Изобретение относится к новым ингибиторам тромбина, в частности к пятичленным гетероциклическим амидинам, соединениям, содержащим производные пятичленного гетероциклического амидина, в качестве составной части ингибиторов серинпротеазы и промежуточным соединением.
Тромбин относится к группе серинпротеаз и играет главную роль в качестве концевого фермента в системе свертывания крови. Как внутренняя, так и внешняя система свертывания крови через несколько стадий усиления приводит к образованию тромбина из протромбина. Катализируемое тромбином расщепление фибриногена до фибрина затем вызывает свертывание крови и агрегацию тромбоцитов, которые со своей стороны за счет связывания тромбоцитного фактора 3 и фактора свертывания XIII, а также целого ряда высокоактивных медиаторов усиливают образование тромбина.
Образование и действие тромбина представляют собой главные факторы при возникновении как белых артериальных, так и красных венозных тромбов и поэтому являются потенциально эффективными точками воздействия для лечения. Ингибиторы тромбина в противоположность гепарину независимо от кофакторов способны полностью подавлять одновременно действия свободного тромбина, так и также связанного с тромбоцитами тромбина. При обострении заболевания они могут предотвращать тромбоэмболию после чрезкожной внутрипросветной коронарной ангиопластики и лизис и служить в качестве антикоагулянтов при экстра-корпоральном кровообращении (аппарат "сердце - легкие", гемодиализ). Они также вообще могут служить для профилактики тромбоза, например, после хирургических вмешательств.
Известно, что синтетические производные аргинина оказывают влияние на ферментативную активность тромбина тем, что они вступают во взаимодействие с активным сериновым остатком тромбинпротеазы. Особенно пригодными оказываются пептиды на основе Phe-Pro-Arg, в которых аминокислота с концевой аминогруппой находится в D-форме. D-Phe-Pro-Arg-изопропиловый сложный эфир описывается как конкурентно действующий ингибитор тромбина (C. Mattson и др., Fo-lia Haematol, Ю9, 43 - 51, 1983).
Превращение карбоксильного конца аргинина в альдегид приводит к усилению ингибирующего действия. Так, описано много аргиналей, которые способны связывать гидроксильную группу "активного" серина с образованием полуацеталя (европейские заявки 185390, 479489,526877, 542525; международные заявки WO 93/15756, 93/18060).
Ингибирующая тромбин эффективность пептидных кетонов, фторированных алкилкетонов, а также сложных кетоэфиров, производных борной кислоты, эфиров фосфорной кислоты и амидов 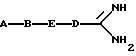 - кетокарбоновых кислот также объясняется с точки зрения этого взаимодействия с серином (европейские заявки 118280,195212, 362002,364344,410411,471651, 589741,293881, 503203, 504064, 530167, международные заявки WO 92/07869, 94 08941).
- кетокарбоновых кислот также объясняется с точки зрения этого взаимодействия с серином (европейские заявки 118280,195212, 362002,364344,410411,471651, 589741,293881, 503203, 504064, 530167, международные заявки WO 92/07869, 94 08941).
В случае описанных J. Oleksyszyn и др., J. Med. Chem., 37. 226 - 231 (1994) пептидных сложных 4-амидинофенил-глицинфосфонат- дифениловых эфиров речь идет о необратимых ингибиторах тромбина с недостаточной селективностью по отношению к другим серинпротеазам.
В патенте ФРГ 3108810, международной заявке WO 93/11152 и европейской заявке 601459 описываются агматин и тем самым производные аргинина, которые не способны взаимодействовать с активным серином серинпротеаз.
К усовершенствованной разработке относятся международная заявка WO 94/29336, европейская заявка 0 601 459 и международная заявка WO 95/23609, при этом агматиновый остаток заменен ариламидиновым остатком.
Кининогеназы являются серинпротеазами, освобождающими из кининогенов вазоактивные пептиды, так называемые кинины (брадикинин, каллидин и Met-Lys-брадикинин). Кининогены представляют собой мультифункциональные протеины, которые встречаются при каскадных реакциях свертывания и воспаления. В качестве ингибиторов они защищают клетки от разрушения цистеинпротеазами (Muller Esterl, 1985. FEBS Lett. 182. 310-314). Важными кининогеназами являются калликреин плазмы, калликреин ткани и трипсин тучных клеток.
Кинины такие, как брадикинин и каллидин являются вазоактивными пептидами, влияющими на множество биологических процессов. Они при воспалительных процессах играют существенную роль. Вследствие повышения сосудистой проницаемости они приводят к гипотензии и отекам. Кроме того, они являются очень могущественными вызывающими боль антацоидами и имеют в качестве клеточных медиаторов в патофизиологии астмы, аллергического ринита и артрита большое значение (K. D. Bhoola, C.D. Figueroa, K. Worthy, Pharmakological Reviews 1992,44 (1), 1-80).
Независимо от механизмов, которые лежат в основе воспалительных процессов, из кровеносных сосудов вытекает жидкость, содержащая все протеиновые системы циркулирующей крови. Это означает, что выход плазменной жидкости из сосудов играет роль при заболеваниях таких, как астма, ринит и при вызываемых воспалительными процессами внутренних болезнях. При этом освобождается в особенности при аллергических процессах трипсин тучных клеток (Salomonsson и др., Am. Rev. Respir. Dis., 1992, 146, 1535-1542).
Хлорометилкетоны аргинина H-(D)-Pro-Phe-Arg-CH2Cl и H-(D)- Phe-Phe-Arg-CH2Cl Kettner и Shaw описывают как ингибитор калликреина (Biochem. 1978, 17. 4778-4784 и Meth. Enzym. 1981, 8Q, 826-842).
Разные синтетические производные бензамидинов и бензиламинов оказались ингибиторами калликреина плазмы, при этом бензамидины обладали существенно более сильным тормозящим действием (F. Markward, S. Drawert, P. Walsmann, Biochemical Pharmacology 1974, 23, 2247-2256).
Эффективным ингибитором для этих кининогеназ является также PKSI-527, т. е. гидрохлорид M-(транс-4-аминометилциклогексилкарбонил) -1-фенилаланин-4-карбоксиметил-анилида (Wanaka, Ohamoto и др., Thromb. Res. 1990, 57 (6), 889-895).
Задачей изобретения является разработка новых ингибиторов тромбина.
Поставленная задача решается предлагаемыми пятичленными гетероциклическими амидинами общей формулы (I)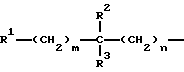
где A, В, D и E имеют следующие значения:
A: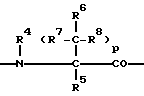
где
m - 0, 1 или 2,
n - 0, 1 или 2,
R1 - группы гидроксил, карбоксил, C1-6-алкил-OOC,
R2 - водород, алкилил с 1-4 атомами углерода,
R3 - водород, алкил с 1-4 атомами углерода,
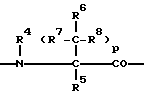
B: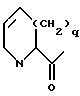
где
R4 - водород, алкил с 1-4 атомами углерода или R1-(CH2)m-, где R1 и m имеют вышеуказанные значения,
p - 0 или 1,
R5 - водород, алкил с 1-4 атомами углерода,
R6 - водород, алкил с 1-6 атомами углерода, циклоалкил с 3-8 атомами углерода,
R4 и R6 вместе могут означать этиленовую группу,
R7 - водород, алкил с 1-8 атомами углерода,
R8 - водород, алкил с 1-4 атомами углерода,
E: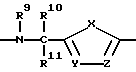
где
q - 0 или 1,
D: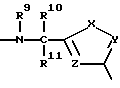
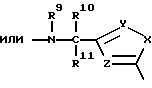
где
R9 - водород, алкил с 1-3 атомами углерода,
R10 - водород, алкил с 1-4 атомами углерода,
R11 - водород, алкил с 1-4 атомами углерода,
X - кислород, сера, NH12, где R12 означает водород, алкил с 1 - 6 атомами углерода,
Y - азот = или -CR13=, где R13 означает водород, алкил с 1 - 4 атомами углерода,
Z - азот = или -CR13=, где R13 имеет вышеуказанное значение, а также их солями с физиологически переносимыми кислотами.
Представленные В производные аминокислоты являются предпочтительно (D)-конфигурированными, в то время как 3,4- дегидропролин и 4,5-дегидропипеколиновая (L)-конфигурированы.
Предпочтительными являются соединения формулы (I), где группы A - E имеют следующие значения:
A:
HOOC-(CH2)t)-(t=1,2 или 3), HOOC-(CH2)2-CH-, HOOC-CH(алкил с 1-4 атомами углерода)-, HOOC-C(алкил с 1-4 атомами углерода)2-,
B: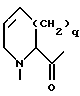
где
p - 0,1
R4 - водород, алкил с 1-4 атомами углерода,
R5 - водород, метил,
R6 - водород, алкил с 1-4 атомами углерода, циклоалкил с 3-8 атомами углерода,
R7 - водород, алкил с 1-8 атомами углерода,
R8 - водород, алкил с 1-4 атомами углерода,
(структурный элемент В является предпочтительно D-конфигурированным),
E:
где q - 0, 1,
D: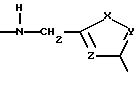
где
X = сера, кислород, NH, NCH3, NC2H5,
Y = CH, C-CH3, и
Z = CH, C-CH3
или X = сера, кислород, NH, N-CH3, Y = азот Z=CH, C-CH3,
или X = сера, кислород, NH, N-CH3, Y = CH, C-CH3, Z=азот
или X = сера, кислород, NH, N-CH3, Y = азот, Z = азот
или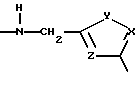
где
X = сера, кислород, NH, NCH3, NC2H5,
Y = CH, C-CH3, и
Z = CH, C-CH3,
или X = кислород, NH, NCH3, Y = азот Z=CH, C-CH3,
или X = кислород, сера, NH, NCH3, Y = CH, C-CH3, Z=азот
или X = кислород, сера, NH, NCH3, Y = Z = азот,
или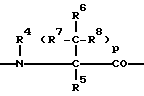
где
X = сера, кислород, NH, NCH3, NC2H5,
Y = CH, C-CH3, и
Z = CH, C-CH3,
или X = кислород, NH, NCH3, Y=азот Z=CH, C-CH3,
или X = кислород, сера, NH, NCH3, Y=CH, C-CH3, Z=азот
или X = кислород, NH, NCH3, Y = Z = азот.
Особенно предпочтительными являются соединения формулы (I), в которых A, В, D и E имеют следующие значения.
A - HOOC-CH2, HOOC-CH2-CH2, HOOC-CH(CH3), HOOC-CH(C2H5)
B: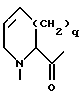
где
p - 0, 1
R4 - водород, метил,
R5 - водород, метил,
R6 - алкил с 1-6 атомами углерода, циклоалкил с 5-8 атомами углерода,
R7 - водород, метил,
R8 - водород, метил,
E: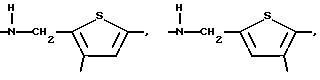
где
q - 0 или 1.
D: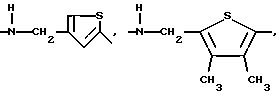
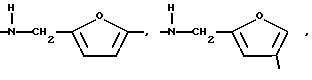
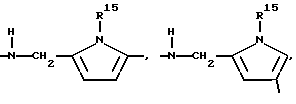

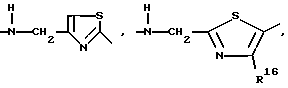
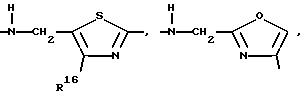
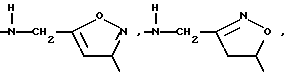
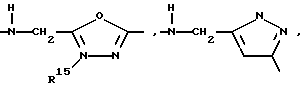

γ
где
R14 - водород, метил, предпочтительно водород,
R15 - водород, метил, этил, предпочтительно метил,
R16 - водород, метил.
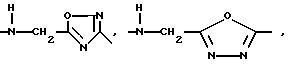
Следующие соединения являются особенно предпочтительными:
tBuOOC-CH2-(D)Chg-Pyr-NH-CH2-5-(2-am)-thioph
HOOC-CH2-(D)Chg-Pyr-NH-CH2-5-(2-am)-thioph
tBuOOC-CH2-(D)Chg-Dep-NH-CH2-5-(2-am)-thioph
HOOC-CH2-(D)Chg-Dep-NH-CH2-5-(2-am)-thioph
HOOC-CH2-(D,L)Cpg-Pyr-NH-CH2-5-(2-am)-thioph
HOOC-CH2-(D,L)Cheg-Pyr-NH-CH2-5-(2-am)-thioph
HOOC-CH2-(D,L)Cog-Pyr-NH-CH2-5-(2-am)-thioph
HOOC-CH2-(D,L)Nog-Pyr-NH-CH2-5-(2-am)-thioph
HOOC-CH2-(D,L)Adaala-Pyr-NH-CH2-5-(2-am)-thioph
HOOC-CH2-(D,L)4-MeCha-Pyr-NH-CH2-5-(2-am)-thioph
HOOC-CH2-(D,L)-MeCha-Pyr-NH-CH2-5-(2-am)-thioph
HOOC-CH2-(D,L)4-MeChg-Pyr-NH-CH2-5-(2-am)-thioph
HOOC-CH2-(D,L)3,3-Me2Chg-Pyr-NH-CH2-5- (2-am)-thioph
HOOC-CH2-(D,L)3,3-Me2Cha-Pvr-NH-CH2-5- (2-am)-thioph
HOOC-CH2-(D,L)4-iPrChg-Pyr-NH-CH2-5-(2-am)-thioph
HOOC-CH2-(D,1)3,4,5(MeO)3Phe-Pyr-NH-CH2-5- (2-am)-thioph
HOOC-CH2-(D,L)Chea-Pyr-NH-CH2-5-(2-am)-thioph
HOOC-CH2-(D)Diphe-Pyr-NH-CH2-5-(2-am)-thioph
HOOC-CH2-(D,L) ββ-Me2Cha-Pyr-NH-CH2- 5-(2-am)-thioph
HOOC-CH2-(D,L)Adagly-Pyr-NH-CH2-5- (2-am)-thioph
HOOC-CH2-(D,L)-1-Tic-Pyr-NH-CH2-5-(2-am)-thioph
HOOC-CH2-(D,L)Dch-Pyr-NH-CH2-5-(2-am)-thioph
HOOC-CH2-(D,L)4-iPrCha-Pyr-NH-CH2-5-(2-am)-thioph
HOOC-CH2-(D,L)α-MeCha-Pyr-NH-CH2-5-(2-am)-thioph
HOOC-CH2-(D,L)a-MeCha-Dep-NH-CH2-5-(2-am)-thioph
HOOC-CH2-(N-Me)(D)Cha-Pyr-NH-CH2-5-(2-am)-thioph
HOOC-CH2-(D)Cha-Pyr-NH-CH2-5-(2-am)-thioph
HOOC-CH2-(D)Cha-Dep-NH-CH2-5-(2-am)-thioph
HOOC-CH2-(D)Cha-Pyr-NH-CH2-4-(2-am)-thioph
HOOC-CH2-(D)Cha-Dep-NH-CH2-4-(2-am)-thioph
HOOC-CH2-(D)Cha-Pyr-NH-CH2-5-(3-am)-thioph
HOOC-CH2-(D)Cha-Dep-NH-CH2-5-(3-am)-thioph
HOOC-CH2-(D)Chg-Pyr-NH-CH2-5-(3-am)-thioph
HOOC-CH2-(D)Chg-Pyr-NH-CH2-4-(2-am)-thioph
HOOC-CH2-(D)Cha-Pyr-NH- CH2-5-(2-am)-fur
HOOC-CH2-(D)Cha-Dep-NH-CH2-5-(2-am)-fur
HOOC-CH2- (D)Cha-Pyr-NH-CH2-4-(2-am)-fur
HOOC-CH2-(D)Cha-Dep-NH-CH2-4-(2-am)-fur
HOOC-CH2-(D)Cha-Pyr-NH-CH2-5-(3-am)-fur
HOOC-CH2-(D)Cha-Dep-NH-CH2-5-(3-am)-fur
HOOC-CH2-(D)Chg-Pyr-NH-CH2-5-(2-am)-fur
HOOC-CH2-(D)Chg-Pyr-NH-CH2-5-(3-am)-fur
HOOC-CH2-(D)Cha-Pyr-NH-CH2-5-(1-Me-2-am)-pyrr
HOOC-CH2-(D)Chg-Pyr-NH- CH2-5-(1-Me-2-am)-pyrr
HOOC-CH2-(D)Cha-Dep-NH-CH2-5-(1-Me-2-am)- pyrr
HOOC-CH2-(D)Cha-Pyr-NH-CH2-4-(1-Me-2-am)-pyrr
HOOC-CH2- (D)Cha-Dep-NH-CH2-4-(1-Me-2-am)-pyrr
HOOC-CH2-(D)Cha-Pyr-NH-CH2-5-(1-Me-3-am)-pyrr
HOOC-CH2-(D)Cha-Dep-NH-CH2-5-(1-Me-3-am)-pyrr
HOOC-CH2-(D)Cha-Pyr-NH-CH2-5-(2-am-3,4- Me2)-thioph
HOOC-CH2-(D)Cha-Pyr-NH-CH2-5-(2-am-3-Me)-thioph
HOOC-CH2-(D)Cha-Pyr-NH-CH2-5-(2-am-4-Me)- thioph
HOOC-CH2-(D)Cha-Pyr-NH-CH2-5-(2-am-3-Me)-fur
HOOC-CH2-(D)Cha-Pyr-NH-CH2-5-(2-am-4-Me)-fur
HOOC-CH2-(D)Cha-Pyr-NH-CH2-5-(3-am-2-Me)-fur
HOOC-CH2-(D)Cha-Pyr-NH-CH2-2-(5-am)-thiaz
HOOC-CH2-(D)Cha-Pyr-NH-CH2-2-(4-am)-thiaz
HOOC-CH2-(D)Cha-Pyr-NH-CH2-4-(2-am)-thiaz
HOOC-CH2-(D)Cha-Pyr-NH-CH2-5-(2-am)-thiaz
HOOC-CH2-(D)Chg-Pyr-NH-CH2-2-(5-am)-thiaz
HOOC-CH2-(D)Chg-Pyr-NH-CH2-2-(4-am)-thiaz
HOOC-CH2-(D)Chg-Pyr-NH-CH2-4-(2-am)-thiaz
HOOC-CH2-(D)Chg-Pyr-NH-CH2-5-(2-am)-thiaz
HOOC-CH2-(D)Cha-Dep-NH-CH2-2-(5-am)-thiaz
HOOC-CH2-(D)Cha-Pyr-NH-CH2-2-(4-am)-oxaz
HOOC-CH2-(D)Chg-Pyr-NH-CH2-2-(4-am)-oxaz
HOOC-CH2-(D)Cha-Pyr-NH-CH2-5-(3-am-1-Me)-pyraz
HOOC-CH2-(D)Cha-Pyr-NH-CH2-3-(5-am)-1,2,4-oxadiaz
HOOC-CH2-(D)Cha-Pyr-NH-CH2-5-(3-am)-1,2,4-oxadiaz
HOOC-CH2-(D)Chg-Pyr-NH-CH2-5-(3-am)-1,2,4-oxadiaz
HOOC-CH2-(D)Cha-Pyr-NH-CH2-5-(3-am-1-Me)-1,2,4-triiaz
HOOC-CH2-CH2-(D)Cha-Pyr-NH-CH2-5-(2-am)-thioph
HOOC-CH2-CH2-(D)Cha-Pyr-NH-CH2-5-(2-am)-fur
HOOC-CH2-CH2-(D)Cha-Pyr-NH-CH2-5-(1-Me-2-am)-pyrr
(HOOC-CH2)2-(D)Cha-Pyr-NH-CH2-5-(2- am)-thioph
(HOOC-CH2)2CH-(D)Cha-Pyr-NH-CH2-5-(2-am)-thioph
HOOC-CH2-(D)Chg-Pyr-NH-CH2-5-(2-am)-1,3,4-thiadaz
HOOC-CH2-(D)Cha-Pyr-NH-CH2-5-(2-am)-1,3,4-thiadaz
HOOC-CH2-(D)Chg-Pyr-NH-CH2-5-(2-am)-1,3,4-oxadiaz
HOOC-CH2-(D)Cha-Pyr-NH-CH2-5-(2-am)-1,3,4-oxadiaz
Перечень условных сокращений, используемых в данной заявке:
Adaala адамантилаланин
Adagly адамантилглицин
AIBN/АИБН азобисизобутиронитрил
Ac ацетил
Ala аланин
am амидино
Asp аспарагиновая кислота
Aze азетидинкарбоновая кислота
Bn бензил
Boc трет.-бутоксикарбонил
Bu бутил
Cbz бензилоксикарбонил
Cha циклогексилаланин
Chea циклогептилаланин
Cheng циклогептилглицин
Chg циклогексилглицин
Cog циклооктилглицин
Cpa циклопентилаланин
Cpg циклопентилглицин
DC/TCX тонкослойная хроматография
DCC/ДЦГК дициклогексилкарбодиимид
Dch дициклогексилаланин
Dcha дициклогексиламин
DCM дихлорметан
Dep 4,5-дегидропипеколиновая кислота
DMF/ДМФА диметилформамид
DIPEA диизопропилэтиламин
Dpa дифенилаланин
Diphe 2,5-дигидрофенилаланин
Et этил
Eq/экв эквивалент
Gly глицин
fur фуран
ham гидроксиамидо
HOSucc гидроксисукцинимид
HPLC/ВЭЖХ высокоэффективная жидкостная хроматография
Hyp гидроксипролин
imi имидазол
2-lnd дигидроиндолкарбоновая кислота
i-Pr изопропил
Leu лейцин
Lsg раствор
Me метил
α -MeCha α -метилциклогексилаланин
ββ -Me2Cha 2-амино-3-циклогексил-3-метил-масляная кислота или- ββ-диметилциклогексилаланин
4-MeCha (4-метилциклогекс-1-ил)аланин
γ -MeCha (1-метилциклогекс-1-ил)аланин
3,3-Me2Cha (3.3-диметилциклогекс-1-ил)аланин
4-MeChg (4-метилциклогексил-1-ил)глицин
3,3-Me2Chg (3,3-диметилциклогекс-1-ил)глицин
MPLC/СЭЖХ среднеэффективная жидкостная хроматография
MTBE простой метил-трет.бутиловый эфир
NBS/БСИ N-бромсукцинимид
Nog норборнилглицин
Oxadias 1,2,4 оксадиазол
Oxas оксазол
Ph фенил
Phe фенилаланин
2Phi 2-пергидроиндолкарбоновая кислота
Pic пипеколиновая кислота
pico пиколил
pim пиперидинилметил
PPA ангидрид пропилфосфоновой кислоты
Pro пролин
Py пиридин
Pyr 3,4-дегидропролин
pyraz пиразол
pyrr пиррол
RT/KT комнатная температура
RP-18 обратная фаза C-18
t/tret./tret. третичный
tBu трет.-бутил
ТВАВ/ТБАБ тетрабутиламмонийбромид
TEA триэтиламин
TFA/ФУК трифторуксусная кислота
TFFA ангидрид трифторуксусной кислоты
thiaz тиазол
thioph тиофен
THP тетрагидропиранил
1Tic 1-тетрагидроизохинолинкарбоновая кислота
3Tic 3-тетрагидроизохинолинкарбоновая кислота
TOTU O-(циан-этоксикарбонилметилен)-амино-1-N, N, N'N',- тетраметилуронийтетрафтороборат
triaz 1,2,3-триазол
Z бензилоксикарбонил
Дальнейшими объектами изобретения являются соединения, содержащие структурный элемент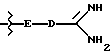
где E и D имеют вышеуказанное значение, а у атома азота структурного элемента E находятся атом водорода, защитная группа, или незамещенный алкильный остаток, в качестве составной части ингибиторов серинпротеазы и в особенности ингибиторов тромбина и калликреина, а также промежуточные соединения формул (IIа) и (IIб)
A-B-E-D-CN (IIа)
A-B-E-D-CSNH2 (IIб)
где A, B, E и D имеют вышеуказанное значение.
Соединения формулы (I) могут иметься в свободном виде или в виде солей с физиологически переносимыми кислотами. Примерами таких кислот являются следующие: соляная кислота, лимонная кислота, винная кислота, молочная кислота, фосфорная кислота, метансульфокислота, уксусная кислота, муравьиная кислота, малеиновая кислота, фумаровая кислота, янтарная кислота, гидрокси-янтарная кислота, серная кислота, глутаровая кислота, аспарагиновая кислота, пировиноградная кислота, бензойная кислота, глюкуроновая кислота, щавелевая кислота, аскорбиновая кислота и ацетилглицин.
Новые соединения формулы (I) можно использовать при следующих показаниях:
- заболевания, патомеханизм которых непосредственно или косвенно основывается на протеолитическом действии тромбина,
- заболевания, патомеханизм которых основывается на тромбинозависимой активации рецепторов и сигнальных трансдукций,
- заболевания, которые сопутствуются стимуляцией [например, PA1-1, тромбоцитарным фактором роста (PDGF), P-селектином, 1 CAM-1, фактором ткани] или ингибированием (например, синтезом моноокиси азота в гладкомышечных клетках) экспрессии генов в соматических клетках,
- заболевания, вызываемые митогенным действием тромбина,
- заболевания, вызываемые тромбинозависимым изменением сократимости и проницаемости эпителиальных клеток (например, клеток эндотелий сосудов),
- тромбинозависимые тромбоэмболические заболевания, такие, как, например, глубокий венозный тромбоз, эмболия легочной артерии, инфаркт миокарда или инсульт, мерцание предсердий, обмурация байпаса,
- диссеминированное внутрисосудистое коагулирование (ДВС-синдром),
- для реокклюзии и в целях сокращения времени реперфузии для комбинированного лечения вместе с тромболитическими средствами, такими, как стрептокиназа, урокиназа, проурокиназа, t-Pa, активатор антитромбоцитарной иммунной сыворотки, плазминогенные активаторы из слюнных желез животных, а также рекомбинантные и мутированные формы всех этих веществ,
- появление ранней реокклюзии и позднего рестеноза после чрезкожной внутрипросветной коронарной ангиопластики,
- тромбинозависимая пролиферация гладкомышечных клеток,
- аккумуляция активного тромбина в центральной нервной системе (например, в случае болезни Альцгеймера),
- опухолевые заболевания и адгезия и метастазирование опухолевых клеток.
Новые соединения можно использовать в особенности для лечения и профилактики тромбинозависимых тромбоэмболических заболеваний, как, например, глубокий венозный тромбоз, эмболия легочной артерии, инфаркт миокарда или инсульт и нестабильная стенокардия, далее для лечения диссеминированного внутрисосудистого коагулирования (ДВС-синдром). Далее они пригодны для комбинированного лечения вместе с тромболитическими средствами, такими, как стрептокиназа, урокиназа, проурокиназа, t-Pa, активатор антитромбоцитарной иммунной сыворотки и другие плазминогенные активаторы, в целях сокращения времени ре-перфузии и удлинения реокклюзии.
Дальнейшими предпочтительными областями применения являются предотвращение тромбинозависимой ранней реокклюзии и позднего рестеноза после чрезкожной внутрипросветной коронарной ангиопластики, предотвращение индуцируемой тромбином пролиферации гладномышечных клеток, предотвращение аккумуляции активного тромбина в центральной нервной системе (например, в случае болезни Альцгеймера), лечение опухолевых заболеваний и нарушение механизмов, приводящих к адгезии и метастазированию опухолевых клеток.
Новые соединения можно также использовать для покрытия искусственных поверхностей, таких, как мембраны гемодиализа и необходимые для этого системы мягких трубок и проводящие линии, а также оксигенаторов внесосудистой циркуляции, расширяющих элементов и сердечных клапанов.
Новые соединения можно применять далее в случае аболеваний, патомеханизм которых непосредственно или косвенно основывается на протеолитическом действии кининогеназ, в особенности калликреина, например, в случае воспалительных заболеваний, таких, как астма, панкреатит, ринит, артрит, крапивная лихорадка и другие воспалительные заболевания.
Предлагаемые согласно изобретению соединения можно вводить обычным образом перорально или парентерально (подкожно, внутривенно, внутримышечно, интраперитонеально, ректально). Их также можно вводить с помощью паров или спреев через носоглотку.
Дозировка зависит от возраста, состояния и массы пациента, а также от способа введения. Как правило, суточная доза активного вещества в расчете на одного субъекта составляет от 10 до 2000 мг при введении парентерально. Эту дозу можно вводить в виде двух-четырех разовых доз или один раз в день в виде лекарственной формы продленного действия.
Новые соединения можно использовать в обычных галеновых, твердых или жидких лекарственных формах, например, как таблетки, таблетки в целлофановой упаковке, капсулы, порошки, грануляты, драже, суппозитории, растворы, мази, кремы или спреи. Их можно получать обычным образом. Активные вещества при этом можно перерабатывать вместе с обычными галеновыми вспомогательными средствами, такими, как связующие таблеток, наполнители, консерванты, порофоры для таблеток, агенты регулирования текучести, мягчители, смачиватели, диспергаторы, эмульгаторы, растворители, средства для придания лекарственной форме пролонгированного действия, антиоксиданты и/или пропелленты (см. Н. Sucker и др. Фармацевтическая технология, изд. Thieme, Штуттгарт, 1978). Таким образом полученные лекарственные формы обычно содержат активное вещество в количестве от 0,1 до 99 мас. %.
Экспериментальная часть
Соединения формулы (I) можно получать согласно схемам I - III.
Структурные элементы A, B, E и D предпочтительно строятся отдельно, а затем их вводят в пригодном защищенном виде (см. схемы I - III в конце описания).
Схема I описывает линейное строение молекулы (I) путем сочетания амина H-D-CN с M-защищенной аминокислотой P-E-OH с получением P-E-D-CN, отщепления N-концевой защитной группы с получением H-E-D-CN, сочетания с N-защищенной аминокислотой P-В-ОH с получением P-B-E- D-CN, отщепления защитной группы P с получением H-B-E-D-CN, последующего алкилирования с также защищенным структурным элементом (P)-A-U (U == уделяемая группа) или восстановительного алкилирования с (P)-A'-U (U = альдегид, кетон) или присоединения по Михаэлу к пригодному производному (P)-A''-C= C с получением (P)-A-B-E-D-CN. Превращение нитрильной функции в амидиногруппу осуществляется либо путем классического синтеза Пиннера (R. Boder, D.G. Neilson, Chem. Rev., 1962, 61. 179) либо путем модифицированного синтеза Пиннера, который протекает через образование соли сложного иминотиоэфира в качестве промежуточной стадии (Н. Vie-weg и др., Pharmazie, 1984, 39. 226) или прямо по методу A. Eschenmoser, Helv. Chimica Acta, 69. (1986), 1224. После этого в молекуле еще имеющиеся защитные группы отщепляют предпочтительно путем гидролиза в кислой среде.
В случае если структурный элемент D вводится в виде H-D-CONHg в синтез, то в одной из защищенных промежуточных стадий осуществляется дегидратация амидной функции до нитрильной функции или превращение до тиоамидных функций. Структурный элемент D можно альтернативно вводить в виде H-D-CSNH2 в синтез.
Схема II описывает линейное строение молекулы 1 путем алкилирования, восстановительного аминирования или присоединения по Михаэлу H-P-В к соответственно пригодным незащищенным или защищенным структурным элементам A с получением (P)-A-В-Р, отщепления C-концевой защитной группы с получением (P)-A-В-ОH, сочетания с H-Е-Р с получением (P)-A-В-Е-Р, отщепления C-концевой защитной группы с получением (P)-A-В-Е-ОН, сочетания с H-D-CN с получением (P)-A-E-C-D-CN и превращения этого промежуточного продукта до конечного продукта аналогично схеме 1.
В случае если при соединениях (P)-A-В-P имеется еще свободная NH-функция у В, то последней еще до отщепления C-концевой защитной группы следует придавать пригодную защитную группу. Используемые в данном случае защитные группы должны быть ортогональными по отношению друг другу.
Вместо структурного элемента H-D-CN можно альтернативно использовать также H-D-CONH2, H-D-CSNH2, H-D-C(NH) NH2, H-D-C(NP)NH2, H-D-C(NP)NHP, причем в первом случае сочтенный промежуточный продукт (P-A-B-E-D-CONH2 подвергается дегидратации до (P)-A-B-E-D-CN или же превращается прямо до (P)-A-B-E-D-CSNH2.
Схема III описывает очень эффективный способ получения соединений 1 путем конвергентного синтеза. Соответственно защищенные структурные элементы (P)-A-В-ОН и H-E-D-CN сочетаются друг с другом, а образуемый промежуточный продукт (P)-A-B-E-D-CN превращается аналогично схеме I до конечного продукта.
Вместо H-E-D-CN можно альтернативно использовать также H-E-D-CONH2 или H-E-D-CSNH2, причем в первом случае сочтенный промежуточный продукт (P)-A-B-E-D-CONH2 подвергается дегидратации до (P)-A-B-E-D-CN или же превращается до (P)-A-B-E-D-CSNH2.
В качестве N-концевых защитных групп используют трет.- бутоксикарбонил, бензилоксикарбонил или 9-флуоренил- метоксикарбонил, предпочтительно трет. -бутоксикарбонил. C-концевыми защитными группами являются метил, трет.-бутил и бензил. В случае, если в молекуле имеются несколько защитных групп, то они должны быть ортогональными по отношению друг другу. Если промежуточные продукты содержат структурный элемент E, то бензилоксикарбонильные и бензиловые защитные группы являются непригодными.
Необходимые реакции сочетания, а также обычные реакции введения и удаления защитных групп осуществляют в стандартных условиях химии пептидов (см. М. Bodanszky, A. Bodanszky "The Practice of Peptide Synthesis", второе издание, изд. Springer, Гейдельберг, 1994 г.).
Трет. -бутоксикарбонильные защитные группы отщепляют с помощью раствора хлороводорода в диоксане или трифторуксусной кислоты в дихлорметане, бензилоксикарбонильные защитные группы отщепляют путем гидрогенолиза или с помощью фтороводорода. Омыление сложноэфирных функций осуществляют с помощью гидроксида лития в спиртовом растворителе или в смеси диоксана с водой. Сложный трет.-бутиловый эфир отщепляют с помощью трифторуксусной кислоты или с помощью смеси диоксана с соляной кислотой.
За протеканием реакций следят с помощью тонкослойной хроматографии, причем обычно используют следующие растворители:
А. Смесь дихлорметана с метанолом в соотношении 95:5.
Б. Смесь дихлорметана с метанолом в соотношении 9:1.
В. Смесь дихлорметана с метанолом в соотношении 8:2.
Г. Смесь дихлорметана с метанолом и 50%-ной уксусной кислотой в соотношении 40:10:5.
Д. Смесь дихлорметана с метанолом и 50%-ной уксусной кислотой в соотношении 35:15:5.
Когда упоминают разделения с помощью колоночной хроматографии, эти разделения осуществляют на силикагеле при использовании вышеуказанных растворителей.
Разделения с помощью высокоэффективной жидкостной хроматографии с обращенной фазой проводят при использовании смеси ацетонитрила с водой и уксуснокислого буфера.
Исходные соединения можно получать следующими способами:
В качестве структурных элементов A используют для алкилирования, например, сложный трет.-бутиловый эфир α - бромуксуной кислоты, сложный трет.-бутиловый эфир β - бромпропионовой кислоты, сложный трет.-бутиловый эфир α - бромпропионовой кислоты, сложный трет.-бутиловый эфир γ -броммасляной кислоты, сложный трет. -бутиловый эфир α - броммасляной кислоты, защищенный ТНР бромметанол, защищенный ТНР γ -бромпропанол, α -бром- γ -бутиролактон, для восстановительного
аминирования, например, дигидроксиацетон, сложный ди-трет.-бутиловый эфир ацетондикарбоновой кислоты, а для присоединения по Михаэлу, например, сложный трет.-бутиловый эфир акриловой кислоты, сложный трет.-бутиловый эфир метакриловой кислоты, сложный ди-трет. -бутиловый эфир фумаровой кислоты. Указанные сложные трет.-бутиловые эфиры, если их нельзя приобретать в торговле, получают аналогично G. Uray, W. Lindner, Tetrahedron 1988, 44 4357-4362.
Структурные элементы B:
Для общего и специального синтеза аминокислот в литературе описываются разнообразные возможности (см. к этому, в частности, обзор в книге Губен-Вейл, том E 16 d, часть 1, стр. 406 и последующие).
Часто используемыми эдуктами являются этиловый эфир бензофенониминоуксусной кислоты, диэтиловый эфир ацетамидомалоновой кислоты и этиловый эфир изонитрилуксусной кислоты.
Различные производные глицина и аланина получают, например, исходя из этилового эфира изонитрилуксусной кислоты и соответствующего кетона или альдегида (см. H. - J. Pratorius, J. Ftossdorf, M.- R. Kula, Chem Ber., 108. 3079 (1975)).
Синтезы циклооктилглицина, 2-норборнилглицина, адамантилаланина, γ--метил-циклогексилаланина, 4- изопропилциклогекс-1-ил-аланина, 4-метилциклогекс-1-ил-аланин и 4- метилциклогекс-1-илглицин осуществляют через соответствующие этиловые эфиры 2-формиламиноакриловой кислоты (U. Schollkopf и R. Meyer, Liebigs Ann. Chem., 1174 (1977)), исходя из этилового эфира изоцианоуксусной кислоты с соответствующими карбонильными соединениями, как циклооктанон, 2-норборнанон, 1-формиладамантан, 1-формил-1-метилциклогексан и 4-метилциклогексанон, по следующим общим методикам:
Общая методика синтеза этилового эфира 2-формиламиноакриловой кислоты
К 100 ммоль трет.-бутилата калия в 150 мл тетрагидрофурана при температуре от 0 до -10oC прикапывают раствор 100 ммоль этилового эфира изоциаоноуксусной кислоты в 50 мл тетрагидрофурана. Спустя 15 мин при той же температуре добавляют 100 ммоль соответствующего карбонильного соединения в 50 мл тетрагидрофурана, реакционную смесь медленно доводят до комнатной температуры и растворитель удаляют на ротационном испарителе. Остаток смешивают с 50 мл воды, 100 мл уксусной кислоты и 100 мл дихлорметана и продукт экстрагируют дихлорметаном. Дихлорметановую фазу сушат над сульфатом натрия и растворитель снимают на ротационном испарителе. Образующиеся почти в чистом состоянии продукты в случае необходимости можно очищать далее с помощью колоночной хроматографии на силикагеле (растворитель: смесь диэтилового эфира с петролейным эфиром).
Общая методика синтеза гидрохлоридов аминокислот из этиловых эфиров 2-формиламиноакриловых кислот
100 ммоль этилового эфира 2-формиламиноакриловой кислоты в присутствии 10%-ного палладия-на-угле в 200 мл ледяной уксусной кислоты гидрируют с помощью водорода вплоть до полного превращения. Затем катализатор отфильтровывают, уксусную кислоту насколько возможно удаляют в ротационном испарителе и остаток в 200 мл полуконцентрированной соляной кислоты кипятят с обратным холодильником в течение 5 ч. Соляную кислоту удаляют на ротационном испарителе, продукт высушивают при 50oC в вакууме и дополнительно промывают его многократно диэтиловым эфиром. Гидрохлориды получают в виде слабоокрашенных кристаллов.
Исходя из 18,9 г (150 ммоль) циклооктанона получают 25,0 г циклооктилглицин-гидрохлорида. Исходя из 16,5 г (150 ммоль) 2-норборнанона получают 26,6 г 2-норборнилглицин-гидрохлорида. Исходя из 19,7 г (120 ммоль) 1-формиладамантана получают 26,0 г адамантилаланин-гидрохлорида. Исходя из 12,6 г (100 ммоль) 1- формил-1-метилциклогексана получают 16,6 г γ-метилциклогексил- аланин-гидрохлорида. Исходя из 16,8 г (150 ммоль) 4- метилциклогексанона получают 25,9 г метилциклогексилглицин- гидрохлорида. Исходя из 15 г транс-1-формил-4-метилциклогексана получают 18 г транс-4-метилциклогекс-1-илаланин-гидрохлорида. Исходя из 9 г 3,3-диметил-1-формилциклогексана получают 10 г 3,3- диметилциклогекс-1-илаланин-гидрохлорида.
Требуемый для синтеза альдегид, 1-формил-3,3- диметилциклогексан получают по методу Moskal и Lensen (Rec. Trav. Chim. Pays-Bas, 137-141, 106. (1987)).
Раствор н. -бутиллития в н.-гексане (72 мл, 115 ммоль) прикапывают при температуре -60oC к перемешанному раствору диэтилизоцианометилфосфоната (17 мл, 105 ммоль) в 280 мл безводного диэтилэфира в течение 10 мин. Образовавшуюся суспензию при -60oC дополнительно перемешивают в течение 15 мин, затем к смеси прибавляют раствор 3,3-диметилциклогексанона (13 г, 105 ммоль) в 100 мл безводного диэтилового эфира в течение 10 мин, при этом температуру держат ниже -45oC. Реакционную смесь доводят до температуры 0oC, перемешивают при той же температуре в течение 90 мин и осторожно добавляют к смеси 150 - 200 мл 38%-ной водной соляной кислоты. Для осуществления полного гидролиза сильно перемешивают при комнатной температуре в течение 15 ч. Реакционную фазу отделяют, промывают водой, насыщенным раствором гидрокарбоната натрия и насыщенным раствором хлористого натрия, взятыми в количестве по 200 мл. Затем сушат над сульфатом магния, отфильтровывают и сгущают на ротационном испарителе с целью удаления растворителей. Получаемый остаток используют без дальнейшей очистки в качестве исходного материала для синтеза аминокислоты.
Трет.-бутилоксикарбонил-(D)- α -метилциклогексилаланин
3,4 г (12,2 ммоль) Вос-(D)- α-метил-Phe-ОH в 100 мл метанола при температуре 50oC в присутствии 250 г 5%-ного родия на оксиде алюминия гидрируют с помощью водорода в течение 20 ч при давлении 10 бар. После отфильтровывания и удаления растворителя получают 2,8 г Вос-(D)- α -метил-Cha-OH.
1H-ЯМР-спектр (дейтерированный диметилсульфоксид, δ в м.д.): 12 (очень широкий сигнал, COOH): 1,7 - 0,8 (25 H (с, Вос), 1,30 (с, метил).
Boc-(3-Ph)-Pro-OH синтезируют согласно J.Y.L Chung и др. (J.Y.L Chung и др., J. Org. Chem. 55, 270 (1990)).
Получение Вос-1-тетралинилглицина
Вос-1-тетралинилглицин получают из 1,2-дигидронафталина. 1,2- дигидронафталин сначала с помощью бромоводорода в 1- тетралилбромид (аналогично J. Med. Chem. 37, 1586 (1996)). Затем бромид вводят во взаимодействие с диэтиловым эфиром ацетамидомалоновой кислоты, проводят гидролитическое расщепление и полученную α -аминокислоту в стандартных условиях переводят в Вос-защищенную форму. Другая возможность получения описывается E. Reimann и D. Voss, Arch. Pharm. 310, 102 (1977).
Получение Boc-1-(D,L)Tic-OH
Boc-1-(D,L)Tic-OH получают по методике R.T. Shuman и др., J. Med. Chem., 36, 314 (1993).
Получение Boc-(D.L)Dch-OH
Boc-(D, L)Dpa-OH (1 ммоль) в 12 мл метанола вместе с каталитическими количествами 5%-ного родия на оксиде алюминия гидрируют при давлении 5 бар. После отфильтровывания и удаления растворителя в вакууме получают с количественным выходом продукт.
Получение циклогептилглицина, циклопентилглицина, 4- изопропилциклогексилглицина и 3,3-диметилциклогексилглицина
Получение этих аминокислот осуществляют путем взаимодействия циклогептанона, циклопентанона, 4-изопропилциклогексанона или 3,3-диметилциклогексанона с этиловым эфиром изонитрилуксусной кислоты согласно методике H.-J. Pratorius, J. Flossdorf, М. Kula, Chem. Ber., 108. 3079 (1985)).
Получение H-D,L-Chea-OH
4,0 г (19,39 ммоль) циклогептилметилметансульфоната, полученного из циклогептилметанола и хлорангидрида метансульфоновой кислоты, вместе с 4,9 г (18,47 ммоль) этилового эфира бензофенониминоглицина, 8,9 г (64,65 ммоль) высушенного тонкоизмельченного карбоната калия и 1 г (3 ммоль) тетрабутиламмонийбромида в 50 мл безводного ацетонитрила в атмосфере инертного газа кипятят с обратным холодильником в течение 10 ч. Затем карбонат калия отфильтровывают, фильтрат выпаривают досуха и сырой продукт непосредственно гидролизуют с помощью 10 мл 2 н. соляной кислоты в 40 мл этанола в течение полутора ч при перемешивании и при комнатной температуре. После разбавления реакционного раствора бензофенон экстрагируют этилацетатом в кислой области pH, затем экстрагируют H-D, L-Chea-OEt с помощью дихлорметана в щелочной области (pH составляет 9), раствор сушат над сульфатом магния и выпаривают в ротационном испарителе. Выход: 3,7 г  95% теории. Получение Boc-(D,L)-(3,4,5-(MeO)3)Phe-OH осуществляют путем алкилирования этилового эфира бензофенониминоглицина с помощью триметоксибензилхлорида, последующего введения трет.- бутоксикарбонильной защитной группы и омыления сложного эфира.
95% теории. Получение Boc-(D,L)-(3,4,5-(MeO)3)Phe-OH осуществляют путем алкилирования этилового эфира бензофенониминоглицина с помощью триметоксибензилхлорида, последующего введения трет.- бутоксикарбонильной защитной группы и омыления сложного эфира.
Получение D-(1,4-циклогексадиен-1-ил)ala-OH осуществляют согласно G. Zivi-lichovsky, V. Gurvich, J. Chem. Soc. , Perkin Trans, 19. 2509-15 (1995)).
Получение H-(D,L)- ββ -Me2Cha-OH осуществляют согласно U. Schollkopf, R. Meyer, L Ann. Chem., 1977. 1174-82.
Указанные аминокислоты по обычным способам с помощью ди- трет.бутил-дикарбоната в смесь воды и диоксана переводят в данном случае в Вос-защищенную форму и затем перекристаллизовывают из смеси этилацетата и гексана или очищают путем колоночной хроматографии на силикагеле с применением в качестве растворителя смеси этилацетата и петролейного эфира.
Вос-защищенные аминокислоты используют в качестве структурных элементов В соответственно схеме I в конце описания.
Указанные аминокислоты частично переводят в качестве структурных элементов В также в соответствующие сложные бензиловые эфиры и сочетают их с соответствующими защищенными структурными элементами A. В случае, если соединения имеют еще свободную N-Н-функцию, то последнюю затем защищают трет. -бутоксикарбонильной группой, группу сложного бензилового эфира удаляют гидрированием и структурный элемент A-В-OH очищают путем кристаллизации, осаждения соли или колоночной хроматографии. Этот способ в нижеследующем описывается примерно для tBuOOC-CH2-(Boc)(D)Cha-OH.
Синтез сложного бензилового эфира D-циклогексилаланина
Суспензию 100 г (481 ммоль) гидрохлорида D-циклогексилаланина, 104 г (962 ммоль) бензилового спирта и 109,7 г (577 ммоль) моногидрата n-толуолсульфокислоты в 2200 мл толуола на водоотделителе медленно нагревают с обратным холодильником. При температуре 80 - 90oC следят за образованием хлороводорода, а также за растворением суспензии до прозрачного раствора. После прекращения выделения воды (спустя 4 ч) отгоняют 500 мл толуола, дают охлаждаться реакционную смесь в течение ночи, отфильтровывают образовавшийся остаток и промывают два раза гексаном, взятым в количестве по 1000 мл. Полученный остаток (195 г) затем взмучивают в 2000 мл дихлорметана, после этого добавляют 1000 мл воды и при перемешивании подщелачивают до значения pH 9 - 9,5 путем последовательной добавки 50%-ного раствора едкого натрия. Органическую фазу отделяют, промывают ее два раза водой, взятой в количестве по 500 мл, сушат над сульфатом натрия, отфильтровывают от осушителя и сгущают фильтрат, после чего получают 115 г (94%) целевого продукта в качестве светлого масла.
Сложный бензиловый эфир N-(трет.-бутилоксикарбонилметилен)-D-циклогексилаланина
115 г (440 ммоль) сложного бензилового эфира D- циклогексилаланина растворяют в 2000 мл ацетонитрила, затем добавляют при комнатной температуре 607,5 г (4,40 моль) сложного трет.-бутилового эфира бромуксусной кислоты и при той же температуре перемешивают в течение 3 сут. Отфильтровывают от карбоната, дополнительно промывают ацетонитрилом, сгущают маточный раствор при температуре 30oC и при давлении 20 мбар, затем остаток прибавляют к 1000 мл простого метил-трет. -бутилового эфира и органическую фазу экстрагируют с помощью 5%-ной лимонной кислоты и насыщенного раствора гидрокарбоната натрия. Органическую фазу сушат над сульфатом натрия, отфильтровывают от осушителя и сгущают. Получаемое масло (168 г) вводят непосредственно в последующую реакцию.
Сложный бензиловый эфир N-Вос-N-(трет.-бутилоксикарбонилметилен) -D-циклогексилаланина
Полученное при предыдущем синтезе масло (168 г, 447 ммоль) растворяют в 1400 мл ацетонитрила, затем прибавляют 618 г (4,47 моль) карбоната калия в виде порошка и 107,3 г (492 ммоль) ди- трет.-бутилдикарбоната и перемешивают при комнатной температуре в течение 6 сут. Карбонат калия отсасывают, дополнительно промывают с помощью ацетонитрила в количестве приблизительно 1000 мл и сгущают фильтрат. Получают 230 г желаемого продукта.
N-Boc-N-(трет. -бутилоксикарбонилметилен)-D-циклогексилаланин- циклогексиламмониевая соль
115 г сложного бензилового эфира N-Вос-N-(трет.- бутилоксикарбонилметилен)-D-циклогексилаланина растворяют в 1000 мл чистого этанола и при температуре 25-30oC в присутствии 9 г 10%-ного палладия на активном угле гидрируют с помощью водорода при нормальном давлении в течение 2 ч. После отфильтровывания и удаления растворителя на ротационном испарителе получают 100 г (260 ммоль) желтого масла, которое добавляют к 1600 мл ацетона и нагревают с обратным холодильником. После удаления нагревательной ванны быстро прибавляют через капельную воронку раствор 27 г (273 ммоль) циклогексиламина в ацетоне. При охлаждении реакционной смеси до комнатной температуры выкристаллизовывается желаемая соль. Отфильтровывают твердое вещество, дополнительно промывают с помощью 200 мл ацетона и для окончательной очистки перекристаллизовывают еще раз из ацетона. После сушки остатка в вакуумном сушильном шкафу при 30oC получают 70,2 г желаемой соли в виде белого порошка.
N-Вос-N-(трет. -бутилоксикарбонилметилен)-D-циклогексилглицин-циклогексиламмониевую соль получают аналогичным образом из циклогексилглицина в качестве эдукта.
N-Bос-N-(трет.-бутилоксикарбонилэтилен)-D-циклогексилаланин- циклогексиламмониевая соль
а) Сложный трет.-бутиловый эфир 3-бромпропионовой кислоты
16,64 г (109 ммоль) бромпропионовой кислоты, 150 мл конденсированного 2-метилпропена и 2 мл концентрированной серной кислоты при -30oC в противотоке азота подают в пригодный для автоклава стеклянный сосуд, который плотно закрывают, и перемешивают при комнатной температуре в течение 72 ч. Для переработки реакционный сосуд снова охлаждают до -30oC и реакционный раствор осторожно подливают в 200 мл имеющего температуру льдя насыщенного раствора гидрокарбоната натрия. При перемешивании дают упариваться избыточный 2-метилпропен, остаток экстрагируют три раза дихлорметаном, взятым в количестве по 50 мл, объединенные органические фазы сушат над сульфатом натрия, отфильтровывают от осушителя и сгущают под вакуумом, получаемым с помощью водоструйного насоса. Маслянистый остаток очищают путем колоночной хроматографии с применением в качестве растворителя н-гексана, позднее смеси н-гексана и диэтилового эфира в соотношении 9 : 1. Получают 18,86 г целевого соединения.
б) Сложный бензиловый эфир N-(трет.-бутилоксикарбонилэтилен) -D-циклогексилаланина
49,4 г (189 ммоль) сложного бензилового эфира D- циклогексилаланина растворяют в 250 мл ацетонитрила, при комнатной температуре добавляют 31,6 г (151 ммоль) трет.-бутилового эфира бромпропионовой кислоты и смесь кипятят с обратным холодильником в течение 5 сут. Отфильтровывают от образовавшегося осадка, дополнительно промывают несколько раз ацетонитрилом, фильтрат сгущают под вакуумом, создаваемым с помощью водоструйного насоса, остаток прибавляют к 350 мл дихлорметана и органическую фазу экстрагируют 5%-ной лимонной кислотой и насыщенным раствором гидрокарбоната натрия. Органическую фазу сушат над сульфатом натрия, отфильтровывают от осушителя и сгущают. Маслянистый остаток очищают путем колоночной хроматографии с применением в качестве растворителя дихлорметана, позднее смеси дихлорметана и метанола в соотношении 95:5. Получают слегка загрязненное масло, которое вводят непосредственно в последующую реакцию.
в) Сложный бензиловый эфир N-Boc-N-(трет.-бутилоксикарбонилэтилен)-D-циклогексилаланина
Полученное в предыдущем синтезе масло (30 г, макс. 70 ммоль) растворяют в 150 мл ацетонитрила, затем добавляют 28 мл (160 ммоль) ди-изопропилэтиламина и 19,2 г (88 ммоль) ди-трет.- бутилдикарбоната и перемешивают при комнатной температуре в течение 3 сут. Реакционную смесь сгущают на ротационном испарителе под вакуумом, создаваемым с помощью водоструйного насоса, остаток прибавляют к н-гексану, промывают пять раз 5%-ным раствором лимонной кислоты, взятым в количестве по 3 мл, затем объединенные органические фазы сушат над сульфатом натрия, отфильтровывают от осушителя и сгущают. Остаток подвергают разделению путем колоночной хроматографии с применением в качестве растворителя смеси гексана и этилацетата в соотношении 95:5. Получают 32,66 г (64 ммоль) желаемого продукта.
г) N-Boc-N-(трет. -бутилоксикарбонилэтилен)-D- циклогексилаланин-циклогексиламмониевая соль
32,66 г (64 ммоль) сложного бензилового эфира N-Вос-N-(трет.- бутилоксикарбонилэтилен)-D-циклогексилаланина растворяют в 325 мл чистого этанола и при температуре 25-30oC в присутствии 3 г 10 %-ного палладия на активном угле гидрируют с помощью водорода при нормальном давлении в течение 14 ч. После фильтрации раствора через Celite®, дополнительной промывки этанолом и удаления растворителя на ротационном испарителе получают 26,7 г желтого масла, которое прибавляют к ацетону и нагревают с обратным холодильником. После удаления нагревательной бани быстро добавляют через капельную воронку раствор 7 г (70 ммоль) циклогексиламина в ацетоне. При охлаждении реакционной смеси до комнатной температуры выкристаллизовывается желаемая соль. Отфильтровывают твердое вещество, дополнительно промывают 25 мл ацетона и для окончательной очистки еще раз перекристаллизовывают из ацетона. После сушки остатка в вакуумном сушильном шкафу при 30oC получают 26,6 г (54 ммоль) желаемой соли в качестве белого порошка.
N-Вос-N-(трет. -бутилоксикарбонилметилен)-(D)-циклогексилаланил- 3.4-дегидропролин
а) N-Boc-Pyr-OH (5 г, 23,45 ммоль) растворяют в метаноле (50 мл), после этого добавляют хлороводород в диоксане (4N, 30 мл). Затем нагревают с обратным холодильником в течение 12 ч. Растворитель удаляют на ротационном испарителе, получают в качестве продукта H-Pyr-OMe гидрохлорид. Выход: 3,84 г (100%).
б) N-(t-BuO2C-CH2)-N-Boc-(D)-Cha-OH (8 г, 20,75 ммоль) растворяют в дихлорметане (78 мл), при температуре -10oC добавляют этилдиизопропиламин (15,5 мл, 89,24 ммоль). Перемешивают при той же температуре в течение 5 мин, а затем прикапывают раствор H-Pyr-OMe гидрохлорида (3,4 г, 20,75 ммоль) в дихлорметане (25 мл). После этого прикапывают 50%-ный раствор ангидрида пропанфосфоновой кислоты в этилацетате (20 мл, 26,96 ммоль) и перемешивают при от -10 до 0oC в течение 2 ч. Исходную смесь разбавляют дихлорметаном и промывают два раза насыщенным раствором гидрокарбоната натрия, взятым в количестве по 80 мл, два раза 5%-ным раствором лимонной кислоты, взятым в количестве по 15 мл, и раз насыщенном раствором хлористого натрия (20 мл). Органическую фазу сушат над сульфатом натрия и растворитель отгоняют на ротационном испарителе. Сырой продукт очищают путем хроматографии на колонне, содержащей силикагель с применением в качестве элюента смеси дихлорметана и метанола в соотношении 95:5. Выход: 6,2 г (60%).
в) N-(t-BuO2C-CH2)-N-Boc-(D)-Cha-Pyr-OMe (5,5 г, 11,12 ммоль) растворяют в диоксане (78 мл), затем прибавляют натровый щелок (1N, 22, 2 мл 22,24 ммоль) и перемешивают при комнатной температуре в течение 2 ч. Диоксан отгоняют на ротационном испарителе, водную фазу промывают этилацетатом и подкисляют с помощью 20%-ного раствора гидросульфата натрия до значения pH 1-2. Водную фазу экстрагируют дихлорметаном и объединенные органические фазы сушат над сульфатом натрия. Выход: 5 г (94%) бесцветной пены. Путем перекристаллизации из насыщенного водой н-гексана получают бесцветные кристаллы (точка плавления: 158 - 160oC).
N-Вос-N-(трет. -бутилоксикарбонилметилен)-(D)- циклогексилглицин-3,4-дигидропролин
Это соединение получают аналогичным образом из N-Вос-N- (трет.-бутилоксикарбонилметилен)-(D)-циклогексилглицина и сложного метилового эфира 3,4-дигидропролина.
Используемый в качестве структурного элемента E (L) 3,4- дегидропролин можно приобретать в торговле, a (D,L)-4,5- дегидропипеколиновую кислоту можно получать согласно A. Burgstahler, C.E. Aiman, J. Org. Chem., 25., 489 (1960) или C. Herdeis, W. Engel, Arch. Pharm. 326,297 (1993), а затем с помощью (Boc)2O переводить в Boc-(D,L)-Dep-OH.
Синтез структурных элементов D осуществляют согласно следующим методам:
5-аминометил-2-цианотиофен
Получение этого структурного элемента осуществляют согласно международной заявке 93/23609.
4-аминометил-2-цианотиофен
а) 2-бром-4-формилтиофен
36 г (320 ммоль) 3-формилтиофена растворяют в 600 мл хлористого метилена и охлаждают до 5oC, затем по порциям прибавляют 100 г (750 ммоль) трихлорида алюминия, после этого реакционную смесь нагревают с обратным холодильником. Затем прикапывают раствор 59 г (19 мл, 360 ммоль) брома в 40 мл хлористого метилена в течение 45 мин и подвергают смесь дополнительной реакции с обратным холодильником в течение 4 ч. После охлаждения реакционный раствор наливают на 600 г ледяной воды и экстрагируют хлористым метиленом, органическую фазу промывают насыщенным раствором гидрокарбоната натрия, сушат над сульфатом магния и отгоняют в вакууме на ротационном испарителе. Получают 64,5 г сырого продукта, который очищают путем колоночной хроматографии на силикагеле с применением в качестве элюента смеси хлористого метилена и петролейного эфира. При этом получают всего 56,5 г слегка загрязненного продукта.
б) 2-циано-4-формилтиофен
К раствору 13,53 г (70,82 ммоль) 2-бром-4-формилтиофена в 25 мл диметилформамида прибавляют 7,6 г (85 ммоль) цианида меди (I), реакционную смесь нагревают с обратным холодильником в течение 3,5 ч, причем суспензия, которая первоначально имеет светло-зеленый цвет, превращается в раствор черного цвета. После добавки воды реакционную смесь несколько раз экстрагируют, органические фазы объединяют, промывают насыщенным раствором поваренной соли, сушат над сульфатом натрия и в легком вакууме перегоняют на ротационном испарителе. К остатку (7 г) прибавляют простой эфир, в результате чего получают 1,6 г чистого продукта. Маточный раствор вместе с сырыми продуктами из других исходных смесей очищают путем хроматографии на силикагеле с применением в качестве элюента смеси хлористого метилена и петролейного эфира в соотношении 1: 1. Всего 56,6 г 2-бром-4-формилтиофена превращают до 2-циано-4- формилтиофена, при этом получают 12,6 г чистого продукта (Выход: 31%).
в) 2-циано-3-гидрометилтиофен
К суспензии 12,6 г (91,8 ммоль) 2-циано-4-формилтиофена в 200 мл этанола прибавляют по порциям 3,47 г (91,8 ммоль) боргидрида натрия и при комнатной температуре перемешивают в течение 2 ч, при этом реакционная смесь медленно превращается в прозрачный раствор. После сгущения к остатку добавляют сложный этиловый эфир уксусной кислоты, затем промывают последовательно насыщенным раствором поваренной соли, 5%-ной лимонной кислотой и насыщенным раствором поваренной соли.
Органическую фазу сушат над сульфатом натрия и сгущают в вакууме. Получают 11,7 г почти чистого продукта с выходом 91,5%.
г) 3-бромметил-2-цианотиофен
11,7 г (84,07 ммоль) 2-циано-3-гидроксиметилтиофена вместе с 24,1 г (91,87 ммоль) трифенилфосфина в 100 мл тетрагидрофурана растворяют при комнатной температуре, по порциям прибавляют при охлаждении ледяной баней 30,47 г (91,87 ммоль) тетрабромметана. Перемешивают при комнатной температуре в течение 3 ч, а затем сгущают в вакууме и очищают путем хроматографии на силикагеле с применением в качестве элюента смеси хлористого метилена и петролейного эфира. Получают 18,8 г содержащего еще петролейный эфир кристаллического светло-желтого продукта.
д) 4-N,N-бис(трет.-бутоксикарбонил)-аминометил-2-цианотиофен
18,81 г 3-бромметил-2-цианотиофена (сырой продукт, макс. 84,07 ммоль) растворяют в 160 мл тетрагидрофурана и охлаждают до 5oС, по порциям добавляют 3,07 г (102,4 ммоль) 80%-ной суспензии гидрида натрия. Затем при 5oC прикапывают 22,25 г (102,4 ммоль) ди-трет.-бутилиминодикарбоксилата, растворенного в 160 мл тетрагидрофурана, после чего смесь перемешивают при комнатной температуре в течение ночи. Так как в результате тонкослойной хроматографии превращение является незаконченным, дают нагреваться до 30-35oC в течение 4,5 ч. После охлаждения до 0 - 5oC медленно прикапывают 33 мл насыщенного раствора хлористого аммония, тетрогидрофуран отгоняют в вакууме, остаток несколько раз экстрагируют этилацетатом, этилацетатные фазы промывают насыщенным раствором поваренной соли, сушат над сульфатом натрия и в вакууме перегоняют на ротационном испарителе. Красный вязкий остаток (34,61 г) используют в качестве сырого продукта в последующей реакции.
е)4-аминометил-2-цианотиофен-гидрохлорид
34,61 г 4-N, N-бис(трет. -бутоксикарбонил)-аминометил-2- цианотиофена (сырой продукт, макс. 84,07 ммоль) растворяют в 600 мл этилацетата, нагревают до 0-5oC, насыщают хлористоводородным газом и нагревают до комнатной температуры. По истечении 3 ч образовавшуюся суспензию перегоняют на ротационном испарителе, осуществляют несколько раз совместную перегонку с хлористым метиленом, декантируют остаток с диэтиловым эфиром и сушат остаток в вакууме. Получают 13,85 г продукта в качестве светлого порошка. Выход через две стадии: 94,3%.
2-аминометил-4-цианотиофен
а) 4-цианотиофен-2-карбальдегид
49,3 г (258,05 ммоль) 4-бром-тиофен-2-карбальдегида и 27,8 г (310,41 ммоль) цианида меди (I) суспендируют в 130 мл абсолютного диметилформамида и нагревают с обратным холодильником в течение 8 ч. Растворитель при 40oC отгоняют на ротационном испарителе в вакууме, остаток суспендируют в этилацетате и вводят в аппарат Сокслета. Остаток экстрагируют в течение ночи, желтый раствор сушат над сульфатом натрия и перегоняют на ротационном испарителе в вакууме. Получаемое твердое вещество желтого цвета перекристаллизуют из диэтилового эфира. Получают 25,3 г продукта (80% теории).
б) 4-цианотиофен-2-карбальдегид-оксим
11,6 г (84,6 ммоль) 4-цианотиофен-2-карбальдегида растворяют в 140 мл метанола, после этого добавляют 12,3 г (116,1 ммоль) карбоната натрия. Затем при охлаждении прибавляют по порциям при 15oC 6,5 г 93,5 ммоль) гидроксиламин-гидрохлорида и перемешивают при 10oC еще в течение 2 ч. После добавки 80 мл воды реакционную смесь экстрагируют, пять раз диэтиловым эфиром, взятым в количестве по 50 мл. Органическую фазу сушат над сульфатом натрия, растворитель удаляют в вакууме. Получают 12,5 г желаемого продукта в качестве желтого кристаллического порошка (96% теории).
в) 2-аминометил-4-цианотиофен-гидрохлорид
11,22 г (171,64 ммоль) мелкой цинковой пыли по нескольким небольшим порциям прибавляют осторожно к охлажденному до 0-5oC раствору 4,65 г (30,60 ммоль) 4-цианотиофен-2-карбальдегид-оксима в 50 мл трифторуксусной кислоты так, что температура не превышает 15oC. После трехчого перемешивания декантируют от избыточного цинка, трифторуксусную кислоту большей частью удаляют под вакуумом, создаваемым с помощью масляного насоса, остаточное масло охлаждают до 0oC, после чего к нему прибавляют по порциям предварительно охлажденную до 0oC смесь 150 мл 3N натрового щелока и 2 л хлористого метилена. После фильтрации нерастворимых компонентов органическую фазу отделяют, водную фазу восемь раз экстрагируют хлористым метиленом, взятым в количестве по 20 мл, объединенные органические фазы сушат над сульфатом натрия и затем к ним добавляют при охлаждении льдом 20 мл 6M метанольной соляной кислоты. Продукт осаждается в качестве гидрохлорида в виде белого твердого вещества, при этом для усовершенствования кристаллизации суспензию охлаждают до 4oC в течение ночи. Получают 2,2 г продукта в качестве бесцветных иголок (50% теории).
Гидрохлорид амида 5-аминометил-3,4-диметил-тиофен-2- карбоновой кислоты
19 г (150,42 ммоль) амида 5-аминометил-3,4-диметил-тиофен-2- карбоновой кислоты суспендируют в 760 мл метанола и 110 мл 2N раствора соляной кислоты, затем прибавляют 9,5 г палладия на угле (10%) и гидрируют при комнатной температуре. После добавки 4,7 л водорода (4 ч) отгоняют метанол в вакууме, водную фазу три раза экстрагируют этилацетатом, а затем водную фазу подвергают сублимационной сушке. Получают 16,3 г желаемого продукта в качестве твердого вещества белого цвета (70,4% теории).
Амид 5-аминометил-изоксазол-3-карбоновой кислоты
а) Сложный этиловый эфир 5-хлорметил-изоксазол-3-карбоновой кислоты
К охлажденной до 10-15oC смеси 30 г (198 ммоль) сложного этилового эфира 2-хлор-2-гидроксимино-уксусной кислоты и 150 мл пропаргилхлорида прикапывают при перемешивании 21,2 г триэтиламина и дополнительно перемешивают при комнатной температуре в течение часа, затем добавляют воду, экстрагируют диэтиловым эфиром, органическую фазу сушат над сульфатом магния и перегоняют на ротационном испарителе в вакууме. Остаток подвергают перегонке в вакууме при 0,5 Торр, при этом продукт перегоняется при 116 - 122oC.
б) 5-хлорметил-изоксазол-3-карбоновая кислота
К 47,3 г (250 ммоль) сложного этилового эфира 5-хлорметил- изоксазол-3-карбоновой кислоты в 150 мл этанола добавляют 14 г (250 ммоль) гидроксида калия, реакционную смесь перемешивают при 60-70oC в течение 6 ч. После охлаждения сгущают в вакууме, остаток подают в воду и экстрагируют диэтиловым эфиром, водную фазу подкисляют с помощью соляной кислоты, затем экстрагируют несколько раз диэтиловым эфиром, эфирную фазу сушат над сульфатом натрия и сгущают в вакууме, создаваемом с помощью масляного насоса (50oC). Получают 31 г желаемого продукта (77% теории).
в) Хлорид 5-хлорметил-изоксазол-3-карбоновой кислоты
120 г (743 ммоль) 5-хлорметил-изоксазол-3-карбоновой кислоты вместе с 500 мл тионилхлорида и 2 каплями пиридина нагревают с обратным холодильником в течение 10 ч, после этого сгущают в вакууме, а затем подвергают перегонке при 20 Торр. Продукт перегоняется при 125 - 133oC. Получают 78 г (58% теории).
г) Амид 5-хлорметил-изоксазол-3-карбоновой кислоты
В раствор 10 г (55,56 ммоль) хлорида 5-хлорметил-изоксазол- 3-карбоновой кислоты в 100 мл метиленхлорида вводят при 10 - 15oC аммиак в течение часа, а затем при комнатной температуре далее перемешивают в течение часа. После охлаждения раствора до 0oC осадок отсасывают, промывают небольшим количеством холодного хлористого метилена, а остаток для удаления солей аммониевого основания два раза декантируют водой. После сушки в вакууме получают 6,58 г чистого продукта в качестве светлого порошка (74% теории).
д) Гидрохлорид амид 5-аминометил-изоксазол-3-карбоновой кислоты
К смеси 100 мл концентрированного аммиачного раствора и 72 мл метанола добавляют 2,44 г (15,2 ммоль) амида 5-хлорметил- иэоксазол-3-карбоновой кислоты, реакционный раствор нагревают до 40oC и при этом постоянно насыщают аммиачным газом. Спустя 6 ч превращение эдукта завершается. Метанол удаляют в вакууме, водную фазу два раза экстрагируют хлористым метиленом, а затем водную фазу осторожно в вакууме отгоняют на ротационном испарителе досуха. Твердый остаток белого цвета вводят в качестве сырого продукта в сочетание с Вос-дегидропролином.
2-аминометил-тиаэол-4-тиокарбоксамид получают согласно G. Videnov, D. Kaier, C. Kompter и G. Jung, Angew. Chemie, (1996) 108, 1604, при этом описанному там же защищенному N-Boc соединению лишают защиту с помощью эфирной соляной кислоты в хлористом метилене.
4-аминометил-тиазол-2-тиокарбоксамид
Используемый в предварительной стадии сложный этиловый эфир 4-аминометил-тиаэол-2-карбоновой кислоты получают согласно патенту США 4 826 816. После введения Вос-защитной группы на аминофункции эфирную группу подвергают омылению, образовавшуюся кислотную функцию переводят через смешанный ангидрид (сложный изобутиловый эфир угольной кислоты) в амид карбоновой кислоты, а затем с помощью реактива Лавессона - в тиоамид. После отщепления защитной группы получают вышеупомянутое промежуточное соединение.
5-аминометил-2-цианофуран
а) 5-цианофуран-2-карбальдегид
К охлажденному до -70oC раствору 26,7 г (264 ммоль) диизопропиламина в 600 мл тетрагидрофурана добавляют 165 мл (264 ммоль) 1,6 молярного раствора н-бутиллития в н-гексане в течение 20 мин. Раствор доводят до -20oC, снова охлаждают до -75oC и при этой же температуре медленно прикапывают раствор 22,3 г (240 ммоль) 2-цианофурана в 100 мл тетрагидрофурана. Дают дополнительно перемешиваться в течение 30 мин, прикапывают медленно 93 мл диметилформамида и снова дают перемешиваться в течение 30 мин. Для обработки добавляют при температуре -70oC раствор 40 г лимонной кислоты в 200 мл воды. Сгущают на ротационном испарителе, добавляют 600 мл насыщенного раствора хлористого натрия и экстрагируют три раза диэтиловым эфиром, взятым в количестве по 200 мл. Объединенные органические экстракты сушат над сульфатом магния. После отфильтровывания осушителя отгоняют растворитель в вакууме, создаваемом при помощи водоструйного насоса, и остаток очищают путем колоночной хроматографии (растворитель: дихлорметан). Элюат сгущают, а остаток подвергают перегонке с водяным паром (температура кипения азеотропа с водой: 60-65oC при давлении 0,1 мм рт. ст.). После экстрагирования дистиллята с помощью диэтилового эфира, сушки органической фазы и сгущения раствора получают 10,6 г (88 ммоль, 36%) целевого соединения.
1H-ЯМР (270 мГц, d6-ДМСО): δ = 7,7 (д, 1H), 7,8 (д, 1H), 9,75 (с, 1H).
б) 5-гидроксиметил-2-цианофуран
К раствору 30 г (0,25 моль) 5-цианофуран-карбальдегида в 500 мл абсолютного метанола при -30oC добавляют по порциям 2,34 г (62 ммоль) боргидрида натрия. Перемешивают при -30oC в течение 2 ч и доводят охлажденный реакционный раствор с помощью 5%-ного раствора лимонной кислоты в воде до значения pH 7. Реакционную смесь сгущают в вакууме, создаваемом при помощи водоструйного насоса, к остатку добавляют насыщенный раствор хлористого натрия, затем экстрагируют несколько раз диэтиловым эфиром, взятым в количестве по 150 мл, объединенные органические фазы сушат над сульфатом магния, осушитель отфильтровывают, а растворитель отгоняют при комнатной температуре в вакууме, создаваемом при помощи водоструйного насоса. Таким образом получают 27 г (22 ммоль) целевого соединения в качестве темно-красного масла, которое без дальнейшей очистки вводят в последующие реакции.
1H-ЯМР (250 мГц, d6-ДМСО): δ = 4,4 (м,2H), 5,6 (бс, 1H), 6,6 (д, 1H), 7,5 (д, 1H).
в) 5-бромметил-2-цианофуран
К раствору 15 г (121 ммоль) 5-гидроксиметил-2-цианофурана в 250 мл тетрагидрофурана добавляют 38 г (145 ммоль) трифенилфосфина. Охлаждают до -10oC и прибавляют раствор 48 г (145 ммоль) тетробромметана в 100 мл тетрагидрофурана. Дают нагреваться до комнатной температуры и перемешивают при той же температуре в течение 3 ч. Реакционный раствор сгущают на ротационном испарителе в вакууме, создаваемом при помощи водоструйного насоса, и остаток очищают путем колоночной хроматографии (растворитель: смесь петролейного эфира и дихлорметана в соотношении 1:1, Rf = 0,5). Получают 11,5 г целевого соединения.
1H-ЯМР (250 мГц, d6-ДМСО): δ = 4,8 (м, 2H), 6,7 (д, 1H), 7,7 (д, 1H).
г) 5-N,N-бис(трет.-бутоксикарбонил)аминометил-2-цианофуран
К охлажденному до 0oC раствору 22,9 г (123 ммоль) 5-бромметил-2-цианофурана в 400 мл тетрагидрофурана прибавляют по порциям 4,0 г (135 ммоль) гидрида натрия (80%-ная суспензия в минеральном масле). После этого прикапывают раствор 29,4 г (135 ммоль) ди-трет.-бутилиминодикарбоксилата в 200 мл тетрагидрофурана, при этом температура не превышает 5oC. Дают нагреваться до комнатной температуры и перемешивают в течение ночи. Так как по данным тонкослойной хроматографии превращение является незаконченным, дополнительно прибавляют тремя порциями всего 1,2 г гидрида натрия в течение 9 ч. Для завершения превращения нагревают до 35oC в течение дальнейших 3 ч, затем дают охлаждаться до комнатной температуры и добавляют медленно 600 мл насыщенного раствора хлористого алюминия. Растворитель отгоняют в вакууме, создаваемом при помощи водоструйного насоса, остаток несколько раз экстрагируют этилацетатом, объединенные органические фазы промывают насыщенным раствором хлористого натрия, сушат над сульфатом магния и сгущают на ротационном испарителе. Получают 37,3 г маслянистого остатка, который содержит еще ди-трет. -бутил- иминодикарбоксилат и который вводят в качестве сырого продукта в последующую реакцию.
1H-ЯМР (250 мГц, d6-ДМСО): δ = 1,40, 1,45 (с, 18H), 4,75 (с, 2H), 6,55 (д, 1H), 7,55 (д, 1H).
д) 5-аминометил-2-цианофурангидрохлорид
37,3 г 5-N,N-бис(трет.-бутоксикарбонил)аминометил-2- цианофурана (сырой продукт из д), макс. 123 ммоль) растворяют в 600 мл этилацетата и охлаждают до 0oC. Насыщают хлороводородным газом, при этом спустя 30 мин осаждается белый осадок. Доводят до комнатной температуры и перемешивают в течение ночи, затем образовавшуюся суспензию сгущают на ротационном испарителе, остаток декантируют диэтиловым эфиром и отфильтровывают от растворителя, твердый остаток сушат при комнатной температуре в вакууме. Получают 15,1 г (77% выхода через две стадии) целевого соединения в качестве порошка светло-охрового цвета.
1H-ЯМР (250 мГц, d6-ДМСО): δ = 4,15 (бс, 2H), 6,85 (д, 1H), 6,65 (д, 1H), 8,8 - 9,0 (бс, 3H).
5-аминометил-3-цианофуран
а) Сложный этиловый эфир 4-оксопентановой кислоты
100 г (0,86 моль) 4-оксопентановой кислоты, 150 г этанола и 1 мл серной кислоты в 200 мл бензола нагревают с обратным холодильником, пока не будет закончено водоотделение в ловушке Деан-Штарка. Охлажденную реакционную смесь промывают водой, раствором карбоната натрия и снова водой, а затем с обратным холодильником сушат при помощи ловушки Деан-Штарка. После завершения отделения водяной фазы растворитель отгоняют и остаток перегоняют при пониженном давлении. Точка кипения: 85 - 87oC/16 мм рт. ст. Выход: 105,5 г (85%).
б) Сложный этиловый эфир 4,4-диэтоксипентановой кислоты
Смесь 171,3 г (1,19 моль) сложного этилового эфира 4- оксопентановой кислоты, 207 мл (184,2 г, 1,24 моль) триэтилортоформиата, 26 мл абсолютного этанола и 1 г n- толуолсульфоновой кислоты при прочном перемешивании нагревают с обратным холодильником в течение 8 ч и затем перегоняют в вакууме. Получают 187,9 г (72,5%) сложного этилового эфира 4,4- ди-этоксипентановой кислоты. Точка кипения: 104 -106oC/14 мм рт. ст.
в) Смесь 106,3 г (0,489 моль) сложного этилового эфира 4,4- диэтоксипентановой кислоты и 80 мл (73,6 г, 0,99 моль) этилформиата при интенсивном перемешивании и 10-15oC добавляют каплями к суспензии 12,7 г (0,55 грамм-атом) (тонкослоистого) натрия в 300 мл безводного бензола в течение 3 ч. Перемешивание продолжают в течение дальнейших 3 ч и реакционной смеси дают стоять в течение ночи. Добавляют 250 мл воды при интенсивном перемешивании, которое продолжают в течение дальнейших 15 мин. Водяной слой отделяют, а бензольный слой экстрагируют 70 мл воды. Объединенные водяные экстракты подкисляют до значения pH 2, затем их экстрагируют пять раз этилацетатом, взятым в количестве по 50 мл, органические экстракты сушат над хлоридом кальция. Раствор этилацетата перегоняют в вакууме, собирают фракцию с точкой кипения 102-110oC/ 1 мм рт. ст. Эта фракция представляет собой смесь сложного этилового эфира 2-формиллевулиновой кислоты и его диэтилкеталя. Соотношение компонентов смеси зависит от интенсивности перемешивания и времени разделения фаз при выделении продуктов формилирования.
Бензольный слой сушат также над хлоридом кальция, растворитель удаляют и остаток перегоняют при пониженном давлении. После этого полученную смесь сложного этилового эфира левулиновой кислоты и кеталя обрабатывают вместо чистого сложного эфира левулиновой кислоты таким же образом, как описывается под б). Стадии (2) и (3а) повторяют, пока не будет получено требуемое количество сложного этилового эфира 2-формиллевулиновой кислоты.
г) Сложный этиловый эфир 5-метилфуран-3-карбоновой кислоты
Указанную выше смесь сложного этилового эфира 2-формиллевулиновой кислоты и его диэтилкеталя растворяют в бензоле, затем прибавляют катализатор, и получаемый раствор оставляют с обратным холодильником в ловушке Деан-Штарка в течение 3-3 1/2 ч, пока не вода будет полностью удалена. После этого реакционную смесь перегоняют при пониженном давлении, при этом получают 15 г (97 ммоль) сложного этилового эфира 5-метилфуран-3-карбоновой кислоты с точкой кипения 97oC/15 мм рт.ст.
д) 5-метилфуран-3-карбоновая кислота
Смесь 31,7 г (206 ммоль) сложного этилового эфира 5-метилфуран-3-карбоновой кислоты, 40 мл 45%-ного гидроокиси калия и 100 мл воды при перемешивании нагревают с обратным холодильником в течение 4 ч, затем ее охлаждают до 10oC и с помощью 15%-ной соляной кислоты подкисляют до значения pH 1. Образовавшуюся реакционную смесь дают стоять при той же температуре в течение 2 ч, преципитат отфильтровывают и при 45-50oC сушат до постоянства веса, при этом получают 23,7 г (188 ммоль, 91%) 5- метилфуран-3-карбоновой кислоты.
е) Хлорид 5-метилфуран-3-карбоновой кислоты
К суспензии 23,7 г (188 ммоль) 5-метилфуран-3-карбоновой кислоты в 100 мл бензола добавляют при перемешивании небольшими количествами 39,2 г (188 ммоль) пентахлорида фосфора, результатом чего являются значительное выделение тепла и образование хлористого водорода. Образовавшуюся смесь оставляют при перемешивании с обратным холодильником в течение 4 ч, а затем ее перегоняют в вакууме. Получают 24,7 г (171 ммоль, 91%) хлорангидрида кислоты. Точка кипения: 79oC/12 мм рт.ст.
ж) Амид 5-метилфуран-3-карбоновой кислоты
24,7 г (171 ммоль) хлорида 5-метилфуран-3-карбоновой кислоты при перемешивании добавляют при 25-40oC каплями к смеси 80 мл 25 %-ного раствора гидроокиси аммония и 80 мл бензола. Образовавшуюся смесь перемешивают в течение 3 ч и дают стоять в течение ночи. В последующий день отфильтровывают белые амидные кристаллы, которые промывают холодной водой и сушат при 40-45oC до постоянства веса. Выход: 19,7 г (158 ммоль, 92%), точка плавления: 158oC.
з) 5-метил-3-цианофуран
К суспензии 19,7 г (158 ммоль) амида 5-метилфуран-3- карбоновой кислоты в 100 мл бензола прибавляют при 30-40oC небольшими количествами 32,9 г пентахлорида фосфора. Образовавшуюся смесь перемешивают с обратным холодильником до осветления (3 1/2-4 ч), а затем перегоняют при пониженном давлении. Собирают фракцию с точкой кипения 79-140oC/15 мм рт.ст. В результате второй перегонки получают 12,7 г (119 ммоль, 75%) целевого соединения, точка кипения: 79-80oC/15 мм рт.ст.
и) 5-бромметил-3-фуранкарбонитрил
12,7 г (119 ммоль) 5-метил-3-цианофурана растворяют в 100 мл тетрахлорметана, прибавляют 22 г (122 ммоль) N-бромсукцинимида и 12 г (73 ммоль) азобисизобутиронитрила. Когда начинается экзотермическая реакция, образовавшуюся смесь при интенсивном перемешивании нагревают до 70oC. После окончания выделения тепла реакционную смесь при 80oC перемешивают в течение 3 ч и затем охлаждают до комнатной температуры. Образовавшийся сукцинимид отфильтровывают и два раза промывают на фильтре тетрахлорметаном, взятым в количестве по 15 мл.
Растворитель удаляют при пониженном давлении, остаток перегоняют в вакууме, при этом получают 12,7 г (86 ммоль, 57%) 5- бромметил-3-фуранкарбонитрила с точки кипения 105oC/1 мм рт.ст. По спектру 1H-ЯМР соединение содержит загрязнения, которые при δ 1,3 и 2,2 создают сигналы. После второй перегонки содержание является более низким и удовлетворительным, однако в результате потеряют примерно 15% продукта. 5-бромметил-3-фуранкарбонитрил представляет собой кристаллическое вещество белого цвета. Точка кипения: 40 - 45oC, 1H-ЯМР (CDCl3, млн.дол.): 4,41 (2H, CH2), 6,85 (1H, H4), 7,92 (1H, H2): 13C-ЯМР (CDCl3, млн.дол.): 20,86 (CH2), 99,03 (CN или меньше, по вероятности C3), 109,97 (C4), 112,27 C3 (или CN)), 149,84 (C2), 152,32 (C5).
Реакционный продукт обладает сильной раздражающей способностью, вследствие чего требуется чрезвычайно осторожное обращение с ним.
Синтез 5-N,N-,бис(трет.-бутоксикарбонил)аминометил-3- цианофурана осуществляют аналогично синтезу 5-N,N-,бис(трет.- бутоксикарбонил)аминометил-2-цианофурана. Последующее отщепление трет.-бутоксикарбонильных групп проводят в насыщенном растворе хлористого водорода в хлороформе.
Гидроацетат 2-амидино-5-(N-Вос-аминометил)-1-метилпиррола
а) 5-циано-1-метилпиррол-2-карбальдегид
1-метилпиррол можно переводить в 2-циано-1-метилпиррол путем взаимодействия с хлорсульфонилизоцианатом и диметилформамидом в ацетонитриле (см., например, C.E. Loader и др., Can. J. Chem. (1981), 59, 2673-6).
К 17,5 мл (124,38 ммоль) диизопропиламина в атмосфере азота в 100 мл тетрагидрофурана прикапывают при -78oC 75,9 мл (124,38 ммоль) 15%-ного раствора н. -бутиллития в гексане. После этого перемешивают при -20oC в течение 45 мин, а затем охлаждают до -78oC. При той же температуре прикалывают раствор 12 г (113,07 ммоль) N-метилпиррол-2-карбонитрила в 50 мл тетрагидрофурана. После перемешивания при -78oC в течение 45 мин прикапывают 43,9 мл (546,46 ммоль) диметилформамида и перемешивают при той же температуре в течение дальнейших двух ч. После добавки 20,56 г моногидрата лимонной кислоты нагревают до комнатной температуры и к смеси прибавляют 112 мл воды. Тетрагидрофуран отгоняют на ротационном испарителе, водную фазу насыщают хлоридом натрия и три раза экстрагируют диэтиловым эфиром, взятым в количестве по 200 мл. Объединенные органические фазы промывают насыщенным раствором хлорида натрия и сушат над сульфатом натрия. Растворитель
отгоняют на ротационном испарителе, а сырой продукт очищают путем хроматографии на силикагеле с применением в качестве элюента дихлорметана. Выход: 8,25 г (54%). 1H-ЯМР (CDСl3) δ = 4,1 (с, 3H), 6,8 (д, 1H), 6,9 (д, 1H), 9,7 (с, 1H).
б) 5-гидроксиметил-1-метилпиррол-2-карбонитрил
Полученные согласно а) продукт (8,2 г, 61,1 ммоль) растворяют в 200 мл этанола, затем при -10oC к нему добавляют 2,31 г (61,13 ммоль) боргидрида натрия. После перемешивания при 0-5oC в течение полтора часа растворитель отгоняют на ротационном испарителе и к остатку прибавляют ледяную воду и 20%-ный раствор гидросульфата натрия. Водную фазу экстрагируют этилацетатом. Объединенные органические фазы подвергают промывке насыщенным раствором гидрокарбоната натрия и водой и сушат над сульфатом натрия. Растворитель отгоняют на ротационном испарителе и сырой продукт очищают путем хроматографии на силикагеле с применением в качестве элюента смеси дихлорметана и метанола в соотношении 97,5: 2,5. Выход: 7,6 г (91%).
1H-ЯМР (CDCl2) δ = 1,9 (т, 1H), 3,75 (с, 3H), 4,6 (д, 2H), 6,1 (д, 1H), 6,7 (д, 1H).
в) 5-азидометил-1-метилпиррол-2-карбонитрил
Полученный согласно б) продукт (7,5 г, 55,08 ммоль) растворяют в 220 мл диметилформамида, затем при 0oC прибавляют 43,34 г (165,25 ммоль) трифенилфосфина. При той же температуре перемешивают в течение 5 мин, после чего добавляют 54,8 г (165,25 ммоль) тетрабромметана. Затем перемешивают при 0oC в течение 30 мин, и при комнатной температуре в течение 90 мин. После охлаждения до 0oC добавляют 4,37 г (67,21 ммоль) азида натрия. После этого перемешивают при комнатной температуре в течение четырех с половиной часов. Прикапывают при 0oC насыщенный раствор хлорида натрия, затем исходную смесь разбавляют этилацетатом. Органическую фазу отделяют, а водную фазу экстрагируют диэтиловым эфиром. Объединенные органические фазы промывают водой и сушат над сульфатом натрия. Растворитель отгоняют на ротационном испарителе и сырой продукт очищают путем хроматографии на силикагеле с применением в качестве элюента смеси этилацетата и гексана в соотношении 1: 20. Выход: 5,6 г (63%).
1H-ЯМР (CDCl3) δ 3,75 (с, 3H), 4,35 (с, 2H), 6,2 (д, 1H), 6,7 (д, 1H).
г) 5-аминометил-1-метилпиррол-2-карбонитрил
Полученный согласно в) продукт (4,71 г, 29,75 ммоль) растворяют в 100 мл метанола, к смеси добавляют 1 г палладия на угле (10%). Затем гидрируют под давлением одной атмосферы с помощью водорода в течение 4 ч. Катализатор фильтруют на кизельгуре марки Celite®, , а фильтрат перегоняют на ротационном испарителе. Остаток декантируют смесью дихлорметана и диэтилового эфира в соотношении 1:1. Продукт отсасывают и сушат при 35oC в вакуумном сушильном шкафу. Выход: 2,7 г (68%).
1H-ЯМР (CDCl3) δ = 3,75 (с, 3H), 3,85 (с, 2H), 6,05 (д, 1H), 6,7 (д, 1H).
д) 5-(N-Вос-аминометил)-1-метилпиррол-2-карбонитрил
Полученный согласно г) продукт (2,7 г, 19,97 ммоль) растворяют в 50 мл дихлорметана, к смеси добавляют 2,8 мл (19,97 ммоль) триэтиламина. После этого прикапывают раствор 4,36 г (19,97 ммоль) ди-трет.-бутилдикарбоната в 30 мл дихлорметана. После перемешивания при комнатной температуре в течение 2 ч прибавляют воду, и водную фазу экстрагируют ди-хлорметаном. Объединенные органические фазы сушат над сульфатом натрия и перегоняют на ротационном испарителе. Сырой продукт без дальнейшей очистки вводят в последующую реакцию. Выход: 4,4 г (94%).
1H-ЯМР (CDCl3) δ = 1,45 и 1,55 (в кажд. случае с, вместе 9H), 3,7 (с, 3H), 4,3 (д, 2H), 4,7 (шир.с, 1H), 6,05 (д. 1H), 6,7 (д, 1H).
е) 5-(N-Вос-аминометил)-1-метилпиррол-2-гидроксиамидин
Полученный согласно д) продукт (4,3 г, 18,27 ммоль) растворяют в 100 мл смеси метанола и дихлорметана в соотношении 1:1, добавляют 3,17 г (45,61 ммоль) гидроксиламингидрохлорида. Затем прикапывают при комнатной температуре 19,1 мл (109,65 ммоль) этилдиизопропиламина. Перемешивают при 40oC в течение 12 ч. После этого растворитель отгоняют на ротационном испарителе, к остатку прибавляют воду, затем его с помощью уксусной кислоты подкисляют до значения pH 5 и экстрагируют дихлорметаном и этилацетатом. Объединенные органические фазы сушат над сульфатом натрия и перегоняют на ротационном испарителе. Сырой продукт очищают путем хроматографии на силикагеле с применением в качестве элюента смеси дихлорметана и метанола в соотношении 95:5. Выход: 3,4 г (69%).
1H-ЯМР (CDCl3) δ = 1,4 (с, 9H), 3,7 (с, 3H), 4,3 (д, 2H), 4,7 - 4,9 (м, 3H), 6,05 (д, 1H), 6,3 (д, 1H), 7,3 (шир.с, 1H).
ж) Гидроацетат 2-амидино-5-(N-Вос-аминометил)-1-метилпиррола
Полученный согласно е) продукт (3,4 г, 12,67 ммоль) растворяют в 150 мл метанола, добавляют 1,45 г (25,31 ммоль) уксусной кислоты и 421 мг никеля Ренея. Затем гидрируют при 50oC под давлением одной атмосферы с помощью водорода в течение 5 ч. После охлаждения до комнатной температуры катализатор фильтруют на кизельгуре марки Celite®, и фильтрат сгущают. Полученный таким образом продукт без дальнейшей очистки вводят в последующую реакцию. Выход: 3,7 г (94%).
Масс-спектр: бомбардировка быстрыми атомами (M+H+):253.
Гидроацетат 2-амидино-4-(N-Вос-аминометил)-1-метилпиррола
а) 5-циано-1-метилпиррол-3-карбальдегид
24,24 г (180,86 ммоль) трихлорида алюминия растворяют в 320 мл смеси нитрометана и дихлорметана в соотношении 1:1 и охлаждают до -20oC, к смеси добавляют 8 г (75,36 ммоль) 1-метилпиррол-2-карбонитрила. Затем прикапывают 10,4 г (90,43 ммоль) простого α,α -дихлордиметилэфира, растворенного в 42 мл дихлорметана. После перемешивания при 0oC в течение 4 ч исходную смесь наливают на лед (200 г). Водную фазу экстрагируют диэтиловым эфиром. Объединенные органические фазы подвергают нейтрализующей промывке с применением насыщенного раствора гидрокарбоната натрия, воды и насыщенного раствора хлорида натрия. После сушки над сульфатом натрия растворитель отгоняют на ротационном испарителе. Сырой продукт без дальнейшей очистки вводят в последующую реакцию. Выход: 9,2 г (91%).
1H-ЯМР (CDCl3) δ = 3,8 (с, 3H); 7,2 (с, 1H); 7,4 (с, 1H): 9,85 (с, 1H).
б) Исходя из 5-циано-1-метилпиррол-3-карбальдегида получают 4-аминометил-1-метилпиррол-2-карбонитрил аналогично синтезу 5- аминометил-1-метилпиррол-2-карбонитрила. Однако восстановление 4-азидометил-1-метилпиррол-2-карбонитрила осуществляют предпочтительно через реакцию Штаудингера (см., напр., S. Nagarajan и др., J. Org. Chem., 1987, 52, 5044-6).
в) Исходя из 4-аминометил-1-метилпиррол-2-карбонитрила получают гидроацетат 2-амидино-4-(N-Вос-аминометил)-1- метилпиррола аналогично синтезу гидроацетата 2-амидино-5-(N-Вос-аминометил)-1-метилпиррола.
Масс-спектр: бомбардировка быстрыми атомами (M+H+): 253.
Гидроацетат 2-амидино-(N-Вос-аминометил)-1-метилпиррола
а) 5-циано-1-метилпиррол-2-карбальдегид
10 г (91,6 ммолль) 1-метилпиррол-2-карбальдегида растворяют в 100 мл ацетонитрила и охлаждают до -45oC. Прикапывают 38,9 г (274,9 ммоль) хлорсульфонилизоцианата в 40 мл ацетонитрила в течение 40 мин. Затем перемешивают при комнатной температуре в течение 12 ч. После добавки каплями 35 мл диметилформамида нагревают до 50oC в течение часа. После охлаждения до комнатной температуры реакционную смесь подают на 200 мл льда и 286 мл 2N натрового щелока. Образовавшийся осадок отсасывают. Фильтрат экстрагируют диэтиловым эфиром. Объединенные эфирные фазы подвергают нейтрализующей промывке с применением разбавленного раствора гидрокарбоната натрия и воды и сушат над сульфатом натрия. Растворитель отгоняют в вакууме, создаваемом при помощи водоструйного насоса, и остаток объединяют с заранее полученным осадком. В результате перекристаллизации из петролейного эфира получают 4,3 г 4-циано-1-метилпиррол-2- карбальдегида (см. , напр., C.E, Loader и др. Can. J. Chem. (1981), 59,2673-6).
1H-ЯМР (CDCl3) δ = 4,0 (с, 3H); 7,2 (с, 1H); 7,3 (с, 1H); 9,6 (с, 1H).
13C-ЯМР (CDCl3) δ = 37,4; 94,1; 114,7; 125,8; 132,2; 135,8; 179,7.
б) Исходя из 4-циано-1-метилпиррол-2-карбальдегида получают 5-амино-метил-1-метилпиррол-3-карбонитрил аналогично синтезу гидроацетата 2-амидино-5-(N-Вос-аминометил)-1-метилпиррола.
1H-ЯМР (ДМСО-d6) δ = 3,6 (с, 3H): 3,8 (с, 2H); 4,2 (шир.с, 2H): 6,4 (с, 1H), 7,6 (с, 1H).
в) Исходя из 5-аминометил-1-метилпиррол-3-карбонитрила получают гидроацетат 3-амидино-5-(N-Вос-аминометил)-1- метилпиррола аналогично синтезу гидроацетата 2-амидино-5-(N-Вос-аминометил)-1-метилпиррола.
Масс-спектр: бомбардировка быстрыми атомами (M+H+):253.
Гидрохлорид 5-аминометил-3-циано-1,2,4-оксадиазола
а) N-Вос-5-аминометил-3-циано-1,2,4-оксадиазол
Сложный этиловый эфир N-Вос-5-аминометил-3-циано- 1,2,4-оксадиазол-2-карбоновой кислоты (S. Borg и др., J. Org. Chem. 1995, 60, 3112-20) растворяют в 50 мл метанола. В этот раствор при -10oC до комнатной температуры подают аммиак до завершения реакции. Растворитель отгоняют на ротационном испарителе. Полученный таким образом сырой продукт растворяют в 70 мл дихлорметана и к этому раствору при -5oC добавляют 2,9 мл (16,55 ммоль) диизопропилэтиламина. Затем прикапывают 1,06 мл (7,61 ммоль) ангидрида трифторуксусной кислоты, растворенного в 10 мл дихлорметана. После перемешивания при 0oC в течение полтора часа исходную смесь разбавляют дихлорметаном, экстрагируют 2 раза насыщенным раствором гидрокарбоната натрия, 2 раза 5%-ным раствором лимонной кислоты и раз насыщенным раствором хлористого натрия, а затем ее сушат над сульфатом натрия. Растворитель отгоняют на ротационном испарителе, а сырой продукт очищают путем хроматографии на силикагеле с применением смеси дихлорметана с метанолом в соотношении 97,5:2,5. Выход 1,2 г (80%).
б) Гидрохлорид 5-аминометил-3-циано-1,2,4-оксадиазола
Полученный согласно а) продукт (0,9 г, 4,0 ммоль) растворяют в 45 мл дихлорметана, к раствору при комнатной температуре добавляют 3,9 мл (15,61 ммоль) 4 М соляной кислоты в диоксане. Перемешивают при комнатной температуре в течение 16 ч, затем растворитель отгоняют на ротационном испарителе. Выход: 645 мг (100%).
1H-ЯМР (ДМСО-d6) δ = 4,6 (с, 2H); 9,2 (с, 3H).
Амид N-метил-5-аминометил-пиразол-3-карбоновой кислоты
а) Сложный метиловый эфир N-метил-5-амидо-пиразол-3-карбоновой кислоты
Хлорид N-метил-3-метоксикарбонил-пиразол-5-карбоновой кислоты (полученный из 3,7 г (20,09 ммоль) N-метил-3-метоксикарбонил-3-карбоновой кислоты, см. J. Org. Chem. 1989, 54, 428) растворяют в толуоле и охлаждают до -10oC. Затем при -10oC до 0oC вводят аммиак до завершения реакции. Растворитель отгоняют на ротационном испарителе. Остаток подают в этанол. Перемешивают в течение 15 мин, затем этанол отгоняют на ротационном испарителе, остаток растворяют в теплой воде и дают осаждаться путем охлаждения до 0oC. Осадок отсасывают, промывают ацетоном и сушат в вакууме при 45oC. Выход: 1,5 г (41%).
б) Сложный метиловый эфир N-метил-5-циано-пиразол-3-карбоновой кислоты
Полученный согласно а) продукт (1,5 г, 8,19 ммоль) подают в 20 мл дихлорметана. При -10oC прибавляют 3,85 мл (22,11 ммоль) диизопропилэтиламина и при той же температуре прикапывают раствор 1,3 мл (9,94 ммоль) ангидрида трифторуксусной кислоты в 5 мл дихлорметана в течение 45 мин. После этого перемешивают при 0oC в течение дальнейшего часа. Исходную смесь разбавляют дихлорметаном и промывают два раза насыщенным раствором гидрокарбоната натрия, 2 раза 5%-ным раствором лимонной кислоты и раз насыщенным раствором хлористого натрия. После сушки над сульфатом натрия растворитель отгоняют на ротационном испарителе. Выход: 1,35 г (100%).
Масс-спектр: электронная ионизация (M+):165.
в) Амид N-метил-5-циано-пиразол-3-карбоновой кислоты
Полученный согласно б) продукт (1,35 г, 8,19 ммоль) подают в 50 мл метанола и охлаждают до -10oC. Затем вводят аммиак в течение 8 ч. После перемешивания при комнатной температуре в течение 12 ч эдукт прореагировал. Осажденный продукт отсасывают, промывают холодным метанолом и сушат в вакууме. Выход: 1,22 г (100%).
1H-ЯМР (ДМСО-d6) δ = 4,0 (с, 3H): 7,4 (с, 1H); 7,5 (с, 1H): 7,8 (с, 1H).
г) Амид N-метил-5-аминометил-пиразол-3-карбоновой кислоты
Полученный согласно в) продукт (0,4 г, 2,66 ммоль) растворяют в 30 мл уксусной кислоты, к раствору добавляют 78 мг 10%-ного палладия на угле. Затем при комнатной температуре и атмосферном давлении гидрируют, пока не будет полностью завершена реакция. Катализатор фильтруют на кизельгуре марки Celite®, и растворитель отгоняют на ротационном испарителе. Выход: 0,4 г (100%).
Масс-спектр: электронная ионизация (M+): 154.
Пример 1:
Гидроацетат N-(гидроксикарбонил-метилен)-(D)- циклогексилаланил-3,4-дегидропролил-5-(2-амидино)-тиенилметиламида
а)3,4-дегидропролил-5-(2-циано)-тиенилметиламид
5 г (23,4 ммоль) Вос-3,4-дегидропролила и 4,5 г (25,8 ммоль) гидрохлорида 5-аминометил-2-цианотиофена растворяют в 25 мл дихлорметана. К раствору при 0oC добавляют 28 мл (163,8 ммоль) этилдиизопропиламина с 50%-ным раствором ангидрида пропанфосфоновой кислоты в 24 мл (117 ммоль) этилацетата. Перемешивают при 0oC в течение часа, затем нагревают до комнатной температуры и дополнительно перемешивают при комнатной температуре в течение 12 ч. Реакционную смесь разбавляют ди-хлорметаном и промывают 4 раза раствором гидросульфата натрия, 3 раза раствором гидрокарбоната натрия и раз насыщенным раствором хлористого натрия. После сушки над сульфатом натрия и отфильтровывания осушителя растворитель отгоняют в вакууме, создаваемом при помощи водоструйного насоса. Для отщепления Вос-группы к остатку прибавляют 95 мл дихлорметана, затем перемешивают при комнатной температуре, упаривают досуха, два раза подвергают совместной перегонке с дихлорметаном, снова сгущают и очищают путем колоночной хроматографии. Получают 6,6 г желаемого продукта, содержащего еще небольшое количество растворителя.
б) N-(трет. -бутоксикарбонил-метилен)-(N-Вос)- (D)-циклогексилаланил-3,4-дегидропролил-5-(2-циано)-тиенилметиламид
7,3 г (18,98 ммоль) t-BuO2C-Boc-(D)-Cha-OH и 5,12 г (18,98 ммоль) H-Pyr-NH-CH2-5-(2-CN)-tioph-гидрохлорида растворяют в 100 мл дихлорметана. К раствору добавляют 12,26 г (94,9 ммоль) этилдиизопропиламина. Реакционную смесь охлаждают до 0oC, затем прикапывают 20 мл 50%-ный раствор ангидрида пропанфосфоновой кислоты в этилацетате. Перемешивают при 0-10oC в течение 3 ч, затем разбавляют 100 мл дихлорметана и промывают 3 раза разбавленным раствором гидросульфата натрия, 2 раза насыщенным раствором гидрокарбоната натрия и раз водой. После сушки над сульфатом натрия и отделения осушителя растворитель отгоняют в вакууме, создаваемом при помощи водоструйного насоса. Получают 12,47 г коричневатого масла.
в) N-(трет. -бутоксикарбонил-метилен)-(N-Boc)-(D)- циклогексилаланил-3,4-дегидропролил-5-(2-амидотиокарбонил) -тиенилметиламид
Полученный согласно б) продукт растворяют в 70 мл пиридина и 12 мл триэтиламина. Реакционную смесь охлаждают до 0oC и насыщают сероводородом (раствор принимает зеленый цвет). После этого при комнатной температуре дополнительно перемешивают в течение 48 ч. Избыточный сероводород вытесняют с помощью азота, и растворитель отгоняют в вакууме, создаваемом при помощи водоструйного насоса. Остаток растворяют в 200 мл диэтилового эфира, промывают 2 раза насыщенным раствором гидросульфата натрия, 2 раза насыщенным раствором гидрокарбоната натрия и раз водой. После сушки над сульфатом натрия растворитель отгоняют в вакууме, создаваемом при помощи водоструйного насоса. Сырой продукт (12,6 г) очищают путем хроматографии на силикагеле. Градиент: дихлорметан до смеси дихлорметана с метанолом в соотношении 40:1. Получают 12,1 г желаемого продукта, содержащего еще небольшое количество растворителя.
г) Гидройодид N-(трет.-бутоксикарбонил-метилен)-(N-Вос)-(D)- циклогексилаланил-3,4-дегидропролил-5-(2-S-метилимидокарбонил)- тиенилметиламида
Полученный согласно в) продукт растворяют в 120 мл дихлорметана, к раствору добавляют 16,24 г (114,38 ммоль) метилйодида. Перемешивают при комнатной температуре в течение 12 ч, затем растворитель отгоняют в вакууме, создаваемом при помощи водоструйного насоса. Получают 14,6 г желтоватого масла.
д) Гидроацетат N-(трет.-бутоксикарбонил-метилен)-(N-Вос)-(D)- циклогексилаланил-3,4-дегидропролил-5-(2-амидино)-тиенилметиламида
Полученный согласно г) сырой продукт растворяют в 90 мл ацетонитрила, к раствору добавляют 2,94 г (38,12 ммоль) ацетата аммония. Перемешивают при комнатной температуре в течение 2 ч и при 40oC в течение полтора часа, затем прибавляют 14,65 г (19,05 ммоль) раствора ацетата аммония в метаноле. При 50oC перемешивают в течение дальнейших четырех с половиной часов и отгоняют растворитель в вакууме, создаваемом при помощи водоструйного насоса. К остатку добавляют дихлорметан, соли отсасывают и фильтрат сгущают. Остаток через ионообменних (фирмы Флюка, зак. N 00402) переводят в ацетатную соль. Таким образом получают 11,15 г желтоватого масла.
е) Гидроацетат N-(гидроксикарбонил-метилен)- (D)-циклогексилаланил-3,4-дегидропролил-5-(2-амидино)-тиенилметиламида
Полученный согласно д) продукт растворяют в 175 мл дихлорметана, затем каплями прибавляют 38,3 мл раствора эфирной соляной кислоты. Перемешивают при комнатной температуре в течение 2 ч, после этого растворитель 2 раза отгоняют в вакууме, создаваемом при помощи водоструйного насоса. Сырой продукт очищают при помощи ионообменника (фирмы Флюка, зак. N 00402) путем последующей хроматографии на силикагеле. Градиент: смесь дихлорметана с метанолом в соотношении 4:1, смесь дихлорметана, метанола и 50%-ной уксусной кислоты в соотношении 40:10:2 до смеси дихлорметана, метанола и 50%-ной уксусной кислоты в соотношении 35:15:5. Полученный таким образом продукт растворяют в воде. Отфильтровывают от нерастворимых составных частей, и фильтрат лиофилизируют. При этом получают 5,55 г аморфного твердого вещества белого цвета.
Масс-спектр; бомбардировка быстрыми атомами (M+H+): 462
Аналогично примеру 1 получают следующие соединения:
Пример 2:
Гидроацетат N-(гидроксикарбонил-этилен)-(D)- циклогексилаланил-3,4-дегидропролил-5-(2-амидино)- тиенилметиламида
Масс-спект: бомбардировка быстрыми атомами (M+H+): 476
Получение осуществляют через несколько стадий аналогично примеру 1, исходя из N-трет. -бутоксикарбонил-этилен)-(N-Вос)-(D)- циклогексилаланина и 3,4-дегидропролил-5-(2-циано)-тиенилметиламида.
Пример 3:
Гидроацетат N-(гидроксикарбонил-метилен)-(D)- циклогексилглицил-3,4-дегидропролил-5-(2-амидино)-тиенилметиламида
Масс-спектр; бомбардировка быстрыми атомами (M+H+): 448
Получение осуществляют через несколько стадий аналогично примеру 1, исходя из N-трет. -бутоксикарбонил-метилен)-(N-Вос)-(D)- циклогексилглицина и 3,4-дегидропролил-5-(2-циано)-тиенилметиламида.
Пример 4:
Гидроацетат N-(гидроксикарбонил-метилен)-(D)- циклогексилаланил-3,4-дегидропролил-5-(2-амидино-3,4-диметил)- тиенилметиламида
Масс-спектр: бомбардировка быстрыми атомами (M+H+): 490
Получение осуществляют аналогично примеру 1, исходя из амида 5-аминометил-3,4-диметил-тиофен-2-карбоновой кислоты путем сочетания с Вос-3,4-дегидропролином до получения Вос-3,4- дегидропролил-5-(2-карбамоил-3,4-диметил)-тиенилметиламида. После отщепления Вос-защитной группы этот структурный элемент аналогично примеру 1 сочетают с N-(трет.-бутоксикарбонил-метилен)-(N-Вос)- (D)-циклогексилаланина. Дегидратизацию амидной функции до нитрильной функции осуществляют по следующему способу:
4,8 г (7,42 ммоль) N-(трет.-бутоксикарбонил-метилен)-(N-Вос)- (D)-циклогексилаланил-3,4-дегидропролил-5-(2-карбамоил-3,4- диметил)-тиенилметиламида растворяют в 60 мл хлористого метилена, к раствору добавляют 3,83 г (29,64 ммоль) диизопропилэтиламина и охлаждают до 0oC. Затем медленно прикапывают 2,8 г ангидрида трифторуксусной кислоты в 3 мл хлористого метилена и дополнительно перемешивают при 0-5oC в течение 2 ч. После этого разбавляют при помощи 60 мл хлористого метилена, и реакционную смесь последовательно промывают 3 раза 20%-ной лимонной кислотой, взятой в количестве по 20 мл, два раза насыщенным раствором гидрокарбоната натрия, взятым в количестве по 20 мл, и два раза насыщенным раствором поваренной соли, фазу хлористого метилена сушат над сульфатом натрия и перегоняют на ротационном испарителе в вакууме. Получают 5,35 г содержащего еще небольшое количество растворителя N-(трет. -бутоксикарбонил-метилен)-(N-Вос)-(D)- циклогексилаланил-3,4-дегидропролил-5- (2-циано-3,4-диметил)-тиенилметиламида, который непосредственно вводят в последующую стадию.
Превращение нитрильной функции до амидиновой группы и последующее отщепление защитной группы осуществляют аналогично примеру 1.
Пример 5:
Гидроацетат N-(гидроксикарбонил-метилен)-(D)-циклогексилаланил- 3,4-дегидропролил-5-(3-амидино)-тиенилметиламида
Масс-спектр; бомбардировка быстрыми атомами (M+H+): 462
Получение осуществляют аналогично примеру 1, при этом вместо гидрохлорида 5-аминометил-2-цианотиофена применяют гидрохлорид 5- аминометил-3-цианотиофена.
Пример 6:
Гидроацетат N-(гидроксикарбонил-метилен)-(D)- циклогексилаланил-3,4-дегидро-пролил-4-(2-амидино)- тиенилметиламида
Масс-спектр: бомбардировка быстрыми атомами (M+H+): 462
Получение осуществляют аналогично примеру 1, при этом вместо гидрохлорида 5-аминометил-2-цианотиофена используют гидрохлорид 4- аминометил-2-цианотиофена.
Пример 7а:
Гидроацетат N-(1-гидроксикарбонил-метилен)-(D)- циклогексилаланил-(D)-4,5-дегидропипецолил-5-(2-амидино)- тиенилметиламида
Масс-спектр: бомбардировка быстрыми атомами (М+H+: 476
Пример 7б:
Гидроацетат N-(гидроксикарбонил-метилен)-(D)- циклогексилаланил-4,5-дегидропипеколил-5-(2-амидино)- тиенилметиламида
Масс-спектр: бомбардировка быстрыми атомами (М+H+): 476
Получение осуществляют аналогично примеру 1, при этом вместо Вос-3,4-дегидропролина используют рацемическую Boc-(D, L)-4,5- дегидропипеколиновую кислоту. В стадии N-(трет.-бутоксикарбонилметилен)-(N-Вос)-(D)-циклогексилаланил-(D, L)-4,5- дегидропипеколил-5-(2-циано)-тиенилметиламида можно разделять оба диастереомерных соединения путем хроматографии на силикагеле с применением в качестве элюента смеси циклогексана со сложным этиловым эфиром уксусной кислоты в соотношении 7:3. Оба диастереомера затем переводят аналогично примеру 1 в целевые продукты.
Пример 8:
Гидроацетат N-(гидроксикарбонил-метилен)-(D)- циклогексилглицил-3,4-дегидропролил-5-(3-амидино)- тиенилметиламида
Масс-спектр: бомбардировка быстрыми атомами (М+H+): 448
Получение осуществляют аналогично примеру 1, при этом исходят из эдуктов 5-аминометил-3-цианотиофен и N-(трет.- бутоксикарбонилметилен)-N-Вос-(D)-циклогексилглицил-3,4- дегидропролин.
Пример 9:
Гидроацетат N-(гидроксикарбонил-метилен)-(D)- циклогексилглицил-3,4-дегидропролил-4-(2-амидино)- тиенилметиламида
Масс-спектр: бомбардировка быстрыми атомами (М+H+): 448
Получение осуществляют аналогично примеру 1, при этом исходят из эдуктов 4-аминометил-2-цианотиофен и N-(трет.- бутоксикарбонилметилен)-N-Вос-(D)-циклогексилглицил-3,4- дегидропролин.
Пример 10:
Гидроацетат N-(гидроксикарбонил-метилен)-(D)- циклогексилаланил-3,4-дегидропролил-5-(2-амидино)-фуранилметиламида
Масс-спектр: бомбардировка быстрыми атомами (М+H+): 446
Получение осуществляют аналогично примеру 1, при этом вместо гидрохлорида 5-аминометил-2-цианотиофена используют гидрохлорид 5- аминометил-2-цианофурана.
Пример 11:
Гидроацетат N-(гидроксикарбонил-этилен)-(D)-циклогексилаланил- 3,4-дегидропролил-5-(2-амидино)-фуранилметиламида
Масс-спектр: бомбардировка быстрыми атомами (М+H+): 460
Получение осуществляют аналогично примеру 1, при этом вместо гидрохлорида 5-аминометил-2-цианотиофена используют гидрохлорид 5-аминометил-2-цианофурана.
Пример 12:
Гидроацетат N-(гидроксикарбонил-метилен)-(D)- циклогексилглицил-3,4-дегидропролил-5-(2-амидино)-фуранилметиламида
Масс-спектр: бомбардировка быстрыми атомами (m-H+): 432
Пример 13:
Гидроацетат N-(гидроксикарбонил-метилен)-(D)-циклогексилаланил- 3,4-дегидропролил-5-(3-амидино)-фуранилметиламида
Масс-спектр: бомбардировка быстрыми атомами (М+H+): 446
Пример 14:
Гидрохлорид N-(1-идроксикарбонил-метилен)-(D)- циклогексилаланил-3,4-дегидропролил-5-(2-амидино-1-метил)- пирролметиламида
а) 1,5 г (4,4 ммоль) гидроацетата 5-(N-Вос-аминометил)-1- метилпиррол-2-амидина растворяют в 70 мл изопропанола, после чего добавляют 4,5 мл (24,0 ммоль) 5,5 М изопропанолевой соляной кислоты и нагревают до 50oС в течение 2 ч. После охлаждения до комнатной температуры растворитель отгоняют на ротационном испарителе, остаток подают в раствор 50 мл t-BuO2C-CH-(Boc)-(D)-Cha-Pyr-OH в диметилформамиде. Раствор охлаждают до 0oC, затем прибавляют 1,92 мл (17,44 ммоль) N-метилморфолина. После этого добавляют по порциям 1,18 г (3,58 ммоль) TOTU. Перемешивают при 0oC в течение 45 мин, затем растворитель отгоняют на ротационном испарителе и сырой продукт очищают путем среднепроизводительной жидкостной хроматографии на RP 18 (элюент: смесь ацетонитрила с водой). Выход: 980 мг (45%).
Масс-спектр: бомбардировка быстрыми атомами (M+H+): 615.
б) Полученный согласно а) продукт (559 мг, 0,845 ммоль) растворяют в 50 мл дихлорметана, и раствор при 0-5oC насыщают хлороводородным газом. Затем перемешивают при 0oC в течение полтора часа. Растворитель отгоняют на ротационном испарителе, и сырой продукт лиофилизируют. Выход: 450 мг (100%).
Масс-спектр: бомбардировка быстрыми атомами (М+H+): 615.
Пример 15:
Гидрохлорид N-(гидроксикарбонил-метилен)-(D)- циклогексилаланил-3,4-дегидропролил-2-(4-амидино-1-метил)- пирролметиламида получают аналогично примеру 14.
Масс-спектр: бомбардировка быстрыми атомами (М+H+): 459.
Пример 16:
Гидрохлорид N-(гидроксикарбонил-метилен)-(D)- циклогексилаланил-3,4-дегидропролил-4-(2-амидино-1-метил)- пирролметиламида получают аналогично примеру 14.
Масс-спектр: бомбардировка быстрыми атомами (М+H+): 459.
Пример 17:
Гидрохлорид N-(трет.-бутоксикарбонил-метилен)-(N-Вос)-(D)- циклогексилаланил-3,4-дегидропролил-2-(4- амидотиокарбонил)оксазолметиламида
а) 2,36 г (4,92 ммоль) t-BuO2C-CH2-(Boc)-(D)-Cha-Pyr-OH растворяют в 60 мл дихлорметана. Прикапывают при -10oC 4,3 мл (24,59 ммоль) диизопропилэтиламина. При той же температуре перемешивают в течение 5 мин, затем прибавляют гидрохлорид 2- аминометил-оксазол-4-тиокарбамида (1 г, 5,16 ммоль, см. G. Videnov и др., Angew. Chem. 1996, 108, 1604-9, Вос-группа описанного в этой литературе N-Вос-2-аминометилоксазол-4-тиокарбамида расщепляют с помощью эфирной соляной кислоты, и соответствующий гидрохлорид получают путем сгущения, а затем прикапывают 5,06 мл (6,39 ммоль) 50%-ного раствора ангидрида пропанфосфоновой кислоты в сложном этиловом эфире уксусной кислоты, разбавленного 10 мл дихлорметана, в течение 20 мин. Перемешивают при 0oC в течение часа, затем нагревают до комнатной температуры в течение 3 ч. Исходную смесь разбавляют дихлорметаном, промывают 2 раза насыщенным раствором гидрокарбоната натрия, 2 раза 5%-ным раствором лимонной кислоты и раз насыщенным раствором хлористого натрия и сушат над сульфатом натрия. Растворитель отгоняют на ротационном испарителе и сырой продукт очищают путем хроматографии с применением в качестве элюента смеси дихлорметана и метанола в соотношении 95:5. Выход: 2,5 г (82%).
1H-ЯМР (ДМСО-d6) δ = 0,5 - 2,0 (м, 31H), 3,1 - 5,5 (м, 8H), 5,8 - 6,2 (м, 2H), 8,5 - 9,3 (м, 3H), 9,8 (шир.с, 1H).
б) Гидроацетат N-трет.-бутоксикарбонил-метилен)-(N-Вос(D)- циклогексилаланил-3,4-дегидропролил-2-(4-амидино)-оксазолметиламида
Полученный согласно а) продукт растворяют в 50 мл ацетона, затем к раствору добавляют 1,97 мл (31,29 ммоль) метилйодида и нагревают с обратным холодильником в течение 2 ч. Растворитель и избыточный метилйодид отгоняют на ротационном испарителе. Полученный таким образом сырой продукт растворяют в 50 мл тетрагидрофурана, после чего добавляют 466 мг (6,05 ммоль) ацетата аммония. Нагревают до 60oC в течение полтора часа. Растворитель отгоняют на ротационном испарителе, и сырой продукт при помощи ионообменника (ацетата на полимерном носителе, Флюка 00402) переводят в ацетат, который затем очищают путем хроматографии на силикагеле с применением в качестве элюента смеси дихлорметана, метанола и уксусной кислоты в соотношении 75:20:5. Выход: 2,0 г (75%).
Масс-спектр: бомбардировка быстрыми атомами (М+H2): 603.
в) Гидроацетат N-(гидроксикарбонил-метилен)-(D)- циклогексилаланил-3,4-дегидропролил-2-(4-амидино)- оксазолметиламида
Полученный согласно б) продукт (1,95 г, 2,94 ммоль) растворяют в 50 мл дихлорметана, затем к раствору добавляют 3,7 мл (14,71 ммоль) 4 М соляной кислоты в диоксане. Перемешивают при комнатной температуре в течение 20 ч, после чего растворитель отгоняют на ротационном испарителе, сырой продукт растворяют в воде и лиофилизируют. Выход: 1,5 г, (100%).
1H-ЯМР (ДМСО-d6) δ = 168.6, 167.8, 166.2, 162.2, 156.4, 144.7, 129.6, 127.7, 125.5, 67.9, 55.0, 53.5, 45.3, 36.4, 35.7, 33.0, 32.5, 32.0, 25.7, 25.4, 25.2.
Пример 18:
Гидрохлорид N-(гидроксикарбонил-метилен)-(D)- циклогексилаланил-3,4-дегидропролил-5-(3-амидино)-1,2,4- оксадиазолметиламида
а) N-(трет. -бутоксикарбонил-метилен)-(N-Вос)-(D)- циклогексилаланил-3,4-дегидропролил-5-(3-циано)-1,2,4- оксадиазолметиламид
1,93 г (4,0 ммоль) N-(t-BuO2C-CH2)-N-Boc-(D)-Cha-Pyr-OH растворяют в 65 мл дихлорметана, затем к раствору при -10oC добавляют 3,1 мл (17,67 ммоль) диизопропилэтиламина. После этого прибавляют 645 мг (4,0 ммоль) гидрохлорида 5-аминометил-3-циано-1,2,4-оксалиазола, растворенного в 30 мл дихлорметана. Перемешивают в течение 5 мин, затем прикапывают 3,9 мл (4,93 ммоль) разбавленного с помощью 15 мл дихлорметана 50%-ного раствора ангидрида пропанфосфоновой кислоты в сложном этиловом эфире уксусной кислоты в течение 30 мин. По истечении часа при температуре 0oC исходную смесь разбавляют дихлорметаном, промывают 2 раза насыщенным раствором гидрокарбоната натрия, 2 раза 5%-ным раствором лимонной кислоты и раз насыщенным раствором хлористого натрия. После сушки над сульфатом натрия растворитель отгоняют на ротационном испарителе и сырой продукт очищают путем хроматографии с применением в качестве элюента смеси дихлорметана и метанола в соотношении 95: 5. Выход: 1,55 г (71%).
Масс-спектр: бомбардировка быстрыми атомами (М+H+): 587.
б) Гидроацетат N-(трет. -бутоксикарбонил-метилен)-(N-Вос)-(D)- циклогексилаланил-3,4-дегидропролил-5-(3-амидино)-1,2,4- оксадиазолметиламида
Полученный согласно а) продукт (1,5 г, 2,56 ммоль) растворяют в 5 мл метанола, затем добавляют 450 мг (2,76 ммоль) ацетилцистеина. После этого при 35oC вводят аммиак до полного завершения реакции. Растворитель отгоняют на ротационном испарителе, а сырой продукт с помощью ионообменника (ацетата на полимерном носителе Флюка 00402) переводят в ацетат. Полученный таким образом сырой продукт очищают путем хроматографии (RP-18, элюент: смесь ацетонитрила с водой). Выход: 300 мг (18%).
Масс-спектр: бомбардировка быстрыми атомами (М+H+): 604.
в) Гидрохлорид N-(гидроксикарбонил-метилен)-(D)- циклогексилаланил-3,4-дегидропролил-5-(3-амидино)-1,2,4- оксадиазолметиламида
Полученный согласно б) продукт (300 мг, 0,45 ммоль) растворяют в 20 мл дихлорметана, затем при комнатной температуре добавляют 0,6 мл (2,48 ммоль) 4 М раствора соляной кислоты в диоксане. Перемешивают при комнатной температуре в течение 20 ч, после чего растворитель отгоняют на ротационном испарителе, продукт растворяют в воде и лиофилизируют. Выход 230 мг (98%).
Масс-спектр: бомбардировка быстрыми атомами (М+H+): 448.
Пример 19:
Гидрохлорид N-(гидроксикарбонил-метилен)-(D)-циклогексилаланил- 3,4-дегидропролил-5-(3-амидино-N-метил)пиразолметиламида
а) N-(трет. -бутоксикарбонил-метилен)-(N-Вос)-(D)- циклогексилаланил-3,4-дегидропролил-5-(3-амидо-N-метил)-1,2,4- пиразолметиламид
1,25 г (2,59 ммоль) N-(t-BuO2C-CH2)-N-Boc-(D)-Cha-Pyr-OH подают в 30 мл дихлорметана. При -10oC прикапывают 1,95 мл (11,16 ммоль) диизопропилэтиламина. После этого прибавляют 0,4 г (2,59 ммоль) раствора амида N-метил-5-аминометил-пиразол-3-карбоновой кислоты в 20 мл тетрагидрофурана. Перемешивают в течение 5 мин, затем прикапывают 2,36 мл (3,11 ммоль) 50%-ного раствора ангидрида пропанфосфоновой кислоты в сложном этиловом эфире уксусной кислоты и 5 мл дихлорметана в течение 5 мин. Перемешивают при 0oC в течение 45 мин, после чего нагревают до комнатной температуры в течение 12 ч. Растворитель отгоняют на ротационном испарителе, остаток подают в дихлорметан и промывают 2 раза насыщенным раствором гидрокарбоната натрия, 2 раза 5%-ным раствором лимонной кислоты и раз насыщенным раствором хлористого натрия. После сушки над сульфатом натрия растворитель отгоняют на ротационном испарителе. Сырой продукт очищают путем хроматографии (RP-18, элюент: смесь ацетонитрила с водой). Выход: 220 мг (14%).
Масс-спектр: бомбардировка быстрыми атомами (М+H+): 617.
б) N-(трет. -бутоксикарбонил-метилен)-(N-Вос)-(D)- циклогексилаланил-3,4-дегидропролил-5-(3-циано-N-метил)-1,2,4- пиразолметиламид
Полученный согласно а) продукт (220 мг, 0,36 ммоль) растворяют в 15 мл дихлорметана, затем при -10oC добавляют 0,17 мл (0,96 ммоль) диизопропилэтиламина. Перемешивают в течение 5 мин, после чего прикапывают 0,057 мл (0,41 ммоль) раствора ангидрида трифторуксусной кислоты в 1 мл дихлорметана. По истечение часа разбавляют при 0oC дихлорметаном. Промывают 2 раза насыщенным раствором гидрокарбоната натрия, 2 раза 5%-ным раствором лимонной кислоты и раз насыщенным раствором хлористого натрия. После сушки над сульфатом натрия растворитель отгоняют на ротационном испарителе. Выход: 180 мг (84%).
в) Гидроацетат N-(трет. -бутоксикарбонил-метилен)-(N-Вос)-(D)- циклогексилаланил-3,4-дегидропролил-5-(3-амидино-N-метил)- пиразолметиламида
Полученный согласно б) продукт (180 мг, 0,3 ммоль) растворяют в 1 мл метанола, затем добавляют 52,8 мг (0,32 ммоль) ацетилцистеина. После этого при 35oC вводят аммиак до полного завершения реакции. Растворитель отгоняют на ротационном испарителе, а сырой продукт при помощи ионообменника (ацетат на полимерном носителе Флюка 00402) переводят в ацетат. Сырой продукт очищают путем хроматографии (RP- 18, элюент: смесь ацетонитрила с водой). Выход: 50 мг (16%). Масс-спектр: бомбардировка быстрыми атомами (М+H+): 616.
г) Гидрохлорид N-(гидроксикарбонил-метилен)-(D)- циклогексилаланил-3,4-дегидропролил-5-(3-амидино-N- метил)пиразолметиламида
Полученный согласно в) продукт (50 мг, 0,081 ммоль) растворяют в 5 мл дихлорметана, затем добавляют 0,147 мл 5 М соляной кислоты в диэтиловом эфире. Перемешивают при комнатной температуре в течение 12 ч, после этого растворитель отгоняют на ротационном испарителе, продукт подают в воду и лиофилизируют. Выход: 40 мг (92%).
Масс-спектр: бомбардировка быстрыми атомами (М+H+): 460.
Пример 20:
Гидрохлорид N-(гидроксикарбонил-метилен)-(D)-циклогексилаланил- 3,4-дегидропролил-5-(3-амидино)-изоксазолметиламида
Получение осуществляют исходя из амида 5-аминометилизоксазол- 3-карбоновой кислоты и Вос-3,4-дегидропролина. После сочетания и отщепления Вос-защитной группы полученный структурный элемент сочетают с N-(трет.-бутоксикарбонил-метилен)-(N-Вос)-(D)- циклогексилаланином до получения N-(трет.-бутоксикарбонил-метилен) -(N-Вос)-(D)-циклогексилаланил-3,4-дегидропролил-5-(3- карбамоил)-изоксазолметиламида. После дегидратизации первичного амида до нитрильной функции аналогично примеру 4 осуществляется получение амидина по описанному ниже способу.
Гидроацетат N-(трет.-бутоксикарбонилметилен)-(N-Вос)-(D)- циклогексилаланил-3,4-дегидропролил-5-(3-амидино-N-метил)- изоксазолметиламида
1,75 г (3,0 ммоль) N-(трет.-бутоксикарбонилметилен)-(N-Вос)- (D)-циклогексилаланил-3,4-дегидропролил-5-(3-циано)- изоксазолметиламида растворяют в 10 мл метанола, к раствору прибавляют 0,54 г (3,28 ммоль) N-ацетилцистеина. При пропускании газообразного аммиака нагревают с обратным холодильником в течение 4 ч. Отщепление N-ацетилцистеина, очистка продукта и перевод в соль ацетата осуществляют путем среднепроизводительной жидкостной хроматографии (RP-18, элюент: смесь ацетонитрила с водой и 0,1 М уксусной кислотой). После сублимационной сушки получают 1,39 г белого порошка (70% теории).
Путем снятия защитной группы с очищенного продукта с помощью эфирной соляной кислоты получают целевое соединение.
Масс-спектр: бомбардировка быстрыми атомами (М+H+): 448.
Пример 21:
Гидроацетат N-(гидроксикарбонил-метилен)-(D)- циклогексилаланил-3,4-дегидропролил-2-(4-амидино)-тиазолметиламида
Масс-спектр: бомбардировка быстрыми атомами (М+H+): 463.
Получение осуществляют аналогично примеру 1, при этом исходят из эдуктов 2-аминометил-тиазол-4-тиокарбоксамид и N-Boc-N-трет.- бутилоксикарбонилметилен)-(D)-циклогексилаланил-3,4- дегидропролин.
Пример 22:
Гидрохлорид N-(гидроксикарбонил-метилен)-(D)- циклогексилглицил-3,4-дегидропролил-2-(4-амидино)-тиазолметиламида
Масс-спектр: бомбардировка быстрыми атомами (М+H+): 449.
Пример 23:
Гидроацетат N-(гидроксикарбонил-метилен)-(D)- циклогексилаланил-3,4-дегидро-пролил-4-(2-амидино)-тиазолметиламида
Масс-спектр: бомбардировка быстрыми атомами (М+H+): 463.
Получение осуществляют аналогично примеру 1, при этом исходят из эдуктов 4-аминометил-тиазол-2-тиокарбоксамид и N-трет. - бутилоксикарбонил-метилен)-N-Вос-(D)-циклогексилаланил-3,4- дегидропролин.
Пример 24:
Гидроацетат N-(гидроксикарбонил-метилен)-(D)- циклогексилглицил-3,4-дегидропролил-4-(2-амидино)-тиазолметиламида
Масс-спектр: бомбардировка быстрыми атомами (М+H+): 449.
Получение осуществляют аналогично примеру 1, при этом исходят из эдуктов 4-аминометил-тиазол-2-тиокарбоксамид и N-трет. - бутилоксикарбонил-метилен)-N-Вос-(D)-циклогексилглицил-3,4- дегидропролин.
Пример 25:
Гидроацетат N-(гидроксикарбонил-метилен)-(D)- циклогексилаланил-3,4-дегидропролил-2-(5-амидино)-тиазолметиламида
Масс-спектр: бомбардировка быстрыми атомами (М+H+): 463.
Получение осуществляют аналогично примеру 21.
Пример 26:
Гидроацетат N-(гидроксикарбонил-метилен)-(D)- циклогексилглицил-3,4-дегидро-пролил-2-(5-амидино)-тиазолметиламида
Масс-спектр: бомбардировка быстрыми атомами (М+H+): 449.
Получение осуществляют аналогично примеру 22.
Пример 27:
Гидроацетат N-(гидроксикарбонил-метилен)-(D)- циклогексилаланил-3,4-дегидропролил-5-(2-амидино)-тиазолметиламида
Масс-спектр: бомбардировка быстрыми атомами (М+H+): 463.
Получение осуществляют аналогично примеру 23.
Пример 28:
Гидроацетат N-(гидроксикарбонил-метилен)-(D)- циклогексил-3,4-дегидропролил-5-(2-амидино)-тиазолметиламида
Масс-спектр: бомбардировка быстрыми атомами (М+H+): 449.
Получение осуществляют аналогично примеру 24.
Пример 29:
Гидрохлорид N-(этилоксикарбонил-метилен)-(D)- циклогексилаланил-3,4-дегидропропил-2-(4-амидино)тиазолметиламида
1,5 г (3,24 ммоль) HO2C-CH2-(D)-Cha-Pyr- NH-CH2-2-(4-am)-thiaz (соединение примера 21) растворяют в 50 мл этанола, смешивают с 10 мл 5N соляной кислоты в диэтиловом эфире и реакционную смесь перемешивают при температуре 60oC в течение 4 ч. Согласно данным тонкослойной хроматографии с применением в качестве элюента смеси хлористого метилена, метанола, уксусной кислоты и воды в соотношении 35:17:3,5:3,5 конверсия полная. После упаривания реакционной смеси в ротационном испарителе многократно перегоняют смесью хлористого метилена и диметилового эфира с тем, чтобы удалить прилипшую соляную кислоту. Затем продукт растворяют в небольшом количестве хлористого метилена, осаждают диэтиловым эфиром, остаток отсасывают и сушат в вакууме. Получают 1,8 г целевого продукта в виде белого, гигроскопического вещества.
Масс-спектр: ионизация электронным ударом (М+H+): 491.
13C-ЯМР (ДМСО; [млн. доли] ): 171.89, 169.02, 166.29, 166.19, 157.26, 141.44, 128.75, 127.93, 125.30, 68.04, 61.60, 55.18, 53.64, 45.29, 40.41, 36.21, 32.93, 32.53, 32.04, 25.69,25.40,25.21,13.85.
Дигидрохлорид N-(изопропилоксикарбонил-метилен)-(D)- циклогексилглицил-3,4-дегидропролил-[2-(4-амидино-3-хлор)- тиофен]метиламида
Масс-спектр: ионизация электронным ударом (М+H+): 524.
Дигидрохлорид N-(н-гексилоксикарбонил-метилен)-(D)- циклогексилаланил-3,4-дегидропролил-[2-(4-амидино)- тиазол]метиламида
Масс-спектр: бомбардировка быстрыми атомами (М+H+): 547.
Дигидрохлорид N-(2,2-диметилпропилоксикарбонил- метилен)-(D)-циклогексилаланил-3,4-дегидропролил-[2-(4-амидино)- тиазол]метиламида
Масс-спектр: ионизация электронным ударом (М+H+): 533.
Ингибирующие тромбин свойства соединений общей формулы (I) иллюстрируются результатами следующего опыта.
Опыт
Кровь отобрали из бедренной вены здоровых добровольцев обоих полов, которые по крайней мере за одну неделю до начала опыта не принимали лекарств. Кровь перемешивали с 0,13 моль/л раствора цитрата натрия в объемном соотношении 9:1. Полученную кровь центрифугировали со скоростью 250 g в течение 10 мин при комнатной температуре с тем, чтобы получить богатую тромбоцитами плазму. Бедную же тромбоцитами плазму получали путем центрифугирования со скоростью 3600 g в течение 20 мин. Богатую и бедную тромбоцитами плазму подавали в герметичные полиэтиленовые трубки, которые хранили при комнатной температуре до проведения опыта (но не больше 3 ч). Агрегацию тромбоцитов, вызываемую тромбином, определяли с помощью микрометода при температуре 37oC в четырехканальном агрегометре типа РАР 4 американской фирмы Биодата Корпорейшн. Перед добавлением тромбина 215,6 мкл богатой тромбоцитами плазмы инкубировали вместе с 2,2 мкл исследуемого соединения или носителя при температуре 37oC в течение 3 мин без перемешивания и затем при постоянном перемешивании со скоростью 1000 об/мин. Добавление 2,2 мкл раствора тромбина вызывает максимальную агрегацию, определенную при температуре 37oC со скоростью перемешивания 1000 об/мин. Ингибирующее действие исследуемых соединений определяли путем сравнения максимальной степени агрегации при заданной концентрации с кривой контрольной агрегации, вызываемой тромбином в присутствии носителя. Определяли концентрацию исследуемого соединения в моль/л, обеспечивающую 50 %-ное ингибирование агрегации тромбоцитов, вызываемой тромбином (обозначенную далее как KT50).
Исследуемые соединения и результаты опыта сведены в таблице.
Соединения согласно изобретению относятся к категории малотоксичных веществ.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ТРОМБИНА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ | 1995 |
|
RU2172741C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 3-ПИРРОЛИН-2-КАРБОНОВОЙ КИСЛОТЫ | 1997 |
|
RU2199529C2 |
| ПЕПТИДНЫЕ ПРОИЗВОДНЫЕ, ИХ СТЕРЕОИЗОМЕРЫ ИЛИ ФИЗИОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ОБЛАДАЮЩИЕ ПРОТИВОТРОМБОЗНОЙ, ПРОТИВОСВЕРТЫВАЮЩЕЙ ИЛИ ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПОДАВЛЕНИЯ ТРОМБИНА, СПОСОБ ПОДАВЛЕНИЯ КИНИНОГЕНАЗ, ПРИМЕНЕНИЕ СОЕДИНЕНИЙ В КАЧЕСТВЕ ИСХОДНЫХ В СИНТЕЗЕ ИНГИБИТОРА ТРОМБИНА | 1994 |
|
RU2142469C1 |
| ИНГИБИТОРЫ ПРОТЕАЗЫ СЕРИНА | 1997 |
|
RU2178419C2 |
| ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ АДГЕЗИИ ЛЕЙКОЦИТОВ И VLA-4-АНТАГОНИСТОВ | 1997 |
|
RU2229296C2 |
| ИНГИБИТОРЫ СЕРИНПРОТЕАЗЫ | 1997 |
|
RU2191193C2 |
| ИНГИБИТОРЫ СЕРИНОВЫХ ПРОТЕАЗ | 1997 |
|
RU2172321C2 |
| ПРОИЗВОДНЫЕ АМИДИНОВ, СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1999 |
|
RU2238939C2 |
| ПРОИЗВОДНЫЕ ДИПЕПТИДНЫХ П-АМИДИНО-БЕНЗИЛАМИДОВ С N-КОНЦЕВЫМИ СУЛЬФОНИЛЬНЫМИ ОСТАТКАМИ И ИХ СОЛИ С ФИЗИОЛОГИЧЕСКИ ПРИЕМЛЕМЫМИ КИСЛОТАМИ | 1995 |
|
RU2152953C1 |
| СОДЕРЖАЩИЙ ИНГИБИТОР ТРОМБИНА ВОДНЫЙ РАСТВОР ДЛЯ ВНУТРИВЕННОГО ВВЕДЕНИЯ, СТАБИЛЬНЫЙ ПРИ ХРАНЕНИИ | 1995 |
|
RU2164803C2 |
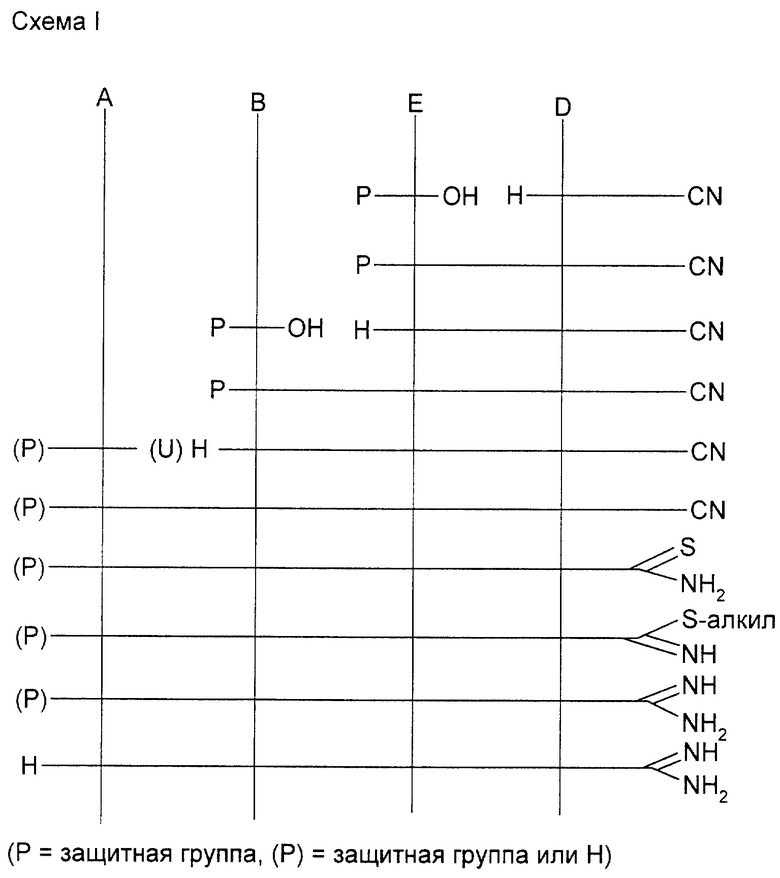
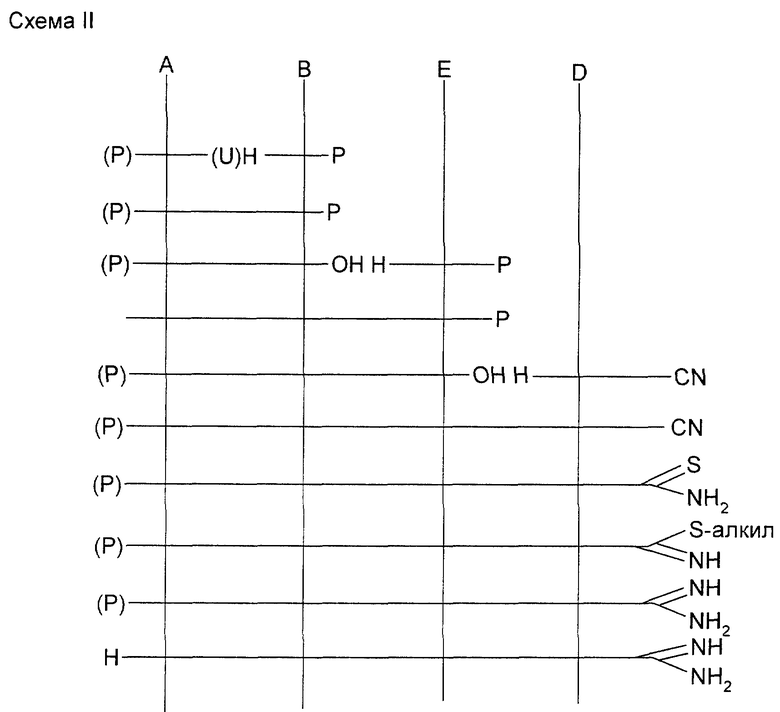
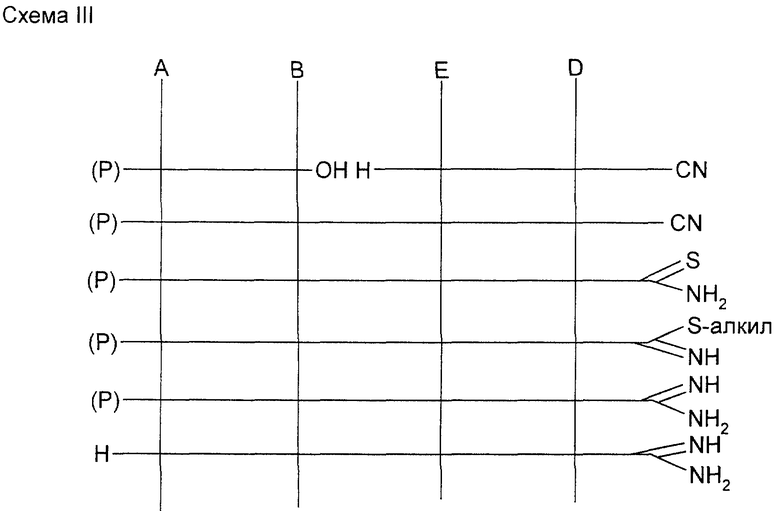
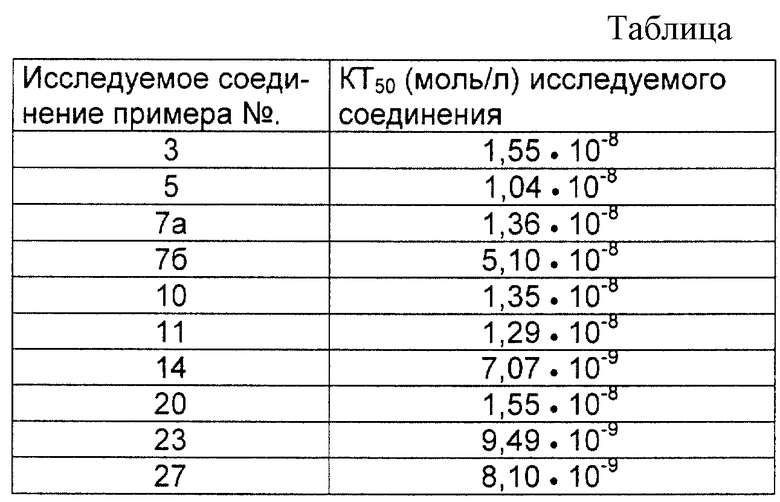
Описываются новые пятичленные гетероциклические амидины формулы (I), где А, В, D и Е имеют следующие значения:
А формулы (II), где m = n = 0,1 или 2; R1 - группы: гидроксил, карбоксил, С1-6-алкил-ООС; R2 = R3 - водород, алкил с 1-4 атомами углерода;
В формулы (III), где R4 - водород, алкил с 1-4 атомами углерода или R1 - (СН)m - (при этом R1 и m имеют вышеуказанные значения); Р - 0 или 1;
R5 - водород или алкил с 1-4 атомами углерода; R6 - водород, алкил с 1-6 атомами углерода, циклоалкил с 3-8 атомами углерода; R4 и R6 вместе могут означать этиленовую группу; R7 - водород, алкил с 1-8 атомами углерода; R8 - водород, алкил с 1-4 атомами углерода;
Е формулы (IV), где q - 0 или 1;
D формул (Va), (Vb), (Cc), где R9 - водород, алкил с 1-3 атомами углерода; R10 = R11 - водород, алкил с 1-4 атомами углерода;
Х - кислород, сера, NR12 (R12 = водород, алкил с 1-6 атомами углерода),
Y - азот = или -CR13= (R13 = водород, алкил с 1-4 атомами углерода);
Z - азот = или -CR13=, где R13 имеет вышеуказанное значение,а также их соли с физиологически переносимыми кислотами. Вышеуказанные соединения обладают свойствами ингибиторов тромбина. Описываются также соединения, содержащие производные пятичленного гетероциклического амидина, в качестве составной части ингибиторов серинпротеазы и промежуточные соединения. 3 с. и 1 з. п.ф-лы, 1 табл.
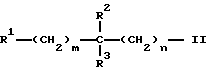
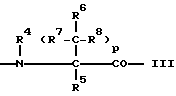
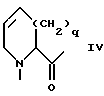
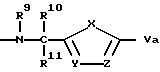
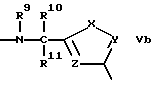

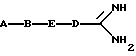
где А, В, D и Е имеют следующие значения:
A: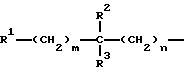
где m - 0, 1 или 2, n - 0, 1 или 2;
R1 - группы: гидроксил, карбоксил, C1-6-алкил-ООС, R2 - водород, алкил с 1 - 4 атомами углерода, R3 - водород, алкил с 1 - 4 атомами углерода,
В: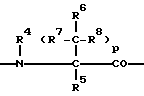
где R4 - водород, алкил с 1 - 4 атомами углерода или R1-(СН2)m, где R1 и m имеют вышеуказанные значения;
р - 0 или 1;
R5 - водород, алкил с 1-4 атомами углерода;
R6 - водород, алкил с 1 - 6 атомами углерода, циклоалкил с 3 - 8 атомами углерода;
R4 и R6 вместе могут означать этиленовую группу;
R7 - водород, алкил с 1 - 8 атомами углерода;
R8 - водород, алкил с 1-4 атомами углерода,
Е: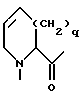
где q - 0 или 1,
D: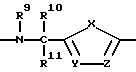
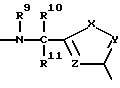
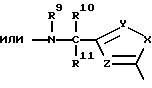
где
R9 - водород, алкил с 1-3 атомами углерода;
R10 - водород, алкил с 1 -4 атомами углерода;
R11 - водород, алкил с 1 -4 атомами углерода;
Х - кислород, сера, NR12, где R12 означает водород, алкил с 1 - 6 атомами углерода;
Y - -азот= или -CR13=, где R13 означает водород, алкил с 1-4 атомами углерода,
Z - -азот= или -CR13=, где R13 имеет вышеуказанное значение,
а также их соли с физиологически переносимыми кислотами.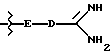
где Е и D имеют указанные в п.1 значения, а у атома азота структурного элемента Е находятся атом водорода, защитная группа или незамещенный алкильный остаток, в качестве составной части ингибиторов серинпротеазы.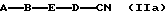
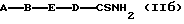
где остатки А, В, Е и D имеют указанное в п.1 значение.

