Изобретение относится к новому способу получения противоопухолевого лекарственного средства - N-[2-(диметиламино) этил] акридин-4-карбоксамида и его производных.
Производное акридина - N-[2-(диметиламино)этил] акридин-4-карбоксамид, известное как DACA, представляет собой новый ДНК-интеркаляционный агент с ингибиторной активностью по отношению к ферментам топоизомераза I и топоизомераза II (Schneider et al, Eur. J. Cancer Clin. Oncol, 1988, 24 1783 и Finlay, et al. Eur J. Cancer 1996, 32A 708). Оно обладает широким спектром активности против твердых опухолей у животных и на него практически не оказывает влияния устойчивость к действию различных лекарств, опосредованная Р-гликопротеином (Atwell et al, J. Med Chem, 1987, 30, 664, Baguley et al, Cancer Chemother. Pharmacol 1995, 36, 244 and Finlay et al Cancer Chemother. Pharmacol. 1993, 31, 401). О некоторых аналогах DACA уже сообщалось и многие из них показали значительную противоопухолевую активность в отношении твердых опухолей мышей (Atwell et al, там же). Известный способ получения DACA, описанный Atwell et al, показан на схеме I.
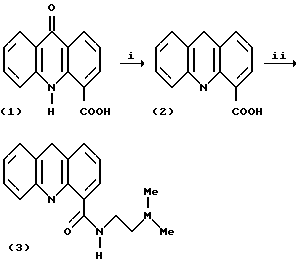
Стадия (i) включает восстановление акридона (1) в результате обработки алюминий-ртутной амальгамой в присутствии КОН в водном растворе этанола при нагревании с обратным холодильником, с последующим повторным окислением полученного акридана с помощью FeCl3 до получения промежуточной акридин-карбоновой кислоты (2). Стадия (ii) включает обработку кислоты (2) 1,1-карбонилдиимидазолом (CDI) и диметилформамидом, а затем N, N-диметилэтилендиамином.
Этот способ имеет ряд недостатков. Один из них заключается в том, что условия восстановления на стадии (i) являются весьма жесткими. Это ограничивает сферу применения способа и делает его непригодным для производства некоторых аналогов DACA, которые имеют в акридиновом ядре заместители, способные окисляться. Например, при использовании этого способа для получения хлорзамещенных производных DACA наблюдалось отщепление хлора. Другой недостаток известного способа состоит в том, что промежуточные акридин-карбоновые кислоты (2) обладают неприятными свойствами, вызывающими слезоточивость и чихание, что ограничивает их использование.
В настоящее время установлено, что DACA и его производные могут быть получены способом, предусматривающим циклизацию альдегидного предшественника, который включает этерифицированную, а не свободную карбоксильную функциональную группу, и затем воздействие на эту этерифицированную группу в циклическом продукте путем непосредственной ее обработки первичным алкиламином. При желании этерифицированная группа в циклическом продукте может быть сначала гидролизована до образования свободной карбоксильной группы, которую затем обрабатывают первичным алкиламином в присутствии подходящего связывающего агента. Альдегидный предшественник легко получают окислением соответствующего спирта, который в свою очередь получают мягким восстановлением соответствующей карбоновой кислоты через промежуточный имидазолид.
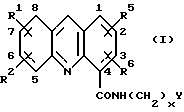
Таким образом, настоящее изобретение обеспечивает способ получения акридин карбоксамида формулы (I):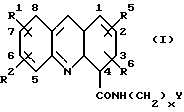
где каждый из R1, R2, R5 и R6, которые могут быть одинаковыми или различными, представляет собой Н, C1-C6 алкил, C1-C6 алкокси, фенилокси, фенил(C1-C3) алкилокси, галоген, фенил, CF3, NO2, NH2, N(R)2, NHCOR, NHCOOR, NHR4, ОН, SH, SR или SR2, где R4 представляет собой Н, COR, SO2R, COPh, SO2Ph или C1-С6 алкил, незамещенный или замещенный ОН или аминогруппой, а R представляет собой C1-C6 алкил; или R1 и R2, или R5 и R5 вместе образуют метилендиоксигруппу; х представляет целое число от 1 до 6, а Y представляет N(R)2, как указано выше; или его фармацевтически приемлемой соли, который включает:
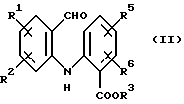
а) циклизацию соединения формулы (II)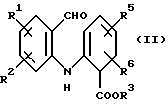
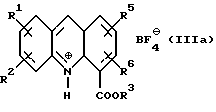
где R1, R2, R5 и R6 имеют значения, определенные выше, а R3 представляет собой C1-C6 алкил, арил или арил-С1-С3-алкил, обработкой трехфтористым бором либо подходящим комплексом на его основе в подходящем растворителе с получением тетрафторборатной соли формулы (IIIа):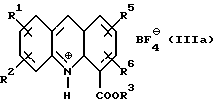
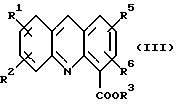
где R1, R2, R3, R5 и R6 определены как указано выше, с последующей обработкой соли неорганическим основанием в EtOAc или CH2Cl2 c образованием соединения формулы (III):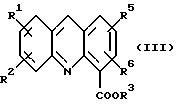
где R1, R2, R3, R5 и R6 определены как указано выше; и (b) обработку либо
(i) соединения формулы (III) первичным алкиламином формулы (IV)
NH2(СН2)xY (IV),
где значения х и Y определены выше, либо
(ii) карбоновой кислоты, получаемой гидролизом соединения формулы (III), в щелочных условиях с помощью первичного алкиламина формулы (IV), в присутствии подходящего связывающего агента с получением соединения формулы (I), определенного выше, и
(с) при желании, превращение одного соединения формулы (I) в другое соединение формулы (I) и/или превращение соединения формулы (I) в его фармацевтически приемлемую соль.
В предпочтительном варианте данного способа R1, R2, R5 и R6 в формуле (II) представляют собой Н, а в формуле (IV) х= 2, а Y= NMe2. Тогда полученное соединение формулы (I) представляет собой N-[(2-диметиламино)этил] акридин-4-карбоксамид (DACA).
Стадию (а) можно осуществлять путем обработки соединения формулы (II) либо трехфтористым бором, либо подходящим комплексом на его основе в подходящем растворителе. Подходящие комплексы включают комплекс с уксусной кислотой. В одном варианте необходимо использовать количество, немного превышающее 1 1/3 моль BF3 (стехиометрическое количество), например, 2 мольных эквивалента. BF3 чаще всего используется в форме его эфирата BF3O(Et)2. Подходящими растворителями для использования вместе с BF3О(Et)2 являются EtOAc или СН2Сl2.
Соединение формулы (III) затем получают в любом случае в виде его тетрафторборатной соли формулы (IIIa):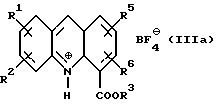
где R1, R2, R3, R5 и R6 имеют вышеуказанные значения.
При использовании BF3 образование тетрафторборатной соли (IIIa) может быть представлено следующим образом: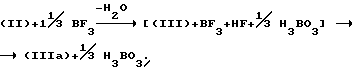
Тетрафторборатная соль формулы (IIIa) осаждается из реакционной смеси и легко может быть удалена фильтрацией. Добавление неорганического основания, например, карбоната натрия и растворителя, такого как этилацетат или дихлорметан, к отфильтрованному твердому осадку приводит к образованию соединения формулы (III). Оно может быть затем обработано амином формулы (IV). Преимущество этой процедуры состоит в том, что тетрафторборатная соль образуется с почти количественным выходом и может быть легко потом подвергнута дальнейшим реакционным взаимодействиям без необходимости отделения избытка реагентов или побочных продуктов. Это облегчает управление процессом при его промышленной реализации, особенно потому, что может быть увеличено количество пропускаемого материала.
Возможно, в некоторых случаях желательно гидролизовать соединение формулы (III) до соответствующей кислоты до обработки амином формулы (IV) на стадии (b). Это может быть, например, в случае, если соединение формулы (III) само неустойчиво к окислению. Гидролиз проводят в мягких щелочных условиях, например, обработкой гидроксидом щелочного металла (например, NaOH или КОН) в растворителе, например, этаноле. Любой связывающий агент может использоваться в реакции кислоты и амина формулы (IV) на стадии (b), например, 1,1'-карбонилдиимидазол.
Соединение формулы (II) получают путем окисления соответствующего спирта формулы (V)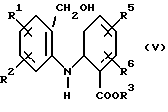
где R1, R2, R3, R5 и R6 имеют значения, определенные выше.
Окисление проводят в любых подходящих окислительных условиях. Оксид марганца (IV) (MnO2) в твердой форме в полярном растворителе, таком как этилацетат или ацетон, является предпочтительным окисляющим агентом. MnO2 можно, например, добавить в виде суспензии в ацетоне к спирту формулы (V) и дать идти реакции при комнатной температуре. Реакция протекает затем, как правило, в течение нескольких дней, например, 2 или 3 дня, пока не завершится. Или же смесь спирта формулы (V) и MnO2 в этилацетате можно вместе перегонять с обратным холодильником, например, в течение ночи.
Спирт формулы (V) получают путем
а) обработки соединения формулы (VI):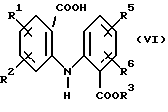
в котором R1, R2, R3, R5 и R6 имеют значения, указанные выше, 1,1'-карбонилдиимидазолом в полярном растворителе с получением соединения формулы (VII):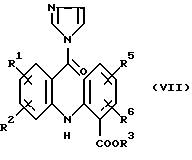
в котором R1, R2, R3, R5 и R6 имеют значения, указанные выше, и (b) восстановлении имидазолида формулы (VII), как указано выше.
На вышеуказанной стадии (а) полярным органическим растворителем может быть, например, ТГФ (тетрагидрофуран). Реагирующие вещества обычно перемешивают при комнатной температуре до тех пор, пока реакция не завершится. На стадии (b) восстановление, как правило, проводят путем обработки соединения формулы (VII) избытком восстанавливающего агента на основе щелочного металла, например, боргидрида натрия. В этом случае раствор, который получают со стадии (а), можно добавить к перемешиваемой суспензии боргидрида натрия в воде.
Использование промежуточного имидазолида формулы (VII) позволяет относительно легко в мягких условиях проводить восстановление карбоновых кислот формулы (VI) в спирты формулы (V).
Соединения формулы (VI) являются известными соединениями или могут быть получены известными способами, например, нагреванием антраниловой кислоты формулы (VIII):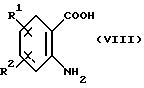
в которой R1 и R2 имеют вышеуказанные значения, вместе с эфиром 2-иодбензойной кислоты формулы (IX):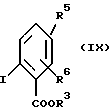
в которой R3, R5 и R6 имеют вышеуказанные значения, в присутствии медного катализатора и основания в среде полярного растворителя.
В качестве медного катализатора подходящими являются галоид меди (I) и порошкообразная медь. Полярным растворителем может быть, например, этиленгликоль или бутан-2,3-диол. Может использоваться любое подходящее основание, например N-этил-морфолин.
В соединениях формулы (I), получаемых способом настоящего изобретения, в трициклическом хромофоре заместители R1 и R2 могут занимать в кольце любое из положений с 5-го по 8-е, а заместители R5 и R6 могут занимать в кольце любое из положений с 1-го по 4-е. Таким образом R1 и R2 могут каждый быть связан с каким-либо одним положением в кольце в исходных и промежуточных соединениях формул (III) и (V)-(VIII), которые соответствуют позициям с 5-й по 8-ю конечных соединений формулы (I). Аналогично, R5 и R6 могут быть связаны с каким-либо одним положением в кольце в исходных и промежуточных соединениях формул (III) и (V)-(VIII), которые соответствуют позициям с 1-й по 4-ю конечных соединений формулы (I).
В некой предпочтительной серии соединений R5 и R6 являются оба водородами. В этой серии соединения имеют общую формулу (Iа):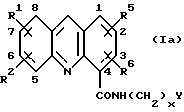
где R1, R2, x и Y имеют значения, указанные выше для формулы (I). Формула (Iа) является таким образом предпочтительным вариантом формулы (I). Как правило, один из R1 и R2 является водородом, а другой - водородом или другим заместителем из указанных выше для формулы (I), связанных в одном любом из положений 5-8 кольца.
В предпочтительной серии соединений формулы (I) каждый из R1 и R2, которые могут быть одинаковыми или различными, является Н, C1-С6 алкилом, C1-С6 алкокси, галогеном, фенилом, CF3, NO2, NH2, N(R)2 в соответствии с определенными ранее значениями, или ОН, x - целое число от 1 до 3, а Y представляет собой N(R)2 в соответствии с определенными ранее значениями.
Чаще всего R1 есть Н, а R2 есть Н или заместитель, отличный от водорода, связанный в положении 5, 6 или 7 в ядре акридина в формуле (I). Например, R1 представляет собой Н, а R2 находится в положении 5 и представляет собой C1-С6 алкил, CF3, фенил, галоген или группу N(R)2; или R1 представляет собой водород, а R2 находится в положении 6 и представляет собой галоген, CF3 или N(R)2 в соответствии с вышеприведенными значениями; или R1 - водород, a R2 - находится в положении 7 и является С1-С6лкилом, фенилом, ОН, галогеном, CF3 или N(R)2.
Или же - R1 не является водородом. Например, когда R2 находится в позиции 5, как определено выше, R1 находится в положении 6, 7 или 8, предпочтительно, 6 или 7 и является C1-C6 алкилом, C1-C6 алкокси, галогеном, фенилом, CF3, NO2, NH2, N(R)2 или ОН. Когда R2 находится в положении 6, R1 в положении 5, 7 или 8, предпочтительно 5 или 7, и является C1-С6 алкилом, C1-С6 алкокси, галогеном, фенилом, CF3, NO2, NH2, N(R)2 или ОН. Когда R2 находится в положении 7, R1 - в положении 5, 6 или 8, предпочтительно 5 или 6, и является C1-C6 алкилом, C1-C6 алкокси, галогеном, фенилом, CF3, NO2, NH2, N(R)2 или ОН.
C1-C6 алкильная группа может быть прямой или разветвленной и представляет собой, например, C1-C6 алкил, такой как метил, этил, н-пропил, изопропил, н-бутил, втор-бутил или трет-бутил. C1-С6 алкоксигруппа также может быть прямой или разветвленной и представляет собой, например, C1-C4 алкокси, такой как метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси или трет-бутокси. Галоген представляет собой, например, фтор, хлор, бром или иод. Арильная группа представляет собой, например, C6-C12 арильную группу, такую как фенильную или нафтильную.
Арильный остаток в арил-C1-С3-алкиле, аралкилокси или арилоксигруппе может быть С6-С12 арильной группой, например, фенилом или нафтилом. Примеры арил-C1-C3 алкильной группы, таким образом, включают фенил-C1-C3-алкильные группы, такие как бензил и фенилэтил.
Соединения формулы (I) можно переводить в фармацевтически приемлемые соли присоединения кислот общеизвестными способами. Например, соли присоединения кислот могут быть получены при взаимодействии свободного основания с соответствующим количеством выбранной кислоты обычным путем. Подходящими являются соли как органических, так и неорганических кислот. Примерами подходящих кислот являются соляная, серная, фосфорная, уксусная, лимонная, щавелевая, малоновая, салициловая, малеиновая, фумаровая, янтарная, аскорбиновая, метан-сульфокислота и им подобные. В зависимости от структуры и условий соединения могут образовывать мультикатионные формы.
Необязательный перевод соединения формулы (I) в другое соединение формулы (I) может осуществляться общепринятыми способами. Например, фторная группа в соединении формулы (I) может быть замещена амино или тиольной группой с получением амина или тиоэфира соответственно; тиольная группа в соединении формулы (I) может быть алкилирована с получением тиоэфира; аминогруппа может быть ацилирована с получением N-ацетильной группы; а нитрогруппа может быть восстановлена с получением амина. Это все представляет собой обычные превращения в органической химии.
Амины общей формулы (IV) являются известными соединениями, коммерчески доступными, или получаются способами, описанными в литературе. Конкретными примерами таких соединений являются NH2(CH2)2NMe2 [х равен 2, а Y представляет N(CH3)2] .
На основе соединений формулы (I) и их солей, полученных заявленным способом, могут быть приготовлены композиции для фармацевтического применения или для использования в ветеринарии. Способ в том виде, как он описан выше, может поэтому дополнительно включать получение композиций на основе соединения формулы (I) или его фармацевтически приемлемой соли с фармацевтически приемлемым или пригодным для применения в ветеринарии носителем или разбавителем. Такую композицию обычно готовят известными способами с тем, чтобы быть пригодной для лечения человека или животных.
Композиция может быть приготовлена в различных лекарственных формах, например для орального введения в форме таблеток, капсул, покрытых оболочкой из сахара или в виде пленки, жидких растворов или суспензий, или для парэнтерального введения, например внутримышечного, внутривенного или подкожного. Соединения формулы (I) могут быть таким образом приготовлены для инъекций или инфузий.
Например, твердые формы для орального применения могут содержать вместе с активным компонентом разбавители, такие как лактоза, декстроза, сахароза, целлюлоза, кукурузный или картофельный крахмал; лубриканты, такие как двуокись кремния, тальк, стеариновая кислота, стеарат магния или кальция и/или полиэтиленгликоли; связывающие агенты, такие как крахмалы, аравийская камедь, желатин, метилцеллюлоза, карбоксиметилцеллюлоза или поливинилпирролидон; дезинтегрирующие агенты, такие как крахмал, альгиновая кислота, альгинаты или крахмал-гликолят натрия; выделяющие газ (шипучие) жидкости, красители; подсластители; смачивающие реагенты, такие как лецитин, полисорбаты, лаурилсульфаты. Такие препараты могут готовиться обычным путем, например, смешивания, гранулирования, таблетирования и путем нанесения покрытия из сахара или пленочного покрытия.
Жидкие дисперсии для орального введения могут представлять собой сиропы, эмульсии и суспензии. Сиропы могут содержать в качестве носителя, например, сахарозу или сахарозу с глицерином и/или маннит и/или сорбит. В частности, сироп может содержать в качестве носителя, например, сахарозу или сахарозу с глицерином и/или маннит и/или сорбит. В частности, сироп для больных диабетом может содержать только, например, сорбит, который не метаболизирует до глюкозы или который метаболизирует только до ее очень малых количеств. Суспензии и эмульсии могут содержать в качестве носителя, например, природную смолу, агар, альгинат натрия, пектин, метилцеллюлозу, карбоксиметилцеллюлозу или поливиниловый спирт.
Суспензии или растворы для внутримышечных инъекций могут содержать, помимо активного соединения фармацевтически пригодный носитель, такой как стерильная вода, оливковое масло, этилолеат, гликоли, например пропиленгликоль, и при желании - подходящее количество гидрохлорида лидокаина. Чаще всего соединения формулы (I) готовятся в виде водных растворов солянокислых или иных фармацевтически приемлемых солей. Растворы для внутривенных инъекций или инфузий могут содержать носитель, например стерильную воду, которая как правило представляет собой воду для инъекций.
Заявленное изобретение дополнительно иллюстрируется следующими примерами.
Пример 1: Получение метил-2-[N-(2-карбоксифенил)амино] бензоата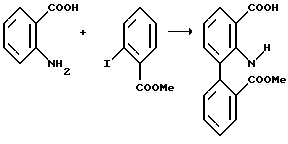
Смесь антраниловой кислоты (16,48 мг, 120 ммоль), метил 2-иодбензоата (39,3 г, 159 ммоль), N-этилморфолина (38,1 мл; 34,5 г, 300 ммоль), этиленгликоля (120 мл), хлористой меди (3 г) и порошкообразной меди (99%, 0,6 г) перемешивали в масляной бане при 140oС в течение 6 часов (внутренняя температура приблизительно 130oС). Реакционную смесь охлаждали и медленно вливали в перемешиваемую смесь этилацетата (300 мл) и 1 М соляной кислоты (200 мл), после чего смесь фильтровали для удаления нерастворимого материала на границе раздела. Полученный на фильтре слой промывали этилацетатом (200 мл).
После отделения органической фазы из фильтрата водный слой экстрагировали последовательно порциями этилацетата (2•100 мл), которые ранее получили после промывки слоя на фильтре. Объединенные органические экстракты перемешивали с активированным углем (3 г) и фильтровали. Фильтрат экстрагировали приблизительно 1,5% водным раствором аммиака (1•400 мл и 2•150 мл). Объединенные аммиачные экстракты медленно добавили к перемешиваемой 1 М соляной кислоте, взятой в избытке, продукт собирали фильтрацией, промывали его горячей водой (3•100 мл) и отбирали для сушки (вес во влажном состоянии около 60 г). После высушивания в вакууме при 55oС получали вышеуказанное соединение (27,8 г, 85,4%). (Чистота по ВЭЖХ ~ 90% а/а; преобладающее загрязнение было дикарбоновой кислотой). Тпл.= 196-198oС.
1Н ЯМР (CDCl3)  3,93 (3, s COOMe), 6,92 (2Н, m J= 7,5, H-4, H-4'), 7,26 (s, растворитель CHCl3), 7,38 (2Н, m, H-5, Н-5'), 7,51 (2Н, br. t, J= 8.9, H-6, Н-6'), 7.98 (1H, dd, J= 7.9 и 1.1, Н-3'), 8.09 (1H, J= 7.9, H-3), 10.82 (1H, br. s, NH).
3,93 (3, s COOMe), 6,92 (2Н, m J= 7,5, H-4, H-4'), 7,26 (s, растворитель CHCl3), 7,38 (2Н, m, H-5, Н-5'), 7,51 (2Н, br. t, J= 8.9, H-6, Н-6'), 7.98 (1H, dd, J= 7.9 и 1.1, Н-3'), 8.09 (1H, J= 7.9, H-3), 10.82 (1H, br. s, NH).
Пример 2: Получение метил-2-[N-(2-гидроксиметил) фенил(амино)] бензоата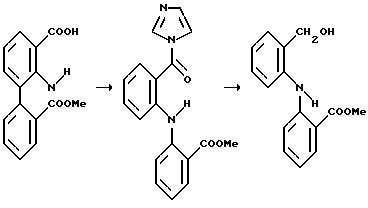
1,1'-Карбонилдиимидазол (19,5 г, 120 ммоль) добавили к продукту, полученному в примере 1 (27,1 г, 100 ммоль) в ТГФ (сортиров. ВЭЖХ, 270 мл) и смесь перемешивали в течение ночи при комнатной температуре до получения светло-коричневого раствора промежуточного имидазолида. ТСХ (SiO2; 10% MeOH/CH2Cl2 с визуализацией в УФ при 254 нм) показала завершение реакции.
Этот раствор через 30 минут добавили к перемешиваемой суспензии боргидрида натрия (12,5 г, 330 ммоль) в воде (375 мл). Осадившаяся смола, имевшая сперва желтый цвет, к моменту окончания превратилась в серовато-желтую суспензию и температура поднялась до 37oС. ТСХ (SiO2; EtOAc с просмотром в УФ при 254 нм) показала завершение реакции. После дополнительного перемешивания суспензии в течение еще 30 минут избыток боргидрида натрия разрушили добавлением конц. НСl (35 мл), сохраняя температуру ниже 30oС с помощью ледяной бани. рН смеси около 7. Добавили EtOAc (300 мл) и насыщенный раствор кислого углекислого натрия (200 мл) и смесь недолго перемешивали, после чего была отделена органическая фаза (объем отброшенной водной фазы - 690 мл). Бледно-желтый органический слой промывали рассолом (100 мл), отделяли и концентрировали в вакууме. Вторичное испарение из EtOAc (3х100 мл) дало вышеуказанное соединение (27,6 г > 100%) в виде желтовато-коричневого масла, ВЭЖХ чистота около 90%. Образец медленно кристаллизовался при хранении. Тпл.= 69-71oС.
1H ЯМР (CDCl3) δ 1,93 (br, s, 1H, ОН), 3,91 (s, 3Н, СООСН3), 4,72 (s, 2H, СН2ОН), 6,74 (ddd, J= 8,0, 7,0, 1,1 Гц, 1Н, Н-5), 7,08-7,44 (m, 6H, Н-3,3', 4,4', 5', 6'), 7,97 (dd, J= 8,0, 1,6 Гц, 1H, H-6), 9,59 (br. s, 1H, NH).
Пример 3: Получение метил-2-[N-(2-формил)фениламино] бензоата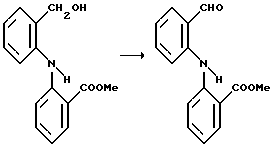
Продукт примера 2 (27,6 г, ~ 100 ммоль) в EtOAc (300 мл) перемешивали с оксидом марганца (IV) (<5 мкм, активированный ~ 85% MnO2), (55 г, 2 веса) и кипятили с обратным холодильником в течение ночи (17 часов). ТСХ (SiO2; EtOAc), просмотр в УФ при 254 нм (при дневном свете альдегид виден в виде желтой точки), показала завершение реакции. Активированный древесный уголь (2,7 г) и кизельгур (2,7 г) добавили к теплой смеси, которую перемешивали в течение 30 минут, и фильтровали через слой кизельгура. Слой затем промывали EtOAc (2•100 мл).
Ярко-желтый фильтрат осторожно концентрировали в вакууме до половины объема, извлекали из испарителя и промывали водой (50 мл). Органическую фазу отделяли, концентрировали в вакууме до малого объема (вес 47 г), пока не началась кристаллизация продукта и осадок не превратился в твердое вещество интенсивного желтого цвета. Для растворения кристаллической массы при перемешивании был добавлен гексан (200 мл), и спустя 1 час продукт отфильтровали, промыли гексаном и высушили в вакууме при 40oС с получением указанного соединения (19,6 г, 76,7%). ВЭЖХ 94,5% а/а т. пл. 110-112oС.
1Н ЯМР (CDCl3) δ 3,95 (s, 3Н, СООСН3), 6,95-7,03 (m, 2Н, Н-4', 5), 7,41-7,45 (m, 2H, Н-5' 6), 7,50 (br d, J= 8,5 Гц, 1Н, H-3 или Н-6'), 7,61 (br. d, J= 8.2 Гц, 1Н, H-6' или H-3), 7.65 (dd, J= 7.7, 1,7 Гц, 1Н, H-3'), 8.01 (dd, J= 7.9, 1,7 Гц, Н-6), 10.00 (s, 1H, СНО), 11,26 (br. s, 1H, NH).
Пример 4: Получение метил акридин-4-карбоксилата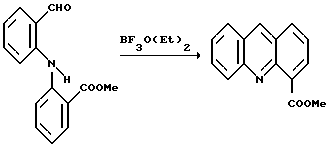
Альдегид, полученный в примере 3 (12.75 г, 50 ммоль), в дегазированном этилацетате (250 мл) перемешивали под азотом и в течение 15 минут добавляли комплекс трехфтористого бора с уксусной кислотой (25 мл, 33,8 г, 180 ммоль). Прежде чем добавление закончилось, соль четырехфтористого бора начала кристаллизоваться в оранжевые кристаллы. Смесь оставили перемешиваться под азотом при комнатной температуре на ночь. Плотный оранжевый осадок удаляли фильтрацией, промывали EtOAc (20 мл), гексаном (50 мл) и отделяли в сухом виде на фильтре (20 г, ~ 92% чистота методом ВЭЖХ).
Этот твердый осадок добавляли к смеси EtOAc (250 мл) и насыщенного раствора карбоната натрия (150 мл). Бледно-желтый органический слой отделяли и промывали насыщенным рассолом (30 мл). ТСХ (SiO2; 10% МеОН в CH2Cl2, просмотр в УФ при 254 нм) показал по существу одну точку. Органический раствор выпаривали в вакууме до получения указанного соединения (11,5 г, 97%) в виде бледно-желтого масла, которое легко кристаллизовалось. ВЭЖХ показала приблизительно 90% чистоту. Продукт использовали без дальнейшей очистки для получения DACA.
1H ЯМР (CDCl3) δ 4,12 (s, 3Н, СООСН3), 7,53-7,58 (m, 2Н, Н-2 и Н-6 или Н-7), 7,79 (ddd, J= 8,8, 6,6, 1,4 Гц, 1Н, Н-7 или Н-6), 8,00 (dd, J= 8,0, 1,0 Гц, 1H, H-1), 8,12-8,14 (m, 2H, Н-5,8), 8,30 (dd, J= 8,7, 0,9 Гц, 1H, Н-3), 8.80 (s, 1H, Н-9).
Пример 5: Получение N-[2-(диметиламино)этил] акридин-4-карбоксамида (DACA)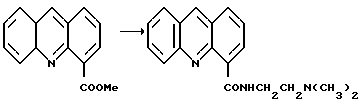
Метилакридин-4-карбоксилат, полученный, как указано в примере 4 (11,2 г, 47 ммоль), разбавили N, N-диметилэтилендиамином (20 мл, 16.2 г, 184 ммоль) и раствор испаряли в роторном испарителе для удаления следов EtOAc (потеря в весе около 1,5 г). Смесь затем нагревали под азотом в масляной бане при 120oС в течение 7 часов и оставляли охлаждаться на ночь в бане. Смесь растворяли в толуоле и концентрировали до состояния смолы в вакууме.
Остаток растворяли в EtOAc (150 мл) и промывали 1 М водным раствором карбоната натрия (2•100 мл). Органический слой отделяли, перемешивали с активированным древесным углем (1,5 г) и кизельгуром (1,5 г) и фильтровали через слой кизельгура. Слой промывали EtOAc, и фильтрат и промывочные воды концентрировали в вакууме до получения желтовато-коричневого масла, которое быстро кристаллизуется в темно-желтый кристаллический осадок, который растирают в порошок с гексаном (100 мл) и фильтруют с получением указанного соединения в виде бледно-желтого осадка. Его промывали гексаном (50 мл) и высушивали в вакууме при 40oС (10,4 г, 75,4%) ВЭЖХ, 90-95% а/а, т. пл. 105-108oС.
1H ЯМР (DMSO) δ 2,34 (6Н, s, N(СН3)2), 2,58 (2Н, t, J= 6,1, CH2N(СН3)2), 3,61 (2Н, m, J= 11,2 и 6,0, CONHCH2), 7,67 (1H, m, J= 7,1, 6,4 и 0,7, Н-7), 7,71 (1H, dd, J= 8,3 и 7,1, H-2), 7,95 (1H, m, J= 7,7, 6,7 и 1,4, Н-6), 8,18 (1H, dd, J= 8,2 и 1,3, Н-8), 8,18 (1H, dd, J= 8,9 и 0,8, Н-5), 8,31 (1H, dd, J= 8,4 и 1,5, Н-1), 8,74 (1H, dd, J= 7.1 и 1,6, Н-3), 9,23 (1H, s, H-9), 11,73 (14, br. t, J= 4,7, CONH).
Пример 6: Получение тригидрата дигидрохлорида [2-(Диметиламино)-этил] акридин-4-карбоксамида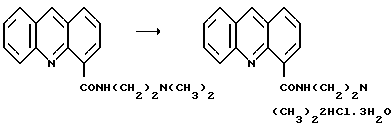
К продукту примера 5 (2,93 г, 100 ммоль), растворенному в смеси толуола (20,7 мл) и EtOH (9 мл), была добавлена по каплям концентрированная НСl (2,0 мл, около 200 ммоль). Произошло осаждение соли, после чего добавили EtOAc (8,6 мл). Смесь охлаждали до 5oС, перемешивали еще 1 час и отфильтровывали кристаллы соли желтоватого цвета, промывали их EtOAc (3•20 мл) и извлекали сухими на фильтр с получением указанного соединения 3,9 г (теоретически для 2НСl•3Н2O, 4,2 г). ВЭЖХ показала чистоту около 98% а/а.
Соль перекристаллизовывали растворением в смеси EtOAc (20 мл) и воды (2 мл) при 70oС. Полученный раствор разбавляли EtOAc (20 мл), поддерживая температуру смеси около 60-70oС. Затем смесь оставили медленно остывать до образования тригидрата дигидрохлорида в виде твердого кристаллического продукта желтоватого цвета, который после охлаждения в ледяной бане в течение 1 часа фильтровали, промывали охлажденной смесью EtOH: EtOAc: вода (10: 10: 1) (2•10 мл) и извлекали сухим на фильтр. Затем продукт оставляли в вытяжном шкафу, доводя до постоянного веса с получением чистой соли (3,6 ~ 85,7%). ВЭЖХ. 99,2% а/а.
1H ЯМР (DMSO) δ 2,90 (6Н, s, N(СН3)2), 3,46 (2Н m, CH2N(СН3)2), 3,98 (2Н, m, CONHCH2), 7,75 и 7,80 (2Н, t и br. t H-7 (7,75) и Н-2(7,80)), 8,02 (1Н, m, Н-6), 8,28 (1Н, d, H-8), 8,46 (1Н, d, Н-1), 8,51 (1Н, d, Н-5), 8.77 (1Н, d, Н-3), 9,43 (1Н, sН-9), 10,65 (1Н, br. s, NH+(СН3)2), 11,45 (1Н, br. t. CONH).
Пример 7: Получение [2-(диметиламино)этил] -акридин-4-карбоксамида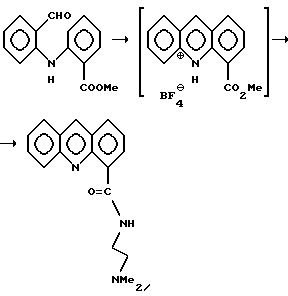
Перемешиваемый раствор альдегида, полученного по примеру 3 (3,0 г, 11,76 ммоль) в СН2Сl2, насыщали азотом и по каплям добавляли в атмосфере азота эфират трехфтористого бора (3,33 г, 23,5 ммоль, 2 эквивалента). Раствор становился оранжевым (выпадал осадок, который растворялся спустя 2-3 мин), затем раствор становился темно-красным, после чего выпадал осадок желтого цвета. Его оставляли перемешиваться в течение 4 часов до завершения реакции, ТСХ (SiO2 СН3ОН: СН2Сl2/1: 40). Добавляли 1М раствор Na2СО3 (15 мл) (рН 7) и перемешивали раствор 5 минут. Нижний органический слой отделяли, затем промывали 1 М Na2СО3 (15 мл). Объединенный водный слой экстрагировали СН2Сl2 (10 мл), органический слой отделяли и добавили к основному органическому слою. Объединенный органический слой промывали рассолом (10 мл), уменьшали примерно до 1/2 объема, затем повторно выпаривали из CH2Cl2 (20 мл). Добавляли N, N-диметилэтилендиамин (NNDMEDA) (5,1 мл, 47,04 ммоль, 4 эквив. ) и реакционную смесь концентрировали, чтобы удалить остатки CH2Cl2.
Остаток нагревали на масляной бане (110-120oС) в течение ночи, ТСХ (SiO2; 10% СН3ОН: СН2Сl2) с получением оранжево-коричневого масла, которое разбавляли толуолом (20 мл), затем концентрировали до малого объема, чтобы удалить избыток NNDMEDA. Остаток разбавили EtOAc (25 мл), затем промыли 1 М раствором NaHCO3 (15 мл). Органический слой отделили, промыли водой (2•10 мл), затем выделили. Объединенный органический слой перемешивали с активированным углем (300 мг), кизельгуром (300 мг) в течение 30 мин, фильтровали через слой сухого кизельгура, промывали EtOAc и фильтрат концентрировали до смолообразного осадка (3,2 г), который быстро кристаллизовался. При растирании в порошок с EtOAc (2 мл) и гексаном (20 мл) получали твердый продукт желтовато-коричневого цвета. Твердый осадок отфильтровывали, промывали гексаном, затем высушивали в вакууме (40oС) с получением указанного соединения (2,65 г, 77%) в виде темно-желтого осадка. Данные, полученные с помощью 1H ЯМР, такие же, как для продукта по примеру 5.
Пример 8: Получение соединений формулы (I) из соединений формулы (VI)
СХЕМА 2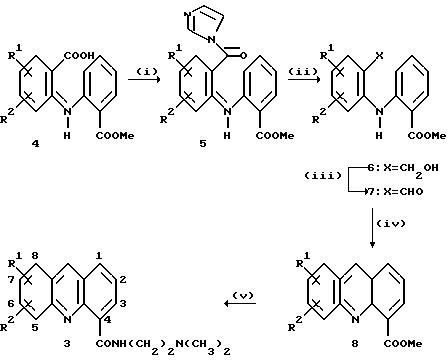
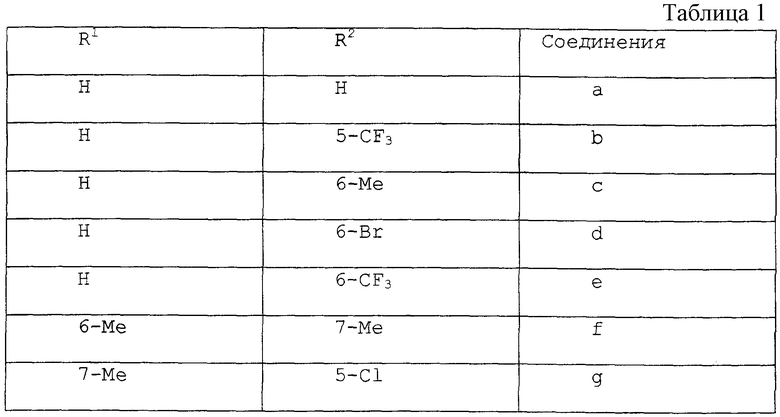
Серию реакций, отраженных на схеме 2, выполняли для получения DACA, соединения 3а и ряда аналогов 3b-3g. Для каждого из этих соединений заместители R1 и R2 по всей схеме 2 имеют следующие значения (см. табл. 1).
Раствор метил-2-[N-(2-карбоксифенил)амино] -бензоата 4а (10 г, 36,9 ммоль) в сухом ТГФ (200 мл) обрабатывали 1,1'-карбонилдиимидазолом (8,97 г, 55,4 ммоль). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 15 часов, затем раствор ТГФ медленно добавляли к суспензии NaBH4 (7,00 г) в Н2O (200 мл) без выделения промежуточного имидазолида 5а. Реакция шла фактически мгновенно и в конце добавления реакционную смесь гасили концентрированной НСl, разделяли между CH2Cl2 (200 мл) и NaHCO3 (200 мл) и органический слой высушивали с Na2SO4. Удаление растворителя и пропускание осадка через воронку с обожженным и отсортированным силикагелем в смеси петролейного эфира и EtOAc (4: 1) дало метил 2-[N-(2-гидроксиметил) фениламино] бензоат 6а (7,85 г, 83%), Т. пл. (СН2Сl2/ петролейный эфир) 69-71oС.
1H ЯМР (СDСl3) δ 1,93 (br. s, 1Н, ОН), 3,91 (s, 3H, СООН СН3), 4,72 (s, 2Н, СН2ОН), 6,74 (ddd, J= 8.0, 7,0, 1,1 Гц, 1Н, Н-5), 7,08-7,44 (m, 6Н, Н-3,3', 4,4'5', 6'); 7,97 (dd, J= 8,1, 1,6 Гц, 1H, H-6), 9,59 (br, s, 1H, NH).
Перемешиваемый раствор 6а (7,74 г, 30 ммоль) в Me2CO (200 мл) обрабатывали суспензией MnO2 (10 г) в течение 3 дней при комнатной температуре. MnO2 отфильтровывали через Целит, а Me2CO удаляли при пониженном давлении с получением метил 2-[N-(2-формил) фениламино] бензоата 7а (7,70 г, 100%). Образец, кристаллизованный из смеси EtOAc/петролейный эфир, имел т. пл. 110-112oС.
1H ЯМР (CDCl3) δ 3,95 (s, 3Н, СООСН3), 6,95-7,03 (m, 2H, Н-4', 5), 7,41-7,45 (m, 2H, H-5', 6), 7,50 (brd, J= 8,5 Гц, 1H, Н-3 или Н-6'), 7,61 (br d, J= 8.2 Гц, 1H, Н-6' или Н-3), 7.65 (dd, J-7,7, 1,7 Гц, 1H, Н-3'), 8,01 (dd J= 7.9, 1,7 Гц, 1H, Н-6), 10,00 (s, 1H, СНО), 11,26 (brs, 1H, NH).
Альдегид 7а (210 мг, 0.82 ммоль) помещали в колбу, которую омывали струей N2, затем добавляли трифторуксусную кислоту и получившийся раствор перемешивали в течение 24 часов при комнатной температуре. Растворитель удаляли при пониженном давлении с получением неочищенного метил акридин-4-карбоксилата 8а (183 мг, 94%). Его разбавили CH2Cl2 (100 мл) и нейтрализовали Еt3N. Растворители удаляли при пониженном давлении и остаток пропускали через небольшую колонку с обожженным силикагелем в смеси EtOAc/петролейный эфир (1: 3) с получением метилакридин-4-карбоксилата (8а) в виде масла оранжевого цвета (1,83 г, 98%).
1H ЯМР (CDCl3) δ 4.12 (s, 3H, СООСН3), 7,53-7,58 (m, 2H, Н-2 и Н-6 или Н-7), 7.79 (ddd, J= 8,8, 6,6, 1,4 Гц, 1Н, Н-7 или Н-6), 8,00 (dd, J= 8,0, 1,0 Гц, 1H, H-1), 8,12-8,14 (m, 2H, Н-5,8), 8,30 (dd, J= 8,7, 0,9 Гц, 1H, Н-3), 8,80 (s, 1H, Н-9).
Раствор 8а (1/83 г, 7,72 ммоль) и N, N-диметилэтилендиамина (3,40 г, 38,6 ммоль) в пропан-1-оле (80 мл) омывали струей азота и смесь нагревали с обратным холодильником 3 дня под азотом. Растворитель удаляли при пониженном давлении, а остаток разделяли между СН2Сl2 (100 мл) и 1М Na2CO3 (100 мл). Органический слой выпаривали, а остаток хроматографировали с помощью окиси алюминия, элюируя смесью CH2Cl2/MeOH (199: 1) и получали N-[2-(диметиламино)-этил] акридин-4-карбоксамид 3а (1,38 г, 61%), т. пл. (diHCl соль) 191-195oС, идентичен с аутентичным образцом.
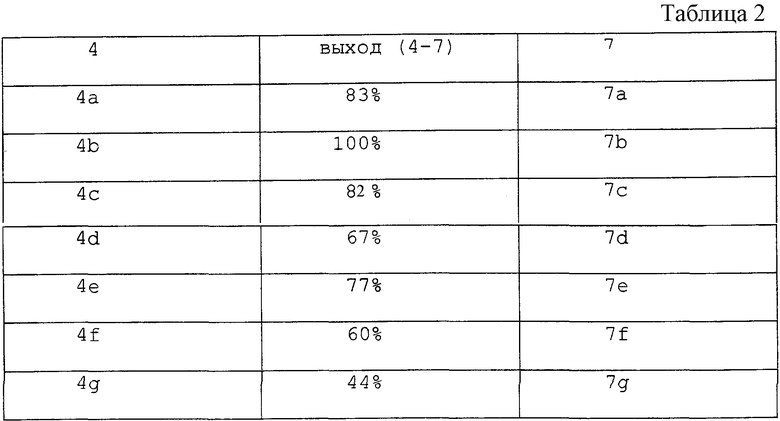
Аналогичные процедуры применяли для получения соединений 3b-3g из исходных соединений 4b-4g, соответственно. Все соединения 3b-3g имели удовлетворительные спектроскопические и аналитические показатели. Значения выходов промежуточных альдегидов 7a-7g из исходных соединений 4a-4g (стадии (i) и (ii)) были следующими (см. табл. 2)
Пример 9: Получение акридин-4-карбоновой кислоты
К метилакридин-4-карбоксилату, полученному в примере 4 (183 мг), добавили дегазированный раствор NaOH в водном EtOH (1: 1, 2М) (35 мл). Смесь перемешивали 3 часа при 50oС, когда раствор стал прозрачным, его нейтрализовали ледяной АсОН. Экстракцией EtOAc (3•50 мл) с последующей хроматографией на силикагеле с элюцией смесью EtOAc/петролейный эфир (1: 4) получили акридин-4-карбоновую кислоту (160 мг, 87%), т. пл. (Мe2СО) 196-197oС (лит. т. пл. 202-204oС).
Тем же самым образом другие соединения формулы (III) гидролизовали в соответствующие акридин-4-карбоновые кислоты.
Пример 10: Получение соединений формулы (I) из предшествующей акридин-4-карбоновой кислоты
Общий метод
Суспензию 7-этилакридин-4-карбоновой кислоты, полученной по способу, описанному в примере 9, из соединения формулы (III), в которой R1 является Н, a R2 представляет собой 7-этиловый заместитель (472 мг, 1,99 ммоль) в сухом ДМФ (10 мл) перемешивали с 1,1'-карбонилдиимидазолом (650 мг, 3,98 ммоль) при 25oС до гомогенного состояния (~ 12 часов). Раствор затем охлаждали до 0oС и обрабатывали N, N-диметилэтилендиамином (0,73 г, 9,96 ммоль) в течение 5 минут. Растворитель затем удаляли при пониженном давлении и остаток разделяли между СН2Сl2 (50 мл) и 1М водным раствором К2СО3 (30 мл). Органический слой промывали водой и выпаривали, а остаток хроматографировали на окиси алюминия. Элюция смесью CH2Cl2/MeOH (19: 1) дала N-[2-(диметиламино) этил] -7-этилакридин-4-карбоксамид (10а) в виде желтого масла (288 мг, 48%).
1H ЯМР (CDCl3) δ 1.35 (t, J= 7,6 Гц, 3Н, СН2СН3), 2,36 (s, 6Н, N(CH3)2), 2,61 (t, J= 6,1 Гц, 2Н, СН2N(CH3)2), 2.89 (g, J= 7,6 Гц, 2Н, СН2СH3), 3,63 (g, J= 5, 6 Гц, 2Н, CH2), 7,73 (dd, J= 8,2, 7,2 Гц, 1Н, Н-2), 7,90 (dd, J= 9,0, 1,9 Гц, 1Н, H-6), 7,99 (br s, 1H, H-8), 8,18 (d, J= 8,9 Гц, 1Н, H-5), 8,34 (dd, J= 8,5, 1,4 Гц, 1H, Н-1), 8,73 (dd, J= 7,1, 1,5 Гц, 1H, Н-3), 9,21 (s, 1H, Н-9), 11,81 (br t, J= 4,7 Гц, 1Н, CONH). Дихлоргидратная соль, т. пл. (EtOAc/MeOH) 173-175oС.
Вышеописанный общий метод был использован для получения следующих соединений формулы (I):
N-[2-(Диметиламино)этил] - 5-этилакридин-4- карбоксамид (соединение 10b), (70%), т. пл. (CH2Cl2/петролейный эфир) 106-108oС; Дихлоргидратная соль, т. пл. (EtOAc/MeOH) 214-217oС.
N-[2-(Диметиламино)этил] - 5-изопропилакридин-4- карбоксамид (соединение 10с) в виде желтого масла (76%), дихлоргидратная соль, т. пл. (EtOAc/MeOH) 213-215oС,
N-[2-(Диметиламино)этил] -5- фторакридин-4- карбоксамид (соединение 1d), (73%), т. пл. (гексан) 95-98,5oС.
N-[2-(Диметиламино)этил] -5- бромакридин-4- карбоксамид (соединение 10е) (52%), т. пл. 149-150oС.
N-[2-(Диметиламино)этил] -5-трифторметилакридин- 4-карбоксамид (соединение 10f) (74%). Хлоргидратная соль, т. пл. (EtOAc/MeOH) 207-211oС.
N-[2-(Диметиламино)этил] -6-фторакридин-4- карбоксамид (соединение 10g) (87%), т. пл. (дихлоргидратная соль из MeOH/EtOAc) 203-204oC (разл. ).
N-[2-(Диметиламино)этил] -6-бромакридин-4- карбоксамид (соединение 10h) (67%), т. пл. (дихлоргидратная соль из MeOH/EtOAc) 161-163oС.
N-[2-(Диметиламино)этил] -7-изопропилакридин- 4-карбоксамид (соединение 10i), в виде желтого масла (97%), дихлоргидратная соль, т. пл. (MeOH/EtOAc) 182-187oС.
N-[2-(Диметиламино)этил] -7-трет-бутилакридин- 4-карбоксамид (соединение 10j) (92%), т. пл. (CH2Cl2 /петролейный эфир) 128-129oС.
N-[2-(Диметиламино)этил] -7-фенилакридин- 4-карбоксамид (соединение 10к) (64%), т. пл. (СН2Сl2/петролейный эфир) - 115-116,5oC, хлоргидратная соль, т. пл. (MeOH/EtOAc) 83-85oС.
N-[2-(Диметиламино)этил] -7-фторакридин- 4-карбоксамид (соединение 10l) (74%), т. пл. (MeOH/EtOAc) 128,5-130oС.
N-[2-(Диметиламино)этил] -7-бромакридин- 4-карбоксамид (соединение 10m) (84%), т. пл. (дихлоргидратная соль из MeOH/EtOAc) 181,5-183oС.
Пример 11: Получение соединений формулы (I) из предшествующего метилакридин-4-карбоксилата
Общий метод
Раствор альдегида метил 2-[N-(2-формилфенил)амино] бензоата (2 г, 7,84 ммоль) в трифторуксусной кислоте (TFA) (20 мл) дегазировали и помещали в двугорлую колбу, которую затем омывали струей азота. Раствор перемешивали в течение 15 часов при комнатной температуре под азотом, а TFA затем удаляли при пониженном давлении. Получившееся масло разбавляли CH2Cl2 (100 мл) и раствор нейтрализовали Et3N. Растворители удаляли при пониженном давлении и остаток фильтровали через небольшую колонку с обожженным силикагелем в EtOAc/Петролейный эфир (1: 3) с получением метилакридин-4-карбоксилата в виде оранжевого масла (1,83 г, 98%).
1H ЯМР (CDCl3), δ 4.12 (s, 3H, СO2СН3), 7,53-7,58 (m, 2H, Н-2 и Н-6 или Н-7), 7,79 (ddd, J= 8,8, 6,6, 1,4 Гц, 1Н, Н-7 или Н-6), 8,00 (dd, J= 8,0, 0,8 Гц, 1H, H-1), 8,12-8,14 (m, 2H, Н-5,8), 8,30 (dd, J= 8,7, 0,8 Гц, 1H, Н-3), 8,80 (s, 1H, Н-9).
Раствор метилакридин-4-карбоксилата (1,83 г, 7,72 ммоль) и N, N-диметилэтилендиамина (3,40 г, 3,86 ммоль) в н-пропаноле (80 мл) омывали струей азота и затем смесь нагревали с обратным холодильником в течение 3 дней под азотом. Затем отгоняли растворитель при пониженном давлении и остаток разделяли между CH2Cl2 (100 мл) и 1М Na2CO3 (100 мл). Органический слой выпаривали и остаток хроматографировали на окиси алюминия с элюцией CH2Cl2/MeOH (199: 1), получая N-[2-(диметиламино)этил] акридин-4-карбоксамид (DACA) (1,47 г, 61%), т. пл. (дихлоргидратная соль из MeOH/EtOAc) 162-165oС.
Вышеописанный общий метод использовали для получения следующего соединения формулы (I):
N-2-(диметиламино) этил-6-трифторметилакридин- 4-карбоксамид (соединение 11а) (92%), т. пл. (MeOH/EtOAC) 188-189,5oС.
Пример 12 - Фармацевтическая композиция
Таблетки весом 0,15 г каждая, содержащие 25 мг одного из соединений формулы (I), получают, как указано ниже:
Композиция, рассчитанная для 10000 таблеток
Соединение формулы (I) - (250 г)
Лактоза - (800 г)
Кукурузный крахмал - (415 г)
Порошок талька - (30 г)
Стеарат магния - (5 г)
Смешивают соединение формулы (I), лактозу и половину количества кукурузного крахмала. Затем смесь продавливают через сито с размером отверстий 0,5 мм. Кукурузный крахмал (10 г) суспендируют в теплой воде (90 мл). Полученную пасту подвергают гранулированию до порошка. Гранулят высушивают и измельчают на сите с размером отверстий 1,4 мм. Добавляют оставшееся количество крахмала, тальк и стеарат магния, тщательно смешивают и формуют в таблетки.
| название | год | авторы | номер документа |
|---|---|---|---|
| БИС(АМИДЫ) АЗОТСОДЕРЖАЩИХ ТРИЦИКЛИЧЕСКИХ КАРБОНОВЫХ КИСЛОТ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1997 |
|
RU2179972C2 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2018 |
|
RU2773444C2 |
| АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1996 |
|
RU2198878C2 |
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
| НОВЫЕ СОЕДИНЕНИЯ КОНДЕНСИРОВАННОГО ИМИДАЗОЛА, ОБЛАДАЮЩИЕ СВОЙСТВАМИ АГОНИСТОВ РЕЦЕПТОРА СВ2 | 2002 |
|
RU2312864C2 |
| КОМПЛЕКСЫ | 2015 |
|
RU2684934C2 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ И СВЯЗАННЫЕ С НИМИ СПОСОБЫ | 2012 |
|
RU2614976C2 |
| Гетероциклические ингибиторы МСТ4 | 2017 |
|
RU2771875C2 |
| Новые соединения | 2016 |
|
RU2734256C2 |
| НОВЫЕ 2-ГЕТЕРОАРИЛ-ЗАМЕЩЕННЫЕ БЕНЗОТИОФЕНЫ И БЕНЗОФУРАНЫ 709 | 2008 |
|
RU2472789C2 |
Изобретение относится к новому способу получения акридин-карбоксамида формулы (I), где R1, R2, R5, R6, x, Y определены в формуле изобретения, который включает циклизацию соединения формулы (II) обработкой трехфтористым бором либо подходящим комплексом на его основе в подходящем растворителе с получением тетрафторборатной соли формулы (IIIa) с последующей обработкой соли неорганическим основанием в EtOAc или CH2Cl2 с образованием соединения формулы (III), и обработку либо (i) соединения формулы (III) первичным алкиламином формулы (IV), либо (ii) карбоновой кислоты, получаемой гидролизом соединения формулы (III) в щелочных условиях, первичным алкиламином формулы (IV) в присутствии подходящего связывающего агента с получением соединения (I) и, при желании, перевод одного соединения формулы (I) в другое соединение формулы (I) и/или в его фармацевтически приемлемую соль. Изобретение позволяет получить полезный продукт, который является противоопухолевым лекарственным средством, с возможностью легкого отделения и дальнейшей переработки тетрафторборатной соли, что облегчает управление процессом при его промышленной реализации, повышает производительность процесса. 2 c. и 6 з. п. ф-лы, 2 табл.
 NH2(CH2)xY (IV)
NH2(CH2)xY (IV)
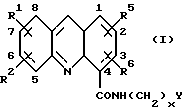
где каждый из R1, R2, R5 и R6, которые могут быть одинаковыми или различными, представляет собой Н, C1-С6 алкил, C1-C6 алкокси, фенилокси, фенил, (C1-C3) алкилокси, галоген, фенил, CF3, NO2, NH2, N(R)2, NHCOR, NHCOOR, NHR4, OH, SH, SR или SR2, где R4 представляет собой H, COR, SO2R, COPh, SO2Ph или C1-С6 алкил, незамещенный или замещенный ОН или аминогруппой;
R представляет собой C1-С6 алкил;
или R1 и R2, или R5 и R6 вместе образуют метилендиоксигруппу;
х представляет целое число от 1 до 6;
Y представляет N(R)2,
или его фармацевтически приемлемой соли, который включает: (а) циклизацию соединения формулы (II)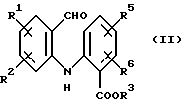
где R1, R2, R5 и R6 имеют значения, определенные выше;
R3 представляет собой C1-С6 алкил, арил или арил-С1-С3-алкил,
обработкой трехфтористым бором либо подходящим комплексом на его основе в подходящем растворителе с получением тетрафторборатной соли формулы (IIIa)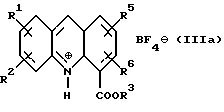
где R1, R2, R3, R5 и R6 определены, как указано выше,
с последующей обработкой соли неорганическим основанием в EtOAc или CH2Cl2 c образованием соединения формулы (III)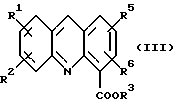
где R1, R2, R3, R5 и R6 определены выше;
и (b) обработку либо (i) соединения формулы (III) первичным алкиламином формулы (IV)
NH2(CH2)хY, (IV)
где значения x и Y определены выше,
либо (ii) карбоновой кислоты, получаемой гидролизом соединения формулы (III) в щелочных условиях, первичным алкиламином формулы (IV) в присутствии подходящего связывающего агента с получением соединения (I), определенного выше, и (c) при желании, превращение одного соединения формулы (I) в другое соединение формулы (I) и/или превращение соединения формулы (I) в его фармацевтически приемлемую соль.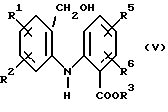
где R1, R2, R3, R5 и R6 определены, как указано в п. 1.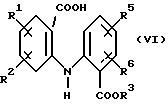
где R1, R2, R3, R5 и R6 определены в п. 1,
1,1'-карбонил-диимидазолом в полярном растворителе с получением соединения формулы (VII)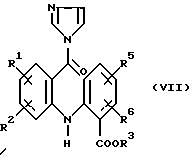
где R1, R2, R3, R5 и R6 определены в п. 1,
и (b) восстановления полученного соединения формулы (VII).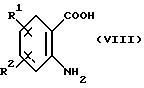
где R1 и R2 определены в п. 1,
и эфира 2-иодобензойной кислоты формулы (IX)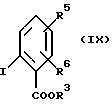
где R3, R5 и R6 определены в п. 1,
в присутствии медного катализатора и основания в полярном растворителе.
который включает (i) обработку соединения формулы (VI)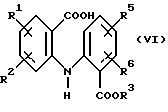
где каждый из R1, R2, R5 и R6 представляет водород;
R3 представляет собой C1-С6 алкил, фенил-С1-С6-алкил или фенил,
1,1'-карбонилдиимидазолом в органическом растворителе с получением соединения формулы (VII)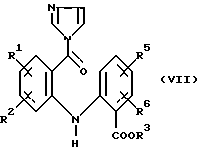
где каждый из R1, R2, R5 и R6 представляет водород;
R3 определен, как указано выше,
(ii) обработку соединения формулы (VII) боргидридом натрия в присутствии воды для получения соединения формулы (V)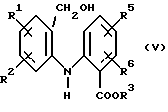
где каждый из R1, R2, R5 и R6 представляет водород;
R3 определен как указано выше,
(iii) окисление соединения формулы (V) с получением соединения формулы (II)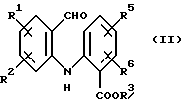
где каждый из R1, R2, R5 и R6 представляет водород;
R3 определен, как указано выше;
(iv) циклизацию соединения формулы (II) обработкой его трехфтористым бором либо подходящим комплексом на его основе в подходящем растворителе с получением тетрафторборатной соли формулы (IIIа)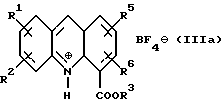
где R1, R2, R3, R5 и R6 определены, как указано выше,
с последующей обработкой соли неорганическим основанием в EtOAc или CH2Cl2 с образованием соединения формулы (III)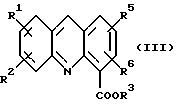
где каждый из R1, R2, R5 и R6 представляет водород;
R3 определен, как указано выше,
и (v) обработку соединения формулы (III) первичным алкиламином формулы (IV)
NH2(CH2)xY, (IV)
где х равен 2;
Y представляет N(CH3)2,
с получением DACA.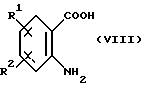
где R1 и R2 определены в п. 6,
с эфиром 2-иодобензойной кислоты формулы (IX)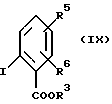
где R3, R5 и R6 определены в п. 6,
в присутствии медного катализатора и основания в полярном растворителе.
Приоритет по пунктам:
18.10.1996 - по п. 6 - стадия (iv);
20.12.1996 - по пп. 1-5, п. 6 (остальная часть, кроме стадии (iv) и пп. 7 и 8.
| ПРОИЗВОДНЫЕ АКРИДИНА, СПОСОБ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННЫХ ОПУХОЛЕЙ | 1992 |
|
RU2119482C1 |
| US 5696131 А, 09.12.1997 | |||
| Устройство для бесконтактного измерения параметров вибрации | 1973 |
|
SU494623A1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| US 5604237 А, 18.02.1997. | |||

