Изобретение относится к 9-N-этенильным производным 9(S)-эритромициламина, новым полусинтетическим антибиотикам класса макролидов, обладающим антибактериальным действием, общей формулы (I):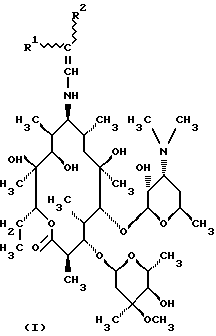
где R1 и R2 одинаковы или различны и представляют собой группу нитрила, карбоксильную группу формулы COOR3, где R3 представляет собой C1-C4-алкильную группу, или кетогруппу формулы COR4, где R4 представляет собой C1-C4-алкильную группу, их фармацевтически приемлемые соли присоединения неорганических или органических кислот, к способу их получения, к способу получения фармацевтических композиций, а также к использованию полученных фармацевтических композиций в борьбе с бактериальными инфекциями.
Уровень техники
Эритромицин A является макролидным антибиотиком, структура которого характеризуется 14-членным макролактоновым кольцом, имеющим карбонильную группу в C-9 положении. Он был обнаружен McGuire в 1952 г. (Antibiot. Chemother. , 1952; 2:281) и в течение 40 лет считался надежным и эффективным антимикробным агентом для лечения болезней, вызываемых грамположительными и некоторыми грамотрицательными микроорганизмами. Однако в кислой среде он легко превращается в ангидроэритромицин, неактивный метаболит C-6/C-12 спирокетальной структуры (Kurath P. et al., Experientia 1971; 27: 362). Хорошо известно, что спироциклизация агликонового кольца эритромицина A успешно ингибируется посредством химической перегруппировки C-9 кетонов или гидроксигрупп в C-6 и/или C-12 позиции. Посредством оксимирования С-9 кетонов (Djokic S. et al., Tetrahedron Lett., 1967; 1945) и последующей модификации полученного 9 (E)-оксима в 9-[O-(2-метоксиэтокси)-метилоксим] эритромицин A (ROXITHROMYCIN) (Ambrieres, G. S., FR 2473525 /1981) или 9(S)-эритромициламин (Egan R. S. et al., J.Org. Chem., 1974; 39:2492) или более сложное производное оксазина, 9-деоксо -11-деокси-9,11-{ имино[2-(2-метоксиэтокси)этилиден] -окси} -9(S)-эритромицин A (DIRITHROMYCIN) (Lugar P. et al., J. Crist. Mol. Struct., 1979; 9:329) были синтезированы новые полусинтетические макролиды, основной характеристикой которых, помимо более высокой стабильности в кислой среде, является лучшая фармакокинетика и более продолжительный биологический период полураспада по сравнению с эритромицином A как исходным антибиотиком.
Первый успешный синтез эритромициламина посредством каталитического восстановления эритромицин-оксима в ледяной уксусной кислоте окисью платины осуществил Massey et al. (Tetrahedron Lett, 1970; 157), и, наряду с 9(S)-изомером, был получен также менее активный 9(R)-изомер (Massey Е. H. et al., J. Med. Chem. 1974, 17, 105).
Kobrehel et al. (J. Med. Chem. 1978, 13, 83) синтезировали серию N-замещенных бензолсульфонилэритромициламинов. Посредством обработки эритромициламина этиленкарбонатом после предварительной защиты 9(S)-аминогруппы (Boyarska-Dahlig H. Et al., Pol. J. Chem., 1979, 53, 2551; Sciavolino F.C., патент США N 4283527/ 1982) были получены 11,12-циклические карбонаты. Посредством синтеза эритромициламина (LeMahieu R. A et al., J. Antib., 1982, 35, 10631) были получены производные эритромициламина без какой-бы то ни было антибиотической активности. Большинство исследований эритромициламина включало реакцию эритромициламина с альдегидами и кетонами в результате чего получали продукты конденсации (Massey Е.H. et al., J. Med. Chem. 1974, 17, 105) или производные 9-N-, 11-O-оксазина (Maier Ret al., патент США 4048306 /1977). Посредством восстановления продукта конденсации с помощью NaBH4 были образованы 9-N-алкил или 9-N-бензил производные (Wildsmith E. et al., J. Med. Chem., 1973; 16; 1059), причем, 9-N, 11-О-оксазин-производные, которые не поддаются восстановлению, были исключением.
В 1989 г. были получены светочувствительные производные эритромициламина посредством присоединения к эритромициламину фотореактивных групп (Arevalo М. A. et al., J. Med. Chem. 1989, 32, 2200). Кроме того, также был получен ряд оснований Шиффа производных эритромициламина (Aries R., заявка на патент FR 2311029-1976; Ewans D., патент Великобритании 1345524/1974; Werner R. G. et al., Biochem. Biophys. Res. Commun. 1978, 83, 1147).
В соответствии с известными и установленными данными предшествующего уровня техники в данной области, до сих пор отсутствовали описания 9-N-этенил-производных 9-(S)-эритромициламина и их фармацевтически приемлемых солей присоединения неорганических или органических кислот, способа их получения, а также методик приготовления и использования в качестве фармацевтических препаратов.
Установлено, и это является предметом настоящего изобретения, что 9-N-этенил-производные 9-(S)-эритромициламина и их фармацевтически приемлемые соли присоединения неорганических или органических кислот можно получить посредством взаимодействия 9(S)-эритромициламина с замещенными производными этоксиметилена и, если это приемлемо, посредством взаимодействия полученных производных 9-N- этенила с неорганическими или органическими кислотами.
Подробное описание изобретения
Установлено, что 9-N-этенильные производные 9-(S)-эритромициламина общей формулы (I):
где R1 и R2 являются одинаковыми или различными и представляют собой нитрил, карбоксильную группу формулы COOR3, где R3 представляет собой C1-C4-алкильную группу или кетогруппу формулы COR4, где R4 представляет собой C1-C4-алкильную группу, и их фармацевтически приемлемые соли присоединения неорганических или органических кислот могут быть получены путем взаимодействия 9-(S)-эритромициламина формулы (II);
с производными этоксиэтилена общей, формулы (III)
где R1 и R2 одинаковы или различны и представляют собой нитрил, карбоксильную группу формулы COOR3, где R3 представляет собой C1-C4-алкильную группу, или кетогруппу формулы COR4, где R4 представляет собой C1-C4-алкильную группу. Реакцию проводят в толуоле, ксилоле или некоторых других ароматических растворителях при температуре от 20 до 80oC.
Фармацевтически приемлемые соли присоединения, которые также являются предметом настоящего изобретения, получают посредством взаимодействия 9-N-этенильного производного 9-(S)-эритромициламина с эквимолярным количеством подходящей неорганической или органической кислоты, такой как соляная кислота, иодистоводородная кислота, серная кислота, фосфорная кислота, уксусная кислота, трифторуксусная кислота, пропионовая кислота, бензойная кислота, бензолсульфокислота, метансульфокислота, лаурилсульфокислота, стеариновая кислота, пальмитиновая кислота, янтарная кислота, этил-янтарная кислота, лактобионовая кислота, щавелевая кислота, салициловая кислота и им подобные кислоты, в растворителе, инертном по отношению к реакции.
Соединения общей формулы (I), где R1, R2, R3 и R4, имеющие значения, как определено здесь выше, демонстрируют антибактериальное in vitro действие, и спектр их действия подобен таковому у эритромицина. Таким образом, они могут быть использованы с той же целью и по такой же методике, что и эритромицин A.
Их действие определяется разбавлением на микропластинках в соответствии с протоколом Национального комитета по Клиническим лабораторным стандартам (NCCLS, M7 - A2). Полученные результаты, выраженные как МИК (MIC) - минимальная ингибирующая концентрация в мкг/мл, наводят на мысль об их возможном использовании в качестве агента для стерилизации, например, комнат и медицинских инструментов, и в качестве промышленных микробных агентов, например, для защиты стен и древесных покрытий.
Способ получения 9-N-этенильных производных 9-(S)-эритромициламина иллюстрируется приведенными ниже примерами, которые никаким образом не ограничивают объем настоящего изобретения.
Пример 1
9 (S)-N-(β,β-дикарбэтоксиэтенил)эритромициламин
Смесь 9(S)-эритромициламина (1,0 г; 1,36 ммоль) и диэтилэтоксиметиленмалоната (3,2 мл; 16,0 ммоль) нагревали с обратным холодильником в течение 90 минут. В реакционную смесь, охлажденную до температуры 0 - 5oC, добавляли простой диэтиловый эфир (14 мл) и полученную суспензию перемешивали в течение 15 минут при той же температуре и в течение 15 минут при комнатной температуре. Было получено 0,480 г 9(S)-N-(β,β-дикарбэтоксиэтенил) эритромициламина.
Пробу для анализа и биологических исследований очистили посредством хроматографии на колонке с силикагелем в системе растворителей CHCl3:MeOH = 9: 1, с выходом 0,27 г 9 (S)-N-(β,β-дикарбэтоксиэтенил)эритромициламина со следующими физико-химическими константами:
ИК (CHCl3) см-1: 3500, 2950, 1725, 1670, 1600, 1450, 1380, 1250, 1225, 1170, 1080;
1H ЯМР (300 МГц, CDCl3) δ: 9.55 ( , 7.79
, 7.79  , 5.09 (1H, H-1''), 4.65 (1H, H-1'), 4.23,
, 5.09 (1H, H-1''), 4.65 (1H, H-1'), 4.23,  , 4.22 (1H, H-3), 4.17
, 4.22 (1H, H-3), 4.17 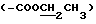 , 3.35 (1H, H-5), 3.34 (3H, 3''-OCH3), 3.28 (1H, H-2'), 3.06 (1H, H-4''), 2.65 (1H, H-9), 2.31 [6H, 3'-N(CH3)2], 2.26 (1H, H-10), 1.96 (1H, H-8), 1.32 и 1.28
, 3.35 (1H, H-5), 3.34 (3H, 3''-OCH3), 3.28 (1H, H-2'), 3.06 (1H, H-4''), 2.65 (1H, H-9), 2.31 [6H, 3'-N(CH3)2], 2.26 (1H, H-10), 1.96 (1H, H-8), 1.32 и 1.28  , 1.16 (3H, 8-CH3), 1.06 (3H, 10-CH3);
, 1.16 (3H, 8-CH3), 1.06 (3H, 10-CH3);
13C ЯМР (75 МГц, CDCl3) δ: 177.7 (C-1), 168.7  , 166.7
, 166.7 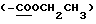 , 160.2
, 160.2  , 132.2 (9-NH-CH=C), 102.2 (C-1'), 95.7 (C-1''), 81.5 (C-5), 79.4 (C-3), 59.2
, 132.2 (9-NH-CH=C), 102.2 (C-1'), 95.7 (C-1''), 81.5 (C-5), 79.4 (C-3), 59.2  , 59.1
, 59.1 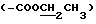 , 77.3 (C-4''), 70.5 (C-2'), 74.7 (C-9), 48.9 (3''-OCH3), 40.0 [3'-N(CH3)2], 32.3 (C-10), 32.3 (C-8), 18.3 (8-CH3), 14.1
, 77.3 (C-4''), 70.5 (C-2'), 74.7 (C-9), 48.9 (3''-OCH3), 40.0 [3'-N(CH3)2], 32.3 (C-10), 32.3 (C-8), 18.3 (8-CH3), 14.1  , 13.9
, 13.9  , 13.0 (10-CH3);
, 13.0 (10-CH3);
FAB-MC m/z 906 (М+H)+
Пример 2
9(S)-N-(β-циано-β-карбэтоксиэтенил)эритромициламин
Смесь 9(S)-эритромициламина (0,5 г; 0,68 ммоль) и этилэтоксиметиленцианоацетата (0,2 г; 1,18 ммоль) в толуоле (20 мл) нагревали с обратным холодильником в течение 60 минут. Реакционную смесь затем быстро охладили и упарили досуха. Полученные желтые кристаллы сырого продукта (0,5 г) очистили посредством хроматографии на колонке с силикагелем в системе растворителей EtOAc: Me2CO = 1: 1, с выходом 0,14 г 9(S)-N-(β-циано-β-карбэтоксиэтенил) эритромициламина со следующими физико-химическими константами:
ИК (CHCl3) см-1: 3500, 2950, 2200, 1730, 1675, 1625, 1450, 1380, 1250, 1225, 1170, 1080;
1H ЯМР (300 МГц, CDCl3) δ: 9.46  , 7.05
, 7.05 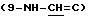 , 5.07 (1H, H-1''), 4.61 (1H, H-1'), 4.22 (1H, H-3), 4.19
, 5.07 (1H, H-1''), 4.61 (1H, H-1'), 4.22 (1H, H-3), 4.19 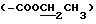 , 3.76 (1H, H-5), 3.34 (3H, 3''-OCH3), 3.25 (1H, H-2'), 2.29 [6H, 3'-N (CH3)2], 2.20 (1H, H-10), 1.96 (1H, H-8), 1.31
, 3.76 (1H, H-5), 3.34 (3H, 3''-OCH3), 3.25 (1H, H-2'), 2.29 [6H, 3'-N (CH3)2], 2.20 (1H, H-10), 1.96 (1H, H-8), 1.31  , 1.14 (3H, 8-CH3), 1.05 (3H, 10-CH3);
, 1.14 (3H, 8-CH3), 1.05 (3H, 10-CH3);
13C ЯМР (75 МГц, CDCl3) δ: 177.5 (C-1), 167.6  , 159.1 (9-NH-CH= C), 119.4 (-CN), 117.5 (9-NH-CH=C), 101.7 (C-1'), 95.2 (C-1''), 80.8 (C-5), 78.9 (C-3), 60.1
, 159.1 (9-NH-CH= C), 119.4 (-CN), 117.5 (9-NH-CH=C), 101.7 (C-1'), 95.2 (C-1''), 80.8 (C-5), 78.9 (C-3), 60.1 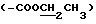 , 77.5 (C-4''), 70.7 (C-2''), 75.5 (C-9), 49.1 (3''-OCH3), 40.1 [3'-N(CH3)2], 32.1 (C-10), 32.7 (C-8), 18.4 (8-CH3), 14.2
, 77.5 (C-4''), 70.7 (C-2''), 75.5 (C-9), 49.1 (3''-OCH3), 40.1 [3'-N(CH3)2], 32.1 (C-10), 32.7 (C-8), 18.4 (8-CH3), 14.2  , 13.2 (10-CH3);
, 13.2 (10-CH3);
FAB-MC m/z 858 (М+H)+.
Пример 3
9(S)-N-(β,β-диацетилэтенил)эритромициламин
В соответствии с процедурой, описанной в примере 2, посредством реакции 9(S)-эритромициламина (0,5 г; 0,68 ммоль) и этоксиметиленацетилацетона (1,0 мл; 6,88 ммоль) в толуоле (20 мл) при нагревании в течение 90 минут при 50oC было получено 0,54 г неочищенного продукта. Посредством хроматографии на колонке с силикагелем в системе растворителей EtOAc:Me2CO = 1:1, получили 0,21 г 9(S)-N-(β,β-диацетилэтенил) эритромициламина со следующими физико-химическими константами:
ИК (CHCl3) см-1: 3500, 2950, 1725, 1610, 1550, 1450, 1375, 1320, 1170, 1080;
1H ЯМР (300 МГц, CDCl3) δ: 10.89  , 5.14 (1H, H-1''), 4.71 (1H, H-1'), 3.84 (1H, H-3), 3.70 (1H, H-5), 3.35 (3H, 3''-OCH3), 3.30 (1H, H-2'), 2.31 [6H, 3'-N (CH3)2] , 2.21 (1H, H-10), 1.99 (1H, H-8), 1.96
, 5.14 (1H, H-1''), 4.71 (1H, H-1'), 3.84 (1H, H-3), 3.70 (1H, H-5), 3.35 (3H, 3''-OCH3), 3.30 (1H, H-2'), 2.31 [6H, 3'-N (CH3)2] , 2.21 (1H, H-10), 1.99 (1H, H-8), 1.96  , 1.87 (3H,-COCH3), 1.20
, 1.87 (3H,-COCH3), 1.20  , 1.06 (3H, 10-CH3);
, 1.06 (3H, 10-CH3);
13C ЯМР (75 МГц, CDCl3) δ: 193.4  , 176.8 (C-1), 163.1
, 176.8 (C-1), 163.1  , 101.9 (C-1'), 95.2 (C-1''), 79.1 (C-5), 78.2 (C-3), 77.6 (C-4''), 70.7 (C-2'), 65.5 (C-9), 48.9 (3''-OCH3), 40.0 [3'-N(CH3)2], 32.9 (C-10), 33.4 (C-8), 28.2
, 101.9 (C-1'), 95.2 (C-1''), 79.1 (C-5), 78.2 (C-3), 77.6 (C-4''), 70.7 (C-2'), 65.5 (C-9), 48.9 (3''-OCH3), 40.0 [3'-N(CH3)2], 32.9 (C-10), 33.4 (C-8), 28.2  , 19.1
, 19.1  , 18.3 (8-CH3), 12.5 (10-CH3).
, 18.3 (8-CH3), 12.5 (10-CH3).
Пример 4
9(S)-N-(β,β-дицианоэтенил)эритромициламин
Смесь 9(S)-эритромициламина (0,5 г; 0,68 ммоль) и этоксиметиленмалондинитрила (0,18 г; 1,47 ммоль) в толуоле (20 мл) перемешивали при комнатной температуре в течение 30 минут. Охлажденную реакционную смесь упарили и полученные желтые кристаллы (0,65 г) очистили посредством хроматографии на колонке с силикагелем в системе растворителей CHCl3:MeOH = 9:1, с выходом 0,26 г 9(S)-N-(β,β-дицианоэтенил)эритромициламина со следующими физико-химическими константами:
ИК (CHCl3) см-1: 3500, 2950, 2200, 1725, 1625, 1550, 1450, 1375, 1320, 1175, 1050, 750;
1H ЯМР (300 МГц, CDCl3) δ: 8.22  , 7.13
, 7.13  , 5.04 (1H, H-1''), 4.59 (1H, H-1'), 3.82 (1H, H-3), 3.68 (1H, H-5), 3.29 (3H, 3''-OCH3), 3.23 (1H, H-2'), 2.31 [6H, 3'-N (CH3)2], 2.22 (1H, H-10), 1.94 (1H, H-8), 1.13 (3H, 8-CH3), 1.05 (3H, 10-CH3);
, 5.04 (1H, H-1''), 4.59 (1H, H-1'), 3.82 (1H, H-3), 3.68 (1H, H-5), 3.29 (3H, 3''-OCH3), 3.23 (1H, H-2'), 2.31 [6H, 3'-N (CH3)2], 2.22 (1H, H-10), 1.94 (1H, H-8), 1.13 (3H, 8-CH3), 1.05 (3H, 10-CH3);
13C ЯМР (75 МГц, CDCl3) δ: 177.1 (C-1), 160.4  , 132.2
, 132.2 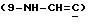 , 116.1 (-CN), 114.6 (-CN), 101.6 (C-1'), 95.4 (C- 1''), 80.8 (C-5), 78.9 (C-3), 77.3 (C-4''), 70.5 (C-2'), 74.4 (C-9), 49.0 (3"-OCH3), 40.0 [3'-N(CH3)2], 31.8 (C-10), 32.5 (C-8), 18.9 (8-CH3), 13.6 (10-CH3).
, 116.1 (-CN), 114.6 (-CN), 101.6 (C-1'), 95.4 (C- 1''), 80.8 (C-5), 78.9 (C-3), 77.3 (C-4''), 70.5 (C-2'), 74.4 (C-9), 49.0 (3"-OCH3), 40.0 [3'-N(CH3)2], 31.8 (C-10), 32.5 (C-8), 18.9 (8-CH3), 13.6 (10-CH3).
FAB-MC m/z 811.5 (M+H)+.
Пример 5
9(S)-N-(β-ацетил-β-карбэтоксиэтенил)эритромициламин
В соответствии с процедурой, описанной в примере 4, посредством реакции 9(S)-эритромициламина (0,5 г; 0,68 ммоль) и этил- α -(этоксиметилен)ацетоацетата (1,0 мл; 5,77 ммоль) в толуоле (20 мл) получили 0,54 г осадка в виде смолы. Посредством хроматографии на колонке с силикагелем в системе растворителей CHCl3: MeOH = 9:1, получили 0,29 г 9(S)-N-(β-ацетил-β-карбэтоксиэтенил)эритромициламина со следующими физико-химическими константами:
ИК (CHCl3) см-1: 3500, 2950, 1725, 1680, 1640, 1570, 1450, 1380, 1250, 1170, 1080;
1H ЯМР (300 МГц, CDCl3) δ: 11.15  , 7.74
, 7.74  , 5.11 (1H, H-1''), 4.74 (1H, H-1'), 4.21 (1H, H-3), 3.71 (1H, H-5), 4.18
, 5.11 (1H, H-1''), 4.74 (1H, H-1'), 4.21 (1H, H-3), 3.71 (1H, H-5), 4.18 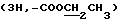 , 3.34 (3H, 3''-OCH3), 3.24 (1H, H-5), 2.45
, 3.34 (3H, 3''-OCH3), 3.24 (1H, H-5), 2.45  , 3.23 (1H, H-2'), 2.34 [6H, 3'-N(CH3)2], 2.22 (1H, H-10), 1.94 (1H, H-8), 1.27
, 3.23 (1H, H-2'), 2.34 [6H, 3'-N(CH3)2], 2.22 (1H, H-10), 1.94 (1H, H-8), 1.27  , 1.15 (3H, 8-CH3), 1.04 (3H, 10-CH3);
, 1.15 (3H, 8-CH3), 1.04 (3H, 10-CH3);
13C ЯМР (75 МГц, CDCl3) δ: 198.5 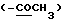 , 177.1 (C-1), 167.7
, 177.1 (C-1), 167.7  , 159.8
, 159.8  , 132.2
, 132.2  , 101.7 (C-1'), 95.3 (C-1''), 80.9 (C-5), 78.8 (C-3), 58.9
, 101.7 (C-1'), 95.3 (C-1''), 80.9 (C-5), 78.8 (C-3), 58.9 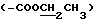 , 77.4 (C-4''), 70.5 (C-2'), 75.1 (C-9), 48.8 (3''-OCH3), 39.9 [3'-N (CH3)2], 32.1 (C-10), 33.2 (C-8), 30.4 (-COCH3), 18.1 (8-CH3), 14.0
, 77.4 (C-4''), 70.5 (C-2'), 75.1 (C-9), 48.8 (3''-OCH3), 39.9 [3'-N (CH3)2], 32.1 (C-10), 33.2 (C-8), 30.4 (-COCH3), 18.1 (8-CH3), 14.0  , 12.9 (10-CH3);
, 12.9 (10-CH3);
FAB-MC m/z 875.2 (М+H)+.
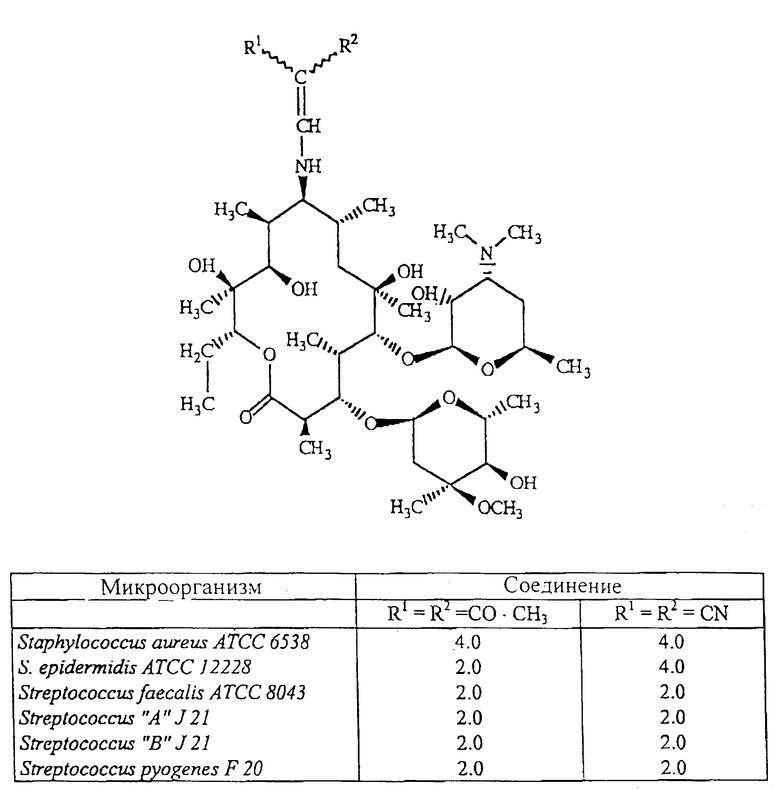
Изобретение относится к новым 9-N-этенильным производным 9 (S)-эритромициламина общей формулы I, где R1 и R2 одинаковы или различны и представляют собой нитрильную группу, карбоксильную группу формулы COOR3, где R3 представляет собой C1-C4-алкильную группу, или кетогруппу формулы COR4, где R4 представляет собой C1-C4-алкильную группу, или к из фармацевтически приемлемым солям присоединения неорганических или органических кислот, а также к способу их получения. Изобретение может быть полезно для приготовления фармацевтических препаратов для борьбы с бактериальными инфекциями. 2 с. и 16 з.п. ф-лы, 1 табл.


где R1 и R2 одинаковы или различны и представляют собой нитрильную группу, карбоксильную группу формулы СООR3, где R3 представляет собой С1 - С4 алкильную группу, или кетогруппу формулы СОR4, где R4 представляет собой С1 - С4 алкильную группу,
или их фармацевтически приемлемые соли присоединения неорганических или органических кислот.
где R1 и R2 одинаковы или различны и представляют собой нитрильную группу, карбоксильную группу формулы СООR3, где R3 представляет собой С1 - С4 алкильную группу, или кетогруппу формулы СОR4, где R4 представляет собой С1 - С4 алкильную группу,
их фармацевтически приемлемых солей присоединения неорганических или органических кислот, отличающийся тем, что 9(S)-эритромициламин общей формулы II
подвергают взаимодействию с производными этоксиэтилена общей формулы III
где R1 и R2 одинаковы или различны и представляют собой нитрил, карбоксильную группу формулы СООR3, где R3 представляет собой С1 - С4 алкильную группу, или кетогруппу формулы СОR4, где R4 представляет собой С1 - С4 алкильную группу,
в толуоле, ксилоле или других ароматических растворителях при температуре от 20 до 80oC, а затем, при необходимости, взаимодействию с неорганическими или органическими кислотами.
| Способ получения 4 @ -эпи-9-дезоксо-9 а-метил-9а-аза-9а-гомоэритромицина А | 1983 |
|
SU1272996A3 |
| Устройство для измерения качества осадка на периодически действующих фильтрах | 1969 |
|
SU292352A1 |
| Способ получения кардовых полиарилатов | 1974 |
|
SU507595A1 |
| 0 |
|
SU345627A1 |

