Настоящее изобретение касается нового способа получения моногидрата ропивакаина гидрохлорида.
Задачей настоящего изобретения является создание нового способа, пригодного для промышленного производства, который обеспечивает воспроизводимый высокий выход энантиомеров и высокую оптическую чистоту целевого продукта.
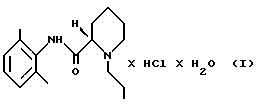
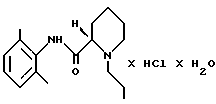
Моногидрат ропивакаина гидрохлорида - это родовое название для соединения моногидрат (S)-(-)-1-пропил-2', 6'- пипеколоксилидида гидрохлорида; это соединение является местным анестезирующим веществом, описанным в европейском патенте 0239710. Его получают путем добавления воды и горячего ацетона к ропивакаину гидрохлориду, после чего кристаллизуется желаемый продукт. Способ получения исходного вещества - ропивакаина гидрохлорида описан в европейском патенте 0151110.
В патенте Великобритании N 1180712 описан способ получения левовращающего 1-н-бутил-2,6-пипеколоксилидида. Указанный способ включал первую стадию - разделение dl- 2',6'-пипеколоксилидида, в ходе которой dl-2',6'-пипеколоксилидид реагировал с O,O-дибензоил-d-винной кислотой, после чего взаимодействием полученной смеси диастереоизомерных O,O-дибензоил-d-тартратов с кипящим ацетоном отделяли нерастворимую в ацетоне соль правовращающего изомера 2', 6'-пипеколоксилидида и выделяли соль левовращающего изомера 2', 6'-пипеколоксилидида из ацетонового раствора. Однако описанный способ сложен, и он включает выделение продукта из горячего ацетона, т.е. он является простым методом для лаборатории, но его нельзя использовать для промышленного производства.
Идея использовать методы разделения, чтобы получить индивидуальные энантиомеры местных анестезирующих веществ мепивакаина и бупивакаина с более длительным анестезирующим действием, появилась в публикации в J.Med.Chem. 14: 891-892, 1971. Смесь 2',6'-пипеколоксилидидов обрабатывали моногидратом дибензоил- L-винной кислоты и путем добавления изопропанола отделяли нерастворимый в изопропаноле энантиомер, после чего выделяли желаемый энантиомер. Использование изопропанола не дает кристаллизационную систему, которая была бы стабильна в течение времени, требуемого для промышленного производства. Это обусловлено тем, что раствор перенасыщен нежелательным энантиомером, и поэтому кристаллизация нежелательной формы может легко начаться в результате малых возмущений в системе; это означает, что изопропанол непригоден для использования в крупномасштабном производстве.
В Acta Chem.Scand B41:757-761, 1987, описано применение изопропанола в смеси с различными количествами воды на стадии разделения. Эти смеси дают различные выходы и качество продукта. Кроме того, комбинация изопропанола и воды дает кристаллизационную систему, недостаточно стабильную для промышленного производства.
Настоящее изобретение направлено на создание способа для промышленного производства моногидрата ропивакаина гидрохлорида, который представляет собой соединение формулы (I)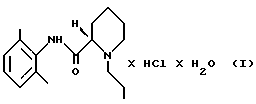
Этот новый способ включает три стадии, причем первая стадия является стадией разделения. Было найдено, что используя разделяющий реагент, образующий стабильную кристаллизационную систему с водой, предпочтительно комбинации кетона и воды, можно отделить нежелательный (R)-пипеколоксилидид и выделить (S)-пипеколоксилидид на этой первой стадии. Таким образом достигается кристаллизационная система, которая стабильна до 24 часов, что достаточно для промышленного производства.
Невозможно увеличить выход энантиомеров на любой из двух последующих стадий; это означает, что эта первая стадия является наиболее важной. Поэтому еще один аспект настоящего изобретения состоял в том, чтобы получить воспроизводимый высокий выход энантиомеров и высокую оптическую чистоту на первой стадии. Это было достигнуто при использовании комбинации кетона, который вместе с водой образует стабильную кристаллизационную систему, и воды.
Новый способ для получения соединения (I) согласно настоящему изобретению включает следующие стадии:
стадию 1: (i) рацемическое исходное вещество - пипеколоксилидид гидрохлорид формулы (II)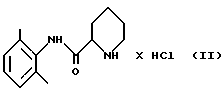
освобождают от, образующей с ним соль, HCl путем экстракции органическим растворителем, содержащим разбавленное основание;
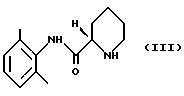
(ii) полученный пипеколоксилидид обрабатывают разбавителем, образующим стабильную кристаллическую систему с водой, и кристаллический продукт (S)-пипеколоксилидид формулы (III)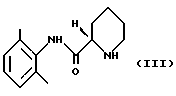
выделяют путем экстракции разбавленным основанием в органическом растворителе, который растворяет минимум около 1 мас.% воды;
стадию 2: (i) S-пипеколоксилидид формулы (III) алкилируют 1-галогенпропаном, предпочтительно 1-бромпропаном или 1-иодпропаном, в присутствии основания и, необязательно, в присутствии катализатора; реакция протекает до конца при нагревании предпочтительно до температуры кипения или, возможно, при более низкой температуре, при которой, однако, реакция протекает более медленно, после чего неорганические соли удаляют путем экстракции водой;
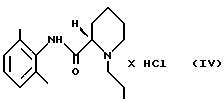
(ii) раствор, полученный на стадии 2 (i), необязательно разбавляют, и полученный продукт осаждают HCl в виде ропивакаина гидрохлорида формулы (IV)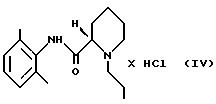
который затем выделяют;
стадию 3: продукт (IV), полученный на стадии 2 (ii), растворяют в водном ацетоне предпочтительно при температуре кипения, продукт (I) осаждают путем добавления ацетона и, наконец, этот продукт выделяют и сушат.
Разбавителями, которые можно использовать на стадии 1(ii), являются L-(-)-дибензоилвинная кислота или L-(-)-дитолуоилвинная кислота, причем L-(-)-дибензоилвинная кислота является предпочтительным разделяющим реагентом.
Разбавленное основание на стадии 1 (i) предпочтительно выбирают из гидроксида натрия, гидроксида калия, карбоната натрия или карбоната калия.
Предпочтительными растворителями для кристаллизации на стадии 1 (ii) являются кетоны, образующие стабильную кристаллизационную систему вместе с водой.
Предпочтительными растворителями для этой кристаллизации являются ацетон или метилэтилкетон, причем наиболее предпочтителен ацетон.
Предпочтительно содержание воды в органическом растворителе, используемом на стадии кристаллизации 1 (ii), составляет 15-25%, наиболее предпочтительно 20%.
Органический растворитель, используемый на стадии экстракции 1 (ii), должен растворять минимум около 1% (вес/вес) воды. Если это не так, реакция идет в двухфазной системе. Кроме того, во время реакции предпочтительно должно присутствовать дополнительное количество воды, около 5%.
Выбор органического растворителя, используемого для экстракции на стадии 1 (ii), будет определяться специалистом в данной области. Однако предпочтительно органический растворитель выбирают из метилизобутилкетона, ацетонитрила, этанола, бутанола или толуола, но могут быть использованы также и другие растворители. Особенно предпочтителен метилизобутилкетон.
Реакция алкилирования на стадии 2 (i) проводится в присутствии основания и предпочтительно в присутствии катализатора. Если в качестве алкилирующего реагента используется 1-иодпропан, нет необходимости применять катализатор для осуществления реакции. Однако, если не применять никакого катализатора, на реакцию может быть затрачено очень много времени.
Основания, которые можно использовать на стадии реакции 2 (i), будут определяться специалистом в данной области. Однако предпочтительными являются карбонаты, в частности карбонат калия или карбонат натрия, или амины, в частности триэтиламин. Наиболее предпочтительным при выборе оснований является карбонат калия.
В качестве катализатора на стадии 2 (i) используют иодид, предпочтительно иодид натрия.
Раствор, полученный на стадии 2 (i), предпочтительно разбавляют ацетоном на стадии 2 (ii).
Изобретение далее будет описано более подробно в следующих примерах.
Пример 1. Стадия 1 - разделение
В реакционный сосуд загружали пипеколоксилидид гидрохлорид (1,0 кг), ацетон (3,75 л) и воду (0,85 л). Добавляли NaOH (водный) до pH > 11. Образовавшиеся при этом фазы разделяли, и органическую фазу разбавляли водой (1,4 л). Добавляли L-(-)-дибензоилвинную кислоту (0,67 кг), растворенную в ацетоне (3,75 л). В раствор вносили затравку для кристаллизации. Суспензию кристаллов охлаждали до 2oC. Кристаллы собирали путем центрифугирования и промывали ацетоном и затем метилизобутилкетоном. Продукт не сушили.
Влажный кристаллический продукт экстрагировали метилизобутилкетоном (3,60 л) и разбавленным NaOH (2,60 л) при pH > 11. Фазы разделяли, органическую фазу промывали водой (0,6 л) и использовали непосредственно на следующей стадии.
Выход (рассчитано по сухому веществу): ~ 0,39 кг (S)-пипеколоксилидида (~ 90%).
Пример 2. Стадия 2 - алкилирование и осаждение соли
Пример 2А
К органической фазе, полученной на предыдущей стадии, добавляли K2CO3 (0,32 кг), NaI (каталитическое количество) и I-бромпропан (0,28 кг), а также около 5% воды. Смесь нагревали путем кипячения с обратным холодильником до завершения реакции. Избыток бромпропана удаляли путем дистилляции. Реакционную смесь экстрагировали водой (1,70 л). К органической фазе добавляли ацетон (1,70 л) и затем HCl (водный) до pH ~ 2. В раствор вносили затравку. Суспензию кристаллов охлаждали до 9oC. Кристаллы собирали путем центрифугирования и промывали ацетоном. Полученный продукт использовали непосредственно на следующей стадии и не сушили.
Выход (рассчитанный по сухому веществу): 0,47 кг ропивакаина гидрохлорида (~ 90%).
Пример 2B
В качестве альтернативной была использована следующая методика.
К органической фазе от предыдущей стадии добавляли K2CO3 (0,32 кг), NaI (каталитическое количество), I-бромпропан (0,28 кг) и воду (1,70 л). Смесь нагревали при кипячении с обратным холодильником до завершения реакции. Избыток бромпропана удаляли путем дистилляции. Реакционную смесь разделяли. К органической фазе добавляли ацетон (1,70л) и затем HCl (водный) до pH ~ 2. В раствор вносили затравку для кристаллизации. Суспензию кристаллов охлаждали до 9oC. Кристаллы собирали путем центрифугирования и промывали ацетоном. Полученный продукт использовали непосредственно на следующей стадии без сушки.
Выход (рассчитан по сухому веществу): 0,47 кг ропивакаина гидрохлорида (~ 90%).
Пример 3. Стадия 3 - перекристаллизация
Ропивакаин гидрохлорид, полученный на предыдущей стадии, суспендировали в ацетоне (1,0 л) при температуре кипения ацетона. Добавляли воду (0,60 л). Полученную смесь фильтровали, и добавляли ацетон (7,6 л) при температуре > 40oC. В раствор вносили затравку для кристаллизации. Суспензию кристаллов охлаждали до 3oC. Кристаллы собирали путем центрифугирования и промывали ацетоном. Кристаллы сушили при 30-40oC в вакууме (< 20 кПа).
Bыxoд: ~ 0,42 кг моногидрата ропивакаина гидрохлорида (~ 80%).
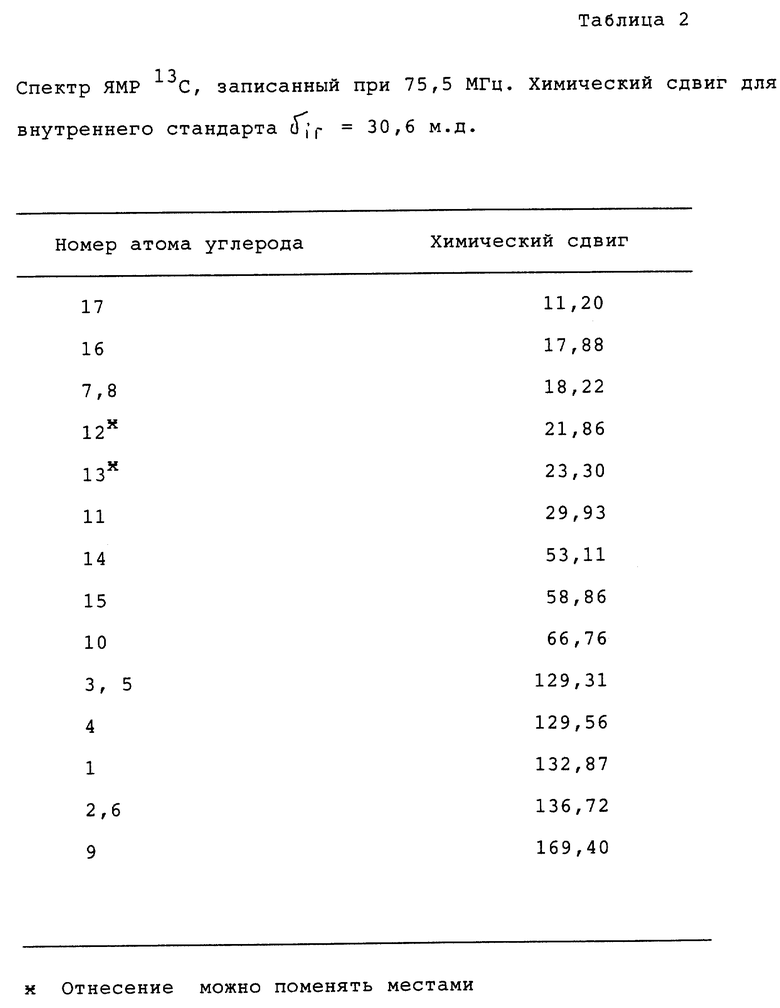
Химический анализ полученного конечного продукта проводили методом ЯМР, как показано ниже.
Спектры ЯМР получали для раствора 22 мг исследуемого вещества в растворе 0,7 мл оксида дейтерия (99,95%) при 23oC. В качестве внутреннего стандарта использовали трет-бутанол. Спектры снимали на приборе Varian Gemini 300.
Номера атомов в таблицах, где дано отнесение спектров, относятся к структуре и нумерации атомов, данной в формуле, приведенной ниже. Результаты приведены как для протонного спектра (таблица 1), так и для спектра ЯМР13C (таблица 2).
 %
%

Изобретение относится к способу получения моногидрата ропивакаина гидрохлорида формулы I, который включает следующие стадии: стадию 1: (i) рацемическое исходное вещество - гидрохлорид пипеколоксилидида формулы II освобождают от НСl, образующего с ним соль, путем экстракции органическим растворителем, содержащим разбавленное основание, (ii) полученный пипеколоксилидид обрабатывают разделяющим реагентом и разбавителем, образующим стабильную кристаллизационную систему с водой, и выделяют кристаллический продукт - (S)- пипеколоксилидид формулы III путем экстракции разбавленным основанием в органическом растворителе, который растворяет минимум около 1 мас. % воды; стадию 2: (i) S-пипеколоксилидид формулы III алкилируют 1-галогенпропаном в присутствии основания и, необязательно, в присутствии иодидного катализатора при нагревании, после чего неорганические соли удаляют путем экстракции водой; (ii) раствор, полученный на стадии 2 (i), в случае необходимости, разбавляют, осаждают соляной кислотой в виде ропивакаина гидрохлорида формулы IV, который затем выделяют; стадию 3: продукт, полученный на стадии 2 (ii), растворяют в водном ацетоне, осаждают продукт формулы I путем добавления ацетона и этот продукт выделяют и сушат. Предлагаемый способ позволяет получить соединение формулы I, являющееся местным анестезирующим средством с высоким выходом и высокой степени чистоты, что необходимо при использовании этого продукта. 18 з.п. ф-лы, 2 табл.
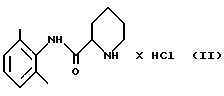
отличающийся тем, что он включает следующие стадии реакции: стадия 1 (i) рацемическое исходное вещество - пипеколоксилидида гидрохлорид формулы II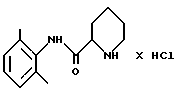
освобождают от HCl, образующего с ним соль, путем экстракции органическим растворителем, содержащим разбавленное основание; (ii) полученный пипеколоксилидид обрабатывают разделяющим реагентом и разбавителем, образующим стабильную кристаллизационную систему с водой, и выделяют кристаллический продукт (S)-пипеколоксилидид формулы III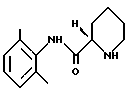
путем экстракции разбавленным основанием в органическом растворителе, который растворяет минимум около 1 мас.% воды, стадия 2 (i) (S)-пипеколоксилидид формулы III алкилируют 1-галогенпропаном в присутствии основания и, необязательно, в присутствии иодидного катализатора при нагревании, после чего неорганические соли удаляют путем экстракции водой; (ii) раствор, полученный на стадии 2 (i), необязательно разбавляют и осаждают продукт в виде ропивакаина гидрохлорида формулы IV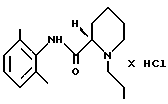
который затем выделяют; стадия 3 продукт IV, полученный на стадии 2 (ii), растворяют в водном ацетоне, осаждают продукт I путем добавления ацетона и этот продукт окончательно выделяют и сушат.
| УСТРОЙСТВО для ЭЛЕКТРОЛОВА РЫБЫ | 0 |
|
SU239710A1 |
| Способ получения оптически чистого моногидрата S-(-)-1-пропил-2 @ ,6 @ -пипеколоксилидид гидрохлорида | 1987 |
|
SU1579455A3 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
