Настоящее изобретение относится к области химии и более конкретно к области терапевтической химии.
Более определенно, предметом настоящего изобретения являются новые производные 2,4-тиазолидиндиона, а именно, 5-феноксиалкил-2,4-тиазолидиндионы, способы их получения, их использование в качестве антидиабетических агентов и при лечении метаболического синдрома инсулиновой устойчивости и содержащие их фармацевтические композиции.
Различные производные 2,4-тиазолидиндионов уже были описаны в качестве антидиабетических агентов (Takeda, Patent EP 193256, Sankyo Patent EP 207581).
Соединения, описанные ранее, были в основном тиазолидиндионовыми, замещенными в положении 5 бензилом, другими словами, соединениями, имеющими алкиленовую цепь, содержащую только один атом углерода между кольцом тиазолидина и арильной группой.
Структура этих соединений содержала в общем варианты заместителя арильного кольца бензильного радикала.
Соединения, обладающие структурами, ранее описанными, и проявляющие значительную гипогликемическую и гипотриглицеридемическую активность, имеют в качестве связи в положении 5 группу R-O-Ar-CH2-.
Эти варианты эксклюзивно влияли на заместитель R у кислорода в пара-положении фенила.
Некоторые из этих соединений, кроме их фармакологических свойств, проявляют феномен гепатотоксичности (Takachi Sohda, Chem. Pharm. Bull 30 (1982) 3580).
Известно, что при инсулин-независимом диабете снижение в эффективности инсулина ведет к гипергликемии.
Снижение в "активности" инсулина связывается, с одной стороны, с панкреатическим дефектом в инсулиновой восприимчивости к глюкозе и, с другой стороны, с печеночной и периферической (мышцы - жировые ткани) инсулиновой устойчивостью.
Некоторые применения в настоящее время антидиабетические препараты стимулируют в основном секрецию инсулина без повышения инсулиновой устойчивости и имеют в качестве основного недостатка ухудшение в течение долгого периода времени диабетического состояния из-за истощения β -панкреатических клеток.
Другие антигипергликемические средства, такие как метформин и соединения, имеющие структуру 2,4-тиазолидиндиона, повышают чувствительность к инсулину.
Эти тиазолидиндионы ослабляют гликемию без стимуляции секреции инсулина и оказываются более активными при инсулиновой устойчивости с гиперинсулинизмом.
Соединения настоящего изобретения являются новыми и отличаются от других производных 2,4-тиозолидиндиона свойствами, которыми соединения известного уровня техники не обладают: отсутствием действия на секрецию инсулина, действием на инсулиновую устойчивость, отсутствием гепатотоксического действия, активностью у больных диабетом в случае диабета без гиперинсулинизма.
Предметом настоящего изобретения являются, в частности, новые 5-феноксиалкил-2,4-тиазолидиндионы, соответствующие общей формуле I: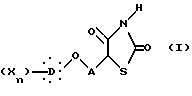
в которой A представляет неразветвленную или разветвленную алкиленовую группу, содержащую от 2 до 16 атомов углерода, D представляет гомо- или гетероуглеродную моно-, ди- или трициклическую ароматическую структуру, которая может включать один или несколько гетероатомов, X представляет заместитель ароматической структуры, выбранный из группы, включающей водород, алкильную группу, имеющую от 1 до 6 атомов углерода, алкоксигруппу, имеющую от 1 до 6 атомов углерода, алкоксиалкильную группу, в которой алкокси и алкильная группы такие, как определено выше, арильную группу, определенную как ароматическую циклическую структуру, содержащую одно или два кольца, необязательно включающую один или два гетероатома в кольце, такую как, например, фенил или α -нафтил или β -нафтил, аралкильную группу, в которой алкильная группа такая, как определяется выше, и арильная группа такая, как определяется выше, и необязательно содержит один или несколько заместителей, аралкильную группу, аралкильная и арильная части которой такие, как определяются выше, галоген, трифторметил, циано, гидрокси, нитро, амино, карбоксил, алкоксикарбонил, карбоксамид, сульфонил, сульфон, сульфонамид, сульфамоил, алкилсульфониламино, ациламино, трифторметокси, n равно целому числу, начиная от 1 и до 3, при условии, что, если A представляет бутильный радикал, то 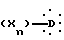 не представляет 4-хлорфенильную группу.
не представляет 4-хлорфенильную группу.
В предыдущем тексте среди ароматических радикалов D могут быть указаны в качестве гомоуглеродных структур фенильный, α -нафтильный, β -нафтильный или флуоренильный радикал.
Среди гетероциклических ароматических радикалов могут быть указаны пиридил, кольцо хинолила или карбазолил.
Что касается изобретения, алкильная группа определяется как имеющая от 1 до 6 атомов углерода и в особенности радикал метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил или пентил и тому подобное, алкоксигруппа определяется как имеющая от 1 до 6 атомов углерода и в особенности радикал метокси, этокси, пропокси, изопропокси, бутокси или изобутокси и тому подобное, группа галоген определяется как фтор, хлор, бром или йод.
Алкиленовая цепь A представляют неразветвленную или разветвленную углеводородную цепь, имеющую от 2 до 16 атомов углерода, которая насыщена или имеет одну или несколько этиленовых связей, необязательно замещена гидроксильным радикалом или фенильным радикалом. Примером такой цепи будет этиленовый или пропиленовый радикал.
Настоящее изобретение относится также к таутомерным формам соединений общей формулы I, энантиомерам, диастереоизомерам и эпимерам этих соединений, а также их сольватам.
Можно предусмотреть, что кетонные функциональные группы, которые присутствуют на кольце тиазолидина, могут енолизироваться и давать моноенолы.
Производные тиазолидиндиона общей формулы I, которые обладают кислотным протоном на азоте кольца тиазолидиндиона, могут в этом случае образовать соль и существовать в форме основных солей.
Примеры основных солей соединений общей формулы I включают фармакологически приемлемые соли, такие как натриевые соли, калиевые соли, магниевые соли, кальциевые соли, соли аминов и другие соли такого же типа (алюминия, железа, висмута и тому подобное). Соли аминов, которые фармакологически неприемлемы, могут служить в качестве средств для идентификации, очистки или разделения на оптические антиподы.
Среди соединений общей формулы I по настоящему изобретению можно указать более конкретно в качестве соединений, в настоящее время предпочтительных:
- 5-[3-(4-фторфенокси)пропил]-2,4-тиазолидиндион,
- 5-(2-феноксиэтил)-2,4-тиазолидиндион,
- 5-[2-(4-фторфенокси)этил]-2,4-тиазолидиндион,
- 5-{[1-гидрокси-2-(4-фторфенокси)]этил}-2,4-тиазолидиндион,
- 5-{[2-гидрокси-3-(4-фторфенокси)]пропил}-2,4-тиазолидиндион,
- 5-[1-метил-2-феноксиэтил]-2,4-тиазолидиндион,
- 5-[2-(4-цианофенокси)этил]-2,4-тиазолидиндион,
- 5-[2-(2-фторфенокси)этил]-2,4-тиазолидиндион,
- 5-[2-(2-нафтилокси)этил]-2,4-тиазолидиндион и их фармакологически приемлемые соли.
Изобретение относится также к способам получения 5-феноксиалкил-2,4-тиазолидиндиона общей формулы I.
Способ синтеза по настоящему изобретению (путь A) является малоновым синтезом, который заключается в том, что соединение формулы II:
в которой X, D и n такие, как определено выше,
подвергают действию дигалоалкилена формулы III:
в которой Hal представляет атом хлора или брома,
A представляет алкиленовый радикал, определяемый как указано выше,
в присутствии основного агента с образованием соединения общей формулы IV: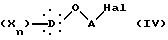
в которой X, D, n и A такие, как определено выше,
которое подвергают действию диалкилмалоната формулы V: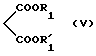
в которой R1 и R'1 представляют алкильные радикалы, в присутствии алкоголята щелочного металла с образованием соединения общей формулы VI: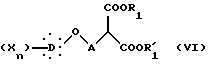
в которой X, D, n, A, R1 и R'1 такие, как определено выше,
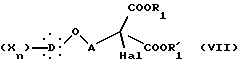
которое подвергают галогенированию действием галогенирующего агента с образованием соединения общей формулы VII: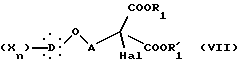
в которой Hal представляет атом хлора или брома, X, D, n, A, R1 и R'1 такие, как определено выше.
Малоновое соединение VI можно галогенировать N-галогенамидом или N-галогенимидом после образования аниона, как, например, действием гидрида натрия в тетрагидрофуране.
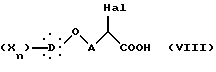
Диалкиловый диэфир общей формулы VII декарбоксилируют и омыляют нагреванием в кислотной смеси, состоящей особенно из хлористоводородной кислоты и уксусной кислоты, получая α -галогенкислоту общей формулы VIII: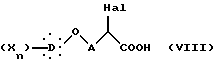
в которой Hal, X, D, n и A такие, как определено выше,
которую подвергают реакции с тиомочевиной с образованием 2-имино-4-тиазолидиндиона общей формулы IX: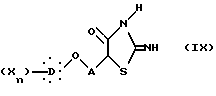
в которой X, D, n и A такие, как определено выше,
который без обязательного выделения гидролизуют в 2,4-тиазолидиндион общей формулы I путем добавления кислотной смеси, такой как хлористоводородная кислота.
Этот гидролиз предпочтительно проводят нагреванием при кипячении с обратным холодильником.
В этом способе основным агентом, используемым для образования соединения общей формулы IV, предпочтительно являются гидроксид щелочного металла и особенно гидроксид натрия. Таким же образом, галогенамидом может быть N-хлорацетамид, N-бромацетамид или N-бромбензамид, и галогенидом может быть N-хлорсукцинимид или N-хлорфталимид.
Другой способ синтеза малоновым путем (путь B) заключается в том, что соединение формулы X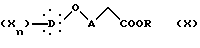
в которой R представляет алкильную группу, X, D, n и A такие, как определено выше,
подвергают галогенированию с образованием α -галогенированного эфира общей формулы XI: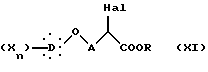
в которой Hal представляет атом хлора или брома, X, D, n и A такие, как определено выше,
и затем последнее соединение подвергают реакции с тиомочевиной в присутствии буферного агента, такого как ацетат натрия, с образованием 2-амино-4-тиазолидиндиона формулы IX: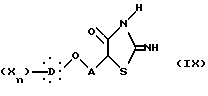
в которой X, D, n и A такие, как описано выше,
которое гидролизуют нагреванием при кипячении с обратным холодильником в хлористоводородной кислоте с образованием тиазолидиндиона общей формулы I: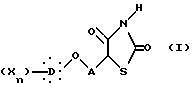
в которой D представляет гомо- или гетероуглеродную моно-, ди- или трициклическую ароматическую структуру, которая может включать один или несколько гетероатомов,
X представляет заместитель ароматической структуры и определяется, как указано выше,
A представляет неразветвленную или разветвленную алкиленовую группу, содержащую от 2 до 16 атомов углерода.
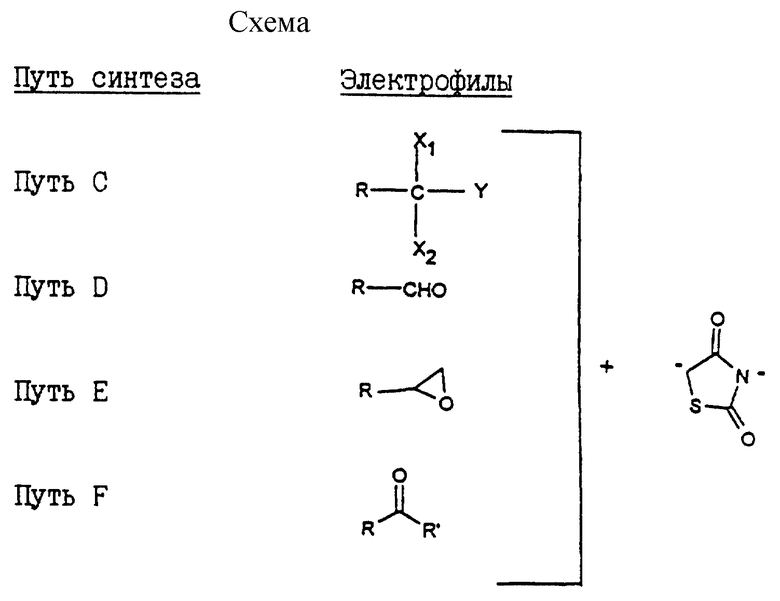
Другой способ синтеза по изобретению (путь C) заключается в том, что галогенированное соединение формулы IV: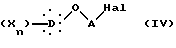
в которой Hal представляет атом хлора или брома, X, D, n и A такие, как определено выше,
подвергают действию дианиона 2,4-тиазолидиндиона, полученного действием производного щелочного металла, такого как бутиллитий, на 2,4-тиазолидиндион с образованием соединения общей формулы I.
В другом способе по данному изобретению (путь D) синтез начинается с арилоксиалкилальдегида и состоит в действии на альдегидное соединение формулы XII: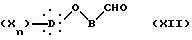
в которой B представляет неразветвленную или разветвленную алкиленовую группу, содержащую от 1 до 15 атомов углерода, X, D и n такие, как определено выше,
дианиона 2,4-тиазолидиндиона, полученного действием производного щелочного металла на 2,4-тиазолидиндион, с образованием соединения общей формулы XIII: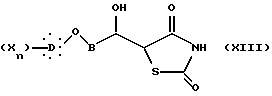
в которой B, X, D и n такие, как определено выше.
Это соединение затем превращают в дегидроксилироанное соединение формулы I дегидратацией и затем селективным гидрированием или, альтернативно, восстановлением спиртового производного в насыщенное производное.
Изобретение относится также к другому способу получения соединений общей формулы I (путь E), в котором оксиран формулы XIV: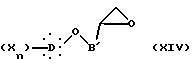
в которой B' представляет неразветвленную или разветвленную алкиленовую группу, содержащую от 1 до 14 атомов углерода, X, D и n такие, как определено выше,
образованный реакцией эпигалогенгидрина с ароматическим производным, подвергают реакции с дианионом 2,4-тиазолидиндиона, полученным действием производного щелочного металла, такого как бутиллитий, с образованием соединения общей формулы XV: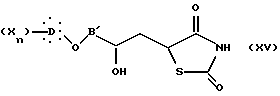
в которой X, D, n и B' такие, как определено выше.
Соединение формулы XV затем превращают в дегидроксилированное соединение общей формулы I путем дегидратации и гидрирования.
Другой способ получения соединений общей формулы I (путь F) заключается, исходя из кетона, в действии на этот кетон общей формулы XVI: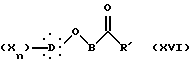
в которой R' представляет собой неразветвленную или разветвленную алкильную группу или арильную или аралкильную группу, замещенную или незамещенную, X, D, n и B такие, как определено выше, 2,4-тиазолидиндиона в присутствии органического основания с образованием, после дегидратации промежуточного карбинола в кислотной среде, соединения общей формулы XVII: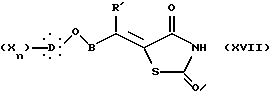
в которой X, D, n, B и R' такие, как определено выше,
и затем в восстановлении двойной связи гидрированием в присутствии катализатора с образованием соединения общей формулы I, в которой алкиленовая цепь является разветвленной цепью: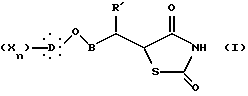
X, D, n, B и R' такие, как определено выше.
Каталитическое гидрирование соединения XVII проводят предпочтительно в присутствии металла семейства платины на инертном носителе, таком, как например, палладированный уголь, платинированный уголь или палладий на карбонате кальция.
Говоря кратко, в способах синтеза C, D, E и F соединения общей формулы I получают действием различных элетрофильных агентов, как описано ниже, неограничивающим образом на дианион 2,4-тиазолидиндиона, предпочтительно при низкой температуре.
Этот дианион можно получить действием сильного основания, такого как диэтиламид лития, амид лития, диизопропиламид лития, н-бутилLi, на 2,4-тиазолидиндион. Далее см. схему в конце описания.
В схеме R представляет: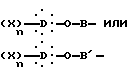
D такой, как определено выше,
X представляет заместитель ароматической структуры и определяется так, как указано выше,
B представляет неразветвленную или разветвленную алкиленовую группу, содержащую от 1 до 15 атомов углерода,
B' представляет неразветвленную или разветвленную алкиленовую группу, содержащую от 1 до 14 атомов углерода,
Y представляет атом брома или хлора или метилсульфонилокси- или п-толуолсульфонилоксирадикал,
R' представляет неразветвленную или разветвленную алкильную группу или, альтернативно, R' представляет собой арильную или аралкильную группу, замещенную или незамещенную,
X1 представляет водород или неразветвленную или разветвленную алкильную группу или, альтернативно, X1 представляет арильную или аралкильную группу, замещенную или незамещенную,
X2 представляет водород или неразветвленную или разветвленную алкильную группу или, альтернативно, X2 представляет арильную или аралкильную группу, замещенную или незамещенную.
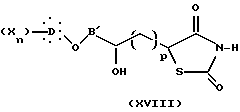
Далее изобретение предлагает также в качестве новых продуктов промежуточные соединения, образованные во время различных способов синтеза и, особенно, соединения общих формул VII, VIII, IX, XVII, XIII и XV, причем два последних могут быть объединены общей формулой: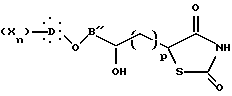
где B'' представляет собой B или B'; p = 0, когда B'' представляет собой B, и p = 1, когда B'' представляет собой B'.
Соединения по данному изобретению проявляют очень ценные фармакологические свойства и, в результате этого, находят использование в терапии.
Соединения изобретения отличаются от других производных 2,4-тиазолидиндиона в интенсивности их антидиабетической активности на моделях диабета без гиперинсулинизма, тогда как оказывается, что соединения известного уровня техники, такие, как например, троглитазон, слабо активны.
Таким образом, соединения изобретения можно использовать при лечении инсулин-независимых диабетических состояний, что делает возможным достижение лучшего подавления гликемии путем достижения пониженного уровня циркулирующего инсулина.
Профилактика этого относительного гиперинсулинизма, связанная с улучшением дислипидемии и антиоксидантной активности, может содействовать снижению микро- и макроангиопатических рисков.
Соединения изобретения можно использовать при лечении метаболического синдрома инсулиновой устойчивости, включая благотворное терапевтическое действие на инсулин-независимый диабет, гипоинсулинизм, гипертензию и дислипидемию, но также инсулин-независимый диабет с гиперинсулинизмом.
Кроме того, эти соединения находят применение при лечении гипертензии у субъектов с инсулиновой устойчивостью, ассоциированной или связанной иначе с другими метаболическими нарушениями.
Диуретическая активность и снижение в захвате Ca2+, наблюдаемые на аорте крысы, могут вызвать антигипертензивную активность некоторых соединений формулы I.
Некоторые из соединений, кроме того, обладают антирадикальной активностью по отношению к гидроксильному и супероксидному аниону, показанной при помощи так называемой модели клеточного исследования.
Для этих целей соединения по данному изобретению используют в форме фармацевтических композиций, которые содержат в качестве активного ингредиента, по меньшей мере, одно соединение общей формулы I в комбинации или смеси с фармацевтически приемлемым нетоксичным инертным наполнителем или носителем.
Фармацевтические композиции по данному изобретению предназначаются для введения парентеральным, дигестивным, ректальным, чрескожным путем или через слизистую оболочку.
Они будут, следовательно, представлены в форме инъецируемых растворов или суспензий, или пузырьков на несколько доз, в форме обычных таблеток или таблеток с покрытием, таблеток с сахарным покрытием, капсул, желатиновых капсул, пилюль, саше, порошков, суппозиториев или реактивных капсул, растворов или суспензий для чрескожного использования в полярном растворителе или для использования через слизистую оболочку.
Подходящими наполнителями для твердых форм являются производные целлюлозы или микрокристаллическая целлюлоза, карбонаты щелочноземельных металлов, фосфат магния, крахмалы, модифицированные крахмалы или лактоза.
Для ректального использования предпочтительными наполнителями являются какао-масло или стеараты полиэтиленгликолей.
Для парентерального использования вода, водные растворы, физиологический солевой раствор, изотонические растворы являются очень удобно используемыми носителями.
Дозировка может варьироваться в пределах широких ограничений в соответствии с терапевтическим показанием и путем введения, а также возрастом и массой субъекта.
Как общее правило, унифицированная доза может составлять от 1 до 200 мг на дозу, и суточная доза может составлять от 2 до 500 мг.
Следующие примеры иллюстрируют данное изобретение, однако, без ограничения его.
Пример 1: 5-[3-(4-фторфенокси) пропил]-2,4-тиазолидиндион (I) (соответственно пути A)
Стадия A
Получение 1-(3-бромпропокси)-4-фторбензола (IV)
Смесь 58,9 г 4-фторфенола, 137 г 1,3-дибромпропана и 65 мл воды нагревают до 60oC. Затем добавляют 58 мл 10 н. раствора гидроксида натрия и смесь нагревают при кипячении с обратным холодильником в течение 72 ч при перемешивании. Реакционную среду (смесь) добавляют к 500 мл воды и 500 мл дихлорметана. Органическую фазу декантируют, промывают 3 раза водой, сушат над сульфатом натрия и концентрируют при сильном вакууме, получая 120 г масла, которое очищают на колонке с диоксидом кремния, элюируя петролейным эфиром. Получают 67,5 г 1-(3-бромпропокси)-4-фторбензола в форме масла.
ЯМР: (CDCl3) δ м.д. 2,25 (2H, м, CH2); 3,55 (2H, т, CH2Br); 4,05 (2H, т, CH2O); 6,7 - 6,9 (4H, м, протоны фенила).
Стадия B:
Получение этилового диэфира [3-(4-фторфенокси)пропил] пропандиовой кислоты (VI)
Раствор этилата натрия (2,68 н. в этаноле) нагревают до 50oC и по каплям добавляют 36 мл диэтилмалоната. Смесь перемешивают в течение 20 мин, затем добавляют 53,6 г 1-(3-бромпропокси)-4-фторбензола. Смесь затем нагревают при кипячении с обратным холодильником в течение 2 ч.
Реакционную среду концентрируют в вакууме и остаток растворяют в 500 мл этилацетата и 500 мл воды. Органическую фазу промывают водой, сушат над сульфатом натрия и концентрируют в сильном вакууме.
Получают 67,5 г диэтилового эфира [3-(4-фторфенокси)пропил]пропандиовой кислоты в форме масла, которое используют без дальнейшей очистки во время следующей стадии:
ИК: 1731 см-1 (C-O эфир)
ЯМР: (CDCl3) δ м. д. 1,2 (6H, т, 2 CH3); 1,4 - 2,2 (4H, м, 2CH2); 3,3 (1H, м, CH); 3,7 - 4,3 (6H, м, 2CH2О + CH2-O-Ar).
Стадия C
Получение этилового диэфира хлор-[3-(4-фторфенокси)пропил]пропандиовой кислоты (VII)
В инертной атмосфере 7,4 г гидрида натрия (80% суспензия в масле) добавляют небольшими порциями к 70,3 г этилового диэфира [3-(4-фторфенокси)пропил]пропандиовой кислоты в 500 мл безводного тетрагидрофурана. Реакция экзотермическая.
Реакционную среду добавляют [sic] в течение 1 ч после того, как ее температура станет опять комнатной, и по частям добавляют 33 г N-хлорсукцинимида. Реакция слабо экзотермическая.
Смесь затем перемешивают в течение 20 ч при 20oC. К реакционной среде добавляют 500 мл воды и 600 мл этилацетата. Органическую фазу декантируют и промывают 3 раза 400 мл воды.
Органическую фазу затем сушат над сульфатом натрия, выпаривают в вакууме, получая 71 г (выход: 91%) этилового диэфира хлор-[3-(4-фторфенокси)пропил] пропандиовой кислоты в форме масла, которое используют без дальнейшей очистки во время следующей стадии.
ИК: 1747 см-1 (C=O эфир)
ЯМР: (DMCO) δ м.д. 1,2 (6H, т, 2CH3-CH2); 1,75 (2H, м, CH2); 2,3 (2Н, м, СН2); 4 (6H, м, CH2 эфиры + CH2).
Стадия D
Получение 2-хлор-5-(4-фторфенокси)пентановой кислоты (VIII)
70 г этилового диэфира хлор-[3-(4-фторфенокси)пропил]пропандиовой кислоты в смеси 200 мл 6 н. хлористоводородной кислоты и 200 мл ледяной уксусной кислоты нагревают при кипячении с обратным холодильником в течение 20 ч.
Затем добавляют 600 мл воды и 400 мл этилацетата. Органическую фазу промывают 5 раз 500 мл воды, сушат над сульфатом натрия и затем выпаривают, получая 56 г 2-хлор-5-(4-фторфенокси)пентановой кислоты в форме масла, которую используют без дальнейшей очистки.
ЯМР: (DMCO) δ м.д. 1,4 - 2,3 (4H, м, 2CH2); 3,9 (2H, т, CH2-O); 4,6 (1H, т, CH); 6,8 - 7 (4H, м, протоны фенила).
Стадия E
Получение 5-[3-(4-фторфенокси)пропил]-2,4-тиазолидиндиона (I)
56 г 2-хлор-5-(4-фторфенокси)пентановой кислоты и 23 г тиомочевины в 410 мл 2-метоксиэтанола нагревают при 110oC в течение 3 ч. Затем добавляют 410 мл 2 н. хлористоводородной кислоты и смесь нагревают при кипячении с обратным холодильником в течение 20 ч. После возвращения к комнатной температуре добавляют 500 мл воды и 500 мл этилацетата. Органическую фазу декантируют и промывают 3 раза 400 мл воды. Органическую фазу сушат над сульфатом натрия и затем выпаривают в вакууме. Получают 48 г масла, которое очищают на диоксиде кремния, элюируя смесью дихлорметан/ацетон (97/3 по объему).
Получают 21 г твердого продукта, который перекристаллизовывают из смеси дихлорметан/гептан. Таким образом, получают 13 г 5-[3-(4-фторфенокси)пропил] -2,4-тиазолидиндиона в форме белого твердого продукта, точка плавления которого 119 - 121oC.
Пример II: 5-(2-феноксиэтил)-2,4-тиазолидиндион (I) (соответственно пути B)
Стадия A
Получение 2-имино-5-(2-феноксиэтил)-4-тиозолидиндиона (IX)
18,5 г этилового эфира 2-бром-4-феноксибутановой кислоты, 6 г тиомочевины и 5 г ацетата натрия в 100 мл 2-метоксиэтанола нагревают при кипячении с обратным холодильником в течение 3 ч. Реакционную среду охлаждают и концентрируют в вакууме и полученный остаток растворяют в 75 мл деминерализованной воды и 75 мл дихлорметана.
Органическую фазу декантируют, промывают и сушат над сульфатом натрия, получая масло. После кристаллизации из ацетонитрила это масло дает 6 г 2-имино-5-(2-феноксиэтил)-4-тиазолидиндиона в форме твердого продукта, который разлагается при температуре выше 250oC.
Стадия B:
Получение 2-имино-5-(2-феноксиэтил)-2,4-тиазолидиндиона (I)
6 г 2-имино-5-(2-феноксиэтил)-4-тиазолидиндиона в 100 мл 2 н. хлористоводородной кислоты нагревают при кипячении с обратным холодильником в течение 8 ч. Кристаллизуется твердый продукт. Этот твердый продукт фильтруют и промывают водой и затем очищают перекристаллизацией из смеси циклогексан/этилацетат, получая 3 г полугидрата 5-(2-феноксиэтил)-2,4-тиазолидиндиона, плавящегося при 105 - 107oC.
5-(2-феноксиэтил)-2,4-тиазолидиндион также получали способом синтеза соответственно пути C. Это соединение получают в безводной форме, оно плавится при 81 - 83oC.
Пример III: 5-[2-(4-фторфенокси)этил]-2,4-тиазолидиндион (I) (соответственно пути C)
33,4 г 2,4-тиазолидиндиона растворяют в 1700 мл тетрагидрофурана в инертной атмосфере и в то время, как температуру реакционной среды поддерживают при -78oC, по каплям добавляют 228 мл бутиллития (2,5 М в гексане). Температуре дают подняться до 20oC и смесь перемешивают в течение 2 ч при этой температуре. Ее охлаждают до -78oC и по каплям добавляют 31,2 г 1-(2-бромэтокси)-4-фторбензола, растворенного в 600 мл тетрагидрофурана. Температуре дают подняться до комнатной температуры и смесь перемешивают в течение 20 ч при этой температуре. Реакционную среду выливают на 2300 мл 2 н. хлористоводородной кислоты.
Органическую фазу декантируют и концентрируют. Остаток растворяют в 800 мл этилацетата и 1000 мл воды. Органическую фазу декантируют, промывают 4 раза водой, сушат над сульфатом натрия и выпаривают, получая масло, которое очищают на диоксиде кремния, элюируя смесью дихлорметан/ацетон (97/3 по объему). Полученный продукт перекристаллизовывают из диизопропилового простого эфира. Получают 8,5 г 5-[2-(4-фторфенокси)этил]-2,4-тиазолидиндиона в форме белого твердого вещества, точка плавления которого 94 - 96oC.
Пример IV: 5-{ [1-гидрокси-2(4-фторфенокси)] этил} -2,4-тиазолидиндион (XIII) (соответственно пути D)
Стадия A
Получение 4-фторфеноксиацетальдегида (XII)
42,9 г трет-бутоксида натрия добавляют к 50 г 4-фторфенола в растворе в 180 мл трет-бутанола, затем добавляют 53 мл диметилацеталя бромацетальдегида и реакционную среду нагревают при кипячении с обратным холодильником в течение 96 ч в инертной атмосфере.
Ее концентрируют в вакууме и остаток растворяют в этиловом простом эфире. Органическую фазу промывают водой и затем 2 н. водным раствором гидроксида натрия до тех пор, пока полностью не исчезнет исходный фенол. Органическую фазу снова промывают водой и затем сушат над сульфатом натрия и затем концентрируют в вакууме. Остаток растворяют в смеси 1000 мл ТГФ и 700 мл 5% водной хлористоводородной кислоты и затем нагревают при кипячении с обратным холодильником в течение 3 ч.
После охлаждения к реакционной среде добавляют 500 мл воды и 500 мл этилацетата. Органическую фазу декантируют, сушат над сульфатом натрия и концентрируют в вакууме, получая масло, которое очищают на колонке с диоксидом кремния, элюируя дихлорметаном. Получают 36 г 4-фторфеноксиацетальдегида в форме масла, которое используют без дальнейшей очистки.
ИК: 1739 см-1 (C=O)
ЯМР: (CDCl3) δ м.д. 4,35 (2H, д, CH2); 6,4 - 7,1 (4H, м, протоны фенила), 9,8 (1H, т, CHO).
Эти данные идентичны с данными, описанными в литературе (J. Med. Chem. 1977, 20, N 4, p. 540-6).
Стадия B:
Получение 5-{ [1-гидрокси-2-(4-фторфенокси)] этил} -2,4-тиазолидиндиона (XIII)
В инертной атмосфере получают раствор 13,7 г 2,4-тиазолидиндиона в 200 мл безводного тетрагидрофурана. По каплям при -78oC добавляют 94 мл бутиллития (2,5 М в гексане).
Реакционную среду затем перемешивают в течение 3 ч при 20oC и затем снова охлаждают до -78oC и осторожно добавляют 36 г 4-фторфеноксиацетальдегида в растворе в 40 мл безводного тетрагидрофурана. Смесь перемешивают в течение 30 мин при -78oC и затем 20 ч при комнатной температуре. Реакционную среду растворяют в 250 мл смеси лед/1 н. хлористоводородная кислота. Органическую фазу декантируют, концентрируют в вакууме и растворяют в 400 мл этилацетата и 400 мл воды.
Органическую фазу промывают водой, сушат над сульфатом натрия и затем выпаривают в вакууме, получая масло, которое очищают на колонке с диоксидом кремния, элюируя смесью дихлорметан/ацетон (90/10 по объему). Получают 12,2 г масла, которое кристаллизуется. Полученный твердый продукт перекристаллизовывают из смеси дихлорметан/гептан, получая 7,6 г 5-{[1-гидрокси-2-(4-фторфенокси)] этил} -2,4-тиазолидиндиона в форме белого твердого продукта, точка плавления которого 131 - 133oC.
Пример V: 5-{ [2-гидрокси-3-(4-фторфенокси)]пропил}-2,4-тиазолидиндион (XV) (соответственно пути E)
Стадия A:
Получение (4-фторфенокси)метилоксирана (XIV)
45 г 4-фторфенола, 94 мл эпихлоргидрина и 56 г карбоната калия в 800 мл безводного ацетонитрила нагревают при кипячении с обратным холодильником в течение 20 ч в инертной атмосфере. К реакционной среде добавляли 400 мл вода и 200 мл этилацетата.
Органическую фазу декантируют, промывают два раза водой, сушат над сульфатом натрия и концентрируют в вакууме. Остаток очищают на колонке с диоксидом кремния с элюированием смесью дихлорметан/гептан (50/50 по объему), получая 40 г (4-фенокси)метилоксирана в форме масла, которое используют в следующей стадии без дальнейшей очистки.
ЯМР (DMCO) δ м.д. 2,8 (2H, м, CH2); 3,3 (1H, м, CH), 3,8 (1H, дд, CH2), 4,3 (1H, дд, CH2), 6,7 - 7,3 (4H, м, протоны фенила).
Эти данные идентичны с данными, описанными в литературе (J. Med. Chem. 1978, 21, N 10, p. 1073-6).
Стадия B:
Получение 5-{ [2-гидрокси-3-(4-фторфенокси)]пропил}-2,4-тиазолидиндиона (XV)
Раствор 28 г 2,4-тиазолидиндиона в 450 мл безводного тетрагидрофурана получают в инертной атмосфере. По каплям при -78oC добавляют 191 мл бутиллития (2,5 М в гексане). Реакционную среду затем перемешивают в течение 3 ч при 20oC и затем снова охлаждают до -78oC и осторожно добавляют 40 г (4-фторфенокси)метилоксирана в растворе в 150 мл безводного тетрагидрофурана. Смесь перемешивают в течение 30 мин при -78oC и затем в течение 20 ч при комнатной температуре.
Реакционную среду растворяют в 600 мл смеси лед/1 н. хлористоводородная кислота. Органическую фазу деканируют и концентрируют в вакууме. Остаток растворяют в этилацетате и полученный раствор промывают несколько раз водой, сушат над сульфатом натрия и затем концентрируют в вакууме. Остаток очищают на колонке с диоксидом кремния с элюированием смесью дихлорметан/ацетон (90/10 по объему), получая 15,2 г масла, которое кристаллизуется.
Этот твердый продукт перекристаллизовывают из смеси дихлорметан/гептан, получая 10,5 г 5-{[2-гидрокси-3-(4-фторфенокси)]пропил}-2,4-тиазолидиндиона в форме белого твердого вещества, точка плавления которого 96 - 98oC.
Пример VI: 5-[1-метил-2-фенокси этил]-2,4-тиазолидиндион (I) (соответственно пути F)
Стадия A:
Получение 5-[1-метил-2-феноксиэтилиден-2,4-тиазолидиндиона (XVII)
29,3 г 2,4-тиазолидиндиона и 34,2 мл фенокси-2-пропанона нагревают при кипячении с обратным холодильником в 500 мл толуола в течение 20 ч в присутствии 2,5 мл пиперидина и 1,3 л уксусной кислоты.
Добавляют 4,75 г моногидрата паратолуолсульфоновой кислоты и смесь снова нагревают при кипячении с обратным холодильником в течение 20 ч, удаляя образованную воду при помощи устройства Дина-Старка. Затем добавляют 1000 мл воды. Органическую фазу декантируют, промывают два раза водой, сушат над сульфатом натрия и выпаривают. Остаток очищают на колонке с диоксидом кремния, элюируя смесью дихлорметан/ацетон (90/10 по объему). Твердый продукт растворяют в диизопропиловом простом эфире и получают после фильтрования 17 г 5-[1-метил-2-феноксиэтилиден] -2,4-тиазолидиндиона в форме желтого твердого продукта, точка плавления которого 153 - 155oC.
Стадия B:
Получение 5-[1-метил-2-феноксиэтил]-2,4-тиазолидиндиона (I)
9 г 5-[1-метил-2-феноксиэтилиден] -2,4-тиазолидиндиона гидрируют при 100oC при давлении 4000 - 5000 кПа (40 - 50 бар) в течение 100 ч в диоксане в присутствии 9 г 10% палладированного угля.
Реакционную среду фильтруют на целите и выпаривают в вакууме, получая 5,5 [lacuna] 5-[1-метил-2-феноксиэтил]-2,4-тиазолидиндиона, точка плавления которого 106 - 108oC.
Пример VII: Фармакологическое исследование соединений изобретения
1 - Цель эксперимента
Для определения антидиабетической активности при пероральном пути введения на экспериментальной модели инсулин-независимого диабета, индуцированного у крыс стрептозотоцином.
2 - Методика
Метод получения крысиной модели nOSTZ
Модель инсулин-независимого диабета получают на крысах путем неонатальной инъекции (в день рождения) стрептозотоцина. Используемые диабетические крысы имеют возраст 8 недель. Животных держат со дня их рождения до дня эксперимента в помещении для животных при регулируемой температуре от 21 до 22oC и подвергают постоянному циклу света (от 7,00 ч до 19,00 ч) и темноты (от 19 ч до 7,00 ч). Их рацион состоял из рациона поддерживания: воду и корм давали по потребности, за исключением 2 часов голодания, предшествующих испытаниям, когда корм убирали.
Метод
В день эксперимента крысам вводят перорально испытуемый продукт и затем через 90' анестезируют нембуталом. Через 2 часа после введения продукта из хвоста отбирают образец 500 мкл крови.
Коллекция образцов
Кровь собирают над гепарином. Все пробирки помещают на лед во время, когда собирают образцы. Их затем центрифугируют в течение 10 минут при скорости вращения 3000 об/мин для отделения организованных элементов так быстро, как возможно.
Полученную плазму распределяют на две аликвоты:
⇒ одну для анализа гликемии и лактатамии; эти анализы проводят сразу,
⇒ другую для анализа инсулинемии - сохраняют в морозильнике при -20oC до дня анализа.
Аналитическая методика
- Глюкоза и лактат
Их определяют методами глюкоза-оксидазы и лактат-оксидазы (Eppendorf Ebio 6666).
Инсулин
Инсулин плазмы анализируют радиоиммунологическим методом.
3 - Результаты
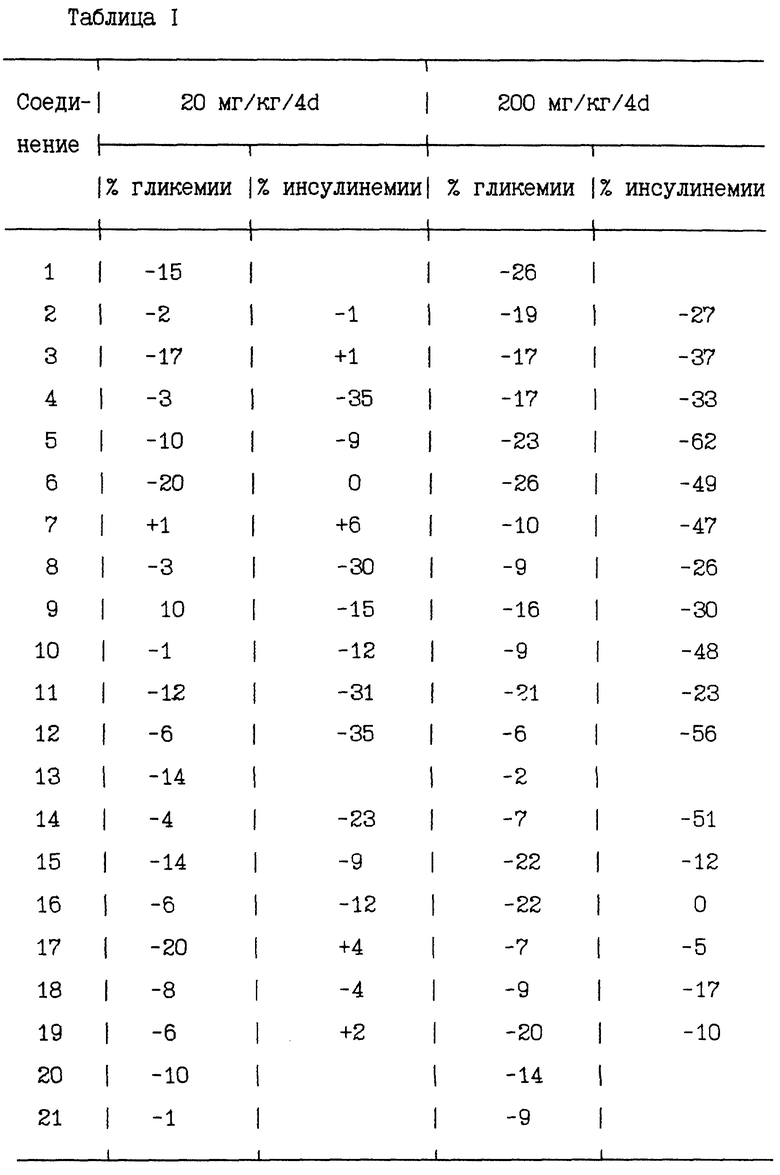
В таблице I приводится сравнение основных полученных результатов
Эти результаты показывают эффективность соединений по настоящему изобретению в снижении спонтанной гликемии и уровня циркулирующего инсулина у животных, сделанных больными диабетом.
Действие на PPAR γ
Данные производные тиазолидиндиона являются антидиабетическими агентами, которые повышают инсулиновую восприимчивость тканей-мишеней в животных моделях инсулин-независимого диабета. Известно, что тиазолидиндионы стимулируют in vitro дифференциацию клеточных линий преадипоцитов и мезенхим в адипоциты; однако, молекулярная основа этого адипогенного эффекта остается точно не известной. Тиазолидиндионы являются сильнодействующими и селективными активаторами γ - рецептора, активированного пролифератором пероксисомы (= PPAR γ : "Пролифератор пероксисомы" - активированный рецептор γ″ ), члена "супер" семейства ядерных рецепторов, действие которых в адипогенезе недавно было показано. Наиболее сильнодействующий из этих агентов, тиазолидиндион BRL 49653, связывается с PPAR γ с константой диссоциации Kd, приблизительно равной 40 нМ. Результатом обработки клеточной линии C3H10T1/2 соединением BRL 49653 является эффективная дифференциация в адипоциты. Это показывает высокую аффинность лиганда PPAR и доказывает, что PPAR γ представляет собой молекулярную мишень для адипогенных действий тиазолидиндионов.
В отличие от других известных тиазолидиндионов (троглитазон, проглитазон, BRL 49653), тиазолидиндионы по настоящему изобретению не обладают активностью по трансактивации PPAR γ. Подобным же образом, тиазолидиндионы по настоящему изобретению не обладают активностью по трансактивации других ядерных рецепторов PPAR, PPAR α и PPAR δ. Тиазолидиндионы по настоящему изобретению обладают слабым адипогенным или не обладают адипогенным действием на клетки 3T3-L1, в отличие от тиазолидиндионов известного уровня техники, которые активируют дифференциацию клеток 3T3-L1 в адипоциты.
Таким образом, соединения по настоящему изобретению обладают свойствами, которыми не обладают соединения известного уровня техники.
Пример VIII
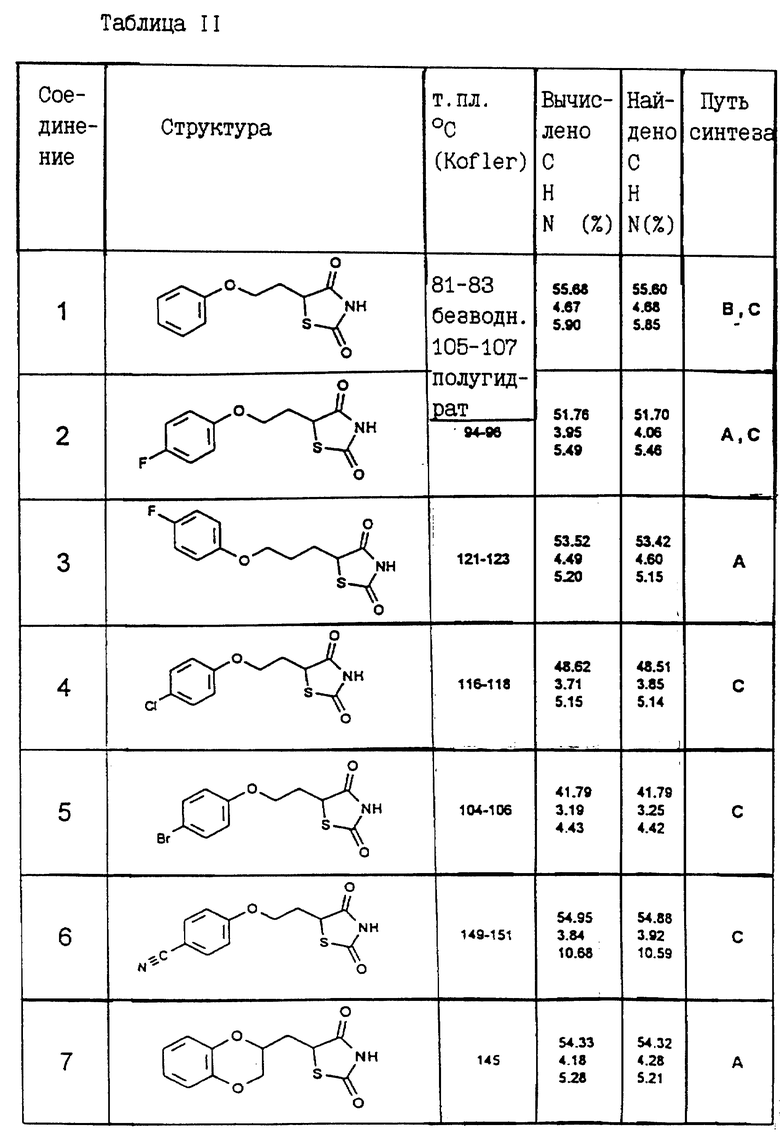
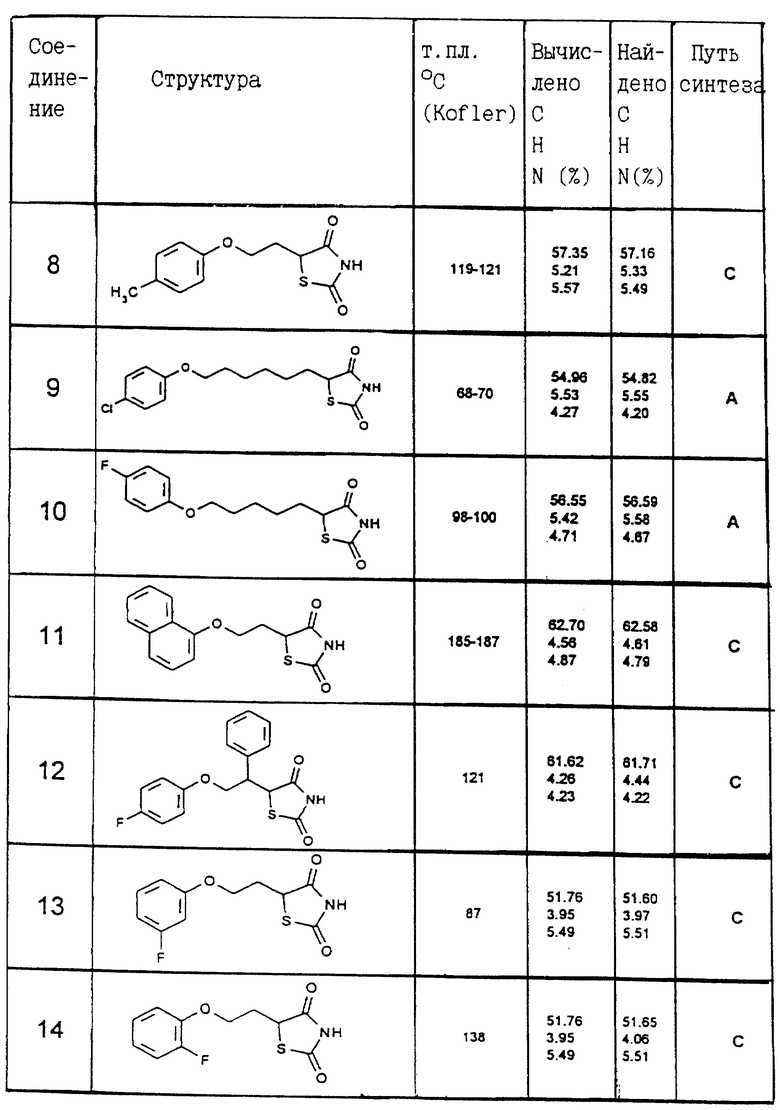
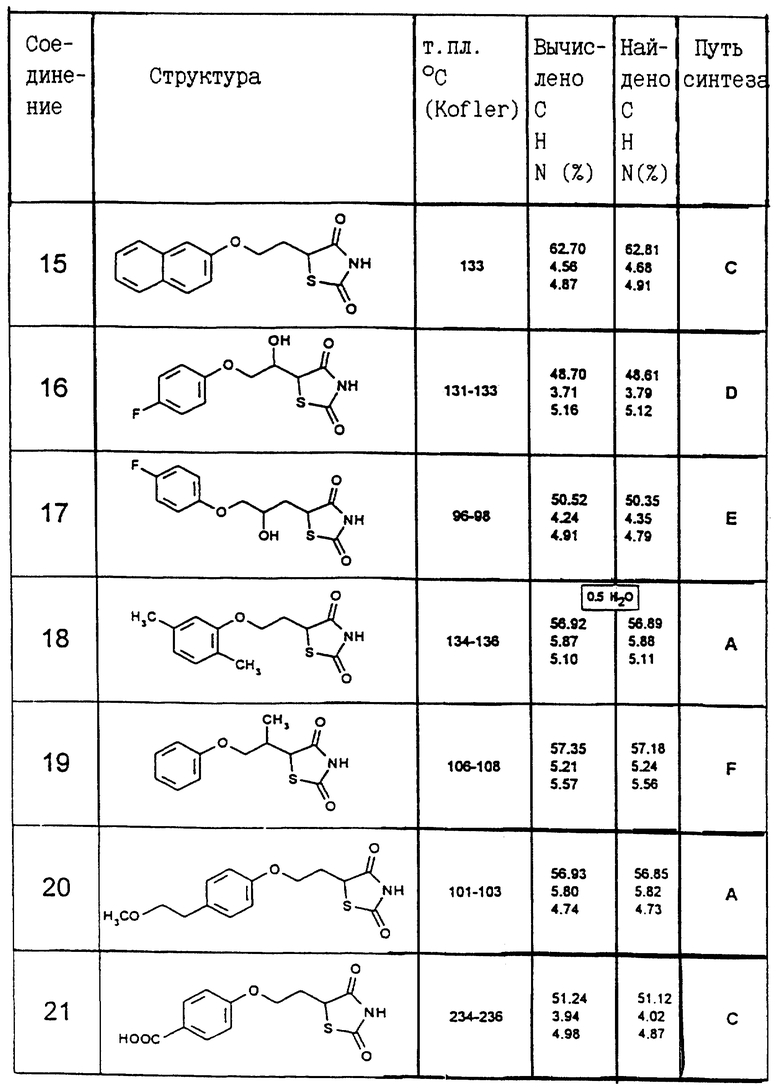
Одним из способов синтеза A, B, C, D, E или F были получены следующие соединения, соответствующие общей формуле I. Их константы перечисляются в приведенной ниже таблице II.
Пример IX:
Таблетки с 100 мг 5-[3-(4-фторфенокси)пропил]-2,4-тиазолидиндиона
Активный ингредиент - 100 г
Пшеничный крахмал - 45 г
Кукурузный крахмал - 55 г
Микрокристаллическая целлюлоза - 12 г
Этилцеллюлоза - 8 г
Стеарат магния - 5 г
для 1000 законченных таблеток со средней массой 0,225 г.
Пример X:
Желатиновые капсулы с 50 мг 5-(2-феноксиэтил)-2,4-тиазилидиндиона
Активный ингредиент - 50 г
Лактоза - 75 г
Стеарат магния - 5 г
для 1000 законченных желатиновых капсул со средней массой 0,130 г.
Пример XI:
Таблетки с покрытием с 75 мг 5-[2-(2-нафтокси)этил]-2,4-тиазолидиндиона
Активный ингредиент - 75 г
Диоксид кремния - 39 г
Лактоза - 112 г
Карбоксиметилкрахмал в виде натриевой соли - 9 г
Тальк - 8 г
Стеарат магния - 7 г
для 1000 сердцевин, весящих в среднем 250 г.
Покрытия:
Тальк
Желатин
Аравийская камедь
Сахароза
Диоксид титана
Пчелиный воск
Карнаубский воск
Этилванилин
для 1000 законченных таблеток с покрытием со средней массой 0,400 г.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 4-(1-ПИПЕРАЗИНИЛ)БЕНЗОЙНОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2178786C2 |
| ПРОИЗВОДНЫЕ БИГУАНИДА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2276136C2 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ГУАНИДИНА И ИХ ТЕРАПЕВТИЧЕСКОЕ ИСПОЛЬЗОВАНИЕ | 2002 |
|
RU2282622C2 |
| ТРИАЗЕПИНОНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1999 |
|
RU2197483C2 |
| ПИПЕРАЗИНОВЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ КОМПОЗИЦИЯ | 1999 |
|
RU2208610C2 |
| ПРОИЗВОДНЫЕ ТРИАЗОЛА | 2006 |
|
RU2416605C2 |
| ТИЕНОПИРИДИНЫ | 2006 |
|
RU2415859C2 |
| 4-ОКСОБУТАНОВЫЕ КИСЛОТЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1997 |
|
RU2175966C2 |
| α-(1-ПИПЕРАЗИНИЛ)АЦЕТАМИДОПРОИЗВОДНЫЕ АРЕНКАРБОНОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СПОСОБЫ ЛЕЧЕНИЯ | 1998 |
|
RU2198881C2 |
| СПОСОБ ПОЛУЧЕНИЯ N-АРИЛМОРФОЛИНОНОВ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2004 |
|
RU2343149C2 |
Изобретение относится к новым производным 5-феноксиалкил-2,4-тиазолидиндионам общей формулы I, где А представляет неразветвленную или разветвленную С2-C16-алкиленовую группу, которая насыщена и необязательно замещена гидрокси или фенилом; D представляет моно- или дициклическую ароматическую группу, которая может содержать один или два атома кислорода; Х обозначает водород, С1-C6-алкил, алкоксиалкильную группу, галоген, циано, карбокси; n равно целому числу 1-3; при условии, что если А представляет бутилен, то 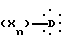 не представляет 4-хлорфенильную группу; в свободной форме или в форме фармакологически приемлемых солей. Способ получения 5-феноксиалкил-2,4-тиазолидиндионов общей формулы I включает взаимодействие соединения формулы II с дигалоалкиленом формулы III в присутствии основного агента, с образованием соединения формулы IV, которое подвергают реакции с диалкилмалонатом формулы V в присутствии алкоголята щелочного металла, с образованием соединения формулы VI, которое подвергают галогенированию, с образованием соединения формулы II, которое нагревают при кипячении с обратным холодильником в кислой среде, получая α-галогенкислоту формулы VIII, которую подвергают реакции с тиомочевиной, получая 2-имино-4-тиазолидинон формулы IX, который далее гидролизуют в кислой среде с получением соединения формулы I. Производное дикарбоновой кислоты формулы VII, производное α-галогенкислоты формулы VIII, производное 2-имино-4-тиазолидинона формулы IX, производное 2,4-тиазолидиндиона формулы XVIII, образованные во время синтеза, где X, D, n. A, Hal, R1, R1', В'' имеют значения, указанные выше, в качестве промежуточных соединений. Фармацевтическая композиция, обладающая антидиабетической активностью, содержит соединение формулы I и фармацевтически приемлемый нетоксичный инертный носитель или наполнитель. 13 с. и 5 з.п.ф-лы, 2 табл.
не представляет 4-хлорфенильную группу; в свободной форме или в форме фармакологически приемлемых солей. Способ получения 5-феноксиалкил-2,4-тиазолидиндионов общей формулы I включает взаимодействие соединения формулы II с дигалоалкиленом формулы III в присутствии основного агента, с образованием соединения формулы IV, которое подвергают реакции с диалкилмалонатом формулы V в присутствии алкоголята щелочного металла, с образованием соединения формулы VI, которое подвергают галогенированию, с образованием соединения формулы II, которое нагревают при кипячении с обратным холодильником в кислой среде, получая α-галогенкислоту формулы VIII, которую подвергают реакции с тиомочевиной, получая 2-имино-4-тиазолидинон формулы IX, который далее гидролизуют в кислой среде с получением соединения формулы I. Производное дикарбоновой кислоты формулы VII, производное α-галогенкислоты формулы VIII, производное 2-имино-4-тиазолидинона формулы IX, производное 2,4-тиазолидиндиона формулы XVIII, образованные во время синтеза, где X, D, n. A, Hal, R1, R1', В'' имеют значения, указанные выше, в качестве промежуточных соединений. Фармацевтическая композиция, обладающая антидиабетической активностью, содержит соединение формулы I и фармацевтически приемлемый нетоксичный инертный носитель или наполнитель. 13 с. и 5 з.п.ф-лы, 2 табл. 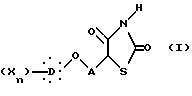

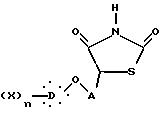
где A представляет неразветвленную или разветвленную алкиленовую группу, содержащую от 2 до 16 атомов углерода, которая насыщена и необязательно замещена гидрокси или фенилом;
D представляет моно- или дициклическую ароматическую группу;
X выбран из группы, включающей водород, алкильную группу, содержащую от 1 до 6 атомов углерода, алкоксиалкильную группу, в которой алкокси и алкильная группы имеют от 1 до 6 атомов углерода, галоген, циано, карбокси; или
D представляет дициклическую ароматическую группу, которая может содержать один или два атома кислорода, и X обозначает водород;
n равно целому числу от 1 до 3, при условии, что если A представляет бутилен, то  не представляет 4-хлорфенильную группу,
не представляет 4-хлорфенильную группу,
в свободной форме или в форме фармакологически приемлемых солей.
5-[3-(4-фторфенокси)пропил]-2,4-тиазолидиндиона,
5-(2-феноксиэтил)-2,4-тиазолидиндиона,
5-[2-(4-фторфенокси)этил]-2,4-тиазолидиндиона,
5-{[1-гидрокси-2-(4-фторфенокси)]этил}-2,4-тиазолидиндинона,
5-{[2-гидрокси-3-(4-фторфенокси)]пропил}-2,4-тиазолидиндиона,
5-[1-метил-2-феноксиэтил]-2,4-тиазолидиндиона,
5-[2-(4-цианофенокси)этил]-2,4-тиазолидиндиона,
5-[2-(2-фторфенокси)этил]-2,4-тиазолидиндиона,
5-[2-(2-нафтилокси)этил]-2,4-тиазолидиндиона,
и их фармакологически приемлемых солей.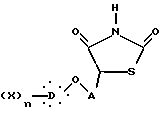
где D представляет моно- или дициклическую ароматическую группу;
X выбран из группы, включающей водород, алкильную группу, содержащую от 1 до 6 атомов углерода, алкоксиалкильную группу, в которой алкокси и алкильная группы имеют от 1 до 6 атомов углерода, галоген, циано, карбоксил; или
D представляет дициклическую ароматическую группу, которая может содержать один или два атома кислорода, и X обозначает водород;
A представляет неразветвленную или разветвленную алкиленовую группу, содержащую от 2 до 16 атомов углерода;
n равно целому числу от 1 до 3,
при условии, что если A представляет бутилен, то  не может представлять 4-хлорфенильную группу,
не может представлять 4-хлорфенильную группу,
отличающийся тем, что соединение формулы II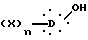
где X, D и n такие, как определено выше,
подвергают взаимодействию с дигалоалкиленом формулы III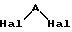
где Hal представляет атом хлора или брома;
A представляет алкиленовую группу, такую, как определено выше,
в присутствии основного агента, с образованием соединения общей формулы IV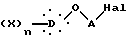
где Hal, X, D, n и A такие, как определено выше,
которое подвергают реакции с диалкилмалонатом формулы V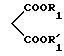
где R1 и R'1 представляет алкил,
в присутствии алкоголята щелочного металла, с образованием соединения общей формулы VI
где X, D, n, A, R1 и R'1 такие, как определено выше,
которое подвергают галогенированию действием галогенирующего агента, с образованием соединения общей формулы VII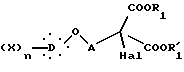
где Hal X, D, n, A, R1 и R'1 такие, как определено выше,
которое нагревают при кипячении с обратным холодильником в кислой среде, получая α-галогенкислоту общей формулы VIII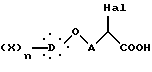
где Hal, X, D, n и A такие, как определено выше,
которую подвергают реакции с тиомочевиной, получая 2-имино-4-тиазолидинон общей формулы IX
где X, D, n и A такие, как определено выше,
который гидролизуют в кислой среде с получением 2,4-тиазолидиндиона общей формулы I.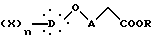
где R представляет алкил;
X, D, n и A такие, как определено выше,
подвергают галогенированию с образованием α-галогенированного сложного эфира общей формулы XI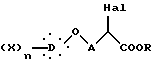
где Hal представляет атом хлора или брома;
X, D, n и A такие, как определено выше,
который подвергают реакции с тиомочевиной с образованием 2-имино-4-тиазолидонона формулы IX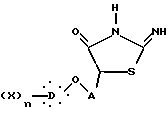
где X, D, n и A такие, как определено выше,
который гидролизуют в кислой среде с образованием соединения общей формулы I.
где B представляет неразветвленную или разветвленную алкиленовую группу, содержащую от 1 до 15 атомов углерода;
X, D и n такие, как определено выше,
подвергают взаимодействию с дианионом 2,4-тиазолидиндиона, полученного реакцией производного щелочного металла с 2,4-тиазолидиндионом, с образованием соединения общей формулы XIII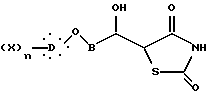
где B, X, D и n такие, как определено выше,
которое превращают в дегидроксилированное производное общей формулы I.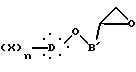
где B' представляет неразветвленную или разветвленную алкиленовую группу, содержащую от 1 до 14 атомов углерода;
X, D и n такие, как определено выше,
полученное реакцией эпигалогенгидрина с соответствующим ароматическим производным, подвергают взаимодействию с дианионом 2,4-тиазолидиндиона, полученным реакцией производного щелочного металла с 2,4-тиазолидиндионом, с образованием соединения общей формулы XV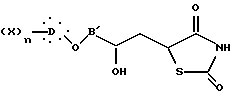
которое превращают в дегидроксилированное производное общей формулы I.
где R' представляет неразветвленную или разветвленную алкильную группу, необязательно замещенную гидрокси, или фенил;
X, D, n и B такие, как определено выше,
подвергают взаимодействию с 2,4-тиазолидиндионом в присутствии органического основания и затем в кислотной среде с образованием соединения общей формулы XVII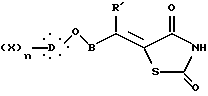
где X, D, n и B такие, как определено выше,
двойную связь которого затем гидрируют действием водорода в присутствии катализатора с образованием соединения общей формулы I, где алкиленовая цепь является разветвленной цепью: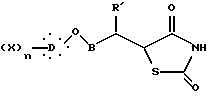
где X, D, n и B такие, как определено выше.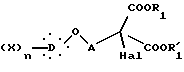
образованные во время синтеза,
где X, D, n, A, Hal, R1 и R'1 имеют значения, указанные выше,
в качестве промежуточных соединений.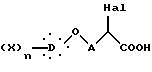
образованные во время синтеза,
где X, D, n, A и Hal имеют значения, указанные выше,
в качестве промежуточных соединений.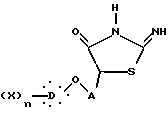
образованные во время синтеза,
где X, D, n и A имеют значения, указанные выше,
в качестве промежуточных соединений.
где B'' представляет собой B или B';
p = 0, когда B'' представляет собой B, и p = 1, когда B'' представляет собой B', при этом B представляет неразветвленную или разветвленную алкиленовую группу, содержащую от 1 до 15 атомов углерода, B' представляет неразветвленную или разветвленную алкиленовую группу, содержащую от 1 до 14 атомов углерода;
X, n и D имеют значения, указанные выше,
в качестве промежуточных соединений.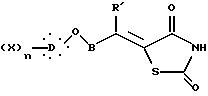
образованные во время синтеза,
где X, n, D, B и R' имеют значения, указанные выше,
в качестве промежуточных соединений.
| Способ получения производных тиазолидиндиона или их фармацевтически приемлемых солей с щелочными металлами | 1987 |
|
SU1611216A3 |
| Способ получения 5-(2-алкоксифенил)тиазолидиндионов или их основных солей с щелочными металлами | 1984 |
|
SU1189341A3 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Роторная машина | 1976 |
|
SU612743A1 |

