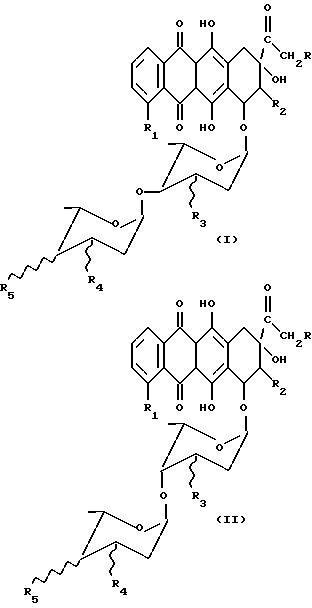
Настоящее изобретение относится к соединениям общей формулы (I) и (II), соответственно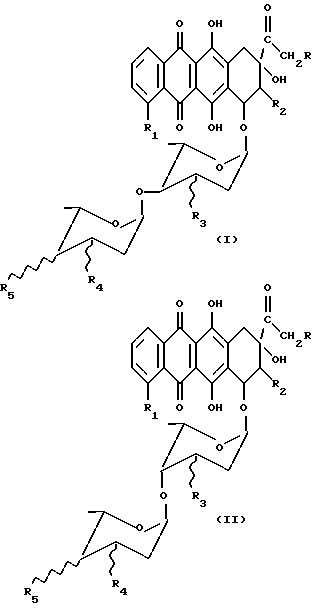
в которых R - H или OH или OR7 группа, где R7 = CHO или COCH3 или ациловый остаток карбоновой кислоты, содержащей до 6 атомов углерода, R1 - H или OH или OCH3, R2 - H или F, R3 - H или OH, R4 и R5 - одинаковые или разные и каждый представляет собой H или OH или NH2, и знак связи  указывает, что заместители R3, R4 и R5 могут быть или в аксиальном или экваториальном положении, и их фармацевтически приемлемым солям, обладающим противораковыми свойствами. Как видно из вышепредставленных формул, соединения (I) и (II) отличаются исключительно пространственным расположением гликозидных групп и поэтому могут быть представлены формулой (A)
указывает, что заместители R3, R4 и R5 могут быть или в аксиальном или экваториальном положении, и их фармацевтически приемлемым солям, обладающим противораковыми свойствами. Как видно из вышепредставленных формул, соединения (I) и (II) отличаются исключительно пространственным расположением гликозидных групп и поэтому могут быть представлены формулой (A)
в которой знак  указывает, что второй остаток углевода может быть связан с атомом углерода 4' первого сахара или в аксиальном или в экваториальном положении.
указывает, что второй остаток углевода может быть связан с атомом углерода 4' первого сахара или в аксиальном или в экваториальном положении.
Настоящее изобретение также относится к способу получения указанных соединений, их фармацевтически приемлемых солей и содержащим их фармацевтическим композициям.
Даунорубицин и доксорубицин представляют собой хорошо известные антибиотики, которые используются в настоящее время в клинической практике при лечении ряда твердых опухолей и лейкоза (F. Arcamone в "Doxorubicin: Anticancer Antibiotics" A. C. Sartorelli Ed. Academic Press. N.Y. 1981).
Соединения, имеющие структуру, схожую со структурой соединений, описанных в настоящей заявке, но представляющие только одну гликозидную группу, описаны в EP-457215, WO 80/00305 и WO 90/07519.
Соединения, имеющие две или несколько частей сахара, в которых сахар, непосредственно связанный с частью агликона, является амино-замещенным, описываются, например, в Thi JournaL of Antibiotics, стр. 1720-1730, Nov. 93, TeTrahedron VoL 37, 24, 4219-4228, DE 2751395, Carbohydrate Ressearch. 228, 171-91 /1992/ и DE 3641833.
Соединения, имеющие три гликозидных фрагмента, по которым не представлены данные об активности, описаны в WO 92/07862. Однако, как известно, острые побочные действия, вызываемые противораковыми агентами, используемыми в настоящее время, ограничивают их применение значительным количеством больных, которые если бы не эти явления, могли бы успешно вылечиваться. Кроме того, добиться существенного прогресса в лечении некоторых важных твердых опухолей, например, легочных и яичниковых, которые плохо поддаются существующим видам лечения.
Из этого следует, что существует острая необходимость в поступлении на рынок лекарств, которые высоко селективны в своем действии торможения пролиферации больных клеток в сравнении со здоровыми.
Цель настоящего изобретения заключается в получении новых противораковых соединений, в частности, аналогов антрациклина, в которых часть углевода состоит из остатка дисахарида.
Неожиданно было выявлено, что заявленные антрациклиновые дисахариды, в которых сахар непосредственно связанный с агликоном, никогда не содержит амино групп, проявляют более высокую противораковую активность и селективность, чем известный антрациклин. Следует отметить, что в известных антрациклинах, содержащих два остатка углевода, сахар, связанный с агликоном, всегда содержит свободную или замещенную амино группу.
Соединения по настоящему изобретению представляют собой соединения общей формулы (I) и (II), как говорилось выше, и их фармацевтически приемлемые соли, в которых R, R1, R2, R3, R4 и R5 имеют те же значения, что указаны выше.
Настоящее изобретение также относится к фармацевтическим композициям, содержащим упомянутые соединения или их соли с фармацевтически приемлемыми кислотами, предпочтительно хлористоводородной кислотой.
Наиболее предпочтительны следующие соединения:
a) 7-O-[2,6-дидеокси-4-O-/2,3,6-тридеокси-3-амино- α -L-ликсогексопиранозил/- α -L-ликсо-гексопиранозил]даунорубицинонахлоргидрат,
b) 7-O-[2,6-дидеокси-4-O-/2,3,6-тридеокси-3-амино- α -L-ликсогексопиранозил/- α -L-арабино-гексопиранозил]даунорубицинонахлоргидрат,
c) 7-O-[2,6-дидеокси-4-O-/2,3,6-тридеокси-3-амино- α -L-ликсогексопиранозил/- α -L-ликсо-гексопиранозил]доксорубицинонахлоргидрат,
d) 7-O-[2,6-дидеокси-4-O-/2,3,6-тридеокси-3-амино- α -L-ликсогексопиранозил/- α -L-арабино-гексопиранозил]доксорубицинона хлоргидрат,
e) 7-O-[2,6-дидеокси-4-O-/2,3,6-тридеокси-3-амино- α -L-ликсогексопиранозил/- α -L-арабино-гексопиранозил]-4-деметокси-даунорубицинона хлоргидрат,
f) 7-O-[2,6-дидеокси-4-O-/2,3,6-тридеокси-3-амино- α -L-ликсогексопиранозил/- α -L-ликсо-гексопиранозил]-4-деметокси-даунорубицинона хлоргидрат,
g) 7-O-[2,6-дидеокси-4-O-/2,3,6-тридеокси-3-амино- α -L-ликсогексопиранозил/- α -L-ликсо-гексопиранозил]-4-деметокси-доксорубицинона хлоргидрат,
h) 7-O-[2,6-дидеокси-4-O-/2,3,6-тридеокси-3-амино- α -L-ликсогексопиранозил/- α -L-арабино-гексопиранозил]-4-деметокси-доксорубицинона хлоргидрат,
i) 7-O-[2,6-дидеокси-4-O-/2,3,4,6-тетрадеокси-4-амино- α -L-эритро-гексопиранозил/- α -L-ликсо-гексопиранозил]даунорубицинона хлоргидрат,
j) 7-O-[2,6-дидеокси-4-O-/2,3,4,6-тетрадеокси-4-амино- α -L-эритро-гексопиранозил/- α -L-ликсо-гексопиранозил]-4-деметоксидаунорубицинона хлоргидрат,
k) 7-O-[2,6-дидеокси-4-O-/2,3,4,6-тетрадеокси-4-амино- α -L-эритро-гексопиранозил/- α -L-ликсо-гексопиранозил]доксорубицинона хлоргидрат,
l) 7-O-[2,6-дидеокси-4-O-/2,3,4,6-тетрадеокси-4-амино- α -L-эритро-гексопиранозил/- α -L-ликсо-гексопиранозил]-4-деметоксидоксорубицинона хлоргидрат,
m) 7-O-[2,6-дидеокси-4-O-/2,3,6-тридеокси-3-амино- α -L-ликсогексопиранозил/- α -L-ликсо-гексопиранозил] -4-деметокси-8-фтородаунорубицинона хлоргидрат.
n) 7-O-[2,6-дидеокси-4-O-/2,3,6-тридеокси-3-амино- α -L-ликсогексопиранозил/- α -L-ликсо-гексопиранозил]-4-деметокси-8-фтородоксорубицинона хлоргидрат,
Соединения общей формулы (I) и (II) можно получить способом, состоящим из следующих стадий:
a) конденсации соединения формулы (III) (III)
(III)
в которой R1 и R2 имеют те же значения, что определены выше, и R6 - H или OR7 группа, где R7 является защитной группой для спиртовой функции, предпочтительно выбранной из ацетиловых, диметилтрибутилсилиловых или п-метоксифенилдифенилметиловых групп, с соединением формулы (IV) или (V):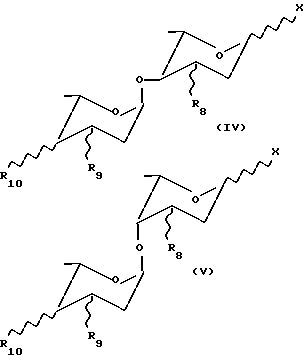
в которых R8 - H или защищенная - OH группа, предпочтительно п-нитробензоат, R9 и R10, одинаковые или разные, каждый представляет собой H или защищенную OH группу, предпочтительно п-нитробензоат или защищенную NH2 группу, предпочтительно трифтороацетамид или аллилкарбоксиамид и X - группа, способная образовывать в условиях конденсации устойчивый карбо-катион, который может связываться с гидроксильной группой в положении C-7 соединения формулы (III), при этом группу X обычно выбирают из групп, используемых в реакциях гликозидирования, например, галогена, такого, как хлор или бром, предпочтительно хлор, или п-нитробензоилокси группы. Таким образом получают соединения формулы (VI) или (VII):
в которых R1, R2, R6, R8, R9, R10 и знак  - те же, что указаны выше,
- те же, что указаны выше,
b) одной или нескольких реакций удаления защитных групп с функциями OH и/или NH2 из соединений формулы (VI) и (VII), чтобы получить соединения формулы (I) и (II), в которых R, R1, R2, R3, R4, R5 и знак  имеют те же значения, что определены выше,
имеют те же значения, что определены выше,
c) превращения вышеупомянутых гликозидов формулы (I) и (II) в их фармацевтически приемлемую соль, предпочтительно хлоргидрат.
Условия реакции для гликозидирования соединения формулы (III) с соединением формулы (IV) или (V) с получением соединения формулы (VI) или (VII), могут быть различными в зависимости от типа заместителей, присутствующих в соединениях формулы (IV) или (V).
Гликозидирование проводят в инертном органическом растворителе в присутствии конденсирующего агента.
Применяемые конденсирующие агенты представляют собой, например, фтороформ сульфонат серебра, хлорнокислое серебро, смеси окиси ртути и бромида ртути, галиды бора, четыреххлористое олово или титан или ионообменные смолы, такие, как Amberlite, трифлат серебра, триметилсилилтрифлат, п-толуолсульфокислота, трифторуксусная кислота.
Предпочтительно гликозидирование проводят с молярным соотношением от 1:1 к 1: 3 в инертном органическом растворителе, таком, как, например, бензол, толуол, этиловый эфир, тетрагидрофуран, диоксан, хлороформ, метиленхлорид или дихлорэтан и их смеси.
Температура реакции может иметь диапазон от -40oC до 40oC, предпочтительно от -20oC до 20oC и время реакции от 15 минут до 3 часов.
Реакционная смесь может включать обезвоживающее вещество, такое, как активированное молекулярное сито. В ходе или в конце реакции реакционную смесь можно добавлять с органическим основанием, таким, как пиридин, коллидин, N, N-диметиламинопиридин, триэтиламин или 1.8-бис-/диметиламино/-нафталин.
В соответствии с настоящим изобретением удаление защитных групп для OH и/или NH2 функций из соединений формулы (VI) и (VII) для получения соединений формулы (I) можно провести в различных условиях в зависимости от типа используемой защитной группы. Если R9 и/или R10 одинаковые или разные и каждый представляет защищенную NH2 группу, такую, как трифторацетамид или защищенную OH группу, такую, как п-нитробензоат, и/или R8 - защищенная OH группа, такая, как п-нитробензоат, и/или R6 - защищенная OH группа, такая, как ацетат, реакции снятия защиты проводят в полярном растворителе, таком, как вода, метанол, этанол, пиридин, диметилформамид или их смеси и в присутствии неорганического основания в стехиометрическом количестве или в его избытке, такого, как гидроксид или карбонат натрия, калия, лития или бария.
Температура реакции может колебаться от 0oC до 50oC и время реакции от 3 часов до 48 часов.
Если R9 и/или R10 каждый представляет собой защищенную NH2 группу, такую, как аллилкарбоксиамид, снятие защиты проводят в инертном растворителе и в присутствии комплекса металла, такого, как тетракис /трифенилфосфин/палладий, как раскрывают, например, в Tetrahedron Letters, 30, 3773 /1989/, или тетракарбонил никель, как раскрывают, например, в J. Org. Chem. 38, 3233 /1973/. Если R6 - защищенная OH группа, такая, как диметилтретбутилсилилэфир, снятие защиты проводят в инертном растворителе и в присутствии фторида тетрабутиламмония, как раскрывают, например, в J. of Antibiot. 37, 853 /1984/.
Если R6 - защищенная OH группа, такая, как п-метоксифенилдифенилметилэфир, снятие защиты проводят в кислотной среде, например, в водной уксусной кислоте, как раскрыто, например, в J. Org. Chem. 42, 3653 /1977/.
Соединения формулы (III) или известны или их можно получить методами и способами, известными в органической химии, как раскрыто, например, в Ga33. Chim. Ital. 114, 517 /1984/, в Bull. Chem. Soc, Jpn. 59, 423 /1986/ и в вышеупомянутой патентной заявке Италии данного заявителя, раскрытие которой включается сюда ссылкой. Соединения формулы (IV) или (V) или известны или их можно получить методами и способами синтеза дисахаридов, известными в органической химии /J. Carbohydr. Chem. 10, 833 /1991/, Carbohydr, Res, 74, 199 /1979/, Carbohydr. Res., 208, 111 /1980/, Tetrahedron, 46, 103, /1990/.
В другом случае при желании антрациклиновые гликозиды формулы (I) и (II), в которых R1, R2, R3, R4, R5 - те же, что указаны выше и R - OH группа, можно получить из гликозидов формулы (I) и (II) или их фармацевтически приемлемых солей, в которых R1, R2, R3, R4, R5 и знак  - те же, что определены выше и R - H, бромированием углерода в положении 14 бромом в хлороформе с последующим гидролизом, при комнатной температуре в течение 48 часов форматом натрия полученных 14-бромопроизводных.
- те же, что определены выше и R - H, бромированием углерода в положении 14 бромом в хлороформе с последующим гидролизом, при комнатной температуре в течение 48 часов форматом натрия полученных 14-бромопроизводных.
При желании гликозиды формулы (I) и (II) можно преобразовать в их фармацевтически приемлемые соли, например, хлоргидраты, обработкой хлористоводородной кислотой в метиловом спирте. Настоящее изобретение также относится к фармацевтическим композициям, содержащим в качестве активного ингредиента соединение формулы (I) или (II) или его фармацевтически приемлемую соль, соединенную с фармацевтически приемлемым разбавителем или носителем.
В соответствии с настоящим изобретением терапевтически эффективная доза соединения формулы (I) или (II) соединяется с инертным носителем. Композиции можно формировать традиционным способом с использованием обычных носителей.
Заявленные соединения полезны для лечения людей и других млекопитающих. В частности, данные соединения являются хорошими противораковыми агентами при назначении в терапевтически эффективных дозах.
Следующие примеры более подробно иллюстрируют настоящее изобретение.
Пример 1
7-O-[2,6-дидеокси-4-O-/2,3,6-тридеокси-3-амино- α -L-ликсо-гексопиранозил/- α -L-ликсо-гексопиранозил] -4-деметокси-даунорубицинона хлоргидрат (соединение формулы II, R=R1=R2=H, R3=R5=OH, R4=NH2). Смесь 4-деметоксидаунорубицинона (соединение формулы III, R1=R2=R6=H) (300 мг, 0,81 мМ) и 2,6-дидеокси-4-O-/2,3,6-тридеокси-4-O-п-нитробензоил-3-трифторацетамидо- α -L-ликсо-гексопиранозил/-3-O-п-нитробензоил- α -L-ликсо-гексопиранозил-п-нитробензоата (соединение формулы IV, R3=R5=п-нитробензоил-окси-, R4=трифторацетамидо-, X=п-нитробензоилокси-/ (600 мг, 0,72 мМ) в метилхлориде (72 мл) и этиловом эфире (24 мл) в присутствии молекулярных сит (A4) при -20oC обрабатывали триметилсилилтрифлатом (266 μ л, 1,44 мМ). Реакционную смесь перемешивали 1 час, затем ее разбавляли метиленхлоридом, промывали насыщенным раствором бикарбоната натрия и выпаривали до сухости. Остаток разделяли хроматографией на силикагеле (элюент CH2Cl2 EtOH), 99/1/ с выходом 360 мг 7-O-[2,6-дидеокси-4-O-/2,3,6-тридеокси-4-O-п-нитробензоил-3- трифторацетамидо-α -L-ликсо-гексопиранозил/-3-O-п-нитробензоил- α -L-ликсо-гексопиранозил] -4-деметоксидаунорубицинона (соединение формулы VII, R1-R2=R6=H, R8=R10=п-нитробензоилокси-, R9-трифторацетамидо-.
Замещенную суспензию дигликозида соединения формулы (VII) /R1=R2=R6=H, R8=R10=п-нитробензоилокси, - R9=трифторацетамидо-/ (120 мг, 0,117 мМ) в 17,6 мл 0,1 М раствора Ba/OH/2 в H2O/MeOH, 1/1, выдерживали с помешиванием при комнатной температуре в течение 3 часов. Реакционную смесь нейтрализовали 0,2 М раствором бисульфата калия и экстрагировали хлороформом, органические экстракты собирали вместе, высушивали над безводным сульфатом натрия, выпаривали до сухости и подвергали поглощению 0,002 М раствором HCl. Кислотный водный раствор промывали хлороформом и высушивали замораживанием с получением 62 мг нужного продукта (соединение формулы II, /R=R1=R2=H, R3=R5=OH, R4=H2/. Выход 39%. Полученные данные ЯМР представлены ниже:
1H-ЯМР /ДМСО-D6/, δ 1,05 /д, 3H/, 1,15 /д, 3H/, 1,5 - 1,95 /м, 4H/, 2,1 /м, 2H/, 2,25 /с, 3H/, 2,95 /дд, 2H/, 3,55 /с, 2H/, 3,8 /м, 1H/, 3,95 /м, 1H/, 4,15 /кв, 1H/, 4,35 /кв, 1H), 4,6 /д, 1H/, 4,9 /шир.с, 2H/, 5,25 /шир. с, 1H/, 5,35 /д. 1H/, 5,55 /с, 1H/, 7,95 /шир.с., 2H/, 8,25 /шир.с., 2H/.
В соответствии с аналогичным способом были также получены следующие соединения формулы (I) и (II):
7-O-[2,6-дидеокси-4-O-/2,3,6-тридеокси-3-амино- α -L-ликсо-гексопиранозил/- α -L-ликсо-гексопиранозил] даунорубицинона хлоргидрат /соединение формулы II, R=R2=H, R1=OCH3, R3=R5=OH, R4=NH2/.
1H-ЯМР /ДМСО-D6/, δ 1,05 /д. 3H/, 1,15 /д, 3H/, 1,35 - 2,15 /м, 6H/, 2,25 /с, 3H/, 2,95 /дд. 2H/, 3,55 /шир.с., 2H/, 3,8 /м, 1H/, 3,95 /с, 3H/, 4,05 - 4,2 /м+кв, 2H/, 4,35 /кв, 1H/, 4,9 /шир.с., 2H/, 5,25 /д, 1H/, 7,65 /м, 1H/, 7,9 /д, 2H/.
7-O-[2,6-дидеокси-4-O-/2,3,6-тридеокси-3-амино- α -L-ликсо-гексопиразонил/- α -L-арабино-гексопиранозил]даунорубицинона хлоргидрат (соединение формулы 1, R=R2=H, R1=OCH3, R3=R5=OH, R4=NH2).
1H-ЯМР (ДМСО-D6) δ 1,13 (д, 3H), 1,15 /д, 3H/, 1,45-1,85 /м, 4H/, 2,05 /м, 2H/, 2,15 /с, 3H/, 2,87 /дд, 2H/, 2,98 /м, 1H/, 3,5 /м, 1H/, 3,6 /м, 1H/, 3,85 /кв, 1H/, 3,9 /кв, 1H/, 3,9 /с, 3H/, 4,84 /м, 2H/, 5,13 /шир. с, 1H/, 5,28 /с, 1H/, 5,32 /д, 1H/, 5,55 /с, 1H/, 7,55 /м, 1H/, 7,8 /м, 2H/.
7-O-[2,6-дидеокси-4-O-/2,3,6-тридеокси-3-амино- α -L-ликсо-гексопиразонил/- α -L-арабино-гексопиранозил]-4-деметокси-даунорубицинона хлоргидрат (соединение формулы 1: R=R1=R2=H, R3=R5=OH, R4=NH2/.
1H-ЯМР /ДМСО-D6/, δ 1,1 /д, 3H/, 1,2 /д, 3H/, 1,5-1,95 /м, 4H/, 2,05 - 2,2 /м, 2H/, 2,25 /с, 3H/, 2,95 /дд, 2H/, 3,1 /т, 1H/, 3,4 /м, 1H/, 3,6 /шир. с. , 1H/, 3,65 /м, 1H/, 3,85-4,00 /кв+кв, 2H/, 3,9 /м, 1H/, 4,95 /д, 1H/, 5,2 /д, 1H/, 5,2 /д, 1H/, 5,4 /шир. с., 2H/, 5,7 /с. 1H/, 7,95 /м, 2H/, 8,25 /м, 2H/.
Данные ЯМР-спектров, относящиеся к полученным соединениям (g) и (1)
Следующие соединения были получены по методике, аналогичной описанной в примерах i в описании.
(1) Соединение (g) формулы II 7-O-[2,6-дидеокси-4-O-(2,3,6-тридеокси-3-амино- α -L-ликсогексопиранозил)- α - L-ликсо-гексопиранозил]-4-деметоксидоксорубицинона хлоргидрат (II, R = OH, R1 = R2 = H, R3eg. = R5ax = OH, R4eg = NH2)
Следующие полосы (d, ppm) были соотнесены следующим образом: 1,05 (d, 3H, CH3''); 1,15 (d, 3H, CH3'); 1,5-1,9 (m, 4H, H-2' и H-2''); 2,1 (m, 2H, H-8 ax и H-8 eg); 3,0 (ds, 2H, H-10 ax и H-10 eg); 3,45 (m, 1H, H-3''); 3,5 (bs, 1H, H-4'); 3,6 (bs, 1H, H-4''); 3,8 (m, 1H, H-3'); 4,1 (g, 1H, H-5'); 4,35 (g, 1H, H-5''); 4,55 (s, 2H, H-14); 4,6 (d, 1H, OH-3'); 4,85 (bs, 1H, OH-14); 4,9 (m, 2H, H-7 и H-1''); 5,2 (d, 1H, H-1'); 5,3 (d, 1H, OH-4''); 5,53 (s, 1H, OH-9); 8,0 и 8,3 (два m, 4H, ароматика)
(2) Соединение (1) формулы II 7-O-[2,6-дидеокси-4-O-(2,3,4,6-тетрадеокси-4-амино- α -L-эритрогексопиранозил)- α -L-ликсогексопиранозил] -4-деметоксидоксорубицинона хлоргидрат (II, R = OH, R1 = R2 = R4 = H, R3eg = OH, R5eg = NH2).
Следующие полосы (d, ppm) были соотнесены следующим образом: 1,1 (d, 3H, CH3''); 1,2 (d, 3H, CH3'); 1,5-2,3 (m, 6H, H-2', H-2'' и H-3''); 2,1 (m, 2H, H-8 ax и H-8 eg); 3,0 (ds, 2H, H-10 ax и H-10 eg); 3,35 (m, 1H, H-4''); 3,5 (bs, 1H, H-4'); 3,65 (bg, 1H, H-5''); 3,8 (m, 1H, H-3'); 4,1 (g, 1H, H-5'); 4,5 (s, 2H, H-14); 4,6 (d, 1H, OH-3'); 4,85 (bs, 1H, OH-14); 4,9 (bt, 1H, H-7); 4,95 (d, 1H, H-1''); 5,2 (d, 1H, H-1'); 5,53 (s, 1H, OH-9); 8,0 и 8,3 (два m, 4H, ароматика).
Цитотоксическая активность (IC50 мг/мл) представлена в опыте 1 (см. в конце описания).
Опыт 2
Биологическая активность соединений:
IIa : 7-O-[2,6-дидеокси-4-O-(2,3,6-тридеокси-3-амино-α-L-ликсогексопиранозил)-α- L-ликсо-гексопиранозил] -4-деметокси-даунорубицинона хлоргидрат (II, R=H, R1=H, R3eg = R5ax = OH, R4eg = NH2);
(Соединение f)
IIc: 7-O-[2,6-дидеокси-4-O-(2,3,6-тридеокси-3-амино-α-L-ликсо-гексопиранозил)-α-L-ликсо-гексопиранозил] -4-деметокси-доксорубицинона хлоргидрат (II, R=OH, R1 =H, R3eg= R5ax=OH, R4eg = NH2);
(Соединение g)
R2 = H во всех указанных соединениях, - проверялась в сравнении с доксорубицином в отношении множества видов рака человека различных гистотипов, включая рак яичника, рак легкого, рак молочной железы, рак шейки матки, рак толстой кишки.
Активность in vitro
Выживаемость клеток после воздействия на них испытуемого соединения оценивалась по увеличению ингибирования, за исключением клеточных линий мелкоклеточного рака легкого (SCLC). Клеточную биомассу в логарифмической фазе роста собирали и размещали в 6 ячейках. Спустя 24 часа после засева клетки подвергали воздействию испытуемых соединений в течение различных периодов времени. После замены среды на среду, не содержащую лекарства, клеточную массу собирали и подсчитывали клетки спустя 72 часа после воздействия соединения. Для SCLC MCF-7 и LOVO использовали метод MTT-анализа и проводили колориметрическую оценку выживаемости в конце инкубационного периода (24 или 96 часов). Результаты, полученные в экспериментах по исследованию цитотоксичности, выражали в виде величины IC50 (т.е. концентрации, требующейся для 50%-ного ингибирования роста клеток).
Испытуемые соединения растворяли в дистиллированной воде и ею разбавляли культуральную среду.
Результаты показаны в табл. 1.
Заявленные соединения IIa-IIc демонстрируют удивительные цитотоксические воздействия, сила которых сопоставима или превышает эффект DXR.
Исследования in vivo
Для исследования in vivo бестимусным мышам подкожно с обеих сторон спины вводили фрагменты опухолей. Рост опухоли сопровождался измерением дважды в неделю ее длины и ширины с помощью кронциркуля. Вес опухоли вычисляли с помощью формулы: мг = объем в мм3 = ширина2 • длина/2 (относительная плотность = 1).
Испытуемые соединения вводились в оптимальных дозах и по графику внутривенно (i.v.) мышам с опухолями, начиная с различных периодов времени в зависимости от использования различных видов рака (см. детально эксперименты). Эффекты, достигнутые лечением, выражали в виде величины ингибирования массы опухоли, % (twi %) у мышей, подвергнутых лечению, по сравнению с контрольными, определяемой спустя 7 - 10 дней после последнего введения лекарства.
Результаты показаны в таблицах 2 и 3.
По отношению к раку яичника A 2780 соединение IIa показывает, по меньшей мере, такую же эффективность, что и доксорубицин, тогда как соединение IIc достигает уровня противоопухолевой активности, значительно более высокого, чем у исходного соединения, снижая рост опухоли на 99%.
В отношении SCLC опухоли POVD, плохо поддающейся лечению DXR доксорубицином, соединение IIa, как и соединение IIc, показывает очень высокую противоопухолевую активность.
Фармацевтическая композиция
Примеры и методики получения
Пример 1.
Сравнительные количества различных компонентов, применяемых в композиции, были такими, как показано ниже (количества даны в расчете на флакон):
Соединение g - 10,00 мг
Лактоза - 50,00 мг
Точное количество (как определено методом ВЭЖХ) соединения g, как активного начала, и лактозы USP (по фармакопее США) последовательно растворяют в воде для инъекций при перемешивании и при комнатной температуре. Раствор деаэрируют барботированием азота и затем добавляют деаэрированную воду для инъекций до получения конечной концентрации активного начала 5 мг/мл.
Полученный в результате раствор фильтруют через фильтр 0,22 мм в стерильных условиях.
Профильтрованный раствор разливают в стерильные стеклянные флаконы (объем 2 мл).
Общая продолжительность лиофилизации составляет 45 часов. Спустя это время обычно давление восстанавливают введением азота, флаконы закрывают стерильными пробками и запечатывают алюминиевыми стерильными колпачками.
Пример 2
Таким же образом получают лиофилизированную композицию на основе соединения g, содержащую 50 кг активного начала:
Соединение g - 50,00 мг
Лактоза - 250,00 мг
Сушка вымораживанием выполнялась в стерильных стеклянных флаконах емкостью 50 мл.
Терапевтическое лечение (протокол)
Противоопухолевая активность соединения IIg 7-O-[2,6-дидеокси-4-O-(2,3,6-тридеокси-3-амино-α-L-ликсо- гексаопиранозил)-α-L-ликсо-гексопиранозил] -4-деметокси доксорубицинона хлоргидрата (II, R-OH, R1=R2=H, R3еg= R5ах= OH, R4eg=NH2) изучалась по отношению к MX-1 опухоли человека (рак молочной железы), привитой бесшерстным мышам.
Изучение действительности: для оценки реакции опухоли на соединение IIg его активность in vivo определяли при 6 мг/кг по графику g3/4dx5, для того, чтобы оптимизировать противоопухолевую активность, учитывая зависимость времени выдержки от его цитотоксического действия в экспериментах in vitro c культурами клеток. Доксорубицин (DXR) использовался как лекарственное средство для сравнения при дозе 7 мг/кг и графике (g7dx3), который был установлен как оптимальный.
Сравнительные подтверждения исследования: для того, чтобы провести всестороннее сравнение двух лекарственных средств, более широкий спектр доз исследовали на опухоли МХ-1 один раз или дважды в неделю. В эту группу экспериментов входило также сравнение соединения IIg и DXR при их максимальных толерантных дозах, для последнего она была определена путем наблюдения над обработанными животными в течение по меньшей мере трех месяцев с момента начала обработки.
Во время исследований in vivo линии опухолевых клеток человека поддерживались серией подкожных (s. c.) пассажей опухолевых фрагментов по обеим сторонам спины взрослым бестимусным бесшерстным мышам. Обычно опухолевые фрагменты гистологически проверялись на предмет человеческого происхождения после окрашивания гематоксилином и эозином и путем электрофореза изоэнзимов дегидрогеназы молочной кислоты.
Бесшерстным мышам (Charles River, Calco, Italy) инъецировали s.c. опухолевые фрагменты в обе стороны спины (изучение действенности) или в один бок (сравнительные подтверждающие исследования). Рост опухоли сопровождался измерением ее длины и ширины с помощью кронциркуля в предопределенные ранее моменты времени (раз в неделю или два раза в неделю). Вес опухоли (TW) вычисляли по формуле: мг = объем в мм3 = ширина2 х длина/2 (относительная плотность = 1). 7/8 дней после последнего введения лекарства. Время удвоения опухоли (ДТ) вычисляли для каждой опухолевой линии по полулогарифмической наиболее соответствующей кривой для каждой контрольной опухоли, вычерченной в сравнении с некоторым уровнем (экспоненциальная фаза роста). Значения величин ДТ были использованы в исследованиях.
Лекарства вводили внутривенно (i.v.) мышам с опухолями в разное время в зависимости от опухоли. Дни первой и последней инъекций были одни и те же для двух лекарств во всех экспериментах. По недельному графику соединение g вводили дважды в один и тот же день с промежутком между инъекциями приблизительно 1 час. В каждой экспериментальной группе было по крайней мере восемь видов рака.
Оценивали следующие эффекты, достигнутые при введении лекарств:
- ингибирование массы опухоли % (TWI %) у мышей, подвергнутых обработке, по сравнению с контрольными, определяемое 7-10 дней спустя после последнего введения лекарства;
- логарифм показателя уничтожения клеток (LCK) у обработанных мышей в соответствии с формулой T-C/ДТ • 3,32, где T и C - время в сутках, необходимое для достижения среднего веса опухоли у обработанных (T) и контрольных (C) мышей, указанного в каждом эксперименте;
- токсическая смерть: количество погибших мышей, у которых отсутствует опухоль измеримой массы, или мышей, погибших до первой смерти в контрольной группе;
- долговременное выживание (LTC):
живые мыши с опухолью или без нее в конце эксперимента (сравнительные подтверждающие исследования).
Результаты
Модель МХ-1 рака молочной железы, полученная от пациентки, не получавшей лечения, была проверена на резистентность к DXR (доксорубицину). По отношению к этой опухоли соединение g было очень активно при различном уровне доз в двух отдельных экспериментах. Достигнутые TWI% и LCK были значительно выше, чем те, которые получали от лечения DXR (Табл.1.1). Второй эксперимент (Табл. 2.1) показал, что противоопухолевая активность соединения по графику g(3/4)dx5 была выше в определениях периода регрессии и продолжительности эффекта.
Таблица 1.1: противоопухолевая активность соединения g и доксорубицина (DXR) в отношении рака молочной железы человека МХ-1. Вес опухоли в начале обработки лекарством составлял 50 мг в обоих экспериментах.
Таблица 2.1: противоопухолевая активность соединения g и доксорубицина (DXR) в отношении рака молочной железы человека МХ-1. Вес опухоли в начале обработки лекарством составил 50 мг в обоих экспериментах.
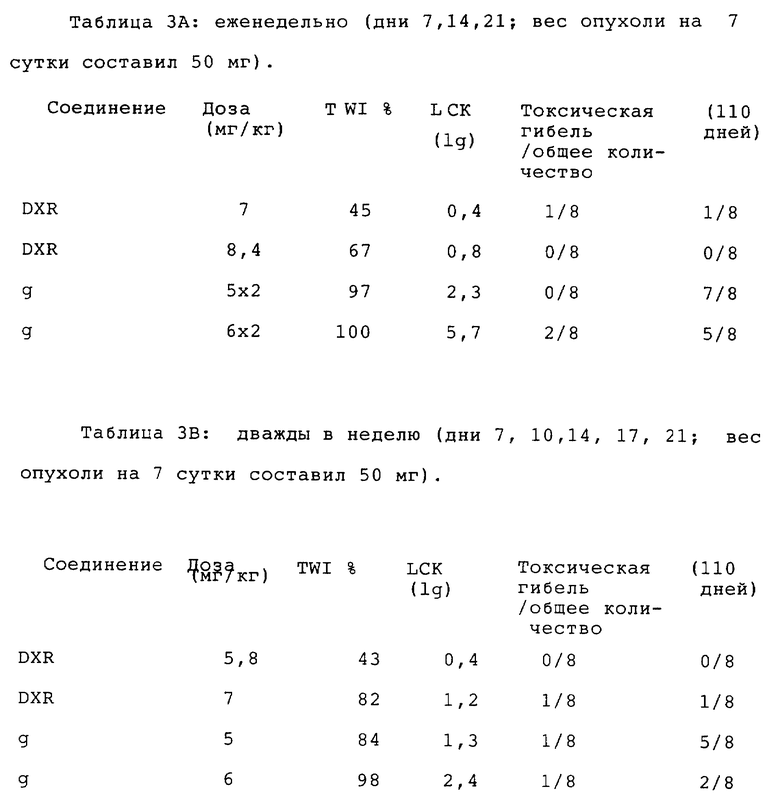
В сравнительных подтверждающих исследованиях соединение g сравнивали с DXR в соответствии с еженедельным (таблица 3A) или 2-недельным (таблица 3B) графиком.
Таблица 3A: еженедельно (дни 7, 14, 21; вес опухоли на 7 сутки составил 50 мг).
Таблица 3B: дважды в неделю (дни 7, 10, 14, 17, 21; вес опухоли на 7 сутки составил 50 мг).
Результаты этих сравнительных подтверждающих исследований свидетельствуют об эффективности соединения g при ингибировании роста этой опухоли, выбранной из-за ее невыразительной реакции на DXR.





Описываются новые антрациклиновые дисахариды общей формулы I и II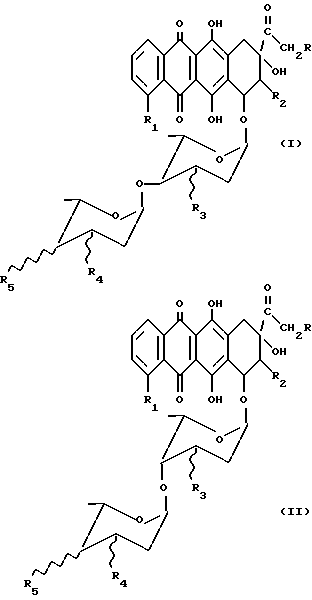 ,
,
где значения радикалов указаны в п.1 формулы. Соединения проявляют более высокую противораковую активность и селективность, чем известный антрациклин. Описывается также способ их получения и содержащие их фармацевтические композиции. 3 с. и 2 з.п ф-лы, 7 табл.
в которых R = H, OH; R1 = H или OCH3, R2 = H, R3 = OH, R4 = H, NH2, R5 = OH, NH2;
и знак связи  указывает, что заместители R3, R4, R5 могут находиться в аксиальном или экваториальном положении,
указывает, что заместители R3, R4, R5 могут находиться в аксиальном или экваториальном положении,
или их фармацевтически приемлемые соли. -L-ликсогексопиранозил] -4-деметокси-доксорубицинона хлоргидрата.
-L-ликсогексопиранозил] -4-деметокси-доксорубицинона хлоргидрата.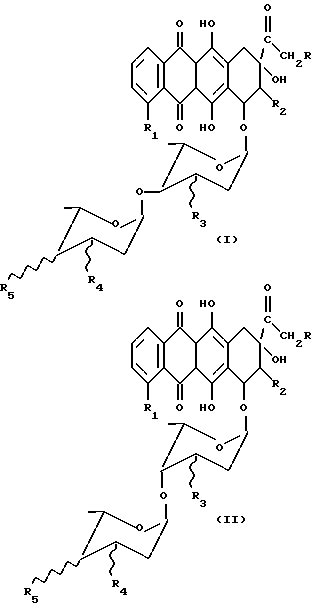
в которых R = H, OH; R1 = H или OCH3, R2 = H; R3 = OH; R4 = H, NH2; R5 = OH, NH2;
и знак связи  указывает, что заместители R3, R4, R5 могут находиться в аксиальном или экваториальном положении,
указывает, что заместители R3, R4, R5 могут находиться в аксиальном или экваториальном положении,
или их фармацевтически приемлемых солей, отличающийся тем, что состоит из следующих стадий:
i) конденсации соединения формулы III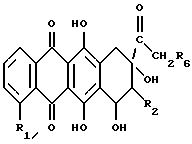
в которой R1 и R2 имеют те же значения, что указаны выше и R6 представляет собой H, или OR7 группу, где R7 является защитной группой для спиртовой функции, выбранной из ацетиловых, диметилтретбутилсилиловых или n-метоксифенилдифениловых групп, с соединением формулы IV или V
в которых R8 - защищенная OH-группа, R9 - H или защищенная NH2 группа, R10 - защищенная OH-группа или защищенная NH2 - группа,
и X - n-нитробензоилокси-группа, с тем, чтобы получить соединения формулы VI или VII
в которых R1, R2, R6, R8, R9, R10 и знак  имеют те же значения, что определены выше,
имеют те же значения, что определены выше,
при этом соединение формулы III растворяют в инертном органическом растворителе, конденсацию проводят в присутствии молекулярных сит в качестве обезвоживающих веществ и триметилсилилтрифлата в качестве конденсирующего реагента,
ii) одной или нескольких реакций удаления защитных групп для функций OH и/или NH2 из соединений формулы VI или VIII, чтобы получить соединения формулы (I) и (II) в которых R, R1, R2, R3, R4, R5 и знак  имеют значения, определенные выше,
имеют значения, определенные выше,
iii) перевода, при необходимости, соединений формулы I и II в их фармацевтически приемлемую соль.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| US 4169142 A, 25.09.79 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Способ получения антрациклингликозидов | 1983 |
|
SU1378784A3 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Пожарный двухцилиндровый насос | 0 |
|
SU90A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| US 5132290 A, 21.07.92 | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| US 4302249 A, 24.10.81 | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| G.B | |||
| Chaires et al | |||
| Dissection of the free energy of Anthracycline antibiotic binding to DNA: electrostatic contributions | |||
| Journal Amer | |||
| Chem | |||
| Soc., 1993, V.115, pp.5360 - 5364 | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| K.Suzuki et al | |||
| Application of Substituent - Controlled Oxidative Coupling of Glycals in a Synthesis an Structural Corroboration of Ciclamycin O: New Possibilities of the Construction of Hybrid Anthracyclines Jornal Amer | |||
| Chem | |||
| Soc., 1990, V.112, pp.8895 - 8902. | |||

