"Настоящее изобретение относится к способу получения 2-галогенметилпенемов, в частности 2-хлорметилпенемов, которые можно использовать для получения антибактериальных пенемов.
Известно что производные пенемов являются соединениями, обладающими широким спектром активности против бактерий [см., например. Wise R. "The Carbapenem and Penem Antibiotics - A. Brief Review" - Antimicrob. Newsl. 7. 73-80 (1990)].
Известно также, что 2-галогенметилпенемы формулы (I), в частности 2-хлорметилпенемы, являются полезными промежуточными продуктами для получения антибактериальных пенемов [G. Pentassuglia et al. J. of Antibiotics Vol. 48.399-407 (1995)] . Способы получения 2-галогенметилпенемов (I), известные вплоть до настоящего времени (см., например, Altamura М. Et al., J. Org. Chem. , 1993, 58, 272-274), содержат стадию, включающую соответствующий 2-гидроксиметилпенем (II):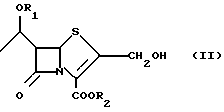
где R1 и R2 такие, как определено ниже.
Получение соединения (II) требует проведения длинного ряда сложных стадий, дающих низкие выходы и включающих использование защитных групп и дорогих реагентов, которые не пригодны для промышленного производства.
Кроме того, синтез соединения (II) требует многочисленных хроматографических разделений для очистки полученных соединений, так как их использование в виде сырых продуктов в последующих кислотных или основных условиях реакции не пригодно из-за их низкой стабильности.
Настоящее изобретение относится к способу получения 2-галогенметилпенемов, в частности 2-хлорметилпенемов, формулы (I)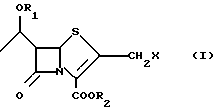
где R1 представляет защитную группу для спиртового гидроксила, R2 представляет защитную группу для карбоксила и X представляет галоген, в частности хлор, содержащему в качестве промежуточной стадии образование соответствующего 2-галогенацетилтиоазетидона.
Задачей настоящего изобретения является создание способа, который позволяет получить 2-галогенпенемы (I), в частности 2-хлорметилпенемы, включающие только три стадии с использованием исходного продукта, который коммерчески легко доступен. Этот путь синтеза, описанный в приложенной схеме, позволяет получить соединения формулы (I), исключая промежуточные стадии, включающие введение защитных групп и удаление защитных групп, и при этом конечный продукт включает все атомы углерода, участвующие в синтезе. Кроме того, способ настоящего изобретения не включает сложные способы разделения или очистки промежуточных продуктов, и поэтому он позволяет получить соединения формулы (I) с высокими выходами. Кроме того, условия реакции позволяют получить соединения (I) с высокой стереоселективностью, поскольку в ходе способа по данному изобретению образуется только нужный оптический изомер, 2-галогенметилпенемы формулы (I) можно превратить непосредственно после удаления защитных групп в нужные конечные продукты.
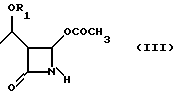
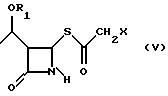
Как показано на схеме в конце описания, способ по настоящему изобретению включает реакцию между соединением формулы (III), (3R,4R)-4-ацетокси-3-[R1O-этил]-2-азетидиноном, где R1 такой, как определен выше, и 2-галогентиоуксусной кислоты (IV), где X представляет галоген, полученной, например, как описано в Arndt. Bekir Berichte. 63В, 2390 (1930). Реакцию проводят в органическом протонном растворителе, предпочтительно диоксане, тетрагидрофуране, хлороформе, при температуре от -10oC до +40oC, в присутствии органического основания, такого как, например, триэтиламин или диизопропилэтиламин и кислоты Льюиса, такой как, например, иодид цинка, бромид цинка, хлорид цинка, хлорид алюминия. 2-Галогенацетилтиоазетидон (V) можно подвергнуть реакции, без дальнейшей очистки, с эфиром оксапилхлорида (VI).
Такую реакцию проводят в протонном органическом растворителе, предпочтительно диоксане, тетрагидрофуране, толуоле, хлороформе, при температуре от -60oC до +20oC, предпочтительно от -20oC до +10oC, в присутствии органического основания, такого как, например, триэтиламин или диизопропилэтиламин.
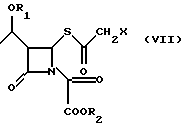
Промежуточное соединение (VII), ацилированное у β-лактамного азота, циклизуют в соответствующий 2-галогенметилпенем (I) под действием органического фосфита, как например, трифенилфосфита, или триметилфосфита, или фосфонита, как например, диметоксиметилфосфина, в органическом растворителе, таком как, например, толуол, ксилол, хлороформ, метиленхлорид, при температуре 20oC - 140oC в течение 1-120 ч.
Реакционную смесь, содержащую сырой 2-галогенметилпенем (I), можно использовать непосредственно, без дальнейшей очистки, для получения требуемых производных пенема (например, как описано в патенте США 4794109).
В частности настоящее изобретение относится к способу получения 2-хлорметилпенемов (соединения формулы (I), где X = Cl, R1 и R2, такие, как описано выше).
По данному изобретению группа R1, защищающая спиртовую гидроксильную группу, предпочтительно представляет три-C1-6-алкилсилил (особенно трет-бутилдиметилсилил и триметилсилил), аллилоксикарбонил, п-нитробензилоксикарбонил; тогда как группа R2, защищающая карбоксильную группу, предпочтительно представляет аллил, бензил (возможно замещенный метокси- или нитрогруппой), CH2OCO(O)mR4, где R4 представляет C1-6-алкильную группу и m равно 0 или 1.
Галоген по настоящему изобретению представляет хлор, бром, иод, особенно хлор.
Для лучшего иллюстрирования изобретения приведены следующие примеры.
ПРИМЕР 1
(3S, 4R)-3-[(R)-трет-Бутилдиметилсилилокси)этил] -4-(2-хлорацетилтио)- 2-азетидон
83,3 г (0,261 моль) Иодида цинка добавляют при 20oC в атмосфере азота в раствор 50 г (0,174 моль) (3R,4R)-4-ацетокси-3- [(R)-трет-бутилдиметилсилилокси)этил] -2-азетидона; через 15 минут добавляют 38,3 г (0,346 моль) 2-хлоруксусной кислоты. Смесь охлаждают до 12oC и затем в течение 1 ч в него добавляют раствор 26,5 мл (0,190 моль) триэтиламина в диоксане (50 мл). Смесь перемешивают в течение 2 ч при той же самой температуре. Добавляют 5,0 мл (0,036 моль) триэтиламина и смесь перемешивают в течение 30 минут.
Раствор выливают в холодный 3% раствор NaHSO3 и экстрагируют этилацетатом. Органическую фазу промывают растворами 3% NaHSO3, 5% NaHCO3, водой, 10% NaCl и сушат над безводным Na2SO4. Выпариванием растворителя в вакууме получают желто-коричневатый продукт. Добавляют диэтиловый эфир, растворитель выпаривают и получают бледно-желтый твердый продукт. Выход: 57 г (97%).
1H NMR (200 MHz) (CDCl3): d 0,07 (3H, s), 0,08 (3H, s), 0,88 (9H, s), 1,21 (3H, D, J = 6,3 Hz), 3,23 (1H, dd, J= 2,3, 4,0 Hz), 4,22 (2H, s), 4,27 (1H, qd, J = 3,7, 6,3 Hz), 5,32 (1H, d, J= 2,3 Hz), 6,4 (1H, br s). 13C NMR (50 MNz) (CDCl3): d -4,3, -5,1, 17,9, 22,3, 25,7, 48,0, 52,4, 64,6, 65,4, 166,1, 194,8. MS TS (m/z): (M+H)+ 338, (M+NH4)+ 355.
ПРИМЕР 2
(3S, 4R)-1-(Аллилоксиоксалил)-3-[(R)-трет-бутилдиметилсилилокси) этил]-4-(2-хлорацетилтио)-2-азетидон
К раствору 57 г (0,169 моль) ((3S,4R)-3-[(R)-трет-бутилдиметилсилилокси) этил] -4-(2-хлорацетилтио)-2-азетидона в безводном тетрагидрофуране (500 мл) при 0 - 3oC в атмосфере азота добавляют 42,3 мл (0,338 моль) аллилоксиоксалилхлорида. Смесь перемешивают в течение нескольких минут и затем добавляют по каплям в течение 45 минут раствор 43,3 мл (0,245 моль) диизопропилэтиламина в тетрагидрофуране (40 мл). Смесь перемешивают 30 минут при такой же температуре. Добавляют 15 мл (0,087 моль) диизопроилэтиламина, раствор перемешивают 30 минут и фильтруют.
Фильтрат выливают в холодный 5% раствор NaHCO3 и экстрагируют н-гексаном, промывая твердый остаток, оставшийся на фильтре, тем же самым растворителем. Органические фазы объединяют, промывают водой и 10% NaCl и сушат над безводным Na2SO4.
Выпариванием растворителя получают коричневатое масло, которое используют в следующей стадии без дальнейшей очистки.
Выход: 73,0 г (96%).
1H NMR (200 MHz) (CDCl3): d - 0,04 (3H, S), -0.09 (3H, S), 0,85 (9H, s), 1,24 (3H, d, J = 6,3 Hz), 3,52 (1H, t, J= 3, Hz), 4,26 (2H, s), 4,38 (1H, qd, J = 3, 6,3 Hz), 4,70-4,82 (2H, m,), 5,22-5,46 (2H, m), 5,80-6,06 (1H, m), 5,97 (1H, d, J = 3 Hz). 13C NMR (50 MNz) (CDCl3): d -5,2, -4,3, 17,8, 21,7, 25,6, 47,9, 53,8, 64,7, 66,3, 67,4, 120,1, 130,5, 154,5, 159,0, 162,9, 190,7. MS TS (m/z): (M+NH4)+ 467.
ПРИМЕР 3
Аллиловый эфир (5R,6S)-2-хлорметил-6-((R)-1- трет-бутилдиметилсилилокси-этил)-пенем-3-карбоновой кислоты
К раствору 73 г (0,162 моль) (3S,4R)-1-(аллилоксиоксалил)-3-[(R)-трет-бутилдиметилсилилокси)этил] -4-(2-хлорацетилтио)-2- азетидона в 730 мл толуола добавляют 59 г (0,356 моль) триэтилфосфита. Раствор кипятят с обратным холодильником в течение 3 ч. Раствор охлаждают и концентрируют в вакууме, получая после колоночной хроматографии (силикагель; циклогексан/этилацетат, 3: 1, об. /об.) целевой аллиловый эфир (5R,6S)-2-хлорметил-6-((R)-1- трет-бутилдиметилсилоксиэтил)-пенем-3-карбоновой кислоты в виде желтого масла. Выход 83%.
ВЭЖХ: 1) колонка: Hypersil 5, внутр. диам. 5 мм, C18, 4,6 x 250 мм;
подвижная фаза: вода/ацетонитрил, 20:80, об./об.; поток = 1 мл/мин, 1 = 220, 320 нм; tR = 8,4 мин. 2) Колонка: BondClone 10, 10 мм, C18 3,9 x 300 мм, подвижная фаза: вода/ацетонитрил, 20:80, об./об., поток = 1 мл/мин, 1 = 205, 245 нм; tR = 9,6 мин.
1H NMR (200 MHz) (CDCl3): d 0,07 (6H, s), 0,87 (9H, s), 1,23 (3H, d, J= 6,2 Hz), 3,73 (1H, dd, J = 1,6, 4,3 Hz), 4,60-4,81 (2H, m), 4,62 and 4,94 (2H, Abq, J = 14 Hz), 5,20-5,47 (2H, m), 5,63 (1H, d, J= 1,6 Hz), 5,81-6,03 (1H, m). 13C NMR (50 MNz) (CDCl3): d -5,3, -4,7, 17,9, 22,3, 25,6, 37,6 (CH2-Cl, 62,4, 64,9, 65,9, 72,0, 118,6, 121,8, 131,2, 150,9, 158,9, 172,3, MS EI: (m/z) 417 (M+).
ПРИМЕР 4
К раствору 19 г (0,056 моль) (3S,4R)-1-(аллилоксиоксалил)-3- [(R)-трет-бутилдиметилсилилокси)этил] -4-(2-хлорацетилтио) -2-азетидона в безводном толуоле (150 мл) в атмосфере азота при 0 - 3oC добавляют 14,1 мл (0,113 моль) аллилоксиоксалилхлорида. Раствор перемешивают в течение нескольких минут и затем по каплям добавляют раствор 11,7 мл (0,084 моль) триэтиламина в толуоле (10 мл). Раствор перемешивают в течение 90 минут при той же температуре. Добавляют 3,9 мл (0,028 моль) триэтиламина, раствор перемешивают в течение 90 минут и фильтруют. Фильтрат промывают холодным 5% водным раствором NaHCO3, водой и 10% раствором NaCl; затем раствор сушат над безводным Na2SO4 и фильтруют. Добавляют 20,4 г (0,123 моль) триэтилфосфита и смесь кипятят с обратным холодильником в течение 3 ч.
Раствор охлаждают, концентрируют в вакууме и очищают колоночной хроматографией (силикагель; циклогексан/этилацетат, 3:1, об./об.), получая целевой аллиловый эфир (5R,6S)-2-хлорметил-6-((R)-1- трет-бутилдиметилсилилоксиэтил)-пенем-3-карбоновой кислоты в виде желтого масла. Выход: 71%.
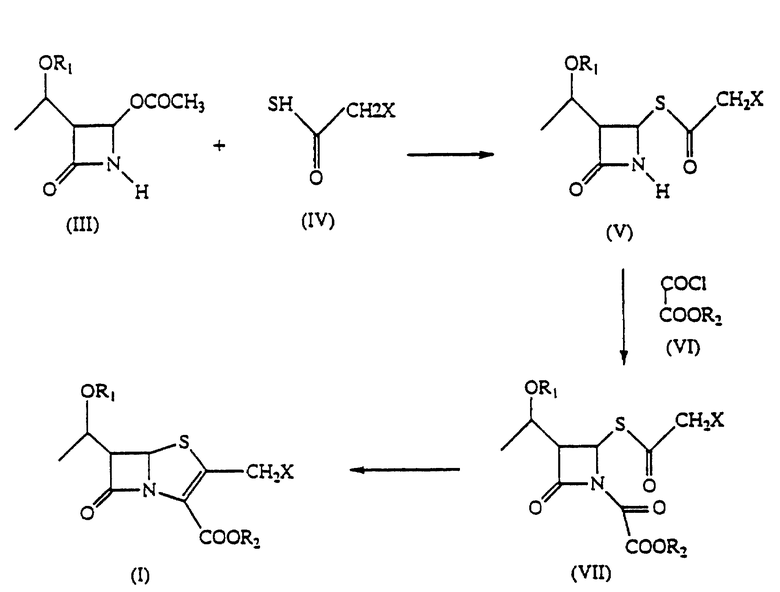
Изобретение относится к способу получения 2-галогенметилпенемов формулы I, где R1 представляет защитную группу для спиртового гидроксила, R2 представляет защитную группу для карбоксила и Х представляет галоген, включающий взаимодействие соединения формулы III с 2-галогентиоуксусной кислотой в органическом растворителе в присутствии органического основания и кислоты Льюиса при температуре от -10 до +40oС с получением соединения формулы V, которое взаимодействует с эфиром оксалилхлорида в органическом растворителе в присутствии органического основания при температуре от -60 до +20oС, предпочтительно от -20 до +10oС с получением соединения формулы VII, которое циклизуют в подходящем растворителе при помощи органического фосфита или фосфонита при температуре 20-140oС. Промежуточные соединения формулы V и формулы VII, где R1 является трет-бутилдиметилсилилом, R2 является аллилом, Х является хлором. Технический результат - упрощение способа получения и увеличение выхода целевого продукта. 3 с. и 3 з.п. ф-лы.
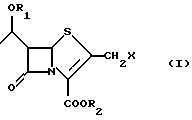
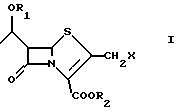
где R1 представляет защитную группу для спиртового гидроксила;
R2 представляет защитную группу для карбоксила;
Х представляет галоген,
отличающийся тем, что включает следующие стадии: а) взаимодействие соединения формулы III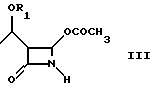
где R1 - как определено выше,
с 2-галогентиоуксусной кислотой в органическом растворителе в присутствии органического основания и кислоты Льюиса при температуре от -10 до +40oС с получением соединения формулы V; б) взаимодействие соединения формулы V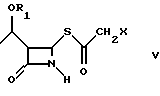
с эфиром оксалилхлорида в органическом растворителе в присутствии органического основания при температуре от -60 до +20oС, предпочтительно, от -20 до +10oС с получением соединения формулы VII; в) циклизацию соединения формулы VII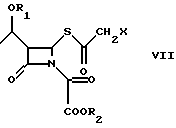
где R1, R2 и Х определены выше,
в подходящем растворителе при помощи органического фосфита или фосфонита при температуре 20 - 140oС в течение 1 - 120 ч.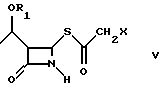
где R1 является трет-бутилдиметилсилилом;
Х - Сl.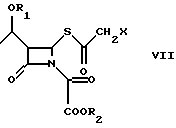
где R1 является трет-бутилдиметилсилилом;
R2 является аллилом;
Х - Сl.
| СПОСОБ НАПРАВЛЕННОГО ВОЗДЕЙСТВИЯ ХИМИЧЕСКИМИ МУТАГЕНАМИ НА РАСТЕНИЯ | 0 |
|
SU399228A1 |
| КОММУНИКАЦИОННАЯ СИСТЕМА МНОГОЭТАЖНОГО ЗДАНИЯ | 1999 |
|
RU2144743C1 |
| Абсорбер | 1976 |
|
SU597401A1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-МЕТОКСИМЕТИЛПЕНЕМОВ | 1990 |
|
RU2049786C1 |

