Изобретение относится к новым ингибиторам тромбина и промежуточным соединениям для их получения.
Тромбин относится к группе серинпротеаз и играет главную роль в качестве концевого фермента в системе свертывания крови. Как внутренняя, так и также внешняя система свертывания крови через несколько стадий усиления приводит к образованию тромбина из протромбина. Катализируемое тромбином расщепление фибриногена до фибрина затем вызывает свертывание крови и агрегацию тромбоцитов, которые со своей стороны за счет связывания тромбоцитного фактора 3 и фактора свертывания XIII, а также целого ряда высокоактивных медиаторов усиливают образование тромбина.
Образование и действие тромбина представляют собой главные факторы при возникновении как белых, артериальных, так и также красных, венозных, тромбов и поэтому являются потенциально эффективными точками воздействия для лечения. Ингибиторы тромбина, в противоположность гепарину, независимо от кофакторов способны полностью подавлять одновременно действия тромбина как в системе свертывания, так и также на тромбоциты. При обострении заболевания они могут предотвращать тромбоэмболию после чрескожной транслюминальной коронарной ангиопластики и лизис и служить в качестве антикоагулянтов при экстракорпоральном кровообращении (аппарат "сердце - легкие", гемодиализ). Они также вообще могут служить для профилактики тромбоза, например, после хирургических вмешательств.
Известно применение в качестве ингибитора тромбина производных метазамещенного фенилаланина (см. заявку WO 92/08709).
Соединения формулы (I) могут быть либо в виде самих соединений, либо в форме их солей с физиологически приемлемыми кислотами. Примерами таких кислот являются следующие: соляная кислоты, лимонная кислота, винная кислота, молочная кислота, фосфорная кислота, метансульфокислота, уксусная кислота, муравьиная кислота, малеиновая кислота, фумаровая кислота, малоновая кислота, янтарная кислота, гидроксиянтарная кислота, серная кислота, глутаровая кислота, аспарагиновая кислота, пировиноградная кислота, бензойная кислота, глюкуроновая кислота, щавелевая кислота, аскорбиновая кислота и ацетилглицин.
Новые соединения можно применять для лечения и профилактики всех заболеваний, при которых играет роль тромбин. Ими являются в особенности тромбоэмболические заболевания, как инфаркт миокарда, периферическая артериальная обтурация, глубокий венозный тромбоз, эмболия легких и инсульт. Сверх того, их можно применять для предотвращения повторной окклюзии после вскрытия артериальных сосудов механическими способами или лизиса.
Далее, вещества пригодны для предотвращения образования тромбина путем прямого ингибирования калликреина.
Их особое преимущество состоит в том, что они эффективны также при оральном введении.
Объектом изобретения являются, далее, следующие вещества формулы (II), которые представляют собой ценные промежуточные продукты для получения соединений формулы (I):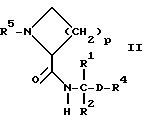
где R4, R2 и D имеют указанные для формулы (I) значения, и
R4 означает амидино-остаток в мета- или пара-положении к C(R1, R2) и
R5 означает водород, алкоксикарбонил, содержащий от одного до четырех атомов углерода, или фенилалкоксикарбонил, содержащий от одного до трех атомов углерода в алкоксильной части;
p означает 1 или 2.
Используемые в описании и в примерах сокращения имеют следующие значения:
Ac = ацетил
Ala = аланин
Am = амидино
(m или р) Amb = (мета- или пара-)амидинобензил
Asp = аспарагиновая кислота
Aze = азетидин-2-карбоновая кислота
Boc = трет-бутоксикарбонил
Bzl = бензил
Cbz = бензилоксикарбонил
Cha = циклогексилаланин
Chg = циклогексилглицин
DCC = дициклогексилкарбодиимид
Dch = дициклогексилаланин
DCM = дихлорметан
DIPEA = диизопропилэтиламин
ДМФ = диметилформамид
Dpa = дифенилаланин
Dpg = дифенилглицин
EDC = N-(3-диметиламинопропил)-N-этилкарбодиимид
Et = этил
Glu = глутаминовая кислота
Gly = глицин
Ham= гидроксиамидино
pHamb = пара-гидроксиамидинобензил
HOBT = гидроксибензотриазол
HOSu = гидроксисукцинимид
Hyp = гидроксипролин
Icс = изохинолинкарбоновая кислота
iPr = изопропил
Leu = лейцин
Man = миндальная кислота
Me = метил
(Me) Val = N-метил-валин
Msu = метионинсульфон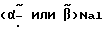 =
=  -нафтилаланин
-нафтилаланин
NBS = N-бромсукцинимид
Ngl = нафтилглицин
n-Pr = н-пропил
Ph = фенил
Phe = фенилаланин
Phg = фенилглицин
2-Phi = 2-пергидроиндолкарбоновая кислота
Pic = пипеколиновая кислота (пиперидин-2-карбоновая кислота)
pico = пиколил
pip = метиллиперидинил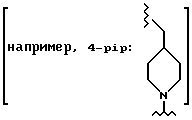
Pro = пролин
pym = метиллиримидил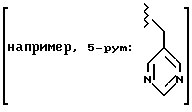
PyBrop = бром-трис(пирролидино)фосфонийгексафторфосфат
Pyr = 3,4-пирролин-2-карбоновая кислота
KT = комнатная температура
Tal = тиенилаланин
TBAB = тетрабутиламмонийбромид
tBu = трет.-бутил
TEA = триэтиламин
TEACl = тетраэтиламмонийхлорид
ТФУК = трифторуксусная кислота
Tgl = тиенилглицин
Tia = тиазолидин-4-карбоновая кислота
Tic = тетрагидроизохинолинкарбоновая кислота
Tol = толил
Trp = триптофан
Val = валин
Z = бензилоксикарбонил (= Cbz)
Примеры
А. Общие методики
A.I. Удаление и введение защитных групп
A. I.а. Защитные группы отщепляют согласно способам, описанным Гроссом и Мейенхофером (Е. Gross, J. Meienhofer "Пептиды; анализ, синтез, биология", первое издание, том 3, Академик Пресс, Нью-Йорк, 1981).
A. I. б. Бензилоксикарбонильные (Cbz) защитные группы отщепляют либо гидрогенолитически в стандартных реакционных условиях, либо с помощью фтороводорода согласно способу, описанному J. М. Stewart, J.D.Young "Синтез пептидов в твердой фазе", второе издание, Пирс Кемикал Компани, 1984).
A. I. в. Если защищенная молекула содержит только трет.-бутоксикарбонильные (Boc) защитные группы, то их отщепляют с помощью хлороводорода в диоксане, соответственно, хлороводорода в метиленхлориде, соответственно, трифторуксусной кислоты в метиленхлориде в стандартных реакционных условиях (см. М. Bodansky и А. Bodansky "Практика синтеза пептидов", изд. Спрингер, 1984).
A.II. Общие методики гидролиза сложноэфирных групп,
A.II.а. 1 ммоль Сложного эфира при 0oC вносят в тетрагидрофуран (4 мл на ммоль). Затем добавляют 1,2 эквивалента гидроксида лития (1 М раствор) и смесь перемешивают в течение ночи при комнатной температуре. После обработки водой получают соответствующую кислоту.
A.II.б. 1 ммоль Сложного эфира при 0oC вносят в метанол (4 мл на ммоль). Затем добавляют 1,2 эквивалента гидроксида лития (1 М раствор) и смесь перемешивают в течение ночи при комнатной температуре. После обработки водой получают соответствующую кислоту.
A. II.в. 1 ммоль Сложного эфира в 2 мл 2 н. соляной кислоты перемешивают в течение ночи при комнатной температуре. Продукт обрабатывают водой.
A.III. Общая методика амидирования
A. III. 1. Следуя методике Vieweg и др. (Н. Vieweg и др., Pharmacia, 39, 226 (1984)), из нитрилов получают амидины, N-гидроксиамидины и N-аминоамидины следующим образом:
1 Эквивалент нитрила растворяют в смеси пиридина с триэтиламином в соотношении 10:1 (примерно 20-30 мл на грамм вещества). Затем раствор насыщают газообразным сероводородом и оставляют стоять в течение ночи при комнатной температуре. После этого реакционную смесь при перемешивании выливают в солянокислую смесь воды со льдом, выпавший осадок отсасывают, дополнительно промывают большим количеством воды и затем высушивают. Вещество растворяют в ацетоне (примерно 20-30 мл на грамм вещества). После добавления метилиодида (1 мл на грамм вещества) раствор оставляют стоять в течение ночи. Путем добавления деэтилового эфира осаждают гидроиодид сложного S-метил-тиоимидоэфира и с целью очистки еще раз переосаждают из метанола с помощью диэтилового эфира.
Соль вносят в абсолютный метанол (примерно 30 мл на грамм вещества). После добавления ацетата аммония (для синтеза N- гидроксиамидинов используют гидроксиаммонийацетат, соответственно, гидроксиаммонийхлорид, а для синтеза N-аминоамидинов применяют гидразинийацетат, соответственно, гидразинийхлорид) смесь перемешивают в течение ночи при комнатной температуре. После отфильтровывания суспензии часть растворителя удаляют в вакууме и амидиногидроиодид осаждают путем добавления эфира и отсасывают. Затем сырой продукт очищают с помощью высокоэффективной жидкостной хроматографии с обращенной фазой.
A.III.2. Амидирование также можно осуществлять с помощью реакции Пиннера (D. Neilson в книге Patai "Химия амидина и имидата", с. 385 - 489, John Wiley and Sons, Нью-Йорк, 1975; R. Roger, D. Neilson, Chem. Rev., 61 179 (1961); далее, см. пример 2).
A. III.3. Другая возможность амидирования состоит в превращении нитрильной группы в гидроксиамидиногруппу с помощью гидроксиламингидрохлорида и последующем гидрировании с помощью водорода в присутствии никеля Ренея (соответственно, с помощью водорода в присутствии палладия-на-угле) с получением амидина. 10 ммоль Нитрильного производного растворяют в 100 мл метанола и после добавления трех эквивалентов гидроксиламингидрохлорида и 4,5 эквивалентов триэтиламина перемешивают при комнатной температуре вплоть до полного превращения. Затем реакционную смесь концентрируют и остаток обрабатывают дихлорметаном. Органическую фазу промывают водой (pH 5-6), сушат на сульфатом натрия и концентрируют.
Остаток растворяют в 100 мл 5%-ного метанольного раствора уксусной кислоты и после добавления никеля Ренея (можно также 10%-ного палладия-на угле) гидрируют в атмосфере водорода. После полного превращения эдукта катализатор отфильтровывают и фильтрат концентрируют. Продукт в случае необходимости очищают либо путем разделения с помощью колоночной хроматографии на силикагеле, либо путем разделения с помощью высокоэффективной жидкостной хроматографии с обращенной фазой.
A.IV. Общая методика гуанидирования аминов
A.IV.1. Получение свободных гуанидинов.
Свободные гуанидины синтезируют, исходя из соответствующих аминов в качестве предшественников, аналогично методике Миллера и др., соответственно, Мошера и др. (A.E. Miller, J.J. Bischoff, Synthesis, 777 (1986); К. Kim, Y. - T. Lin, H.S. Mosher, Tetrahedron Letters, 29, 3183 (1988)).
A.IV.1.a. 1 Эквивалент карбоната калия и 1 эквивалент амина растворяют в 10 мл воды. Затем порциями при интенсивном перемешивании добавляют 1 эквивалент аминоиминометансульфокислоты. После перемешивания в течение двадцати четырех часов реакционную смесь фильтруют. Отфильтрованным твердым веществом является гуанидин.
A.IV.1.б. Эквимолярные количества амина и аминоиминометансульфокислоты в абсолютном метаноле (1 мл на ммоль) перемешивают при комнатной температуре вплоть до образования прозрачного раствора. Затем растворитель удаляют в вакууме и сырой продукт очищают с помощью высокоэффективной жидкостной хроматографии с обращенной фазой.
A.IV.2. Получение алкоксикарбонилгуанидинов
Превращение в алкоксикарбонилгуанидины осуществляют по аналогии со следующими, описанными в литературе методиками:
1. R.J. Bergeron. J.S. MeManis J. Org. Chem. 1987, 52, 1700
2. R. Dubey, S. Abuzar. S. Sharma, R.K. Chatterjee J. Hed. Cem. 1985, 28, 1748
3. S. Shawkat, S. Sharma Synthesis 1992, 664
4. A.S. Vendrini, P. Lucietto, G. Fossati, C. Giordani Tetrahedron Lett. 1992, 33, 6541
5. Z.P. Tian, P. Edwards, R.W. Roeske int. J. Pept. Prot. Res. 1192, 40, 119
A.V. Общие способы этерификации с образованием сложного эфира
A. V. 1. 1 Эквивалент карбоновой кислоты вместе с 1,1 эквивалента гидрохлорида N'-(3-диметиламинопропил)-N- этилкарбодиимида, двумя эквивалентами спирта и каталитическим количеством диметиламинопиридина в метиленхлориде перемешивают в течение ночи при комнатной температуре. Затем раствор разбавляют метиленхлоридом, экстрагируют 20%-ным раствором гидросульфата натрия, сушат и концентрируют в вакууме.
A. V.2. Кипятят 1 эквивалент карбоновой кислоты вместе с соответствующим спиртом и каталитическим количеством п-толуолсульфокислоты в хлороформе (можно также в толуоле). После полного превращения (контроль с помощью тонкослойной хроматографии) раствор промывают с помощью насыщенного раствора гидрокарбоната натрия и раствора хлорида натрия, сушат и выпаривают в ротационном испарителе.
Б. Общая стратегия синтеза
Соединения согласно изобретению можно получать несколькими путями:
1. Исходя из соответствующих защищенных A-производных W-A-OH можно последовательно, согласно известным способам синтеза, вводить в реакцию сочетания структурные элементы H-B-COOW' (W' = алкил) и H-N(H)-C(R1R2)-(CH2)m-D-(NH)n-C(NH)NHR3′ , которые возможно находятся в пригодным образом защищенной форме (см. схему 1). Как обычно в химии пептидов, между отдельными стадиями сочетания нужно целенаправленно отщеплять защитные группы реакционных центров последующей реакции сочетания. Все используемые структурные элементы либо имеются в продаже, либо их можно синтезировать по известным из литературы методикам, соответственно, по аналогии с
2. Синтезы можно осуществлять также в обратной последовательности путем сочетания H-N(H)-C(R1R2)-(CH2)m-D-(NH)n-C(NH)NHR3′ (где R3′ означает пригодную защитную группу) с пригодным образом защищенными W-B-COOH и затем A-производными W-A-OH (см. схему II).
3. Гуанидино-, амидино-, N-гидроксиамидино-, соответственно, N-амино- амидино-группы либо вводят в защищенной форме (протонированными или снабженными пригодными защитными группами) со структурным элементом H-N(R1)-C(R1R2)-(CH2)m-D-(NH)n-C(NH)NHR3′ получении активных веществ и затем защитные группы удаляют, либо, однако, получают после сочетания структурных элементов в стадии образования W-A-B-CO-N(H)-C(R1R2)-(CH2)m-D-NH2 путем гуанидирования, соответственно, W-A-B-CO-N(H)- C(R1R2)-(CH2)m-D-CN путем амидирования, N-гидроксиамидирования или N-аминоамидирования.
Список литературы, относящейся к химии пептидов:
1. Е. Gross, J. Meienhofer "Пептиды; анализ, синтез, биология", первое издание, том 1, Академик Пресс, Нью-Йорк, 1979
2. Е. Gross, J. Meienhofer "Пептиды; анализ, синтез, биология", первое издание, том 2, Академик Пресс, Нью-Йорк, 1980
3. E. Gross, J. Meienhofer "Пептиды: анализ, синтез, биология", первое издание, том 3, Академик Пресс, Нью-Йорк, 1981
4. M. Deffner, E. Jaeger, P. Stelzel, P. Thamm, G. Wendenberg, E. Wbnsch в книге Губен-Вейл "Методы химии", четвертое издание том XV/1, редактор E. Wbnsch, изд. Georg Thieme, Штутгарт, 1974
5. M. Deffner, E. Jaeger, P. Stelzel, P. Thamm, G. Wendenberg, E. Wunsch в книге Губен-Вейл "Методы химии", четвертое издание том XV/2, редактор E. Wunsch, изд. Georg Thieme, Штутгарт, 1974
6. М. Bodansky и A. Bodansky "Практика синтеза пептидов", изд. Спрингер, 1984.
Б.1. Соединение структурных элементов (W-) A-ОН H-B-CO-N(H)-C(R1R2)-(CH2)m-D-(NH)n-C(NH)NHR3′ или H-B-CO-N(H)-C(R1R2)-(CH2)m-D-CN согласно формуле (I) (R1, R2, означают водород, алкил) и схеме II:
Сначала из W-B-COOH (W означает защитную группу, предпочтительно Boc или Cbz) и амина H-N(H)-C(R1R2)-(CH2)m-D-(NH)n-C(NH)NHR3′ соответственно H-N(H)-C(R1R2-(CH2)m-D-CN получают гидрохлорид 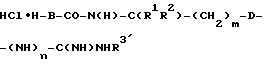 соответственно HCl • H-B-CO-N(H)-C(R1R2)-(CH2)m-D-CN в стандартных для сочетания пептидов условиях (см. М. Bodansky и A. Bodansky "Практика синтеза пептидов", изд. Спрингер, 1984) с последующим отщеплением защитных групп. Гидрохлорид затем превращают в индивидуальные соединения (согласно общей формуле) следующим образом:
соответственно HCl • H-B-CO-N(H)-C(R1R2)-(CH2)m-D-CN в стандартных для сочетания пептидов условиях (см. М. Bodansky и A. Bodansky "Практика синтеза пептидов", изд. Спрингер, 1984) с последующим отщеплением защитных групп. Гидрохлорид затем превращают в индивидуальные соединения (согласно общей формуле) следующим образом:
Б.I.a.W-A-OH означает защищенную аминокислоту (согласно общей формуле)
1 Эквивалент защищенной аминокислоты W-A-OH и 1,1 эквивалента гидрохлорида 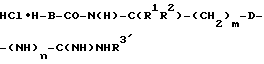 , соответственно, HCl•H-B-CO-N(H)-C(R1R2)-(CH2)m-D-CN (согласно общей формуле (I)) по стандартным для сочетания пептидов методикам превращают в целевой продукт. Если E означает циангруппу, то согласно A. III.1-3 переводят в амидино- или гидроксиамидиногруппу. Затем имеющиеся защитные группы отщепляют по стандартным способам.
, соответственно, HCl•H-B-CO-N(H)-C(R1R2)-(CH2)m-D-CN (согласно общей формуле (I)) по стандартным для сочетания пептидов методикам превращают в целевой продукт. Если E означает циангруппу, то согласно A. III.1-3 переводят в амидино- или гидроксиамидиногруппу. Затем имеющиеся защитные группы отщепляют по стандартным способам.
Б. I. б. A-ОН означает N-ацил-AK (AK представляет собой указанные в значении A в общей формуле (I) аминокислоты: ацил означает карбоксиалкилкарбонил, содержащий от одного до шести атомов углерода в алкильной части, алкилкарбонил, содержащий от одного до двенадцати атомов углерода в алкильной части, фенилалкилкарбонил, содержащий от одного до четырех атомов углерода в алкильной части, 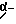 , соответственно, β- нафтилалкилкарбонил, содержащий от одного до четырех атомов углерода в алкильной части).
, соответственно, β- нафтилалкилкарбонил, содержащий от одного до четырех атомов углерода в алкильной части).
Б. I. б. 1. Сначала защищенную аминокислоту W-A-OH как описано в Б.I.а. сочетают с описанным там гидрохлоридом R3′ должен представлять собой обычную защитную группу). Затем удаляют защитную для концевой амино-функции группу аминокислоты (защитную группу нужно отщеплять ортогонально к R3′ и последнюю вводят во взаимодействие с карбоновыми кислотами
(согласно общей формуле) в стандартных для сочетания пептидов условиях с получением целевого продукта. Для высвобождения амидиногруппы, N-аминоамидиногруппы, N-гидроксиамидиногруппы или гуанидиногруппы защитную группу (если желательно) удаляют в стандартных условиях (см. раздел A.I.). Если E означает циангруппу, то ее, согласно A.III.1-3, переводят в амидиногруппу или гидроксиамидиногруппу.
Б.I.б.2. Сначала аминокислоту с концевой аминогруппой H-A-OCH3 амино-концом вводят во взаимодействие с карбоновой кислотой (согласно общей формуле (I)) в стандартных для сочетания пептидов условиях с получением N-ацилированного сложного эфира аминокислоты и затем сложноэфирную группу гидролизуют. После этого ацилированную амино кислоту, как описано в Б.I.б.1., сочетают с гидрохлоридом;  соответственно, HCl • H-B-CO- N(H)-C(R1R2)-(CH2)m-D-CN в стандартных условиях.
соответственно, HCl • H-B-CO- N(H)-C(R1R2)-(CH2)m-D-CN в стандартных условиях.
Если E означает циангруппу, то ее согласно A.III.1-3, переводят в амидиногруппу, соответственно, гидроксиамидиногруппу. Затем защитные группы отщепляют.
Б. I.в. A-OH означает N-алкил-AK (AK представляет собой указанные в значении A в общей формуле (I) аминокислоты: алкил означает алкил, содержащий от одного до двенадцати атомов углерода, фенилалкилен, содержащий от одного до четырех атомов углерода в алкиленовой части, карбоксиалкилен, содержащий от одного до шести атомов углерода в алкиленовой части, α- , соответственно, β- нафтилалкилен, содержащий от одного до четырех атомов углерода в алкиленовой части):
Один путь синтеза (путь синтеза 1) представляет собой получение алкилированного структурного элемента A-OH (или A-B-COOH) с последующим сочетанием со структурным элементом H-B-CO-N(H)-C(R1R2)-(CH2)m-D-(NH)n-C(NH)NHR3′ (соответственно, H-N(H)-C(R1R2)-(CH2)m-D-(NH)n-C(NH)NHR3′ или H-B-CO-N(H)-C(R1R2)-(CH2)m-D-CN (соответственно, H-N(H)-C(R1R2)-(CH2)m-D-CN). Альтернативный путь (путь синтеза 2) представляет собой синтез структурного элемента H-AK-B-CO-N(H)-C(R1R2)-(CH2)m-D-(NH)n-C(NH)NHR3′ соответственно, H-AK-B-CO-N(H)-C(R1R2)-(CH2)m-D-CN с последующим алкилированием концевой аминогруппы. Имеющаяся амидиногруппа, соответственно, гуанидиногруппа должна быть пригодным образом защищена.
Если E означает циангруппу, то ее, согласно A.III.1-3, переводят в амидиногруппу, соответственно, гидроксиамидиногруппу.
Путь синтеза 2:
Б. I. в. 1. 1 ммоль H-AS-B-CO-N(H)-C(R1R2)-(CH2)m-D-(NH)n-C(NH)NHR3′ соответственно, H-AS-B-CO-N(H)-C(R1R2-(CH2)m-D-CN растворяют в 10 мл метанола. После добавления 1 ммоль тетраэтиламмонийхлорида, 0,7 ммоль цианборгидрида натрия и 1,05 ммоль альдегида реакционную смесь перемешивают в течение ночи. Растворитель удаляют в вакууме и остаток обрабатывают этилацетатом. Органическую фазу промывают два раза водой и один раз насыщенным раствором хлорида натрия и сушат над сульфатом натрия. После удаления растворителя сырой продукт очищают с помощью высокоэффективной жидкостной хроматографии с обращенной фазой.
Если E означает циангруппу, то ее, согласно A.III.1-3, переводят в амидиногруппу, соответственно, гидроксиамидиногруппу.
Б. I.в.2. 1 ммоль H-AK-B-CO-N(H)-C(R1R2)-(CH2)m-D-(NH)n-C(NH)NHR3′ соответственно, H-AK-B-CO-N(H)-C(R1R2)-(CH2)m-D-CN вводят в ацетонитрил вместе с 2,5 эквивалентами карбоната калия. После добавления алкилирующего агента реакционную смесь перемешивают при 60oC вплоть до полного превращения эдукта. После охлаждения смесь обрабатывают водой и продукт очищают с помощью высокоэффективной жидкостной хроматографии с обращенной фазой.
Если E означает циангруппу, то ее, согласно A.III.1-3, переводят в амидиногруппу, соответственно, гидроксиамидиногруппу.
Путь синтеза 1:
Алкилированный структурный элемент A-OH (где A означает N- алкил-AK) получают аналогично известной из литературы методике Ивасаки и др. (G. Iwasaki и др. , Chem. Pharm. Bull., 31, 280 (1989), и Chem. Lett., 1691 (1988)). Нижеследующая методика основана на этой, известной из литературы методике:
Б. I. в. 3. 1,5 Эквивалента H-AK-OCH3 или H-AK-B-COOCH3 вместе одним эквивалентом алкилирующего агента и двумя эквивалентами карбоната аммония в смеси нитрометана с водой перемешивают в течение четырех дней при 60oC. Смесь обрабатывают водой и продукт очищают с помощью хроматографии. После защиты N-алкиламинофункции с помощью пригодной защитной группы омыляют сложноэфирную функцию аминокислоты и продукт превращают аналогично Б.I.а.
Б.I.в.4. Структурный элемент N-алкил-AK-OCH3 или N-алкил-AK-B-COOCH3 можно также получать путем восстановительного аминирования из H-AK-OCH3 или H-AK-B-COOCH3 и альдегида.
Б. I. г. A-OH означает N-замещенный аминокарбонил-AK (AK представляет собой указанные для A в общей формуле (I) аминокислоты; замещенный аминокарбонил означает алкиламинокарбонил, содержащий от одного до шести атомов углерода в алкильной части, фенилалкиламинокарбонил, содержащий от одного до четырех атомов углерода в алкильной части).
1 Эквивалент H-AK-B-CO-N(H)-C(R1R2)-(CH2)m-D-(NH)n-C(NH)NHR3′ соответственно, H-AK-B-CO-N(H)-C(R1R2)-(CH2)m-D-CN вводят во взаимодействие с различными изоцианатами в стандартных реакционных условиях (Arnold и др., Chem. Rev. , 57, 47 (1957)) с получением соответствующих производных мочевины.
Если E означает циангруппу, то ее, согласно A.III.1-3, переводят в амидиногруппу, соответственно, гидроксиамидиногруппу.
Б. II. Связывание амина H-N(H)-C(R1R2)-(CH2)m-D-(NH)n-C(NH)NHR3′ соответственно, H-N(H)-C(R1R2)-(CH2)m-D-CN со структурным элементом A-B-COOH (согласно общей формуле (I) и схеме 1).
Сначала синтезируют структурный элемент A-B-COOH аналогично общей методике, описанной в разделе Б.1. Затем амин H-N(H)-C(R1R2)-(CH2)m-D-(NH)n-C(NH)NHR3′ сочетают со структурным элементом A-B-CO-OH согласно нижеописанному:
Б. II. a. R2 означает водород, алкил, содержащий от одного до четырех атомов углерода, фенилалкилен, содержащий от одного до четырех атомов углерода в алкиленовой части; n означает ноль.
Б. II. а. 1. Структурный элемент (возможно замещенный) A-B-COOH в стандартных для сочетания пептидов условиях вводят во взаимодействие с амином H-N(H)-C(R1R2)-(CH2)m-D-(NH)n-C(NH)NHR3′ (где R3′ означает алкоксикарбонил, содержащий от одного до двадцати атомов углерода в алкоксильной части). После обработки водой сырой продукт очищают с помощью высокоэффективной жидкостной хроматографии с обращенной фазой. Вещества, где R3 означает водород, можно получать путем стандартных способов удаления защитных групп.
Б. II. а. 2. Структурный элемент (возможно замещенный) A-B-COOH в стандартных для сочетания пептидов условиях вводят во взаимодействие с амином H-N(H)-C(R1R2)-(CH2)m-D-CN (где R1, R2, D, m имеют значения, указанные в случае общей формулы (I)). После обработки водой сырой продукт очищают с помощью высокоэффективной жидкостной хроматографии с обращенной фазой. Полученный нитрил, как описано в разделе A.III., переводят в амидин (R3 означает водород), N-гидрокси-амидин (R3 означает гидроксил), N-аминоамидин (R4 означает аминогруппу).
Б. II. б. R1 означает водород, алкил, содержащий от одного до четырех атомов углерода; R2 означает водород, алкил, содержащий от одного до четырех атомов углерода, фенилалкилен, содержащий от одного до четырех атомов углерода в алкиленовой части: n означает 1.
Б. II. б. 1. Структурный элемент (возможно защищенный) A-B-COOH в стандартных для сочетания пептидов условиях вводят во взаимодействие с амином H-N(H)-C(R1R2)-(CH2)m-D-(NH)n- C(NH)NHR3 (где R3 означает алкоксикарбонил, содержащий от одного до двадцати атомов углерода в алкоксильной части). После обработки водой сырой продукт очищают с помощью высокоэффективной жидкостной хроматографии с обращенной фазой. Вещества, где R3 означает водород, можно получать путем стандартных способов удаления защитных групп.
Б. II. б. 2. Структурный элемент (возможно защищенный) A-B-COOH в стандартных для сочетания пептидов условиях вводят во взаимодействие с амином H-N(H)-C(R1R2)-(CH2)m-D-NHW (где W означает ортогональную к одновременно имеющимся защитным группам защитную группу). После обработки водой сырой продукт очищают с помощью высокоэффективной жидкостной хроматографии с обращенной фазой. Полученный защищенный амин высвобождают с помощью стандартных способов удаления защитных групп и, как описано в разделе A.IV, переводят в гуанидин (где R3 означает водород).
Б.II.в. 1. R3 означает -CO-X14 (где X14 означает алкил, аралкил)
Структурный элемент A-B-CO-N(H)-CR1(COOH-(CH2)m-D-(NH)n-C(NH)NHR3′ получают согласно стандартным способам COX14-Функцию затем получают по реакции Дакин-Веста в стандартных реакционных условиях (W. Steglich, G. Hofle, Angew. Chem. Int. Ed., 8, 981 (1969); W. Steglich, G. Hofle, Chem. Ber., 102. 883 (1969)). В случае этого превращения R3' должен представлять собой алкоксикарбонильную группу (предпочтительно Вое или Cbz). Альтернативно этому в реакции Дакин-Веста можно использовать также A-B-CO-N(H)-CR1(COOH)-(CH2)m-D-CN. В этом случае затем нитрильную функцию, согласно A.III.1-3, переводят в амидиногруппу, соответственно, гидроксиамидиногруппу. Общая методика для реакции Дакин-Веста следующая:
1 Эквивалент защищенного на амино-конце трипептида вместе с 2,5 эквивалентами триэтиламина, 5 эквивалентами ангидрида [X14- C(O)-O-(O)C-X14, согласно общей формуле (I)] и 0,1 эквивалента диметиламинопиридина перемешивают при 50 - 60oC вплоть до полного прекращения выделения диоксида углерода. Затем реакционную смесь вместе с насыщенным раствором карбоната натрия перемешивают в течение двух часов при 60oC. Смесь распределяют между этилацетатом и насыщенным раствором гидрокарбоната натрия и органическую фазу после этого экстрагируют два раза с помощью насыщенного раствора гидрокарбоната натрия и один раз с помощью 20%-ного раствора гидросульфата натрия, сушат над сульфатом натрия и выпаривают в ротационном испарителе. Сырой продукт очищают с помощью высокоэффективной жидкостной хроматографии с обращенной фазой.
В. Получение эдуктов
B.1. Получение N-трет.-бутоксикарбонил-(D)-фенилаланил-O-сукцинимида
1 Эквивалент N-трет.-бутоксикарбонил-(D)-фенилаланина вместе с 1,05 эквивалента гидроксисукцинимида и 1,05 эквивалента дициклогексилкарбодиимида в ацетонитриле (2,5 мл на ммоль) перемешивают в течение ночи при комнатной температуре. Затем суспензию отфильтровывают и фильтрат выпаривают в ротационном испарителе. Получают продукт почти с количественным выходом.
В.2. Получение N-трет.-бутоксикарбонил-(D,L)-дифенилаланина N-трет.-бутоксикарбонил-(D, L)-дифенилаланин получают по методике Каккара и др. (L. Cheng, C. A. Goodwin, M. F. Schully, V.V. Kakkar, J. Med. Chem., 35, 3364 (1992)).
В.3. Получение N-трет.-бутоксикарбонил-(D,L)-дициклогкесилаланина
1 ммоль N-трет. -бутоксикарбонил-(D,L)-дифенилаланина в 12 мл метанола вместе с каталитическим количеством 5%-ного родия на оксиде алюминия гидрируют при давлении 5 бар. После фильтрации и удаления растворителя в вакууме получают продукт с количественным выходом.
В. 4. Получение трет.-бутоксикарбонил-1-(D,L)- тетрагидроизохинолинкарбоновой кислоты трет. -Бутоксикарбонил-1- (D,L)-тетрагидроизохинолинкарбоновую кислоту получают по методике Р.Т. Шумана и др. (R.T. Shuman и др., J. Med. Chem., 36, 314 (1993)).
В.5. Получение N-бензилоксикарбонил-(D)-фенилаланилпролил-O- сукцинимида
I. 30 г N-Бензилоксикарбонил-(D)-фенилаланина, 11,54 г гидроксисукцинимида, 20,68 г дициклогексилкарбодиимида и 300 мл диметоксиэтана перемешивают в течение ночи при комнатной температуре. Затем суспензию отфильтровывают, фильтрат выпаривают в ротационном испарителе и остаток растворяют в 200 мл ацетонитрила. Выпавшую в осадок дициклогексилмочевину отфильтровывают и фильтрат концентрируют. Получают 40 г сложного сукцинимидоэфира (твердое вещество белого цвета).
II. 40 г N-Бензилоксикарбонил-(D)-фенилаланил-O-сукцинимида, 17,31 г пролина, 12,63 г гидрокарбоната натрия, 225 мл воды и 225 мл диметоксиэтана перемешивают в течение ночи при комнатной температуре (выделение газа). Затем диметоксиэтан удаляют в вакууме и в оставшемся водном растворе устанавливают pH-значение, равное двум, с помощью 1 н. соляной кислоты. Выделившееся масло экстрагируют метиленхлоридом. Объединенные метиленхлоридные экстракты промывают насыщенным раствором хлорида натрия и после высушивания над сульфатом натрия выпаривают в ротационном испарителе. Получают 39,7 г N-Бензил-оксикарбонил-(D)- фенилаланилпролина (твердое вещество белого цвета).
III. 39,7 г N-Бензилоксикарбонил-(D)-фенилаланилпролина, 11,53 г гидроксисукцинимида, 20,66 г дициклогексилкарбодиимида и 400 мл диметоксиэтана перемешивают в течение ночи при комнатной температуре. На следующий день отфильтровывают дициклогексилмочевину, фильтрат концентрируют и остаток обрабатывают с по мощью 300 мл ацетонитрила. Еще раз выпавшую в осадок дициклогексилмочевину отфильтровывают и фильтрат концентрируют в вакууме. Получают 48,92 r сложного гидроксисукцинимидоэфира.
В. 6. Получение защищенных с помощью трет.-бутоксикарбонила производных фенилаланина
Поскольку аминокислот H-A-OH, соответственно, Boc-A-OH нет в продаже, их получают по аналогии с известными из литературы способами (обзор: Губен-Вейл, том E 16d/часть 1 S 406 и последующие).
Зачастую используемыми эдуктами для производных аланина являются этиловый эфир бензофенонаминоуксусной кислоты, диэтиловый эфир ацетамидомалоновой кислоты и этиловый эфир изонитрилуксусной кислоты.
Например, следует упомянуть:
1 Эквивалент дифенилглицинимина, 3 эквивалента карбоната калия и соответствующий бензилбромид (возможно также бензилхлорид или бензилиодид) в ацетонитриле кипятят в течение ночи. После охлаждения реакционную смесь отфильтровывают и фильтрат концентрируют. Затем остаток перемешивают в 1 н. соляной кислоте вплоть до полного расщепления имина. После этого водную фазу экстрагируют этилацетатом, подщелачивают ее с помощью карбоната натрия и экстрагируют этилацетатом. Объединенные органические экстракты сушат над сульфатом натрия и выпаривают в ротационном испарителе. Остаток, соответствующее производное фенилаланина, в стандартных условиях защищают на амино-конце с помощью защитной группы (предпочтительно трет.-бутоксикарбонильной или бензилоксикарбонильной группы).
В. 7. Получение защищенных с помощью трет.-бутоксикарбонила производных глицина
Различные производные глицина получают, например, исходя из этилового эфира изонитрилуксусной кислоты и соответствующего кетона (см. Н. - J. Prgtorius, J. Fiossdorf, М. - R. Kula, Chem. Ber., 108. 3079 (1995)).
N-трет. -Бутоксикарбонил-суберилглицин синтезируют аналогично известному из литературы способу (О.Р. Goel и др., Tetrahedron Lett., 34, 953 (1993)).
N-трет.-Бутоксикарбонил-(3-фенил)-пролин синтезируют аналогично методике J.Y.L. Chung и др. (J.Y.L. Chung и др., J.Org. Chem., 55, 270 (1990)).
Тетралинилглицин получают исходя из 1,2-дигидронафталина, причем 1,2-дигидронафталин сначала с помощью бромоводорода переводят в
1-тетралилибромид (аналогично J. Med. Chem., 37, 1586 (1994)). Затем бромид вводят во взаимодействие с диэтиловым эфиром ацетамидомалоновой кислоты, гидролитически расщепляют и полученную α- аминокислоту в стандартных условиях переводят в защищенную трет.-бутоксикарбонильной группой форму.
В. 8. Получение N-трет.-бутоксикарбонил-(D)- α- метил)- циклогексилаланина N-трет.-Бутоксикарбонил-(D)- α- метил)- циклогексилаланин получают путем гидрирования (D)-( α- метил)- фенилаланина и последующего введения трет. -бутоксикарбонильной защитной группы. Другими возможностями синтеззамещенных аминокислот являются синтез по способу Бухерера, при котором исходят из кетонов, а также  алкилирование
алкилирование  аминокислот.
аминокислот.
В.9. Получение производных гидроксиуксусной кислоты
Производные гидроксиуксусной кислоты получают либо аналогично S.Bajusz (международная заявка на патент 93/18060), либо исходя из соответствующих производных метилового эфира уксусной кислоты путем  гидроксилирования с помощью реагента Дэвиса (F. A. Davis, L.C. Vishwakarma, J.M. Billmers, J. Org. Chem., 49, 3241 (1984)).
гидроксилирования с помощью реагента Дэвиса (F. A. Davis, L.C. Vishwakarma, J.M. Billmers, J. Org. Chem., 49, 3241 (1984)).
В. 10. n-(2-Аминоэтил)бензонитрил:
Получают согласно европейскому патенту 445796
В.11. п-Цианобензиламин:
В. 11. a. 200 г 4-Цианобензилбромида (1,02 моль), 700 мл толуола, 200 г азида натрия (3,07 моль), 32,9 г тетрабутиламмонийбромида и 700 мл воды перемешивают в течение ночи при комнатной температуре. Затем обе фазы разделяют и толуольную фазу еще раз дополнительно промывают водой. Объем растворителя уменьшают до 1/5 в вакууме.
В. 11. б. 267,6 г Трифенилфосфина (1,02 моль) при 10oC вносят в 500 мл тетрагидрофурана. К этому раствору медленно прикапывают растворенный в 165 мл тетрагидрофурана азид (выделение азота). По окончании добавления медленно добавляют 27,6 мл воды (1,53 моль) и реакционную смесь перемешивают при комнатной температуре в течение сорока восьми часов. Раствор затем выпаривают в ротационном испарителе и остаток обрабатывают охлажденной 3 н. соляной кислотой (1 литр). Выпавшее в осадок твердое вещество отсасывают и фильтрат промывают толуолом вплоть до полного удаления трифенилфосфиноксида. Затем кислую водную фазу подщелачивают до pH-значения, равного девяти, с помощью твердого карбоната натрия, и выпавшее в осадок твердое вещество отфильтровывают и фильтрат экстрагируют диэтиловым эфиром. Твердое вещество растворяют в диэтиловом эфире и полученный раствор вместе с эфирными экстрактами сушат. Затем объем эфира уменьшают и гидрохлорид осаждают путем пропускания газообразного хлороводорода. Соль отфильтровывают, промывают диэтиловым эфиром и высушивают на воздухе (соль сублимируется в высоком вакууме).
Выход составляет 137,6 г.
В. 11. в. п-Цианобензиламин получают из п-цианобензилбромида также через фталимид с последующим расщеплением с помощью гидразингидрата при получении хороших выходов.
Также пригоден синтез через соль уротропиния (W. Walter и др., Апп., 660, 60 (1962)).
В.12. м-Цианобензиламин:
Получают согласно литературным данным (Pharmacia, 33, 15 (1978)).
В.13. (D,L)-1-(4-Цианофенил)этиламин:
В.13.а. N-(п-Цианобензил)бензофенонимин
К раствору из 150 г (0,8 моль) 97%-ного бензофенонимина и 144,8 г (0,74 моль) п-циано-бензилбромида в 450 мл ацетонитрила добавляют 270 r (2,0 моль) безводного карбоната калия и перемешивают при комнатной температуре в течение шести часов. После отсасывания неорганических солей отгоняют растворитель, остаток смешивают с 300 мл воды и многократно экстрагируют этилацетатом. Органическую фазу промывают два раза водой, сушат над сульфатом натрия и концентрируют досуха. После настаивания с эфиром получают 180 г белых кристаллов с температурой плавления 101 - 102oC.
В.13.б. 1-(4-Цианофенил)этиламин:
К раствору диизопропиламида лития, полученному из 8,15 г (0,08 моль) диизопропиламина и 48,3 мл (0,08 моль) 15%-ного раствора бутиллития в гексане, в 100 мл абсолютного тетрагидрофурана при температуре -70oC прикапывают 20,7 г (0,07 моль) N-(п-цианобензил)-бензофенонимина и дополнительно перемешивают в течение пятнадцати минут. После этого прикапывают 9,94 г (0,07 моль) метилиодида и температуру реакционной смеси оставляют повышаться до комнатной. После добавления 100 мл воды многократно экстрагируют эфиром, эфирную фазу промывают с помощью 5%-ного раствора лимонной кислоты, 5%-ного раствора гидрокарбоната натрия и воды, сушат над сульфатом натрия и эфир отгоняют. Остаток растворяют в 150 мл тетрагидрофурана, добавляют 100 мл 1 н. соляной кислоты и перемешивают в течение ночи при комнатной температуре. Из реакционной смеси в вакууме отгоняют тетрагидрофуран, остающуюся кислую фазу для удаления бензофенона многократно экстрагируют эфиром, затем кислую фазу подщелачивают при охлаждении льдом с помощью водного раствора карбоната калия и маслянистое основание экстрагируют метиленхлоридом. Экстракт сушат над карбонатом калия. После удаления метиленхлорида получают 9,7 г (95%) желтоватого масла, которое без дальнейшей очистки используют в последующей реакции.
B.14. 4-Аминометил-3-метокси-бензонитрил:
В.14.а. 3-Нитро-4-метил-бензонитрил:
К одному литру дымящей азотной кислоты при -10oC в течение полутора часов добавляют 399 г (2,56 моль) п-толунитрила. Спустя один час после добавления смесь выливают в 2,5 л воды со льдом, причем осаждается твердое вещество, которое отделяют с помощью нутч-фильтра и промывают водой до нейтральной реакции. Выход продукта составляет 363 г (88 %).
1H-ЯМР (дейтерохлороформ; δ в м. д. (миллионные доли)):
8,3 (д, 1H); 7,8 (дд, 1H): 7,5 (дд, 1H); 2,7 (с, 3H).
В.14.б. 3-Амино-4-метил-бензонитрил:
120 г 3-Нитро-4-метил-бензонитрила суспендируют в 1,2 л этанола и в присутствии 7 г 10%-ного палладия-на-угле гидрируют с помощью водорода при комнатной температуре. После отделения катализатора через целит растворитель удаляют и получают 95 г очищенного продукта (97%).
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.):
7,1 г (дд, 1H); 6,90 (д, 1H): 6,85 (дд, 1H); 5,35 (с, 2H, NH2); 2.15 (с, 3H).
В. 14.в. 3-Гидрокси-4-метил-бензонитрил:
К 85 г (0,72 моль) 3-амино-4-метил-бензонитрила в 1,8 л 6 н. соляной кислоты при температуре от 0oC до 5oC в течение тринадцати минут прикапывают раствор из 49,2 г (0,72 моль) пнитрита натрия в 217 мл воды. После этого перемешивают следующие тридцать минут при температуре от 0oC до 5oC и затем еще в течение одного часа кипятят при перемешивании. После охлаждения раствора продукт можно экстрагировать с помощью этилацетата и из полученного экстракта извлекать в виде фенолята с помощью охлажденного льдом 5 н. раствора гидроксида натрия. Водную фазу затем подкисляют с помощью 6 н. соляной кислоты до pH-значения, равного трем, и продукт экстрагируют этилацетатом. Получают 41 г (43%) фенола.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.):
10,3 (с, ОН): 7,25 (дд, 1H), 7,15 (д, 1H); 7,1 (дд, 1H); 2,20 (с, 3H).
В.14.г. 3-Метокси-4-метил-бензонитрил:
15 г (0,11 моль) 3-Гидрокси-4-метил-бензонитрила, растворенные в 30 мл диметилформамида, прикапывают к суспензии из 0,11 моль гидрида натрия в 30 мл диметилформамида и перемешивают вплоть до полного прекращения выделения водорода. После этого прикапывают 10,6 мл (0,17 моль) метилиодида и перемешивают в течение одного часа при комнатной температуре. Раствор выливают в воду со льдом и продукт экстрагируют смесью эфира с этилацетатом в соотношении семь к одному. После удаления растворителя начинает медленно выкристаллизовываться продукт. Получают 14,8 г (89%) продукта.
1Н-ЯМР (дейтерохлороформ: δ в м.д.): 7,2 (м, 2H); 7,02 (с, 1H): 3,85 (с, 3H): 2,25 (с, 3H).
В.14.д.
4-Бромметил-3-метокси-бензонитрил: 14,7 r (0,1 моль) 3-Метокси-4-метил-бензонитрила растворяют в 210 мл 1,2- дихлорэтана, в течение одного часа бромируют путем добавления порциями 19,1 г (0,11 моль) N-бромсукцинимида в присутствии каталитического количества 2,2'-азобисизобутиронитрила при 82oC и по окончании добавления перемешивают следующие тридцать минут при температуре 82oC. После добавления н-гептана отделяют выпавший в осадок сукцинимид и растворитель удаляют. Наряду с небольшим количеством эдукта, продукт содержит еще следовые количества соответствующего бензальбромида.
1Н-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 7,60 (дд, 1H); 7,50 (д, 1H); 7,40 (дд, 1H); 4,68 (с, 2H): 3,96 (с, 3H).
В.14.е. 4-Фталимидометил-3-метокси-бензонитрил:
24,4 г (108 моль) 4-Бромметил-3-метокси-бензонитрила, растворенные в 125 мл диметилформамида, и 20,0 г фталимида калия перемешивают в течение двадцати четырех часов при комнатной температуре и затем еще в течение одного часа при 50oC. Смесь выливают в воду, причем продукт осаждается в виде твердого вещества. Получают 21,5 г (68%) продукта.
1Н-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.):
7,9 (м, 4H); 7,5 (д, 1H); 7,35 - 7,25 (м, 2H); 7,78 (с, 2H); 3,92 (с, 3H).
В.14.ж. 4-Аминометил-3-метокси-бензонитрил:
К 21,2 г (73 ммоль) 4-фталимидометил-3-метокси-бензонитрила, растворенным в 290 мл тетрагидрофурана, добавляют 10,6 мл гидразингидрата и перемешивают в течение двадцати часов при комнатной температуре. После этого прикалывают 180 мл 2 н. соляной кислоты и спустя полтора часа растворитель полностью удаляют. Остаток растворяют в трет.-бутил-метиловом эфире, экстрагируют с помощью 1 н. соляной кислоты, подщелачивают до pH-значения от 9 до 10 с помощью 2 н. раствора гидроксида натрия и экстрагируют дихлорметаном. Получают 8,0 г (68%) продукта.
1Н-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.):
7,55 (дд, 1H); 7,40 (дд, 1H); 7,37 (д, 1H); 3,85 (с, 3H); 3,70 (с, 2H); 2,5 - 1,6 (NH2).
В.15. 4-Аминометил-3-изопропокси-бензонитрил:
В.15.а. 3-Изопропокси-4-метил-бензонитрил:
7,0 г 3-Гидрокси-4-метил-бензонитрила (52,6 ммоль) депротонируют с помощью 57,8 моль гидрида натрия в 100 мл диметилформамида и при 0oC смешивают с 7,4 мл 2-бромпропана. Спустя сорок пять минут температуру повышают до 50oC и перемешивают в течение следующих пяти часов. Реакционную смесь выливают в воду и продукт экстрагируют эфиром. Продукт очищают с помощью колоночной хроматографии на силикагеле (растворитель: дихлорметан с 10% гептана). Получают 6,3 г (68%). Температура плавления составляет 60 - 61oC.
В.15.б. 4-Бромметил-3-изопропокси-бензонитрил:
6,1 г 3-Изопропокси-4-метил-бензонитрила (33,4 ммоль) бромируют аналогично примеру (В.14.д.) с помощью N-бромсукцинимида и 2,2'- азобисизобутиронитрала. Продукт получают почти с количественным выходом.
1Н-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.):
7,65 - 7,30 (3H, аром. Н); 4,85 (1H, CH); 4,63 (2H, CH2); 1,40 - 1,25 (6H, 2-CH3).
В.15.в. 4-Аминометил-3-изопропокси-бензонитрил (гидрохлорид):
8,8 г Бромида (33,4 ммоль) растворяют в 100 мл метанола и при 40oC медленно прикапывают к 150 мл насыщенного аммиаком метанола. Растворитель удаляют, продукт растворяют в дихлорметане, промывают с помощью 1 н. раствора гидроксида натрия и осаждают с помощью эфирного раствора хлороводорода в виде гидрохлорида. Получают 2,6 г.
1Н-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.):
8,6 (3H, МН3 +); 7,65 - 7,40 (3H, аром. Н): 4,80 (1H, CH); 4,00 (2H, CH2); 1,4-1,3 (6H, 2 CH3).
В.16. 4-Аминометил-3-хлор-бензонитрил:
В. 16.а. 4-Бромметил-3-хлор-бензонитрил:
3-Хлор-4-метил-бензонитрил бромируют аналогично примеру (В. 14.д.) с помощью N-бромсукцинимида и 2,2'-азобисизобутиронитрила.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.):
8,10 и 7,85 (3H, аром. Н): 4,80 (2H, CH2).
В.16.б. 4-Аминометил-3-хлор-бензонитрил:
10,0 г Бромида вводят во взаимодействие с фталимидом калия аналогично примеру (В.14.е.). Получают 9,6 г фталимидометил-3-хлор-бензонитрила, который расщепляют с помощью гидразингидрата аналогично примеру (В.14.ж.). Свободный амин (4,0 г) получают путем экстракции с помощью дихлорметана из водной фазы, pH-значение которой устанавливают равным 9 - 10 с помощью раствора гидроксида натрия.
1Н-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.):
7,95 - 7,78 (3H, аром. Н); 3,85 (2H, CH2); 2,1 (уширенный сигнал, 2H, NH2).
Г. Примеры
Пример 1
Boc-(D)-Phe-Pro-NH-(4-Am)-2-фенетил
К раствору 10 ммоль Boc-(D)-Phe-Pro-OH и 11 ммоль N-метилморфолина в 10 мл диметилформамида при температуре -15oC в течение двух минут добавляют 10 ммоль изобутилового эфира хлормуравьиной кислоты, перемешивают дополнительно в течение 10 минут и после этого добавляют раствор 10 ммоль п-цианобензиламина и 11 ммоль N-метилморфолина в 3 мл диметилформамида. После перемешивания дополнительно в течение трех часов при -15oC, согласно контролю с помощью тонкослойной хроматографии (смесь дихлорметана с метанолом в соотношении 9: 1), более не обнаруживают присутствия исходного соединения. Для выделения реакционную смесь выливают в 200 мл воды, причем выделяется масло, которое спустя непродолжительное время затвердевает, и после размельчения продукт отсасывают. Еще влажный остаток растворяют в смеси из 250 мл этилацетата и 50 мл эфира и промывают последовательно с помощью 5%-ного водного раствора лимонной кислоты, раствора гидрокарбоната натрия и насыщенного раствора хлорида натрия. После высушивания над сульфатом натрия растворитель отгоняют в вакууме, остаток смешивают с н-гексаном и затем отсасывают. Перекристаллизация из 50 мл этилацетата дает 7,4 ммоль чистого, согласно тонкослойной хроматографии, продукта, который согласно методике A.III.1. по способу с использованием сероводорода превращают в амидингидроиодид. Получают желтоватые кристаллы; температура плавления составляет 158 - 165oC. Масс-спектр (бомбардировка быстрыми атомами): 508 (М+H+).
Пример 2
H-(D)-Phe-Pro-NH-(4-Am)-2-фенетил
Отщепление Boc-группы от соединения примера 1 осуществляют по методике A. I. в. В качестве смеси растворителей в данном случае используют смесь дихлорметана с этилацетатом в соотношении 1:1. Получают дигидрохлорид в виде белых кристаллов; температура плавления составляет 203 - 206oC (разложение): масс-спектр (бомбардировка быстрыми атомами): 408 (М+H+).
Пример 3
Boc-(L)-Phe-Pro-NH-pAmb
Соединение получают исходя из Boc-(L)-Phe-OH и хлорангидрида H-Pro-п-цианобензиламида по методике Б.I. и путем последующего превращения нитрила в амидин согласно методике A.III.1. Получающийся при этом амидингидроиодид с помощью содержащей ацетатные группы ионообменной смолы (1RA 420) превращают в амидингидроацетат.
1Н-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 8,4 (м, 1H, NH); 7,75 (д, 2H, аром. протоны); 7,45 (д, 2H, аром. протоны); 7,2 (м, 5H, аром. протоны); 7,18/7,02 (2д, 1H, NH): 4,48 - 4,18 (м, 4H, CH2/2-□-H 3,6 (м, 2H, Pro): 3,0 - 2,7 (м, 2H, CH2-фенил): 2,18 - 1,8 (м, 4H, Pro): 1,3 - 1,2 (2с, 9H, Boc).
Масс-спектр: 494 (М+H+); 394(-Boc); температура плавления составляет 142oC.
Пример 4
H-(L)-Phe-Pro-NH-pAmb
Соединение получают путем отщепления Boc-группы от соединения примера 3 согласно методике A.I.в. Полученный дигидрохлорид с помощью колоночной хроматографии на силикагеле за счет добавки уксусной кислоты превращают в дигидроацетат. Температура плавления составляет 69oC; масс- спектр (бомбардировка быстрыми атомами): 394 (М+H+).
Пример 5
Boc-(L)-Phe-Pro-NH-pAmb
К раствору 5,1 г (14,2 ммоль) Boc-(D)-Phe-Pro-OH и 1,53 г (15,2 ммоль) N-метил-морфолина в 15 мл диметилформамида при -15oC в течение двух минут добавляют 2,0 г (14,6 ммоль) изобутилового эфира хлормуравьиной кислоты, перемешивают дополнительно в течение десяти минут и затем добавляют раствор 1,9 г (14,2 ммоль) п- цианобензиламина и 1,53 г N-метилморфолина в 3 мл диметилформамида. После дополнительного перемешивания в течение трех часов при -15oC согласно контролю с помощью тонкослойной хроматографии (смесь дихлорметана с метанолом в соотношении 9: 1) более не обнаруживают присутствия исходного соединения.
Для выделения, реакционную смесь выливают в 200 мл воды, причем выделяется масло, которое спустя непродолжительное время затвердевает, и после размельчения продукт отсасывают. Еще влажный остаток растворяют в смеси из 250 мл этилацетата и 50 мл эфира и промывают последовательно с помощью 5%-ного водного раствора лимонной кислоты, раствора гидрокарбоната натрия и насыщенного раствора хлорида натрия. После высушивания над сульфатом натрия растворитель отгоняют в вакууме, остаток смешивают с н-гексаном и затем отсасывают. Перекристаллизация из 50 мл этилацетата дает 5,6 г чистого, согласно тонкослойной хроматографии, Boc-(D)-Phe- Pro-п-циано-бензиламида; температура плавления составляет 156- 157oC.
Получение тиоамида: 4,1 г Вышеполученного соединения и 4 мл триэтиламина растворяют в 40 мл пиридина, при 0oC насыщают раствор сероводородом и выдерживают в течение ночи при комнатной температуре. Согласно контролю с помощью тонкослойной хроматографии (смесь дихлорметана и метанолом в соотношении 9:1) превращение в тиоамид происходит полностью. Для выделения далее пиридин отгоняют в вакууме, остаток растворяют в 250 мл этилацетата и полученный раствор промывают с помощью раствора хлорида натрия, 5%-ного раствора лимонной кислоты и раствора гидрокарбоната натрия. После высушивания и отгонки растворителя получают 4,1 г чистого кристаллического тиоамида.
Получение амидина: Тиоамид растворяют в 150 мл ацетона и после добавления 7 мл метилиодида выдерживают в течение ночи при комнатной температуре. После удаления растворителя аморфный остаток перемешивают с безводным эфиром и затем высушивают. Гидроиодид метилового эфира S-метилтиоаминокислоты растворяют в 50 мл этанола, смешивают с 15 мл 10%-ного раствора ацетата аммония и в течение трех часов нагревают при 60oC. Для выделения удаляют растворитель, остаток растворяют в 100 мл дихлорметана, отфильтровывают нерастворимые составные части и затем дихлорметан отгоняют. Путем настаивания со смесью этилацетата с диэтиловым эфиром отделяют растворяющиеся в этой смеси примеси. Остающуюся смешанную соль иодид-ацетат растворяют в смеси ацетона с водой в соотношении 3:2 и с помощью содержащей ацетатные группы ионообменной смолы (IRA) переводят в чистый ацетат. Раствор концентрируют досуха и остаток подвергают сушке вымораживанием. Выделяют 3,8 г хроматографически чистого (тонкослойная хроматография; смесь дихлорметана с метанолом и 50%-ной уксусной кислотой в соотношении 20 : 5 : 1) Boc-(D)-Phe-Pro-NH-pAmb в виде ацетата. Температура плавления составляет 195 - 200oC (разложение).
Пример 6
Ac-(D)-Phe-Pro-NH-pAmb
10,4 г (0,05 моль) Ac-(D)-Phe-OH, 6,3 г (0,055 моль) N- гидроксисукцинимида и 11,4 г (0,055 моль) дициклогексилкарбодиимида растворяют в 150 мл ацетонитрила и перемешивают в течение ночи при комнатной температуре. Выпавший осадок отфильтровывают, растворитель отгоняют, остаток высушивают в вакууме и без дальнейшей очистки используют в следующей реакции.
13,3 г (0,05 моль) (4-Цианобензил)пролиламид-гидрохлорида (см. пример 10) растворяют в 100 мл метиленхлорида и при 0oC последовательно смешивают с 15 мл триэтиламина и раствором вышеполученного Ac-(D)-Phe-O-сукцинимида в 70 мл дихлорметана. Реакционную смесь перемешивают в течение ночи при комнатной температуре и промывают последовательно с помощью воды, 5%-ного раствора лимонной кислоты, 5%-ного раствора гидрокарбоната натрия и раствора хлорида натрия. После высушивания и отгонки растворителя остаток очищают при использовании силикагеля (элюирующее средство: смесь дихлорметана с метанолом в соотношении 50:2) и затем аналогично примеру 5 переводят в амидин.
Температура плавления ацетата составляет 220 - 224oC (разложение); масс-спектр (бомбардировка быстрыми атомами): 436 (М+H+).
Пример 7
H-(D)-Phe-Pro-NH-pAmb
4,9 г (10 ммоль) Полученного в примере 5 соединения растворяют в смеси растворителей из 100 мл хлороформа и 100 мл этилацетата и при -15oC в отсутствие влаги насыщают газообразным хлороводородом. Спустя один час, согласно тонкослойной хроматографии (смесь дихлорметана с метанолом и 50%-ным раствором уксусной кислоты в соотношении 20:5:1), более не обнаруживают никакого соединения с Boc-группой.
Далее, путем пропускания азота при температуре -15oC удаляют избыточный газообразный хлороводород, причем выделяется дигидрохлорид в виде мелких кристаллов. После добавления 50 мл эфира для полноты осаждения осадок отсасывают и дополнительно промывают смесью этилацетата с эфиром в соотношении 1: 1. Осадок растворяют в воде, обрабатывают активным углем и лиофилизируют. Получают 3,7 г (95% от теоретически рассчитанного количества дигидрохлорида в виде белых кристаллов; температура плавления составляет 215oC (разложение): масс-спектр (бомбардировка быстрыми атомами): 394 (М+H+).
Пример 8
H-(D)-Phe-Pro-N(Me)-pAmb
Соединение получают аналогично примеру 5 путем введения во взаимодействие Boc-(D)-Phe-Pro-OH с М-метил-4-цианобензиламином. Затем Boc-группу отщепляют согласно методике A.I.в. Получают дигидрохлорид в виде аморфного твердого вещества; масс-спектр (бомбардировка быстрыми атомами): 408 (М+H+).
Пример 9
Me-(D)-Phe-Pro-NH-pAmb
4,0 г Нижеполученного соединения (пример 10) растворяют в 25 мл этанола, смешивают с 1,55 г 32%-ной соляной кислоты и после добавления 0,6 г 10%-ного палладия-на-угле гидрируют. Спустя один час превращение количественное (согласно тонкослойной хроматографии: смесь дихлорметана с метанолом и 50%-ным раствором уксусной кислоты в соотношении 35:15:5). После отсасывания катализатора и отгонки растворителя остаток обрабатывают с помощью 100 мл этилацетата с получением белого порошка и после растворения его в воде лиофилизируют. Выделяют 3,1 г дигидрохлорида, который после агломерации при 100oC разлагается выше 215oC.
Пример 10
Z-Me-(D)-Phe-Pro-NH-pAmb
(4-Цианобензил)пролиламид-гидрохлорид:
276 г Boc-Pro-O-сукцинимида (0,88 моль) при 0oC растворяют в 2 л дихлорметана. К этому раствору добавляют последовательно 163,9 г 4-цианобензилами-нгидрохлорида (0,97 моль) и 230 мл диизопропилэтиламина (1,34 моль). Суспензию перемешивают в течение сорока восьми часов на бане с тающим льдом и затем отфильтровывают. Фильтрат экстрагируют четырехкратно с помощью 20%-ного раствора сульфата натрия, трехкратно с помощью насыщенного раствора гидрокарбоната натрия и двухкратно с помощью насыщенного раствора хлорида натрия, сушат и выпаривают в ротационном испарителе. Получают 299 г продукта, который после перекристаллизации из метил-трет.-бутилового эфира плавится при 124 - 125oC.
299 г Соединения с защитной Boc-группой растворяют в одном литре диэтилового эфира. После добавления эфирного раствора хлороводорода (хлороводород в избытке) перемешивают в течение ночи. Осадившуюся соль отфильтровывают, промывают диэтиловым эфиром и высушивают в вакууме. Сырой продукт перекристаллизуют из этанола.
Выход составляет 200 г; температура плавления составляет 209 - 213oC (разложение).
Z-Me-(D)-Phe-Pro-п-циано-бензиламид:
3,1 г (0,01 моль) Z-Me-(D)-Phe-OH и 1,49 г (0,011 моль) гидроксибензотриазола растворяют в 50 мл диметилформамида и при 0oC смешивают с 2,1 г (0,01 моль) дициклогексилкарбодиимида. Спустя тридцать минут добавляют 2,7 г (0,01 моль) (4- цианобензил)пролиламидгидрохлорида и 2,2 мл N-метилморфолина. Реакционную смесь перемешивают в течение ночи при комнатной температуре, выпавшую в осадок мочевину отфильтровывают и растворитель отгоняют в вакууме. Остаток растворяют в 100 мл этилацетата и промывают последовательно с помощью воды, 5%-ного раствора лимонной кислоты, 5%-ного раствора гидрокарбоната натрия и раствора хлорида натрия. После высушивания и отгонки растворителя получают 4,7 г (90% от теоретически рассчитанного количества) вязкого масла, которое используют в последующей реакции.
Амидирование при помощи реакции Пиннера
К раствору из 8,9 г абсолютного этанола в 25 мл дихлорметана при 0oC прикапывают 12,3 г ацетилхлорида и дополнительно перемешивают в течение 40 минут. Затем при 0oC прикапывают раствор из 4,7 г вышеполученного вещества в 30 мл абсолютного дихлорметана. Реакционную смесь выдерживают в течение четырех дней при 0oC. После концентрирования раствора в вакууме остаток разбавляют с помощью 100 мл дихлорметана и этот раствор встряхивают с охлажденным льдом 15%-ным раствором карбоната калия. Высушивание и отгонка растворителя дают сырой имидоэфир в виде основания, которое растворяют в 30 мл метанола и смешивают с 0,8 г ацетата аммония. Раствор выдерживают в течение двух дней при комнатной температуре.
После отфильтровывания растворителя остаток очищают при использовании колонки с силикагелем (смесь дихлорметана с метанолом и 50%-ной уксусной кислотой в соотношении 40 : 10 : 2,5). Выпаренные элюаты растворяют в толуоле и снова выпаривают в ротационном испарителе. Остаток растворяют в воде, обрабатывают активным углем и затем лиофилизируют. Получают 4,1 г (76% от теоретически рассчитанного количества) белых кристаллов, которые агломерируются при 83oC и плавятся при 178 - 184oC.
Пример 11
HOOC-CH2-(D)-Phe-Pro-NH-pAmb
2,4 г Гидроацетата t-BuOOC-CH2-(Boc)-(D)-Phe-Pro-NH-pAmb (из примера 13) в 80 мл абсолютного дихлорметана и 15 мл эфирного раствора хлороводорода перемешивают в течение ночи при комнатной температуре. Растворитель удаляют в вакууме, остаток перемешивают в смеси дихлорметана с ацетоном в соотношении 2: 1 и отфильтровывают. Получают 1,6 г продукта в виде гидрохлорида, соответственно, дигидрохлорида, соответственно, в виде смеси обеих солевых форм, в виде твердого вещества белого цвета. Температура плавления составляет 210 - 220oC.
Пример 12
MeOOC-CH2-(D)-Phe-Pro-NH-pAmb
0,5 г Соединения из примера 11 вместе с 2 мл эфирного раствора хлороводорода, 3 мл дихлорметана и 3 мл метанола перемешивают в течение тридцати часов при комнатной температуре. Растворитель удаляют и остаток многократно перемешивают с эфиром. Получают 0,5 г продукта в виде гидрохлорида, соответственно, дигидрохлорида, соответственно, в виде смеси обеих солевых форм. Температура плавления составляет 104 - 120oC.
Пример 13
t-BuOOC-CH2-(Boc)-(D)-Phe-Pro-NH-pAmb
а) H-(D)-Pbe-Pro-п-цианобензиламид:
5,6 г Boc-(D)-Phe-Pro-п-цианобензиламида (из примера 5) расщепляют согласно методике A. I. в. Получают 4,6 г (95%) продукта в виде гидрохлорида в форме белых кристаллов.
б) t-BuOOC-CH2-(Boc)-(D)-Phe-Pro-п-цианобензиламид:
6,19 г H-(D)-Pbe-Pro-п-цианобензиламида (15 ммоль) вместе с 0,98 г трет. -бутилового эфира бромуксусной кислоты (5 ммоль) и 0,63 г карбоната аммония в смеси из 35 мл воды и 8 мл нитрометана в течение двух часов нагревают при 50oC. Затем экстрагируют этилацетатом, органическую фазу многократно промывают с помощью 0,1 н. соляной кислоты, водные фазы экстрагируют дихлорметаном и объединенные органические фазы сушат над сульфатом магния. После удаления растворителя продукт осаждают в виде гидрохлорида с помощью эфирного раствора хлороводорода. Получают 2,6 г (98%) гидрохлорида. Избыточный H-(D)-Phe-Pro-п-цианобензиламид рекуперируют путем экстракции водных фаз при pH-значении, равном 10, с помощью дихлорметана.
в) t-BuOOC-CH2-(Boc)-(D)-Phe-Pro-п-цианобензиламид:
2,6 г Вышеполученного гидрохлорида (4,9 ммоль) вместе с 1,2 г (Boc)2O (5,5 ммоль) и 1,87 мл диизопропилэтиламина (11 ммоль) в 95 мл абсолютного дихлорметана перемешивают в течение ночи при комнатной температуре. Затем растворитель удаляют, остаток растворяют в эфире, промывают 0,1 н. соляной кислоты, затем водой, сушат над сульфатом магния и растворитель удаляют в вакууме. После перемешивания остатка в гексане получают 2,8 г продукта в виде твердого вещества белого цвета.
г) t-BuOOC-CH2-(Boc)-(D)-Phe-Pro-NH-pAmb
Превращение нитрильной функции в амидиногруппу осуществляют согласно методике A.III.1. с общим выходом 96%.
Переведение гидроиодида в гидроацетат осуществляют с помощью содержащей ацетатные группы ионообменной смолы (IRA); температура плавления составляет 116-121oC.
Пример 14
EtOOC-(D)-Phe-Pro-NH-pAmb
Соединение получают путем взаимодействия N-(этоксикарбонил)- (D)-фенил-аланина (J. Org. Chem., 45, 4519 (1980)) с (4- цианобензил)пролиламидгидрохлоридом (из примера 10) и последующего образования амидина (аналогично примеру 5). Получают белые кристаллы гидроацетата; температура плавления составляет 105 - 107oC: масс-спектр (бомбардировка быстрыми атомами): 466 (М+H+).
Пример 15
Boc-(D)-Phe-Pro-NH-pAmb
Соединение получают из Boc-(D)-Phe-Pro-OH с м-цианобензиламином (аналогично примеру 5). Гидроацетат получают в форме белых кристаллов; температура плавления составляет 130 - 133oC.
Пример 16
H-(D)-Phe-Pro-NH-pAmb
Отщепление Boc-группы от соединения примера 15 осуществляют согласно методике A. I. в. Белые кристаллы дигидрохлорида плавятся при 155 - 160oC; масс-спектр (бомбардировка быстрыми атомами): 394 (М+H+).
Пример 17
Z-(D)-Phe-Pro-(D,L)-(4-Am)-PhgOH
а) 39,7 (100,1 ммоль) Z-(D)-Phe-Pro-OH, 11,53 г (100,1 ммоль) гидроксисукцинимида и 20,66 г (100,1 ммоль) N,N'- дициклогексилкарбодиимида в 400 мл диметоксиэтана перемешивают в течение восемнадцати часов при комнатной температуре. Затем твердое вещество отфильтровывают, фильтрат концентрируют и остаток обрабатывают ацетонитрилом. Снова выпавшее в осадок твердое вещество отфильтровывают и органический раствор выпаривают в вакууме досуха. Выход сырого Z-(D)-Phe-Pro-0-сукцинимида составляет 48,9 г.
б) 24,53 г Гидрохлорида H-Phg(4-CN)-Oet, 34,9 мл диизопропилэтиламина и 41,9 г Z-(D)-Phe-Pro-O-сукцинимида растворяют в 200 мл диметилформамида и перемешивают в течение восемнадцати часов при комнатной температуре. Для обработки диметилформамид удаляют в вакууме и полученный остаток обрабатывают дихлорметаном. Органическую фазу экстрагируют с помощью 1 н. соляной кислоты, сушат над сульфатом натрия и концентрируют в вакууме. Получают 54,04 г Z-(D)-Phe-Pro-NH- Phg(4-CN)-Oet.
в) 54,04 г Z-(D)-Phe-Pro-NH-Phg(4-CN)-Oet растворяют в 340 мл смеси тетрагидрофурана с этанолом и водой в соотношении 3:1:1 и после добавления 3,05 г гидроксида лития перемешивают в течение восемнадцати часов при комнатной температуре. Затем реакционную смесь концентрируют в вакууме, оставшийся водный раствор подкисляют до pH-значения, равного двум, и экстрагируют этилацетатом. Объединенные органические экстракты промывают насыщенным раствором хлорида натрия, сушат над сульфатом натрия и концентрируют досуха в ротационном испарителе. Выход сырого Z-(D)- Phe-Pro-NH-Phg(4-CN)-OH составляет 40,94 г.
г) 2,78 г Z-(D)-Phe-Pro-NH-Phg(4-CN)-OH, 18,9 мл пиридина и 8,7 мл триэтиламина смешивают в реакционной колбе и насыщают газообразным сероводородом. Раствор выдерживают в течение восемнадцати часов при комнатной температуре. Затем реакционную смесь выливают в два литра воды со льдом и водную фазу подкисляют до pH-значения, равного трем, с помощью 1 н. соляной кислоты. Выпавший в осадок продукт отфильтровывают, растворяют в этилацетате и раствор сушат над сульфатом натрия. После удаления этилацетата в вакууме остаток смешивают с 20 мл ацетона и 3,5 мл метилиодида и перемешивают в течение восемнадцати часов при комнатной температуре. После этого летучие составные части удаляют в вакууме и сырой тиометилимингидрохлорид в 8 мл метанола и 8 мл 10%-ного метанольного раствора ацетата аммония перемешивают в течение восемнадцати часов. Затем реакционную смесь концентрируют, остаток обрабатывают дихлорметаном и осадившееся твердое вещество отфильтровывают. После концентрирования фильтрата получают 3,75 г сырого продукта. Полученный продукт очищают с помощью высокоэффективной жидкостной хроматографии с обращенной фазой. Выход составляет 1,5 г.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 8,4 - 8,0 (2м, 1H, NH): 7,7 - 7,4 (м, 4H, аром. протоны); 7,3 - 7,1 (м, 10H, аром. протоны); 7,0 (уш. с. , 1H, NH): 5,2 - 4,8 (м, 3H, OCH2/ α- Phg): 4,6 - 4,2 (м, 2H, 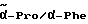 ); 3,6 - 3,2 (2H, δ- Pro); 3,0 - 2,6 (м, 2H, CH2-Ph); 2,2 - 1,6 (м, 4H, α/β- Pro).
); 3,6 - 3,2 (2H, δ- Pro); 3,0 - 2,6 (м, 2H, CH2-Ph); 2,2 - 1,6 (м, 4H, α/β- Pro).
Масс-спектр (бомбардировка быстрыми атомами): 572 (М+H+).
Температура плавления составляет 155 - 158oC.
Пример 18
Z-(D)-Phe-Pro-(D,L)-(4-Am)-Phg-OMe
а) К 14 мл дихлорметана при 0oC добавляют 5, 14 мл абсолютного метанола и 6,16 мл ацетилхлорида. Затем к этому раствору добавляют 2,78 г Z-(D)-Phe-Pro-NH-Phg(4-CN)-OH в 10 мл дихлорметана и выдерживают при комнатной температуре в течение сорока восьми часов. Для обработки реакционную смесь концентрируют, остаток растворяют в этилацетате и промывают холодным 5%-ным раствором карбоната калия. После высушивания органической фазы над сульфатом натрия растворитель удаляют в ротационном испарителе и сырой простой иминометиловый эфир в 6,5 мл метанола и 6,5 мл 10%-ного метанольного раствора ацетата аммония выдерживают в течение восемнадцати часов. После выпаривания раствора получают 2,4 г сырого продукта, который очищают с помощью высокоэффективной жидкостной хроматографии с обращенной фазой.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,6 - 9,2 (уш. с., NH), 8,75 / 8,5 (2д, 1H, NH); 7,8 (м, 2H, аром. протоны): 7,6 (м, 2H, аром. протоны): 7,35 - 7,2 (м, 10H, аром. протоны): 7,05 (уш. с., 1H, NH); 5,6 (2д, 2H, OCH2): 5,0 - 4,2 (3м, 4H, 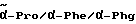 ); 3,6 (2с, 3H, OCH3); 3,5 - 3,2 (2H, δ-Pro): 3,0 - 2,6 (м, 2H, CH2 - фенил): 2,2-1,6 (м, 4H, β/γ-Pro).
); 3,6 (2с, 3H, OCH3); 3,5 - 3,2 (2H, δ-Pro): 3,0 - 2,6 (м, 2H, CH2 - фенил): 2,2-1,6 (м, 4H, β/γ-Pro).
Масс-спектр (бомбардировка быстрыми атомами): 586 (М+H+).
Температура плавления гидрохлорида составляет 129-131oC.
Пример 19
H-(D)-Phe-Pro-(D,L)-(4-Am)-Phg-OH
Соединение получают путем отщепления Z - группы от продукта примера 17.
1H-ЯМР (дейтерированный диметилсульфоксид; δ м.д.): 9,0 (уш. с., NH): 8,7 / 8,4 / 8,05 (3д, 1H, NH): 7,8 - 7,0 (м, 9H, аром. протоны); 5,1 / 4,9 (д, 1H, α- Phg); 4,45 - 4,0 (м, 2H, 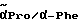 ), 3,8 - 3,0 (м, 2H, δ-Pro); 3,0 - 2,7 (м, 2H, CH2 - фенил); 2,2-1,4 (м,4H, β/γ-Pro).
), 3,8 - 3,0 (м, 2H, δ-Pro); 3,0 - 2,7 (м, 2H, CH2 - фенил); 2,2-1,4 (м,4H, β/γ-Pro).
Масс-спектр (бомбардировка быстрыми атомами): 438 (М+H+).
Температура плавления дигидрохлорида составляет 149 - 150oC.
Пример 20
Boc-(D)-Phe-Pro-(4-Am)-Phg-CH2Ph
а) N-(Дифенилметилен)-4-циано-бензиламин:
33,73 г (0,2 моль) 4-Цианобензиламина и 36,25 г (0,2 моль) бензофенонимина при комнатной температуре растворяют в 540 мл дихлорметана и перемешивают в течение ночи. Затем реакционную смесь промывают два раза по 90 мл воды, сушат над сульфатом натрия и растворитель удаляют в ротационном испарителе. Получают 55,59 г (93,7%) сырого продукта. После перекристаллизации из 550 мл изопропанола получают 97,67 г (80,4%) чистого продукта.
1H-ЯМР (дейтерохлороформ; δ в м.д.): 7,7 - 7,15 (м, 9H, аром. протоны): 4,65 (с, 2H, CH2N).
б) N-(Дифенилметилен)- α-(β- фенилацетил)-4-циано-бензиламин:
63,3 моль Диизопропиламида лития при -30oC вносят в 45 мл тетрагидрофурана. Затем медленно прикапывают раствор 15 г (50,61 ммоль) N-(дифенилметилен)-4-циано-бензиламина в 75 мл тетрагидрофурана. После перемешивания в течение следующих десяти минут при температуре -30oC медленно добавляют при -78oC раствор 8,6 г (55,7 ммоль) хлорангидрида кислоты в 7,5 мл тетрагидрофурана. После перемешивания реакционной смеси в течение восемнадцати часов (реакционная температура в течение ночи повышается до комнатной) охлаждают до -20oC, смешивают с 3,6 мл уксусной кислоты и 17,25 мл воды. После того, как температура реакционной смеси повышается до комнатной, тетрагидрофуран удаляют в вакууме, остаток обрабатывают эфиром, органическую фазу промывают насыщенным раствором хлорида натрия, сушат и эфир удаляют в ротационном испарителе. Получают 26,18 г сырого продукта, который без дальнейшей очистки используют в следующей стадии.
в) α-(β- Фенилацетил)-4-циано-бензиламин:
26,19 г Полученного сырого кетона в 250 мл 0,25 н. соляной кислоты перемешивают в течение ночи при комнатной температуре. Затем реакционную смесь экстрагируют дихлорметаном и водную фазу лиофилизируют. После разделения с помощью высокоэффективной жидкостной хроматографии
с обращенной фазой получают 2,52 г α-(β- фенилацетил)-4-цианобензиламина.
1H-ЯМР (дейтерированный диметилсульфоксид; β в м.д.): 9,0 (с, 3H, NH3); 8,0 / 7,75 (2д, 4H); 7,25 (м, 3H), 7,0 (дд, 2H); 5,7 (с, 1H, NCH): 3,9 / 3,6 (2д, фенил-CH2-); масс-спектр (бомбардировка быстрыми атомами) (М+): 250.
г) 2,51 г (6,92 ммоль) Boc-(D)-Phe-Pro-OH растворяют при -20oC в 40 мл абсолютного дихлорметана. Затем к раствору добавляют 0,80 мл N-метилморфолина и 0,90 мл (6,92 ммоль) изобутилового эфира хлормуравьиной кислоты и реакционную смесь перемешивают в течение двадцати минут при -20oC. После добавления следующих 0,80 мл N-метилморфолина и 2,52 г (6,92 ммоль) α-(β- фенилацетил)-4-циано-бензиламина реакционную смесь перемешивают в течение следующего часа. Для обработки реакционную смесь разбавляют с помощью 50 мл дихлорметана и органический раствор промывают трехкратно по 40 мл соляной кислотой, двухкратно по 40 мл 5%-ным раствором гидрокарбоната натрия и один раз 40 мл насыщенного раствора хлорида натрия. После высушивания и концентрирования раствора получают 3,97 г сырого продукта. Его очищают с помощью колоночной хроматографии (смесь гексана с этилацетатом). Выход составляет 3,43 г.
д) 3,43 г (5,77 ммоль) Boc-(D)-Phe-Pro-Phg(4-CN)-CH2-Ph-HOAc растворяют в 21,9 мл пиридина и 9,85 мл триэтиламина. Этот раствор при комнатной температуре насыщают газообразным сероводородом и перемешивают в течение ночи. Затем с помощью азота удаляют сероводород и раствор выливают в 5%-ную лимонную кислоту. Водную фазу экстрагируют многократно этилацетатом. Объединенные органические фазы промывают с помощью насыщенного раствора хлорида натрия и после высушивания над сульфатом натрия концентрируют в ротационном испарителе. Выход сырого продукта составляет 4,13 г.
е) 4,13 г Сырого тиоамида вместе с 23,3 мл ацетона и 4,05 мл метилиодида перемешивают в течение восемнадцати часов. Затем реакционный раствор концентрируют в ротационном испарителе. Остаток растворяют в смеси из 9,1 мл абсолютного метанола и 9,1 мл 10%-ного раствора ацетата натрия в абсолютном метаноле и перемешивают в течение восемнадцати часов. После этого реакционную смесь концентрируют досуха и остаток очищают с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (смесь ацетонитрила с водой). Выход гидроацетата составляет 680 мг.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,4 / 9,2 / 8,8 / 8,6 / (4д, 1H, NH): 7,9 - 7,5 (м, 4H, аром. протоны): 7,3 - 7,0 (м, 11H, аром. протоны / NH); 6,0 / 5,7 (2м, 1H α- Phg); 4,4 - 2,6 (м, 8H); 2,2 - 1,6 (м, 4H, β/γ- Pro): 1,3-1,2 (2 уш. с., 9H, Boc).
Масс-спектр: 612 (М+H+), 512 (-Boc), 365, 161, 120.
Пример 21
H-(D)-Phe-Pro-(4-Am)-Phg-CH2Ph
Соединение получают путем отщепления Boc-группы (хлороводород в диоксане) от соединения примера 20.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,5 / 9,4 (2д, 1H, NH); 9,4 / 9,2 (2 уш. с., 3H, NH); 7,8 (д, 2H, арильные протоны), 7,6 (д, 2H, аром. протоны): 7,4 - 7,0 (м, 10H, аром. протоны): 5,8 / 5,6 (2д, 1H α-Phg); 4,35 (м, 2H, 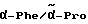 ); 4,1 / 3,9 (4д, 2H, CH2-Ph): 3,7 (м, 2H, CH2); 3,2 - 3,0 (2м, 2H, CH2); 1,8-1,6 (2м, 4H, β/γ-Pro).
); 4,1 / 3,9 (4д, 2H, CH2-Ph): 3,7 (м, 2H, CH2); 3,2 - 3,0 (2м, 2H, CH2); 1,8-1,6 (2м, 4H, β/γ-Pro).
Масс-спектр дигидрохлорида: 512 (М+H+), 393, 252, 161.
Пример 22
H-(D)-Phe-Pro-NH-pAm-[(D,L)- α- Me]-бензил
а) N-(п-Цианобензил)-бензофенонимин:
К раствору 150 г (0,8 моль) 97%-ного бензофенонимина и 144,8 г (0,74 моль) п-циано-бензилбромида в 450 мл ацетонитрила добавляют 270 г (2,0 моль) безводного карбоната калия и перемешивают при комнатной температуре в течение шести часов. Далее, после отсасывания неорганических солей растворитель отгоняют, остаток смешивают с 300 мл воды и многократно экстрагируют этилацетатом. Органическую фазу промывают два раза водой, сушат над сульфатом натрия и концентрируют досуха. После настаивания в эфире получают 180 г белых кристаллов; температура плавления составляет 101 - 102oC.
б) 1-(4-Цианофенил)этиламин:
К раствору диизопропиламида лития, полученному из 8,15 г (0,08 моль) диизопропиламина и 48,3 мл (0,08 моль) 15%-ного раствора бутиллития в гексане, в 100 мл абсолютного тетрагидрофурана при -70oC прикапывают 20,7 г (0,07 моль) N-(п-цианобензил)бензофенонимина и дополнительно перемешивают в течение пятнадцати минут. Затем прикапывают 9,94 г (0,07 моль) метилиодида и реакционную смесь оставляют стоять для повышения ее температуры до комнатной. После добавления 100 мл воды многократно экстрагируют эфиром, эфирную фазу промывают с помощью 5%-ного раствора лимонной кислоты, 5%-ного раствора гидрокарбоната натрия и воды, сушат над сульфатом натрия и эфир отгоняют. Остаток растворяют в 150 мл тетрагидрофурана, добавляют 100 мл 1 н. соляной кислоты и перемешивают в течение ночи при комнатной температуре. Из реакционной смеси в вакууме удаляют тетрагидрофуран, оставшуюся кислую фазу с целью удаления бензофенона многократно экстрагируют эфиром, затем кислую фазу подщелачивают при охлаждении льдом с помощью водного раствора карбоната калия и маслянистое основание экстрагируют дихлорметаном. Экстракт сушат над карбонатом калия. После удаления дихлорметана получают 9,7 г (95%) желтоватого масла, которое без дальнейшей очистки вводят в следующую реакцию.
в) Boc-(D)-Фенилаланил-пролин-(D,L)- α- метил-4-цианобензиламид:
К раствору 3,65 г (25 ммоль) 1-(4-цианофенил)этиламина и 9,1 г (25 ммоль) Boc-(D)-Phe-Pro-OH в 150 мл дихлорметана при -5oC прикапывают 16,2 г диизопропиламина и 22 мл (30 ммоль) 50%-ного раствора ангидрида пропанфосфоновой кислоты в этилацетате. Перемешивают дополнительно в течение двух часов, причем температуру повышают от -5oC до 20oC. Органическую фазу промывают водой, 5%-ным раствором гидрокарбоната натрия и 5%-ным раствором лимонной кислоты, сушат над сульфатом натрия и концентрируют досуха. Получают слегка желтоватый кристаллический остаток, который без дальнейшей очистки вводят в следующую реакцию.
г) (D)-Фенилаланил-пролин-(D,L)- α- метил-4-амидино-бензиламид:
4,1 г Вышеполученного соединения и 4 мл триэтиламина растворяют в 40 мл пиридина, при 0oC раствор насыщают сероводородом и выдерживают при комнатной температуре в течение ночи. Согласно контролю с помощью тонкослойной хроматографии (смесь дихлорметана с метанолом в соотношении 9:1) превращение в тиоамид за это время протекает полностью. Для выделения, далее, пиридин отгоняют в вакууме, остаток растворяют в 250 мл этилацетата и промывают раствором хлорида натрия, 5%-ным раствором лимонной кислоты и раствором гидрокарбоната натрия. После высушивания и отгонки растворителя получают 4,1 г чистого кристаллического тиоамида.
Тиоамид растворяют в 150 мл ацетона и после добавления 7 мл метилиодида оставляют стоять в течение шести часов. После удаления растворителя аморфный остаток перемешивают с безводным эфиром и после этого высушивают. Гидроиодид метилового эфира S- метил-тиоиминокислоты растворяют в 50 мл этанола, смешивают с 15 мл 10%-ного раствора ацетата аммония и нагревают при 60oC в течение трех часов. Для выделения растворитель удаляют, остаток растворяют в 100 мл дихлорметана, отфильтровывают нерастворимые составные части и после этого отгоняют дихлорметан. Путем настаивания со смесью этилацетата с диэтиловым эфиром отделяют растворимые в этой смеси примеси. Полученную смешанную соль иодидацетат растворяют в смеси ацетона с водой в соотношении 3:2 и с помощью содержащей ацетатные группы ионообменной смолы (IRA) переводят в чистый ацетат и затем подвергают сушке вымораживанием. Выделяют порошок белого цвета. Температура плавления составляет 110-115oC; масс-спектр (бомбардировка быстрыми атомами): 508 (М+H+).
д) H-(D)-Phe-Pro-NH-pAm[(D,L)- α- Me]-бензил:
Вышеполученное соединение растворяют в 70 мл дихлорметана и смешивают с 80 мл насыщенного хлороводородом этилацетата. Спустя непродолжительное время выпадает осадок, причем полноты осаждения достигают путем добавления эфира. Полученный осадок отсасывают, промывают не содержащим хлороводорода эфиром и высушивают в вакууме. Получают белые кристаллы. Температура плавления дигидрохлорида составляет 190 - 195oC: масс-спектр (бомбардировка быстрыми атомами): 407 (М+).
Пример 23
Me-(D)-Phe-Pro-(D или L)(4-Am)-Ph ψ (CH2OH)(диастереомер (а))
N-(п-Цианобензил)бензофенонимин в ацетонитриле гидроксиметилируют с помощью безводного карбоната калия, тетрабутиламмонийиодида и параформальдегида. Получают белого цвета кристаллы с температурой плавления 115-117oC. Затем спиртовую группу этерифицируют до простой эфирной группы с помощью трет.-бутилдиметилсилилхлорида и с помощью 0,1 н. метансульфокислоты в тетрагидрофуране расщепляют до защищенного (D,L)-α-гидроксиметил-п- цианобензиламина. Этот амин в стандартных условиях сочетают с Z-Me-(D)-Phe-Pro-OH и путем воздействия сероводорода продукт переводят в тиоамид, который с помощью колоночной хроматографии разделяют на диастереомеры. Чистый диастереомер (а) амидируют и затем путем катализа кислотой или гидрогенолитически отщепляют защитные группы. Полученные после сушки вымораживанием белые кристаллы плавятся при 175 - 180oC. Масс-спектр (бомбардировка быстрыми атомами): 438 (М+H+).
Пример 24
Me-(D)-Phe-Pro-(D или L)(4-Am)-Ph ψ (CH2OH)(диастереомер (б))
Из находящегося в виде чистого диастереомера тиоамида (б) путем амидирования и удаления защитных групп получают белые кристаллы.
Масс-спектр (бомбардировка быстрыми атомами): 438 (М+H+).
Пример 25
Boc-(D)-Phe(4-фтор)-Pro-NH-pAmb
Соединение получают путем взаимодействия Boc-(D)-Phe (4- фтор)-ОН с N-(4-цианобензил)пролинамидом и последующего амидирования. Кристаллы белого цвета: температура плавления гидроацетата составляет 184-187oC.
Пример 26
H-(D)-Phe(4-фтор)-Pro-NH-pAmb
Соединение получают отщеплением Boc-группы от продукта примера 25 в стандартных условиях. Дигидрохлорид представляет собой кристаллы белого цвета с температурой плавления 225 - 230oC; масс-спектр (бомбардировка быстрыми атомами): 412 (М+H+).
Пример 27
Boc-(D)-Phe(4-фтор)-Pro-NH-pAmb
Соединение получают согласно примеру 3 из Boc-(D)-Phe(4- фтор)-ОН. Температура плавления гидроацетата составляет 124 - 137oC.
Пример 28
H-(D)-Phe(4-фтор)-Pro-NH-pAmb
Согласно методике A.I.в. отщепляют Boc-группу от соединения примера 27. Получают соединение в виде дигидрохлорида, температура плавления которого составляет 221 - 234oC.
Пример 29
Boc-(D,L)-Phe(4-бром)-Pro-NH-pAmb
Соединение получают исходя из Boc-(D,L)-Phe(4-бром)-ОН и гидрохлорида H-Pro-п-циано-бензиламида согласно примеру 3.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 8,4/8,1 (т, 1H, NH): 7,78 (2д, 2H, аром. протоны): 7,2 (м, 2H, аром. протоны), 7,0 (2д, 1H, NH), 4,5 - 4,2 (м, 4H, CH2 / 2 α- H); 3,6 (м, 2H, Pro); 3,0 - 2,6 (м, 2H, CH2-фенил); 2,15 - 1,7 (м, 4H, β/γ- Pro); 1,3/1,2 (2д, 9H, Boc).
Масс-спектр (бомбардировка быстрыми атомами): 575 / 574 (М+H+).
Температура плавления гидроацетата составляет 171oC (разложение).
Пример 30
H-(D,L)-Phe(4-бром)-Pro-NH-pAmb
Соединение получают путем отщепления Boc-группы от продукта примера 29.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,2 (уш. с., 4H); 8,6 / 8,5 (2т, 1H, NH); 7,8 (2д, 2H, аром. протоны); 7,4 (м, 4H, аром. протоны); 7,2 (м, 2H, аром. протоны); 7,2 (уш. с., 3H, NH); 4,38 (2дд, 3H, CH2 / α- Н); 4,2 (м, 1H, α- Н); 3,8 - 3,6 (м, 2H, Pro); 3,1 - 2,8 (м, 2H, CH2); 2,2 - 1,7 (м, 4H, Pro); масс-спектр (бомбардировка быстрыми атомами): 474 (М+H+); температура плавления дигидроацетата составляет 56oC (разложение).
Пример 31
H-(D)-Phe(4-OH)-Pro-NH-pAmb
Соединение получают путем гидрогенолитического отщепления бензильной группы в присутствии палладия-на-угле от соединения примера 37 согласно обычному отщеплению защитной группы Z по методике A.I.б. После отфильтровывания катализатора и удаления растворителя получают дигидрохлорид путем осаждения с помощью раствора хлороводорода в эфире. Температура плавления составляет 129-140oC.
Пример 32
Boc-(D)-Phe(4-MeO)-Pro-NH-pAmb
Соединение получают из Boc-(D)-Phe(4-MeO)-OH согласно примеру 3. Температура плавления гидроацетата составляет 67 - 83oC.
Пример 33
H-(D)-Phe(4-MeO)-Pro-NH-pAmb
Отщепление Boc-группы от соединения примера 32 осуществляют согласно методике A.I.в. Температура плавления дигидрохлорида составляет 215 - 227oC.
Пример 34
Boc-(D,L)-Phe(4-EtO)-Pro-NH-pAmb
Соединение получают из Boc-(D,L)-Phe(4-EtO)-OH согласно методике примера 3. Температура плавления гидроацетата составляет 115 - 145oC.
Пример 35
H-(D,L)-Phe(4-EtO)-Pro-NH-pAmb
Отщепление Boc-группы от соединения примера 34 осуществляют согласно методике A.I.в. Температура плавления дигидрохлорида составляет 218 - 230oC.
Пример 36
Boc-(D)-Phe(4-BzlO)-Pro-NH-pAmb
Соединение получают из Boc-(D)-Phe(4-BzlO)-OH согласно методике примера 3. Температура плавления гидроацетата составляет 111 - 125oC.
Пример 37
H-(D)-Phe(4-BzlO)-Pro-NH-pAmb
Отщепление Boc-группы от соединения примера 36 осуществляют согласно методике A.I.в. Температура плавления дигидрохлорида составляет 201 -210oC.
Пример 38
Boc-(D,L)-Phe(4-3THA)-Pro-NH-pAmb
Соединение получают в виде гидроацетата согласно примеру 3, исходя из Boc-(D,L)-Phe(4-этил)-ОН.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,25 (2H, амидин); 8,85 (2H, амидин): 8,42 и 8,09 (вместе 1H, NH); 7,80 - 6,95 (9H, аром. протоны и NH); 4,45 - 4,20 (4H, 2 CH и CH2; 3,75 - примерно 3,0 (2H, CH2); 3,0 - 2,6 (2H, CH2); 2,6 - примерно 2,5 (2H, CH2); 2,3 - 1,5 (4H, 2 CH2); 1,3 - примерно 1,2 (9H, Boc): примерно 1,2 - 1,5 (3H, CH3).
Пример 39
H-(D,L)-Phe(4-3THA)-Pro-NH-pAmb
Дигидрохлорид получают путем отщепления Boc-группы от соединения примера 38 согласно методике A.I.в.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,55 - 9,40 (2H, амидин): 9,32 - 9,20 (2H, амидин); 8,90 и 8,70 (вместе 1H, NH); 8,85 - 8,75 и 8,40 и 8,30 (вместе 3H, NH3); 7,90 - 7,05 (8H, аром. протоны): 4,50 - 4,10 (4H, CH2 и 2 CH); 2,6 - примерно 2,5 (2H, CH2; 1,9-1,3 (4H, 2 CH2) 1,2/1,1 (3H, CH3).
Пример 40 Boc-(D,L)-Phe(4-изопропил)-Pro-NH-pAmb
Гидроацетат получают путем превращения Boc-(D,L)-Phe(4- изопропил)-ОН согласно примеру 3.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,25 (2H, амидин): 8,50 (2H, амидин); 8,43 и 8,09 (вместе 1H, NH): 7,80 - 6,95 (9H, аром. протоны и NH); 4,45 - 4,20 (4H, 2 CH и CH2); 3,7 - примерно 3,2 (2H, CH2); 3,0 - 2,6 (3H, CH2 и CH); 2,2 - 1,5 (4H, 2 CH2); 1,3 - примерно 1,2 (9H, Boc); примерно 1,2 - 1,05 (6H, 2CH3).
Пример 41
H-(D,L)-Phe(4-изопропил)-Pro-NH-pAmb
Дигидрохлорид получают путем отщепления Boc-группы от соединения примера 40 согласно методике A.I.в.
1H-ЯМР (дейтерированный диметилсульфоксид: δ в м.д.): 9,50 - 9,40 (2H, амидин); 9,30 - 9,20 (2H, амидин); 8,90 и 8,70 (вместе 1H, NH); 8,75 и 8,30 (вместе 3H, NH3 +); 7,85 - 7,10 (8H, аром. протоны); 4,50 - 4,10 (4H, CH2 и 2CH); примерно 3,8 - 3,3 (2H, CH2); 3,3 - 2,8 (3H, CH2 и CH); 2,4 - 1,3 (4H, 2 CH2); 1,20 (6H, 2 CH3).
Пример 42
Z-(D)-Phe(4-tBuO)-Pro-NH-pAmb
Гидроацетат получают путем превращения Z-(D)-Phe(4-tBuO)-OH согласно примеру 3. Температура плавления составляет 92 - 104oC.
Пример 43
H-(D)-Phe(4-tBuO)-Pro-NH-pAmb
От соединения примера 42 гидрогенолитически в присутствии палладия-на-угле, согласно методике A.I.б., отщепляют защитную Z - группу. Получают продукт в виде дигидроацетата с температурой плавления 94-102oC.
Пример 44
Boc-(D,L)-Phe(4-tBu)-Pro-NH-pAmb
Гидроацетат получают путем превращения Boc-(D,L)-Phe(4-tBu)- OH согласно примеру 3. Температура плавления составляет 151 - 158oC.
Пример 45
H-(D,L)-Phe(4-tBu)-Pro-NH-pAmb
Гидрохлорид получают путем отщепления Boc-группы от соединения примера 44 согласно методике A.I.в. Температура плавления составляет 96-110oC.
Пример 46
H-(D,L)-Phe(4-фенил)-Pro-NH-pAmb
Согласно примеру 3, Boc-(D,L)-Phe(4-фенил)-ОН превращают в гидроацетат Boc-(D, L)-Phe(4-фенил)-Pro-NH-pAmb и затем Boc-группу отщепляют согласно методике A.I.в.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,50 - 9,40 (2H, амидин); 9,30 - 9,20 (2H, амидин): 8,90 и 8,70 (вместе 1H, NH): 8,80 и 8,35 (вместе 3H, NH3 +): 7,85 - 7,25 (13H, аром. протоны); 4,50 - 4,15 (4H, CH2 и 2 CH); примерно 3,8 - 3,2 (2H, CH2); 3,10 - 2,95 (2H, CH2); 2,6 - 1,3 (4H, 2CH2).
Пример 47
Boc-(D,L)-Phe(4-H-бутил)-Pro-NH-pAmb
Соединение настоящего примера получают согласно примеру 3, исходя из Boc-(D,L)-Phe(4-н-бутил)-ОН и гидрохлорида H-Pro-п- цианобензиламида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.) гидроацетата: 10,0 - 9,2 (уш. с., NH); 8,4 / 8,0 (2т, 1H, NH): 7,78 (2д, 2H, аром. протоны); 7,42 (м, 3H, аром. протоны); 7,25 - 7,0 (м, 4H, аром. протоны / NH): 4,4 - 4,2 (м, 4H,(CH2 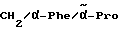 ; 3,7 - 3,0 (м, 2H, δ- Pro / CH2; 3,0 - 2,6 (м, 4H, 2 CH2-фенил); 2,2 - 1,7 (м, 5H, β/γ- Pro); 1,5 (м, 3H, β/γ- Pro / CH2; 1,25 (м, 9H, Boc); 0,95 (т, 3H, CH3). Масс-спектр: 550 (М+H+); 4,50 (-Boc); 247, 185, 134.
; 3,7 - 3,0 (м, 2H, δ- Pro / CH2; 3,0 - 2,6 (м, 4H, 2 CH2-фенил); 2,2 - 1,7 (м, 5H, β/γ- Pro); 1,5 (м, 3H, β/γ- Pro / CH2; 1,25 (м, 9H, Boc); 0,95 (т, 3H, CH3). Масс-спектр: 550 (М+H+); 4,50 (-Boc); 247, 185, 134.
Пример 48
H-(D,L)-Phe(4-H-бутил)-Pro-NH-pAmb
Соединение получают путем отщепления Boc-группы от продукта примера 47.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,4 (д, 2H, NH); 9,2 (д, 2H, NH): 8,9 / 8,6 (2т, 1H, NH); 7,75 (2д, 2H, аром. протоны): 7,45 (д, 2H, аром. протоны); 7,23 (д, 1H, NH): 7,15 (м, 4H, аром. протоны); 4,4 - 4,2 (3м, 4H, CH2 / 2 α-Н): 3,6 (м, 2H, γ-Pro / CH2); 3,1 / 2,9 (2м, 2H, CH2-фенил); 2,56 / 2,4 (2м, 2H, CH2); 2,1 / 1,9 /1,6 / 1,3 (4м, 11H, β/γ-Pro / CH2)); 0,95 (2т, 3H, CH3).
Масс-спектр: 450 (M+H+); 247, 176; соединение примера 48 находится в виде дигидроацетата.
Пример 49
Boc-(D)-Phe(4-COOMe)-Pro-NH-pAmb
а) Boc-(D)-Phe(4-COOMe)-Pro-NH-п-цианобензил:
12,5 г Boc-(D)-Tyr(Bzl)-OH согласно примеру 3 превращают в 18,9 г Boc-(D)-Tyr(Bzl)-Pro-п-цианобензил, который растворяют в 780 мл метанола и гидрируют с присутствии палладия-на-угле при комнатной температуре. Спустя 6 часов катализатор отфильтровывают и растворитель удаляют. Получают с количественным выходом Boc-(D)- Tyr-Pro-п-цианобензиламид. 12,5 г Вышеполученного соединения растворяют в 50 мл пиридина и вместе с 4,7 мл ангидрида трифторметансульфокислоты перемешивают в течение одного часа при 0oC. После выдерживания в течение следующего часа при комнатной температуре смесь выливают в воду и экстрагируют этилацетатом. Органическую фазу промывают водой и сушат над сульфатом натрия. Получают 14 г Boc-(D)-Tyr(SO2CF3)-Pro-п-цианобезиламида. 4 г Трифлата вносят в смесь из 1,93 мл триэтиламина, 134 мг бисдифенилфосфинпропана, 73 мг ацетата палладия-(II), 1 мл метанола и 40 мл диметилформамида и при температуре 60 - 70oC обрабатывают газообразным монооксидом углерода вплоть до прекращения поглощения газа. Смесь выливают в насыщенный раствор хлорида натрия и экстрагируют этилацетатом. После промывки органической фазы 10%-ным раствором лимонной кислоты, высушивания над сульфатом натрия и удаления растворителя получают 3,3 г Boc-(D)-Tyr(4-COOMe)-Pro-п-цианобензиламида.
б) Boc-(D)-Phe(4-COOMe)-Pro-NH-pAmb:
1,7 г Вышеполученного нитрила превращают в амидингидрохлорид согласно примеру 3. Получают 650 мг соединения после очистки с помощью колоночной хроматографии на силикагеле.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,4 - 8,7 (4H, амидин); 8,9 - 8,1 (1H, NH (2 геометрических изомера)); 7,9 - 7,2 (9H, аром. протоны и NH): 3,85 (3H, COOMe); 1,3 - 1,2 (9H, Boc).
Пример 50
H-(D)-Phe(4-COOMe)-Pro-NH-pAmb
1,3 г Соединения примера 49 превращают в амидингидроацетат с помощью содержащей ацетатные группы ионообменной смолы и согласно методике A.I.в. отщепляют Boc-группу. Получают 1,0 г продукта в виде дигидрохлорида с температурой плавления 204 - 207oC (разложение).
Пример 51
H-(D)-Phe(4-нитpo)-Pro-NH-pAmb
а) Boc-пролин-(п-амидинобензил)амид:
Boc-пролин-(п-цианобензил)амид (получение описывается в примере 5) согласно методике A.III.1. путем обработки сероводородом переводят в тиоамид и затем превращают в амидин. Амидин получают в форме белых кристаллов с температурой плавления 237 - 239oC. Масс-спектр (бомбардировка быстрыми атомами): 347 (М+H+).
б) N-(п-Амидинобензил)пролинамиддигидрохлорид:
Согласно методике A.I.в., от вышеполученного соединения отщепляют защитную Boc-группу. Дигидрохлорид выделяют в виде очень гигроскопичного порошка с температурой плавления 130 - 140oC. Масс-спектр (бомбардировка быстрыми атомами): 247 (М+H+).
в) Boc-(D)-Phe(4-нитро)-Pro-NH-pAmb;
Раствор 3,9 г (12,6 ммоль) Boc-(D)-Phe(4-нитро)-OH в 40 мл тетрагидрофурана после добавления 1,9 г (12,6 ммоль) 1-гидрокси- бензотриазола и 3,3 г (25 ммоль) дициклогексилкарбодиимида перемешивают в течение четырех часов при комнатной температуре. Выпавшую в осадок мочевину отсасывают и промывают дополнительно небольшим количеством тетрагидрофурана.
К полученному фильтрату при 5oC добавляют раствор 4,1 г (12,6 ммоль) N-(п-амидинобензил)пролинамиддигидрохлорида и 1,6 г гидрокарбоната натрия в 6 мл воды. После перемешивания в течение сорока восьми часов при комнатной температуре растворитель отгоняют, остаток обрабатывают этанолом, отфильтровывают от нерастворимых составных частей и снова концентрируют. Остаток очищают путем колоночной хроматографии на силикагеле при использовании смеси дихлорметана с метанолом и 50%-ной уксусной кислотой в соотношении 45 : 5 : 1,5. Из элюата однородных фракций удаляют путем отгонки растворитель, под конец с толуолом в качестве добавки, и остаток перекристаллизуют из 50 мл ацетона при добавлении небольшого количества воды. Выделяют 3,3 r (48% от теоретически рассчитанного количества) амидинацетата в форме кристаллов белого цвета; температура плавления составляет 162 - 165oC; масс-спектр (бомбардировка быстрыми атомами): 539,5 (M+H+).
г) H-(D)-Phe(4-нитро)-Pro-NH-pAmb:
Отщепление Boc-группы осуществляют согласно методике A.I.в. Соединение получают в виде дигидрохлорида в форме кристаллов белого цвета; температура плавления составляет 218 - 225oC (разложение); масс-спектр (бомбардировка быстрыми атомами): 439 (М+H+).
Пример 52
Boc-(D,L)-Phe(3-фтор)-Pro-NH-pAmb
Соединение получают согласно примеру 3, исходя из Boc-(D,L)- Phe(3-фтор)-ОН и гидрохлорида H-Pro-п-цианобензиламида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,4 - 9,0 (уш. с., NH); 8,9 / 8,4 / 8,15 (3т, 1H, NH); 7,8 (уш. с., 2H, аром. протоны), 7,45 (2д, 2H, аром. Н); 7,35 - 6,9 (2м, 5H, аром. Н / NH); 4,5 - 4,2 (м, 4H, 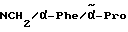 ); 3,7 / 3,5 / 3,2 (3м, 2H, δ-Pro); 3,0 - 2,7 (2м, 2H, арил-CH2); 2,2 - 1,7 (3м, 4H, β/γ- Pro); 1,25 (2c, 9H, Boc).
); 3,7 / 3,5 / 3,2 (3м, 2H, δ-Pro); 3,0 - 2,7 (2м, 2H, арил-CH2); 2,2 - 1,7 (3м, 4H, β/γ- Pro); 1,25 (2c, 9H, Boc).
Масс-спектр(бомбардировка быстрыми атомами): 511 (M+H+).
Пример 53
H-(D,L)-Phe(3-фтор)-Pro-NH-pAmb
Соединение получают путем отщепления Boc-группы от соединения примера 52.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,4 / 9,2 (2д, 4H, NH); 8,9 / 8,55 (1H, NH); 8,75 / 8,3 (2 уш. c., 3H, NH3); 7,8 (уш. с., 2H, аром. протоны): 7,45 (2д, 2H, аром. протоны): 7,4 - 7,05 (м, 4H, аром. протоны); 4,45 - 4,2 (м, 4H, 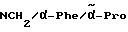 ); 3,9 - 3,3 (2H, δ-Pro); 3,2 - 2,8 (м, 2H, арил-CH2); 2,2 - 1,8 (4H, β/γ-Pro).
); 3,9 - 3,3 (2H, δ-Pro); 3,2 - 2,8 (м, 2H, арил-CH2); 2,2 - 1,8 (4H, β/γ-Pro).
Масс-спектр дигидрохлорида (бомбардировка быстрыми атомами): 411 (М+H+).
Пример 54
Boc-(D,L)-Phe(3-xлop)-Pro-NH-pAmb
Гидроацетат получают из Boc-(D,L)-Phe(3-хлор)-OH аналогично примеру 3.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,4 - 8,6 (4H, амидин); 8,45 / 8,15 (1H, NH); 7,8-7,0 (9H, аром. H и NH); 1,3- 1,2 (9H, Boc).
Пример 55
H-(D,L)-Phe(3-хлор)-Pro-NH-pAmb
Отщепление Boc-группы от соединения примера 54 осуществляют согласно методике A.I.в. Получают продукт в виде дигидрохлорида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,5 - 9,2 (4H, амидин): 8.95 (1H, NH); 8,8 / 8,4 (3H, NH3 +); 7,9 - 7,2 (8H, аром. протоны); 4,50 - 4,15 (4H, CH2 и 2CH): 3,8 - примерно 3,3 (2H, CH2); 3,25 - 2,95 (2H, CH2); 2,2 - 1,5 (4H, 2 CH2).
Пример 56
H-(D,L)-Phe(3-OH)-Pro-NH-pAmb
Соединение получают путем отщепления Boc-группы от Boc-(D,L)- Phe(3-OH)-Pro-NH-pAmb.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,6 / 9,0 (уш. с., NH); 8,8 / 8,5 (2т, 1H, NH); 8,2 (уш. с., NH); 7,8 (2д, 2H, аром. протоны); 7,5 (2д, 2H, аром. протоны): 7,1 (м, 1H, аром. протоны); 6,8 - 6,6 (м, 3H, аром. протоны); 4,4 - 4,1 (м, 4H, CH2 / 2 α-Н); 3,8 - 3,6 (м, 2H, Pro), 3,0 / 2,8 (2м, 2н, CH2); 2,1 - 1,5 (м, 4H, β/γ-Pro).
Масс-спектр (бомбардировка быстрыми атомами): 410 (М+H+).
Температура плавления дигидроацетата составляет 168oC (разложение).
Пример 57
Boc-(D,L)-Phe(3-MeO)-Pro-NH-pAmb
Гидроацетат получают из Boc-(D,L)-Phe(3-MeO)-OH согласно примеру 3.
1H-ЯМР (дейтерированный диметилсульфоксид;  в м.д.): 9,4 - 8,7 (4H, амидин); 8,4 / 8,1 (1H, NH); 7,8 - 6,7 (9H, аром. протоны и NH): 3,7 (3H, OCH3); 1,3-1,2 (9H, Boc).
в м.д.): 9,4 - 8,7 (4H, амидин); 8,4 / 8,1 (1H, NH); 7,8 - 6,7 (9H, аром. протоны и NH): 3,7 (3H, OCH3); 1,3-1,2 (9H, Boc).
Пример 58
H-(D,L)-Phe(3-MeO)-Pro-NH-pAmb
Отщепление Boc-группы от соединения примера 57 осуществляют согласно методике A.I.в. Получают продукт в виде дигидрохлорида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,55 -9,15 (4H, амидин); 8,9 / 8,7 (1H, NH): 8,75 / 8,30 (3H, NH3 +); 7,9 - 6,7 (8H, аром. протоны): 4,5 - 4,1 (4H, CH2 и 2 CH); 3,75 / 3,72 (3H, OCH3); 3,3 - 2,9 (2H, CH2); 2,2 - 1,4 (4H, 2 CH2).
Пример 59
Boc-(D,L)-Phe(3-PhO)-Pro-NH-pAmb
Соединение получают из Boc-(D,L)-Phe(3-PhO)-OH. Получают амидингидроацетат согласно примеру 3.
Пример 60
H-(D,L)-Phe(3-PhO)-Pro-NH-pAmb
Дигидрохлорид получают путем отщепления Boc-группы от соединения примера 59.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,5 - 9,2 (4H, амидин): 8,9 / 8,2 (1H, NH); 8,75 / 8,35 (3H, NH3 +); 7,85 - 6,80 (8H, аром. протоны), 4,50 - 4,10 (4H, CH2 и 2CH): 3,8 - 2,9 (4H, 2 CH2); 2,8 - 1,5 (4H, CH2).
Пример 61
Boc-(D,L)-Phe(3-Me)-Pro-NH-pAmb
Соединение получают согласно примеру 3, исходя из Boc-(D,L)- Phe(3-Me)-OH и гидрохлорида H-Pro-п-цианобензиламида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 8 / 8,4 / 8,05 (3т, 1H, NH), 7,74 (2д, 2H, аром. протоны); 7,45 (2д, 2H, аром. протоны): 7,2 - 6,9 (м, 5H, аром. протоны / NH); 4,4 - 4,2 (м, 4H, NCH2, 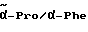 ); 3,7 - 3,1 (2м, 2H, δ-Pro): 2,9 - 2,7 (2H, арил-CH2); 2,5 (2с, 3H, CH3); 2,1 - 1,6 (м, 4H, β/γ-Pro); 1,25 (2c, 9H, Boc).
); 3,7 - 3,1 (2м, 2H, δ-Pro): 2,9 - 2,7 (2H, арил-CH2); 2,5 (2с, 3H, CH3); 2,1 - 1,6 (м, 4H, β/γ-Pro); 1,25 (2c, 9H, Boc).
Масс-спектр гидроацетата: 508 (М+H+), 408 (-Boc), 277, 247.
Пример 62
H-(D,L)-Phe(3-Me)-Pro-NH-pAmb
Соединение получают путем отщепления Boc-группы от соединения примера 61.
Масс-спектр дигидроацетата: 408 (М+H+), 247, 185, 134, 93.
Пример 63
H-(D,L)-Phe(3-фенил)-Pro-NH-pAmb
Соответствующее, защищенное Boc-группой соединение получают согласно примеру 3 из Boc-(D, L)-Phe(3-фенил)-ОН и затем согласно методике A.I.в. расщепляют до дигидрохлорида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,5 - 9,2 (4H, амидин); 8,9 / примерно 8,7 (1H, NH); 8,8 / 8,35 (3H, NH3 +); 7,85 - 7,25 (13H, аром. протоны); 4,5 - 4,15 (4H, CH2 и 2 CH): 3,2 - 3,00 (2H, CH2); 2,2 - 1,4 (4H, 2 CH2).
Пример 64
Boc-(D,L)-Phe(3-CF3)-Pro-NH-pAmb
Соединение получают аналогично примеру 3, исходя из Boc- (D,L)-Phe(3-CF3)-OH и гидрохлорида H-Pro-п-цианобензиламида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 10,0 - 9,2 (уш. с. , NH); 8,9 / 8,4 / 8,15 (3т, 1H, NH); 7,8 - 7,4 (8H, аром. протоны); 7,22 / 7,05 (2д, 1H, NH): 4,6 - 4,2 (4H, 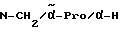 ); 3,8 - 3,4 (2H, δ-Pro): 3,1 / 2,8 (2H, арил-CH2); 2,1 - 1,6 (4H, β/γ-Pro): 1,2 (2с, 9H, Boc).
); 3,8 - 3,4 (2H, δ-Pro): 3,1 / 2,8 (2H, арил-CH2); 2,1 - 1,6 (4H, β/γ-Pro): 1,2 (2с, 9H, Boc).
Масс-спектр гидроацетата: 562 (М+H+), 462 (-Boc), 247, 188, 134.
Пример 65
H-(D,L)-Phe(3-CF3)-Pro-NH-pAmb
Соединение получают путем отщепления Boc-группы от соединения примера 64.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,4 (2с, 2H, NH); 9,2 / (2с, 2H, NH); 8,9 / 8,8 (2т, 1H, NH); 8,8 / 8,6 / 8,4 (3 уш. с., 3H, NH3 +); 7,8 - 7,4 (8H, аром. протоны); 4,42 - 4,1 (4H, 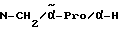 Pro); 3,8 (м, 1H, δ-Pro / арил- CH2); 2,2-1,5 (4H, β/γ-Pro).
Pro); 3,8 (м, 1H, δ-Pro / арил- CH2); 2,2-1,5 (4H, β/γ-Pro).
Температура плавления дигидроацетата составляет 195-197oC.
Пример 66
Boc-(D,L)-Phe(2-фтор)-Pro-pAmb
Соединение получают согласно примеру 3, исходя из Boc-(D,L)- Phe(2-фтор)-OH и гидрохлорида H-Pro-NH-п-цианобензиламида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,8 - 9,2 (уш. с., NH); 8,5 / 8,2 (2т, 1H, NH); 7,75 (2д, 2H, аром. протоны): 7,45 (2д, 2H, аром. протоны); 7,4 - 7,0 (м, 5H, аром. протоны / NH); 4,6 - 4,2 (м, 4H, 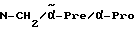 ); 3,6 - 3,0 (4м, 2H, □-Pro ; 2,9 - 2,7 (2м, 2H, арил-CH2); 2,2 - 1,7 (3м, 4H, β/γ-Pro): 1,2 (2с, 9H, Boc).
); 3,6 - 3,0 (4м, 2H, □-Pro ; 2,9 - 2,7 (2м, 2H, арил-CH2); 2,2 - 1,7 (3м, 4H, β/γ-Pro): 1,2 (2с, 9H, Boc).
Масс-спектр гидроацетата: 512 (М+H+), 412 (-Boc), 247, 134.
Пример 67
H-(D,L)-Phe(2-фтор)-Pro-NH-pAmb
Соединение получают путем отщепления Boc-группы от соединения примера 66.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,4 (2с, 2H, NH); 9,15 (2с, 2H, NH): 8,9 / 8,6 (2 уш. с., 3H, NH); 7,8 (2д, 2H, аром. протоны): 7,45 (2д, 2H, аром. протоны): 7,4 - 7,1 (м, 4H, аром. протоны); 4,5 - 4,2 (3м, 4H, 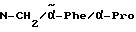 ); 3,6 - 3,2 (2м, 2H, δ-Pro): 3,0 / 2,7 (2м, 2H, арил-CH2); 2,2 - 1,5 (5м, 4H, β/γ-Pro).
); 3,6 - 3,2 (2м, 2H, δ-Pro): 3,0 / 2,7 (2м, 2H, арил-CH2); 2,2 - 1,5 (5м, 4H, β/γ-Pro).
Масс-спектр дигидрохлорида: 412 (M+H+), 247, 134.
Пример 68
Boc-(D,L)-Phe(2-хлоp)-Pro-NH-pAmb
Гидроацетат получают из Boc-(D,L)-Phe(2-XAOp)-OH согласно примеру 3.
Пример 69
H-(D,L)-Phe(2-хлор)-Pro-NH-pAmb
Дигидрохлорид получают из соединения примера 68 согласно методике A.I.в.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,5 - 9,2 (4H, амидин): 8,9 / 8,7 (1H, NH): 8,85 / 8,45 (3H, NH3 +), 7,9 - 7,2 (8H, аром. протоны); 4,5 - 4,1 (4H, CH2 и 2 CH); 3,8 - 3,0 (4H, 2 CH2; 2,2 - 1,4 (4H, 2 CH2).
Пример 70
Boc-(D,L)-Phe(2-OH)-Pro-NH-pAmb
Соединение получают согласно примеру 3, исходя из Boc-(D,L)- Phe(2-OH)-OH и гидрохлорида H-Pro-NH-п-цианобензиламида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 8,4 / 8,0 (2т, 1H, NH): 7,7 (2д, 2H, аром. протоны); 7,4 (2д, 2H, аром. протоны); 7,2 / 7,15 (2д, 1H, NH): 7,0 - 6,6 (4H, аром. протоны): 4,45 - 4,2 (м, 4H, 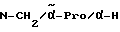 ); 3,8 - 3,2 (2H, δ Pro); 3,0 / 2,8 (2м, 2H, арил-CH2); 2,1 - 1,6 (4H, β/γ- Pro); 1,2 (2с, 9H, Boc).
); 3,8 - 3,2 (2H, δ Pro); 3,0 / 2,8 (2м, 2H, арил-CH2); 2,1 - 1,6 (4H, β/γ- Pro); 1,2 (2с, 9H, Boc).
Масс-спектр гидроацетата: 510 (М+H+), 410 (-Boc), 247, 134.
Пример 71
H-(D,L)-Phe(2-OH)-Pro-NH-pAmb
Соединение получают путем отщепления Boc-группы от соединения примера 70.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,4 - 9,0 (уш. с., NH); 8,85 / 8,5 (2т, 1H, NH): 7,8 (2д, 2H, аром. протоны); 7,5 (2д, 2H, аром. протоны): 7,2 - 6,7 (4H, аром. протоны): 4,4 - 3,7 (м, 4H, 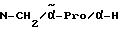 ); 3,4 - 3,2 (2H, δ-Pro); 3,1 - 2,75 (2H, арил-CH2): 2,1 -1,4 (4H, β/γ-Pro).
); 3,4 - 3,2 (2H, δ-Pro); 3,1 - 2,75 (2H, арил-CH2): 2,1 -1,4 (4H, β/γ-Pro).
Масс-спектр дигидроацетата: 410 (М+H+): 369, 277, 247.
Пример 72
Boc-(D,L)-Phe(2-MeO)-Pro-NH-pAmb
Гидроацетат получают согласно примеру 3 из Boc-(D,L)-Phe(2- MeO)-OH.
Пример 73
H-(D,L)-Phe(2-MeO)-Pro-NH-pAmb
Дигидрохлорид получают из соединения примера 72 согласно методике A.I.в.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,55 - 9,25 (4H, амидин): 8,90 / 8,65 (1H, NH): 8,7 / 8,2 (3H, NH3 +); 7,9 - 6,8 (8H, аром. протоны); 4,5 - 4,1 (4H, CH2 и 2 CH); 3,80 (3H, OCH3); примерно 3,8 - 3,3 (2H, CH2); 3,2 - 2,6 (2H, CH2); 2,2 - 1,4 (4H, 2 CH2).
Пример 74
Boc-(D,L)-Phe(2-Me)-Pro-NH-pAmb
Соединение получают согласно примеру 3, исходя из Boc-(D,L)- Phe(2-Me)-OH и гидрохлорида H-Pro-п-цианобензиламида.
1H-ЯМР (дейтерированный диметилсульфоксид: δ в м.д.): 8,4 / 8,05 (2т, 1H, NH); 7,75 (2д, 2H, аром. протоны); 7,4 (2д, 2H, аром. протоны): 7,2 - 7,0 (м, 5H, аром. протоны / NH); 4,6 - 4,2 (м, 4H, CH2 / 2 α-H); 3,7 - 3,55 (2м, 2H, δ-Pro); 3,0 - 2,6 (м, 2H, CH2); 2,3 (2с, 3H, CH3); 2,2-1,6 (м, 4H, β/γ-Pro); 1,35 - 1,2 (1с, 9H, Boc).
Масс-спектр (бомбардировка быстрыми атомами) гидроацетата: 508 (М+H+).
Пример 75
H-(D,L)-Phe(2-Me)-Pro-NH-pAmb
Соединение получают путем отщепления Boc-группы от соединения примера 74.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.) дигидрохлорида: 9,35 (c, 2H, NH); 9,05 (c, 2H, NH); 8,8 / 8,5 (2т, 1H, NH); 7,8 / 7,75 (2д, 2H, аром. H); 7,5 / 7,45 (2д, 2H, аром. H); 7,2-7,0 (м, 4H, аром. H), 4,4-4,2 (3м, 4H, CH2 / 2 α-H); 3,6 / 3 (2M, 2H, δ-Pro); 3,1 / 3,0 (2м, 2H, CH2); 2,38 (м, 3H, CH3); 2,2 - 1,3 (4H, β/γ-Pro).
Масс-спектр: 408 (М+H+), 247, 185, 134.
Пример 76
Boc-(D,L)-Phe(2-изопропил)-Pro-NH-pAmb
Гидроацетат получают из Boc-(D,L)-Phe(2-изопропил)-OH согласно примеру 3.
Пример 77
H-(D,L)-Phe(2-изопропил)-Pro-NH-pAmb
Дигидрохлорид получают из соединения примера 76 согласно методике A.I.в. Температура плавления составляет 220 - 221oC.
Пример 78
Boc-(D,L)-Phe(2-фенил)-Pro-NH-pAmb
Гидроацетат получают из Boc-(D,L)-Phe(2-фенил)-OH согласно примеру 3.
Пример 79
H-(D,L)-Phe(2-фенил)-Pro-NH-pAmb
Дигидрохлорид получают из соединения примера 78 согласно методике A.I.в.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,5 - 9,2 (4H, амидин): 8,85 / 8,67 (1H, NH); 8,6 / 8,2 (3H, NH3 +); 7,85 - 7,15 (13H, аром. протоны); 4,4 - 3,0 (8H, 3 CH2 и 2 CH); 2,2 - 1,4 (4H, 2 CH2).
Пример 80
Boc-(D,K)-Phe(3,4-дифтор)-Pro-NH-pAmb
Гидроацетат получают из Boc-(D,L)-Phe(3,4-дифтор)-OH (J. Med. Chem., 10, 64 (1967)) согласно примеру 3 в виде белых кристаллов с температурой плавления 110-114oC.
Масс-спектр (бомбардировка быстрыми атомами): 530 (М+H+).
Пример 81
H-(D,L)-Phe(3,4-дифтор)-Pro-NH-pAmb
Дигидрохлорид получают путем отщепления Boc-группы от соединения примера 80 согласно методике A.I.в.; белые кристаллы с температурой плавления 190-195oC (разложение).
Масс-спектр (бомбардировка быстрыми атомами): 430 (М+H+).
Пример 82
Boc-(D,L)-Phe(3,4-дихлор)-Pro-NH-pAmb
Гидроацетат получают путем превращения Boc-(D,L)-Phe(3,4- дихлор)-ОН (Int. J. Pept. Protein Res., 30, 13 (1987)) согласно примеру 3 в виде кристаллов белого цвета; температура плавления составляет 135 - 138oC.
Масс-спектр (бомбардировка быстрыми атомами): 562 (М+H+).
Пример 83
H-(D,L)-Phe(3,4-дихлор)-Pro-NH-pAmb
Дигидрохлорид получают путем отщепления Boc-группы от соединения примера 82 согласно методике A. I. в. в виде кристаллов белого цвета; температура плавления составляет 208 - 212oC.
Масс-спектр (бомбардировка быстрыми атомами): 462 (М+H+).
Пример 84
Boc-(D,L)-Phe(3-хлор-4-MeO)-Pro-NH-pAmb
Гидроацетат получают из Boc-(D,L)-Phe(3-хлор-4-MeO)-OH согласно примеру 3.
Пример 85
H-(D,L)-Phe(3-хлор-4-MeO)-Pro-NH-pAmb
Дигидрохлорид получают путем отщепления Boc-группы от соединения примера 84 согласно методике A.I.в.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,58 - 9,30 (4H, амидин): 8,98 / 8,85 (1H, NH): 8,8 / 8,35 (3H, NH3 +); 7,9 - 7,0 (7H, аром. протоны): 4,50 - 4,20 (4H, CH2 и 2 CH); 3,85 / 3,82 (3H, OCH3); 3,2 -2,9 (2H, CH2); 2,2 - 1,5 (4H, 2 CH2).
Пример 86
Boc-(D,L)-Phe(3-хлор-4-этокси)-Pro-NH-pAmb
Соединение получают в виде гидроацетата согласно примеру 3, исходя из Boc-(D,L)-Phe(3-хлор-4-этокси)-OH и гидрохлорида H-Pro-п-цианобензиламида.
Масс-спектр (бомбардировка быстрыми атомами): 573 (М+H+).
Пример 87
H-(D,L)-Phe(3-xлop-4-этокси)-Pro-NH-pAmb
Соединение получают в виде дигидрохлорида путем отщепления Boc-группы от соединения примера 86.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,4 (д, 2H, NH); 9,2 (д, 2H, NH); 8,9 / 8,7 (2т, 1H, NH); 8,4 - 8,2 (уш. с., 3H, NH3 +); 7,8 (2д, 2H, аром. протоны); 7,45 (2д, 2H, аром. протоны); 7,3 (м, 1H, аром. протоны): 7,2 / 7,0 (2H, аром. протоны); 4,5 - 4,2 (4H, 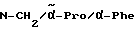 ): 4,1 (м, 2H, OCH2); 3,7 - 3,1 (м, 2H, δ-Pro); 3,1 - 2,7 (м, H, арил-CH2); 2,2 - 1,6 (м, 4H, β/γ- Pro); 1,35 (к, 3H, CH3).
): 4,1 (м, 2H, OCH2); 3,7 - 3,1 (м, 2H, δ-Pro); 3,1 - 2,7 (м, H, арил-CH2); 2,2 - 1,6 (м, 4H, β/γ- Pro); 1,35 (к, 3H, CH3).
Масс-спектр: 472 (М+H+), 247, 134, 70.
Пример 88
H-(D,L)-Phe(3-4-диметокси)-Pro-NH-pAmb
Соответствующее соединение с защитной Boc-группой получают согласно примеру 3 из Boc-(D,L)-Phe(3-4-диметокси)-OH и затем расщепляют до дигидрохлорида согласно методике A.I.в.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,55 - 9,25 (4H, амидин); 8,95 / примерно 8,8 (1H, NH); 8,8 / 8,35 (3H, МН3 +); 7,9 - 6,7 (7H, аром. протоны): 4,50 - 4,15 (4H, CH2 и 2 CH); 3,75 - 3,68 (6H, 2 OCH3); 3,2 - 2,8 (2H, CH2); 2,2 - 1,4 (4H, 2 CH2).
Пример 89
Boc-(D,L)-Phe(3-4-диметил)-Pro-NH-pAmb
Гидроацетат получают согласно примеру 3 из Boc-(D,L)-Phe(3-4- диметил)-OH в виде кристаллов белого цвета с температурой плавления 108 - 112oC.
Масс-спектр (бомбардировка быстрыми атомами): 522 (М+H+).
Пример 90
H-(D,L)-Phe(3-4-диметил)-Pro-NH-pAmb
Дигидрохлорид получают путем отщепления Boc-группы от соединения примера 89 согласно методике A.I.в. в виде кристаллов белого цвета с температурой плавления 195 - 200oC.
Масс-спектр (бомбардировка быстрыми атомами): 422 (М+H+).
Пример 91
Boc-(D,L)-Phe(3-метил-4-изопропил)-Pro-NH-pAmb
Гидроацетат получают из Boc-(D,L)-Phe(3-метил-4-изопропил)-OH согласно примеру 3.
Пример 92
H-(D,L)-Phe(3-метил-4-изопропил)-Pro-NH-pAmb
Дигидрохлорид получают путем отщепления Boc-группы от соединения примера 91 согласно методике A.I.в.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,5 - 9,2 (4H, амидин): 8,9 / 9,6 (1H, NH): 8,85 / 8,40 (3H, NH3 +); 7,9 - 6,85 (7H, аром. протоны); 4,5 - 4,0 (4H, CH2 и 2 CH); 3,2 - 2,9 (2H, CH2); 2,32 / 2,30 (3H, CH3); 2,2 - 1,4 (4H, 2 CH2); 1,2-1,1 (6H, 2 CH3).
Пример 93
Boc-(D,L)-Phe(2,3-диметокси)-Pro-NH-pAmb
Гидроацетат получают из Boc-(D,L)-Phe(2,3-диметокси)-OH согласно примеру 3.
Пример 94
H-(D,L)-Phe(2,3-диметокси)-Pro-NH-pAmb
Дигидрохлорид получают путем отщепления Boc-группы от соединения примера 93. Смесь диастереомеров в соотношении 1,3 : 1 имеет область плавления от 138 до 140oC (разложение).
Пример 95
Boc-(D,L)-Phe(2,5-диметокси)-Pro-NH-pAmb
Гидроацетат получают из Boc-(D,L)-Phe(2,5-диметокси)-OH согласно примеру 3.
Пример 96
H-(D,L)-Phe(2,5-диметокси)-Pro-NH-pAmb
Дигидрохлорид получают путем отщепления Boc-группы от соединения примера 95.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,55 - 9,25 (4H, амидин); 8,95 / примерно 8,7 (1H, NH); 8,7 / 8,2 (3H, NH3 +); 7,9 - 7,4 и 7,0 - 6,7 (7H, аром. протоны); 4,50 - 4,1 (4H, CH2 и 2 CH); 3,8 / 3,7 (6H, 2 OCH3); 3,25 - 2,65 (2H, CH2); 2,2 - 1,5 (4H, 2 CH2).
Пример 97
Boc-(D,L)-Phe(3,5-диметокси)-Pro-NH-pAmb
Гидроацетат получают из Boc-(D,L)-Phe(3,5-диметокси)-OH согласно примеру 3.
Пример 98
H-(D,L)-Phe(3,5-диметокси)-Pro-NH-pAmb
Дигидрохлорид получают путем отщепления Boc-группы от соединения примера 97 согласно методике A.I.в.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,5 - 9,2 (4H, амидин): 8,95 / примерно 8,7 (1H, NH): 8,7 / 8,3 (3H, NH3 +); 7,85 - 7,40 и 6,60 - 6,35 (7H, аром. протоны); 4,50 - 4,15 (4H, CH2 и 2 CH); 3,75 / 3,72 (6H, 2 OCH3); 3,2 - 2,8 (2H, CH2); 2,2 - 1,4 (4H, 2 CH2).
Пример 99
Boc-(D,L)-Phe(3,4,5-триметокси)-Pro-NH-pAmb
Соединение получают согласно примеру 3, причем эдукт Boc- (D,L)-Phe(3,4,5-триметокси)-ОН получают путем алкилирования бензофенониминоглицинового сложного эфира с помощью триметоксибензилхлорида с последующим введением защитной Boc-группы и омылением сложного эфира. Температура плавления дигидроацетата составляет 109-121oC.
Пример 100
H-(D,L)-Phe(3,4,5-триметокси)-Pro-NH-pAmb
Соединение получают из полученного в примере 99 продукта. Температура плавления дигидрохлорида составляет 180 - 239oC.
Пример 101
Boc-(D,L)-Phe(2,4,6-триметил)-Pro-NH-pAmb
Гидроацетат получают из Boc-(D,L)-Phe(2,4,6-триметил)-OH согласно примеру 3.
Пример 102
H-(D,L)-Phe(2,4,6-триметил)-Pro-NH-pAmb
Дигидрохлорид получают путем отщепления Boc-группы от соединения примера 101 согласно методике A.I.в.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,5 - 9,15 (4H, амидин); 8,85 / примерно 8,7 (1H, NH); 8,8 / 8,5 (3H, NH3 +); 7,9 - 7,4 и 6,9 - 6,75 (6H, аром. протоны); 4,5 - 4,0 (4H, CH2, и 2 CH): примерно 3,7 - 3,3 (2H, CH2); 3,2 - 3,0 (2H, CH2); 2,25 - 2,10 (6H, 3 CH3); примерно 2,1 -1,4 (4H, 2 CH2).
Пример 103
Boc-(D)-α-Nal-Pro-NH-pAmb
Соединение получают согласно примеру 3. Температура плавления гидроацетата составляет 136 - 178oC.
Пример 104
H-(D)-α-Nal-Pro-NH-pAmb
Соединение получают из полученного в примере 103 продукта. Температура плавления дигидрохлорида составляет 228 - 234oC.
Пример 105
H-(D)-α-Nal-Pro-NH-pAmb
Соединение получают путем отщепления Boc-группы от Boc-(D)-β-Nal-Pro-NH-pAmb; температура плавления дигидрохлорида составляет 223 - 229oC.
Пример 106
Boc-(D,L)-α-Ngl-Pro-NH-pAmb
Соединение получают согласно примеру 3, исходя из Boc-(D,L)-α-Ngl-OH и гидрохлорида H-Pro-п-цианобензиламида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 10,0 (уш. с., NH); 8,7 / 8,5 (2т, 1H, NH); 8,3 - 7,3 (12H, аром. протоны / NH); 6,2/6,1 (2д, 1H, α-NgI); 4,4 (3H, 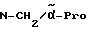 ); 3,8 - 2,8 (2H, δ-Pro); 2,2 - 1,7 (4H, β/γ-Pro); 1,3 (2с, 9H, Boc).
); 3,8 - 2,8 (2H, δ-Pro); 2,2 - 1,7 (4H, β/γ-Pro); 1,3 (2с, 9H, Boc).
Масс-спектр гидроацетата: 530 (M+H+), 247, 134.
Температура плавления гидроацетата составляет 183 - 185oC (разложение).
Пример 107
H-(D,L)-α-Ngl-Pro-NH-pAmb
Соединение получают путем отщепления Boc-группы от продукта примера 106.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,5 / 9,3 (2д, 4H, NH); 9,0 / 8,8 (2т, 1H, NH); 8,7 (уш. с., 3H, NH3 +); 8,4 (м, 1H, аром. протоны); 8,1 (2д, 2H, аром. протоны); 7,9 (2д, 2H, аром. протоны); 7,7 - 6,7 (6H, аром. протоны): 6,25 / 6,18 (2с, 1H, α-Ngl); 4,6 - 4,35 (м, 3H, 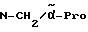 ); 3,85 / 3,6 / 3,4 (3м, 2H, δ-Pro); 2,2 - 1,6 (4H, β/γ-Pro).
); 3,85 / 3,6 / 3,4 (3м, 2H, δ-Pro); 2,2 - 1,6 (4H, β/γ-Pro).
Масс-спектр дигидрохлорида: 430 (M+H+), 369, 277.
Пример 108
Boc-(D,L)-β-Ngl-Pro-NH-pAmb
Соединение получают согласно примеру 3, исходя из Boc-(D,L)- β- Ngl-OH и гидрохлорида H-Pro-п-цианобензиламида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,8 - 9,2 (уш. с., NH); 8,6 / 8,4 (2 уш. с., 1H, NH); 8,0 - 7,75 (6H, аром. протоны); 7,6 - 7,5 (5H, аром. протоны); 7,35 / 7,18 (2 уш. с., 1H, NH); 5,6 / 5,45 / 5,35 (3 уш. c. , 1H,  Ngl); 4,4 (3H,
Ngl); 4,4 (3H, 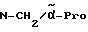 ); 3,9 / 3,7 (2 уш. с., 1H, δ- Pro); 3,2 (уш. с. , 1H, δ- Pro); 2,2 - 1,85 (4H, β/γ- Pro); 1,4 (2c, 9H, Boc).
); 3,9 / 3,7 (2 уш. с., 1H, δ- Pro); 3,2 (уш. с. , 1H, δ- Pro); 2,2 - 1,85 (4H, β/γ- Pro); 1,4 (2c, 9H, Boc).
Масс-спектр гидроацетата: 530 (M+H+), 430 (-Boc), 247, 185, 134.
Температура плавления гидроацетата составляет 183 - 185oC (разложение).
Пример 109
H-(D,L)-β-Ngl-Pro-NH-pAmb
Соединение получают путем отщепления Boc-группы от продукта примера 108 в виде дигидроацетата.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,6 - 9,0 (уш. с., NH); 8,75 / 8,6 (2т, 1H, NH); 8,0 - 7,8 (6H, аром. протоны): 7,6 - 7,4 (м, 5H, аром. протоны): 5,2 (с, 1H, β-Ngl); 4,5 - 4,3 (м, 3H, 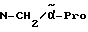 ); 3,9 - 3,0 (2H, δ- Pro); 2,2 - 1,7 (4H, β/γ-Pro).
); 3,9 - 3,0 (2H, δ- Pro); 2,2 - 1,7 (4H, β/γ-Pro).
Масс-спектр: 430 (M+H+), 247.
Пример 110
H-(D,L)-1-Tic-Pro-NH-pAmb
Соединение получают в виде дигидроацетата путем отщепления Boc-группы от продукта примера 255.
1H-ЯМР (дейтерированный диметилсульфоксид; / δ в м.д.): 9,4 / 9,0 (2 уш. с. , 4H, NH); 8,7 (уш. с., 1H, NH); 7,75 (2д, 2H, аром. протоны); 7,45 (2д, 2H, аром. протоны); 7,4 - 6,8 (4H, аром. протоны); 5,2 (2с, 1H α-Tic); 4,8 - 4,4 (3H, 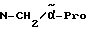 ); 3,6 - 3,2 (4H, арил-CH2 / δ-Pro): 3,0 - 2,7 (2H, N-CH2; 2,2 - 1,8 (4H, β/γ-Pro).
); 3,6 - 3,2 (4H, арил-CH2 / δ-Pro): 3,0 - 2,7 (2H, N-CH2; 2,2 - 1,8 (4H, β/γ-Pro).
Пример 111
Boc-(D)-3-Tic-Pro-NH-pAmb
Соединение получают согласно примеру 3, исходя из Boc-(D)-3- Tic-OH и гидрохлорида H-Pro-п-цианобензиламида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.); 8,44 / 8,2 (2 уш. с. , 1H, NH); 7,8 / 7,65 (2д, 2H, аром. протоны); 7,5 (2д, 2H, аром. протоны); 7,4 / 7,2 (м, 4H, аром. протоны); 4,8 - 4,6 (м, 2H, CH2); 4,4 - 4,2 (м, 4H, CH2 / 2 α-Н); 3,62 (м, 2H, Pro); 3,1 -2,6 (м, 2H, CH2-фенил); 2,2 - 1,75 (м, 4H, Pro): 1,3 (2c, 9H, Boc).
Масс-спектр: 506 (М+H+); 406 (-Boc); температура плавления гидроацетата составляет 143oC.
Пример 112
H-(D)-3-Tic-Pro-NH-pAmb
Соединение получают путем отщепления Boc-группы от продукта примера 111.
Масс-спектр (бомбардировка быстрыми атомами): 406 (М+H+). Температура плавления дигидроацетата составляет 204oC.
Пример 113
1-Icc-Pro-NH-pAmb
Соединение получают согласно примеру 3, исходя из 1-Icc-OH и гидрохлорида H-Pro-п-цианобензиламида.
Масс-спектр (бомбардировка быстрыми атомами): 402 (М+H+).
Пример 114
Boc-(D,L)-2-Tgl-Pro-NH-pAmb
Соединение получают в виде гидроацетата согласно примеру 3, исходя из Вос-(D,L)-2-Tgl-OH и гидрохлорида H-Pro-n-цианобензиламида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 8,6/8,4 (2т, 1H, NH): 7,75 (2д, 2H, аром. протоны); 7,45 - 6,9 (6H, аром. протоны): 5,7 - 5,4 (1H, α-Tgl): 4,4 (м, 3H, 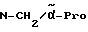 ); 3,8 - 3,2 (2H, δ-Pro); 2,1 - 1,7 (4H, β/γ-Pro); 1,35 (2c, 9H, Boc).
); 3,8 - 3,2 (2H, δ-Pro); 2,1 - 1,7 (4H, β/γ-Pro); 1,35 (2c, 9H, Boc).
Масс-спектр: 486 (M+H+), 386 (-Boc), 247, 185, 134.
Пример 115
H-(D,L)-2-Tgl-Pro-NH-pAmb
Соединение получают путем отщепления Boc-группы от продукта примера 114.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,4/9,2 (2 уш. с., 4H, NH), 8,9/8,75 (2т, 1H, NH); (уш. с., 3H, NH): 7,8 (2д, 2H, аром. протоны): 7,62 (2д, 2H, аром. протоны); 7,5 (2д, 2H, аром. протоны): 7,4 (уш. с., 1H, аром. протоны): 7,1 (м, 1H, аром. протоны): 5,65/5,6 (2c, 1H, α-Tgl); 4,5-4,4 (м, 3H, 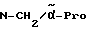 ); 3,95-3,75 (2м, 1H, δ-Pro); 3,2/3,0 (2 дд, 1H, δ-Pro); 2,2-2,0 (1H, β-Pro); 1,9-1,7 (3H, β/γ-Pro).
); 3,95-3,75 (2м, 1H, δ-Pro); 3,2/3,0 (2 дд, 1H, δ-Pro); 2,2-2,0 (1H, β-Pro); 1,9-1,7 (3H, β/γ-Pro).
Масс-спектр (бомбардировка быстрыми атомами) дигидроацетата: 386 (М+H+).
Пример 116
Boc-(D)-2-Tal-Pro-NH-pAmb
Соединение получают согласно примеру 3, исходя из Boc-(D)-2-Tal-OH и гидрохлорида H-Pro-п-цианобензиламида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 8,85/8,15 (2т, 1H, NH); 7,75 (2д, 2H, аром. протоны): 7,45 (2д, 2H, аром. протоны): 7,35 (д, 1H, аром. протоны); 7,25 (уш. с., 1H, аром. протоны); 7,0 - 6,7 (2H, аром. протоны); 4,82 - 4,3 (4H, 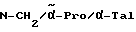 ); 4,05/3,6 (2м, 1H, δ-Pro): 3,5 - 2,9 (м, 3H, арил-CH2/ δ-Pro); 2,2-1,7 (4H, β/γ-Pro); 1,25 (2с, 9H, Boc).
); 4,05/3,6 (2м, 1H, δ-Pro): 3,5 - 2,9 (м, 3H, арил-CH2/ δ-Pro); 2,2-1,7 (4H, β/γ-Pro); 1,25 (2с, 9H, Boc).
Масс-спектр гидроацетата: 500 (М+H+), 400 (-Boc), 247, 134.
Пример 117
H-(D)-2-Tal-Pro-NH-pAmb
Соединение получают путем отщепления Boc-группы от продукта примера 116.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,4 - 9,0 (4H, NH): 8,85 (уш. с., 3H, NH3 +); 7,8 (д, 2H, аром. протоны): 7,5 (д, 2H, аром. протоны): 7,45 (д, 1H, аром. протоны): 7,0 (дд/с, 2H, аром. протоны); 4,4 - 4,15 (4H, 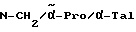 ); 3,8 - 2,9 (3H, арил-CH/ δ-Pro); 2,8 (дц, аром. H); 1,8 (м, 2H, β-Pro): 1,75-1,55 (2м, 2H, γ-Pro).
); 3,8 - 2,9 (3H, арил-CH/ δ-Pro); 2,8 (дц, аром. H); 1,8 (м, 2H, β-Pro): 1,75-1,55 (2м, 2H, γ-Pro).
Масс-спектр (бомбардировка быстрыми атомами) дигидроацетата: 400 (М+H+).
Пример 118
Boc-(D)-Phg-Pro-NH-pAmb
Гидроацетат получают из Boc-(D)-Phg-OH согласно примеру 3.
Пример 119
H-(D)-Phg-Pro-NH-pAmb
Дигидрохлорид получают из соединения примера 118.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,6 - 9,3 (4H, амидин): 9,1 - 8,7 (4H, NH и NH3 +); 8,0 - 7,3 (9H, аром. протоны); 5,4 (1H, CH): 4,6 - 4,3 (3H, CH2 и CH); 3,1 - 2,7 (2H, CH2; 2,2-1,6 (4H, 2 CH2).
Пример 120
Boc-(D,L)-Phg(4-MeO)-Pro-NH-pAmb
Гидроацетат получают из Boc-(D,L)-Phg(4-MeO)-OH согласно примеру 3.
Пример 121
H-(D,L)-Phg(4-MeO)-Pro-NH-pAmb
Дигидрохлорид получают из соединения примера 120.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,6 - 9,3 (4H, амидин): 9,0/8,9 (1Н, NH): 8,8/8,6 (3H, NH3 +); 7,9 - 7,8 и 7,55 - 7,45 и 7,05 - 6,90 (8H, аром. протоны): 5,3 (1H, CH); 4,5 - 4,3 (3H, CH2 и CH): 3,75 (3H, OCH3); 2,2 - 1,6 (4H, 2 CH2).
Пример 122
Boc-(D)-Chg-Pro-NH-pAmb
а) При 0oC в колбе смешивают 8 г (31, 1 моль) Boc-(D)-Chg-OH, 9,88 г (37,3 ммоль) гидрохлорида H-Pro-п-цианобензиламида, 32 мл (186, 54 ммоль) диизопропилэтиламина и 108 мл 50%-ной полифосфорной кислоты в этилацетате и перемешивают в течение восемнадцати часов при температуре от 0oC до комнатной. 3атем реакционную смесь разбавляют этилацетатом и экстрагируют пятикратно 20%-ным раствором гидросульфата натрия, однократно 5%-ным раствором гидрокарбоната натрия и насыщенным раствором хлорида натрия. После высушивания и концентрирования органического раствора получают 13,8 г чистого Boc-(D)-Chg-n-цианобензиламида.
б) 13,8 г Boc-(D)-Chg-Pro-п-цианобензиламида растворяют в 113 мл пиридина и 53 мл триэтиламина. Раствор насыщают газообразным сероводородом и выдерживают в течение ночи при комнатной температуре. Для обработки реакционную смесь сначала продувают азотом и затем выливают в 1 л 5%-ного раствора лимонной кислоты. Выпавший осадок отфильтровывают и фильтрат трехкратно экстрагируют этилацетатом. Осадок затем растворяют в этилацетате и объединяют с органическими экстрактами. Объединенные фазы промывают 5%-ным раствором лимонной кислоты, сушат над сульфатом натрия и концентрируют. Сырой продукт без дальнейшей очистки используют в следующей реакции.
в) Сырой тиоамид растворяют в смеси из 120 мл ацетона и 21 мл метилиодида и перемешивают в течение ночи при комнатной температуре. Затем реакционную смесь концентрируют в вакууме досуха и остаток растворяют в 48 мл метанола. После добавления 48 мл 10%-ного метанольного раствора ацетата аммония раствор перемешивают в течение восемнадцати часов при комнатной температуре. Для обработки амидина растворитель удаляют в ротационном испарителе, остаток обрабатывают дихлорметаном, осадок отсасывают и фильтрат концентрируют в вакууме. Получают 24,6 г сырого продукта, который очищают с помощью высоко эффективной жидкостной хроматографии с обращенной фазой. Выход гидроацетата составляет 7 г.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,6 - 9,2 (уш. с., NH); 8,7/8,1 (2 т, 1H, NH); 7,75 (2д, 2H, аром. протоны); 7,45 (2д, 2H, аром. протоны): 7,0/6,9 (2 д, 1H, NH); 4,4 (м, 3H, 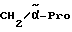 ); 4,0 (т, 1H,
); 4,0 (т, 1H, 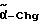 ); 3,6 - 3,0 (2H,
); 3,6 - 3,0 (2H, 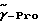 ); 2,1 - 1,5 (м, 11H,
); 2,1 - 1,5 (м, 11H, 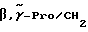 ); 1,4/1,3 (2с, 9H, Boc); 1,1 - 0,9 (м, 4H).
); 1,4/1,3 (2с, 9H, Boc); 1,1 - 0,9 (м, 4H).
Масс-спектр: 486 (M+H+): 386 (-Boc), 247, 134.
Пример 123
H-(D)-Chg-Pro-NH-pAmb
Соединение получают путем отщепления Boc-группы от продукта примера 122.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 9,4 - 9,0 (уш. с. , NH); 8,9 (т, 1H, NH); 8,4 (уш. с., NH); 7,8 (д, 2H, аром. протоны); 7,5 (д, 2H, аром. протоны); 4,4 (м, 3H, 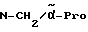 ); 3,9 - 3,6 (2м, 2H,
); 3,9 - 3,6 (2м, 2H, 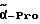 ); 3,8 (д, 1H,
); 3,8 (д, 1H, 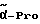 ); 2,0 - 1,5 (м, 10H, Chg/ β/γ-Pro); 1,2 - 1,0 (м, 4H, Chg).
); 2,0 - 1,5 (м, 10H, Chg/ β/γ-Pro); 1,2 - 1,0 (м, 4H, Chg).
Масс-спектр: 386 (М+H+), 247, 185; температура плавления дигидрохлорида составляет 133oC.
Пример 124
EtOOC-(D)-Chg-Pro-NH-pAmb
Сначала из Boc-(D)-Chg-OH и гидрохлорида H-Pro-n-цианобензиламида согласно примеру 3 получают гидрохлорид H-(D)-Chg-Pro-п-цианобензиламида. Затем при комнатной температуре к 25 мл дихлорметана последовательно добавляют 2,5 г (6,17 ммоль) гидрохлорида H-(D)-CHg-Pro-п-цианобензиламида, 2,33 мл (13,58 ммоль) диизопропилэтиламина, 0,075 г (0,617 ммоль) диметиламино-пропиламина и 0,652 мл этилового эфира хлормуравьиной кислоты и реакционный раствор перемешивают в течение восемнадцати часов при комнатной температуре. После этого реакционную смесь разбавляют дихлорметаном, промывают 20%-ным раствором гидросульфата натрия, сушат и концентрируют. Выход сырого EtOOC-Н-(D)-CHg-Pro-п-цианобензиламида составляет 2,51 г. Таким образом полученный промежуточный продукт переводят в соответствующий амидин согласно методике A.III. 1. Сырой продукт очищают путем высокоэффективной жидкостной хроматографии с обращенной фазой (смесь акрилонитрила с водой). Выход составляет 0,483 г.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 8,7/8,1 (2т, 1H, NH); 7,8 (2д, 2H, аром. протоны); 7,4 (2д, 2H, аром. протоны); 7,4 (2д, 2H, аром. протоны); 7,39/7,3 (2д, 1H, NH); 4,9/4,4 (2м, 3H, 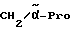 ); 4,0 (2т, 1H,
); 4,0 (2т, 1H, 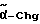 ); 3,8 (т, 2H, OCH); 3,7-3,3 (3м, 2H, δ-Pro); 2,1 (м, 1H, β-Pro); 1,9-1,5 (м, 11H, CH2/ β/γ-Pro): 1,2 - 0,9 (м, 9H, CH2/CH3).
); 3,8 (т, 2H, OCH); 3,7-3,3 (3м, 2H, δ-Pro); 2,1 (м, 1H, β-Pro); 1,9-1,5 (м, 11H, CH2/ β/γ-Pro): 1,2 - 0,9 (м, 9H, CH2/CH3).
Масс-спектр гидроацетата: 458 (М+H+), 247, 134, 70.
Пример 125
HOOC-CH2-(D)-Chg-Pro-NH-pAmb
Соединение получают путем отщепления сложноэфирной трет.-бутильной группы от продукта примера 126.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 8,5/8,3 (2т, 1H, NH); 7,8 (2д, 2H, аром. протоны); 7,6/7,45 (2д, 2H, аром. протоны); 4,4 - 4,2 (м, 3H, 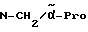 ); 4,1 (м, 1H,
); 4,1 (м, 1H, 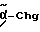 ); 3,8 - 3,2 (4H, HOOCCH2/ δ-Pro); 2,1 - 1,4 (м, 11H); 1,2-0,9 (м, 4H).
); 3,8 - 3,2 (4H, HOOCCH2/ δ-Pro); 2,1 - 1,4 (м, 11H); 1,2-0,9 (м, 4H).
Масс-спектр гидрохлорида: 444 (М+H+), 386, 247.
Пример 126
tBuOOC-CH2-(D)-Chg-Pro-NH-pAmb
Соединение получают согласно получению сложного трет.-бутилового эфира в качестве предшественника продукта примера 246.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 8,4 (т, 1H, NH); 7,75 (д, 2H, аром. протоны): 7,4 (д, 2H, аром. протоны); 4,4 (м, 4H, 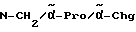 ); 3,8 - 2,9 (4H, HOOCCH2/ δ-Pro); 2,1 - 0,9 (м, 15H): 1,3 (с, 9H, трет.-бутил).
); 3,8 - 2,9 (4H, HOOCCH2/ δ-Pro); 2,1 - 0,9 (м, 15H): 1,3 (с, 9H, трет.-бутил).
Масс-спектр гидроацетата: 500 (М+H+), 444, 247, 170.
Пример 127
Boc-(D)-Cha-Pro-NH-pAmb
Соединение примера 127 получают согласно примеру 3.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,4 (уш. с., 4H, NH); 8,8/8,15 (2т, 1H, NH); 7,75 (2д, 2H, аром. протоны); 7,45 (2д, 2H, аром. протоны); 7,05 (д, 1H, NH); 4,8/4,35 (д/м, 3H, 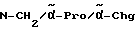 ); 3,75/3,5 - 3,2 (2H,
); 3,75/3,5 - 3,2 (2H, 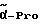 ); 2,1 - 1,85 (4H, β/γ-Pro); 1,7 - 1,3 (м, 6H): 1,3 (2д, 9H, Boc): 1,4 - 0,9 (м, 7H).
); 2,1 - 1,85 (4H, β/γ-Pro); 1,7 - 1,3 (м, 6H): 1,3 (2д, 9H, Boc): 1,4 - 0,9 (м, 7H).
Масс-спектр: 500 (M+H+), 400 (-Boc), 247, 134; температура плавления гидроацетата составляет 125 - 127oC.
Пример 128
Me-(D)-Cha-Pro-NH-pAmb
Соединение получают путем отщепления Z - группы от продукта примера 129.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,3/8,9 (2с, 4H, NH); 8,85/8,8 (2 уш. с., 2H, NH); 8,7 (т, 1H, NH); 7,8 (2д, 2H, аром. протоны); 7,5 (2д, 2H, аром. протоны): 4,4 (м, 3H, 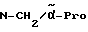 ); 4,25 (уш. д., 1H,
); 4,25 (уш. д., 1H, 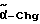 ); 3,9/3,4 (2м, 2H, δ-Pro); 2,5 (с, 3H, N-CH2); 2,2 (м, 1H, β-Pro); 2,5 (с, 3H, N-CH2; 2,2 (м, 1H, β-Pro); 2,0 - 1,8 (м, 4H); 1,8 - 1,5 (м, 6H); 1,4 - 0,9 (6H).
); 3,9/3,4 (2м, 2H, δ-Pro); 2,5 (с, 3H, N-CH2); 2,2 (м, 1H, β-Pro); 2,5 (с, 3H, N-CH2; 2,2 (м, 1H, β-Pro); 2,0 - 1,8 (м, 4H); 1,8 - 1,5 (м, 6H); 1,4 - 0,9 (6H).
Масс-спектр гидроацетата: 414 (М+H+), 247, 140.
Пример 129
Me-(Z)-(D)-Cha-Pro-NH-pAmb
Соединение получают согласно примеру 3, исходя из Me-(Z)-(D)-Cha-OH и гидрохлорида H-Pro-п-цианобензиламида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,8 - 9,2 (уш. с., 4H, NH), 8,8/8,5 (2т, 1H, NH): 7,8 (2д, 2H, аром. протоны); 7,5 (2д, аром. протоны): 7,4 (м, 5H, Ph-H); 5,2 - 5,0 (2H, OCH2); 4,95 - 4,5 (1H, 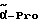 ); 4,4 (м, 3H,
); 4,4 (м, 3H, 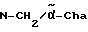 ); 3,6 - 3,0 (2H, δPro); 2,82/2,75/2,7 (3с, 3H, NCH3); 2,1 (м, 1H, β-Pro); 1,9 - 1,4 (м, 11H, β/γ-Pro/CH2); 1,2-0,8 (м, 5H).
); 3,6 - 3,0 (2H, δPro); 2,82/2,75/2,7 (3с, 3H, NCH3); 2,1 (м, 1H, β-Pro); 1,9 - 1,4 (м, 11H, β/γ-Pro/CH2); 1,2-0,8 (м, 5H).
Масс-спектр (бомбардировка быстрыми атомами) гидроацетата: 548 (М+H+).
Пример 130
N,N-Me-(D)-Cha-Pro-NH-pAmb
Соединение получают согласно примеру 3, исходя из N,N-диметилциклогексилаланина и гидрохлорида H-Pro-n-цианобензиламида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 8,8/8,4 (2т, 1H, NH); 7,8 (2д, 2H, аром. протоны); 7,45 (2H, аром. протоны): 4,45 - 4,3 (д/м, 3H, 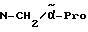 ); 3,9 (м, 1H,
); 3,9 (м, 1H, 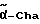 ); 3,6 - 3,2 (2H, δ-Pro); 2,2 (2с, 6H, NCH3); 2,1-1,5 (м, 13H); 1,3-0,8 (м, 4H).
); 3,6 - 3,2 (2H, δ-Pro); 2,2 (2с, 6H, NCH3); 2,1-1,5 (м, 13H); 1,3-0,8 (м, 4H).
Масс-спектр (бомбардировка быстрыми атомами) гидроацетата: 428 (М+H+).
Пример 131
Boc-(D)-Trp(Boc)-Pro-NH-pAmb
Соединение получают согласно примеру 3, исходя из Boc-(D)-Trp(Boc)-OH и гидрохлорида H-Pro-n-цианобензиламида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 9,8 - 9,2 (уш. с. , NH): 8,8 - 8,5 (2 уш. с., 1H, NH); 8,25/8,0/7,8-7,2 (м, 10H, аром. протоны/NH); 4,85/4,5-4,2 (д/м, 4H, CH2-H); 3,6/3,5 (2м, 2H, CH2, Pro), 3,1 - 2,8 (м, 2H, CH2; 2,2 - 1,6 (м, 4H, Pro); 1,3 (2с, 18H, Boc).
Масс-спектр (бомбардировка быстрыми атомами) гидроацетата: 633 (М+H+).
Пример 132
H-(D)-Trp-Pro-NH-pAmb
Соединение получают путем отщепления Boc-групп от продукта примера 131.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 11,1 (с, 1H, NH); 9,4/9,15 (2с, 4H, NH); 8,8 (т, 1H, N-Z); 8,6 (с, 3H, NH); 7,75 (д, 2H, аром. протоны):
7,45 (д, 3H, аром. протоны); 7,35 (д, 1H, аром. протоны); 7,25 (с, 1H, аром. протоны); 7,0 (2т, 2H, аром. протоны); 4,3 (м, 2H, CH2); 4,18 (уш. с., 1H,  ; 3,5 (м, 2H, CH2, Pro); 3,3 - 3,1 (м, 2H, CH2; 2,15 (дц, 1H, Pro): 1,6/1,4 (2м, 3H, β/γ- Pro).
; 3,5 (м, 2H, CH2, Pro); 3,3 - 3,1 (м, 2H, CH2; 2,15 (дц, 1H, Pro): 1,6/1,4 (2м, 3H, β/γ- Pro).
Масс-спектр (бомбардировка быстрыми атомами) дигидрохлорида: 433 (М+H+).
Пример 133
Boc-(D,L)-Dpa-Pro-NH-pAmb
Соединение получают согласно примеру 3, исходя из Boc-(D)-Dpa-OH и гидрохлорида H-Pro-п-цианобензиламида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 8,6/8,1 (2т, 1H, NH); 7,75 (2д, 2H, аром. протоны): 7,45 - 7,0 (м, 13H, аром. протоны/NH); 5,25/5,1 (2т, 1H, α- Dpa); 4,4 - 4,1 (3H, 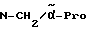 ); 3,75 (м, 1H, CH); 3,6 - 2,95 (2H, δ- Pro); 2,0 - 1,5 (4H, β/γ- Pro); 1,2 (2 д. с., 9H, Boc).
); 3,75 (м, 1H, CH); 3,6 - 2,95 (2H, δ- Pro); 2,0 - 1,5 (4H, β/γ- Pro); 1,2 (2 д. с., 9H, Boc).
Масс-спектр: 570 (М+H+), 470 (-Boc), 247, 196, 134.
Температура плавления гидроацетата составляет 156oC.
Пример 134
H-(D или L)-Dpa-Pro-NH-pAmb (диастереомер(а))
Соединение примера 134 получают путем отщепления Boc-группы от продукта примера 133 и последующего разделения диастереомеров с помощью высокоэффективной жидкостной хроматографии с обращенной фазой.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 9,3 (2с, 4H, NH): 8,9/8,2 (2т, 1H, NH): 8,4 (уш. с., 3H, NH); 7,8 (2д, 2H, аром.протоны); 7,6 (2д, 2H, аром. протоны); 7,5-7,1 (10H, аром. протоны); 5,1/4,6 (2д, 1H, α- Dpa); 4,4-4,1 (4H, 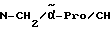 ); 3,8 - 3,0 (2H, δ- Pro); 2,1 - 1,1 (4H, β/γ- Pro).
); 3,8 - 3,0 (2H, δ- Pro); 2,1 - 1,1 (4H, β/γ- Pro).
Масс-спектр (бомбардировка быстрыми атомами) дигидроацетата: 470 (М+H+).
Пример 135
H-(D или L)-Dpa-Pro-NH-pAmb (диастереомер (б))
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 9,3/9,2 (2с, 4H, NH); 8,4 (т, 1H, NH); 8,35 (уш. с., 3H, NH); 7,8/7,65 (2д, 4H, аром. протоны); 7,4 - 7,1 (10H, аром. протоны); 5,0 (д, 1H, α- Dpa); 4,4/3,9 (м, 4H, 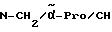 ); 3,6/2,9 (2м, 2H, δ- Pro): 1,7 - 1,3 (4H, β/γ- Pro).
); 3,6/2,9 (2м, 2H, δ- Pro): 1,7 - 1,3 (4H, β/γ- Pro).
Масс-спектр (бомбардировка быстрыми атомами) дигидроацетата: 470 (М+H+).
Пример 136
EtOOC-(D или L)-Dpa-Pro-NH-pAmb (диастереомер (а))
Для получения вышеуказанного соединения Boc-(D,L)-Dpa-Pro-п-цианобензиламид (промежуточный продукт синтеза соединения примера 133) сначала путем обработки раствором хлороводорода в диоксане переводят в соответствующий гидрохлорид H-(D,L)-Dpa-Pro-п-цианобензиламида. Затем соль, согласно примеру 124, переводят в диастереомерную пару и оба диастереомера разделяют путем высокоэффективной жидкостной хроматографии с обращенной фазой (смесь ацетонитрила с водой).
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 8,6/6,6 (2т, 1H, NH); 7,8 - 7,0 (м, 15H, аром. протоны, NH); 5,3/5,1 (2т, 1H, α- Dpa); 4,4 (2д, 1H, 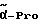 ); 4,3/4,1 (2т, 2H, CH2); 4,0 (м, 1H, CH), 3,85 (т, 2H, OCH2; 3,6/3,3/3,0 (3м, 2H, δ- Pro); 2,0-1,4 (м, 4H, β/γ- Pro), 1,0 (м, 3H, CH3).
); 4,3/4,1 (2т, 2H, CH2); 4,0 (м, 1H, CH), 3,85 (т, 2H, OCH2; 3,6/3,3/3,0 (3м, 2H, δ- Pro); 2,0-1,4 (м, 4H, β/γ- Pro), 1,0 (м, 3H, CH3).
Масс-спектр гидроацетата: 542 (М+H+, 268, 134, 70.
Пример 137
EtOOC-(D или L)-Dpa-Pro-NH-pAmb (диастереомер (б))
Соединение получают согласно примеру 3.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 8,2 (2т, 1H, NH); 7,75 (д, 2H, аром. протоны); 7,6 (д, 2H, аром. протоны); 7,4 - 7,2 (м, 12H, аром. протоны); 5,15 (м, 1H, α- Dpa); 4,4 (м, 3H, 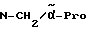 ); 3,95 (м, 1H, CH); 3,8/3,1 (2м, 2H, δ- Pro); 3,7 (м, 2H, OCH2); 1,8 - 1,4 (м, 4H, β/γ- Pro); 1,0 (м, 3H, CH3).
); 3,95 (м, 1H, CH); 3,8/3,1 (2м, 2H, δ- Pro); 3,7 (м, 2H, OCH2); 1,8 - 1,4 (м, 4H, β/γ- Pro); 1,0 (м, 3H, CH3).
Масс-спектр гидроацетата: 542 (М+H+), 268, 134, 70.
Пример 138
HOOC-CH2-(D или L)-Dpa-Pro-NH-pAmb (диастереомер (a))
Сначала с помощью хлороводорода в диоксане отщепляют Boc-группу от Boc-(D, L)-Dpa-Pro-п-цианобензиламида (промежуточный продукт синтеза соединения примера 133). 3,42 г (7 ммоль) Полученного таким образом гидрохлорида растворяют в 20 мл метанола и после добавления 0,6 г (6,65 ммоль) гидрата глиоксиловой кислоты и 1,75 г (28 ммоль) цианоборгидрида натрия перемешивают в течение ночи. Для обработки реакционную смесь концентрируют, остаток обрабатывают дихлорметаном и полученный таким образом раствор экстрагируют водой. Полученный после высушивания и концентрирования органической фазы остаток растворяют в 5 мл метанола и целевой продукт осаждают путем прикапывания полученного раствора в диизопропиловый эфир. Выход сырого продукта составляет 3,7 г. Его без дальнейшей очистки переводят в соответствующий амидин согласно методике примера A.III.1. Смесь диастереомеров разделяют путем высокоэффективной жидкостной хроматографии с обращенной фазой (смесь ацетонитрила с водой).
Масс-спектр гидроацетата: 528 (М+H+), 254.
Пример 139
HOOC-CH2-(D или L)-Dpa-Pro-NH-pAmb (диастереомер (б))
Масс-спектр гидроацетата: 528 (М+H+), 254, 134, 83.
Пример 140
Boc-(D или L)-Dpa-(4,4'-дихлор)-Pro-NH-pAmb (диастереомер (а))
Соединение получают согласно примеру 3, исходя из Boc-(D,L)-Dpa-(4,4'-дихлор)-ОН и гидрохлорида H-Pro-n-цианобензиламида. Полученную пару диастереомеров разделяют путем высокоэффективной жидкостной хроматографии с обращенной фазой.
Масс-спектр гидроацетата: 638 (М+H+), 538 (-Boc), 303, 277, 247.
Пример 141
Boc-(D или L)-Dpa-(4,4'-дихлор)-Pro-NH-pAmb (диастереомер (б))
Масс-спектр гидроацетата: 638 (М+H+), 538 (-Boc), 303, 247, 134, 70.
Пример 142
H-(D или L)-Dpa (4,4'-дихлор)-Pro-NH-pAmb (диастереомер (а))
Соединение получают путем отщепления Boc-группы от соединения примера 140.
Масс-спектр гидроацетата: 538 (М+H+), 303, 247, 134, 70.
Пример 143
H-(D или L)-Dpa(4,4'-дихлор)-Pro-NH-pAmb (диастереомер (б))
Соединение получают путем отщепления Boc-группы от продукта примера 141.
Масс-спектр гидроацетата: 538 (М+H+), 303, 264, 247, 134, 70.
Пример 144
EtOOC-(D или L)-Dpa(4,4'-дихлор)-Pro-NH-pAmb (диастереомер (а))
Для получения вышеуказанного соединения сначала Boc-(D,L)-Dpa (4,4'-дихлор)-Pro-п-цианобензиламид (промежуточный продукт синтеза соединения примера 141) путем обработки хлороводородом в диоксане переводят в соответствующий гидрохлорид H-(D, L)-Dpa (4,4'-дихлор)-Pro-п-цианобензиламида. Затем соль, согласно примеру 124, переводят в смесь диастереомеров продукта. Оба диастереомера разделяют путем высокоэффективной жидкостной хроматографии с обращенной фазой (смесь ацетонитрила с водой).
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 8,6 (2т, 1H, NH); 7,75 (2д, 2H, аром. протоны); 7,6 - 7,1 (11H, аром. протоны/NH); 5,2/5,0 (2т, 1H, α- Dpa); 4,4/4,38 (2д, 1H, CH); 4,3 (м, 1H, 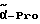 ; 4,0 (м, 2H, N-CH2; 3,75 (м, 2H, OCH2); 3,7 - 3,3 (2H, δ-Pro); 2,0 (м, 1H, β-Pro); 1,95 - 1,4 (м, 4H, β/γ-Pro); 1,0 (2т, 3H, CH3).
; 4,0 (м, 2H, N-CH2; 3,75 (м, 2H, OCH2); 3,7 - 3,3 (2H, δ-Pro); 2,0 (м, 1H, β-Pro); 1,95 - 1,4 (м, 4H, β/γ-Pro); 1,0 (2т, 3H, CH3).
Масс-спектр гидроацетата: 610 (М+H+), 247, 134, 70.
Пример 145
EtOOC-(D или L)-Dpa(4,4'-дихлор)-Pro-NH-pAmb (диастереомер (б))
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 8,2 (2т, 1H, NH); 7,75 - (2д, 2H, аром. протоны); 7,6 -7,1 (11H, аром. протоны/NH); 5,1 (2т, 1H, α-Dpa); 4,4 (2д, 1H, CH); 4,3 (м, 2H, N-CH2); 4,0/4,39 (м, 1H, 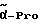 ); 3,85 (м, 2H, OCH2); 3,7 (2H, δ-Pro); 1,9 - 1,5 (4H, β/γ-Pro); 1,0 (2т, 3H, CH3).
); 3,85 (м, 2H, OCH2); 3,7 (2H, δ-Pro); 1,9 - 1,5 (4H, β/γ-Pro); 1,0 (2т, 3H, CH3).
Масс-спектр гидроацетата: 610 (М+H+), 247, 185, 134, 93.
Пример 146
HOOC-CH2-(D или L)-Dpa(4,4'-дихлор)-Pro-NH-pAmb (диастереомер (а))
Сначала с помощью хлороводорода в диоксане отщепляют Boc-группу от Boc-(D, L)-Dpa(4,4'-дихлор)-Pro-п-цианобензиламида (промежуточный продукт синтеза соединения примера 140). Затем целевой продукт получают согласно примеру 138.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 11,2 (уш. с., COOH): 8,9/8,6 (2 уш. с., 1H, NH); 7,8 - 7,2 (14H, аром. протоны NH); 4,4 - 4,0 (5H, 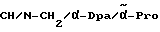 ); 3,8 - 3,0 (2H, δ-Pro); 2,8 (2д, 2H, HOOC-CH2), 2,0 - 1,4 (4H, β/γ-Pro).
); 3,8 - 3,0 (2H, δ-Pro); 2,8 (2д, 2H, HOOC-CH2), 2,0 - 1,4 (4H, β/γ-Pro).
Масс-спектр гидроацетата: 596 (М+H+), 247, 134, 93, 70.
Пример 147
HOOC-CH2-(D или L)-Dpa (4,4'-дихлор)-Pro-NH-pAmb (диастереомер (б))
Масс-спектр (бомбардировка быстрыми атомами): 596 (М+H+).
Пример 148
H-(D или L)-Dch-Pro-NH-pAmb (диастереомер (а))
Соединение примера 148 получают согласно примеру 3, исходя из Boc-(D, L)-Dch-OH и гидрохлорида H-Pro-п-цианобензиламида. Синтезированную пару диастереомеров разделяют путем высокоэффективной жидкостной хроматографии с обращенной фазой.
1H-ЯМР (дейтерированный диметилсульфоксид: δ в м. д.): 9,3 - 9,0 (уш. с. , NH); 8,9/8,5 (2т, 1H, NH); 7,75/7,5 (2д, 4H, аром. протоны); 4,5 - 4,0 (4H, 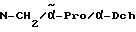 ); 3,7 - 3,0 (2H, δ-Pro); 2,2 - 1,0 (4H, β/γ-Pro).
); 3,7 - 3,0 (2H, δ-Pro); 2,2 - 1,0 (4H, β/γ-Pro).
Масс-спектр (бомбардировка быстрыми атомами): 481 (М+H+).
Температура плавления дигидроацетата составляет 127oC.
Пример 149
H-(D или L)-Dch-Pro-NH-pAmb (диастереомер (б))
Масс-спектр (бомбардировка быстрыми атомами): 481 (М+H+).
Температура плавления дигидроацетата составляет 127oC.
Пример 150
Boc-(D)-Val-Pro-NH-pAmb
Соединение получают согласно примеру 3.
Температура плавления гидроацетата составляет 132-145oC.
Пример 151
H-(D)-Val-Pro-NH-pAmb
Соединение получают из продукта примера 150.
Температура плавления дигидрохлорида составляет 60 - 80oC.
Пример 152
Boc-(D)-Leu-Pro-NH-pAmb
Соединение получают согласно примеру 3.
Температура плавления гидроацетата составляет 68 - 82oC.
Пример 153
H-(D)-Leu-Pro-NH-pAmb
Соединение получают из продукта примера 152.
Температура плавления дигидрохлорида составляет 228 - 233oC.
Пример 154
Boc-(D)-Gly(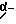 tBu)-Pro-NH-pAmb
tBu)-Pro-NH-pAmb
Соединение получают согласно примеру 3.
Температура плавления гидроацетата составляет 211 - 220oC.
Пример 155
H-(D)-Gly(α-tBu)-Pro-NH-pAmb
Соединение получают из продукта примера 154.
Температура плавления дигидрохлорида составляет 236 - 239oC.
Пример 156
Boc-(D)-Ala(β-tBu)-Pro-NH-pAmb
Соединение получают согласно примеру 3.
Температура плавления гидроацетата составляет 185 - 192oC.
Пример 157
H-(D)-Ala(β-tBu)-Pro-NH-pAmb
Соединение получают из продукта примера 156.
Температура плавления дигидрохлорида составляет 225 - 231oC.
Пример 158
H-(D или L)-Msu-Pro-NH-pAmb (диастереомер (а))
Дигидрохлорид получают согласно примеру 3 из Boc-(D,L)-Msu-OH и затем отщепляют Boc-группы согласно методике A.I.в. Диастереомеры разделяют путем высокоэффективной жидкостной хроматографии.
1H-ЯМР (дейтерированный диметилсульфоксид: δ в м. д.): 9,40/9,20 (4H, амидин); 8,9 (1Н, NH): 8,55 (3H, NH3 +); 7,85/7,50 (4H, аром. протоны); 4,50 - 4,35 (4H, CH2 и 2 CH): 3,85 - примерно 3,3 (4H, 2 CH2; 2,95 (3H, CH3): 2,3 - 1,8 (6H, 3 CH2).
Пример 159
H-(D или L)-Msu-Pro-NH-pAmb (диастереомер (б))
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.) дигидрохлорида: 9,45/9,30 (4H, амидин); 8,95 (1Н, NH); 8,85 (3H, NH3 +); 7,80/7,45 (4H, аром. протоны): 4,4 - 4,2 (4H, CH2 и 2CH); 3,85 - примерно 3,3 (4H, 2 CH2; 3,00 (3H, CH3); 2,3 - 1,7 (6H, 3 CH2).
Пример 160
Boc-(цикло)Leu-Pro-NH-pAmb
Соединение примера 160 получают согласно примеру 3, исходя из Boc-(цикло)Leu-ОН и гидрохлорида H-Pro-п-цианобензиламида.
Масс-спектр гидроацетата: 472 (М+H+), 372 (-Boc), 247, 185, 140.
Пример 161
H-(цикло)Leu-Pro-NH-pAmb
Соединение получают путем отщепления Boc-группы от продукта примера 160.
Масс-спектр (бомбардировка быстрыми атомами) дигидроацетата: 372 (М+H+).
Пример 162
H-Gly-Pro-NH-pAmb
Дигидрохлорид получают путем отщепления Boc-группы от Boc-Gly-Pro-NH-pAmb, который синтезируют исходя из Boc-Gly-OH согласно примеру 3.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 9,50/9,25 (4H, амидин); 8,85 (1Н, NH); 8,30 (3H, NH3 +); 7,80/7,45 (4H, аром. протоны); 4,5 - 4,2 (3H, CH2 и CH); 3,9 - примерно 3,3 (4H, 2 CH2; 2,2 - 1,7 (4H, 2 CH2).
Пример 163
Ph-CH2-Gly-Pro-NH-pAmb
Дигидрохлорид получают путем отщепления Boc-группы от Ph-CH2-(Boc)Gly-Pro-NH-pAmb-гидроацетата.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 9,6 (2H, NH2 +); 9,4 - 9,2 (4H, амидин): 8,80 (1 H, NH); 7,80 - 7,35 (9H, аром. Н); 4,40 - 4,25 (3H, CH2 и CH); 4,10 (2H, CH2); 3,95 (2H, CH2); 3,6 - 3,4 (2H, CH2); 2,2 - 1,8 (4H, 2 CH2).
Пример 164
β- Нафтил-CH2-Gly-Pro-NH-pAmb
[(2-нафтилметил)-амино] уксусную кислоту подвергают взаимодействию с гидрохлоридом (4-цианобензил)пролиламида аналогично примеру 10 и затем амидируют аналогично примеру 5.
Получают белый, аморфный дигидрохлорид.
Масс-спектр (бомбардировка быстрыми атомами): 444 (M-H+).
Пример 165
Аналогично получают: [3,4-(MeO)2-фенил]-CH2-Gly-Pro-NH-Amb
Исходное сырье: [(3,4-диметоксибензил)амино]-уксусная кислота.
Белый, аморфный дигидрохлорид.
Масс-спектр (бомбардировка быстрыми атомами): 452 (M-H+).
Путем взаимодействия фенилуксусной кислоты, 3-фенилпропионовой кислоты, 4-фенилмасляной кислоты, нафтилин-1-карбоновой кислоты, нафтилин-2-карбоновой кислоты и 1- или 2- нафтилуксусной кислоты с гидрохлоридом (4-циано-бензил)пролиламида аналогично примеру 10 и последующего амидирования аналогично примеру 5 получают следующие соединения:
Пример 166
PhCH2-CO-Pro-NH-pAmb
Белый, аморфный гидрохлорид.
Масс-спектр (бомбардировка быстрыми атомами): 365 (M-H+).
Пример 167
PhCH2CH2-CO-Pro-NG-pAmb
Желтоватый, аморфный гидройодид.
Масс-спектр (бомбардировка быстрыми атомами): 379 (M-H+).
Пример 168
PhCH2CH2CH2-CO-Pro-NH-pAmb
Желтоватый, аморфный гидройодид.
Масс-спектр (бомбардировка быстрыми атомами): 393 (M-H+).
Пример 169
α-нафтил-CO-NH-Pro-NH-pAmb
Белый, аморфный гидрохлорид.
Масс-спектр (бомбардировка быстрыми атомами): 401 (M-H+).
Пример 170
β-нафтил-CO-NH-Pro-NH-pAmb
Желтоватый, аморфный гидройодид.
Масс-спектр (бомбардировка быстрыми атомами): 401 (M-H+).
Пример 171
α-нафтил-CH2-CO-Pro-NH-pAmb
Белый, аморфный гидрохлорид.
Масс-спектр (бомбардировка быстрыми атомами): 415 (M-H+).
Пример 172
β-нафтил-CH2-CO-Pro-NH-pAmb
Белый, аморфный гидрохлорид.
Масс-спектр (бомбардировка быстрыми атомами): 415 (M-H+).
Пример 173
β-Нафтилсульфонил-Pro-NH-pAmb
Соединение получают путем сочетания β- нафтилсульфонилхлорида с H-Pro-OCH3, последующего омыления сложного эфира, сочетания с п-цианобензиламином и переведения нитрильной функции в амидиногруппу. Температура плавления гидроацетата составляет 66-72oC.
Пример 174
п-Толилсульфонил-Pro-NH-pAmb
Соединение получают согласно примеру 173. Температура плавления гидроацетата составляет 89 - 95oC.
Пример 175
Ph-CH2-CH2-SO2-Pro-NH-pAmb
Соединение получают согласно примеру 173. Температура плавления гидроацетата составляет 61 - 69oC.
Пример 176
H-Asp-Pro-NH-pAmb
Согласно методике A.I.в. от Boc-Asp (OBzI)-Pro-NH-pAmb отщепляют Boc-группу и сложный бензиловый эфир в присутствии палладия-на-угле гидрируют до образования кислоты. Дигидрохлорид получают путем обработки эфирным раствором хлороводорода.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 9,4/9,2 (4H, амидин); 8,6 (1Н, NH); 8,45 (3H, NH3 +); 7,80/7,45 (4H, аром. протоны); 7,80/7,45 (4H, аром. протоны); 4,45 - 4,30 (4H, CH2 и 2CH); 3,8 - примерно 3,5 (2H, CH2); 3,2 - примерно 2,6 (2H, CH2; 2,2 - 1,7 (4H, 2 CH2).
Пример 177
Boc-Asp(OMe)-Pro-NH-pAmb
Boc-Asp(OMe)-OH и гидрохлорид (4-цианобензил)пролиламида превращают аналогично примерам 5 и 10 до амидинового соединения. Соединение выделяют в качестве желтоватого, аморфного гидройодида.
Масс-спектр (бомбардировка быстрыми атомами): 476 (M-H+).
Пример 178
H-Asp(OMe)-Pro-NH-pAmb
Соединение получают путем отщепления Вос аналогично A.I.в из соединения примера 177 и переводят посредством ацетатного ионита в гидроацетат.
Получают белый, аморфный порошок.
Масс-спектр (бомбардировка быстрыми атомами): 376 (M-H+).
Пример 179
PhCH2-CO-Asp(OMe)-Pro-NH-pAmb
a) Boc-Asp (OMe)-OH подвергают взаимодействию с гидрохлоридом (4-цианобензил)пролиламида аналогично примеру 10 и затем отщепляют Boc-группу (см. данные по A.I.в.).
б) Ацилирование: К раствору 2,5 г (6,3) ммоль) соединения стадии а) и 1,76 мл триэтиламина в 80 мл хлористого метилена каплями добавляют при температуре -20oC раствор 0,98 г (6,3 ммоль) фенацетилхлорида в 20 мл хлористого метилена. Реакционную смесь перемешивают в течение ночи, в результате чего температура реакции повышается. Метиленхлоридную фазу последовательно промывают растворами бисульфата натрия, бикарбоната натрия и хлористого натрия, сушат над сульфатом натрия и сгущают в вакууме. Остаток очищают хроматографией на колонке (элюент: хлористый метилен и метанол в соотношении 9:1).
Выход: 1,6 г вязкого масла.
Масс-спектр (бомбардировка быстрыми атомами): 477 (M-H+).
в) Амидирование проводят аналогично примеру 5 и остаток переводят в гидроацетат.
Получают белые аморфные кристаллы.
Масс-спектр (бомбардировка быстрыми атомами): 494 (M-H+).
Нижеследующие соединения получают аналогично, причем кислотную функцию получают путем каталитического дебензилирования сложных бензиловых эфиров.
Пример 180
PhCH2CH2-CO-Asp(OMe)-Pro-NH-pAmb
Белый, аморфный гидроацетат.
Масс-спектр (бомбардировка быстрыми атомами): 508 (M-H+).
Пример 181
(n-Pr)2CH-CO-Asp-Pro-NH-pAmb
Белый, аморфный гидроацетат.
Масс-спектр (бомбардировка быстрыми атомами): 488 (M-H+).
Пример 182
(n-Pr)2CH-CO-Asp(OBzl)-Pro-NH-pAmb
Белый, аморфный гидроацетат.
Масс-спектр (бомбардировка быстрыми атомами): 578 (M-H+).
Пример 183
PhCH2-CO-Asp-Pro-NH-pAmb
Белый, аморфный гидрохлорид.
Масс-спектр (бомбардировка быстрыми атомами): 480 (M-H+).
Пример 184
PhCH2CH2-CO-Asp-Pro-NH-pAmb
Белый, аморфный гидрохлорид.
Масс-спектр (бомбардировка быстрыми атомами): 494 (M-H+).
Пример 185
(n-Pr)2CH-CO-Asp(OMe)-Pro-NH-pAmb
Белый, аморфный гидрохлорид.
Масс. спектр (бомбардировка быстрыми атомами): 502 (M-H+).
Пример 186
H-(D)-Asp-Pro-NH-pAmb
Дигидрохлорид получают согласно примеру 176.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 9,45/9,30 (4H, амидин); 9,05 (1H, NH): 8,9 (3H, NH3 +); 7,80/7,45 (4H, аром. протоны); 4,45 - 4,15 (4H, CH2 и 2 CH); 2,2 - 1,7 (4H, 2CH2).
Масс-спектр (бомбардировка быстрыми атомами): 362 (М+H+).
Пример 187
H-(D)-Asp(OtBu)-Pro-NH-pAmb
Дигидроацетат получают из Z-(D)-Glu(OtBu)-Pro-NH-pAmb путем гидрирования в присутствии палладия-на-угле.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 9,3 (4H, амидин); 8,5 (1Н, NH): 8,3 (3H, MH3 +); 7,75/7,25 (4H, аром. протоны); 4,4 - 4,3 (4H, CH2 и 2 CH); 2,9 - 2,6 (2H, CH2); 2,2 - 1,8 (4H, 2 CH2); 1,4 (9H, tBu).
Масс-спектр (бомбардировка быстрыми атомами): 418 (М+H+).
Пример 188
Z-(D)-Asp(OMe)-Pro-NH-pAmb
Получают аналогично примерам 5 и 10.
Исходное сырье: Z-(D)-Asp(OMe)-OH
Белый, аморфный гидроацетат.
Масс. спектр (бомбардировка быстрыми атомами): 510 (M-H+).
Пример 189
Z-(D)-Asp(OtBu)-Pro-NH-pAmb
Исходное сырье: Z-(D)-Asp(OtBu)-OH
Белый, аморфный гидроацетат.
Масс-спектр (бомбардировка быстрыми атомами): 552 (M-H+).
Пример 190
Boc(D)-Asp(OBzl)-Pro-NH-pAmb
Исходное сырье: Boc-(D)-Asp(OBzI)OH
Белый, аморфный гидроацетат.
Масс-спектр (бомбардировка быстрыми атомами): 552 (M-H+).
Пример 191
H-(D)-Asp(OBzl)-Pro-NH-pAmb
Соединение получают аналогично A. I.в из соединения примера 190 в качестве белого, аморфного дигидрохлорида.
Масс-спектр (бомбардировка быстрыми атомами): 452 (M-H+).
Пример 192
H-Asp(OBzl)-Pro-NH-pAmb
Исходное сырье: Boc-Asp(OBzl)-Pro-NH-pAmb
Белый, аморфный гидрохлорид.
Масс-спектр (бомбардировка быстрыми атомами): 452 (M-H+).
Пример 193
Z-(D)-Glu(OtBu)-Pro-NH-pAmb
Z-(D)-Glu(OtBu)-OH подвергают взаимодействию аналогично примеру 10 с гидрохлоридом (4-цианобензил)-пролиламида и затем аналогично примеру 5 переводят в амидиновое соединение. Посредством ацетатного ионита продукт реакции переводят в гидроацетат амидина и после лиофилизации выделяют в качестве белого, аморфного порошка.
Масс-спектр (бомбардировка быстрыми атомами): 566 (M-H+).
Пример 194
H-(D)-Glu(OtBu)-Pro-NH-pAmb
Из соединения примера 193 аналогично примеру 9 удаляют гидрированием защитную группу Z. После очистки хроматографиней (элюент: смесь хлористого метилена, метанола и уксусной кислоты в соотношении 16/4/1) и лиофилизации выделяют белый, аморфный порошок.
Масс-спектр (бомбардировка быстрыми атомами): 432 (M-H+).
Пример 195
H-(D)-Glu-Pro-NH-pAmb
Путем введения хлористоводородного газа в раствор хлористого метилена соединения примера 194 отщепляют трет.-бутильную группу. После удаления растворителя получают белый, аморфный порошок.
Масс-спектр (бомбардировка быстрыми атомами): 376 (M-H+).
Пример 196
(O-Ph-CH2-CHOH-CO-Pro-NH-pAmb
а) 3-Фенил-(D)-лактил-пролин-(п-цианобензил)амид:
5,5 г (20,4 ммоль) О-Тетрагидропиранил-3-фенил-D-молочной кислоты (международная заявка на патент 93/18060) растворяют в 30 мл диметилформамида и последовательно смешивают с 5,4 г (20,4 ммоль) N-(п-цианобензил)пролинамида, 3,3 г (20,4 ммоль) N-гидроксибензотриазола, 3,0 г диизопропилэтиламина и 4,33 г (20,6 ммоль)дициклогексилкарбодиимида. Дополнительно перемешивают в течение сорока восьми часов при комнатной температуре. После отсасывания выпавшей в осадок мочевины далее растворитель удаляют в вакууме, остаток смешивают с 50 мл воды и экстрагируют этилацетатом. После промывки водой, раствором гидрокарбоната натрия и высушивания над сульфатом натрия этилацетат отгоняют, полученный маслянистый остаток растворяют в метаноле и подкисляют до значения pH, равного двум, с помощью п-толуолсульфокислоты. Этот раствор выдерживают в течение шести часов при комнатной температуре. Затем метанол отгоняют, остаток растворяют в этилацетате, полученный раствор промывают водой, 5%-ным раствором лимонной кислоты и раствором гидрокарбоната натрия. Полученный после высушивания над сульфатом натрия и отгонки растворителя остаток очищают с помощью колоночной хроматографии (элюирующее средство: смесь дихлорметана с ацетоном и метанолом в соотношении 45:5:2). Получают 2,5 г белых кристаллов, которые после кристаллизации из смеси эфира с гексаном плавятся при 108 - 110oC.
б) 3-Фенил-D-лактил-пролин-(п-амидинобензил)амид-ацетат
2,0 г Вышеполученного соединения и 3 мл триэтиламина растворяют в 30 мл пиридина, при 0oC насыщают сероводородом и выдерживают в течение ночи при комнатной температуре. Согласно контролю с помощью тонкослойной хроматографии (смесь дихлорметана с метанолом в соотношении 9:1) превращение в тиоамид происходит полностью. Для выделения далее пиридин отгоняют в вакууме, остаток обрабатывают 250 мл этилацетата и полученный раствор промывают раствором хлорида натрия, 5%-ным раствором лимонной кислоты и раствором гидрокарбоната натрия. После высушивания и отгонки растворителя получают 2,3 г аморфного тиоамида.
Тиоамид растворяют в 40 мл ацетона и после добавления 4 мл метилиодида выдерживают в течение шести часов при комнатной температуре. Остающийся после удаления растворителя аморфный остаток перемешивают с безводным эфиром и после этого высушивают. Гидроиодид метилового эфира S-метил-тиоимидокислоты растворяют в 50 мл этанола, смешивают с 15 мл 10%-ного раствора ацетата аммония и нагревают в течение трех часов при 60oC. Для выделения растворитель удаляют, остаток растворяют в 100 мл дихлорметана, отфильтровывают нерастворимые составные части и затем отгоняют дихлорметан. Путем настаивания со смесью этилацетата с диэтиловым эфиром отделяют растворимые в этой смеси примеси. Полученную смешанную соль иодид-ацетат растворяют в смеси ацетона с водой в соотношении 3:2 и с помощью содержащей ацетатные группы ионообменной смолы (IRA) переводят в чистый ацетат, после чего очищают путем колоночной хроматографии (элюирующее средство: смесь дихлорметана с метанолом и 50%-ной уксусной кислотой в соотношении 40:10:1,5). Однородные фракции после удаления растворителей подвергают сушке вымораживанием. Получают 1,1 г порошка белого цвета с температурой плавления 185-187oC. Масс-спектр (бомбардировка быстрыми атомами): 395 (М+H+).
Пример 197
(D)-Man-Pro-NH-pAmb
Гидроацетат получают согласно примеру 196, исходя из О-тетрагидропиранил-(D)-миндальной кислоты (международная заявка на патент 93/18060), в виде кристаллов белого цвета. Температура плавления составляет 211 -213oC. Масс-спектр (бомбардировка быстрыми атомами): 381 (М+H+).
Пример 198
Boc-(D)-Phe-Aze-NH-pAmb
Boc-Aze-OH подвергают взаимодействию с гидрохлоридом (4-цианобензил)пролиламида аналогично примеру 10, затем отщепляют Boc-группу, снова сочетают с Boc-D-Phe-PH и затем амидируют. Получают желтоватый, аморфный гидрохлорид.
Масс-спектр (бомбардировка быстрыми атомами): 480 (M+H+).
Пример 199
Boc-(D)-Phe-Pro-(D,L)-Pic-NH-pAmb
Исходя из Boc-(D,L)-Pic-OH получают аналогично примеру 198 вышеуказанное соединение.
Белый аморфный гидроацетат.
Масс-спектр (бомбардировка быстрыми атомами): 508 (M+H+).
Пример 200
H-(D)-Phe-Aze-NH-pAmb
Смешанную соль гидроиодид/гидрохлорид получают путем взаимодействия Boc-(D)-Phe-OH с H-Aze-п-цианобензиламидом согласно примеру 3 вплоть до образования амидина и последующего отщепления Boc-группы.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 9,3/9,1 (4H, амидин); 9,0 (1Н, NH); 8,7 (3H, NH3 +); 7,8 - 7,2 (9H, аром. протоны); 4,5 - примерно 3,3 (6H, 2CH2 и 2CH); 3,2 - 2,8 (2H, CH2; 2,2 - 1,8 (2H, CH2).
Пример 201 и Пример 202
H-(D)-Phe-(D или L)-Pic-NH-pAmb (диастереомер (а)) и H-(D)-Phe-(D или D-Pic-NH-pAmb (диастереомер (б))
Дигидрохлорид пары диастереомеров получают из Boc-(D)-Phe-OH и H-(D, L)-Pic-п-цианобензиламида вплоть до образования амидина согласно примеру 3.
Затем Boc-группу отщепляют.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,6 - 9,3 (4H, амидин); 9,1 - 8,7 (4H, NH и NH3 +); 7,8 - 7,2 (9H, аром. протоны); 4,6 - 4,3 (4H, CH2 и 2 CH); 3,3 - 2,8 (2H, CH2); 2,3 - 0,9 (6H, 3 CH2).
Масс-спектр (бомбардировка быстрыми атомами): 408 (М+H+).
Оба диастереомера затем разделяют путем высокоэффективной жидкостной хроматографии, получая соединения примера 201 и примера 202.
Пример 203
Boc-(D)-Phe-(D,L/тpaнс)-Pic(4-Me)-NH-pAmb
Исходное сырье: Boc-(D,L/транс-Pic(4-Me)-OH
Белый, аморфный гидроацетат.
Масс-спектр (бомбардировка быстрыми атомами): 522 (М+H+).
Пример 204
H-(D)-Phe-(D,L/транс)-Pic(4-Me)-NH-pAmb
Дигидрохлорид получают согласно примерам 201 и 202, исходя из Boc-(D)-Phe-ОН и H-(D,L/транс)-Pic(4-Me)-п-цианобензиламида. Температура плавления составляет 160-170oC.
Пример 205
Boc-(D)-Phe-Pyr-NH-pAmb
Данное соединение синтезируют согласно примеру 3, исходя из гидрохлорида Boc-(D)-Phe-OH и H-Pyr-п-цианобензиламида.
Масс-спектр: 492,5 (М+H+), 392, 245, 133.
Пример 206
H-(D)-Phe-Pyr-NH-pAmb
Соединение получают путем отщепления Boc-группы от продукта примера 205.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 9,4 - 9,2 (уш. с. , NH); 8,8 - 8,2 (уш. с., NH); 8,6 (2т, 1H, NH); 7,75 (2д, 2H, аром. протоны); 7,45 (2д, 2H, аром. протоны); 7,35 - 7,1 (м, 5H, аром. протоны); 6,15/6,0/5,85/5,75 (4 уш. с., 2H, CH=CH); 5,5/4,9 (уш. с., 1H, α-Pyr); 4,4 -4,2 (м, 4H, CH2/α-Phe/δ-Pyr ; 3,6 (д, 1H, δ-Pyr); 3,1 - 3,0 (м, 2H, CH2-фенил).
Масс-спектр (бомбардировка быстрыми атомами): 392 (М+H+).
Пример 207
Boc-(D)-Phe-Hyp(OtBu)-NH-pAmb
Соединение получают согласно примеру 1, исходя из Boc-(D)-Phe-Hyp(OtBu)-ОН и гидрохлорида п-цианобензиламина.
Масс-спектр: 566 (М+H+), 466 (-Boc), 319 (-Phe).
Пример 208
H-(D)-Phe-Hyp-NH-pAmb
Соединение получают путем отщепления Boc-группы и трет.-бутильной группы от соединения примера 207.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 9,35 (с, 2H, NH); 9,1 (с, 2H, NH); 8,8 (т, 1H, NH); 8,5 (уш. с., 3H, NH); 7,75 (2д, 2H, аром. протоны); 7,45 (2д, 2H, аром. протоны); 7,35 - 7,2 (м, 5H, аром. протоны); 4,4 - 4,2 (м, 5H, CH2/ 2 α- H/CHOH); 3,8 (м, 1H, Pro); 3,0 (м, 2H, CH2); 2,75 (м, 1H, Pro); 1,95 (м, 1H, Pro); 1,8 (м, 3H, Pro).
Масс-спектр (бомбардировка быстрыми атомами): 410 (М+H+).
Пример 209
Boc-(D)-Phe-(Me)Val-NH-pAmb
Исходное сырье: Boc-(Me)Val-OH
Желтоватый, аморфный гидроиодид.
Масс-спектр (бомбардировка быстрыми атомами): 510 (М+H+).
Пример 210
Boc(D)-Phe-Val-NH-pAmb
Получают аналогично примеру 1.
Белый, аморфный гидрохлорид.
Масс-спектр (бомбардировка быстрыми атомами): 496 (М+H+).
Пример 211
H-(D)-Phe-Val-NH-pAmb
Из соединения примера 210 отщепляют Boc-группу.
Белый, аморфный гидрохлорид.
Масс-спектр (бомбардировка быстрыми атомами): 396 (М+H+).
Пример 212
H-(D)-Phe-(Me)Val-NH-pAmb
Дигидрохлорид получают согласно примеру 3, исходя из Boc-(D)-Phe-OH и H-(Me)Val-п-цианобензиламида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,45/9,25 (4H, амидин); 8,8 (1H, NH); 8,6 (3H, NH3 +); 7,8/7,5/7,3 (9H, аром. протоны); 4,65 (1H, CH); 4,60 (2H, CH2); 4,45 - 4,20 (2H, CH2; 3,20 - 2,95 (2H, CH2; 2,85 (3H, N-CH3); 2,0 (1Н, CH); 0,8/0,45 (6H, 2CH3).
Пример 213
Boc-(D)-Phe-Tia-NH-pAmb
Соединение получают согласно примеру 1, исходя из Boc-(D)-Phe-Tia-OH и п-цианобензиламингидрохлорида.
Масс-спектр: 512 (М+H+), 412 (-Boc), 265, 204, 133.
Пример 214
H-(D)-Phe-Tia-NH-pAmb
Соединение получают путем отщепления Boc-группы от продукта примера 213.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 9,4/9,2 (2 уш. с. , 4H, NH); 9,0 (т, 1H, NH); 7,75 (2д, 2H, аром. протоны); 7,45 (2д, 3H, аром. протоны); 7,4 - 7,2 (м, 5H, аром. протоны); 4,8 (д, 1H, α-Tia); 4,7/3,7 (2д, 2H, N-CH2-S); 4,4 - 4,2 (м, 3H, CH2/ α-Phe); 3,2 - 3,1 (2м, 2H, SCH2); 3,0/2,7 (м, 3H, CH2-фенил).
Масс-спектр (бомбардировка быстрыми атомами): 412 (М+H+).
Пример 215
H-(D)-Phe-Pro-NH-3-(6-Am)-pico
а) 2-Циано-5-(азидометил)пиридин:
К раствору 8,8 г (0,07 ммоль) 2-циано-5-(гидроксиметил)пиридина (международная заявка на патент 83/01446) и 6,9 г триэтиламина в 200 мл дихлорметана при комнатной температуре прикапывают раствор 14,5 г (0,07 моль) ангидрида трифторуксусной кислоты в 20 мл дихлорметана и затем перемешивают дополнительно в течение двух часов. Полученный после отгонки дихлорметана остаток растворяют в смеси из 50 мл толуола и 50 мл диметилсульфоксида, смешивают с 11,2 г (0,17 моль) азида натрия и 0,7 г тетрабутиламмонийбромида и дополнительно перемешивают в течение ночи при комнатной температуре.
Реакционную смесь выливают в 300 мл воды и многократно экстрагируют эфиром. После высушивания над сульфатом натрия и отгонки эфира получают 6,8 г желтоватых кристаллов с температурой плавления 62 - 64oC, которые без дальнейшей очистки вводят в следующую реакцию.
б) 2-Циано-5-(аминометил)пиридин:
Полученное в стадии а) соединение растворяют в смеси из 45 мл тетрагидрофурана и 1,2 мл воды и при перемешивании порциями смешивают с 11,2 г трифенилфосфина. Реакционную смесь выдерживают в течение ночи при комнатной температуре. Полученный после отгонки растворителя остаток обрабатывают 100 мл эфира, выпавший в осадок трифенилфосфиноксид отсасывают и фильтрат подкисляют до pH-значения, равного двум, с помощью эфирного раствора хлороводорода. Осадившийся гидрохлорид отсасывают, промывают эфиром и настаивают последовательно с толуолом и горячим изопропанолом. Получают 4,7 г (40%) гидрохлорида с температурой плавления 253 - 256oC (разложение).
в) Boc-(D)-фенилаланил-пролин-(6-циано-3-пиколил)амид:
К раствору 2,11 г (12,5 ммоль) 2-циано-5-(аминометил)пиридина и 4,5 г (12,5 ммоль) Boc-(D)-Phe-Pro-OH в 70 мл дихлорметана при -5oC прикапывают 8,12 г диизопропилэтиламина и затем 11 мл (15 ммоль) 50%-ного раствора в этилацетате ангидрида пропанфосфоновой кислоты. Дополнительно перемешивают в течение двух часов, причем температура повышается от -5 до 20oC. Органическую фазу промывают водой, 5%-ным раствором гидрокарбоната натрия и 5%-ным раствором лимонной кислоты, сушат над сульфатом натрия и концентрируют досуха. Получают слегка желтоватый кристаллический остаток с температурой плавления 167 - 170oC, который без дальнейшей очистки вводят в следующую реакцию.
г) Boc-(D)-фенилаланил-пролин-(6-амидино-3-пиколил)амид: 1,15 г (16,5 ммоль) Гидроксиамингидрохлорида суспендируют в 5 мл этанола, смешивают с 1,2 г 25%-ного раствора аммиака и перемешивают в течение десяти минут. После добавления 45 мл этанола отсасывают выпавшую в осадок соль, а к раствору добавляют 3,14 г (6,6 ммоль) полученного в стадии в) соединения. Спустя непродолжительное время осаждается гидроксиамидин, который после дополнительного перемешивания в течение тридцати минут отсасывают и промывают небольшим количеством холодной воды и этанолом. Увлажненный этанолом осадок растворяют в 40 мл этанола и 8 мл ледяной уксусной кислоты, смешивают с 250 мг 10%-ного палладия-на-угле и гидрируют примерно при 50oC. Спустя пять часов согласно контролю с помощью тонкослойной хроматографии (смесь дихлорметана с метанолом и 50%-ной уксусной кислотой в соотношении 20:5:1) более не обнаруживают исходного соединения.
После отсасывания катализатора через слой целита отгоняют растворитель, с добавлением под конец отгонки толуола. После добавления 50 мл ацетона выкристаллизовывается амидинацетат, который отфильтровывают. Получают кристаллы белого цвета с температурой плавления 130 -134oC.
Масс-спектр (бомбардировка быстрыми атомами): 495 (М+H+).
д) H-(D)-Phe-Pro-NH-3-(6-Am)-pico:
От соединения, полученного в стадии г), в стандартных условиях отщепляют Boc-группу. Получают дигидрохлорид в виде кристаллов белого цвета с температурой плавления 235 - 240oC.
Масс-спектр (бомбардировка быстрыми атомами): 395 (М+H+).
Пример 216
Boc-(D)-Chg-Pro-NH-3-(6-Am)-pico
а) Boc-(D)-циклогексилглицилпролин:
29 г (0,113 моль) Boc-(D)-циклогексилглицина и 18,7 г (0,113 моль) гидрохлорида метилового эфира пролина суспендируют в 300 мл дихлорметана и путем прикапывания 58,3 г (0,45 моль) диизопропилэтиламина суспензию переводят в раствор. После охлаждения до температуры -15oC прикапывают 113 мл (0,147 моль) 50%-ного раствора ангидрида пропанфосфоновой кислоты в этилацетате и дополнительно перемешивают в течение одного часа.
После добавления 200 мл воды отделяют органическую фазу и промывают ее водным раствором карбоната калия, 0,5 н. соляной кислотой и 5%-ным раствором гидрокарбоната натрия. После высушивания над сульфатом натрия отгоняют растворитель, маслянистый остаток в количестве 41 г растворяют в 400 мл этанола, смешивают со 120 мл 1 н. раствора гидроксида натрия и перемешивают в течение двух часов при комнатной температуре.
После отгонки спирта разбавляют водную фазу водой и многократно экстрагируют метил-трет. -бутиловым эфиром. Водную фазу подкисляют раствором гидросульфата калия и экстрагируют трехкратно дихлорметаном. Полученный после высушивания и отгонки дихлорметана маслянистый остаток кристаллизуют из смеси диизопропилового эфира с н-гексаном в соотношении 1:3. Выделяют 28 г кристаллов белого цвета с температурой плавления 145 - 148oC.
б) Boc-(D)-циклогексилглицил-пролин-(6-циано-3-пиколил)амид:
26,6 г (0,075 моль) Boc-(D)-циклогексилглицилпролина и 12,7 г (0,075 моль) 6-циано-3-пилиламингидрохлорида суспендируют в 300 мл дихлорметана и смешивают с 47 г (0,364 моль) диизопропилэтиламина. 3атем при температуре -10oC прикапывают 66 мл 50%-ного раствора в этилацетате ангидрида пропанфосфоновой кислоты, дополнительно перемешивают в течение часа при 0oC, смешивают с 200 мл воды и отделяют дихлорметановую фазу. После промывки органической фазы 0,1 н. раствором гидроксида натрия и водой ее сушат и растворитель отгоняют. Остаток обрабатывают 100 мл этилацетата, причем быстро начинается кристаллизация, которая происходит полностью за счет добавления 150 мл н-гексана. После отсасывания и высушивания выделяют 31,4 г (89% от теоретически рассчитанного количества) кристаллов белого цвета с температурой плавления 150-151oC.
в) Boc-(D)-циклогексилглицил-пролин-(6-амидино-3-пиколил)амид:
Амидин получают согласно примеру 215, стадия г). Ацетат представляет собой кристаллы белого цвета с температурой плавления 160 - 168oC (разложение).
Масс-спектр (бомбардировка быстрыми атомами): 487 (М+H+).
Пример 217
H-(D)-Chg-Pro-NH-3-(6-Am)-pico
Соединение получают путем отщепления Boc-группы от продукта стадии в) вышеуказанного соединения в стандартных условиях. Дигидрохлорид представляет собой кристаллы белого цвета с температурой плавления 235 - 238oC (разложение).
Масс-спектр (бомбардировка быстрыми атомами): 387 (М+H+).
Пример 218
HOOC-CH2-(D)-Chg-Pro-NH-3-(6-Am)-pico
а) H-(D)-Циклогексилглицилпролин-(6-циано-3-пиколил)амид: 46,9 (0,1 моль) Boc-(D)-Циклогексилглицилпролин-(6-циано-3- пиколил)-амида (соединение 219, стадия б)) суспендируют в 300 мл эфира, при перемешивании при комнатной температуре смешивают с 600 мл насыщенного хлороводородом эфира и дополнительно перемешивают в течение ночи. Затем суспензию при перемешивании и охлаждении льдом выливают в 1,5 л 15%-ного раствора гидроксида натрия. После добавления 80 мл дихлорметана отделяют органическую фазу, а щелочную фазу экстрагируют шестикратно смесью эфира с дихлорметаном в соотношении 7:3. Объединенные органические фазы сушат над сульфатом натрия и концентрируют досуха. Получают 27,2 г аморфного белого порошка, который согласно тонкослойной хроматографии (смесь дихлорметана с метанолом в соотношении 4:1) содержит еще примерно 5-10% образовавшегося за счет гидролиза циангруппы амида.
б) N-(трет. -Бутоксикарбонилметил)-(D)- циклогексилглицилпролин-(6-циано-3-пиколил)амид:
К раствору 27,2 г (0,074 моль) вышеполученного соединения (стадия а)) и 28,6 г (0,22 моль) диизопропилэтиламина в 150 мл дихлорметана при перемешивании и при комнатной температуре прикапывают 14 г (0,072 моль) трет.-бутилового эфира бромуксусной кислоты и перемешивают в течение ночи.
Реакционный раствор промывают водой, сушат над сульфатом натрия и остаток после отгонки растворителя хроматографируют при использовании колонки с силикагелем, элюируя смесью дихлорметана с ацетоном и метанолом в соотношении 45:5:1. Выделяют 28,6 г (80% от теоретически рассчитанного количества) аморфного белого порошка. Образец кристаллизуют из диизопропилового эфира при добавлении небольшого количества диэтилового эфира: он плавится при 89 - 91oC.
в) N-трет. -Бутоксикарбонилметил-(D)-циклогексилглицилпролин- (6-амидино-3-пиколил)амид:
Вышеполученное соединение, согласно примеру 215, стадия г), переводят в амидин. Ацетат представляет собой белый аморфный порошок: масс-спектр (бомбардировка быстрыми атомами): 501 (М+H+).
г) N-(Карбоксиметил)-(D)-циклогексилглицилпролин-(6-амидино- 3-пиколил)амид:
2,4 г Вышеполученного амидинацетата растворяют в 50 мл смеси дихлорметана с трифторуксусной кислотой в соотношении 1:1 и выдерживают в течение ночи при комнатной температуре. Раствор концентрируют в вакууме, остаток обрабатывают дихлорметаном, еще раз растворитель отгоняют при добавлении толуола и после этого хроматографируют на колонке с силикагелем, элюируя смесью метанола с 25%-ным водным аммиаком в соотношении 50:2. После отгонки растворителей продукт растворяют в воде и после обработки активным углем лиофилизируют.
Лиофилизат (1,45 г) имеет температуру плавления 202-205oC.
Масс-спектр (бомбардировка быстрыми атомами): 445 (М+H+).
Пример 219
HOOC-CH2-(D)-Chg-Pyr-NH-3-(6-Am)-pico
а) 5,2 г (14,75 ммоль) Boc-(D)-Chg-Pyr-OH, 2,88 г (17 ммоль) 6-циано-3-амино-метилпиридина, 12,2 мл диизопропилэтиламина и 17 мл 50%-ного раствора в этилацетате ангидрида пропанфосфоновой кислоты при 0oC вносят в 50 мл дихлорметана. Затем при перемешивании в течение полутора часа реакционную смесь доводят до комнатной температуры. Для обработки раствор разбавляют 250 мл этилацетата и промывают трехкратно насыщенным раствором гидрокарбоната натрия, трехкратно 20%-ным раствором гидросульфата натрия и однократно насыщенным раствором хлорида натрия. После высушивания раствора над сульфатом натрия этилацетат удаляют в ротационном испарителе. Выход сырого продукта составляет 7,8 г. Сырой продукт без дальнейшей очистки используют в следующей реакции.
б) Boc-(D)-Chg-Pyr-NH-3-(6-CN)-pico вносят в 10 мл дихлорметана. После охлаждения раствора до 0oC добавляют 20 мл трифторуксусной кислоты в виде 50%-ного раствора в дихлорметане. Затем реакционную смесь в течение трех часов оставляют нагреваться до комнатной температуры и после этого раствор концентрируют в ротационном испарителе. Остаток растворяют в толуоле и раствор еще раз концентрируют в вакууме. Этот процесс повторяют еще раз. Выход сырого продукта составляет 13,5 г.
в) 13,5 г H-(D)-CHg-Pyr-NH-3-(6-CN)-pico-гидротрифторацетата вносят в 100 мл ацетонитрила. После добавления 2,69 г иодида калия, 6,11 г карбоната калия и 2,87 г трет.-бутилового эфира бромуксусной кислоты суспензию перемешивают при комнатной температуре в течение пяти часов. Затем карбонат калия и иодид калия отфильтровывают, ацетонитрил удаляют в вакууме в ротационном испарителе и остаток растворяют в этилацетате. Раствор промывают два раза водой и один раз насыщенным раствором хлорида натрия, сушат над сульфатом натрия и концентрируют. Выход сырого продукта составляет 6,4 г.
г) 6 г tBuOOC-CH2-(D)-Chg-Pyr-NH-3-(6-CN)-pico растворяют в 42 мл пиридина и 19,4 мл триэтиламина и насыщают газообразным сероводородом. После выдерживания в течение восемнадцати часов при комнатной температуре раствор сначала продувают азотом и затем выливают в 2 л воды со льдом. Водный раствор экстрагируют шестикратно этилацетатом и объединенные органические экстракты промывают 5%-ным раствором гидросульфата натрия. После высушивания и концентрирования получают 6,1 г tBuOOC-CH2-(D)-Chg-Pyr-NH-3-(6-CSNH2)-pico в виде сырого продукта.
д) 6,1 г Сырого tBuOOC-CH2-(D)-Chg-Pyr-NH-3-(6-CSNH2)-pico растворяют в 7,4 мл метилиодида и 70 мл ацетона и перемешивают в течение четырех с половиной часов при комнатной температуре. Затем раствор концентрируют, обрабатывают толуолом и еще раз выпаривают досуха в ротационном испарителе. Выход сырого продукта составляет 6,1 г.
е) 6,1 г tBuOOC-CH2-(D)-Chg-Pyr-NH-3-(6-CSMe=NH)-pico в одногорлой колбе смешивают с 30 мл метанола и 10 мл 20%-ного метанольного раствора ацетата аммония и выдерживают в течение восемнадцати часов при комнатной температуре. Раствор концентрируют и остаток растворяют в дихлорметане. Органический раствор промывают 3 раза по 20 мл водой, сушат над сульфатом натрия и концентрируют в ротационном испарителе. После переосаждения сырого продукта из смеси этилацетата с диизопропиловым эфиром получают 2,7 г сырого продукта. Сырой продукт очищают путем высокоэффективной жидкостной хроматографии с обращенной фазой. Выход составляет 0,364 г.
ж) 0,28 г tBuOOC-CH2-(D)-Chg-Pyr-NH-3-(6-Am)-pico при 0oC вносят в 5 мл диоксана и после добавления 5 мл раствора хлороводорода в диоксане перемешивают в течение сорока восьми часов при комнатной температуре. Полученный после концентрирования раствора сырой продукт очищают с помощью колоночной хроматографии (метанол с 3% концентрированного раствора аммиака). Выход составляет 180 мг.
Масс-спектр (бомбардировка быстрыми атомами): 443 (М+H+).
Пример 220
HOOC-CH2-(D)-Chg-2-Phi-NH-3-(6-Am)-pico
Исходя из Boc-(D)-Chg-2-Phi-OH и 3-аминометил-6-цианопиридина согласно примеру 219 получают tBuOOC-CH2-(D)-Chg-2-Phi-NH-3-(6- CN)-pico. Этот промежуточный продукт превращают в соединение примера 220 следующим образом:
а) 8 г (14,9 ммоль) tBuOOC-CH2-(D)-Chg-2-Phi-NH-3-(6-CN)-pico вместе с 8 мл триэтиламина, 2,58 г гидроксиламингидрохлорида и 90 мл этанола перемешивают в течение восемнадцати часов при 70oC. Затем суспензию концентрируют, остаток растворяют в дихлорметане и раствор промывают три раза по 5 мл 30%-ной уксусной кислотой. После высушивания над сульфатом натрия дихлорметан удаляют в ротационном испарителе. N-гидроксиамидин без дальнейшей очистки используют в следующей реакции.
б) В реакционной колбе смешивают 5 г N-гидроксиамидина с 6 г никеля Ренея, 40 мл этанола и 9 мл уксусной кислоты и гидрируют в атмосфере водорода при 60oC. Сырой продукт разделяют путем высокоэффективной жидкостной хроматографии с обращенной фазой (смесь ацетонитрила с водой). Выход составляет 0,7 г.
в) 0,7 г tBuOOC-CH2-(D)-Chg-2-Phi-NH-3-(6-Am)-pico согласно примеру 219 переводят в свободную кислоту. Масс-спектр (бомбардировка быстрыми атомами): 499 (М+H+).
Пример 221
HOOC-CH(Me)-(D)-Chg-Pro-NH-3-(6-Am)-pico
а) 7,4 г (15,22 ммоль) H-(D)-Chg-Pro-NH-3-(6-CN)-pico- гидротрифторацетата, 6,3 г карбоната калия и 3,69 г бензилового эфира 2-бромпропионовой кислоты в 100 мл ацетонитрила перемешивают в течение двенадцати часов при 50oC. После полного превращения эдукта отфильтровывают твердое вещество и фильтрат концентрируют. После этого остаток растворяют в этилацетате и промывают два раза водой. После высушивания органического раствора этилацетат удаляют в ротационном испарителе. Выход сырого продукта составляет 5 г. После колоночной хроматографии на силикагеле получают 3 г продукта.
б) 3 г BzlOOC-CH(Me)-(D)-Chg-Pro-NH-3-(6-CN)-pico согласно примеру 220 превращают в соответствующий амидин. Выход составляет 0,8 г.
в) Свободную кислоту получают путем гидрирования сложного бензилового эфира в стандартных условиях. Сырой продукт очищают путем высоко эффективной жидкостной хроматографии с обращенной фазой. Выход составляет 0,4 г.
Масс-спектр (бомбардировка быстрыми атомами): 459 (М+H+).
Пример 222
Boc-(D)-Phe-Pro-NH-3-(2-Me-6-Am)-pico
Соединение получают согласно примеру 224. Температура плавления гидроацетата составляет 130 - 140oC.
Пример 223
H-(D)-Phe-Pro-NH-3-(2-Me-6-Am)-pico
Соединение получают в виде дигидрохлорида согласно примеру 225.
13C-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 170,79; 167,62; 161,85; 156,34; 140,72; 138,41; 135,87; 134,53; 129,30; 128,49; 127,29; 120,70; 60,23; 52,18; 46,61; 39,1; 36,48; 29,22; 23,33; 21,72.
Пример 224
Boc-(D)-Chg-Pro-NH-3-(2-Me-6-Am)-pico
а) Получение Boc-(D)-Chg-Pro-NH-3-(2-Me)-pico:
6,4 г Boc-(D)-Chg-Pro-OH (18,05 ммоль) вместе с 4,0 г 2-метил-3-пиколил-амина (20,5 ммоль; получение см. Arch. Pharm., 308. 969 - 976 (1975)) и 14 мл диизопропилэтиламина (81,8 ммоль) вносят в 200 мл дихлорметана, охлаждают до 5oC и при этой температуре прикалывают 18,8 мл 50%-ного раствора ангидрида пропанфосфоновой кислоты в этилацетате (23,92 ммоль). После нагревания до комнатной температуры дополнительно перемешивают в течение часа и после этого концентрируют в вакууме. Остаток обрабатывают этилацетатом, этилацетатную фазу экстрагируют примерно 10 раз водой, сушат над сульфатом магния и выпаривают в ротационном испарителе. Путем перемешивания остатка с диизопропиловым эфиром получают 7,2 г (87%) Boc-(D)-Chg-Pro-NH-3- (2-Me)-pico в виде белого твердого вещества.
б) Получение Boc-(D)-Chg-Pro-NH-3-(2-Me-1-оксо)-pico:
5,3 г Boc-(D)-Chg-Pro-NH-3-(2-Me)-pico (11,58 ммоль) вместе с 3,1 г 98%-ной м-хлорнадбензойной кислоты (18,14 ммоль) в 150 мл дихлорметана перемешивают два часа при комнатной температуре. Затем пропускают газообразный аммиак вплоть до насыщения, перемешивают в течение часа при комнатной температуре, осадок отсасывают, промывают дихлорметаном, а фильтрат еще раз насыщают аммиаком. После этого дихлорметановую фазу промывают три раза водой, сушат над сульфатом магния и концентрируют в вакууме. Получают 5,5 г Boc-(D)-Chg-Pro-NH-3-(2-Me-1-оксо)-pico в виде белого твердого вещества.
в) Получение Boc-(D)-Chg-Pro-NH-3-(2-Me-1-MeO)-pico:
3,6 г Boc-(D)-Chg-Pro-NH-3-(2-Me-1-оксо)-pico (7,58 ммоль) растворяют в 10 мл дихлорметана, смешивают с 2,0 мл диметилсульфата (21,1 ммоль) в 20 мл дихлорметана, перемешивают в течение ночи при комнатной температуре, раствор концентрируют в вакууме и остаток перемешивают 3 раза с эфиром. Получают 4,55 г (100%) Boc-(D)-Chg-Pro-NH-3-(2-Me-1-MeO)-pico⊕CH3OSO
г) Получение Boc-(D)-Chg-Pro-NH-3-(2-Me-6-CN)-pico:
4,55 Boc-(D)-Chg-Pro-NH-3-(2-Me-1-MeO)-pico⊕CH3OSO
д) Получение Boc-(D)-Chg-Pro-NH-3-(2-Me-6-Ham)-pico:
3,63 г Boc-(D)-Chg-Pro-NH-3-(2-Me-6-CN)-pico (7,51 ммоль) вместе с 1,9 г гидроксиламмонийхлорида (18,76 ммоль) и 6,4 мл диизопропилэтиламина (37,525 ммоль) в 50 мл дихлорметана перемешивают в течение четырех часов при комнатной температуре, затем выпаривают в вакууме в ротационном испарителе, остаток обрабатывают этилацетатом, промывают 6 раз разбавленной соляной кислотой (pH-значение равно четырем), органическую фазу сушат над сульфатом магния и выпаривают в вакууме в ротационном испарителе. Получают 3,8 г (98%) Boc-(D)-Chg-Pro-NH-3-(2-Me-6-Ham)-pico в виде белого твердого вещества.
е) Получение Boc-(D)-Chg-Pro-NH-3-(2-Me-6-Am)-pico:
3,8 г Boc-(D)-Chg-Pro-NH-3-(2-Me-6-Ham)-pico (7,35 ммоль) с добавлением два раза на кончике шпателя 10%-ного палладия-на-угле в смеси из 80 мл этанола и 15 мл уксусной кислоты гидрируют при 60oC в течение восьми часов при слегка повышенном давлении, катализатор отфильтровывают через фильтр из стекловолокна, промывают этанолом и фильтрат концентрируют в вакууме (1 мбар). После двухкратного перемешивания остатка с эфиром получают 4,0 г (97%) Boc-(D)-Chg-Pro-NH-3-(2-Me-6-Am)-pico в виде белого твердого вещества. Температура плавления гидроацетата составляет 144 - 153oC.
Пример 225
H-(D)-Chg-Pro-NH-3-(2-Me-6-Am)-pico
2,8 г Boc-(D)-Chg-Pro-NH-3-(2-Ме-6-Am)-Pico-гидроацетата (4,99 ммоль) в 10 мл дихлорметана и 15 мл метанола с 25 мл эфирного раствора хлороводорода (концентрация выше 3М) перемешивают в течение четырех часов при комнатной температуре. Раствор концентрируют в вакууме, многократно обрабатывают дихлорметаном, метанолом, каждый раз отгоняя растворитель после обработки, и остаток перемешивают со смесью эфира с дихлорметаном и эфира с метанолом. Получают 2,5 г Н-(D)-Chg-Pro-NH-3-(2-Ме-6-Am)-pico- дигидрохлорида в виде белого твердого вещества. Температура плавления дигидрохлорида составляет 128-135oC.
13C-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 170,96; 167,72; 161,86; 156,30; 140,76; 138,53; 135,85; 120,72; 60,56; 55,17; 47,43; 39,20; 38,78; 29,66; 27,75; 25,40; 25,31; 25,20; 23,71; 21,76.
Пример 226
tBuOOC-CH2-(D)-Chg-Pro-NH-3-(2-Me-6-Am)-pico
а) Получение H-(D)-Chg-Pro-NH-3-(2-Me)-pico:
7,8 г Boc-(D)-Chg-Pro-NH-3-(2-Me)-pico (17,0 ммоль) в 35 мл дихлорметана и 35 мл эфирного раствора хлороводорода (концентрация выше 3М) перемешивают в течение двух часов при комнатной температуре, концентрируют в ротационном испарителе в вакууме, многократно обрабатывают метанолом и дихлорметаном, каждый раз отгоняя растворитель после обработки, и остаток перемешивают с эфиром. Получают 7,3 г (100%) H-(D)-Chg-Pro-NH-3-(2-Ме)-pico- дигидрохлорида в виде белого твердого вещества.
б) Получение tBuOOC-CH2-(D)-Chg-Pro-NH-3-(2-Me)-pico:
9,4 г H-(D)-Chg-Pro-NH-3-(2-Ме)-pico-дигидрохлорида (21,79 ммоль) вместе с 11,26 г (14,9 мл) диизопропилэтиламина (81,16 ммоль) и 4,89 г (3,69 мл) трет. -бутилового эфира бромуксусной кислоты (25,0 ммоль) в 150 мл дихлорметана (высушенного над молекулярным ситом) перемешивают в течение шестнадцати часов при комнатной температуре. Так как согласно тонкослойной хроматографии обнаруживают еще наличие эдукта, то добавляют еще 0,4 мл трет.-бутилового эфира бромуксусной кислоты и 1,5 мл диизопропилэтиламина и перемешивают следующие три часа при комнатной температуре. Затем реакционную смесь концентрируют сначала в вакууме водоструйного насоса, затем при давлении 1 мбар максимально при 40oC. Остаток перемешивают с эфиром, отфильтровывают и промывают эфиром. После растворения кристаллизата в воде многократно экстрагируют при pH-значении, равном 7,5, этилацетатом, эти этилацетатные экстракты объединяют с вышеполученным эфирным фильтратом, сушат и концентрируют в вакууме в ротационном испарителе. После обработки остатка эфиром добавляют эфирный раствор хлороводорода вплоть до pH-значения, равного трем, образовавшийся осадок отсасывают, хорошо промывают эфиром и еще два раза перемешивают с эфиром. Получают 9,1 г (82%) t(BuOOC-CH2-(D)-Chg-Pro-NH-3-(2-Me)-pico-гидрохлорида в виде белого твердого вещества.
в) Получение tBuOOC-CH2-(Boc)(D)-Chg-Pro-NH-3-(2-Me)-pico:
9,5 г tBuOOC-CH2-(D)-Chg-Pro-NH-3-(2-Ме)-pico-гидрохлорида (18,66 ммоль) вместе с 18,66 г (Boc)2O (18,66 ммоль) вносят в 160 мл дихлорметана, в течение пяти минут смешивают с 5,3 г (7,03 мл) диизопропилэтиламина (41,05 ммоль) и затем перемешивают в течение ночи при комнатной температуре. После следующего добавления дихлорметана промывают 0,5 М раствором соляной кислоты до тех пор, пока в дихлорметане не будут более обнаруживать никаких следов диизопропилэтиламина (контроль с помощью тонкослойной хроматографии), сушат над сульфатом магния и концентрируют в ротационном испарителе в вакууме. Путем колоночной хроматографии на силикагеле при элюировании с помощью дихлорметана с 0-5% метанола получают 5,8 г tBuOOC-CH2-(Boc)(D)-Chg-Pro-NH-3-(2-Me)-pico в виде белого твердого вещества.
г) Получение tBuOOC-CH2-(Boc)-(D)-Chg-Pro-NH-3-(2-Me-1-оксо)- pico:
5,8 г tBuOOC-CH2-(Boc)-(D)-Chg-Pro-NH-3-(2-Me)-pico (10,12 ммоль) вместе с 99,9 г 70%-ной м-хлорнадбензойной кислоты (40,5 ммоль) в 200 мл дихлорметана перемешивают в течение двух часов при комнатной температуре. Затем пропускают газообразный аммиак вплоть до насыщения, перемешивают в течение часа при комнатной температуре, осадок отсасывают, промывают дихлорметаном и фильтрат еще раз насыщают аммиаком. После этого дихлорметановую фазу промывают три раза водой, сушат над сульфатом магния и концентрируют в вакууме. Получают 5,95 г (100%) продукта.
д) Получение 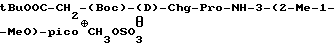
5,95 г tBuOOC-CH2-(Boc)-(D)-Chg-Pro-NH-3-(2-Me-1-оксо)-pico (10,12 ммоль) растворяют в 25 мл дихлорметана и смешивают с 28 мл 5%-ного раствора диметилсульфата в дихлорметане. После перемешивания в течение пяти часов при 40oC и выдерживания в течение ночи при комнатной температуре разбавляют 100 мл дихлорметана, быстро промывают три раза водой, сушат над сульфатом магния и концентрируют в ротационном испарителе в вакууме. Полученный 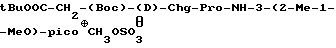 в виде неочищенного продукта используют в последующем превращении.
в виде неочищенного продукта используют в последующем превращении.
е) Получение tBuOOC-CH2-(Boc)-(D)-Chg-Pro-NH-3-(2-Me-6-CN)-pico:
Полученный в вышеприведенной стадии в виде сырого продукта 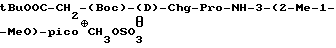 в течение 20 минут прикаgывают к раствору 1,1 г цианида натрия (21,3 ммоль) в 50 мл диметилформамида, причем температуру поддерживают при 23 - 25oC путем охлаждения. Спустя следующие двадцать минут диметилформамид отгоняют в вакууме (1 мбар), остаток обрабатывают эфиром, эфирный раствор промывают последовательно водой, раствором гидросульфата калия (pH-значение равно двум), водой и насыщенным раствором хлорида натрия, эфирную фазу сушат над сульфатом магния и концентрируют в ротационном испарителе в вакууме. После очистки с помощью колоночной хроматографии на силикагеле (элюирующее средство: дихлорметан с 0-2% метанола) получают 4,1 г твердого вещества, которое перемешивают с эфиром. Выход tBuOOC-CH2-(Boc)(D)-Chg-Pro-NH-3-(2-Me-6-CN)-pico составляет 4,0 г (66%)
в течение 20 минут прикаgывают к раствору 1,1 г цианида натрия (21,3 ммоль) в 50 мл диметилформамида, причем температуру поддерживают при 23 - 25oC путем охлаждения. Спустя следующие двадцать минут диметилформамид отгоняют в вакууме (1 мбар), остаток обрабатывают эфиром, эфирный раствор промывают последовательно водой, раствором гидросульфата калия (pH-значение равно двум), водой и насыщенным раствором хлорида натрия, эфирную фазу сушат над сульфатом магния и концентрируют в ротационном испарителе в вакууме. После очистки с помощью колоночной хроматографии на силикагеле (элюирующее средство: дихлорметан с 0-2% метанола) получают 4,1 г твердого вещества, которое перемешивают с эфиром. Выход tBuOOC-CH2-(Boc)(D)-Chg-Pro-NH-3-(2-Me-6-CN)-pico составляет 4,0 г (66%)
ж) Получение tBuOOC-CH2-(Boc)-(D)-Chg-Pro-NH-3-(2-Me-6-Ham)-pico: 3,95 г tBuOOC-CH2-(Boc)-(D)-Chg-Pro-NH-3-(2-Me-6-CN)-pico (6,6 ммоль) вместе с 1,15 г гидроксиламингидрохлорида (16,52 ммоль) и 5,12 г (6,78 мл) диизопропилэтиламина (39,6 ммоль) в 75 мл дихлорметана (высушенного над молекулярным ситом) в течение двух часов кипятят с обратным холодильником и затем перемешивают в течение ночи при комнатной температуре. После добавления дальнейшего количества дихлорметана промывают разбавленной соляной кислотой (pH-значение составляет четыре), органическую фазу сушат над сульфатом магния и концентрируют в ротационном испарителе в вакууме. Полученный в виде сырого продукта tBuOOC-CH2-(Boc)-(D)-Chg-Pro-NH-3-(2-Me-6-Ham)-pico в количестве 4,2 г используют без дальнейшей очистки в последующем превращении.
з) Получение tBuOOC-CH2-(Boc)-(D)-Chg-Pro-NH-3-(2-Me-6-Am)-pico:
4,2 г tBuOOC-CH2-(Boc)-(D)-Chg-Pro-NH-3-(2-Me-6-Ham)-pico в виде сырого продукта в смеси из 15 мл уксусной кислоты и 80 мл этанола в присутствии 10%-ного палладия-на-угле гидрируют водородом в течение пяти часов при 50oC. Затем катализатор отфильтровывают, промывают этанолом, фильтрат концентрируют в вакууме в ротационном испарителе, остаток многократно обрабатывают толуолом и дихлорметаном, каждый раз отгоняя растворитель после обработки, растворяют в 100 мл эфира и промывают три раза по 4 мл водой. Объединенные водные фазы концентрируют в вакууме (1 мбар) в ротационном испарителе максимально при 35 - 40oC и остаток обрабатывают этанолом, отгоняя затем последний. Получают 4,2 г почти чистого tBuOOC-CH2-(Boc)-(D)-Chg- Pro-NH-3-(2-Me-6-Am)-рiсо-гидроацетата (94% после стадий ж) из)) в виде белого твердого вещества.
и) Получение tBuOOC-CH2-(D)-Chg-Pro-NH-3-(2-Me-6-Am)-pico
2,0 г tBuOOC-CH2-(Boc)-(D)-Chg-Pro-NH-3-(2-Me-6-Am)-pico- гидроацетата (2,96 ммоль) в 10 мл дихлорметана вместе с 10 мл эфирного раствора хлороводорода (эфир, насыщенный хлороводородом) перемешивают в течение одного часа двадцати минут при комнатной температуре, затем концентрируют в вакууме в ротационном испарителе, остаток обрабатывают водой и многократно экстрагируют этилацетатом. Водную фазу концентрируют в вакууме (1 мбар) в ротационном испарителе максимально при 35-40oC и многократно обрабатывают ацетоном, каждый раз отгоняя ацетон после обработки. После разделения с помощью колоночной хроматографии полученной смеси при использовании силикагеля (элюирующее средство: смесь дихлорметана с метанолом и уксусной кислотой в соотношении 100: 10: 2 до 100: 20: 5) получают 0,7 г tBuOOC-CH2-(D)-Chg-Pro-NH-3-(2- Me-6-Am)-pico (HX)1,2 (где Xθ означает хлор и/или ацетил) в виде белого твердого вещества, которое плавится с разложением начиная с 205oC.
Пример 227
HOOC-CH2-(D)-Chg-Pro-NH-3-(2-Me-6-Am)-pico
2,2 г tBuOOC-CH2-(Boc)-(D)-Chg-Pro-NH-3-(2-Me-6-Am)-pico- гидроацетата (3,25 ммоль) в 30 мл дихлорметана вместе с 15 мл эфирного раствора хлороводорода перемешивают в течение нескольких часов при комнатной температуре, причем в течение этого времени медленно осаждается твердое вещество. Твердое вещество отсасывают, многократно перемешивают с горячим дихлорметаном и после этого хроматографируют на силикагеле (элюирующее средство: метанол с 25%-ным водным раствором аммиака в соотношении 95:5). Получают 1,3 г HOOC-CH2-(D)-Chg-Pro-NH-3-(2-Me-6-Am)-pico в виде белого твердого вещества, которое плавится с разложением начиная с 210oC.
Пример 228
MeOOC-CH2-(D)-Chg-Pro-NH-3-(2-Me-6-Am)-pico
0,45 г HOOC-CH2-(D)-Chg-Pro-NH-3-(2-Me-6-Am)-pico (0,1 ммоль) вносят в 30 мл метанола (высушенного над молекулярным ситом), смешивают путем прикапывания с 1 мл тионилхлорида и перемешивают в течение двух часов при кипячении с обратным холодильником. После дальнейшего добавления 0,3 мл тионилхлорида и перемешивания в течение часа при кипячении с обратным холодильником раствор концентрируют в вакууме в ротационном испарителе, многократно обрабатывают метанолом и дихлорметаном, каждый раз отгоняя растворитель после обработки, и остаток очищают путем колоночной хроматографии
на силикагеле (элюирующее средство: смесь дихлорметана с метанолом и уксусной кислотой в соотношении 100:20:5). После многократной обработки толуолом и каждый раз отгонки последнего получают 0,38 г MeOOC-CH2-(D)-Chg-Pro-NH-3-(2-Me-6-Am)-pico (HX)1,2 (где Xθ означает хлор и/или ацетил) в виде белого твердого вещества, которое плавится при 155 - 160oC.
Пример 229
Boc-(D)-Chg-Pro-NH-2-(5-Am)-pico
а) 5-Карбоксамидо-2-пиколиламин:
К раствору 3,5 г (24 ммоль) 2-циано-5-карбоксамидопиридина в 80 мл метанола и 20 мл концентрированного аммиака добавляют 3 г никеля Ренея и гидрируют при комнатной температуре. Спустя примерно семь часов полностью происходит поглощение водорода. После отсасывания катализатора фильтрат концентрируют и остаток растворяют в 20 мл 2 н. соляной кислоты и 20 мл метанола. Путем добавления 150 мл этилацетата осаждают гидрохлорид, который отсасывают и высушивают (3,7 г). Свободное основание плавится при 198 - 202oC.
б) 5-Циано-2-пиколиламин:
41 г (0,22 моль) 5-Карбоксамидо-2-пиколиламина суспендируют в 150 мл метанола и 300 мл дихлорметана, охлаждают до 10oC и путем добавления 150 мл триэтиламина получают раствор. Затем прикапывают раствор 47,6 г (0,22 моль) (Boc)2O и дополнительно перемешивают в течение четырех часов при комнатной температуре. Полученный после удаления растворителя остаток смешивают с насыщенным раствором карбоната калия и экстрагируют пять раз дихлорметаном. Объединенные экстракты сушат и растворитель, под конец при добавлении толуола, отгоняют.
5,4 г Остатка суспендируют в 40 мл диоксана и 15 мл дихлорметана, смешивают с 4,3 г пиридина и затем при 0oC прикапывают 5,2 г ангидрида трифторуксусной кислоты, причем образуется прозрачный раствор.
После добавления 100 мл воды экстрагируют этилацетатом, органическую фазу промывают разбавленным раствором лимонной кислоты, раствором гидрокарбоната натрия и водой. После высушивания и удаления растворителя получают желтого цвета масло (примерно 5 г), которое растворяют в 15 мл изопропанола с 30 мл этилацетата и смешивают с 35 мл эфирного раствора хлороводорода. После выдерживания в течение ночи отсасывают осадившийся гидрохлорид и высушивают его. Выделяют 4 г белых кристаллов. Температура плавления составляет 230 - 234oC.
в) Boc-(D)-Циклогексилглицилпролин-(5-циано-2-пиколил)амид:
Получают согласно примеру 216, стадия б), путем сочетания Boc-(D)-циклогексилглицилпролина с 5-циано-2-пиколиламином. Соединение представляет собой кристаллы белого цвета с температурой плавления 128 -129oC.
г) Boc-(D)-Циклогексилглицилпролин-(6-амидино-2-пиколил)амид:
Амидирование вышеполученного соединения осуществляют согласно примеру 215, стадия г). Ацетат представляет собой белые кристаллы с температурой плавления 98 - 100oC (разложение).
Масс-спектр (бомбардировка быстрыми атомами): 487 (М+H+).
Пример 230
H-(D)-Chg-Pro-NH-2-(5-Am)-pico
В стандартных условиях от соединения примера 229, стадия г), отщепляют защитную группу. Дигидрохлорид представляет собой белые кристаллы с температурой плавления 233 - 235oC (разложение).
Масс-спектр (бомбардировка быстрыми атомами): 386 (М+H+).
Пример 231
HOOC-CH2-(D)-Chg-Pro-NH-2-(5-Am)-pico
Согласно примеру 218, стадии а), б), в) и г), путем отщепления Boc-группы от Boc-(D)-циклогексилглицилпролин-(5-циано- 2-пиколил)амида, причем не происходит никакого образования амида, N-алкилирования с помощью трет.-бутилового эфира бромуксусной кислоты, образования амидина и кислотного омыления сложного трет.-бутилового эфира получают целевое соединение в виде кристаллов белого цвета.
Температура плавления составляет 162 - 164oC.
Масс-спектр (бомбардировка быстрыми атомами): 445 (М+H+).
Пример 232
HOOC-CH2-(D)-Chg-Pro-NH-5-(2-Am)-pym
а) 2-Тиометил-5-аминометилпиримидингидрохлорид:
28,1 г (182,2 ммоль) 2-Тиометил-5-формил-пиримидина (Z. Amold и др., J. Heterocyclic Chem. , 28, 1281 (1991)) при -23oC вносят в 880 мл смеси метанола с тетрагидрофураном в соотношении 1:1. После добавления 12,8 г (34,3 ммоль) гептагидрата хлорида перия порциями добавляют 5,19 г (137,2 ммоль) боргидрида натрия. Спустя полтора часа времени реакции реакционный раствор смешивают с 1,5 л насыщенного раствора хлорида натрия и эту смесь экстрагируют четыре раза по 130 мл дихлорметаном. Объединенные органические фазы сушат и концентрируют в вакууме. Выход составляет 26,9 г.
26,89 г (172,14 ммоль) 2-Тиометил-5-гидроксиметил-пиримидина растворяют в 390 мл абсолютного дихлорметана и после добавления одной капли диметилформамида и 27 мл (370,37 ммоль) тионилхлорида перемешивают в течение сорока пяти минут при 0oC. Для обработки реакционный раствор концентрируют досуха.
Таким образом полученный 2-тиометил-5-хлорметил-пиримидин вместе с 16,79 г (258,2 ммоль) азида натрия в 84 мл диметилсульфоксида перемешивают в течение ночи при комнатной температуре. Вследствие неполного превращения добавляют следующие 4,2 г азида натрия. После дальнейшего времени реакции, равного двум часам, хлорпроизводное полностью превращается. Для обработки реакционную смесь выливают в 300 мл воды и водную фазу экстрагируют пятикратно по 100 мл диэтиловым эфиром. Объединенные органические экстракты промывают 3 раза по 25 мл водой, сушат и затем эфир почти полностью удаляют в вакууме.
Остаток после концентрирования эфирного раствора 2-тиометил-5-азидо-метилпиримидина растворяют в 28 мл тетрагидрофурана и при охлаждении льдом осторожно добавляют к раствору 45,15 г (172,1 ммоль) трифенилфосфина в 84 мл тетрагидрофурана. Спустя пятнадцать минут прекращают охлаждение льдом, добавляют к реакционной смеси 4,65 мл воды и реакционный раствор перемешивают в течение восемнадцати часов при комнатной температуре. Для обработки реакционную смесь концентрируют в вакууме досуха и полученный остаток растворяют в 70 мл 3 н. соляной кислоты. Водный раствор промывают четыре раза по 50 мл смесью этилацетата с диэтиловым эфиром в соотношении 1:1. Затем в растворе устанавливают pH-значение, равное девяти, с помощью карбоната натрия и экстрагируют двенадцать раз по 50 мл дихлорэтаном. Объединенные органические экстракты сушат и концентрируют. Остаток растворяют в смеси дихлорметана с этилацетатом и свободный амин осаждают в виде гидрохлорида с помощью хлороводорода в диоксане. Выход составляет 30,48 г.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 2,55 (с, 3H, CH3); 4,1 (к, 2H, N-CH2); 8,8 (с, 2H, аром. протоны); 10,8 (уш. с., NH).
б) Boc-Pro-NH-5-(2-Sme)-pym:
12,9 г (60 ммоль) Boc-Pro-ОН вместе с 15 г (65,8 ммоль) 2-тиометил-5-аминометилпиримидингидрохлорида и 61,4 мл (359 ммоль) диизопропилэтиламина при 0oC вносят в 150 мл дихлорметана. После добавления 63,4 мл 50%-ного раствора ангидрида пропанфосфоновой кислоты в этилацетате реакционную смесь перемешивают в течение шести часов при температуре от 0oC до комнатной температуры. После того, как Boc-Pro-OH полностью провзаимодействовал (контроль с помощью тонкослойной хроматографии: смесь дихлорметана с метанолом в соотношении 95:5), реакционную смесь растворяют в 300 мл этилацетата. Органическую фазу промывают 2 раза 20%-ным раствором гидросульфата натрия, два раза водой и один раз насыщенным раствором хлорида натрия. После высушивания органической фазы над сульфатом натрия этилацетат удаляют в вакууме. Получают 16,7 г целевого продукта.
в) Boc-Pro-NH-5-(2-метилсульфонил)-pym:
20,5 г (58,1 ммоль) Boc-Pro-NH-5-(2-Sme)-pym при комнатной температуре вносят в 700 мл дихлорметана. Затем в течение тридцати минут к раствору порциями добавляют 42,94 г (174 ммоль) м-хлорпероксибензойной кислоты. После протекания времени реакции в целом два часа реакционную смесь экстрагируют два раза 20%-ным раствором гидросульфата натрия, шесть раз 5%-ным раствором гидрокарбоната натрия и три раза 20%-ным раствором мононадсернокислого натрия. После высушивания раствора и удаления дихлорметана получают 21,7 г сульфона Boc-Pro-NH-5-(2-метилсульфонил)-pem.
г) Boc-Pro-NH-5-(2-CN)-pym:
21,7 г (56,4 ммоль) Boc-Pro-NH-5-(2-метилсульфонил)-pym растворяют в 30 мл диметилсульфоксида и после добавления 2,84 г цианида натрия перемешивают в течение ночи при комнатной температуре. Затем раствор выливают в 150 мл воды и водный раствор экстрагируют пять раз по 100 мл дихлорметаном. Объединенные органические фазы промывают пять раз насыщенным раствором хлорида натрия и два раза водой. После высушивания и концентрирования органического раствора получают 15,3 г целевого цианида.
д) H-Pro-NH-5-(2-CN)-pym тригидротрифторацетат:
13,98 г (42,1 ммоль) Boc-Pro-NH-5-(2-CN)-pym вносят в дихлорметан. После добавления 13 мл (170 ммоль) трифторуксусной кислоты раствор перемешивают при комнатной температуре вплоть до полного превращения эдукта (контроль с помощью тонкослойной хроматографии). После концентрирования раствора в вакууме получают целевую соль, которую без дальнейшей очистки вводят во взаимодействие далее в следующей реакции.
е) H-(D)-Chg-Pro-NH-5-(2-CN)-pym тригидротрифторацетат:
10 ммоль H-Pro-NH-5-(2-CN)-pym тригидротрифторацетата, 2,44 г (9,5 ммоль) Boc-(D)-Chg-OH и 9,8 мл (57 ммоль) диизопропилэтиламина смешивают при 0oC. После добавления 10,1 мл 50%-ного раствора ангидрида пропанфосфоновой кислоты в этилацетате реакционную смесь при перемешивании в течение шести часов доводят до комнатной температуры. Для обработки ее разбавляют 300 мл этилацетата и органическую фазу промывают два раза 20%-ным раствором гидросульфата натрия, два раза водой и один раз насыщенным раствором хлорида натрия. После высушивания органической фазы над сульфатом натрия этилацетат удаляют в вакууме. Получают 4,74 г целевого продукта. Таким образом полученный сырой продукт переводят в соль трифторуксусной кислоты как описано выше.
ж) tBuOOC-CH2-(D)-Chg-Pro-NH-5-(2-CN)-pym:
4,1 г (11,07 ммоль) H-(D)-Chg-Pro-NH-5-(2-CN)-pym тригидротрифторацетата вместе с 1,68 г (12,17 ммоль) карбоната калия и 1,63 мл (11,07 ммоль) трет. -бутилового эфира бромуксусной кислоты перемешивают при комнатной температуре. После полного превращения отфильтровывают карбонат калия и фильтрат концентрируют в ротационном испарителе. Остаток растворяют в этилацетате и органический раствор промывают 5%-ным раствором гидрокарбоната натрия и насыщенным раствором хлорида натрия. Затем растворитель удаляют в вакууме (выход сырого продукта составляет 3,66 г). Сырой продукт очищают с помощью колоночной хроматографии (смесь дихлорметана с метанолом в соотношении 98:2 с 0,5% концентрированного раствора аммиака). Получают 1,3 г чистого продукта.
з) tBuOOC-CH2-(D)-Chg-Pro-NH-5-(2-Am)-pym:
1,3 г (2,68 ммоль) tBuOOC-CH2-(D)-Chg-Pro-NH-5-(2-SMe)-pym растворяют в 15 мл этанола и после добавления 0,5 г (6,71 ммоль) гидроксиламмонийхлорида и 2,5 мл диизопропилэтиламина перемешивают в течение четырех часов при 60oC. Реакционную смесь концентрируют в ротационном испарителе и растворяют в дихлорметане. Полученный после промывки органического раствора небольшим количеством воды, высушивания и концентрирования раствора сырой продукт снова растворяют в этаноле и после добавления никеля Ренея гидрируют в атмосфере водорода в течение четырех часов при 60oC. После отфильтровывания никеля Ренея этанольный раствор концентрируют и сырой продукт очищают путем разделения на колонке с силикагелем (смесь дихлорметана и метанолом и 50%-ной уксусной кислоты в соотношении 40:10:2). Выход составляет 250 мг.
и) HOOC-CH2-(D)-Chg-Pro-NH-5-(2-Am)-pym:
250 мг tBuOOC-CH2-(D)-Chg-Pro-NH-5-(2-Am)-pym с помощью трифторуксусной кислоты в дихлорметане расщепляют до кислоты и сырой продукт очищают путем колоночной хроматографии (метанол с 3% концентрированного раствора аммиака). Выход составляет 108 мг. Масс-спектр: 446 (М+H+), 369.
Пример 233
(D)-Man-Pro-NH-4-(1-Am)-pip
Раствор 4,2 г (12,6 ммоль) O-тетрагидропиранил-(D)-2-фенил-2- гидроксиацетил-(1)-пролина (международная заявка на патент 93/18060) в 40 мл тетрагидрофурана после добавления 1,9 г (12,6 ммоль) 1-гидроксибензотриазола и 3,3 г (25 ммоль) дициклогексилкарбодиимида перемешивают в течение четырех часов при комнатной температуре. Выпавшую в осадок мочевину отсасывают и дополнительно промывают небольшим количеством тетрагидрофурана.
К этому фильтрату при 5oC добавляют раствор 2,9 г (12,6 ммоль) 1-амидино-4-аминометил-пиперидин-дигидрохлорида и 1,6 г гидрокарбоната натрия в 6 мл воды. После перемешивания в течение сорока восьми часов при комнатной температуре далее растворитель отгоняют в вакууме, остаток обрабатывают этанолом, нерастворимые составные части отфильтровывают и раствор снова концентрируют. Остаток очищают с помощью колоночной хроматографии на силикагеле при использовании смеси дихлорметана с метанолом и 50%-ной уксусной кислотой в соотношении 45: 5: 1,5. Из однородных фракций отгоняют растворители, под конец с толуолом в качестве добавки, и остаток перекристаллизуют из 50 мл ацетона при добавлении небольшого количества воды. Получают 3,5 г ацетата в форме белого цвета кристаллов.
Температура плавления составляет 199 - 202oC (разложение).
Масс-спектр (бомбардировка быстрыми атомами): 388 (М+H+).
Пример 234
Boc-(D)-Phe-Pro-NH-pHamb
Boc-(D)-Phe-Pro-p-цианобензиламид (получение см. пример 5) подвергают взаимодействию аналогично примеру 215, стадия г) с гидрохлоридом гидроксиламина.
Белые кристаллы.
Масс-спектр (бомбардировка быстрыми атомами): 510 (М+H+).
Пример 235
H-(D)-Phe-Pro-NH-pHamb
От соединения примера 234 отщепляют по стандартным условиям Boc-группу.
Белые кристаллы.
Масс-спектр (бомбардировка быстрыми атомами): 410 (М+H+).
Пример 236
Boc-(D)-Phe-Pro-NH-(2-MeO)-pAmb
а) Boc-Pro-(4-циано-2-метокси)-бензиламид:
16,0 г Boc-Пролина (50 ммоль), растворенных в 80 мл тетрагидрофурана, вместе с 5,7 г гидросисукцинимида и 10,2 г дициклогексилкарбодиимида перемешивают при 0oC в течение тридцати минут. Затем при 0oC прикапывают раствор 8,0 г (50 ммоль) 4-аминометил-3-метокси-бензонитрила в 50 мл тетрагидрофурана и перемешивают в течение двадцати часов при комнатной температуре. Твердое вещество отфильтровывают, фильтрат смешивают с таким же объемом этилацетата и промывают холодным 5%-ным раствором гидросульфата натрия, а также насыщенным раствором хлорида натрия. Получают 11,5 г (65%) продукта.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 8,38 (м, NH); 7,50 - 7,35 (м, 3H); 4,40 - 4,05 (м, 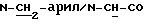 ); 3,87 (с,
); 3,87 (с,  ); 3,50-3,25 (м, 2H,
); 3,50-3,25 (м, 2H,  ); 2,25 - 2,00 (м, 1H): 1,90 - 1,65 (м, 3H): 1,40 и 1,30 (2с, 9H).
); 2,25 - 2,00 (м, 1H): 1,90 - 1,65 (м, 3H): 1,40 и 1,30 (2с, 9H).
б) H-Pro-(4-циано-2-метокси)бензиламид:
11,4 г (31,7 ммоль) Boc-пролин-(2-метокси-4-циано)бензиламида растворяют в 130 мл дихлорметана и при температуре от 0 до 5oC насыщают газообразным хлороводородом. Спустя два часа Boc-группы полностью отщепляется. Растворитель удаляют в вакууме и продукт без дальнейшей очистки используют в следующей реакции.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 10,25 (с, 1H); 8,60 (с, 1H); 7,50 (д, 1H): 7,42 (дд, 1H); 7,39 (д, 1H); 4,40 - 4,20 (м, 3H); 3,88 (с, 3H); 3,20 (м, 2H); 2,35 (м, 1H); 2,00 - 1,80 (м, 3H).
в) Boc-(D)-Phe-Pro-(4-циано-2-метокси)бензиламид:
3,54 г (13,35 ммоль) Boc-(D)-Phe-Pro-OH, 9,9 мл диизопропилэтиламина и 4,80 г (13,35 ммоль) H-Pro-(4-циано-2- метокси)бензиламидгидрохлорида при -5oC смешивают с 11,1 мл (15,0 ммоль) 50%-ного этилацетатного раствора ангидрида пропанфосфоновой кислоты в 100 мл дихлорметана и перемешивают при 0oC в течение двух часов. Реакционную смесь промывают последовательно 1 н. раствором гидроксида натрия, 1н соляной кислотой и насыщенным раствором хлорида натрия и сушат над сульфатом натрия. После удаления растворителя получают 6,5 г (96%) продукта.
1Н-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 8,75/7,88 (1H, NH (2 геометрических изомера)); 7,5-7,1 (9H, аром. протоны и NH); 4,4 - 4,1 (4H, CH2 и 2 CH); 3,85 (3H, OCH3); 3,7 - 3,4 (2H, CH2; 3,0 - 2,7 (2H, CH2); 2,3 -1,5 (4H, 2 CH2); 1,3-1,1 (9H, Boc).
г) Boc-(D)-Phe-Pro-(4-амидино-2-метокси)бензиламид:
Согласно методике A.III.1, полученный в предыдущей стадии нитрил превращают в 4,6 г амидингидроиодида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,25/8,85 (4H, амидин); 8,75/7,95 (1Н, NH (2 геометрических изомера)); 7,4 - 7,1 (9H, аром. протоны и NH): 4,45 - 4,10 (4H, CH2 и 2CH); 3,90 (3H, OCH3): 3,65 - примерно 3,4 (2H, CH2; 3,0 - 2,7 (2H, CH2); 1,95 - 1,55 (4H, 2CH2; 1,3 - 1,2 (9H, Boc).
Пример 237
H-(D)-Phe-Pro-NH-(2-MeO)-pAmb
Амидингидроиодид (пример 236) с помощью содержащей ацетатные группы ионообменной смолы (IRA 420) превращают в амидингидроацетат, затем растворяют в 50 мл дихлорметана и при 0oC насыщают газообразным хлороводородом. Спустя час растворитель удаляют. Получают 3,0 г амидина в виде дигидрохлорида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 9,50/9,27 (4H, амидин); 8,80 (3H, NH3 +); 8,75 (2H, NH); 7,50 - 7,20 (8H, аром. протоны); 4,35 -4,10 (4H, CH2 и 2 CH): 3,90 (3H, OCH3); примерно 1,9 - 1,35 (4H, 2 CH2).
Пример 238
Boc-(D)-Phe(4-MeO)-Pro-NH-(2-MeO)-pAmb
а) Boc-(D)-Phe(4-МеО-Pro-(4-циано-2-метокси)бензиламид:
1,55 г (5,25 ммоль) Boc-(D)-Phe(4-OMe)-OH, 3,9 мл диизопропилэтиламина и 1,55 г (5,25 ммоль) пролин-(2-метокси-4- циано)бензиламидгидрохлорида при -5oC смешивают с 4,4 мл (5,9 ммоль) 50%-ной этилацетатного раствора ангидрида пропанфосфоновой кислоты в 35 мл дихлорметана и перемешивают в течение часа при 0oC. Реакционную смесь промывают последовательно 1 н. раствором гидроксида натрия, 1 н. соляной кислотой и насыщенным раствором хлорида натрия и сушат над сульфатом натрия. После удаления растворителя получают 2,4 г твердого вещества.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 8,72 и 7,87 (т, 2H): 7,42 (1H); 7,35 (м, 3H); 7,15 (д, 2H); 6,85 (д, 2H); 7,00/6,70 (2д, 1H (2 геометрических изомера)); 4,40 - 4,10 (м, 4H); 3,85 (с, 3H); 3,70 (с, 3H); 3,05 - 2,55 (м, 2H); 1,95 - 1,55 (м, 4H); 1,2 (c, 9H).
б) Boc-(D)-Phe(4-MeO)-Pro-NH-(2-MeO)-pAmb:
2,4 г Нитрила (пример 238, стадия а)), согласно методике A.III.1, после очистки с помощью колоночной хроматографии на силикагеле (растворитель: смесь дихлорметана с метанолом в соотношении 9:1) превращают в 1,7 г амидингидроиодида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 9,25/8,85 (4H, амидин): 7,95 (1H, NH): 7,4 - 6,8 (8H, аром. протоны и NH): 4,4 - 4,1 (4H, CH2 и 2 CH); 3,90/3,70 (6H, 2 OCH3): примерно 3,7 - 2,9 (2H, CH2); 3,0 - 2,6 (2H, CH2); 1,9 - 1,5 (4H, 2 CH2); 1,3 - 1,2 (9H, Boc).
Пример 239
H-(D)-Phe-(4-MeO)-Pro-NH-(2-MeO)-pAmb
1,7 г Амидингидроиодида (пример 238) с помощью содержащей ацетатные группы ионообменной смолы (IRA 420) превращают в гидроацетат и затем отщепляют Boc-группу согласно примеру 237. Получают 1,0 г дигидрохлорида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,50/0,25 (4H, 16 амидин): 8,85 - 8,65 (4H, NH и NH3 +), 7,50 - 7,30 и 7,15/6,90 (7H, аром. протоны); 4,28 - 4,05 (4H, CH2 и 2 CH); 3,90/3,75 (6H, 2 OCH3); 3,20 - 2,85 (2H, CH2); 1,95 - 1,40 (4H, 2 CH2.
Масс-спектр (бомбардировка быстрыми атомами): 454 (М+H+).
Пример 240
HOOC-CH2-(D)-Phe(4-MeO)-Pro-NH-(2-MeO)-pAmb
От соединения примера 238, стадия а), согласно примеру 237 отщепляют Boc-группу. 3,5 г Полученного продукта расщепления растворяют в 80 мл дихлорметана и вместе с 4,45 мл диизопропилэтиламина и 1,09 мл трет.-бутилового эфира бромуксусной кислоты перемешивают в течение трех дней при комнатной температуре. Продукт обрабатывают как описано в примере 246, стадия а). 3,0 г Полученного tBuOOC-CH2-(D)-Phe(4-МеО)-Pro-(2-метокси-4-циано)бензиламида согласно примеру 246, стадия 6), вводят во взаимодействие с гидроксиламингидрохлоридом 3,1 г полученного гидроксиамидина в присутствии 185 мг никеля Ренея в 65 мл метанола, к которому добавлено 0,31 мл ледяной уксусной кислоты, гидрируют при 50oC до получения амидингидроацетата. Катализатор отфильтровывают и tBuOOC-CH2-(D)-Phe(4-MeO)-Pro-(2-MeO)-pAmb-гидроацетат очищают путем колоночной хроматографии на силикагеле (растворитель: дихлорметан с 10% метанола и 2% 50%-ной уксусной кислоты). Получают 1,3 г сложного трет. -бутилового эфира (масс-спектр (бомбардировка быстрыми атомами): 568 (М+H+)) и 1,15 г этого эфира согласно примеру 243, стадия г), превращают в 850 мг HOOC-CH2-(D)-Phe(4-MeO)-Pro-NH-(2-MeO)-pAmb.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,9 - 9,7 и 9,2 - 9,0 (2H, NH2 +); 9,60/9,35 (4H, амидин); 7,50 - 6,73 (5H, аром. протоны); 4,50 - 3,45 (8H, 3 CH2 и 2 CH); 3,90 (3H, OCH3); 3,73 (3H, OCH3); 3,40 - 3,27 и 3,06 - 2,87 (2H, CH2); 2,43 - 1,25 (4H, 2 CH2).
Пример 241
Boc-(D)-Chg-Pro-NH-(2-MeO)-pAmb
а) Boc-(D)-Циклогексилглицил-пролин-(2-метокси-4-циано)бензиламид:
К 10,0 г (28,2 ммоль) Boc-(D)-Chg-Pro-OH в 70 мл абсолютного дихлорметана при -5oC добавляют 20,8 мл диизопропилэтиламина (121 ммоль), 4,58 г (28,2 ммоль) 2-метокси-4-циано-бензиламина и 25 мл 50%-ного раствора ангидрида пропанфосфоновой кислоты в эталацетате и перемешивают в течение двух часов при 0oC. Затем раствор промывают последовательно 0,5 н. раствором гидроксида натрия, 1 н. соляной кислотой и насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель полностью удаляют в вакууме. Осадившийся почти с количественным выходом продукт без дальнейшей очистки используют в следующей стадии.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 7,9 (1H, NH); 7,45 и 7,35 (3H, аром. протоны); 7,1 (1H, NH); 4,45 - 3,50 (6H, 2 CH2 и 2 CH); 3,86 (3H, OCH3); 2,2 - 1,0 (24H, циклогексил и 2 CH3 и Boc).
б) Boc-(D)-Циклогексилглицилпролин-(2-метокси-4- гидроксиамидино)бензиламид:
12,0 г (24 ммоль) Содержащего циангруппу эдукта из стадии а) согласно примеру 243, стадия б), вводят во взаимодействие с гидроксиламингидрохлоридом. Продукт почти количественно осаждается в виде объемистого осадка.
1H-ЯМР (дейтерированный диметилсульфоксид: δ в м.д.): 9,7 - 9,5 (1H, ОН); 5,8 (2H, NH2).
в) Boc-(D)-Циклогексилглицил-пролин-(2-метокси-4- амидино)бензиламид:
Гидроксиамидин из стадии б) гидрируют в присутствии никеля Ренея согласно примеру 240. Продукт очищают с помощью колоночной хроматографии на силикагеле (растворитель: дихлорметан с 10%-20% метанола и 2% 50%-ной уксусной кислоты). Получают 10,5 г амидина в виде гидроацетата (выход составляет 75% в расчете на нитрил стадии а)).
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): амидиногруппа в виде ацетатной соли дает очень уширенный сигнал примерно при 10-8 м. д.; 7,95 (1Н, NH); 7,4 - 7,3 (3H, аром. протоны); 7,05 (1Н, NH), 4,4 - 3,4 (6H, 2 CH2 и 2 CH): 3,89 (3H, OCH3); 2,2 - 1,0 (24H, циклогексил, 2CH2 и Boc).
Пример 242
H-(D)-Chg-Pro-NH-(2-MeO)-pAmb
10,5 г Boc-(D)-Chg-Pro-(2-метокси-4-амидино)бензиламидарастворяют в 200 мл абсолютного дихлорметана с 10 мл метанола и при температуре 0-5oC в течение часа пропускают газообразный хлороводород. Перемешивают следующий час при 0oC, растворитель полностью удаляют в вакууме и получают 7,6 г (86%) продукта в виде дигидрохлорида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,60 и 9,33 (4H, амидин): 8,87 (1Н, NH); 8,62 (3H, NH3 +); 7,5 - 7,3 (3H, аром. протоны); 4,45 - 4,15 (4H, CH2 и 2 CH); 3,95 (3H, OCH3); 3,95 - 3,82 (1Н, CH2); 3,65 - 3,55 (1Н, CH2); 2,2 - 1,0 (15H, циклогексил и 2 CH2).
Пример 243
HOOC-CH2-(D)-Chg-Pro-NH-(2-MeO)-pAmb
а) N-трет. -Бутилоксикарбонилметил-(D)-циклогексилглицил- пролин-(2-метокси-4-циано)бензиламид:
0,72 г (1,65 ммоль) H-(D)-Chg-Pro-(2-метокси-4-циано)- бензиламидгидрохлорида вносят в 30 мл абсолютного дихлорметана. После добавления 1 мл (5,8 ммоль) диизопропилэтиламина при комнатной температуре в течение сорока минут прикалывают раствор из 1,65 ммоль трет.-бутилового эфира бромуксусной кислоты и 15 мл дихлорметана. Реакционную смесь перемешивают в течение трех дней при комнатной температуре, затем промывают последовательно 0,5 н. раствором гидроксида натрия, 0,5 н. соляной кислотой и насыщенным раствором хлорида натрия. После высушивания получают 0,7 г сырого продукта, который без дальнейшей очистки используют в следующей стадии.
б) N-трет.-Бутилоксикарбонилметил-(D)-циклогексилглицил-пролин- (2-метокси-4-гидроксиамидино)бензиламид:
1,45 г (2,8 ммоль) 2-Метокси-4-циано-бензиламида из стадии а) растворяют в 20 мл смеси дихлорметана с метанолом в соотношении 1:1 и вместе с 0,49 г (7,1 ммоль) гидроксиламингидрохлорида и 2,8 мл диизопропилэтиламина перемешивают в течение двадцати часов при комнатной температуре. После удаления растворителя продукт растворяют в дихлорметане, промывают водой и насыщенным раствором хлорида натрия и сушат. Получают 1,2 г сырого продукта, который непосредственно используют дальше.
в) N-трет.-Бутилоксикарбонилметил-(D)-циклогексилглицилпролин- (2-метокси-4-амидино)бензиламид:
Гидроксиамидиновое соединение из стадии б) гидрируют в присутствии никеля Ренея согласно примеру 240. После очистки с помощью колоночной хроматографии на силикагеле (растворитель: дихлорметан с 10-20% метанола и 2% 50%-ной уксусной кислоты) получают 0,5 г продукта в виде гидроацетата.
г) N-Гидроксикарбонилметил-(D)-циклогексилглицил-пролин-(2- метокси-4-амидино)бензиламид:
0,5 г (0,85 ммоль) Амидиногидроацетата из стадии в) растворяют в 25 мл абсолютного дихлорметана. При температуре от 0oC до 5oC пропускают газообразный хлороводород вплоть до насыщения раствора. Спустя сорок минут перемешивают при комнатной температуре в течение следующего часа. После удаления растворителя в вакууме получают 370 мг чистого продукта в виде дигидрохлорида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.); 9,57 и 9,33 (4H, амидин); 9,8 - 9,1 (2H, NH2 +); 7,6 - 7,3 (3H, аром. протоны); 4,5 - 4,1 (4H, CH2 и 2 CH); 3,93 (3H, OCH3); 3,9 - 3,4 (4H, 2 CH2); 2,3 - 1,0 (15H, циклогексил и 2 CH2).
Пример 244
Boc-(D)-Chg-Aze-NH-(2-MeO)-pAmb
а) (2-Метокси-4-циано)-бензиламид Boc-(L)-aзетидин-2- кapбoнoвoй кислоты:
К 2,12 г Boc-(L)-азетидин-2-карбоновой кислоты (10,5 ммоль) в 50 мл тетрагидрофурана при температуре от 0 до 5oC добавляют 1,22 г (10,5 ммоль) гидроксисукцинимида и 2,18 г (10,5 ммоль) дициклогексилкарбодиимида и перемешивают в течение тридцати минут. Затем при температуре от 0oC до 5oC добавляют 2,10 г (10,5 ммоль) 2-метокси-4-цианобензиламингидрохлорида и, наконец, 1,48 мл триэтиламина. Реакционную смесь перемешивают в течение ночи при комнатной температуре. Выпавшую в осадок мочевину отделяют на нутч-фильтре, фильтрат растворяют в этилацетате и промывают последовательно 0,5 н. соляной кислотой, 0,5 н. раствором гидроксида натрия и насыщенным раствором хлорида натрия. Органическую фазу сушат над сульфатом натрия и затем растворитель полностью удаляют в вакууме. Получают 3,1 г (85%) продукта, который используют без дальнейшей очистки.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 8,5 (1H, NH); 7,48 и 7,40 - 7,25 (3H, аром. протоны); 4,55 (дд, 1H, CH); 4,45 - 4,15 (2H, CH2; 3,88 (3H, OCH3): 3,9 - 3,7 (2H, CH2; 2,5 - 2,3 (1H, CH2; 2,15 - 1,95 (1H, CH2): 1,35 (9H, Boc).
б) (2-Метокси-4-циано)-бензиламид(L)-азетидин-2-карбоновой кислоты:
3,0 г (8,7 ммоль) Boc-Aze-(2-метокси-4-циано)бензиламида вводят во взаимодействие согласно примеру 236, стадия б), с получением почти с количественным выходом целевого продукта.
1H-ЯМР (дейтерированный диметилсульфоксид: δ в м.д.): 10,0 - 9,85 (1H, NH2 +); 7,50 и 7,45 - 7,35 (3H, аром. протоны); 5,10 - 4,95 (1H, CH): 4,35 (д, 2H, CH2); 4,05 - 3,65 (2H, CH2); 3,89 (3H, OCH3); 2,8 - 2,6 (1H, CH2); 2,5 - 2,3 (1H, CH2).
в) (2-Метокси-4-циано)-бензиламид Boc-(D)-циклогексилглицил-(L)-азетидин- 2-карбоновой кислоты:
2,2 г Boc-(D)-Chg-OH вводят во взаимодействие с 2,4 г H-Aze-(2-метокси-4-циано)-бензиламидгидрохлорида согласно примеру 238, стадия а). Получают 3,5 г продукта.
г) (2-Метокси-4-амидино)-бензиламид Boc-(D)-циклогексилглицил-(L)-азетидин-2-карбоновой кислоты:
3,4 г Нитрила из стадии в) вводят во взаимодействие с гидроксиламингидрохлоридом согласно примеру 243, стадия б), и образовавшийся гидроксиамидин гидрируют в присутствии никеля Ренея согласно примеру 240. Получают 3,1 г амидина в виде гидроацетата.
Пример 245
H-(D)-Chg-Aze-NH-(2-MeO)-pAmb
3,1 г Соединения с Boc-группой (пример 244) согласно примеру 242 расщепляют до получения дигидрохлорида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,45/9,20 (4H, амидин); 9,0 (1H, NH); 8,55 (3H, NH3 +); 7,45/7,40 (3H, аром. протоны); 4,75 - 4,10 (4H, CH2 и 2CH); 3,90 (3H, OCH3); 2,7 - 1,0 (13H, циклогексил и CH2).
Пример 246
Boc-(D)-Chg-Pro-NH-(2-изопропокси)-pAmb
4,1 г (11,5 ммоль) Boc-(D)-Chg-Pro-OH согласно примеру 236, стадия а), вводят во взаимодействие с одним эквивалентом гидроксисукцинимида, одним эквивалентом дициклогексилкарбодиимида, одним эквивалентом 4-аминометил-3-изопропоксибензонитрилгидрохлорида и одним эквивалентом триэтиламина. Получают 5,7 г (94%) сырого продукта, который используют в следующей стадии без дальнейшей очистки.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 7,85 (1Н, NH); 7,43 и 7,30 (3H, аром. протоны); 7,08 (1H, NH); 4,80 - 3,50 (7H, 2 CH2, 3 CH); 2,2 - 1,0 (30Н, Boc, циклогексил, 2 CH3 и 2 CH2.
Пример 247
H-(D)-Chg-Pro-NH-(2-изопропокси)-pAmb
5,7 г Соединения с Boc-группой (пример 246) согласно примеру 243, стадия б), вводят во взаимодействие с гидроксиламингидрохлоридом и образовавшийся гидроксиамидин гидрируют в присутствии никеля Ренея согласно примеру 240. Полученный при этом амидингидроацетат расщепляют согласно примеру 242. Получают 3,1 г продукта в виде дигидрохлорида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,40 /9,15 (4H, амидин); 8,75 (1H, NH); 8,55 (3H, NH3 +); 7,40 - 7,25 (3H, аром. протоны); 4,80 (1H, CH); 4,4 - 3,5 (6H, 2 CH2, 2 CH); 2,3 - 1,0 (15H, циклогексил и 2 CH2; 1,3 (6H, 2 CH3).
Пример 248
Boc-(D)-Chg-Pro-NH-(2-хлор)-pAmb
а) Boc-Пролин-(2-хлор-4-циано)бензиламид:
5,4 г (24 ммоль) Boc-Pro-OH согласно примеру 241, стадия а), вводят во взаимодействие с 20 мл ангидрида пропанфосфоновой кислоты, 17,9 мл диизопропилэтиламина и 4,0 г (24 ммоль) 2-хлор-4-цианобензиламина с образованием Boc-Pro-(2-хлор-4- циано)бензиламида. Получают 7,0 г (80%) продукта.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 8,57 (1Н, NH); 8,05 - 7,45 (3H, аром. протоны); 4,50 - 4,10 (3H, CH2 и CH); 3,4 - 3,2 (2H, CH2); 2,25 - 1,70 (4H, 2 CH2; 1,4 - 1,3 (9H, Boc).
б) Пролин-(2-хлор-4-циано)-бензиламидгидрохлорид:
Согласно примеру 236, стадия б), отщепляют Boc-группу. Получают 5,6 г (97%) гидрохлорида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 10,2 и 8,6 (NH2 +); 9,45 (1Н, NH); 8,05 - 7,50 (3H, аром. протоны); 4,45 (д, 2H, CH2); 4,28 (1Н, CH); 3,20 (2H, CH2); 2,40 - 1,80 (4H, 2CH2).
в) Boc-(D)-Циклогексилглицилпролин-(2-хлор-4-циано)бензиламид:
4,76 г (18,7 ммоль) Boc-(D)-Циклогексилглицина согласно примеру 238, стадия а), вводят во взаимодействие с 15,5 мл ангидрида пропанфосфоновой кислоты, 14 мл диизопропилэтиламина и 5,6 г (18,7 ммоль) гидрохлорида из стадии б) с образованием Boc-(D)-Chg-Pro-(2-хлор-4-циано)-бензиламида. Получают 8,7 г (92%) продукта.
г) Boc-(D)-Циклогексилглицилпролин-(2-хлор-4-гидроксиамидино) бензиламид:
Циангруппу соединения из стадии в) согласно примеру 243, стадия б), вводят во взаимодействие с гидроксиамином, получая почти с количественным выходом Boc-(D)-Chg-Pro-(2-хлор-4- гидроксиамидин)-бензиламид. Получают 4,6 г продукта.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,75 (1Н, ОН); 5,90 (2H, NH2).
д) Boc-(D)-Циклогексилпролин-(2-хлор-4-амидино)бензиламид:
4,6 г Гидроксиамидина из стадии г) согласно примеру 240 гидрируют в присутствии никеля Ренея до образования амидина. После очистки с помощью колоночной хроматографии на силикагеле (растворитель: дихлорметан с 15% метанола и 2% 50%-ной уксусной кислоты) получают 5,4 г амидина в виде ацетата.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): сигнал амидина в виде гидроацетата невозможно локализовать из-за его уширения: 8,15 (1Н, NH); 7,9 - 7,5 (3H, аром. протоны), 7,05 (1Н, NH); 4,5 - 3,4 (6H, 2 CH2 и 2 CH); 2,2 - 1,0 (24H, циклогексил, 2 CH2 и Boc).
Пример 249
H-(D)-Chg-Pro-NH-(2-хлор)-pAmb
От соединения примера 251 отщепляют Boc-группу согласно примеру 242. Получают 3,0 г (65%) продукта в виде дигидрохлорида.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 9,55 и 9,34 (4H, амидин); 9,05 (1Н, NH); 8,60 (3H, NH3 +); 7,95 - 7,48 (3H, аром. протоны); 4,5 - 3,5 (6H, 2 CH2, 2 CH); 2,25 - 1,0 (15H, циклогексил и 2 CH2).
Пример 250
H-(D)-Phe-Pro-(D,L)(4-Am)-PhgOMe
Соединение получают путем отщепления Z - группы от продукта примера 18.
1H-ЯМР (дейтерированный диметилсульфоксид; δ в м. д.): 9/9,2/8,85/8,8 (4д, 1H, NH); 7,8 (м, 2H, аром. протоны); 7,6 (м, 2H, аром. протоны); 7,3 - 7,0 (м, 5H, аром. протоны); 5,7/5,6 (2д, 1H, α-Н); 4,8/4,4 (2дд, 1H, α-Phe); 3,9 (м, 1H, α-Pro); 3,0 - 2,6 (м, 2H, CH2-фенил); 2,2 - 1,6 (м, 4H, β/γ-Pro).
Масс-спектр: 452 (M+H+), 305, 192, температура плавления дигидроацетата составляет 71 - 73oC.
Пример 251
H-(D)-Chg-Pro-NH-3-(2-MeO-6-Am)-pico
а) Гидрохлорид 2-метокси-2-пиколилового спирта:
20,0 г 2-Метоксиникотиновой кислоты (130,59 ммоль) вместе с 28,7 мл N-метилморфолина (261,18 ммоль) при -10oC вносят в тетрагидрофуран и быстро прикапывают 25,4 мл изобутилового эфира хлормуравьиной кислоты (195,89 ммоль) в 50 мл тетрагидрофурана, причем выпадает осадок. После перемешивания в течение часа при 0oC порциями добавляют 16,3 г боргидрида натрия (430,95 ммоль) и затем медленно прикапывают 250 мл метанола (сильное газовыделение и экзотермическая реакция). После отсасывания осадившейся соли фильтрат концентрируют в вакууме в ротационном испарителе, остаток растворяют в этилацетате, промывают водой, разбавленной соляной кислотой (pH-значение составляет два), насыщенным раствором хлорида натрия (используя очень малые количества
водных фаз), сушат над сульфатом магния и концентрируют в ротационном испарителе. Остаток обрабатывают эфиром, смешивают с эфирным раствором хлороводорода, осадок отсасывают и перемешивают с эфиром. Получают 16,3 г (71%) гидрохлорида 2-метокси-3-пиколилового спирта в виде соли белого цвета.
б) 2-Метокси-3-пиколилхлоридгидрохлорид:
К 17 г Гидрохлорида 2-метокси-3-пиколилового спирта (96,76 ммоль), суспендированным в 60 мл дихлорметана, прикапывают 51 мл тионилхлорида (696,67 ммоль), раствор перемешивают в течение одного часа при комнатной температуре, растворитель и избыточный тионилхлорид удаляют в вакууме, остаток четырехкратно обрабатывают метанолом, каждый раз отгоняя его в вакууме, и затем перемешивают с эфиром. Получают 15,2 г (81%) 2-метокси-3-пиколилхлоридгидрохлорида в виде белой кристаллической соли.
в) 2-Метокси-3-пиколиламиндигидрохлорид:
15,1 г 2-Метокси-3-пиколилхлоридгидрохлорида (77,76 ммоль) при 35oC медленно прикапывают к смеси из 600 мл 30%-ного раствора аммиака в воде и 250 мл метанола, причем постоянно пропускают газообразный аммиак. После перемешивания в течение одного часа при 35oC раствор концентрируют в вакууме в ротационном испарителе, остаток подщелачивают с помощью 20%-ного раствора гидроксида натрия (используя небольшое количество водной фазы), многократно экстрагируют дихлорметаном, органические фазы сушат над сульфатом магния и концентрируют в вакууме в ротационном испарителе. После обработки остатка эфиром, добавления эфирного раствора хлороводорода, отсасывания и промывки осадка эфиром получают 11,0 (67%) 2-метокси-3-пиколиламин-дигидрохлорида в виде белого кристаллизата.
г) Boc-(D)-Chg-Pro-NH-3-(2-MeO)-pico:
Получают по аналогии с Boc-(D)-Chg-Pro-NH-3-(2-Me)-pico (см. пример 224). Выход составляет 92%.
д) Boc-(D)-Chg-Pro-NH-3-(2-MeO-1-оксо)-pico:
4,9 г Boc-(D)-Chg-Pro-NH-(2-MeO)-pico (10,32 ммоль) растворяют в 100 мл дихлорметана, смешивают с 3,6 г м-хлорбензойной кислоты (20,164 ммоль) и перемешивают в течение нескольких дней при комнатной температуре (согласно тонкослойной хроматографии происходит только частичное превращение). Раствор затем разбавляют дихлорметаном, сушат над сульфатом магния, насыщают газообразным аммиаком, выпавший осадок отфильтровывают и фильтрат концентрируют в вакууме. Смесь продуктов обрабатывают эфиром, экстрагируют 1М раствором гидросульфата калия (pH-значение равно двум), водную фазу подщелачивают карбонатом калия, многократно встряхивают с дихлорметаном, сушат над сульфатом магния и концентрируют в вакууме в ротационном испарителе. Получают 1,6 г Boc-(D)-Chg-Pro-NH-3-(2-MeO-1-оксо)- pico в виде твердой пены.
3,1 г Эдукта можно рекуперировать из кислого эфирного экстракта и снова использовать для получения N-оксида. Путем многократных повторений в целом получают 4,2 г продукта.
е) Boc-(D)-Chg-Pro-NH-3-(2-MeO-6-CN)-pico:
4,2 г Boc-(D)-Chg-Pro-NH-3-(2-MeO-6-CN)-pico (8,56 ммоль) смешивают с 16 мл тетраметилсилилцианида, 5 мл 1,2-дихлорэтана и 10 мл хлорангидрида диметилкарбаминовой кислоты, тотчас нагревают до 70oC и перемешивают в течение десяти минут при этой температуре. После концентрирования в вакууме смесь продуктов (2 основных компонента, тонкослойная хроматография: Rf = 0,46, соответственно, 0,26; растворитель: смесь дихлорметана с метанолом в соотношении 95:5) разделяют путем колоночной хроматографии на силикагеле (элюирующее средство: дихлорметан с возрастающей от 0,5% до 1,5% добавкой метанола).
Первая основная фракция содержит 1,17 г Boc-(D)-Chg-Pro-NH-3- (2-MeO-6-CN)-pico.
ж) Получение Boc-(D)-Chg-Pro-NH-3-(2-MeO-6-Ham)-pico:
1,17 г Boc-(D)Chg-Pro-NH-3-(2-MeO-6-CN)-pico (2,34 ммоль) вместе с 0,41 г гидроксиламмоний хлорида и 2 мл диизопропилэтиламина (11,7 ммоль) в 10 мл дихлорметана перемешивают в течение четырех часов при комнатной температуре, затем концентрируют в вакууме в ротационном испарителе, остаток обрабатывают этилацетатом, многократно экстрагируют разбавленной соляной кислотой (pH-значение равно четырем), органическую фазу сушат над сульфатом магния и концентрируют в вакууме в ротационном испарителе. Полученную смесь продуктов (2 компонента; тонкослойная хроматография: Rf = 0,54, соответственно, 0,42; растворитель: дихлорметан с метанолом в соотношении 9:1) разделяют с помощью колоночной хроматографии на силикагеле (элюирующее средство: дихлорметан с возрастающей от 0,5% до 1,5% добавкой метанола).
Первая основная фракция содержит 300 мг Boc-(D)Chg-Pro-NH-3- (2-MeO-6-Ham)-pico.
13C-ЯМР (дейтерированный диметилсульфоксид; δ в м.д.): 171,16; 170,46; 159,13; 155,69; 148,89; 145,44; 135,84; 120,76; 111,53; 77,80; 59,55; 56,66; 52,84; 46,56; 38,4; 36,32; 28,87; 28,38; 28,17; 27,72; 27,57; 25,37; 25,29; 25,09; 23,65.
з) Boc-(D)Chg-Pro-NH-3-(2-MeO-6-Am)-pico:
300 мг Boc-(D)Chg-Pro-NH-3-(2-MeO-6-Ham)-pico (0,56 ммоль) в 10 мл этанола и 2 мл уксусной кислоты в присутствии 10%-ного палладия-на-угле гидрируют в течение четырех часов при 60oC. После отфильтровывания катализатора и концентрирования реакционного раствора в вакууме получают 260 мг Boc-(D)Chg-Pro-NH- 3-(2-MeO-6-Am)-pico в виде сырого продукта, который без дальнейшей очистки используют в следующей реакции.
и) H-(D)Chg-Pro-NH-3-(2-MeO-6-Am)-pico:
260 мг Полученного в виде сырого продукта Boc-(D)Chg-Pro-NH- 3-(2-MeO-6-Am)-pico в 5 мл дихлорметана и 5 мл метанола вместе с 5 мл эфирного раствора хлороводорода перемешивают в течение ночи при комнатной температуре, реакционную смесь концентрируют в вакууме в ротационном испарителе, остаток многократно обрабатывают толуолом и метанолом, отгоняя каждый раз используемый растворитель, и затем перемешивают с эфиром. Получают 210 мг Boc-(D)Chg-Pro-NH-3-(2-MeO- 6-Am)-pico-гидрохлорида или -дигидрохлорида в виде белого кристаллического твердого вещества. Температура плавления составляет 205 - 212oC.
Пример 252
Boc-(D,L)-Phe(3-OH)-Pro-NH-pAmb
Boc-(D, L)-Phe(3-OBzI) подвергают взаимодействию с дигидрохлоридом (4-амидинобензил)-пролинамида (получение см. стадии а и б, пример 51) аналогично примеру 51 и затем гидролизом отщепляют бензильную группу.
Белый, аморфный гидроацетат.
Масс-спектр (бомбардировка быстрыми атомами): 510 (М+H+).
Пример 253
Boc-(D,L)-1-Tic-Pro-NH-pAmb
Исходное сырье: Boc-(D,L)-1-Tic-OH
Белый, аморфный гидроацетат.
Масс-спектр (бомбардировка быстрыми атомами): 506 (М+H+).

Описываются соединения формулы (I), а также их соли с физиологически приемлемыми кислотами и их стереоизомеры, где заместители имеют указанные в описании значения. Также в изобретении описываются промежуточные продукты формулы (II) для их получения. Соединения пригодны для борьбы с заболеваниями в системе свертывания крови, а также могут служить для профилактики тромбоза, например после хирургических вмешательств. 2 с.п. ф-лы.
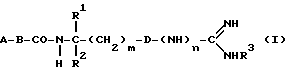
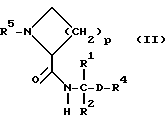
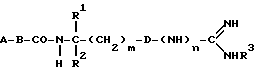
в которой А означает группу формул
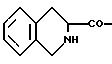
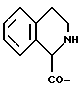
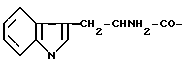
где Х1 означает водород или алкоксикарбонил с 1 - 4 атомами углерода в алкоксильной части,
еще А означает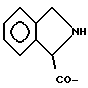
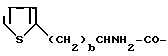
где b = 0 или 1,
еще А означает
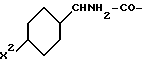
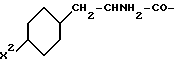
где Х2 означает водород или метокси,
еще А означает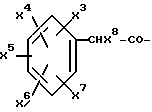
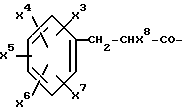
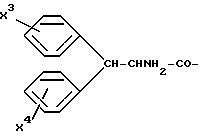
где Х3 означает водород, хлор, алкил с 1 - 4 атомами углерода, алкоксил с 1 - 4 атомами углерода, алкоксикарбонил с 1 - 4 атомами углерода;
Х4 - водород, хлор, алкил с 1 - 4 атомами углерода, алкоксил с 1 - 4 атомами углерода, фенилалкоксил с 1 - 4 атомами углерода в алкоксильной части;
Х5 - водород, фтор, хлор, бром, гидроксил, алкил с 1 - 4 атомами углерода, алкоксил с 1 - 4 атомами углерода, гидроксил;
Х6 - водород, фтор, хлор;
Х7 - водород, фтор, хлор;
Х8 - аминогруппа, гидроксил,
еще А означает
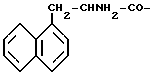
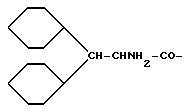
еще А означает алкилкарбонил с 1 - 12 атомами углерода, Х9-SО2-СН2-СН2-СНNН2-СО-, Н2N-СНХ9-СО-, где Х9 означает алкил с 1 - 8 атомами углерода, Н2N-СН2-СО-, Х10-(СН2)f-SО2-, где f = 0, 1 или 2, а Х10 - фенил, незамещенный или замещенный 1 - 3 метильными группами, α- или β-нафтил, при этом во всех вышеуказанных остатках А α-иминогруппа или α-аминогруппа может быть монозамещена алкилом с 1 - 12 атомами углерода, фенилалкиленом с 1 - 4 атомами углерода в алкиленовой части, Х11ОС-алкиленом с 1 - 6 атомами углерода в алкиленовой части или Х11ОС-алкилкарбонилом с 1 - 4 атомами углерода в алкильной части, где Х11 означает гидроксил или алкоксил с 1 - 6 атомами углерода,
В - группа формул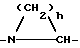
где h = 2 или 3, причем одна метиленовая группа может быть заменена на атом серы, а в случае h = 3 одна метиленовая группа может быть также заменена на гидроксиметиленовую группу,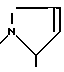
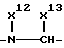
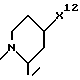
где Х12 означает водород или алкил с 1 - 4 атомами углерода;
Х13 - алкил с 1 - 6 атомами углерода, фенил, бензил;
R1 - водород или алкил с 1 - 4 атомами углерода;
R2 - водород, алкил с 1 - 4 атомами углерода, фенилалкилен с 1 - 4 атомами углерода в алкиленовой части, гидроксиметил, -СО-Х14, где Х14 означает водород, алкил с 1 - 4 атомами углерода, гидроксиметил, фенилалкилен с 1 - 4 атомами углерода в алкиленовой части;
m = 0 или 1;
D - фенилен, с которым связаны находящиеся в пара- или мета-положении друг по отношению к другу (СН2)m и (NН)n и который в орто-положении к (СН2)m может быть замещен алкоксилом с 1 - 4 атомами углерода, или СОХ15, где Х15 означает алкиламиногруппу с 1 - 4 атомами углерода или пиримидинилен;
n = 0 или 1;
R3 - водород, гидроксил, алкоксикарбонил с 1 - 20 атомами углерода в алкоксильной части,
а также их соли с физиологически приемлемыми кислотами и их стереоизомеры.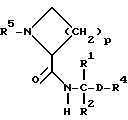
где R1, R2 и D имеют указанные в п.1 значения;
R4 - амидино остаток в мета- или пара-положении к С(R1, R2);
R5 - водород, алкоксикарбонил с 1 - 4 атомами углерода или фенилалкоксикарбонил с 1 - 3 атомами углерода;
р = 1, 2.
| 0 |
|
SU168342A1 | |
| ПРОИЗВОДНЫЕ ГЛИЦИНА ИЛИ ИХ ФИЗИОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ОБЛАДАЮЩИЕ СПОСОБНОСТЬЮ ТОРМОЗИТЬ СВЯЗЫВАНИЕ ФИБРИНОГЕНА У ФИБРИНОГЕННОГО РЕЦЕПТОРА ТРОМБОЦИТОВ | 1990 |
|
RU2024549C1 |
| Круглая отсадочная машина | 1973 |
|
SU479489A1 |
| Теплообменное устройство | 1974 |
|
SU504064A1 |

