Изобретение относится к новым 8-арил-1,7-нафтиридинам, к способам их получения, к их применению в качестве лекарственных средств и к содержащим их фармацевтическим композициям.
В настоящем изобретении предложены 8-арил-1,7-нафтиридины в свободной форме или в форме фармацевтически приемлемой соли. Под понятием "арил" подразумевают моно- или бициклический ароматический или гетероароматический фрагмент, который имеет до 10 ароматических атомов, отличных от водорода, и который связан с 1,7-нафтиридином либо непосредственно (например, фенил, пиридил, тетразолил, бензофуразанил или бензотиофуразанил), либо через метиленовый мостик (например, бензил или пиридилметил); предпочтителен при этом моноциклический ароматический фрагмент, имеющий до шести ароматических атомов углерода, из которых до 2 атомов могут быть замещены азотом, например фенил, бензил, 4-пиридил или 4-пиридилметил, каждый из которых необязательно включает карбоксигруппу, эфир карбоновой кислоты или гидроксигруппу. 8-арильный фрагмент необязательно может быть дополнительно замещен, предпочтительно заместителем, являющимся акцептором электрона, например нитро-, нитрило-, иминогруппой, галогеном или галогенсодержащим заместителем (например, трифторметилом) или цианогруппой, предпочтительно, в мета-положении. Например, 8-арильный фрагмент может представлять собой цианфенил, нитрофенил, тетразолилфенил (например, тетразол-1-илфенил) или хлорфенил. Необязательно двойное кольцо в нафтиридиновом фрагменте молекулы может также быть дополнительно замещено одним или двумя заместителями, предпочтительно замещено в положении 6 гидроксигруппой, алкилом, алкенилом, алкинилом, алкоксигруппой, арилом, арилокси-, амино-, ариламино-, диариламино-, алкиламино-, диалкиламино-, ариламидо- или алкамидогруппой, где "алк" обозначает алифатический фрагмент, имеющий до 8 атомов углерода и необязательно включающий карбоксигруппу, эфир карбоновой кислоты либо гидроксигруппу и/или необязательно содержащий простую эфирную связь и/или сложноэфирную связь.
В частности, в изобретении предложены новые 8-фенил- и 8-бензил-1,7-нафтиридины, в которых двойная связь 1,7-нафтиридина необязательно замещена в положении 6, как, в частности, указано ниже в примерах, а фенильное кольцо необязательно замещено заместителем, являющимся акцептором электронов, таким как нитрогруппа; например, соединения формулы I:
где R1 обозначает фенил, бензил, 3-нитрофенил, 3-хлорфенил, 3- цианфенил, 3-(тетразолил) фенил, бензофуразанил или бензотиофуразанил;
R2 обозначает гидроксигруппу, трифторметансульфонилокси, аллил, алкил, алкенил, алкинил, алкоксигруппу, арил, аралкил, арилокси-, амино-, ариламино-, диариламино-, алкамино-, диалкаминогруппу, алкарил, ариламидо- или алкамидогруппу;
и их сложные эфиры и амиды в свободной форме или в форме фармацевтически приемлемой соли.
В формуле I и далее "алк" и "арил" имеют значения, указанные выше для 8-арил-1,7-нафтиридинов по изобретению.
Предпочтительно R2 выбирают из группы, включающей гидрокси-, амино-, ариламино- (например, фениламиногруппу), арил (например, фенил), алкарил [например, (низш.)алкилфенил], алкенил (например, винил), алкинил (например, этинил), алкоксигруппу, содержащую простую эфирную и/или сложноэфирную связь (например, метоксикарбонилметоксигруппу) и алкамидо-(например, ацетамидогруппу).
Неожиданно было установлено, что совершенно новый класс 6,8-арил-1,7-нафтиридинов пригоден для применения в качестве лекарственных средств, в частности в качестве активных при оральном введении ингибиторов PDE 4, например, для лечения астмы.
Таким образом, предпочтительными согласно изобретению являются 6-(карбоксифенил- или -карбоксиметилфенил)-8-(фенил,бензо [с] тиадиазолил или бензо[с] фуразанил)-1,7-нафтиридины и их сложные эфиры и амиды в свободной форме или в форме фармацевтически приемлемой соли.
Более предпочтительно изобретение относится к 6-(4-карбоксифенил- или -4-карбоксиметилфенил)-8- (фенил, 4-бензо [с] тиадиазолил или 4-бензо [с] фуразанил)-1,7- нафтиридинам и к их сложным эфирам и амидам в свободной форме или в форме фармацевтически приемлемой соли.
Под бензо [с] тиадиазолилом и бензо [с] фуразанилом понимают радикалы формул А и Б соответственно: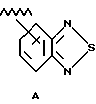

Так, в особенно предпочтительном варианте изобретение относится к соединению формулы II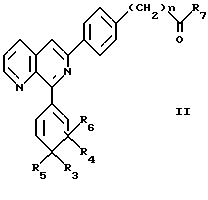
где n равно 0 или 1;
R7 обозначает гидрокси-, амино-, C1- C4алкиламино- или C1-C4алкоксигруппу, предпочтительно гидрокси- или аминогруппу;
и либо
R3 обозначает H и R4 обозначает нитрогруппу, галоген (например, хлор), цианогруппу или тетразолил (например, 1-тетразолил) и R5 и R6 вместе образуют дополнительную связь, либо
R3, R4, R5 и R6 вместе обозначают =N-O-N= или =N-S-N=;
и к их сложным эфирам и амидам в свободной форме или в форме фармацевтически приемлемой соли.
Пригодные фармацевтически приемлемые соли 8-арил-1,7-нафтиридинов, например, формулы I или II, для применения в качестве лекарственных средств получают общепринятыми способами. Например, соединения, имеющие свободную группу карбоновой кислоты, например соединения формулы II, в которых R7 обозначает ОН, могут быть подвергнуты взаимодействию с пригодным основанием, например аминосахаром, таким как N-метилглюкамин, с получением соответствующей соли присоединения основания. Как правило, такие соли присоединения оснований являются водорастворимыми.
8-арил-1,7-нафтиридины по изобретению, например, формулы I могут быть получены взаимодействием 2-циан-3- пиридилацетонитрила
а) с кислотой, например H-A, где А обозначает галоген, например бром, с получением соединения формулы III: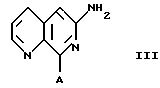
которое может быть подвергнуто последующей дериватизации с получением cоединений по изобретению, например, с помощью реакции сопряжения с использованием металлсодержащих реагентов в присутствии палладиевого или никелевого катализатора с получением углерод-углеродной связи, например, по реакции Стилла, Сузуки или Хека, т.е. взаимодействием соединения формулы III с соединением Y-R8, где Y обозначает металлсодержащую уходящую группу, например В(ОН)2, (CH2)3Sn-, (CH3(CH2)3)3Sn-, a R8 обозначает 8-арильный фрагмент, как указано выше, например бензил или 3-нитрофенил, с получением соответствующего соединения формулы IV
или
б) с реагентом Гриньяра, например R8-MgBr, где R8 обозначает 8-арильный фрагмент, как указано выше, например бензил или 3-нитрофенил, с получением соответствующего соединения формулы IV, или
в) со смесью щелочного металла-спирта, например с натрий-метанолом, с получением соединения формулы III'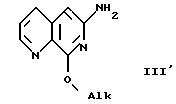
где Alk обозначает C1-C8алкильную группу, например метил. Заместитель O-Alk может быть превращен в галоген А и после этого соединение может быть подвергнуто дериватизации в положении 8, как описано выше для соединения формулы III.
Аминогруппу соединений формулы III, III' или IV делают пригодной для последующего взаимодействия, например,
(I) активацией с помощью пригодного активирующего реагента, например трифторметансульфоновой кислоты и NaNO2, с получением трифлата формулы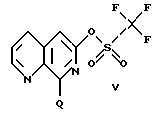
где Q обозначает либо галоген А, как указано выше, заместитель -O-Alk, как указано выше для соединения формулы III', либо 8-арильный фрагмент R8, как указано выше, который является новым и очень важным промежуточным продуктом для получения соединений по изобретению, например, когда радикал R2 в соединении формулы I связывают с остальной частью молекулы через углерод-углеродную связь, замещение может быть осуществлено, например, с помощью реакции сопряжения с использованием металлсодержащих реагентов в присутствии палладиевого или никелевого катализатора, с получением углерод-углеродной связи, например, по реакции Стилла, Сузуки или Хека, т.е. взаимодействием соединения формулы V, где Q обозначает R8, с соединением формулы Y-R2, где Y обозначает металлсодержащую уходящую группу, как указано выше, и R2 обозначает требуемый углеродный заместитель, например алкил, алкенил, алкинил, арил или алкарил, как указано выше для формулы I, необязательно в защищенной форме, при необходимости с последующим удалением защитной группы, или
(II) алкильным или арильным замещением (например, путем взаимодействия с соответствующим алкилгалогенидом или металлорганическим соединением) с получением требуемого вторичного или третичного амина, или
(III) ацилированием (например, путем взаимодействия с карбоновой кислотой или с ангидридом карбоновой кислоты) с получением соответствующего амида, используя общепринятые методики, или
(IV) превращением в гидроксигруппу, например, взаимодействием с NaNO2 в присутствии разбавленной кислоты, например серной кислоты, и необязательно последующей дериватизацией, например, О-алкилированием, например, путем взаимодействия с алкилгалогенидом в пригодных реакционных условиях.
Предпочтительные 6-(карбоксифенил- или карбоксиметилфенил)-8-(фенил, бензо[с] тиадиазолил или бензо[с]фуразанил)-1,7-нафтиридины или их сложные эфиры или амиды, например, формулы II, как правило, получают следующим способом:
(А) для получения 6-(карбоксифенил- или карбоксиметилфенил)-8- (фенил, бензо[с] тиадиазолил или бензо[с]фуразанил)-1,7- нафтиридинов или их сложных эфиров или амидов осуществляют взаимодействие 6-Х-8-(фенил, бензо[с]тиадиазолил или бензо[с]фуразанил)-1,7-нафтиридина с Х'-(карбокси-или карбоксиметил-фенилом) или его сложным эфиром или амидом либо осуществляют взаимодействие 6-(карбокси- или карбоксиметил-фенил)- 8-Х-1,7-нафтиридина или его сложного эфира или амида с X'- (фенилом, бензо[с]тиадиазолилом или бензо[с] фуразанилом), где X и X' обозначают уходящие группы, способные принимать участие в реакции сопряжения, например, где X обозначает трифторметансульфонил или галоген, например, бром или хлор, а X' обозначает металлсодержащую уходящую группу, например, замещенный бор (например, -В(ОН)2, -B(OAlk)2 или BAlk2, где Alk обозначает алкил, например, метил или этил), или триалкилстаннил (например, (CH3(CH2)3)3Sn- или (CH3)3Sn, или радикал Гриньяра (например, MgBr), и/или
(Б) осуществляют необязательное взаимодействие 6-(карбокси- или карбоксиметил-фенил)-8-(фенил, бензо[с] тиадиазолил или бензо[с]фуразанил)-1,7-нафтиридина с пригодным амином, например, с аммиаком или с C1-C4алкиламином, с получением соответствующего амида, и/или
(В) осуществляют необязательное взаимодействие 6-(карбокси- или карбоксиметил-фенил)-8-(фенил, бензо [с] тиадиазолил или бензо [с] фуразанил)-1,7- нафтиридина с пригодным спиртом, например, с C1-C4спиртом, с получением соответствующего сложного эфира и выделяют образовавшийся 6-(карбокси- или карбоксиметил-фенил)-8-(фенил, бензо [с] тиадиазолил или бензо [с] фуразанил)-1,7- нафтиридин или его сложный эфир или амид в свободной форме или в форме соли.
Реакционные условия для реакций стадии (А) являются известными по реакциям сопряжения, осуществляемым с использованием металлсодержащих реагентов, например, в присутствии палладиевого или никелевого катализатора, с получением углерод-углеродной связи, например, как в случае реакций Стилла, Сузуки или Хека. Реакционные условия для реакций стадий (Б) и (В) являются известными по технологии получения амидов с помощью взаимодействия карбоновых кислот с аминами или получения эфиров из карбоновых кислот и спиртов, например, в кислых или щелочных условиях. Выделение и очистку проводят обычными методами, например хроматографией или кристаллизацией.
Таким образом, в предпочтительном варианте изобретение относится к способу получения соединения формулы II, как описано выше, включающему взаимодействие соединения формулы VI
где R3, R4, R5, R6 и X имеют указанные выше значения, с соединением формулы VII
где R7 и X' имеют указанные выше значения, и выделение полученного таким путем соединения формулы II в свободной форме или в форме соли.
6-Х-8-(фенил, бензо [с] тиадиазолил или бензо[с]фуразанил)-1,7-нафтиридины и 6-(карбоксифенил или карбоксиметилфенил)-8-Х-1,7-нафтиридины или их сложные эфиры или амиды, предназначенные для использования в указанных выше реакциях, как правило, получают взаимодействием 2-циан-3-пиридилацетонитрила с кислотой, например с H-A, где А обозначает галоген, например бром, с получением соединения формулы III, которое при необходимости может быть подвергнуто последующей дериватизации, как описано выше.
Описанные выше способы получения 8-арил-1,7-нафтиридинов по изобретению, в частности предпочтительных 6-(карбоксифенил или карбоксиметилфенил)-8-(фенил, бензо [с] тиадиазолил или бензо [с] фуразанил)-1,7-нафтиридинов и их сложных эфиров и амидов, являются новыми, поскольку новыми являются промежуточные продукты формул IV, V и VI, и эти новые способы и промежуточные продукты включены в объем настоящего изобретения. Очевидно, что соединения формулы V, в которых Q обозначает R8, и соединения формулы IV подпадают под определение 8-арил-1,7-нафтиридинов по изобретению.
Таким образом, другими объектами изобретения являются промежуточные продукты формулы V'
в которых Q' обозначает галоген или -O-Alk, где Alk обозначает C1-С8алкильную группу, и формулы VI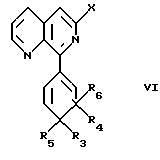
где R3, R4, R5, R6 и X имеют указанные выше значения.
Ниже изобретение проиллюстрировано на примерах (cм. табл. 2).
ПРИМЕРЫ
Пример 1: 6-амино-8-(3-нитрофенил)-1,7-нафтиридин
А. 2-циан-З-пиридилапетонитрил
К перемешиваемой суспензии 3-цианметилпиридин-N-оксида (30 г, 0,22 моля; синтез см. у Shigenobu Okuda, Michael М. Robison, J. Am. Chem. Soc. 81, 704 (1959)) в дихлорметане (200 мл) добавляют триметилсиланкарбонитрил (26 г, 0,26 моля). К этой суспензии добавляют диметилкарбамилхлорид (28 г, 0,26 моля). Смесь перемешивают в течение 45 ч. Растворитель удаляют и остаток растворяют в этилацетате. Раствор промывают 1н. NaOH и водой и концентрируют в вакууме. Продукт очищают с помощью колоночной экспресс-хроматографии на силикагеле (толуол/ацетон в соотношении 15:2), получая указанное в заголовке соединение. МС: М+H 144,1; tпл 62-63oC.
Б. 6-амино-8-бром-1,7-нафтиридин
Перемешиваемый раствор 2-циан-3-пиридилацетонитрила (3,6 г, 0,025 моля) в толуоле (80 мл) барботируют HBr в течение 5 ч. Затем осторожно добавляют 4н. NaOH и суспензию интенсивно перемешивают. Смесь фильтруют и продукт промывают водой и сушат. Кристаллизацией из толуола получают указанное в заголовке соединение. МС: М+H 225; tпл 188oC, разложение.
В. 6-амино-8-(3-нитрофенил)-1.7-нафтиридин
К перемешиваемому раствору 6-амино-8-бром-1,7-нафтиридина (4 г, 0,018 моля) в смеси тетрагидрофурана (80 мл) и водного Na2CO3 (34 мл, 2н.) добавляют бис(дибензилиденацетон)палладий (0,40 г, 0,0007 моля), трифенилфосфен (0,37 г, 0,0014 моля) и 3-нитрофенилбороновую кислоту (3,7 г, 0,022 моля). Смесь перемешивают в течение 16 ч при 80oC. Смесь фильтруют, добавляют этилацетат и смесь промывают 2н. NaOH и водой. Органический растворитель удаляют и остаток суспендируют в простом эфире. После фильтрации получают указанное в заголовке соединение. МС: М+H 267; tпл 221-223oC.
Пример 2: 6-амино-8-(4-бензо[с]фуразанил)-1,7-нафтиридин
К перемешиваемому раствору 6-амино-8-бром-1,7-нафтиридина (3,0 г, 13,4 ммоля) в ДМФ (50 мл) добавляют бис(дибензилиденацетон)палладий (308 мг, 0,54 ммоля), трифенилфосфин (565 мг, 2,15 ммоля) и 4-триметилстаннил- бензо[с]фуразанил (4,92 г, 16,0 ммолей). Смесь выдерживают при 125oC в течение 4 ч. Добавляют этилацетат (500 мл), затем водный KF (40%, 100 мл). Смесь интенсивно перемешивают в течение 45 мин и фильтруют. Органическую фазу отделяют, промывают водой и концентрируют. Остаток растворяют в простом эфире (20 мл), перемешивают в течение 30 мин (0oC) и фильтруют, получая указанное в заголовке соединение (2,8 г). МС: М+H 264; tпл 244-250oC.
Пример 3: 6-гидрокси-8-(3-нитрофенил)-1,7-нафтиридин
К перемешиваемому раствору 6-амино-8-(3-нитрофенил)-1,7-нафтиридина (500 мг, 1,87 ммоля; полученного согласно примеру 1) в концентрированной серной кислоте и воде (3 мл, 2:1), добавляют при 4oC нитрат натрия (155 мг, 2,25 ммоля). Через 30 мин ледяную баню удаляют и смесь нагревают до 70oC в течение 30 мин. Реакционную смесь сливают на лед и раствор нейтрализуют, добавляя бикарбонат натрия. Осадок фильтруют и промывают водой, получая указанное в заголовке соединение, tпл 262-265oC.
Пример 4: 6-метоксикарбонилметокси-8- (3-нитрофенил)-1,7-нафтиридин
Суспензию, содержащую 6-гидрокси-8-(3-нитрофенил)-1,7-нафтиридин (107 мг, 0,40 ммоля, полученный согласно предыдущему примеру, необязательно без дополнительной очистки), карбонат калия (55 мг, 0,40 ммоля) и метиловый эфир бромуксусной кислоты (37 мкл, 0,4 ммоля), перемешивают в течение 2 ч в смеси ацетона и диметилформамида (2 мл, 1:1) при комнатной температуре. Добавляют этилацетат и органическую фазу промывают 2н. NaOH. Растворитель удаляют в вакууме и неочищенный продукт очищают препаративной тонкослойной хроматографией, получая указанное в заголовке соединение. МС: М+H 340; tпл 160-162oC.
Пример 5: Гидрохлорид 6-ацетамидо-8-(3-нитрофенил)-1,7-нафтиридина
Суспензию, содержащую 6-амино-8-(3-нитрофенил)-1,7-нафтиридин (300 мг, 1,1 ммоля; полученный согласно примеру 1) и ангидрид уксусной кислоты (0,12мл, 1,2 ммоля) в пиридине (1 мл) и диметилформамиде (3 мл), выдерживают при 80oC в течение 3 ч. Добавляют воду и осадок фильтруют и тщательно промывают водой. После фильтрации получают указанный в заголовке продукт. МС: М+H 308; tпл 235-238oC.
Пример 6: 6-амино-8-бензил-1,7-нафтиридин
К раствору 2-циан-3-пиридилацетонитрила (2 г, 0,014 моля) в толуоле (20 мл) добавляют при температуре окружающей среды бензилмагнийбромид (8,4 мл, 2М в тетрагидрофуране, 0,17 моль). Через 1 ч реакцию прекращают с помощью насыщенного раствора хлорида аммония. Продукт экстрагируют этилацетатом и органический слой промывают 2н. NaOH и водой. Продукт очищают с помощью колоночной экспресс-хроматографии на силикагеле (10:3 толуол/ацетон), получая указанное в заголовке соединение. МС: М+H 236; tпл 113-116oC.
Пример 7: 6-фениламино-8-(3-нитрофенил)-1,7-нафтиридин
К раствору трифенилвисмута (182 мг, 0,41 ммоля) в дихлорметане (0,5 мл) и тетрагидрофуране (0,5 мл) добавляют перуксусную кислоту (0,083 мл, 40%). Смесь перемешивают в течение 1 ч. Затем добавляют раствор 6-амино-8-(3-нитрофенил)-1,7-нафтиридина (100 мг, 0,37 ммоля; получен согласно примеру 1) в дихлорметане (0,5 мл) и ТГФ (0,5 мл). Добавляют медь (30 мг, 0,47 ммоля) и суспензию перемешивают в течение 40 ч. Суспензию разбавляют этилацетатом и фильтруют. Фильтрат промывают 2н. Na2CO3 и водой и органический слой концентрируют в вакууме. С помощью препаративной тонкослойной хроматографии (дихлорметан/н-гексан в соотношении 8:2) получают указанное в заголовке соединение. МС: М+Н 343,1; tпл 158-160oC.
Пример 8: 6-трифторметансульфонилокси-8-(3-нитрофенил)-1,7-нафтиридин
К раствору 6-амино-8-(3-нитрофенил)-1,7-нафтиридина (1,24 г, 0,0047 моля; полученного согласно примеру 1) в трифторметансульфоновой кислоте (12 мл) добавляют в виде нескольких порций нитрит натрия (0,64 г, 0,0093 моля). Раствор нагревают до 60oC и перемешивают в течение ночи. Раствор сливают на смесь этилацетата и льда. Затем добавляют 2н. NaOH до тех пор, пока значение pH водной фазы не станет щелочным. Органическую фазу промывают водой и концентрируют в вакууме. Продукт очищают с помощью колоночной экспресс-хроматографии на силикагеле (гексан/этилацетат в соотношении 7:3), получая указанное в заголовке соединение. МС: М+H 400; tпл 106-108oC.
Пример 9: 6-фенил-8-(3-нитрофенил)-1,7-нафтиридин
К раствору 6-трифторметансульфонил-8-(3-нитрофенил) -1,7-нафтиридина (300 мг, 0,75 ммоля; полученного согласно примеру 8) в тетрагидрофуране (5 мл), добавляют фенилбороновую кислоту (118 мг, 0,97 ммоля), бис(дибензилиденацетон)палладий (18 мг, 0,03 ммоля), трифенилфосфин (16 мг, 0,06 ммоля) и водный раствор Na2CO3 (2н., 1,44 мл). Раствор выдерживают при 60oC в течение 20 ч. Раствор разбавляют этилацетатом, фильтруют и промывают 1н. NaOH и водой. Растворитель выпаривают в вакууме, получая чистый продукт. MC: M+H 328; tпл 172-175oC.
Пример 10: 6-винил-8-(3-нитрофенил)-1,7-нафтиридин
К раствору 6-трифторметансульфонил-8-(3-нитрофенил)- 1,7-нафтиридина (250 мг, 0,62 ммоля; получен согласно примеру 8) в тетрагидрофуране (3 мл) добавляют винилтрибутилстаннан (218 мг, 0,68 ммоля), бис(дибензилиденацетон)палладий (14 мг, 0,025 ммоля), трифенилфосфин (13 мг, 0,049 ммоля) и хлорид лития (78 мг, 1,86 ммоля). Смесь выдерживают при 70oC в течение ночи. Раствор разбавляют этилацетатом, фильтруют и промывают водой. С помощью препаративной тонкослойной хроматографии (дихлорметан/н-гексан в соотношении 8:2) получают указанное в заголовке соединение. МС: М+H 278; tпл 145-151oC.
Пример 11: 6-этинил-8-(3-нитрофенил)-1,7-нафтиридин
А. 6-триметилсилилэтинил-8-(3-нитрофенил)-1,7-нафтиридин получают следующим образом:
К раствору 6- трифторметансульфонил-8-(3-нитрофенил)-1,7-нафтиридина (200 мг, 0,50 ммоля; получен согласно примеру 8) в диметилформамиде (1 мл) и триэтиламине (0,5 мл) добавляют этинилтриметилсилан (0,078 мл, 0,56 ммоля), бис(дибензилиденацетон)палладий (5,8 мг, 0,020 ммоля), трифенилфосфин (5,3 мг, 0,020 ммоля) и йодид меди (3,8 мг, 0,020 ммоля). Смесь выдерживают при 60oC в течение 2 ч. Раствор разбавляют этилацетатом и промывают водой. Продукт очищают колоночной экспресс-хроматографией на силикагеле (толуол/ацетон в соотношении 20:0,2), получая 6-триметилсилилэтинил-8-(3-нитрофенил)-1,7-нафтиридин. МС: М+H 348; tпл 160-163oC.
Б. К раствору 6-триметилсилилэтинил-8-(3-нитрофенил)-1,7- нафтиридина (77 мг, 0,22 ммоля) в метаноле (0,5 мл) и толуоле (0,5 мл) добавляют 1н. гидроксид калия (0,22 мл). Смесь перемешивают в течение 2 ч. Суспензию фильтруют и продукт промывают водой и простым эфиром, получая указанное в заголовке соединение. МС: М+H 276; tпл 215oC, разложение.
Пример 12: 6-(4-карбоксифенил)-8-(3-цианфенил)-1,7-нафтиридин
К раствору 6-трифторметансульфонил-8-(3-цианфенил) -1,7-нафтиридина (2,15 г, 5,67 ммоля) в ДМФ (21,5 мл) добавляют 4-карбоксифенилбороновую кислоту (1,13 г, 6,81 ммоля), бис(дибензилиденацетон)палладий (131 мг, 0,23 ммоля), трифенилфосфин (95 мг, 0,36 ммоля) и водный К2СО3 (2н., 17 мл). Реакционную смесь перемешивают при 80oC в течение 2,5 ч. Горячий раствор фильтруют через целит и неочищенный продукт осаждают, осторожно добавляя воду (10 мл) и водный раствор HCl (2н., 8 мл). Суспензию фильтруют и неочищенный продукт перемешивают в горячем ТГФ (30 мл). Холодную суспензию еще раз фильтруют, получая указанное в заголовке соединение (1,12 г). М+H 352; tпл > 300oC. Время удерживания при использовании ЖХВР составляет 7,58 мин (колонка LiChroCart 125-4, Supersphere 60 RP-select В, 40oC; элюент ацетонитрил/вода (0,1% ТФК) в соотношении 45:55; скорость потока 1 мл/мин, обнаружение при 254 нм).
Пример 13: 6-(4-карбамоилфенил)-8-(3-цианфенил)-1,7-нафтиридин
К суспензии 6-(4-карбоксифенил)-8-(3-цианфенил) -1,7-нафтиридина (100 мг, 0,28 ммоля) в толуоле (2 мл) добавляют тионилхлорид (0,1 мл, 1,37 ммоля). Реакционную смесь выдерживают при температуре дефлегмации в течение 3 ч. Растворитель выпаривают и остаток растворяют в ТГФ (2 мл). Добавляют водный аммиак и раствор перемешивают в течение 2 ч при температуре окружающей среды. Добавляют этилацетат и органический слой промывают водой, получая чистое указанное в заголовке соединение с tпл 237-240oC.
Пример 14: 6-(4-карбоксифенил)-8-(4-бензо[с]фуразанил)- 1,7-нафтиридин
А. 6-амино-8-метокси-1,7-нафтиридин
К раствору натрия (3,2 г, 0,139 моля) в метаноле (1400 мл) добавляют 2-циан-3-пиридилацетонитрил (20 г, 0,139 моля) и реакционную смесь перемешивают в течение 17 ч при температуре окружающей среды. Затем добавляют воду (700 мл) и после упаривания большей части метанола получают путем кристаллизации указанное в заголовке соединение, tпл 178-180oC.
Б. 6-трифторметансульфонилокси-8-метокси-1,7-нафтиридин
К раствору 6-амино-8-метокси-1,7-нафтиридина (19 г, 0,108 моля) в смеси 1: 1 воды и трифторметансульфоновой кислоты (380 мл) осторожно добавляют раствор нитрита натрия (11,2 г, 0,162 моля) в воде (40 мл) при 0oC. Через 1 ч охлаждающую баню удаляют и реакционную смесь перемешивают еще в течение 1 ч при температуре окружающей среды. Затем добавляют этилацетат и раствор нейтрализуют, добавляя бикарбонат натрия (4н., 1 л). Водную фазу повторно экстрагируют этилацетатом (3х500 мл). Органический растворитель выпаривают и неочищенный продукт очищают колоночной экспресс-хроматографией на силикагеле (толуол/ацетон в соотношении 20:3), получая указанное в заголовке соединение. М+308; tпл 99-101oC.
В. 6-(4-карбоксифенил)-8-метокси-1,7-нафтиридин
К раствору 6-трифторметансульфонил-8-метокси-1,7-нафтиридина (1,5 г, 4,86 ммоля) в ДМФ (40 мл) добавляют 4-карбоксифенилбороновую кислоту (0,866 г, 5,34 ммоля), бис(дибензилиденацетон)палладий (112 мг, 0,167 ммоля), три-орто-толилфосфин (96 мг, 0,32 ммоля) и водный раствор Na2CO3 (14,6 мл, 2н.). Реакционную смесь перемешивают при 110oC в течение 3 ч. Горячий раствор фильтруют через целит и раствор упаривают досуха. Неочищенный продукт растворяют в горячей воде (60 мл) и водную фазу промывают этилацетатом (3х50 мл). Продукт осаждают из водной фазы, осторожно добавляя водный раствор HCl (2н. , 6 мл). Суспензию фильтруют и неочищенный продукт еще раз перемешивают в горячем этилацетате (50 мл). Холодную суспензию фильтруют, получая указанное в заголовке соединение (1,10 г). М+280; tпл > 300oC.
Г. 6-(4-карбоксифенил)-8-бром-1,7-нафтиридин.
К раствору 6-(4-карбоксифенил)-8-метокси-1,7-нафтиридина (250 мг, 0,892 ммоля) в ДМФ (10 мл) добавляют PBr3 (0,63 мл, 6,63 ммоля). Реакционную смесь нагревают до 100oC в течение 30 мин. Затем суспензию сливают на воду (50 мл) и раствор промывают этилацетатом (2х50 мл). Органический растворитель удаляют и неочищенный продукт перемешивают в простом эфире. Суспензию фильтруют, получая указанное в заголовке соединение (215 мг). tпл 248-250oC, разложение.
Д. 4-бензо[с]фуразанилбороновая кислота
К раствору 4-бензо[с] фуразанилбромида (4 г, 0,020 моля) в тетрагидрофуране (80 мл) и н-пентане (20 мл) добавляют триэтилборат (3,8 мл, 0,022 моля) и N,N,N,N-тетраэтилендиамин (3 мл, 0,02 моля). Затем по каплям при -100oC добавляют н-BuLi (8,8 мл, 2,5н. в гексане, 0,022 моля) и раствор перемешивают в течение еще 5 мин. Реакционную смесь сливают на водный раствор насыщенного хлорида аммония и водную фазу экстрагируют этилацетатом. Органический растворитель удаляют и неочищенный продукт растворяют в смеси дихлорметан/н-гексан (в соотношении 5:6). Суспензию фильтруют, получая указанное в заголовке соединение (1,05 г), которое используют без дополнительной очистки.
Е. 6-(4-карбоксифенил)-8-(4-бензо[с]фуразанил)-1,7-нафтиридин
К раствору 6-(4-карбоксифенил)-8-бром-1,7-нафтиридина (100 мг, 0,304 ммоля) в ДМФ (2,5 мл) добавляют 4-бензо [с] фуразанилбороновую кислоту (60 мг, 0,36 ммоля), бис(дибензилиденацетон)палладий (7 мг, 0,0122 ммоля), три-орто-толилфосфин (7,4 мг, 0,024 ммоля) и водный раствор Na2CO3(0,9 мл, 2н.). Реакционную смесь перемешивают при 80oC в течение 2 ч. Горячий раствор фильтруют через целит и раствор упаривают досуха. Остаток перемешивают в этилацетате (20 мл), суспензию фильтруют и органический слой удаляют. Остаток растворяют в горячем ДМФ (7 мл) и неочищенный продукт осаждают, осторожно добавляя водный раствор HCl (2н., 1 мл). Неочищенный продукт перекристаллизовывают из горячего ДМФ, получая указанное в заголовке соединение (68 мг). MH+369; tпл > 300oC. М+Н=369. Время удерживания при использовании ЖХВР составляет 10,40 мин (колонка LiChroCart 125-4, Supersphere 60 RP-select В, 40oC; элюент ацетонитрил/вода (0,1% ТФК) в соотношении 45:55; скорость потока 1 мл/мин, обнаружение при 254 нм).
По методике, описанной выше в соответствующем примере, и с использованием соответствующих исходных продуктов получают следующие соединения формулы 1, указанные в таблице 1.
Соли присоединения оснований соединений, имеющих свободные карбоксильные группы, например соединений, описанных выше, могут быть получены взаимодействием соединения с соответствующим аминосахаром, например, N-метил-О-глюкамином. Следующий пример иллюстрирует получение таких солей присоединения оснований.
Пример 34: 6-(4-карбоксифенил)-8-(4-бензо[с]фуразанил)-1,7-нафтиридин: соль с N-метил-D-глюкамином
К горячему раствору 6-(4- карбоксифенил)-8-(4-бензо[с]фуразанил)-1,7-нафтиридина (1 г, 2,72 ммоля) в ДМФ (100 мл) добавляют N-метил-D-глюкамин (0,53 г, 2,72 ммоля). Растворитель удаляют при пониженном давлении и остаток перекристаллизовывают из горячего метанола (примерно 50 мл), получая чистый продукт (1,11 г). tпл 230oC, разложение. Время удерживания при использовании ЖХВР составляет 6,65 мин (колонка LiChroCart 125-4, Supersphere 60 RP-select В, 50oC; элюент: градиент от 0% Б/100% А до 70% Б/30% А в течение 15 мин; А обозначает 2,7 г KHPO4/0,027 г Na2HPO4 в 900 мл воды/100 мл ацетонитрила, а Б обозначает 100 мл воды/900 мл ацетонитрила; обнаружение при 220 нм). Продукт растворим в воде.
8-Арил-1,7-нафтиридины по изобретению, например, формулы I, в частности предпочтительно 6-(карбоксифенил или карбоксиметилфенил)-8- (фенил,бензо[с] тиадиазолил или бензо[с] фуразанил)-1,7-нафтиридины и их сложные эфиры и амиды в свободной форме или в форме соли (обозначенные далее в настоящем описании как "агенты по изобретению") проявляют фармакологическую активность и пригодны в качестве лекарственных средств, например, для терапии при лечении указанных ниже в настоящем описании болезней и состояний.
В частности, агенты по изобретению проявляют ингибирующую активность в отношении изофермента циклической нуклеотидной фосфодиэстеразы (PDE), избирательную в отношении изофермента типа 4.
Агенты по изобретению обладают противовоспалительными, препятствующими повышенной реактивности дыхательных путей, и бронхолитическим свойствами. Кроме того, они обладают иммунодепрессивной, ингибирующей секрецию TNF α и другими фармакологическими активностями, что может быть продемонстрировано на стандартных методах тестирования, приведенных в последующих примерах.
А. Ингибирование PDE4: Анализ ингибирования рекомбинантного изофермента PDE4A, PDE4B, PDE4C и PDE4D.
Клонирование и экспрессия: кДНК PDE4, кодирующую четыре изофермента: человеческую PDE4A (как описано у Sullivan и др., Cell Signal 1994; 6: 793-812), крысиную PDE4B (как описано у Colicelli и др., Proc. Natl. Acad. Sci. USA 1989; 86: 3599-3903), человеческую PDE4C (как описано у Engels и др., FEBS Lett. 1995; 358: 305-310) и человеческую PDE4D (как описано у Baecker и др. , Gene, 1994; 138: 253-256), клонируют либо в экстрахромосомном экспрессирующем векторе дрожжей (PDE4C, PDE4D), либо путем интеграции (PDE4A, PDE4B; одна копия) в локусе рер4 штамма Saccharomyces cerevisiae, лишенного обоих генов PDE дрожжей дикого типа. Штаммы дрожжей, экспрессирующие изоферменты PDE4, выращивают в 1 л культур при 30oC, пеллетируют (осаждают центрифугированием) и хранят в замороженном состоянии до гомогенизации.
Гомогенизация: Пеллетированные дрожжи (5 мл) суспендируют в 50 мл буфера (10 мМ трис-гидроксиметиламинометан, 1 мМ этилендиамин-тетрауксусная кислота, по 1 мг/мл лейпептина и пепстатина А, 175 мг/мл фенилметилсульфонилфторида, 1 мМ дитиотреитола, pH 7,4 с HCl). После центрифугирования к дебрису добавляют 15 г промытых буфером стеклянных гранул (диаметром 425-600 мм, промытых кислотой, фирмы Sigma Chemical Co.). К этой суспензии добавляют 1 мл буфера и 60 мг холамидопропансульфоновой кислоты и суспензию интенсивно перемешивают в течение 4 ч при 4oC. Дрожжевые клетки разлагают (дезинтегрируют), оценивая с помощью микроскопии (фазово-контрастная оптика), когда количество темных клеток превысит 30% (обычно 50%). Суспензию переносят в крупнозернистую стеклянную воронку, гомогенат собирают с помощью отсоса и стеклянные гранулы промывают буфером общим объемом 15 мл. Клеточные фрагменты отделяют от цитозоля центрифугированием (2000xg, 10 мин, 4oC). Дебрис ресуспендируют в 15 мл буфера и оценивают в отношении PDE-активности совместно с цитозолем.
Другие препараты изоферментов получают, используя в качестве источников клетки и органы человека. Препараты изоферментов типа 3 и 4 получают по описанной ниже методике, используя тот факт, что в тромбоцитах преобладают изоферменты типа 3, а в нейтрофилах - изоферменты типа 4.
Клетки и ткани гомогенизируют на льду в 10 мМ трис-HCI буфере, pH 7,4, содержащем сахарозу (250 мМ), ЭДТК (1 мМ), дитиотреитол (1 мМ), лейпептин и пепстатин А (по 1 мкг/мл каждого) и фенилметилсульфонилфторид (ФМСФ, 0,17 мг/мл, который добавляют непосредственно перед гомогенизаций).
Нейтрофилы (тип 4) и тромбоциты (типы 2 и 3) получают из крови человека и обрабатывают ультразвуком (зонд типа Branson, 4х15 с). Полученные при операции больных людей образцы легкого (типы 1 и 5) гомогенизируют, используя гомогенизатор Polytron (два импульса по 30 с). Препараты изоферментов:
Препараты PDE3 и 4 (субстрат цАМФ, 1 мкМ) состоят соответственно из надосадочных гомогенатов тромбоцитов и нейтрофилов, полученных при небольшой скорости вращения. Изоферменты типа 1 (субстрат цАМФ, 1 мкМ, Ca2+ 0, 5 мМ, калмодулин, 125 нМ), типа 2 (цАМФ, 100 мкМ) и типа 5 (цГМФ, 1 мкМ) отделяют с помощью анионообменной хроматографии (Q-Sepharose), используя градиент NaCI, в буфере для гомогенизации без сахарозы и ФМСФ (от 0 до 0,1М NaCI в 2,5 объемах колонки, от 0,1 до 0,45М в 24 объемах колонки). PDE1: фракции, в которых гидролиз 1 мкМ цАМФ может стимулироваться Ca2+ калмодулином (0,5 мМ и 125 нМ соответственно); элюирование при концентрации NaCI 0,17-0,18М. PDE2: фракции, в основном проявляющие гидролитическую активность в отношении цАМФ при концентрации субстрата 100 мкМ, но неактивные при 1 мкМ; элюирование при концентрации NaCI 0,31-0,32М. PDE5: фракции, избирательно гидролизирующие цГМФ, 1 мкМ, но не обладающие активностью в отношении цАМФ, 1 мкМ; элюирование при концентрации NaCI 0,20-0,24М.
Анализ PDE: Протокол анализа основан на двухстадийном методе, описанном Thompson и др. (Adv. Second Messenger Phosphoprotein Res. 1979; 10:69-92), модифицированном для 96-луночных титрационных микропланшетов. Этот метод в основном состоит в том, что фермент растворяют в буфере для гомогенизации (см. выше) для того, чтобы общий гидролиз субстрата в процессе анализа составлял 10-30%. Для инициации реакции 25 мл разбавленного фермента добавляют к 25 мл субстрата ([3H]-цАМФ, 1,25 мМ, 740 Бк) и 75 мл раствора ингибитора (см. ниже). После инкубации в течение 30 мин при 37oC реакцию прекращают с помощью бани с горячей водой (65oC, 5 мин). Планшеты охлаждают на льду и инкубируют в течение 10 мин при 37oC с 25 мл 5'-нуклеотидазы (из яда змеи, из oiophaghus hannah, фирма Sigma Chemical Co., 0,1 мг/мл в воде). Непрореагировавший субстрат отделяют от [3H] - аденозина путем последовательного добавления аликвотных объемов (100+50+50 мл с 5-минутными интервалами) 30 об. % суспензии Dowex 1х2 (ацетатная форма) в 0,2 об.%-ной уксусной кислоте. Dowex осаждают центрифугированием (150xg, 5 мин). Аликвоты надосадочных жидкостей переносят на 96-луночные планшеты со сцинтилляционной твердой фазой (LumaPlate, Canberra Packard), используя автоматические пипетки (типа Hamilton MicroLab 2200), сушат (по крайней мере 4 ч при 50oC) и подсчитывают радиоактивность (счетчик типа Canberra Packard TopCount).
Ингибиторы: Маточные растворы ингибиторов готовят в диметилсульфоксиде (ДМСО) и разбавляют водой/ДМСО, получая 7 концентраций, выбранных таким образом, чтобы они вызывали ингибирование в диапазоне от 30% до 70%. Концентрацию ДМСО в процессе анализа поддерживают на постоянном уровне в 50 мл/мл.
Определение параметров ингибирования: Концентрацию, при которой происходит ингибирование на 50% (IC50), и угол наклона кривой доза-ответ (коэффициент Хилла) определяют из графиков зависимости ингибирования от концентрации с помощью нелинейного выравнивания методом наименьших квадратов с использованием логарифмического уравнения с двумя параметрами. Результаты выражают в виде отрицательного десятичного логарифма концентрации ингибитора, при которой наблюдается 50%-ное ингибирование (IC50) (в молях/л; pIC50). Оценивают 95%-ные доверительные интервалы и выражают в виде pL и pU (отрицательные десятичные логарифмы нижнего и верхнего доверительных пределов соответственно). Концентрации, которые вызывают заметное осаждение при исследовании, исключают из анализируемых данных.
При исследовании этим методом агенты по изобретению в основном ингибируют изоферменты PDE типа 4, проявляя незначительное действие в отношении изоферментов типов 1, 2, 3 и 7. В группе изоферментов PDE типа 4 (т.е. изоферменты PDE типа 4 от А до D) агенты по изобретению в основном проявляют избирательное ингибирование изофермента D PDE типа 4 по сравнению с ингибированием изоферментов 4A, 4B и 4C PDE типа 4.
Противовоспалительная активность: Ингибирование активации эозинофилов формил-MetLeuPhe (fMLP)
Очищенные эозинофилы человека (104/лунку в 0,2 мл HBSS) стимулируют fMLP (1 мкМ) в присутствии люцигенина (25 мкМ). Ингибирование окислительного потенциала (измеренного по изменениям хемилюминисценции) определяют по кривым доза-ответ, используя логарифмическое уравнение.
Агенты по изобретению проявляют активность при тестировании указанным выше методом в концентрациях порядка от 0,001 до 5 мкМ, обычно в концентрациях, находящихся в диапазоне небольших наномолярных концентраций. Например, при оценке этим методом значение IC50 соединения из примера 2 составляет 0,006 мкМ.
Учитывая их противовоспалительную активность, влияние на повышенную реактивность дыхательных путей и профиль в отношении ингибирования изоферментов PDE, в частности их активность в качестве селективных ингибиторов PDE типа 4, агенты по изобретению пригодны для лечения, в частности для профилактического лечения, обструктивного или воспалительного заболевания дыхательных путей. Так, при постоянном и регулярном введении в течение продолжительных периодов времени агенты по изобретению пригодны для осуществления успешной защиты от повторяющегося бронхостеноза или других симптоматических воздействий, сопровождающих обструктивное или воспалительное заболевание дыхательных путей, или для контроля, уменьшения интенсивности или обратного развития основного статуса такого заболевания.
Учитывая их бронхолитическую активность, агенты по изобретению пригодны в качестве бронхолитических средств, например, для лечения хронического или острого бронхостеноза, например, для симптоматического лечения обструктивного или воспалительного заболевания дыхательных путей.
Понятия "лечение" или "процесс лечения" в контексте настоящего описания и формулы изобретения относится к обструктивному или воспалительному заболеванию дыхательных путей и поэтому они включают и профилактический, и симптоматический виды терапии.
В соответствии с вышеизложенным объектами настоящего изобретения также являются следующие.
А. Способ
а) лечения повышенной реактивности дыхательных путей,
б) осуществления увеличения просвета бронхов или бронхиол или, в частности,
в) лечения обструктивного или воспалительного заболевания дыхательных путей у пациента, нуждающего в таком лечении, причем этот способ включает введение пациенту эффективного количества агента по изобретению.
Обструктивные или воспалительные заболевания дыхательных путей, которые подпадают под объем настоящего изобретения, включают астму, пневмокониоз, хроническое обструктивное заболевание дыхательных путей или легких (ОЗДП или ОЗЛ) и респираторный дистресс-синдром взрослых (ARDS), а также обострение повышенной реактивности дыхательных путей в результате лечения другим лекарством, например аспирином, или терапии с использованием β-агонистов.
Настоящее изобретение применимо для лечения астмы любого типа или происхождения, включая врожденную и прежде всего приобретенную астму. Оно применимо для лечения аллергической (атопической/опосредованной lgE) астмы. Оно также применимо для лечения неатопической астмы, включая, например, бронхиальную, приобретенную и профессиональную астму, астму, индуцированную бактериальной инфекцией, и другие типы неаллергической астмы. Оно также применимо для лечения синдрома стерторозного дыхания детей (детская, ранняя астма).
Изобретение применимо для лечения пневмокониоза любого типа или происхождения, включая, например, алюминоз, антракоз, асбестоз, халикоз, птилоз, сидероз, силикоз, табакоз и биссиноз.
Изобретение применимо для лечения OЗДП или ОЗЛ, включая хронический бронхит, эмфизему легких или связанную с ними одышку.
Изобретение также применимо для лечения бронхита любого типа или происхождения, включая острый, арахиноидный, катаральный, хронический, крупозный или гнойный туберкулезный бронхит.
Учитывая их активность в качестве селективных ингибиторов выделения TNF-α, агенты по изобретению также пригодны для понижающей регуляции или ингибирования выделения TNF-α, например, для лечения заболеваний или состояний, которые обусловлены выделением TNF-α или в которых TNF-α выполняет роль медиатора, например заболеваний или состояний, этиология которых включает или содержит патологию, например, нежелательное, избыточное или нерегулируемое выделение TNF-α, в частности, для лечения кахексии или эндотоксического шока и для лечения СПИДа [ср. данные Sharief и др., Mediators of Inflammation, 1, 323-338 (1992)].
Способ по изобретению применим для лечения кахексии, связанной с патологическим выделением TNF-α или с патологическими уровнями TNF-α в сыворотке крови, независимо от происхождения, включая кахексию, являющуюся следствием, например, бактериальной, вирусной или паразитарной инфекции или следствием лишения или истощения гуморальной или другой органической, например почечной, функции. Он, например, применим для лечения раковой, малярийной и вермальной кахексии, кахексии, являющейся результатом дисфункции гипофиза, щитовидной железы или тимуса, а также уремической кахексии. Он, в частности, применим для лечения связанной со СПИДом кахексии, например, кахексии, являющейся следствием или связанной с ВИЧ-инфекцией.
Способ по изобретению также применим для лечения септического шока, например шоковых состояний, являющихся результатом бактериальной инфекции. В этой связи следует отметить, что настоящее изобретение относится к способу лечения септического шока, а также состояний, являющихся следствием или имеющих симптомы септического шока, например ARDS (респираторный дистресс-синдром взрослых).
Способ по изобретению, кроме того, применим для лечения заболевания, являющегося следствием ВИЧ-инфекции, например СПИДа, с целью уменьшения интенсивности или контроля за развитием такого заболевания.
Учитывая их профиль в отношении ингибирования изоферментов PDE и/или ингибирования выделения TNF-α , а также их иммунодепрессивную активность, агенты по изобретению также пригодны в качестве иммунодепрессантов, например, для лечения аутоиммунных заболеваний, в частности для лечения аутоиммунных заболеваний, при которых имеют место воспалительные процессы или которые имеют воспалительный компонент или этиологию, или в качестве противовоспалительных агентов для лечения воспалительного заболевания, в частности для лечения воспалительного заболевания, при котором имеют место аутоиммунные реакции или которое имеет аутоиммунный компонент или этиологию.
Примеры таких заболеваний, к которым применимо настоящее изобретение, включают аутоиммунные гематологические нарушения (например, гемолитическую анемию, гипопластическую анемию, настоящую анемию эритроцитов и идиопатическую тромбоцитопению), системную красную волчанку, полихондрит, склеродерму, грануломатоз Вегенера, дерматомиозит, хронический активный гепатит, тяжелую псевдопаралитическую миастению, синдром Стивенса-Джонсона, идиопатическую спру, аутоиммунное воспалительное заболевание кишечника (например, язвенный колит и болезнь Крона), эндокринную офтальмопатию, болезнь Грейвса, саркоидоз, альвеолит, хроническую гиперчувствительную пневмонию, рассеянный склероз, первичный билиарный цирроз печени, юношеский диабет (сахарный диабет типа I), увеит (передний и задний), сухой кератоконъюнктивит и вернальный кератоконьюнктивит, интерстициальный фиброз легкого, псориатический артрит и гломерулонефрит (с нефротическим синдромом или без него, например, включая идиопатический нефротический синдром или нефропатию с минимальным изменением), а также воспалительные и/или гиперпролиферативные заболевания кожи, такие как псориазный атопический дерматит, пемфигоид и, в частности, контактный дерматит, например, аллергический контактный дерматит.
Агенты по изобретению особенно пригодны для лечения артрита и других ревматоидных или воспалительных заболеваний, прежде всего для лечения ревматоидного артрита.
Соединения по изобретению также показаны для применения в качестве иммунодепрессантов для предотвращения отторжения трансплантата, например, для поддержания трансплантатов аллогенных органов или т.п., например, трансплантатов почки, печени, легкого, сердца, сердца-легкого, кишечника, костного мозга, кожи или роговицы.
Учитывая их противовоспалительную активность, в частности? в отношении ингибирования активации эозинофилов, агенты по изобретению также пригодны для лечения связанных с эозинофилами заболеваний, например эозинофилии, и прежде всего связанных с эозинофилией заболеваний дыхательных путей (например, включающих патологическую эозинофильную инфильтрацию тканей легкого), в том числе гиперэозинофилию, оказывающую воздействие на дыхательные пути и/или на легкие, а также, например, связанных с эозинофилией заболеваний дыхательных путей, которые являются следствием или сопровождают синдром Леффлера, эозинофильную пневмонию, паразитическое (в частности метазоальное) заражение (включая тропическую эозинофилию), бронхопульмональный аспергиллоз, полиартрит nodosa (включая синдром Churg-Strauss), эозинофильную гранулему и связанные с эозинофилией нарушения дыхательных путей, вызванные реакцией на лекарство.
Учитывая их профиль в отношении ингибирования изоферментов PDE, в частности их профиль в качестве избирательных ингибиторов PDE типа 4, агенты по изобретению, кроме того, пригодны в качестве ингибиторов PDE типа 4, например, для лечения заболевания, связанного со снижением кальция в тканях, прежде всего дегенеративных заболеваний костей и суставов, вызванных снижением кальция, в частности остеопороза. В этом отношении они также пригодны для лечения аллергических воспалительных заболеваний, таких как ринит, конъюнктивит, атопический дерматит, крапивница и желудочно-кишечные аллергии, в качестве сосудорасширяющих агентов, например, для лечения стенокардии, гипертензии, застойной сердечной недостаточности и мультиинфарктной деменции и для лечения других состояний, при которых показано ингибирование PDE типа 4, например депрессии, состояний и заболеваний, характеризующихся нарушением когнитивной функции, включая болезнь Альцгеймера, болезнь Паркинсона и удар.
Учитывая их способность синергетически взаимодействовать с иммунодепрессантами и/или противовоспалительными лекарственными средствами, агенты по изобретению также пригодны в качестве сотерапевтических агентов, предназначенных для использования в сочетании с указанными лекарствами, например, в качестве усилителей терапевтической активности указанных лекарств или в качестве средств, понижающих необходимую величину дозы или потенциальные побочные эффекты таких лекарств. Лекарственные средства, которые могут применяться совместно с агентами по изобретению включают, например, циклопептидные, циклопептолидные или макролидные иммунодепрессанты или противовоспалительные лекарственные средства, например лекарства из класса циклоспоринов, например циклоспорины А или G, такие лекарственные средства, как такролимус (также известное как FK 506), аскомицин и рапамицин, и различные известные родственные им средства и их производные, а также лекарства из класса глюкокортикостероидов. Заболевания, в отношении которых может применяться такая совместная терапия, включают, например, любое заболевание или состояние, для которого необходима иммунодепрессивная или противовоспалительная лекарственная терапия, например, приведенное выше в настоящем описании. В частности, агенты по изобретению пригодны для использования в вышеуказанной совместной терапии, например, для целей иммунодепрессивного, противовоспалительного или антиастматического лечения, например, для достижения воздействия, оказываемого циклоспорином, например, умеренного воздействия, обусловленного циклоспорином А, макролидами или стероидами.
В соответствии с вышеизложенным еще одним объектом настоящего изобретения является следующий.
Б. Способ
а) понижающей регуляции или ингибирования выделения TNF-α,
б) ингибирования активности изофермента PDE типа 4,
в) осуществления иммунодепрессии,
г) лечения воспалительного заболевания или
д) лечения любого конкретного состояния или заболевания, указанного выше,
у пациента, нуждающегося в таком лечении, причем этот способ включает введение пациенту эффективного количества агента по изобретению.
Настоящее изобретение также относится к:
В. агенту по изобретению, предназначенному для применения в качестве лекарственного средства, например, для применения в соответствии с любым указанным выше методом или для лечения любого указанного выше заболевания или состояния, например, указанного выше в разделах А и Б.
Дозы, применяемые при практическом осуществлении настоящего изобретения, как очевидно, варьируются в зависимости, например, от конкретной болезни или состояния, подлежащего лечению, в частности? от конкретно примененного агента по изобретению, пути введения и требуемой терапии. В целом, однако, удовлетворительные результаты, например, при лечении указанных выше заболеваний, могут быть получены при оральном введении доз, составляющих от 0,01 до 2,0 мг/кг. Для более крупных млекопитающих, например людей, рекомендованная суточная доза для орального введения должна соответствовать диапазону от приблизительно 0,75 до 150 мг, и она обычно применяется однократно или разделена на суточные дозы для двукратного или четырехкратного введения или находится в форме с непрерывным высвобождением. Таким образом, пригодные стандартные дозируемые формы для орального введения включают приблизительно от 0,2 до 75 или 150, например, от приблизительно 0,2 или 2,0 до 50, 75 или 100 мг агента по изобретению вместе с фармацевтическим приемлемым разбавителем или носителем.
При применении для лечения хронического или обструктивного заболевания дыхательных путей, например астмы, агенты по изобретению также могут вводиться таким путем, как ингаляция. И в этом случае применяемые дозы должны варьироваться, например, в зависимости от конкретного заболевания или состояния, конкретного используемого агента по изобретению, конкретного пути введения (например, либо с помощью сухой порошкообразной формы для ингаляции, либо иного метода) и требуемого действия. Однако в целом рекомендуемая суточная доза для ингаляции составляет от приблизительно 2,5 до приблизительно 130,0 мкг/кг/день, например, от приблизительно 13,0 до приблизительно 60,0 мкг/кг/день. Для более крупных млекопитающих, например людей, рекомендованная суточная доза для введения путем ингаляции, например, для лечения астмы должна находиться в диапазоне от приблизительно 0,2 до 10,0 мг, например от 1 до 5 мг, и она обычно применяется для однократного введения или разделена на 2 или 3 отдельные дозы для введения в течение суток. Приемлемая доза для введения, таким образом, должна составлять от приблизительно 200 мкг до приблизительно 3,3 мг при введении 3 раза в день, предпочтительно при введении с помощью приспособления для ингаляции сухого порошка в виде серий, состоящих из 2-8 вдуваний при каждом введении.
Агенты по изобретению также могут вводиться другим пригодным для этой цели методом, например путем инфузии, например, для лечения эндотоксического шока; назально, например, для лечения ринита; закапыванием в глаз, например, для лечения аутоиммунных заболеваний глаза; дермально, т.е. местно на кожу, например, для лечения дерматозов или псориаза; или ректально, например, с помощью клизмы или суппозитория, например, для лечения воспалительного заболевания кишечника. Приемлемые дозы для введения такими путями обычно должны быть в 10-100 раз ниже доз, необходимых при оральном введении.
Фармацевтические композиции, включающие агенты по изобретению, могут быть получены с использованием общепринятых разбавителей или эксципиентов или с помощью методов, хорошо известных в области приготовления галеновых препаратов. Так, например, дозируемые формы для орального введения могут представлять собой таблетки, капсулы и т.п. Композиции для дермального нанесения могут иметь форму кремов, мазей, гелей или чрескожно действующих терапевтических систем, например бляшек, и в дополнение к инертным разбавителям или носителям также могут содержать агенты, усиливающие проникновение через кожу, также известные в данной области.
Композиции для ингаляции могут представлять собой аэрозоли или другие предназначенные для распыления формы, а также формы в виде предназначенных для ингаляции сухих порошков, с разбавителем или без него, для введения с помощью любой известной в данной области приемлемой системы ингаляции сухого порошка. Для получения форм для ингаляции в виде сухого порошка агенты по изобретению можно применять в виде фармацевтически приемлемой кислотно-аддитивной соли. Это соединение в форме соли обычно измельчают, например, с использованием воздушного сопла или керамической мельницы с получением тонкоизмельченного порошка для ингаляции, например, со средним диаметром частиц приблизительно 2-3 мкм. Предпочтительно, чтобы по крайней мере 90% продукта имело средний диаметр частиц менее 7,8 мкм, более предпочтительно менее 4,8 мкм. Для получения приемлемого и соответствующего конкретного продукта, пригодного для введения с помощью ингаляции в форме сухого порошка, может оказаться предпочтительным осуществлять измельчение действующего вещества, предварительно смешанного с пригодным носителем, который можно применять при ингаляции, например с лактозой, при пониженной температуре.
В соответствии с вышеизложенным настоящее изобретение также относится к фармацевтической композиции, содержащей агент по изобретению вместе с фармацевтически приемлемым разбавителем или носителем, например, для использования в любом из описанных выше методов.



Изобретение относится к новым производным нафтиридина формулы I, где R1 обозначает фенил, бензил, 3-нитрофенил, 3-хлорфенил, 3-цианфенил, 3-(тетразолил)фенил или бензофуразанил; R2 обозначает гидроксигруппу, трифторметансульфонилокси, аллил, алкил, алкенил, алкинил, алкоксигруппу, фенил, фенилокси, карбоксифенил, карбоксиметилфенил, карбамоилфенил, фениламино-, диафениламино-, амино-, алкамино-, алкамидогруппу, где "алк" обозначает алифатический фрагмент, имеющий до 8 атомов углерода и необязательно включающий карбоксигруппу, эфир карбоновой кислоты либо гидроксигруппу и/или необязательно содержащий простую эфирную и/или сложноэфирную связь, или к его N-оксиду в свободной форме или в форме фармацевтически приемлемой соли. Также описана фармацевтическая композиция, обладающая противовоспалительной активностью на основе указанных соединений. Изобретение может быть использовано в медицине в качестве лекарственных средств, например, для лечения астмы. 2 с. и 4 з.п. ф-лы, 2 табл.

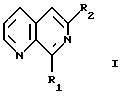
где R1 обозначает фенил, бензил, 3-нитрофенил, 3-хлорфенил, 3-цианфенил, 3-(тетразолил)фенил или бензофуразанил;
R2 обозначает гидроксигруппу, трифторметансульфонилокси, аллил, алкил, алкенил, алкинил, алкоксигруппу, фенил, фенилокси, карбоксифенил, карбоксиметилфенил, карбамоилфенил, фениламино-, диафениламино-, амино-, алкамино- или алкамидогруппу, где "алк" обозначает алифатический фрагмент, имеющий до 8 атомов углерода и необязательно включающий карбоксигруппу, эфир карбоновой кислоты либо гидроксигруппу и/или необязательно содержащий простую эфирную связь и/или сложноэфирную связь,
или его N-оксид в свободной форме или в форме фармацевтически приемлемой соли.
где n равно 0 или 1;
R7 обозначает гидрокси-, амино-, С1-С4алкиламино- или С1-С4алкокси-группу, предпочтительно гидрокси- или аминогруппу;
либо R3 обозначает водород и R4 обозначает нитрогруппу, хлор, цианогруппу или тетразолил (например, 1-тетразолил) и R5 и R6 вместе образуют дополнительную связь,
либо R3, R4, R5 и R6 вместе обозначают =N-O-N=,
или его сложный эфир в свободной форме или в форме фармацевтически приемлемой соли.
6-амино-8-(3-нитрофенил)-1,7-нафтиридин,
6-амино-8-(4-бензо[с]фуразанил)-1,7-нафтиридин,
6-гидрокси-8-(3-нитрофенил)-1,7-нафтиридин,
6-метоксикарбонилметокси-8-(3-нитрофенил)-1,7-нафтиридин,
гидрохлорид 6-ацетамидо-8-(3-нитрофенил)-1,7-нафтиридина,
6-амино-8-бензил-1,7-нафтиридин,
6-фениламино-8-(3-нитрофенил)-1,7-нафтиридин,
6-трифторметансульфонилокси-8-(3-нитрофенил)-1,7-нафтиридин,
6-фенил-8-(3-нитрофенил)-1,7-нафтиридин,
6-винил-8-(3-нитрофенил)-1,7-нафтиридин,
6-этинил-8-(3-нитрофенил)-1,7-нафтиридин,
6-(4-карбоксифенил)-8-(3-цианфенил)-1,7-нафтиридин,
6-(4-карбамоилфенил)-8-(3-цианфенил)-1,7-нафтиридин,
6-(4-карбоксифенил)-8-(4-бензо[с]фуразанил)-1,7-нафтиридин,
N-мeтил-D-глюкaминoвaя соль 6-(4-карбоксифенил)-8-(4-бензо[с] фуразанил)-1,7-нафтиридина
или соединения формулы I, в которых
R1 обозначает 3-хлорфенил и R2 обозначает амино,
R1 обозначает 3-цианфенил и R2 обозначает амино,
R1 обозначает 4-(5-тетразолил)фенил и R2 обозначает амино,
R1 обозначает бензил и R2 обозначает фениламино,
R1 обозначает бензил и R2 обозначает дифениламино,
R1 обозначает бензил и R2 обозначает трифторметансульфонилокси,
R1 обозначает 3-хлорфенил и R2 обозначает трифторметансульфонилокси,
R1 обозначает 3-цианфенил и R2 обозначает трифторметансульфонилокси,
R1 обозначает 4-бензо[с] фуразанил и R2 обозначает трифторметансульфонилокси,
R1 обозначает 4-(5-тетразолил)фенил и R2 обозначает трифторметансульфонилокси,
R1 обозначает бензил и R2 обозначает фенил,
R1 обозначает бензил и R2 обозначает винил,
R1 обозначает бензил и R2 обозначает этинил,
R1 обозначает 3-хлорфенил и R2 обозначает 4-карбоксифенил,
R1 обозначает 3-нитрофенил и R2 обозначает 4-карбоксифенил,
R1 обозначает 3-хлорфенил и R2 обозначает 4-аминокарбонилфенил,
R1 обозначает 3-нитрофенил и R2 обозначает 4-аминокарбонилфенил,
R1 обозначает 3-хлорфенил и R2 обозначает 4-метоксикарбонилфенил или
R1 обозначает 3-нитрофенил и R2 обозначает 4-метоксикарбонилфенил.
| ПРОИЗВОДНЫЕ ХИНОЛОН- И НАФТИРИДОНКАРБОНОВОЙ КИСЛОТЫ В ВИДЕ СМЕСИ ИЗОМЕРОВ ИЛИ ОТДЕЛЬНЫХ ИЗОМЕРОВ, ИХ ГИДРАТЫ И СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2105770C1 |
| СПОСОБ ДИАГНОСТИКИ МИОПАТИИ В ЭКСПЕРИМЕНТЕ | 2016 |
|
RU2625743C1 |
| US 5466697 A, 14.11.1995 | |||
| US 4956371 A, 11.09.1990 | |||
| ПРИБОР ДЛЯ КОНТРОЛЯ ПРЯМОЛИНЕЙНОСТИ НАПРАВЛЯЮЩИХ ПОВЕРХНОСТЕЙ | 0 |
|
SU172058A1 |

