOH, при медленном увеличении концентрации метанола в следующем порядке: 2%, 3%, 4%, 5%, 6%, 10%). В результате получают целевой продукт в виде белой пены.
Спектр 1H ЯМР (400 МГц, CDCl3) δ : 7.53 (синглет, 1H), 7.29-7.09 (мультиплет, 9H), 6.52 (дублет, J=8.3 Гц, 1H), 5.24 (двойной дублет J=8.2 и 4.9 Гц, 1H), 4.23 (двойной дублет, J=4.7 и 4.03 Гц, 1H), 4.25-4.00 (широкий синглет, 1H), 3.83-3.81 (мультиплет, 1H), 3.03-2.88 (мультиплет, 4H), 2.82-2.73 (мультиплет, 7H), 2.50-1.60 (широкий синглет, 2H), 2.45 (дублет, J = 6.2 Гц, 2H), 2.32-2.79 (мультиплет, 1H), 1.98 (мультиплет, 1H), 1.51 (мультиплет, 1H), 1.33 (синглет, 9Н).
Стадия 10: Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-гидрокси-5(1-(4-(3-пиридилметил-2(S)-N'- (трет. -бутилкарбоксамидо)-пиперазинил))-пентанамида
К смеси 10,0 г (0,019 моля) N-(2(R)-гидрокси-1(S)-инданил- 2(R)-фенилметил-4(S)-гидрокси-5-(1-(2-(S)-трет. бутил- карбоксамидо)пиперазинил)-пентанамида и 3,45 г (0,021 моля) 3-пиколилхлорида, растворенного в 40 мл ДМФ, добавляют 5,85 мл (0,042 моля) триэтиламина. Через 3 часа добавляют дополнительное количество 3-пиколилхлорида (0,313 г). Через 2 часа реакционную смесь разбавляют 400 мл EtOAc и промывают водой (3х75 мл), рассолом (1х100 мл), сушат над MgSO4 и концентрируют. Остаток обрабатывают 30 мл EtOAc и затем собирают полученный в результате белый осадок. В результате последующей перекристаллизации из EtOAc получают целевой продукт (т.пл. 167,5 -168oC).
Пример 16.
Следуя методике, описанной в примере 15, со следующими изменениями, включающими обработку N-(2(R)-гидрокси-1(S)-инданил- 2(R)-фенилметил-4(S)-гидрокси-5-(1-(2(S)-N'-(трет. бутилкарбоксамидо)пиперазинил))-пентанамида, используемого ниже (см. соединение (i), описанное ниже), алкилирующим агентом (ii), указанным ниже, вместо 3-пиколилхлорида, используемого на стадии 10, получают следующие продукты, отвечающие формуле (iii),приведенной в конце описания.
Пример 17. Получение дигидро-5(S)-(трет. бутилдиметилсилилоксиметил)-3(2Н)-фуранона
К раствору 3,00 г (25,8 моля) дигидро-5(S)-(гидроксиметил)- 2-(3Н)-фуранона в 25 мл дихлорметана добавляют 3,51 (51,6 ммоля) имидазола и затем добавляют 4,67 г (31,0 ммоля) трет. бутилдиметилсилилхлорида. Реакционную смесь перемешивают при комнатной температуре в течение 8 часов и реакцию прекращают добавлением 2 мл метанола. Полученную смесь концентрируют до состояния масла и затем разбавляют 150 мл эфира, после чего промывают 5% HCl (2х10 мл), насыщенным раствором NaHCO3 (1х10 мл), водой (1х10 мл), рассолом (1х10 мл), сушат над MgSO4 и концентрируют. Остаток очищают методом хроматографии мгновенного испарения (колонка 40х150 мм, градиентное элюирование, смесь гексаны: этилацетат от 5:1 до 4:1) с получением продукта в виде прозрачного масла.
Спектр 1H ЯМР (300 МГц, CDCl3) δ : 4.68-4.60 (мультиплет, 1H), 3.89 (двойной дублет, J =3.3 и 11.3 Гц, 1H), 3.71 (двойной дублет, J=3.2 и 5411,3 Гц, 1H), 2.71-2.45 (мультиплет, 2H), 2.35-2.16 (мультиплет, 2H), 0,91 (синглет, 9H), 0.10 (синглет, 3H), 0,09 (синглет, 3Н).
Хотя приведенное выше описание включает принципиальные моменты настоящего изобретения, причем примеры приведены в целях иллюстрации, следует иметь в виду, что практическая реализация изобретения охватывает все обычные варианты, адаптации или модификации, входящие в сферу следующей ниже формулы изобретения, а также их эквиваленты.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНИЛПЕНТАМИДА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ИНГИБИРОВАНИЯ ВИЧ-ПРОТЕАЗЫ | 1992 |
|
RU2131416C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПИПЕРАЗИНИЛПЕНТАНАМИДА И ПРОИЗВОДНОЕ ПИПЕРАЗИНИЛПЕНТАНАМИДА | 1992 |
|
RU2171254C2 |
| ИНГИБИТОРЫ HIV ПРОТЕАЗЫ | 1994 |
|
RU2137768C1 |
| ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ СИНТЕЗА ИНГИБИТОРОВ ВИЧ-ПРОТЕАЗ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2125561C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ, ИСПОЛЬЗУЕМЫХ ДЛЯ СИНТЕЗА ИНГИБИТОРОВ ВИЧ-ПРОТЕАЗЫ | 1994 |
|
RU2134263C1 |
| СОЧЕТАНИЕ ИНГИБИТОРОВ ВИЧ-ПРОТЕАЗЫ | 1995 |
|
RU2194506C2 |
| СПОСОБ РАЦЕМИЗАЦИИ ОПТИЧЕСКИ ЧИСТОГО ИЛИ ОБОГАЩЕННОГО ПИПЕРАЗИН-2- ТРЕТ.БУТИЛКАРБОКСАМИДНОГО СУБСТРАТА И РАЦЕМИЧЕСКИЙ 2-ТРЕТ-БУТИЛКАРБОКСАМИД-4-(3- ПИКОЛИЛ)ПИПЕРАЗИН | 1995 |
|
RU2135482C1 |
| СПОСОБ ЛЕЧЕНИЯ РЕТРОВИРУСНЫХ ИНФЕКЦИЙ У МЛЕКОПИТАЮЩИХ (ВАРИАНТЫ) | 1995 |
|
RU2166317C2 |
| СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ ПРОТЕАЗЫ ВИЧ | 1994 |
|
RU2139280C1 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ БЛОКАДЫ РЕЦЕПТОРОВ НЕЙРОКИНИНА-1 У МЛЕКОПИТАЮЩИХ, СПОСОБ ДЛЯ БЛОКАДЫ РЕЦЕПТОРОВ НЕЙРОКИНИНА-1 У МЛЕКОПИТАЮЩИХ | 1993 |
|
RU2140914C1 |
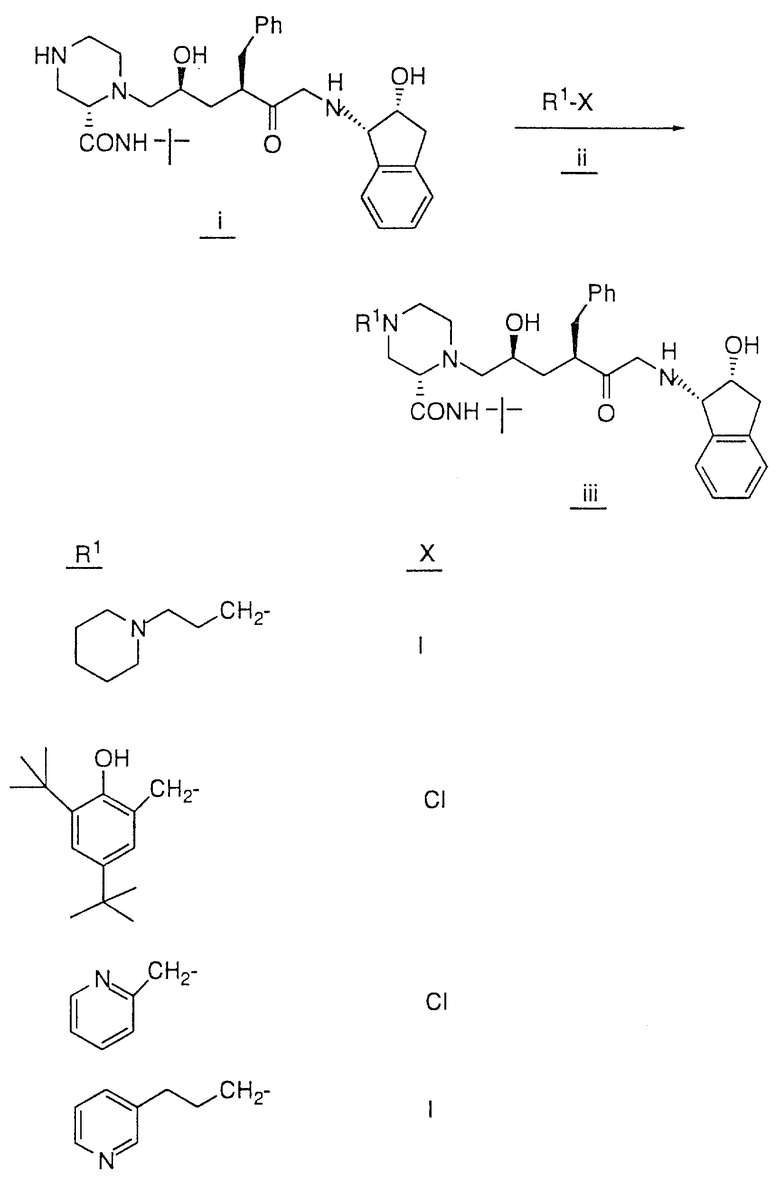
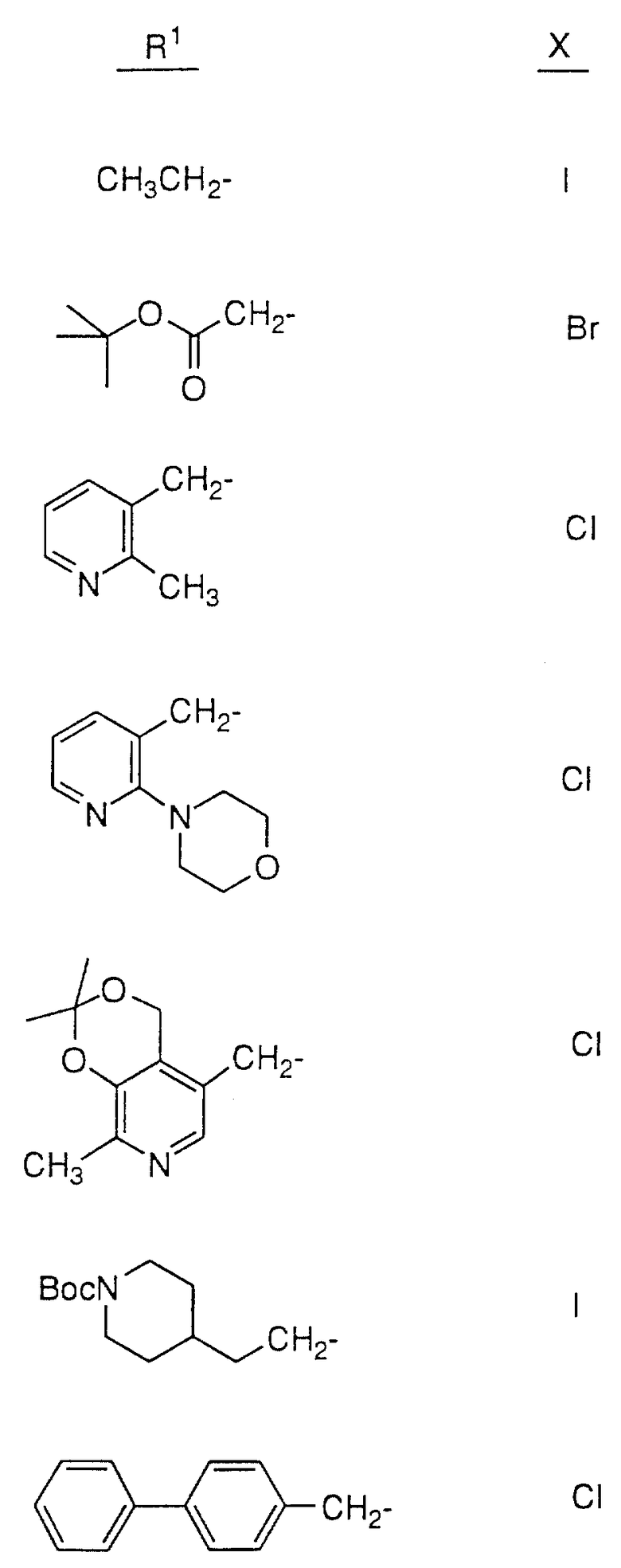
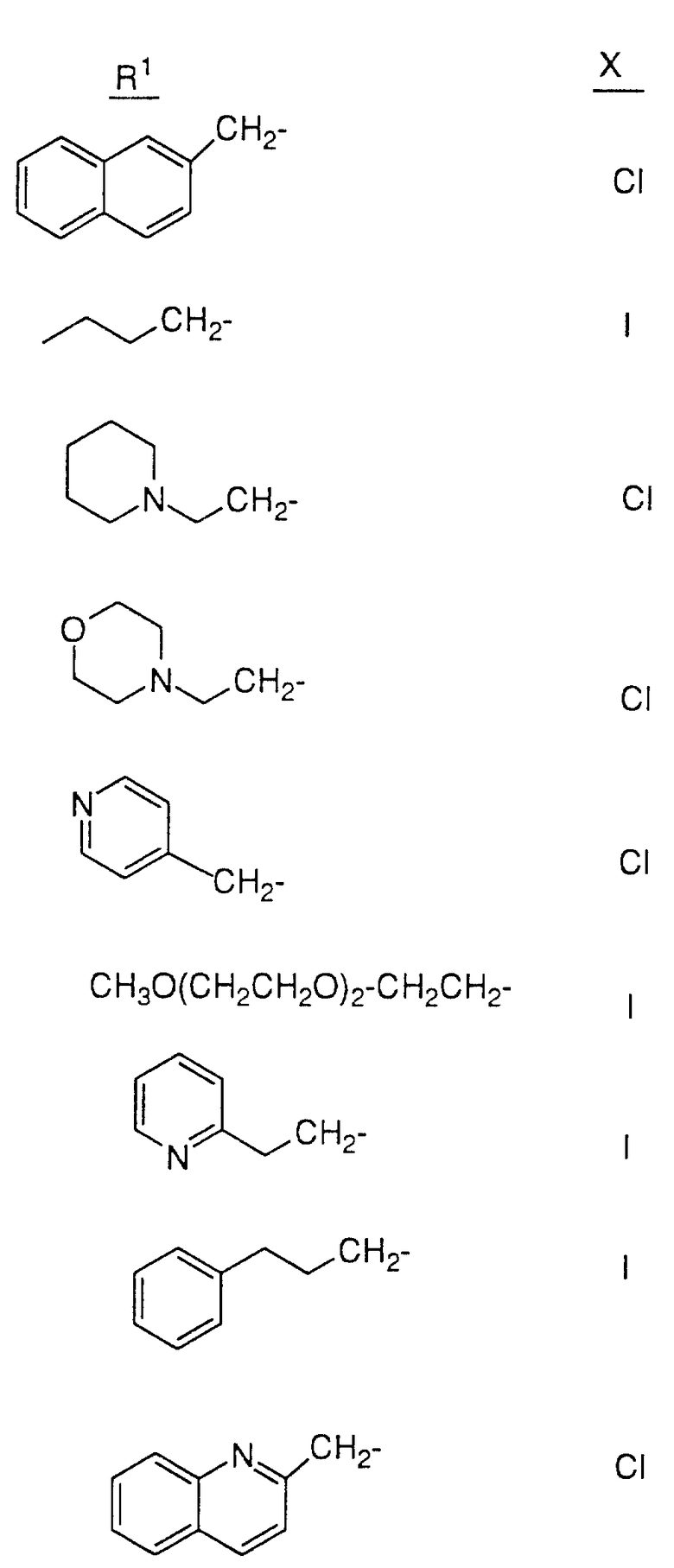
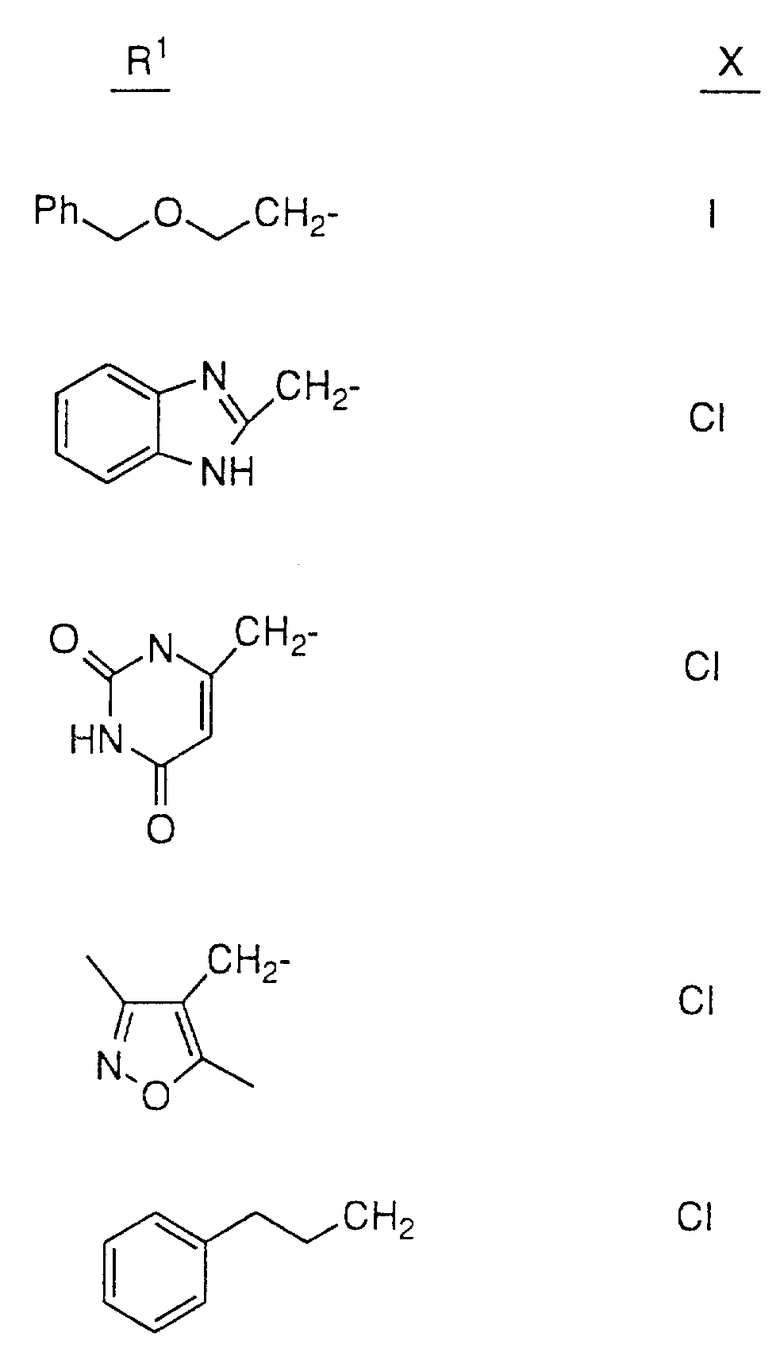
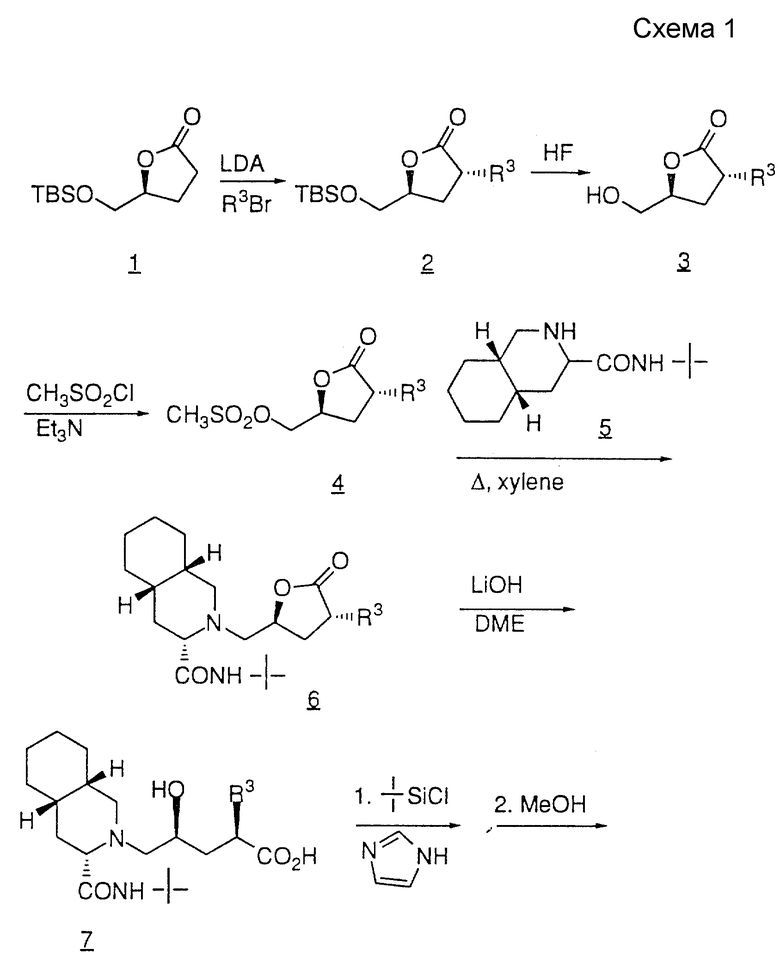
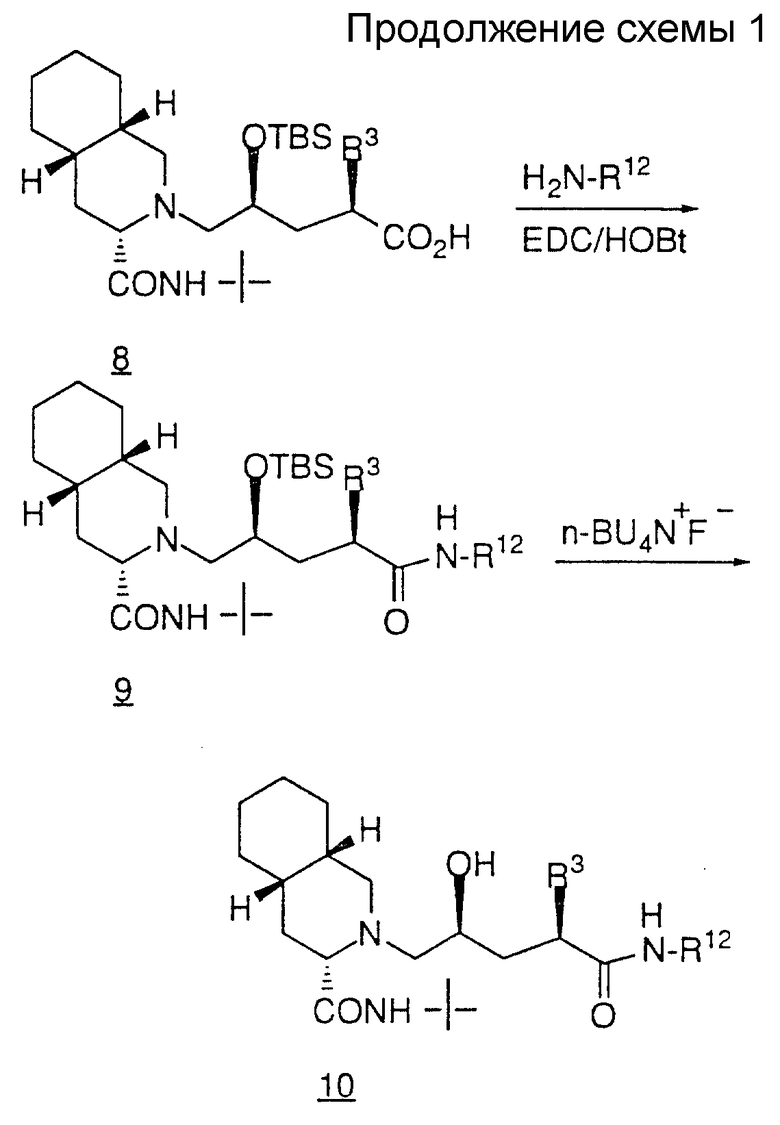
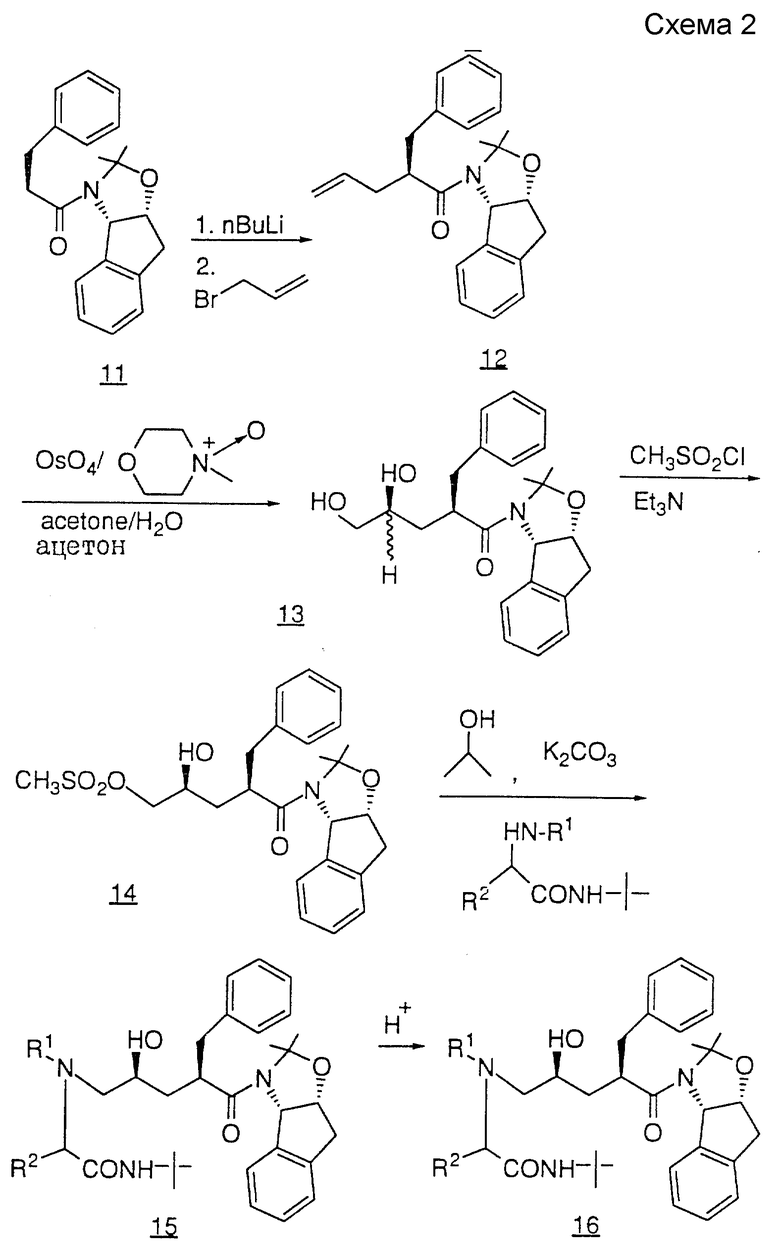
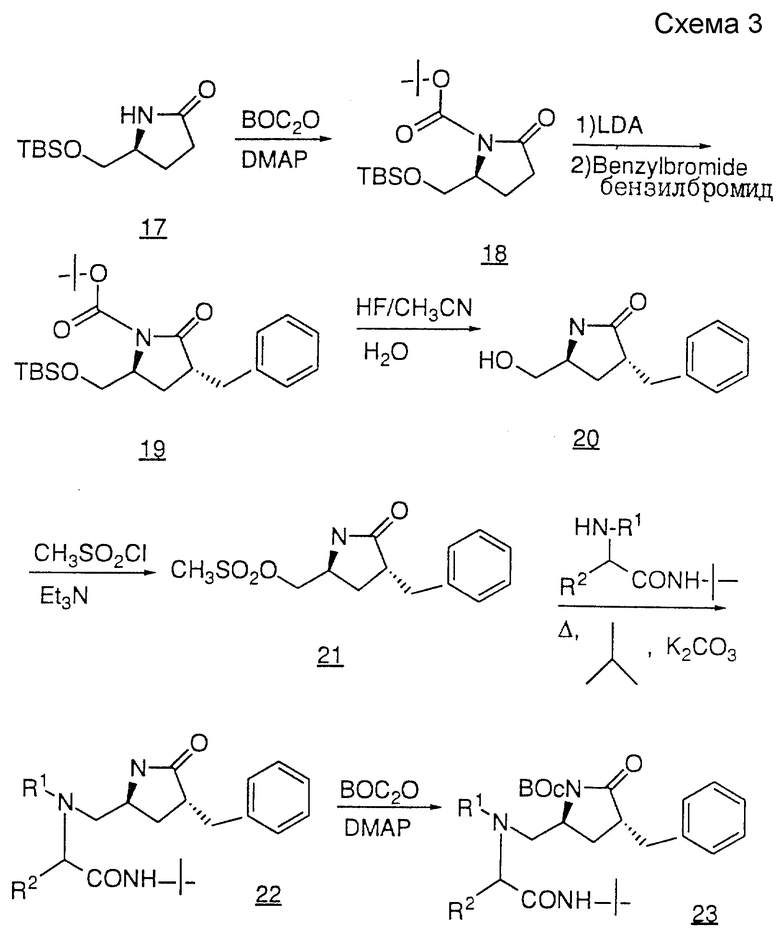
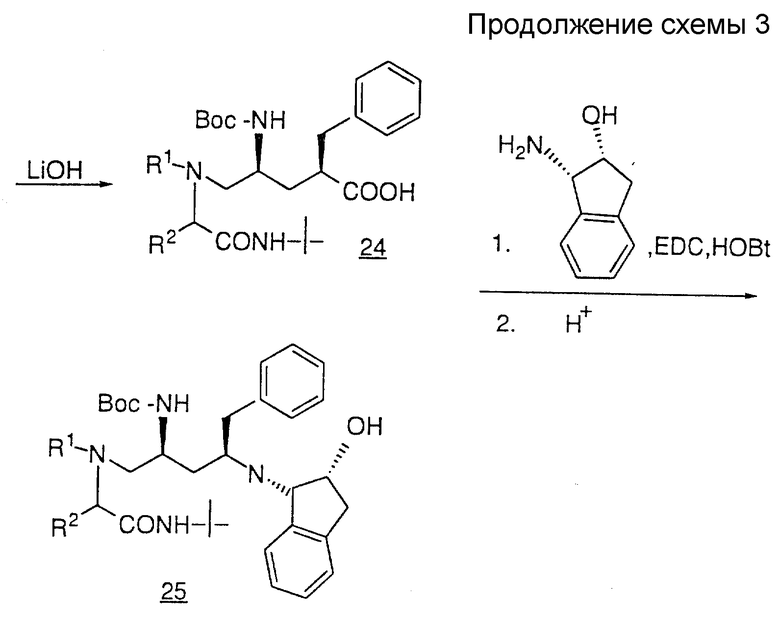
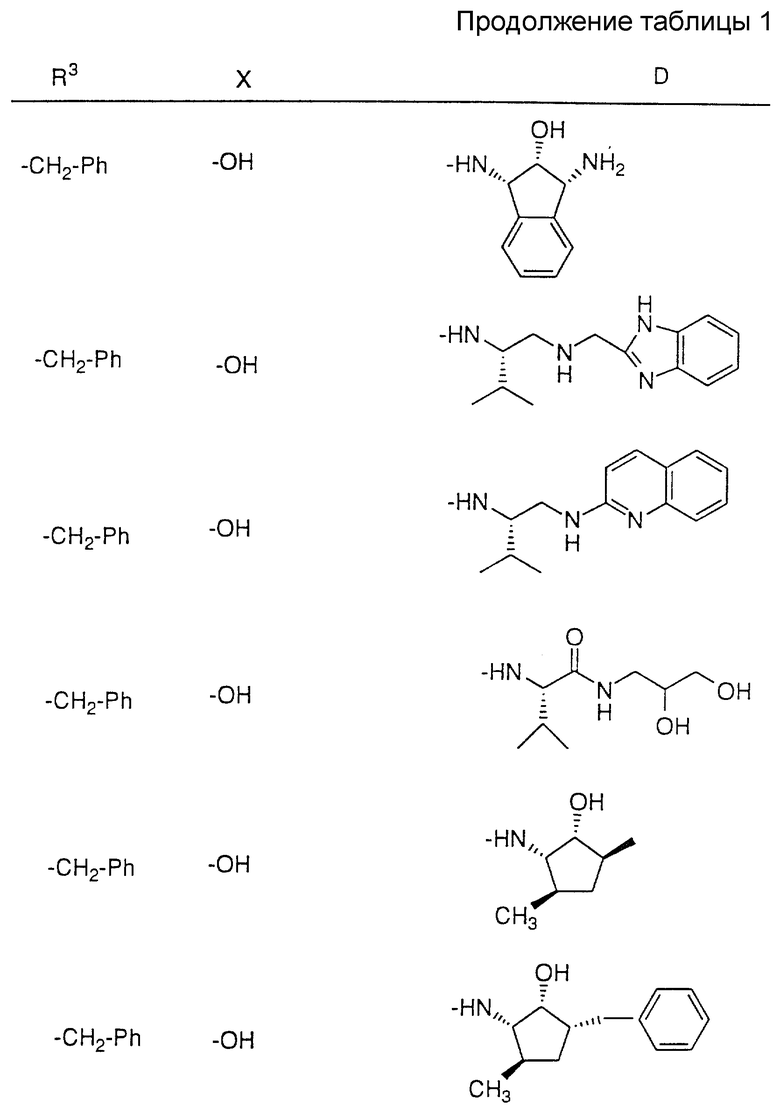
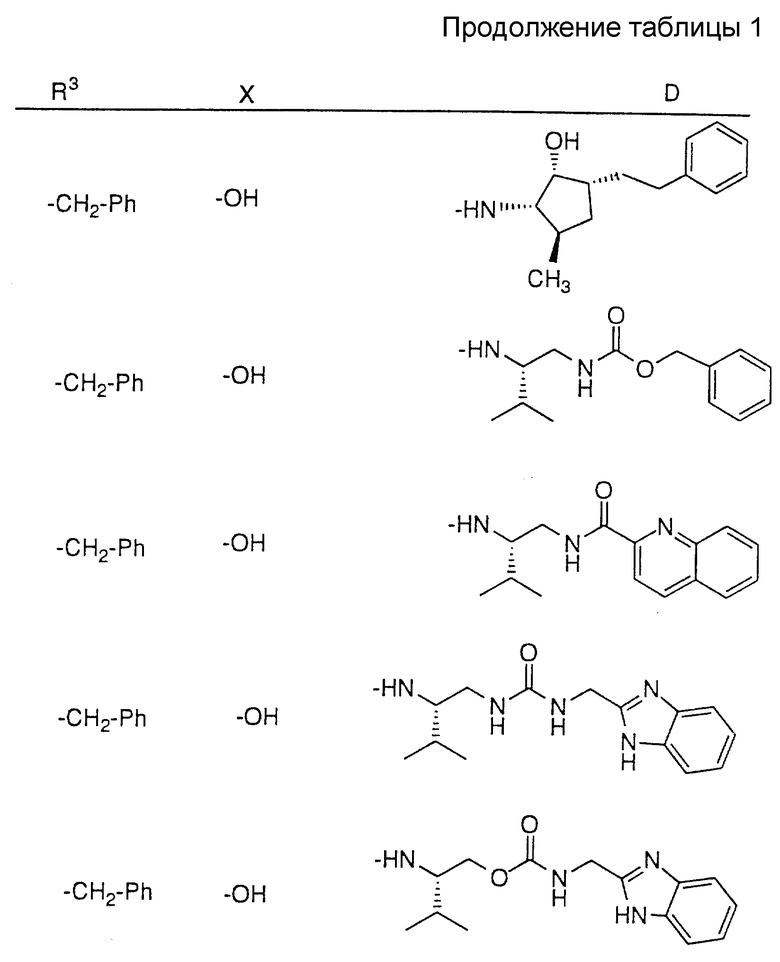

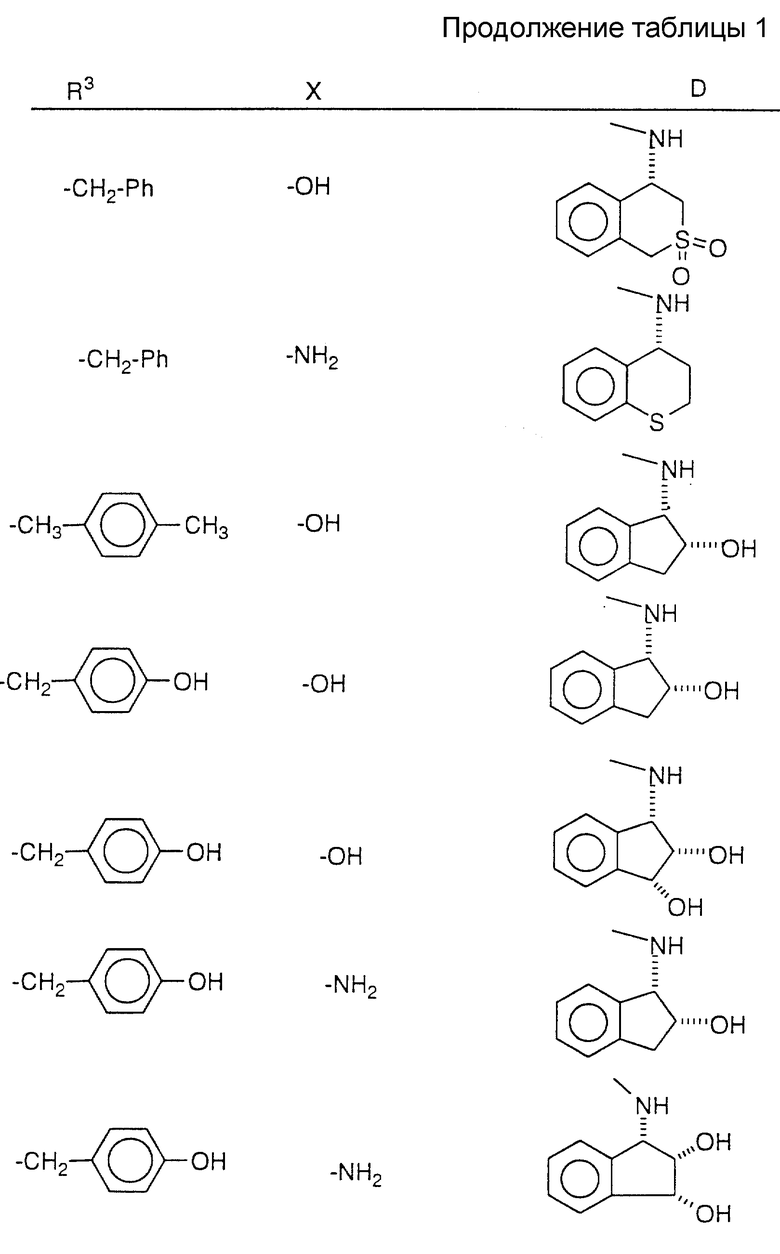
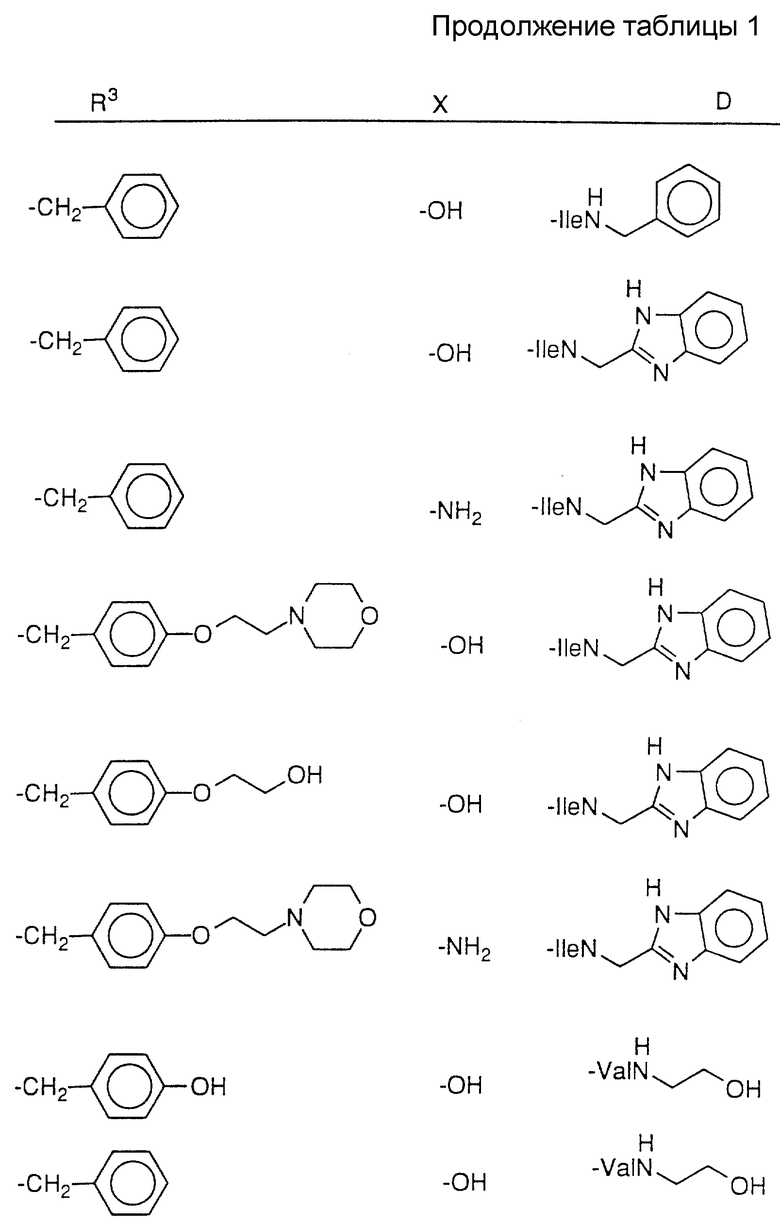
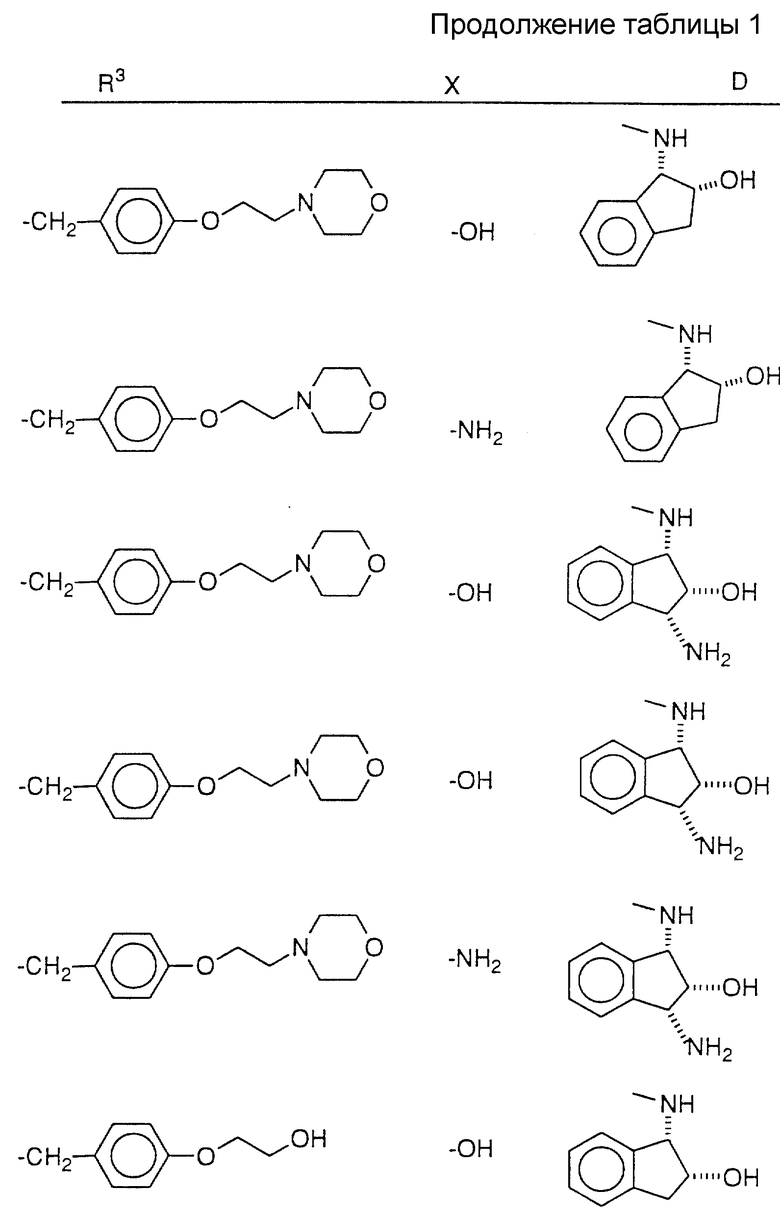
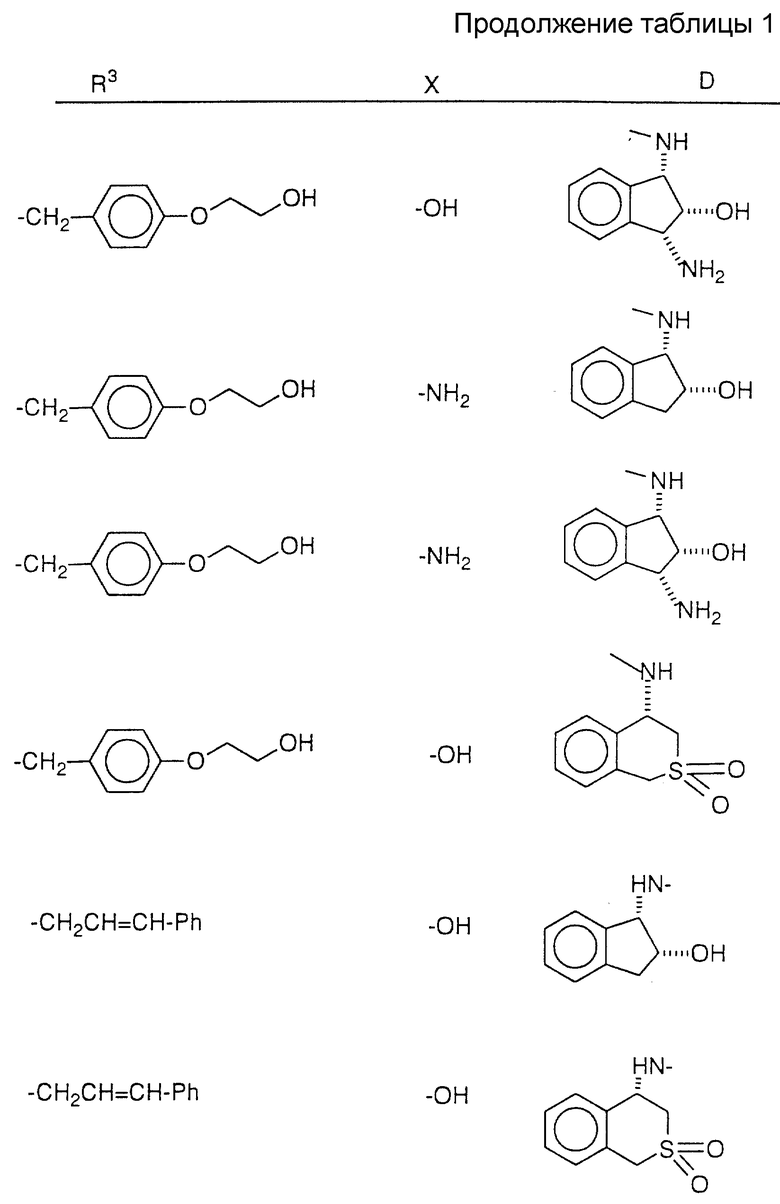
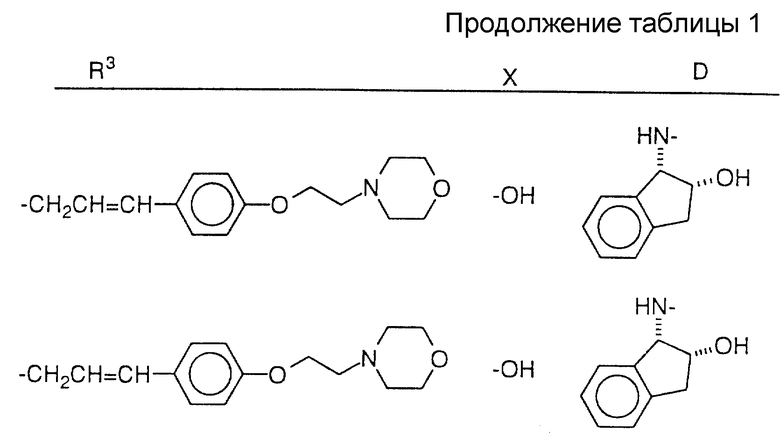
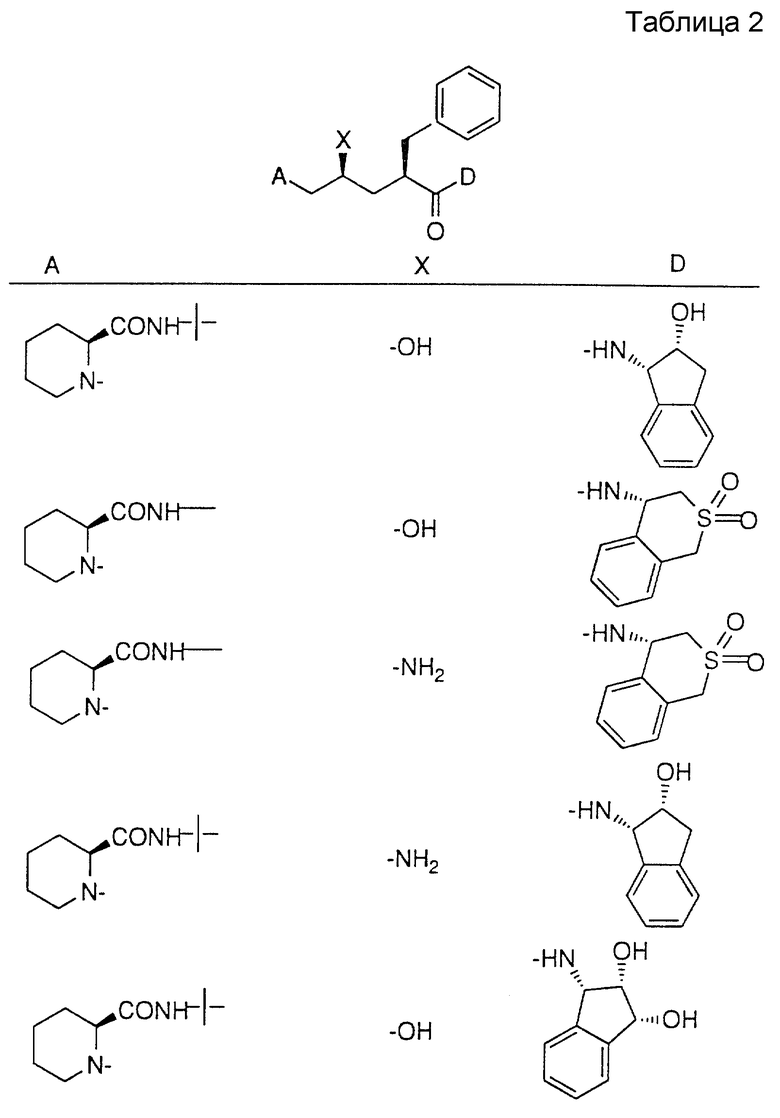

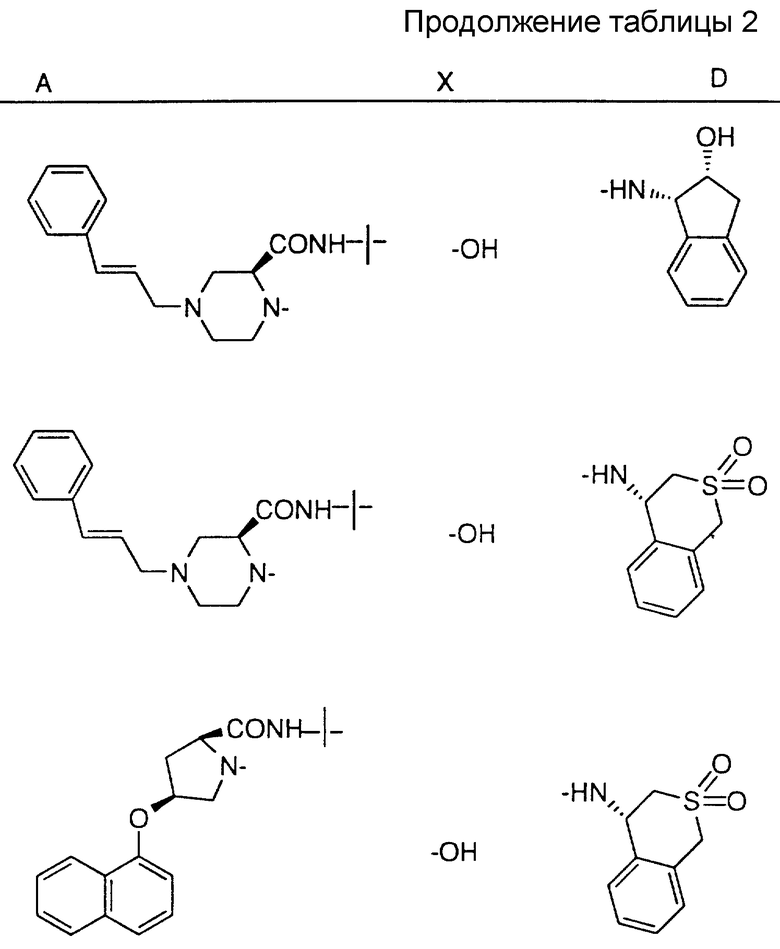
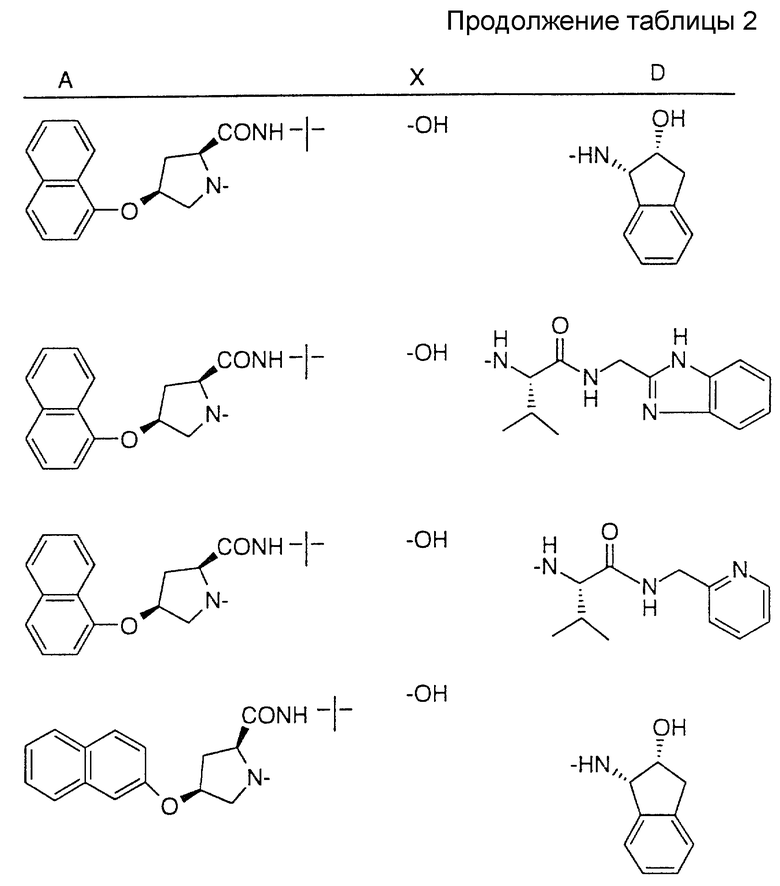

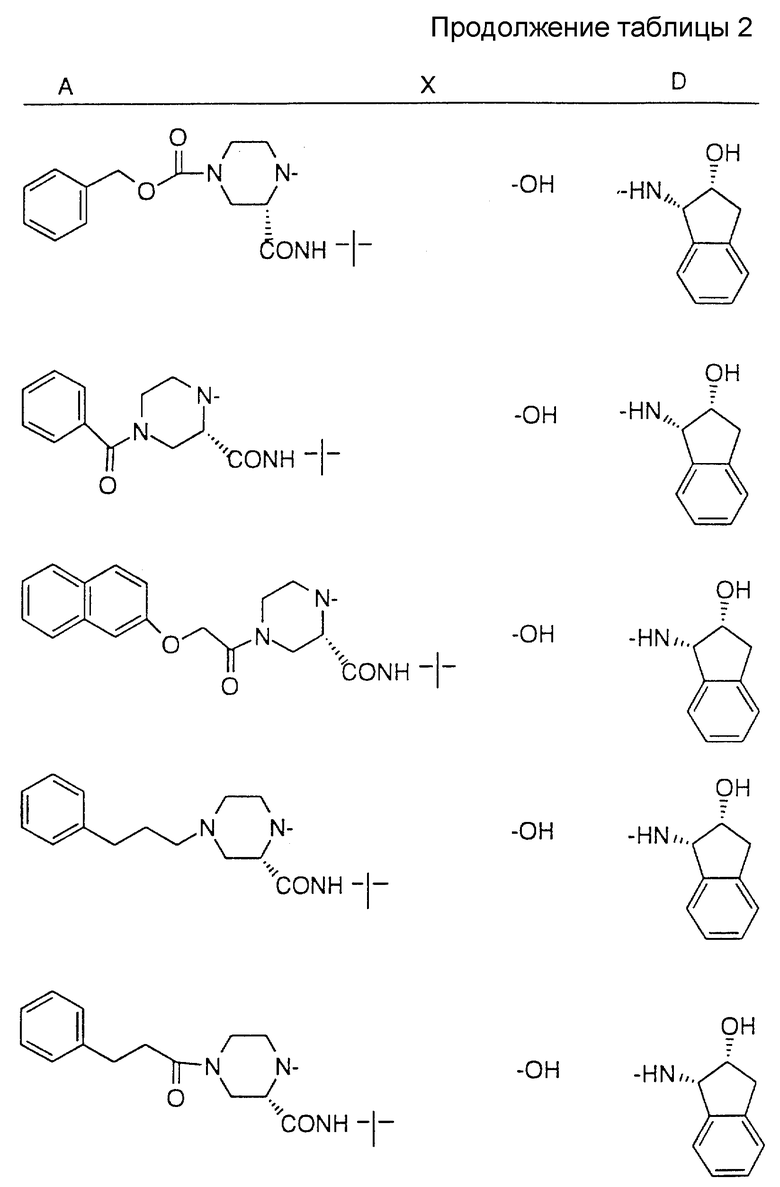
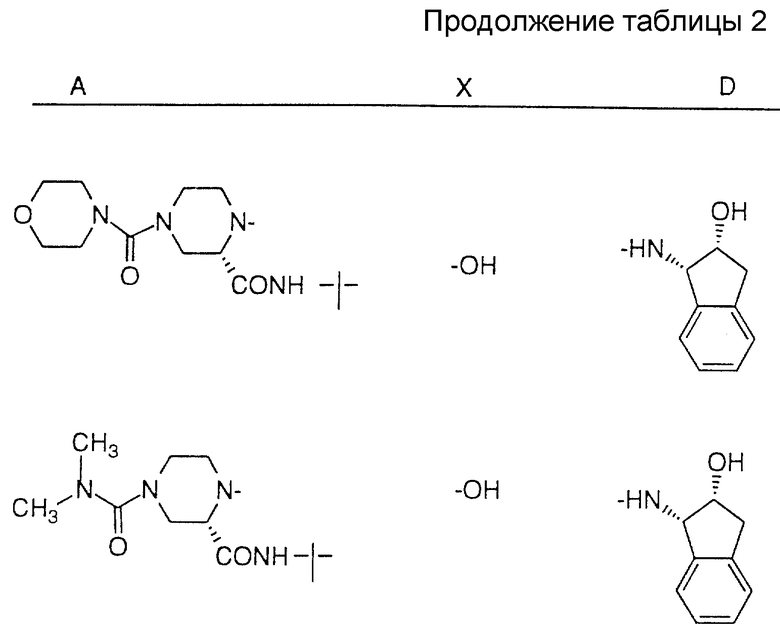
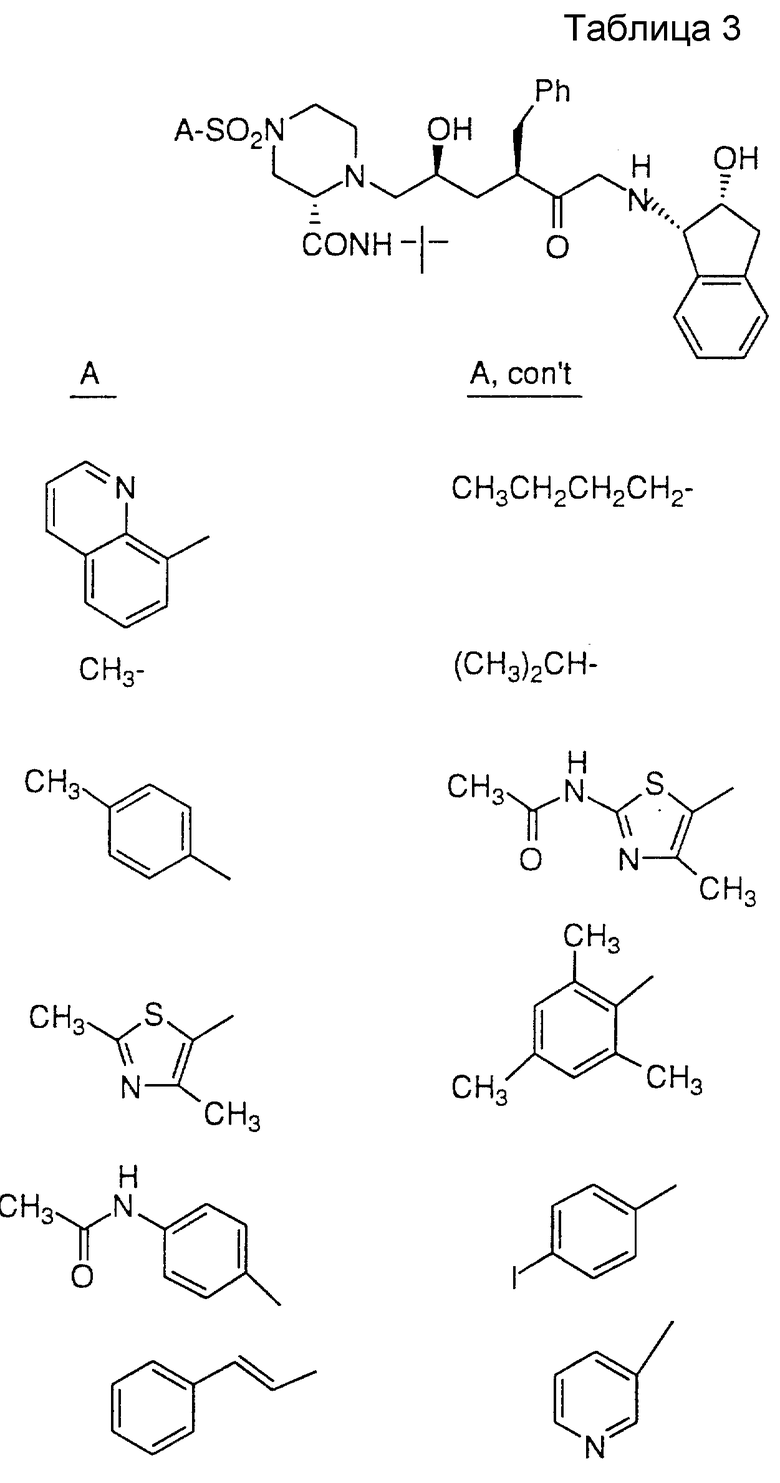
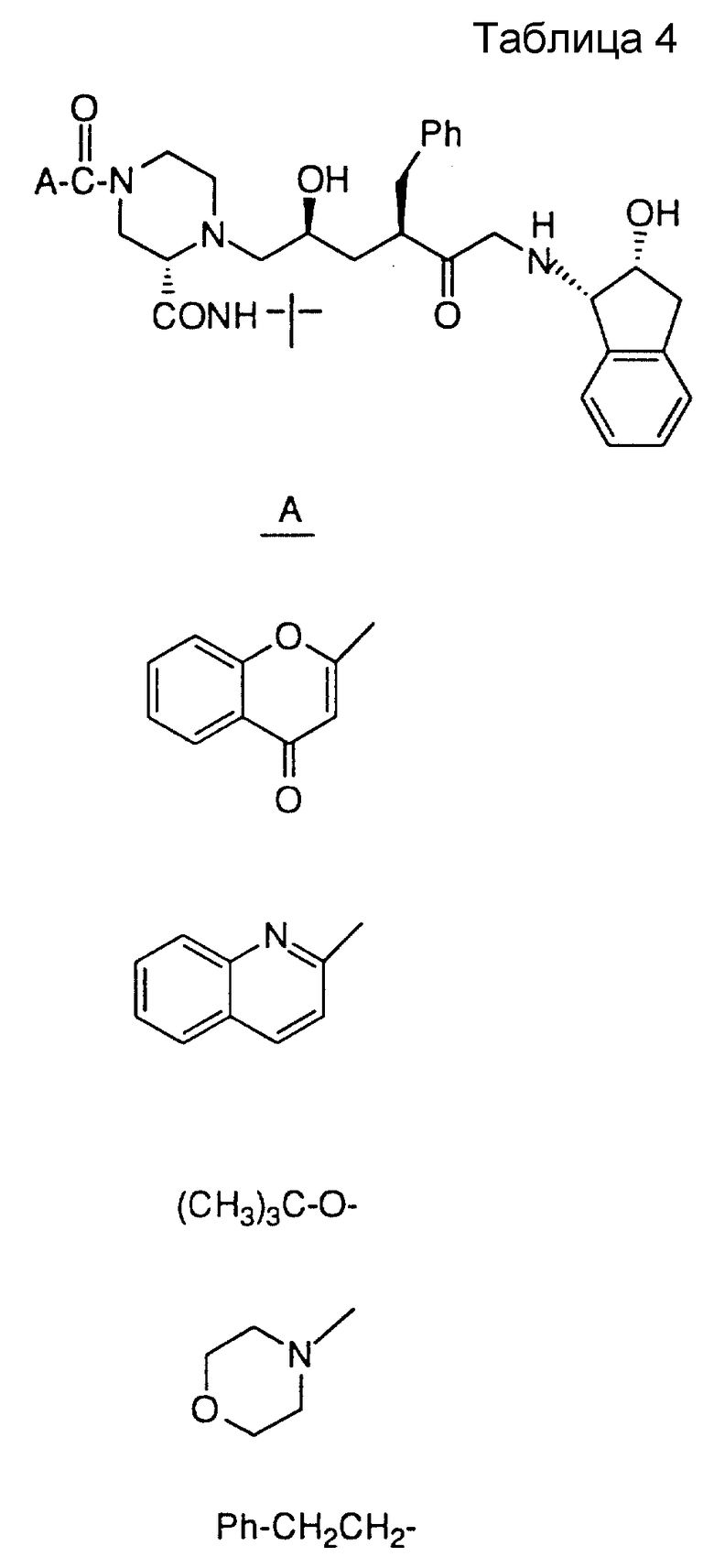





Изобретение относится к медицине. Комбинация соединений включает N-(2(R)-гидрокси-1(S)-инданил-2(R)-фенилметил-4(S)-гидрокси-5-(1-(4-(3-пиридилметил)-2(S)-N'(трет-бутилкарбамоил) пиперазинил)пентанамид или его фармацевтически приемлемую соль и любое из соединений, выбранных из 3'-азидо-3'-деокситимидина, 2',3'-дидеоксиинозина и 2',3'-дидеоксицитидина. Указанная комбинация соединений может быть использована как таковая либо в виде фармацевтической композиции в сочетании с фармацевтически приемлемым носителем для ингибирования ВИЧ-протеазы. Технический результат заключается в реализации указанного назначения. 3 с. и 2 з.п.ф-лы, 6 табл.
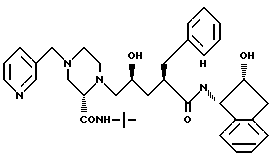
или его фармацевтически приемлемую соль и любое из соединений, выбранных из 3'-азидо-3'-деокситимидина, 2', 3'-дидеоксиинозина и 2', 3'-дидеоксицитидина.
| УСТРОЙСТВО ДЛЯ ПРОЯВЛЕНИЯ ФОРМАТНЫХ ФОТОМАТЕРИАЛОВ__В__П^Т БФОНЛ | 1972 |
|
SU434365A1 |
| СПОСОБ НЕРАЗРУШАЮЩЕГО КОНТРОЛЯ МЕТАЯЗГОВ' | 0 |
|
SU337714A1 |
| Способ отделочной обработки | 1973 |
|
SU484071A1 |
| US 4724232 A, 1988 | |||
| ПОЛИПЕПТИДЫ ИЛИ ИХ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ПРОТИВОВИРУСНОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ ВИРУСА ИММУНОДЕФИЦИТА ЧЕЛОВЕКА | 1991 |
|
RU2073685C1 |

