Данная заявка является частичным продолжением патентных заявок США N N 08/356.462 и 08/356.653, обе из которых были поданы 15 декабря 1994 г., и их описания включены сюда как ссылки.
Предпосылки создания изобретения
Область техники, к которой относится изобретение
Данное изобретение относится к неводным печатным краскам и покрытиям, которые содержат продукт из модифицированного углерода.
Соответствующий уровень техники
Неводные печатные краски и покрытия используются во многих применениях, в которых водные наполнители не подходят. Например, печатные краски, которые должны быть напечатаны на гидрофобных непористых подложках, таких как металл, стекло или пластики, должны быстро сохнуть. Поэтому вместо воды часто используют такие растворители, как кетоны, сложные эфиры, спирты или углеводороды. Такие печатные краски на основе растворителей широко используются для промышленного маркирования картонных коробок и различных металлических и пластиковых контейнеров и деталей. Конкретные примеры включают в себя композиции газетных печатных красок и композиции глянцевых красок, закрепляющихся при нагревании, для рулонной офсетной печати.
В определенных ситуациях также требуется, чтобы печатные краски и покрытия были водонепроницаемыми. Поэтому в неводных растворителях для композиций печатных красок и покрытий часто растворяют смолы, чтобы обеспечить требуемую водонепроницаемость после сушки. Такие неводные покрытия в основном применяют на металлических и пластмассовых частях автомобилей.
Краткое описание сущности изобретения
Настоящее изобретение относится к композиции неводного покрытия или печатной краски, имеющей внесенный в нее продукт из модифицированного углерода, содержащий уголь с присоединенной к нему замещенной или незамещенной ароматической группой. Используемый здесь углерод способен реагировать с солью диазония с образованием вышеупомянутого продукта из модифицированного углерода. Углерод может быть кристаллического или аморфного типа. Примеры включают в себя графит, сажу, стеклообразный углерод, а также активированный древесный уголь или активированный уголь, но не ограничены ими. Предпочтительны мелкодисперсные формы вышеуказанных (материалов); кроме того, можно использовать смеси различных углеродов.
Описание предпочтительных вариантов воплощения изобретения
Используемый здесь углерод способен реагировать с солью диазония с образованием вышеупомянутых продуктов из модифицированного углерода. Углерод может быть кристаллического или аморфного типа. Примеры включают в себя графит, сажу, стеклообразный углерод, а также активированный древесный уголь и активированный уголь и их смеси, но не ограничены ими. Предпочтительны мелкодисперсные формы вышеуказанных /материалов/; кроме того, можно использовать смеси различных углеродов. Продукты из модифицированного углерода могут быть получены путем реакции углерода с солью диазония в жидкой реакционной среде, для того чтобы присоединить по меньшей мере одну органическую группу к поверхности углерода. Соль диазония может содержать органическую группу, которую присоединяют к углероду. В соответствии с изобретением соль диазония является органическим соединением, имеющим одну или несколько диазониевых групп. К предпочтительным реакционным средам относится вода, любая среда, содержащая воду, и любая среда, содержащая спирт. Наиболее предпочтительной средой является вода. Эти продукты из модифицированного углерода, в которых углерод является сажей, а также различные способы их получения описаны в патентной заявке США с порядковым номером 08/356.600 с названием "Реакция сажи с солями диазония, конечные продукты из сажи, и их применения", поданной 15 декабря 1994 г. и в ее частичном продолжении, поданном параллельно с данной заявкой, причем обе из них включены сюда как ссылки. Данные продукты из модифицированного углерода, в которых углерод не является сажей, и различные способы их получения описаны в патентной заявке с порядковым номером 08/356.653 с названием "Реакция углеродных материалов с солями диазония и образующиеся продукты из углерода", поданной 15 декабря 1994 г. и также включенной сюда как ссылка.
Для того чтобы получить вышеуказанные продукты из модифицированного углерода, необходимо только, чтобы соль диазония была достаточно устойчива, чтобы позволить провести реакцию с углеродом. Таким образом, реакцию можно провести с некоторыми солями диазония, которые в иных случаях полагают неустойчивыми и способными разлагаться. Некоторые процессы разложения могут конкурировать с реакцией между углеродом и солью диазония и могут снизить общее число органических групп, присоединенных к углероду. Кроме того, реакцию можно проводить при повышенных температурах, когда многие соли диазония могут быть подвержены разложению. Повышенные температуры также могут выгодно повысить растворимость соли диазония в реакционной среде и улучшить работу с ней во время процесса. Однако повышенные температуры могут привести к некоторой потере соли диазония вследствие других процессов разложения. Соли диазония по настоящему изобретению можно приготовить in situ. Предпочтительно, чтобы продукты из модифицированного углерода по настоящему изобретению не содержали побочных продуктов или солей.
Сажа может реагировать с солью диазония тогда, когда она присутствует в виде разбавленной, легко перемешиваемой водной суспензии или же в присутствии надлежащего количества воды для образования гранул из сажи. Если требуется, гранулы сажи можно образовать путем использования обычной технологии грануляции. Другие углероды могут аналогичным образом реагировать с солью диазония. Кроме того, тогда когда в продуктах из модифицированного углерода используют углерод, отличный от сажи, для применения в неводных печатных красках и покрытиях, то перед реакцией с солью диазония углерод предпочтительно размалывают до малого размера частиц, для того чтобы предотвратить нежелательное осаждение в печатных красках и покрытиях.
Органическая группа может быть алифатической группой, циклической органической группой или органическим соединением, имеющим алифатическую часть и циклическую часть. Как обсуждалось выше, соль диазония, которую используют в реакции с углеродом, может быть произведена из первичного амина, имеющего одну из таких групп и способного образовывать, даже промежуточно, соль диазония. Органическая группа может быть замещенной или незамещенной, разветвленной или неразветвленной. К алифатическим группам относятся, например, группы, произведенные из алканов, алкенов, спиртов, простых эфиров, альдегидов, кетонов, карбоновых кислот и углеводов. Циклические органические группы включают алициклические углеводородные группы (например циклоалкилы, циклоалкенилы), гетероциклические углеводородные группы (например пирролидинил, пирролинил, пиперидинил, морфолинил и т.п.), арильные группы (например фенил, нафтил, антраценил и т.п.) и гетероарильные группы (имидазолил, пиразолил, пиридинил, тиенил, тиазолил, фурил, триазинил, индолил и т.п.), но не ограничены ими. Когда стерическое затруднение замещенной органической группы увеличивается, число органических групп, присоединенных к углероду в реакции между солью диазония и углеродом, может уменьшиться.
Когда органическая группа является замещенной, она может содержать любую функциональную группу, совместимую с образованием соли диазония. Предпочтительные функциональные группы представляют собой R, OR, COR, COOR, OCOR, галоген, CN, NR2 SO2NR(COR), SO2NR2, NR(COR), CONR2, NO2 и N=NR', но не ограничены ими. R независимо является водородом, C1-C20 - замещенным или незамещенным алкилом (разветвленным или неразветвленным), C3-C20-замещенным или незамещенным алкенилом, (C2-C4 алкиленокси)xR'', или замещенным или незамещенным арилом, R' независимо является водородом, C1-C20-замещенным или незамещенным алкилом (разветвленным или неразветвленным) или замещенным или незамещенным арилом. R'' является водородом, C1-C20-замещенным или незамещенным алкилом, C3-C20-замещенным или незамещенным алкенилом, C1-C20 замещенным или незамещенным алканоилом, или замещенным или незамещенным ароилом. Целое число n находится в интервале от 1-8 и предпочтительно от 2-4. Целое число x находится в интервале от 1 до 40 и предпочтительно от 2 до 25. Анион X- является галогенидом или анионом, произведенным из минеральной или органической кислоты.
Предпочтительной органической группой является ароматическая группа формулы AyAr-, которая соответствует первичному амину формулы AyArNH2. В этой формуле переменные имеют следующие значения: Ar является ароматическим радикалом, выбираемым из группы, состоящей из фенила, нафтила, антраценила, фенантренила, дифенила, пиридинила и триазинила; A является заместителем в ароматическом радикале, независимо выбираемым из вышеописанной предпочтительной функциональной группы, или же A является линейным, разветвленным или циклическим углеводородным радикалом (предпочтительно содержащим от 1 до 20 углеродов), незамещенным или замещенным одной или несколькими этими функциональными группами; и y является целым числом от 1 до 5, когда Ar представляет собой фенил, от 1 до 7, когда Ar представляет собой нафтил, от 1 до 9, когда Ar представляет собой антраценил, фенантренил или дифенил, или от 1 до 4, когда Ar представляет собой пиридинил, или от 1 до 2, когда Ar представляет собой триазинил. Когда A представляет собой (C2-C4 алкиленокси)x-R'' - группу, то она предпочтительно является полиэтоксилатной группой, полипропоксилатной группой или статистической или блочной смесью этих двух групп. Особенно предпочтительные органические группы приведены ниже в примерах.
Предпочтительный продукт из модифицированного углерода содержит углерод и присоединенную органическую группу, имеющую а) ароматическую группу и б) по меньшей мере одну группу формулы SO2NR2 или SO2NR(COR). R предпочтительно независимо является водородом, C1-C20-замещенным или незамещенным алкилом, C3-C20-замещенным или незамещенным алкенилом, (C2-C4 алкиленокси)xR', или замещенным или незамещенным арилом; R' является водородом, C1-C20-замещенным или незамещенным алкилом, C3-C20-замещенным или незамещенным алкенилом, C1-C20-замещенным или незамещенным алканоилом, или замещенным или незамещенным ароилом; и x находится в интервале от 1 до 40. К особо предпочтительным ароматическим группам относятся п-C6H4SO2NH2, п-C6H4SO2NHC6H13, п-C6H4SO2NHCOCH3, п-C6H4SO2NHCOC5H11 и п-C6H4SO2NHCOC6H5. Вышеуказанные продукты из модифицированного углерода можно использовать в композициях неводных печатных красок. Таким образом, в изобретении предлагается усовершенствованная композиция печатных красок, которая содержит растворитель и продукт из модифицированного углерода, имеющий присоединенную замещенную или незамещенную ароматическую группу. В состав печатной краски можно ввести другие известные добавки к краскам. Кроме того, в рамки настоящего изобретения попадает применение состава для печатной краски, содержащего смесь немодифицированного углерода и продукта из модифицированного углерода.
Как правило, печатная краска состоит из четырех основных компонентов: (1) краситель или пигмент, (2) наполнитель или лак, который действует как носитель во время печати, (3) добавки для улучшения печатных свойств, высыхания и т.п. и (4) растворители для того, чтобы регулировать вязкость, высыхание и совместимость остальных компонентов печатной краски. Общее обсуждение свойств, получения и применений печатных красок изложено в The Printing Manual (Руководство по печати), 5 изд., ред. R.H.Leach и др. (Chapman & Hall, 1993).
Продукты из модифицированного углерода по изобретению можно ввести в состав печатной краски, используя стандартные методики, либо в виде предварительной дисперсии, либо в виде твердого вещества. Применение продуктов из модифицированного углерода по настоящему изобретению может дать значительное преимущество и экономию затрат путем уменьшения стадий размалывания, которые обычно используют в случае обычных углеродистых материалов, таких как сажи. Кроме того, продукты из модифицированного углерода по настоящему изобретению могут обеспечить улучшенную черноту.
Как будет отражено более подробно ниже, составы неводной печатной краски и покрытий по настоящему изобретению могут проявлять улучшенные оптические свойства.
Продукты из модифицированного углерода также можно использовать в композициях неводных покрытий, таких как краски или отделочные покрытия. Таким образом, одним из вариантов изобретения является усовершенствованная композиция покрытия, содержащая растворитель, связующее и продукт из модифицированного углерода, имеющего присоединенную к нему замещенную или незамещенную ароматическую группу. В композиции для неводных покрытий можно ввести другие обычные добавки для покрытий.
Составы для покрытий изменяются в широких пределах в зависимости от условий и требований при конечном применении. Как правило, системы покрытий содержат до 30 вес.% углерода. Содержание смолы может изменяться в широких пределах до примерно 100%. К примерам относятся акриловые, алкидные, уретановые, эпоксидные, целлюлозные и подобные смолы. Содержание растворителя может изменяться от 0 до 80%. К их примерам относятся ароматические углеводороды, алифатические углеводороды, спирты, многоатомные спирты, кетоны, сложные эфиры и т.п. Двумя другими общими классами добавок являются наполнители и модификаторы. Примерами наполнителей являются другие окрашивающие пигменты, глины, тальки, кремнеземы и карбонаты. Наполнители можно добавлять до 60%, в зависимости от требований при конечном применении. Примерами модификаторов являются вспомогательные средства, способствующие текучести и выравниванию, а также биоциды, которые обычно добавляют в количестве менее 5%.
Продукты из модифицированного углерода по изобретению можно ввести в композицию неводного покрытия, используя стандартные методики, либо в виде предварительной дисперсии или в виде твердого вещества. Так же как и в случае композиций неводных печатных красок, применение продукта из модифицированного углерода, имеющего прикрепленную замещенную или незамещенную ароматическую группу, может дать значительное преимущество и экономию затрат путем уменьшения стадий размалывания, которые обычно используют в случае обычных углеродистых материалов. Кроме того, продукты из модифицированного углерода по настоящему изобретению могут обеспечить улучшенную черноту.
Следующие примеры предназначены для того, чтобы проиллюстрировать, но не ограничить заявляемое изобретение.
Если не указано иначе, для измерений площади поверхности используют площади поверхности по азоту согласно БЭТ в соответствии с ASTM D-4820. Поверхность по СТАВ была получена согласно ASTM D-3765. Данные по DBPA были получены согласно ASTM D-2414.
ПРИМЕРЫ
Пример 1
Получение метансульфоната метилового эфира PEG 750
В данном примере описано получение метансульфоната метилового эфира PEG 750, который используют в примере 2. К смеси метилового эфира полиэтиленгликоля со средним молекулярным весом 750 (метиловый эфир PEG 750) (30.4 г) и 20 мл хлористого метилена в атмосфере азота добавляли пиридин (6.42 г). После того как все материалы растворились, в течение примерно 3 минут добавляли метансульфонилхлорид (5.58 г). Реакцию оставляли перемешиваться в течение как минимум 4 часов и даже до 18-48 часов для достижения завершения.
Реакцию заканчивали путем разбавления 100 мл хлористого метилена и промывания деионизированной водой (ДИ водой), разбавленным раствором HCl и раствором бикарбоната натрия. Раствор в хлористом метилене сушили над безводным сульфатом натрия, фильтровали, а затем проводили испарение при пониженном давлении с получением продукта в виде масла, 31.01 г (92.3% от теории). Анализ посредством 1Н ЯМР показал чистоту 82%.
Пример 2
Получение сульфаниламида метилового эфира PEG 750
В данном примере описано получение сульфониламида метилового эфира PEG 750, который используют в примерах 11, 12 и 30. Сульфаниламид (3.74 г), 85% гидроксид калия (1.2 г) и 30 мл ацетонитрила соединяли в атмосфере азота. Смесь нагревали до 60-70oC и добавили небольшое количество (<1 мл) воды. После перемешивания при 60-70oC в течение примерно одного часа за 10 минут добавили раствор метансульфоната метилового эфира PEG 750 (15 г) в 15 мл ацетонитрила. Перемешивание данной смеси при 60-70oC продолжали по меньшей мере 14 часов.
Реакцию заканчивали путем первоначального удаления растворителя при пониженном давлении. Этот материал разбавляли 75 мл хлористого метилена и промывали ДИ водой и насыщенным раствором хлорида натрия. Раствор в хлористом метилене сушили над безводным сульфатом натрия, фильтровали, а затем концентрировали в вакууме с получением продукта в виде масла, 13.45 г (82% выход сырого вещества). Анализ посредством 1Н ЯМР показал чистоту примерно 80%.
Пример 3
Получение метансульфоната метилового эфира PEG 350
В данном примере описано получение метансульфоната метилового эфира PEG 350, который используют в примере 4. Данная процедура аналогична таковой из примера 1, за исключением того, что метиловый эфир PEG 750 был заменен эквимолярным количеством метилового эфира полиэтиленгликоля со средним молекулярным весом 350 (14.21 г). В результате процедуры получено 16.55 г продукта (95% выход сырого вещества). Анализ посредством 1Н ЯМР показал чистоту примерно 85%.
Пример 4
Получение сульфаниламида метилового эфира PEG 350
В данном примере описано получение сульфаниламида метилового эфира PEG 350, который используют в примере 13. Сульфаниламид (2.41 г), 85% гидроксид калия (0.78 г) и 20 мл ацетонитрила соединяли в атмосфере азота. Смесь нагревали до 60-70oC и добавили небольшое количество (<1 мл) воды. После перемешивания при 60-70oC в течение примерно одного часа за 5 минут добавили раствор метансульфоната метилового эфира PEG 350 (5 г) в 5 мл ацетонитрила. Перемешивание данной смеси при 60-70oC продолжали по меньшей мере 4 часа.
Реакцию заканчивали путем доведения pH смеси до примерно 5 с помощью уксусной кислоты и удаления растворителя при пониженном давлении. Этот материал разбавляли 75 мл хлористого метилена и промывали ДИ водой и насыщенным раствором хлорида натрия. Раствор в хлористом метилене сушили над безводным сульфатом натрия, фильтровали, а затем концентрировали в вакууме с получением продукта в виде геля, 4.93 г (84% выход сырого вещества). Анализ посредством 1Н ЯМР показал чистоту 68%.
Пример 5
Получение метансульфоната полиоксиэтилен(2)-цетилового эфира
В данном примере описано получение метансульфоната полиоксиэтилен(2)цетилового эфира, который используют в примере 6. К смеси полиоксиэтилен(2)цетилового эфира 30.4 г и 10 мл хлористого метилена в атмосфере азота добавляли пиридин (4.78 г). После того как все материалы растворились, добавляли метансульфонилхлорид (3.80 г). Через 3 часа реакционную смесь разбавляли 15 мл хлористого метилена и перемешивание продолжали 18 часов.
Реакцию заканчивали путем разбавления 50 мл хлористого метилена и промывания ДИ водой, разбавленным раствором HCl и разбавленным раствором бикарбоната натрия. Раствор в хлористом метилене сушили над безводным сульфатом натрия, фильтровали, а затем проводили испарение при пониженном давлении с получением продукта, 11.78 г (95% выход сырого вещества). Анализ посредством 1Н ЯМР показал чистоту 90%.
Пример 6
Получение N1-сульфаниламида полиоксиэтилен(2)-цетилового эфира
В данном примере описано получение N1-сульфаниламида полиоксиэтилен(2)цетилового эфира, который используют в примере 14. Сульфаниламид (2.0 г) и 85% гидроксид калия (0.66 г) соединяли в атмосфере азота и разбавляли 15 мл ацетонитрила. Смесь нагревали до 60-70oC и затем добавили примерно 3 капли ДИ воды и гидроксид тетрабутиламмония (1.18 мл 1.0 М раствора в метаноле). Смесь перемешивали при 60-70oC в течение 22 часов.
Реакцию заканчивали путем добавления около 2 капель уксусной кислоты и последующего удаления растворителя при пониженном давлении. Остаток разбавляли 95 мл хлористого метилена и промывали ДИ водой и насыщенным раствором хлорида натрия. Фазу в хлористом метилене сушили над безводным сульфатом натрия, фильтровали, а затем выпаривали при пониженном давлении с получением продукта, 4.19 г (88% выход сырого вещества). Анализ посредством 1Н ЯМР показал чистоту 90%.
Пример 7
Получение продукта из сажи
Сульфаниламид (5.0 г), 14.4 мл 6 N HCl и 25 мл деионизированной воды соединили и смесь нагревали, если это было необходимо для растворения сульфаниламида. Раствор охлаждали в бане с ледяной водой. Сажу с площадью поверхности 58 м2/г и DBPA 46 мл/100 г (100 г) суспендировали в 400 мл ДИ воды, и перемешиваемую суспензию охлаждали в бане с ледяной водой. В приблизительно 20 мл ДИ воды растворили нитрит натрия (2,2 г), и этот раствор в течение нескольких минут добавляли к раствору сульфаниламида.
Через 10-20 минут к суспензии сажи в воде при 0-5oC добавляли раствор соли диазония. В течение примерно 5 минут было очевидно выделение газа. Суспензию удалили из ледяной бани и продолжали перемешивание до тех пор, пока выделение газа больше не наблюдалось. Продукт может быть выделен путем выпаривания воды либо при пониженном давлении при примерно 110oC, либо в лабораторной печи при примерно 125oC. В альтернативном случае продукт может быть выделен путем фильтрования с вакуумом и промывания водой для удаления побочных солей. Продукт имеет присоединенные группы п-C6H4SO2NH2.
Пример 8
Получение продукта из сажи
Повторяли процедуру из примера 7 с заменой сульфаниламида из предыдущей методики на эквимолярное количество натриевой соли ацетилсульфаниламида. Это привело к продукту, имеющему присоединенные группы п-C6H4SO2NHCOCH3.
Пример 9
Получение продукта из сажи
Обработку проводили в условиях, изменяющихся от нейтральных до основных, путем повторения процедуры из примера 7 и добавления к смеси сажи и воды концентрированного гидроксида аммония в небольшом избытке, примерно 5%, по отношению к количеству HCl, используемому на стадии образования диазония. (Обработанная таким образом сажа является несколько более диспергируемой в воде и перед выделением фильтрованием должна быть вначале суспендирована в разбавленном растворе HCl). Продукт имел присоединенные группы п-C6H4SO2NH2.
Пример 10
Получение продукта из сажи
Обработку проводили в условиях, изменяющихся от нейтральных до основных, путем повторения процедуры из примера 8 и добавления к смеси сажи и воды концентрированного гидроксида аммония в небольшом избытке, примерно 5%, по отношению к количеству HCl, используемому на стадии образования диазония. (Обработанная таким образом сажа является несколько более диспергируемой в воде и перед выделением фильтрованием должна быть вначале суспендирована в разбавленном растворе HCl). Это привело к продукту, имеющему присоединенные группы п-C6H4SO2NHCOCH3.
Пример 11
Получение продукта из сажи
Процедура обработки была подобна таковой для примера 7. К раствору 13.1 г сульфаниламида метилового эфира PEG 750 из примера 2 в 100 мл ДИ воды добавили 3.6 мл концентрированной HCl и 50 мл ДИ воды для промывки. Раствор охладили в бане с ледяной водой. Суспензию 25 г сажи с площадью поверхности 58 м2/г и DBPA 46 мл/100 г в 100 мл охладили в бане с ледяной водой.
К раствору сульфаниламида метилового эфира PEG 750 добавили раствор 1.00 г нитрита натрия в 10 мл ДИ воды для образования соли диазония. Было добавлено всего 0.14 г сульфаминовой кислоты (0.1 эквивалент), чтобы снизить избыток азотистой кислоты. Этот раствор разделили на две равные части. Одну часть добавили к водной суспензии сажи в воде. Из суспензии выделялся газ, и перемешивание продолжали до тех пор, пока не перестало обнаруживаться газовыделение. В этом случае сухой продукт из сажи извлекали путем выпаривания в вакуумной печи при 110oC, с получением 30.1 г (примерно 98% от ожидаемого веса). Это привело к продукту, который имел присоединенные группы п-C6H4SO2NH(C2H4O)16CH3.
Пример 12
Получение продукта из сажи
Методика была аналогична таковой из примера 11, в которой снижен уровень обработки. В этом случае для обработки 10 г сажи использовали диазоний, образованный из 1.0 г сульфаниламида PEG 750 (пример 2), 0.27 мл концентрированной HCl и 0.08 г NaNO2. Это привело к продукту, который имел присоединенные группы п-C6H4SO2NH(C2H4O)16CH3.
Пример 13
Получение продукта из сажи
Методика была аналогична таковой из примера 11, осуществленного в половинном масштабе, с использованием диазония, полученного из 3.66 г сульфаниламида PEG 350 из примера 4, 1.82 мл концентрированной HCl и 0.50 г NaNO2. Одна половина данного раствора была использована для того, чтобы обработать 12.5 г сажи. Это привело к продукту, который имел присоединенные группы п-C6H4SO2NH(C2H4O)7CH3.
Пример 14
Получение продукта из сажи
Методика была аналогична таковой из примера 13 с использованием 3.52 г N'-сульфаниламида полиоксиэтилен(2)цетилового эфира из примера 6 вместо сульфаниламида PEG 350. Это привело к продукту, который имел присоединенные группы п-C6H4SO2NH(C2H4O)2C16 H33.
Пример 15
Получение продукта из сажи
Использовали сажу с площадью поверхности 58 м2/г и DBPA, равной 46 мл/100 г. Суспензию 50 г этой сажи получали путем перемешивания ее в 450 г воды. Раствор 0.85 г NaNO2 в 4 г холодной воды медленно добавляли к раствору 0.94 г анилина и 1.98 г концентрированной азотной кислоты в 5 г воды, которую охлаждали в ледяной бане. Образовался нитрат фенилдиазония. После 15 минут перемешивания смесь добавляли к перемешиваемой суспензии сажи. Выделялись пузырьки. Когда выделение пузырьков прекратилось, продукт собирали фильтрованием, дважды промывали водой и сушили в печи при 125oC. Продукт из сажи имел присоединенные фенильные группы.
Пример 16
Получение продукта из сажи
Использовали сажу с площадью поверхности 58 м2/г и DBPA, равной 46 мл/100 г. Суспензию 50 г этой сажи получали путем перемешивания ее в 450 г воды. Раствор 5.12 г NaNO2 в 15 г холодной воды медленно добавляли к раствору 1.40 г п-фенетидина и 1.98 г концентрированной азотной кислоты в 5 г воды, которую охлаждали в ледяной бане. Образовался нитрат 4-этоксифенилдиазония. После 15 минут перемешивания смесь добавляли к перемешиваемой суспензии сажи. Выделялись пузырьки. Через 30 минут продукт собирали фильтрованием, дважды промывали водой и сушили в печи при 125oC. Продукт из сажи имел присоединенные группы п-C6H4OC2H5.
Пример 17
Получение продукта из сажи
Использовали сажу с площадью поверхности 58 м2/г и DBPA, равной 46 мл/100 г. Суспензию 50 г этой сажи получали путем перемешивания ее в 450 г воды. Раствор 0.85 г NaNO2 в 4 г холодной воды медленно добавляли к дисперсии 2.98 г 4-тетрадециланилина, 1.98 г концентрированной азотной кислоты и 4 мл ацетона в 15 г воды, которую охлаждали в ледяной бане. Образовался нитрат 4-тетрадецилфенилдиазония. После 15 минут перемешивания смесь добавляли к перемешиваемой суспензии сажи. Выделялись пузырьки. Через 30 минут продукт собирали фильтрованием, промывали водой и тетрагидрофураном (ТГФ) и сушили в печи при 125oC. Продукт из сажи имел присоединенные группы п-C6H4C14H29.
Пример 18
Получение сульфаниламида полиоксиэтилен(4)лаурилового эфира
В данном примере описано получение сульфаниламида полиоксиэтилен(4)лаурилового эфира, который используют в примерах 20 и 21. К смеси полиоксиэтилен(4)лаурилового эфира (10 г) и 10 мл хлористого метилена в атмосфере азота добавляли пиридин (4.37 г). После того как все материалы растворились, добавляли метансульфонилхлорид (3.48 г) и температуру поддерживали ниже 40oC. Через примерно 1 час реакционную смесь разбавили 10 мл хлористого метилена и перемешивание продолжали 21 час.
Реакцию заканчивали путем разбавления 50 мл хлористого метилена и промывания ДИ водой, разбавленным раствором HCl и разбавленным раствором гидроксида натрия. Раствор в хлористом метилене сушили над безводным сульфатом натрия, фильтровали, а затем проводили испарение при пониженном давлении с получением продукта, 10.49 г (86% выход сырого продукта). Анализ посредством 1Н ЯМР показал чистоту 83%. Продукт - метансульфонат полиоксиэтилен(4)лаурилового эфира использовали в следующей процедуре.
Сульфаниламид (1.88 г) и 85% гидроксид калия (0.61 г) соединяли в атмосфере азота и разбавляли 15 мл ацетонитрила. Смесь нагревали до 60-70oC и затем добавили примерно 3 капли ДИ воды и гидроксид тетрабутиламмония (1.1 мл 1.0 М раствора в метаноле). Добавили метансульфонат полиоксиэтилен(4)лаурилового эфира (4.0 г), растворенного в 5 мл ацетонитрила. Смесь перемешивали при 60-70oC в течение 22 часов. Через 3 часа добавили дополнительное количество ацетонитрила (20 мл).
Реакцию заканчивали путем добавления около 6 капель уксусной кислоты и последующего удаления растворителя при пониженном давлении. Остаток разбавляли 75 мл хлористого метилена и промывали ДИ водой и насыщенным раствором хлорида натрия. Фазу в хлористом метилене сушили над безводным сульфатом натрия, фильтровали, а затем выпаривали при пониженном давлении с получением продукта, 4.27 г (91% выход сырого вещества). Анализ посредством 1Н ЯМР показал чистоту 88%.
Пример 19
Получение N1-гексилсульфаниламида
В данном примере описано получение N1-гексилсульфаниламида, который используют в примерах 22 и 23. Сульфаниламид (10.0 г) и 85% гидроксид калия (2.79 г) соединяли в атмосфере азота и разбавляли 100 мл ацетонитрила. Смесь нагревали до 60-70oC и затем добавили гидроксид тетрабутиламмония (5.6 мл 1.0 М раствора в метаноле). К реакционной смеси добавили н-гексилбромид (9.12 г), после чего промывали 20 мл ацетонитрила. Смесь перемешивали при 60-70oC в течение 23 часов.
Реакцию заканчивали путем удаления растворителя при пониженном давлении с последующим разбавлением 100 мл этилацетата. Этот раствор промывали ДИ водой, насыщенным [раствором] бикарбоната натрия и насыщенным раствором хлорида натрия. Этилацетатную фазу сушили над безводным сульфатом натрия, фильтровали, а затем выпаривали при пониженном давлении с получением продукта, 12.93 г (87% выход сырого вещества). Этот материал перекристаллизовывали из 50% водного этанола с получением 9.90 г продукта (выход 69.8%).
Пример 20
Получение продукта из сажи
Соединяли сульфаниламид полиоксиэтилен(2)лаурилового эфира из примера 18 (3.38 г), 1.63 мл концентрированной HCl, 10 мл ацетона и 50 мл деионизированной воды. Раствор охлаждали в бане с ледяной водой. Сажу с площадью поверхности 58 м2/г и DBPA 46 мл/100 г (11.25 г) суспендировали в 50 мл ДИ воды и перемешиваемую суспензию охлаждали в бане с ледяной водой. В приблизительно 20 мл ДИ воды растворили нитрит натрия (0.50 г), и этот раствор в течение нескольких минут добавляли к раствору сульфаниламида.
Через 10-20 минут при 0-10oC раствор соли диазония был разделен на две равные порции. Одну порцию добавили к суспензии сажи в воде. В течение примерно 5 минут было очевидно выделение газа. Суспензию удалили из ледяной бани и продолжали перемешивание до тех пор, пока выделение газа больше не наблюдалось. Продукт выделяли путем выпаривания воды при пониженном давлении при примерно 110oC. Продукт имеет присоединенные группы п-C6H4SO2NH(C2H4O)4C12 H25.
Пример 21
Получение продукта из сажи в присутствии гидроксида аммония
Сажу с площадью поверхности 58 м2/г и DBPA 46 мл/100 г (11.25 г) суспендировали в смеси 0.68 мл концентрированного гидроксида аммония и 50 мл ДИ воды, и перемешиваемую суспензию охлаждали в бане с ледяной водой. К суспензии сажи в воде добавили оставшуюся порцию раствора соли диазония из примера 20. В течение примерно 5 минут было очевидно выделение газа. Суспензию удалили из ледяной бани и продолжали перемешивание до тех пор, пока выделение газа больше не наблюдалось. Продукт был выделен путем выпаривания воды при пониженном давлении при примерно 110oC. Продукт имеет присоединенные группы п-C6H4SO2NH(C2H4O)4C12 H25
Пример 22
Получение продукта из сажи
Соединяли N1-гексилсульфаниламид из примера 19 (7.43 г), 7.2 мл концентрированной HCl, 20 мл ацетона и 100 мл деионизированной воды. Раствор охлаждали в бане с ледяной водой. Сажу с площадью поверхности 58 м2/г и DBPA 46 мл/100 г (50 г) суспендировали в 200 мл ДИ воды и перемешиваемую суспензию охлаждали в бане с ледяной водой. В приблизительно 20 мл ДИ воды растворили нитрит натрия (2.20 г), и этот раствор в течение нескольких минут добавляли к раствору сульфаниламида.
Через 10-20 минут при 0-10oC раствор соли диазония был обработан 0.28 г сульфамовой кислоты и разделен на две равные порции. Одну порцию добавили к суспензии сажи в воде. В течение примерно 5 минут было очевидно выделение газа. Суспензию удалили из ледяной бани и продолжали перемешивание до тех пор, пока выделение газа больше не наблюдалось. Продукт был выделен путем фильтрования и промывания несколькими порциями ДИ воды. Влажный продукт сушили при пониженном давлении при примерно 110oC. Продукт имел присоединенные группы п-C6H4SO2NHC6H13.
Пример 23
Получение продукта из сажи в присутствии гидроксида аммония
Сажу с площадью поверхности 58 м2/г и DBPA 46 мл//100 г (50 г) суспендировали в смеси 3.0 мл концентрированного гидроксида аммония и 200 мл ДИ воды, и перемешиваемую суспензию охлаждали в бане с ледяной водой. К суспензии сажи в воде добавили оставшуюся порцию раствора соли диазония из примера 22. В течение примерно 5 минут было очевидно выделение газа. Суспензию удалили из ледяной бани и продолжали перемешивание до тех пор, пока выделение газа больше не наблюдалось. Продукт был выделен путем фильтрования и промывания несколькими порциями ДИ воды. Влажный продукт сушили при пониженном давлении при примерно 110oC. Продукт имел присоединенные группы п-C6H4SO2NHC6H13.
Пример 24
Оценка продуктов из сажи в системах глянцевых печатных красок
Продукты из сажи из примеров 7-14 оценивали в стандартном составе глянцевых печатных красок, закрепляющихся при нагревании, полученном на трехвальцовой краскотерке.
Образцы сажи были получены для размалывания на трехвальцовой краскотерке путем ручного смешивания 15 г сажи с 35 г размолотой маточной смеси, состоящей из 9 частей LV-3427XL (связующее для растирания, закрепляющееся при нагревании Lawter International, Northbrook, IL на 1 часть масла MAGIESOL 47), до тех пор, пока весь образец не был равномерно смочен. Смесь размалывали в трехвальцовой краскотерке Kent при 70oF (21.11oC). Образцы разбавляли путем смешения с равным количеством размолотой маточной смеси, а затем для оценки размола подавали в калиброванный прибор для измерения степени перетира производства NIPRI. Стандарты обычно пропускали через краскотерку по четыре раза. Если показание калиброванного прибора для измерения степени перетира было более 20, то проводили дополнительные прогоны. Конечная печатная краска была получена путем смешивания размолотого материала с равным количеством истертой маточной смеси.
MAGIESOL является зарегистрированной торговой маркой масел, доступных от Magie Brothers, Franklin Park, IL.
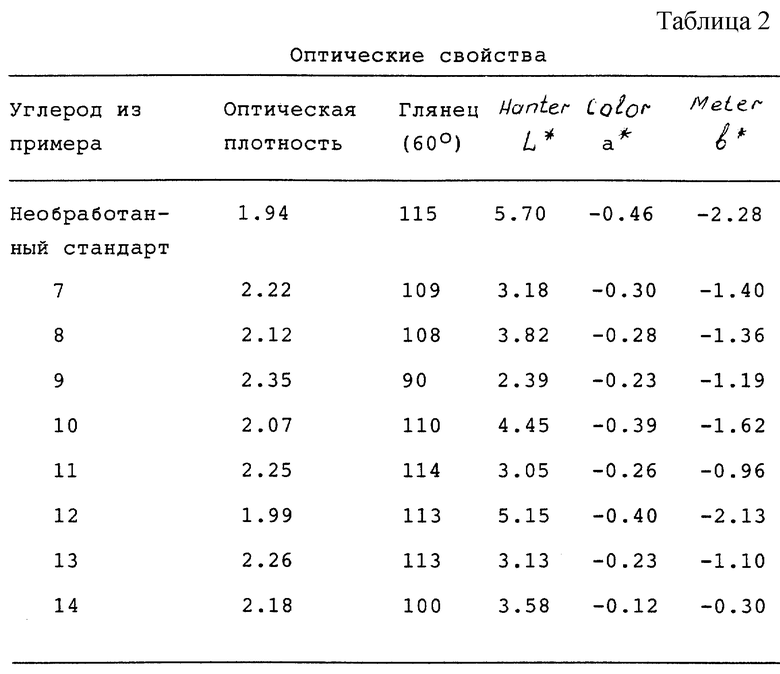
Оптические свойства определяли на мазках в 3 мил (76.2 мкм), которые сушили при 285oF (140.56oC) в течение 3 минут. Для измерения L*, a* и b* использовали [прибор] Hunter Color Meter. Оптическую плотность измеряли с помощью денситометра MacBeth RD 918. Глянец измеряли на приборе для определения глянца BYK Garbner модели 4527.
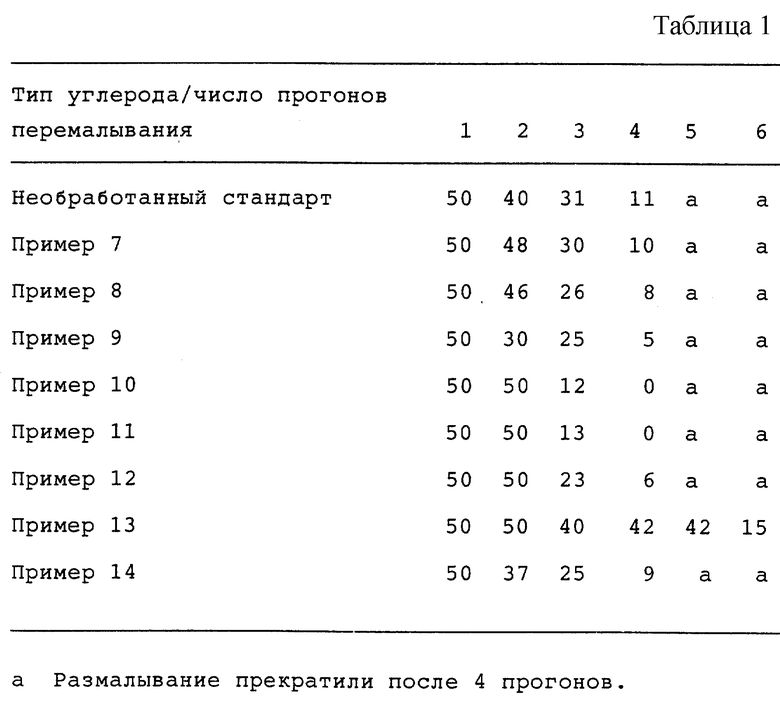
Степень дисперсности помола для продуктов из сажи из примеров 7-14 и оптические свойства полученных печатных красок приведены ниже. Числа в данных по помолу в таблице приведены в микронах, как они были измерены на калиброванном приборе для измерения степени перетира G2, и указывают на уровень, при котором на калиброванном приборе для измерения степени перетира обнаруживаются 3 дефектных зерна.
Эти данные показывают, что все продукты из сажи, описанные в примерах 7-14, можно использовать для получения печатных красок. Большинство продуктов из сажи из примеров приводит к более черным печатным краскам (меньшее значение L*), а некоторые из этих продуктов из сажи диспергируются в связующем при размалывании более быстро, чем стандартная сажа. Улучшенная скорость диспергирования и качество цвета являются очень желательными свойствами для полиграфической промышленности.
Данные по размалыванию (см. табл. 1).
Оптические свойства (см. табл. 2).
Пример 25
Оценка продуктов из сажи в системах глянцевых печатных красок
Продукты из сажи из примеров 15-17 оценивали в таких же композициях глянцевых печатных красок, что и в примере 24, с применением тех же методик. Данные в таблицах показывают, что продукты из сажи по примерам 15-17 также можно использовать для получения печатных красок.
Данные по размалыванию (см. табл. 3).
Оптические свойства (см. табл. 4).
Пример 26
Результаты размалывания в трехвальцовой краскотерке для примеров 20-23
Продукты из сажи из примеров 20-23 оценивали в таких же композициях глянцевых печатных красок, что и в примере 24, с применением тех же методик. Данные в таблицах показывают, что продукты из сажи по примерам 20-23 также можно использовать для получения печатных красок.
Данные по размалыванию (см. табл. 5).
Оптические свойства (см. табл. 6).
Пример 27
Получение продукта из сажи
Соединяли моногидрат натриевой соли ацетилсульфаниламида (44.2 г) и 43.5 мл концентрированной HCl в 300 мл деионизированной воды. Смесь нагревали, если это требовалось для растворения ацетилсульфаниламина, а затем охлаждали в бане с ледяной водой. В отдельном стакане 50 г сажи с площадью поверхности по CTAB 350 м2/г и DBPA 120 мл/100 г (50 г) суспендировали в 400 мл ДИ воды и перемешиваемую суспензию охлаждали в бане с ледяной водой. В приблизительно 20 мл ДИ воды растворили нитрит натрия (6.6 г), и этот раствор добавили к раствору ацетилсульфаниламида. Через 10-20 минут при 0-5oC к суспензии сажи в воде добавили половину раствора соли диазония. В течение примерно 5 минут было очевидно выделение газа. Суспензию удалили из ледяной бани и продолжали перемешивание до тех пор, пока выделение газа больше не наблюдалось. Продукт может быть выделен путем выпаривания воды либо при пониженном давлении при примерно 110oC, либо в лабораторной печи при примерно 125oC. В альтернативном случае продукт можно выделить путем фильтрования с вакуумом и промывания водой для удаления побочных солей. Высушенные в печи сажи можно очистить путем экстракции в аппарате Сокслета с применением 90% этанола в воде. Это привело к продукту, который имел присоединенные группы п-C6H4SO2NHCOCH3.
Пример 28
Получение продукта из сажи
Получение было таким же, как в примере 27, за исключением того, что его проводили в условиях, изменяющихся от нейтральных до основных, путем добавления к смеси сажи и воды концентрированного гидроксида аммония в небольшом избытке, примерно 5%, по отношению к количеству HCl, используемому на стадии образования диазония. (Обработанная таким образом сажа является несколько более диспергируемой в воде и перед выделением фильтрованием должна быть вначале суспендирована в разбавленном растворе HCl). Это привело к продукту, который имел присоединенные группы п-C6H4SO2NHCOCH3.
Пример 29
Получение продукта из сажи
Методика была идентична таковой из примера 28, за исключением того, что в этом примере натриевую соль ацетилсульфаниламида заменили на эквимолярное количество сульфониламида. Это привело к продукту, который имел присоединенные группы п-C6H4SO2NH2.
Пример 30
Получение продукта из сажи
Методика обработки была подобна таковой из примера 28. К раствору 221 г сульфаниламида метилового эфира PEG 750 (из примера 2) чистоты 62% по весу в 2.1 л ДИ воды добавляли 61 мл концентрированной HCl и 100 мл ДИ воды для промывки. Раствор охладили в бане с ледяной водой. К этому раствору добавили раствор 8.0 г (0.95 эквивалента) нитрата натрия в 30 мл ДИ воды для образования соли диазония. Для снижения избытка азотистой кислоты добавили в общей сложности 2.2 г сульфамовой кислоты (0.1 эквивалент).
Сажу с площадью поверхности по CTAB 350 м2/г и DBPA 120 мл/100 г (50 г) суспендировали в смеси 450 мл ДИ воды и 25.5 мл концентрированного гидроксида аммония, а затем охлаждали в бане
с ледяной водой. К суспензии сажи добавили половину раствора соли диазония. При реакции выделялся газ, и перемешивание продолжали до тех пор, пока выделение газа больше не наблюдалось. В этом случае сухую сажу выделяли путем выпаривания в вакуумной печи при 125oC, после чего проводили экстракцию в аппарате Сокслета с применением 90% этанола в воде. Это привело к продукту, который имел присоединенные группы п-C6H4SO2NH(C2H4O)16CH3.
Пример 31
Получение продукта из сажи
Использовали сажу с площадью поверхности по CTAB 350 м2/г и DBPA 120 мл/100 г. Суспензию 50 г этой сажи получали путем перемешивания ее в 450 г воды, которая содержала несколько капель изопропанола. Раствор 5.12 г NaNO2 в 10 г холодной воды медленно добавляли к раствору 5.62 г анилина и 11.88 г концентрированной азотной кислоты в 10 г воды, которая охлаждалась на ледяной бане. Образовался нитрат фенилдиазония. После 15 минут перемешивания смесь добавляли к перемешиваемой суспензии сажи. Выделялись пузырьки. Когда выделение пузырьков закончилось, продукт собирали фильтрованием, дважды промывали водой и сушили в печи при 125oC. Продукт из сажи имел присоединенные фенильные группы.
Пример 32
Получение продукта из сажи
Использовали сажу с площадью поверхности по CTAB 350 м2/г и DBPA 120 мл/100 г. Суспензию 50 г этой сажи получали путем перемешивания ее в 450 г воды, которая содержала несколько капель изопропанола. Раствор 5.12 г NaNO2 в 15 г холодной воды медленно добавляли к раствору 8.40 г п-фенетидина и 11.88 г концентрированной азотной кислоты в 10 г воды, которая охлаждалась на ледяной бане. Образовался нитрат 4-этоксифенилдиазония. После 15 минут перемешивания смесь добавляли к перемешиваемой суспензии сажи. Выделялись пузырьки. Через 30 минут продукт собирали фильтрованием, дважды промывали водой и сушили в печи при 125oC. Продукт из сажи имел присоединенные группы п-C6H4OC2H5.
Пример 33
Получение продукта из сажи
Использовали сажу с площадью поверхности по CTAB 350 м2/г и DBPA 120 мл/100 г. Суспензию 50 г этой сажи получали путем перемешивания ее в 450 г воды, которая содержала несколько капель изопропанола. Раствор 5.12 г NaNO2 в 10 г холодной воды медленно добавляли к дисперсии 17.91 г 4-тетрадециланилина, 11,88 г концентрированной азотной кислоты и 20 мл ацетона в 100 г воды, которая охлаждалась на ледяной бане. Образовался нитрат 4-тетрадецилфенилдиазония. После 15 минут перемешивания смесь добавляли к перемешиваемой суспензии сажи. Выделялись пузырьки. Через 30 минут продукт собирали фильтрованием, промывали водой и тетрагидрофураном (ТГФ) и сушили в печи при 125oC. Продукт из сажи имел присоединенные группы п-C6H4C14H29.
Пример 34
Применение продуктов из сажи в композициях покрытий
В данном примере иллюстрируется применение продуктов из сажи в термореактивных акриловых композициях. В качестве эталонов сажи использовали поверхностно обработанную сажу с площадью поверхности 560 м2/г и DBPA 100 мл/100 г, а также сажу с площадью поверхности по CTAB 350 м2/г и DBPA 120 мл/100 г.
Композиции покрытий получали следующим образом. В каждую из стальных шаровых краскотерок объемом в половину галлона (1.893 л) загрузили: 2.1 кг стальных шариков размера 1/4" (0.635 см), 3.3 кг стальных шариков размера 1/2" (1.27 см), 282 г растирочной маточной смеси (64 части смолы ACRYLOID AT 400 : 30 частей н-бутанола : 6 частей метил-н-амилкетона) и 30 г сажи. Мельничные сосуды вращали при 44 об/мин на краскотерке с вращающимися сосудами, работающей при 82 об/мин (Paul O.Abbe, модель 96806 или эквивалентная) в течение указанного времени. Из краскотерок отбирали пробы непосредственно в калиброванный прибор для определения степени перетира Hegman в указанные моменты времени. Величины степени перетира более 7 обычно принимают как таковые для полного измельчения. Результаты приведены ниже.
Законченную композицию покрытия получали путем проведения первоначального разбавления в каждой краскотерке с помощью 249 г смолы AT-400 и вращения в течение одного 4 часа. Второе разбавление производили путем добавления 304 г смеси 33 частей смолы AT-400; 35.3 частей меламиноформальдегидной смолы CYMEL 303, 7,2 частей метил-н-амилкетона; 8.5 частей целлозольвацетата; 1.8 части CYCAT 4040 (кислотный катализатор из толуолсульфокислоты и изопропилена); 0.3 части добавки FLUORAD FC 431; 14 частей н-бутанола и проводили вращение в течение одного часа.
ACRYLOID является зарегистрированной торговой маркой смол, доступных от Rohm & Haas, Philadelphia, PA. CYMEL и CYCAT являются зарегистрированными торговыми марками продуктов, доступных от American Cyanamid, Stamford, CT. FLUORAD является зарегистрированной торговой маркой добавки к покрытиям, доступной от 3М, St. Paul, MN.
Оптические свойства определяли для пленки толщиной 3 мила (76,2 мкм) на герметизированной шкале Leneta, которую сушили на воздухе в течение 30 минут и затем подвергали термической обработке при 250oF (121.11oC) в течение 30 минут. Данные по растиранию соответствуют величинам по Хегману, когда 5 "песочных" частиц собраны вместе. Величина "0" для песка указывает на то, что он присутствует во время всего проведения измерений.
Эти данные показывают (см. табл. 7), что продукты из сажи из примеров 27-33 можно использовать в термореактивной акриловой композиции. При равных временах перемалывания некоторые из этих продуктов приводят к более черному блестящему покрытию, чем стандарт.
Пример 35
Применение продуктов из сажи в композициях покрытий
В данном примере иллюстрируется применение продуктов из сажи в эмалевых акриловых композициях. В качестве эталона сажи использовали поверхностно обработанную сажу с площадью поверхности 560 м2/г и DBPA 100 мл/100 г. Продукты из сажи из примеров 32 и 33 перед применением подвергали экстракции тетрагидрофураном в аппарате Сокслета.
Размалывание производили на виброгрохоте для красок с помощью смеси 200 г шариков из хромистой стали размером 3/16" (0.476 см), 2.19 г образца сажи и связующего для измельчения, состоящего из 19.9 г смеси (80/20) DMR-499 Acrylic Mixing Enamel [PPG Finishes, Strongsville, OH] и ксилола. Общее время размалывания составляло 2 часа. Образцы оценивали на приборе Хегмана при нижеуказанных интервалах времени. После того как цикл размалывания был завершен, получали конечную композицию путем добавления в каждую краскотерку 23.3 r DMR-499, 17.3 г ксилола и 1.4 г уретанового отвердителя DXR-80 /PPG Finishes, Strongsville, OH] и встряхивания в течение еще 15 минут.
На герметизированной шкале Leneta был сделан мазок 3 мила (76.2 мкм) для каждой из этих композиций. Пленку сушили на воздухе в течение 30 минут и затем подвергали термической обработке при 140oF (60.00oC) в течение 30 минут. Эту пленку использовали для оценки оптических свойств, представленных ниже. Данные по растиранию соответствуют величинам по Хегману, когда 5 "песочных" частиц собраны вместе. Величина "0" для песка указывает на то, что он присутствует во время всего проведения измерений. Эти данные показывают, что продукты из сажи из примеров 29, 32 и 33 можно использовать для получения акриловых эмалевых композиций с хорошим черным блестящим цветом.
Данные по растиранию (см. табл. 8).
Измельчение прерывали на 18 часов, после чего возобновляли.
Оптические свойства (см. табл. 9).
Пример 36
Получение N1-гексаноилсульфаниламида
N1-гексаноилсульфаниламид получали следующим образом. К смеси N4-ацетилсульфаниламида (55.0 г) и пиридина (54 мл) при 95oC добавляли гексаноилхлорид (28.8 г) в течение 20 минут. Через еще один час реакционную смесь охлаждали до комнатной температуры и реакцию заканчивали путем разбавления 600 мл ДИ воды и подкисления водной HCl. Продукт реакции, N4-ацетил-N1-гексаноилсульфаниламид, выделяли фильтрованием и промывали ДИ водой (700 мл) с получением не совсем белого твердого вещества. Этот материал непосредственно переносили на следующую стадию.
N4-ацетил-N1-гексаноилсульфаниламид с предыдущей стадии обрабатывали гидроксидом натрия (24.6 г) в ДИ воде (200 мл) при нагревании до появления флегмы. Через 3 часа реакции нагревание прекращали и раствор pH доводили до 8 с помощью 2 N HCl. Раствор охлаждали в ледяной бане и затем фильтровали для удаления сульфаниламида. Фильтрат подкисляли водной HCl до pH 2, чтобы осадить продукт, N1-гексаноилсульфаниламид, 37.9 г. Этот материал перекристаллизовывали из горячего этанола (75 мл) и осаждали путем добавления 40 мл ДИ воды, получая перекристаллизованный N1-гексаноилсульфаниламид, 24.8 г (суммарный выход 43%). 1H ЯМР показал чистоту > 90%.
Пример 37
Получение N1,N1-бис(2-оксиэтил)сульфаниламида
N1, N1-бис(2-оксиэтил)сульфаниламид был получен следующим образом. (Данный способ частично взят из [работы] G. DiModica и E. Angeletti, Gazz. chim. ital. , 1960, 90, 434-9 [CA 55:11344d]). Смесь карбоната натрия (55.6 г), ДИ воды (120 мл) и диэтаноламина (57.8 г) перемешивали и нагревали до 60-70oC. В течение часа добавляли ацетилсульфанилхлорид (116.8 г) в виде твердого вещества. Во время добавления ацетилсульфанилхлорида порциями добавляли ДИ воду (225 мл) для того, чтобы поддерживать смесь перемешиваемой. Смесь перемешивали еще 3 часа при 60-70oC, после чего оставляли охлаждаться до комнатной температуры в течение примерно 16 часов. Твердые вещества выделяли фильтрованием и промывали 200 мл холодной ДИ воды.
Неочищенный продукт (170 г) гидролизовали путем обработки 5% раствором NaOH (675 мл) при 60oC в течение 4 часов. Водный раствор экстрагировали 3 порциями этилацетата (всего 1.5 л). Экстракты сушили над безводным сульфатом натрия, фильтровали и выпаривали с получением продукта, N1,N1-бис(2-оксиэтил)сульфаниламида в виде не совсем белого твердого вещества, 72.1 г (выход 55.5% неочищенного продукта).
Пример 38
Получение продукта из сажи
Данный пример отражает альтернативный способ, в котором не требуется применение добавленной кислоты. Ацетилсульфаниламид (10 г) растворяли в 500 мл ДИ воды при 90oC. К данному раствору добавили сажу с площадью поверхности 58 м2/г и DBPA 46 мл/100 г (100 г). После того как сажа была введена в смесь, к суспензии добавили нитрит натрия (3.24 мл) в ДИ воде (10 мл). Немедленно выделялся газ. Нагрев был приостановлен и смесь перемешивали и оставляли охлаждаться до температуры окружающей среды. Суспензию сажи выпаривали досуха при 65oC. Продукт имел присоединенные группы п-C6H4SO2NHCOCH3.
Пример 39
Получение продукта из сажи
Сульфабензамид (26.0 г), 23.5 мл концентрированной HCl, 25 мл ацетона и 100 мл ДИ воды соединяли и перемешивали. Сажу с площадью поверхности 58 м2/г и DBPA 46 мл/100 г (100 г) суспендировали в 500 мл ДИ воды и перемешиваемую суспензию охлаждали в бане с ледяной водой. Нитрит натрия (6.50 г) растворили в примерно 25 мл ДИ воды и этот раствор в течение нескольких минут добавляли к раствору сульфабензамида.
Через 10-20 минут раствор соли диазония разделили на две равные порции. Одну порцию добавили к суспензии сажи в воде. Сразу же было очевидно выделение газа. Перемешивание продолжали до тех пор, пока выделение газа больше не наблюдалось. Продукт выделяли путем фильтрования и промывали ДИ водой. Продукт сушили при 75oC в течение 16 часов. Продукт имел присоединенные группы п-C6H4SO2NHCOC6H5.
Пример 40
Получение продукта из сажи в присутствии гидроксида аммония
Сажу с площадью поверхности 58 м2/г и DBPA 46 мл/100 г (100 г) суспендировали в смеси 9.8 мл концентрированного гидроксида аммония и 500 мл ДИ воды и перемешивали. Оставшуюся порцию раствора соли диазония из примера 39 добавили к суспензии сажи в воде. Сразу же было очевидно выделение газа. Перемешивание продолжали до тех пор, пока выделение газа больше не наблюдалось. Продукт выделяли путем фильтрования и промывания ДИ водой, а затем сушили при 75oC в течение 16 часов. Продукт имел присоединенные группы п-C6H4SO2NHCOC6H5.
Пример 41
Получение продукта из сажи
Соединяли N1-гексаноилсульфаниламид (12.7 г) из примера 36, 11.8 мл концентрированной HCl, 40 мл ацетона и 200 мл ДИ воды. Раствор охлаждали в бане с ледяной водой. Сажу с площадью поверхности 58 м2/г и DBPA 46 мл/100 г (50 г) суспендировали в 200 мл ДИ воды и перемешиваемую суспензию охлаждали в бане с ледяной водой. Нитрит натрия (3.24 г) растворили в примерно 20 мл ДИ воды и этот раствор в течение нескольких минут добавляли к раствору N1-гексаноилсульфаниламида.
Через 10-20 минут при 0-10oC раствор соли диазония разделили на две равные порции. Одну порцию добавили к суспензии сажи в воде. Сразу же было очевидно выделение газа. Смесь удаляли из ледяной бани и перемешивание продолжали до тех пор, пока выделение газа больше не наблюдалось. Продукт выделяли путем фильтрования и промывания несколькими порциями ДИ воды. Влажный продукт сушили при 75oC. Продукт имел присоединенные группы п-C6H4SO2NHCOC5H11.
Пример 42
Получение продукта из сажи в присутствии гидроксида аммония
Сажу с площадью поверхности 58 м2/г и DBPA 46 мл/100 г (50 г) суспендировали в смеси 4.9 мл концентрированного гидроксида аммония и 200 мл ДИ воды, и перемешиваемую суспензию охлаждали в бане с ледяной водой. Оставшуюся порцию раствора соли диазония из примера 41 добавили к суспензии сажи в воде. Сразу же было очевидно выделение газа. Суспензию удаляли из ледяной бани и перемешивание продолжали до тех пор, пока выделение газа больше не наблюдалось. Продукт выделяли путем фильтрования и промывания несколькими порциями ДИ воды. Влажное твердое вещество сушили при 75oC. Продукт имел присоединенные группы п-C6H4SO2NHCOC5H11.
Пример 43
Получение продукта из сажи
Соединяли N1, N1-бис(2-оксиэтил)сульфаниламид (66.3 г) из примера 37, 66.8 мл концентрированной HCl и 350 мл ДИ воды. Раствор охлаждали в бане с ледяной водой. Сажу с площадью поверхности по CTAB 350 м2/г и DBPA 120 мл/100 г (150 г) суспендировали в 1,500 мл ДИ воды и перемешиваемую суспензию охлаждали в бане с ледяной водой. Нитрит натрия (17.6 г) растворили в примерно 60 мл ДИ воды и этот раствор в течение нескольких минут добавляли к раствору N1, N1-бис(2-оксиэтил)сульфанилмида.
Через 10-20 минут при 0-10oC раствор соли диазония добавили за одну порцию к суспензии сажи в воде. Сразу же было очевидно выделение газа. Смесь удаляли из ледяной бани и перемешивание продолжали до тех пор, пока выделение газа больше не наблюдалось. Продукт выделяли путем фильтрования и промывания несколькими порциями ДИ воды. Влажный продукт сушили при 60oC. Продукт имел присоединенные группы п-C6H4SO2N(C2H4OH)2.
Пример 44
Получение продукта из сажи
Сульфабензамид (93.9 г) растворили в растворе 340 мл 1 N гидроксида натрия и 1,700 мл ДИ воды. К этому раствору добавили сажу с площадью поверхности по CTАB 350 м2/г и DBPA 120 мл/100 г (200 г). Через 15 минут к суспензии медленно добавили 85 мл концентрированной HCl. Нитрит натрия (23.5 г) растворили в примерно 75 мл ДИ воды и этот раствор в течение нескольких минут добавляли к раствору сульфабензамида. Сразу же было очевидно выделение газа. Смесь удаляли из ледяной бани и перемешивание продолжали до тех пор, пока выделение газа больше не наблюдалось. Продукт выделяли путем фильтрования и промывания несколькими порциями ДИ воды. Влажный продукт сушили при 60oC. Продукт имел присоединенные группы п-C6H4SO2NHCOC6H5.
Пример 45
Результаты размалывания на трехвальцовой краскотерке для примеров 39-42
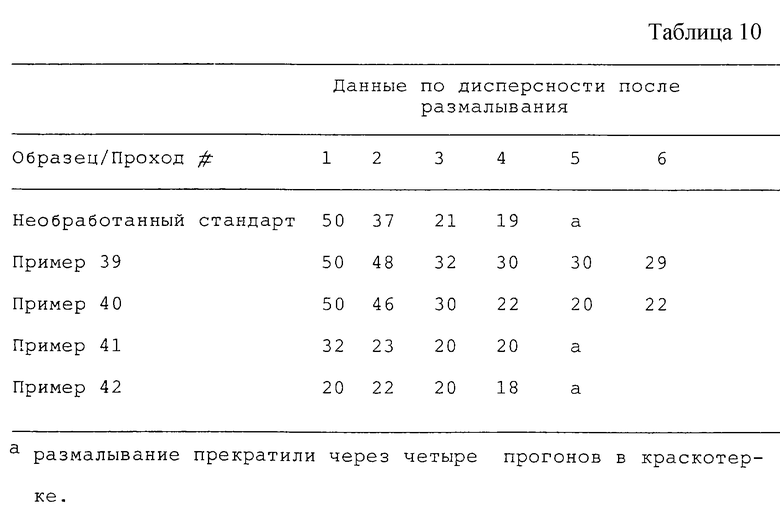
Продукты из сажи по примерам 39-42 (см. табл. 10) оценивали на скорость внедрения и оптические свойства в соответствии с методами, описанными в примере 24. Данные из нижеприведенных таблиц показывают, что продукты по примерам 39-42 можно использовать для получения печатной краски. Продукты по примерам 41 и 42 продемонстрировали усиленное внедрение в наполнитель для размалывания.
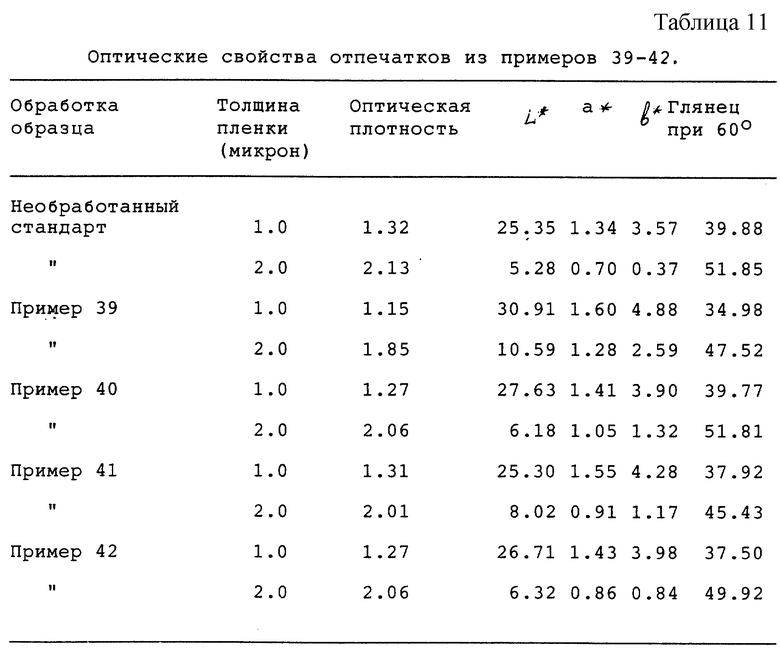
Оптические свойства печатных красок, изготовленных из продуктов из сажи по примерам 39-42, и необработанного стандарта определяли из отпечатков, изготовленных с использованием пробопечатного устройства RNA-52 (Research North America Inc.), и они приведены ниже в табл. 11. Величины для толщины пленок 1.0 и 2.0 микрон рассчитывали из линейной регрессии данных для отпечатков, характеризующихся интервалом толщины пленок.
Оптические свойства отпечатков из примеров 39-42 (см. табл.11).
Пример 46
Применение продуктов из сажи в композициях покрытий
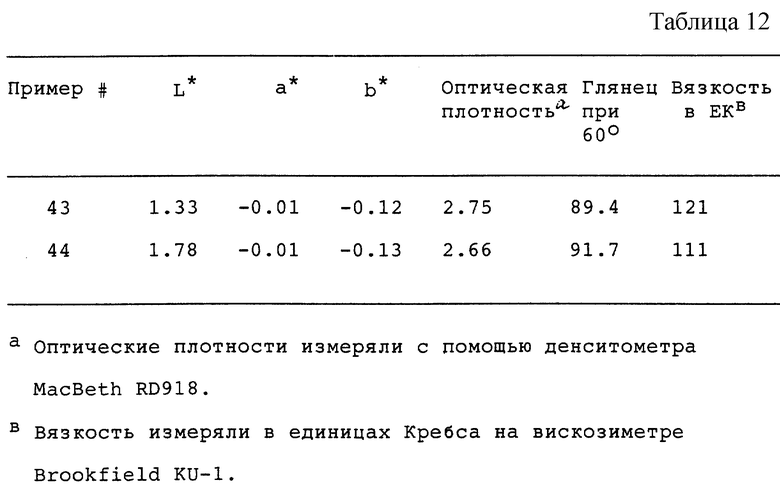
Продукты из сажи из примеров 43 и 44 (см. табл. 12) использовали в термореактивной акриловой композиции так, как описано в примере 34. Ниже обобщены оптические свойства этих покрытий в результате размалывания в шаровой краскотерке в течение 96 часов. Эти данные показывают, что продукты из сажи по примерам 43 и 44 можно использовать в термореактивной акриловой композиции.




Описывается новая композиция неводного покрытия, содержащая продукт из модифицированного углерода. При этом указанный продукт содержит углерод с присоединенной к нему замещенной или незамещенной ароматической группой. Описывается также продукт из модифицированного углерода. Описывается также способ улучшения оптических свойств композиции. Технический результат - создание водонепроницаемых покрытий с улучшенными характеристиками. Такие неводные покрытия применяют на металлических и пластмассовых частях автомобилей. 4 с. и 35 з.п. ф-лы, 12 табл.
| US 2867540 A, 06.01.1959 | |||
| Черная печатная краска | 1972 |
|
SU507609A1 |
| Устройство для вытягивания из расплава монокристаллической нити | 1974 |
|
SU475075A1 |

