Настоящее изобретение относится к немедленно связывающимся ингибиторам тромбина, процессу получения названных ингибиторов, фармацевтическим композициям, содержащим их, и использованию этих ингибиторов тромбина в качестве антитромботических агентов.
Большое внимание было уделено ингибированию тромбина в качестве возможности потенциальной антикоагуляции. Ингибиторы фермента тромбина, ключевой сериновой протеиназы в каскаде реакций свертывания крови, некоторое время считали потенциальными кандидатами на роль профилактических и терапевтических антикоагулянтов. В частности, многоплановая роль тромбина, выражающаяся в его воздействии на факторы свертывания, циркулирующие компоненты крови и клетки стенок кровеносных сосудов делает его особенно выраженной мишенью в условиях различных патологических состояний. Более того, ограничения, связанные с применением обычно используемых антикоагулянтов, в частности, возникающие при осложнениях в виде кровотечений, диктуют необходимость поиска агентов, действующих более специфично.
Обнаружено много пептидо(подобных) ингибиторов сериновых протеиназ и среди них - ингибиторы промежуточного состояния тромбина. Однако многие из них являются медленно связывающимися ингибиторами. Использование медленно связывающихся ингибиторов тромбина весьма уязвимо. In vivo тромбин постоянно вырабатывается в плазме, и первоочередная функция ингибиторов тромбина - замедление процесса выработки тромбина путем ингибирования опосредованных тромбином ступеней каскадного процесса. Для замедления каскадных реакций предпочтительнее было бы использовать не-медленно связывающийся ингибитор тромбина. Такого же результата можно достичь и повышая дозу медленно связывающегося ингибитора, однако соответственно увеличится и риск побочных реакций.
Соответствующие ингибиторы тромбина были обнаружены Brady et al. , Bioorganic & Medicinal Chemistry, 3 (1995), 1063-78; они являются производными D-Phe-Pro-Arg-амида и D-Phe-Pro-Lys-X, где Х - кетосложный эфир или амин. Было показано, что эти соединения являются медленно связывающимися ингибиторами тромбина, и в связи с этим они исключаются из рассмотрения в настоящем изобретении. В поисках немедленно связывающихся ингибиторов тромбина Jones et al. , J. Enzyme Inhibition, 9 (1995), 43-60, сделали попытку добиться успеха, используя производные D-Cha-Pro-Lys-COOH. Однако несмотря на то, что эти производные оказались более сильными ингибиторами тромбина, они все-таки обладали свойствами медленно связывающихся ингибиторов тромбина.
Среди недавних попыток получить сильные не-медленно связывающиеся ингибиторы тромбина следует отметить работу Levis et al. , Thrombosis and Haemostasis, 74 (4) (1995), 1107, в которой получили производные Ме-D-Phe-Pro-Lys-X, где X - карбоксиамид или карбоновая кислота. Эти соединения, и в особенности Me-D-Phe-Pro-Lys-COOH, классифицированы как медленно связывающиеся ингибиторы. Следовательно, они не удовлетворяют требованиям настоящего изобретения и поэтому должны быть изъяты из рассмотрения.
Ингибитор тромбина с алкил-замещенным лизином описан в патенте США N 5523308.
В более ранних работах описаны другие последовательности, например, в работе Iwanowicz et al. In Bioorganic & Medicinal Chemistry Letters, 2 (1992), 1607-12, изучали производные D-Phe-Pro-Lys-X, где Х является, например, кетосложным эфиром. Эти соединения также относятся к медленно связывающимся ингибиторам.
Обнаружены и другие типы пептидов, ингибирующих различные сериновые протеиназы. Tsutsumi et al. in J. Med/ Chem. , 37 (1994), 3492-3502 описаны пептидоподобные соединения, содержащие на С-конце тиазол и бензотиазол. Было показано, что такие производные тиазола в 300 раз эффективнее соответствующих тиофеновых аналогов. Позже было установлено, что С-концевые гетероциклические группы могут обеспечить критическое взаимодействие посредством водородной связи с гистамином протеазы пролилэндопептидазы. Хотя в дальнейшем и высказывалось предположение, что это явление можно перенести и на другие сериновые протеиназы, однако конкретно тромбин не был указан. Механистическое объяснение Tsutsumi было в дальнейшем продолжено Edwards et al. In J. Med. Chem. , 38 (1995), 76-85, однако эти авторы обнаружили еще, что ингибиторы эластазы типа D-Phe-Val-Pro-Val-X, где Х тиазол или бензотиазол, представляют собой немедленно связывающиеся ингибиторы указанной сериновой протеиназы. Ими было также высказано предположение, что пептидил-альфа-кетогетероциклы могут оказаться подходящими ингибиторами и для других сериновых протеиназ.
Настоящее изобретение связано с неожиданным обнаружением того, что результаты, полученные Edwards, Tsutsumi и другими, могут быть применимы также и к ингибиторам тромбина. Как обнаружили Lewis, Jones и Brady, приложение С-концевых гетероциклов к этим соединениям обеспечивает потенциальным ингибиторам тромбина свойство их немедленного связывания с тромбином. Более того, многие из этих соединений характеризуются повышенным временем биологической полужизни и полезными свойствами при оральном применении.
Таким образом, изобретение связано с немедленно связывающимися ингибиторами тромбина, имеющими формулу
A-B-C-Lys-D,
где А является Н, 2-гидрокси-3-циклогексилпропионил-, R1, R1-О-СО-, R1-CO-, R1-SO2, -(CHR2)nСООR3 или N-защищающую группу, где R1 выбран из-(1-6С)алкилен-СООН, (1-12С)алкила, (2-12С)алкенила, (6-14С)арила (7-15С)аралкила и (8-16С)аралкенила, арильная группа которого может быть заменена на (1-6С)алкил, (2-12С)алкокси, гидрокси или галоген; R2 является Н или имеет то же значение, что и R1.
R3 выбран из Н, (1-12С)алкила, (2-12С)алкенила, (6-14С)арила, (7-15С)аралкила и (8-16С)аралкенила, арильная группа которого может быть заменена на (1-6С)алкил, (2-12С)алкокси, гидрокси или галоген;
n - целое число от 1 до 3;
В - связь, L-ASp или производная его сложного эфира, Leu, norLeu, -N(бензил)-СН2-СО-, -N(2-индан)-СН2-СО-, D-l-Piq. D-3-Piq, D-Tiq, Atc или D-аминокислота, имеющая гидрофобную ароматическую боковую цепь;
С - это Azt, Pro, Pec, norLeu(цикло)Gly, аминокислота одной из формул -N-[(3-8С)циклоалкил] -СН2-СО- или -N(бензил)-СН2-СО-;
D выбран из СООН, тетразола, оксазола, тиазола и бензотиазола; или А и С имеют вышеуказанные значения, В является D-(3-8C)циклоалкилаланином и D является тетразолом, оксазолом, тиазолом или бензотиазолом;
или его пролекарством;
или его фармацевтически приемлемой солью;
за исключением соединения Me-D-Phe-Lys-COOH.
Соединения согласно настоящему изобретению полезны при лечении и профилактике опосредованных тромбином и связанных с ним заболеваний. Сюда входит множество тромботических и протромботических состояний, в которых активирован каскад свертываемости; такие состояния включают следующий перечень, но не ограничены им: тромбоз глубоких вен, легочная эмболия, тромбофлебиты, закупорка артерий от тромбозов или эмболии, артериальная реокклюзия во время или после ангиопластики или растворения тромба, послеоперационный тромбоз вен или эмболия, рестеноз в результате повреждения сосудов или инвазивных кардиологических процедур, острый или хронический атеросклероз, удар, инфаркт миокарда, рак и метастазирование и нейродегенеративные заболевания. Соединения согласно изобретению могут быть использованы также в качестве антикоагулянтов при экстракорпоральном кровообращении, как это бывает необходимо при диализе и операциях. Соединения согласно изобретению могут быть использованы также и в качестве антикоагулянтов in vitro.
Предпочтительными соединениями согласно изобретению являются соединения, в которых D является СООН. Кроме того, предпочтительно, чтобы А представляло собой Н, (1-12С)алкил, -СО-(7-15С)аралкил, -SO2-(1-12C)алкил, -SO2-(6-14С)арил или -SO2-(7-15C) аралкил; В представляет собой связь, L-Asp, norLeu, D-1-Piq или D-Phe, С является Pro, norLeu(цикло)Gly или -N-циклопентил-СН2-СО-. Более предпочтительными являются немедленно связывающие тромбин ингибиторы, в которых А представляет собой -SO2-бензил, В - связь, а С -norLeu-(цикло)-Gly, или где А является -SO2-этилом, В является D-Phe, а С является Pro; или же где А - водород, В является D-1-Piq, а С представляет собой Pro.
Остальными предпочтительными соединениями согласно изобретению являются такие, в которых D является оксазолом или тиазолом. Далее, предпочтительно, чтобы А представляло собой Н, (1-12С)алкил, 2-гидрокси-3-циклогексил-пропионил-, -СО-(СН2)nСООН, -СО-(7-15C)аралкил, -SO2-(6-14С)арил, -SO2-(7-15С)аралкил, -SO2-(1-12C)алкил, -(CHR2)nCOOR3, R2 является Н или (1-12С) алкилом, а R3 являлся Н, (1-12С)алкилом или бензилом; и чтобы С являлся Pro, norLeu(цикло)Gly или -N[(3-8С)циклоалкил] -СН2-СО-. Особенно предпочтительными являются не-медленно связывающиеся ингибиторы тромбина, в которых А является -(СН2)nCOOR3, R3 является Н, (1-12С)алкилом или бензилом; В является D-(3-8C)циклоалкилаланином или D-Phe, факультативно однозамещенным алкокси или галогеном; и С является Pro. Наиболее предпочтительными соединениями согласно изобретению являются соединения, в которых D является тиазалом. Определенно предпочтительным является немедленно связывающийся ингибитор HOOC-CH2-D-Cha-Pro-Lys-(2-тиазолил).
N-защищающей группой, определяемой при изучении подвижности А, может являться какая-нибудь N-защищающая группа, используемая в пептидах. Подходящие N-защищающие группы можно найти в работе Т. W. Green and P. G. M. Wuts: Protective Groups in Organic Synthesis, Second Edition (Wiley, NY, 1991) and in The Peptides, Analysis, Synthesis, Biology, Vol. 3 E. Gross and J. Meienhofer, Eds. , (Academic Press, New York. 1981).
Используемый здесь алкил является разветвленной или неразветвленной алкильной группой, имеющей от 1 до 12 атомов углерода, такой как метил, этил, изопентил, додецил и другие им подобные.
Обозначение (1-6С)алкилен означает разветвленную или неразветвленную алкиленовую группу, содержащую от 1 до 6 атомов углерода, такую как -(СН2)m-, где m принимает значения от 1 до 6, -СН(СН3)-, -СН(СН3)-(CH2)- и т. д. Предпочтительной алкиленовой группой является метилен.
Алкенил представляет собой разветвленную или неразветвленную ненасыщенную алкенильную группу, имеющую от 2 до 12 атомов углерода.
Примерами являются этенил, аллил, пропенил и им подобные.
Аралкильные и аралкенильные группы представляют собой, соответственно, алкильные и алкенильные группы, замещенные одной или более арильными группами с общим количеством атомов углерода от 7 до 15 и от 8 до 16, соответственно. Предпочтительными аралкильными группами являются, например, группы согласно формуле
-(СН2)p-СН-(С6Н5)2,
где р принимает значения 1 или 2, или -(СН2)q-C6H5, факультативно замещенные галогеном, и где q принимает значения 1, 2 или 3.
Арил в вышеназванном определении и в определении, использованном в соединении согласно изобретению, является ароматической частью, содержащей от 6 до 14 атомов углерода. Арильная группа может в дальнейшем содержать один или более гетероатомов, таких как азот, сера или кислород. Примерами арильных групп являются фенил, нафтил, (изо)хинолил, инданил и им подобные. Наиболее предпочтительной является фенильная группа. Арильная группа может быть замещена одной или более алкильной группой, предпочтительно метильной, алкокси-группами, предпочтительно метокси, гидрокси или галогеном. Под галогеном подразумеваются фтор, хлор, бром или йод. Хлор является наиболее предпочтительным галогеном.
Обозначения D-1-Piq и D-3-Piq означают 1- и 3-карбоксипергидроизохинолин, соответственно. Tiq означает 1,2,3,4-тетрагидроизохинолин-карбоновую кислоту. Atc является 2-амино-тетралин-2-карбоновой кислотой. Azt и Реc означают 2-азетидин-карбоновую кислоту и пипеколиновую кислоту, соответственно.
Обозначение norLeu(цикло)Gly означает структурный фрагмент формулы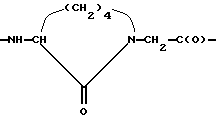
Под гидрофобной ароматической боковой цепью подразумевается (1-12С)алкил, замещенный одной или более (6-14С)арильными группами (которые могут содержать гетероатом, например, азот), например, фенилом, пиридинилом, нафтилом, тетрагидронафтилом и им подобными, гидрофобные боковые цепи которых могут быть замещены гидрофобными заместителями, такими как галоген (предпочтительно хлор), трифторметил, низший алкил (например, метил или этил), низший алкокси (например, метокси), фенилокси, бензилокси и им подобными.
Обозначение (3-8С)циклоалкил означает циклопропил, циклобутил, циклопентил, циклогептил или циклооктил.
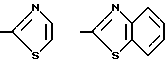
Тетразол, оксазол, тиазол и бензотиазол имеют, соответственно, следующие формулы: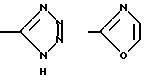
Настоящее изобретение включает в себя также и пролекарства соединений общей формулы, которые после приема (вовнутрь) метаболизируются в активные соединения. Пригодными для этого пролекарствами являются, например, N-алкоксикарбонил-защищенные (предпочтительно N-этоксикарбонил) производные общей формулы.
Использованное здесь понятие фармацевтически допустимой соли относится к солям, которые сохраняют требуемую биологическую активность исходного соединения и которые не обладают нежелательным токсическим действием. Примерами таких солей являются соли присоединения кислот, образованные неорганическими кислотами, например, хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты, фосфорной кислоты, азотной кислоты и им подобных. Соли могут быть образованы также и из органических кислот, таких как, например, уксусная кислота, щавелевая кислота, винная кислота, янтарная кислота, малеиновая кислота, фумаровая кислота, глюконовая кислота, лимонная кислота, яблочная кислота, аскорбиновая кислота, бензойная кислота, дубильная кислота, памоевая кислота, альгиновая кислота, полиглутаминовая кислота и им подобные. Соли могут быть сформированы с поливалентными ионами металлов, таких как цинк, кальций, висмут, барий, магнезий, алюминий, медь, кобальт, никель и тому подобное, или с органическим катионом, образованным из N, N'-дибензилэтилендиамина или этилендиамина или же их комбинаций (например, цинковая соль дубильной кислоты).
Соединения согласно изобретению обладают одним или несколькими хиральными атомами углерода и могут, следовательно, быть получены в виде чистого энантиомера или в виде смеси энантиомеров, или в виде смеси, содержащей диастереомеры. Способы получения чистых энантиомеров хорошо известны, например, кристаллизация солей, полученных из оптически активных кислот, и рацемически смешанных, или хроматография с использованием хиральных колонок. Для диастереомеров можно использовать колонки с прямой или обратимой фазой.
Изобретение включает далее процесс получения соединения формулы, процесс, включающий попарное соединение защищенных должным образом аминокислот или аминокислотных аналогов, после чего протекторные группы убираются.
Соединения согласно общей формуле можно получить общепринятым для таких соединений способом. Для этого соответствующие Nα-защищенные (и с защищенными боковыми цепями, если они реактивны) производные аминокислот или пептидов активируются и попарно связываются с подходящей защищенной с С-конца аминокислотой или пептидными производными либо в растворе, либо на твердом носителе. Защиту α-амино-функций обычно производят с помощью уретана, такого как кислотолабильная трет. -бутилоксикарбонильная группа (Boc), бензилоксикарбонильная (Z) группа и замещенные аналоги или щелочелабильная 9-фторенил-метилоксикарбонильная (Fmoc) группа, Z-группа может также быть удалена путем каталитической гидрогенизации. Другие подходящие защитные группы включают Nps, Bmv, Врос, Aloc, MSC и т. д. Хороший обзор амино-защищающих групп приведен в книге: The Peptides, Analysis, Synthesis. Biology. Vol. 3 E. Gross and J. Meienhofer, Eds. , (Academic Press, New York, 1981). Защиту карбоксильных групп можно произвести путем образования сложного эфира, например, щелочелабильных сложных эфиров наподобие метила или этила, кислотолабильных сложных эфиров, таких как трет. бутил или гидрогенолитически-лабильных сложных эфиров, таких как бензил. Защита функций боковых групп, например, лизина может быть обеспечена с помощью указанных выше групп. Активацию карбоксильной группы защищенных аминокислот или пептидов можно произвести с помощью азида, смешанного ангидрида, активного сложного эфира или карбодиимидным способом, особенно с добавлением каталитических и тормозящих рацемизацию соединений наподобие 1-гидроксибензотриазола, N-гидроксисукцинимида, 3-гидрокси-4-оксо-3,4-дигидро-1,2,3-бензотриазина, N-гидрокси-5-норборнен-2,3-дикарбоксимида. Могут быть использованы также ангидриды фосфорных кислот. См. , например, The Peptides, Analysis. Synthesis, Biology, supra Pure and Applied Chem. 59(3), 331-344 (1987).
Эти соединения можно получить также и твердофазным способом по Merrifield. Известны различные твердые носители и разные способы - см. , например, Barany and Merrifield in The Peptides, Analysis, Synthesis, Biology. Vol. 2, E. Gross and J. Meienhofer, Eds. , (Acad. Press, N. Y. , 1980), Kneib-Cordonier and Mullen Int. J. Peptide Protein Res. , 30, 705-739 (1987) and Fields and Noble Int. J. Peptide Protein Res. , 35, 161-214 (1990).
Удаление защитных групп и, в случае твердофазного синтеза пептидов, отделение от твердой подложки производят разными способами, в зависимости от природы этих защитных групп и типа линкера к твердой подложке. Обычно удаление защиты происходит в кислых условиях и в присутствии утилизаторов супероксидных радикалов. См. , например, тома 3, 5 и 9 серии The Peptides Analysis, Synthesis, Biology, supra.
Другой возможностью применения ферментов в синтезе таких соединений является, например, способ, описанный в обзоре H. D. Jakubke in The Peptides, Analysis. Synthesis Biology. Vol. 9. S. Udenfriend and J. Meienhofer. Eds. , (Acad. Press. N. Y. , 1987).
Полученные тем или иным способом, такие соединения полезны в изготовлении медикаментов, используемых при лечении патологических состояний, при которых имеет место нежелательная свертываемость крови. В таком случае конкретное синтезированное соединение будет соответствующим образом ассоциировано с фармацевтическим носителем. Фармацевтические носители могут быть разными, начиная от относительно простых, таких как стерильная вода для инъекций, до относительно сложных, таких как микросферы и биодеградируемые имплантаты.
В качестве медикаментов предпочтительно применять эти соединения перорально, подкожно, местно, в нос, внутривенно, внутримышечно или локально (например, в виде имплантата). Возможно также применение с целью депонирования.
Точная доза и режим применения этих соединений и композиций непременно будут зависеть от индивидуальных потребностей больного, которому назначается медикамент, от степени нарушений, имеющихся у больного, и, конечно же, от точки зрения лечащего врача. Обычно парентеральный способ введения требует меньших доз, чем другие, которые больше зависят от степени абсорбции. Однако ориентировочная доза составляет от 0,001 до 100 мг/кг веса тела, предпочтительнее - от 0,01 до 10 мг/кг веса тела.
Медикаменты, изготовленные с применением этих соединений, могут быть использованы также в качестве адъювантов в острой антикоагуляционной терапии. В таком случае препарат назначают совместно с другими соединениями, необходимыми при лечении подобных патологических состояний.
Эти соединения могут использоваться также и в имплантационных фармацевтических приемах, таких как описано в патенте США 4767628, содержание которого использовано в настоящей работе. Затем в этих приемах должно быть использовано достаточное количество соединения для его медленного высвобождения в течение длительного периода (например, более месяца).
Способы изготовления медикаментов, содержащих это соединение, для внутреннего и парентерального применения описан в работе Gennaro et al. , Remington's Pharmaceutical Sciences, (18th ed. , Mack Publishing Company, 1990, см. , в частности. Часть 8: Фармацевтические препараты и их изготовление), стр. 1519-1580. В смеси с фармацевтически необходимыми добавками, указанные соединения могут быть спрессованы с получением твердых дозированных единиц, таких как драже, таблетки, а также капсулы или суппозитории. С использованием фармацевтически приемлемых жидкостей эти соединения могут быть также получены и применимы в виде раствора, суспензии, эмульсии, например, для использования в виде инъекций, или в виде аэрозоля, например, для распыления в нос.
Для изготовления дозированных единиц, например, таблеток, подразумевается использование традиционных добавок, таких как красители, наполнители, полимерные связующие вещества и другие. Обычно используют какую-нибудь фармацевтически приемлемую добавку, которая не влияет на функциональные свойства активных соединений.
Приемлемыми носителями, в сочетании с которыми могут назначаться эти соединения, являются такие как лактоза, крахмал, производные целлюлозы и им подобные, либо их смеси, используемые в приемлемых количествах.
Дальнейшие разъяснения в связи с настоящим изобретением будут представлены в виде иллюстративных примеров.
Пример 1.
3,3-Дифенилпропионил-Pro-Lys-(2-тиазолил)
3,3-Дифенилпропионил-пролил-метил-сложный эфир
К холодному раствору (0oС) 3,3-дифенилпропионовой кислоты (5,0 г) в этилацетате (100 мл) последовательно добавляли DCCI (1,3-дициклогексилкарбодиимид; 5,03 г), HOBt. (1-гидроксибензотриазолгидрат; 3,28 г), Н-Pro-ОМе. НСl (3,66 г) и триэтиламин (3,1 мл). Реакционную смесь перемешивали в течение 1 часа при 0oС, а затем выдерживали в течение ночи при комнатной температуре. Затем реакционную смесь охлаждали до -20oС и путем фильтрации удаляли DCU (1,3-дициклогексилмочевина). Фильтрат последовательно промывали 5%-ным бикарбонатом натрия, водой, 5%-ным бисульфатом калия и насыщали водным хлоридом натрия, высушивали над сульфатом натрия и концентрировали в вакууме. Осадок хроматографически очищали на кремнеземе (элюэнт: дихлорметан/этилацетат; 9/1 по объему) с выходом 5,68 г 3,3-дифенилпропионил-пролилметил-сложного эфира в виде кристаллического порошка. ТСХ (тонкослойная хроматография) Rf = 0,75, силикагель, дихлорметан/этилацетат = 7/3 по объему.
3,3-Дифенилпропионил-пролил-ОН
3,3-Дифенилпропионил-пролил-метил-сложный эфир (5,6 г) растворяли в диоксане/воде в соотношении 7/3 объем на объем (60 мл) медленно, в течение 30 минут при комнатной температуре добавляли 4 М раствор NaOH (6,2 мл), доведя рН до 10-10,5. Через 30 минут реактивную смесь разбавляли водой (60 мл), а 4 М раствором HCl доводили рН раствора до 2,0, и водный слой экстрагировали этилацетатом. Объединенные органические фазы промывали водой, насыщали водным хлористым натрием и высушивали над сульфатом натрия, растворитель удаляли путем выпаривания, в результате получался 3,3-дифенилпропионил-пролил-ОН в виде сиропа (5,18 г). ТСХ: Rf = 0,65, силикагель, EPAW (этилацетат/пиридин/уксусная кислота/вода) 63/20/6/11 по объему.
Boc-Lys(Cbz)-NМeOMe
Boc-Lys(Cbz)-OH. DCHA (10 г) суспендировали в дихлорметане (200 мл). Суспензию дважды промывали охлажденным раствором 0,1 N HCl. К полученной органической фазе добавляли 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурониумтетрафторборат (6,0 г) и O, N-диметил-гидроксиламин-хлористоводородная кислота (1,82 г), и добавлением триэтиламина рН доводили до 8. Реакционную смесь перемешивали в течение часа при комнатной температуре. Эту смесь промывали последовательно холодным раствором 2 N соляной кислоты, водой, 5%-ным бикарбонатом натрия и водой. Органический слой высушивали над сульфатом натрия, фильтровали и выпаривали. Осадок хроматографически очищали на кремнеземе (элюент: дихлорметан/метанол; 5/5 по объему) с получением Вос-Lys-(Cbz)-NMeOMe (7,2 г), ТСХ: Rf = 0,55, силикагель, дихлорметан/метанол 95/5 по объему.
Boc-Lys(Cbz)-(2-тиазолил)
К холодному (-78oС) перемешиваемому раствору n-бутиллития (63,9 ммоль) в диэтиловом эфире (58 мл) по каплям добавляли раствор 2-бромотиазола (10,5 г) в диэтиловом эфире (30 мл). Раствор перемешивали в течение 30 минут при -78oС, затем медленно добавляли раствор Boc-Lys(Cbz)-NMeOMe (8,2 г) в сухом THF (тетрагидрофуран; 75 мл). Смесь перемешивали при -78oС в течение 1 часа, затем добавляли 5%-ный водный гидрокарбонат натрия. Смесь выдерживали при комнатной температуре и образовавшиеся слои разделяли. Водный слой экстрагировали диэтиловым эфиром. Объединенные органические фракции промывали водой, высушивали над сульфатом натрия, фильтровали и выпаривали. Осадок хроматографически очищали на кремнеземе (элюент: этилацетат/гептан; 3/1 по объему) с выходом Boc-Lys(Cbz)-(2-тиазола) (8,6 г). ТСХ: Rf = 0,77, силикагель, этилацетат/гептан = 3/1 по объему.
H-Lys(Cbz)-(2-тиазолил). TFA
Boc-Lys(Cbz)-(2-тиазолил) (500 мг) растворяли в смеси 50%-ной TFA (трифторуксусная кислота)/дихлорметан (5 мл) и перемешивали в течение часа при комнатной температуре. Сырой H-Lys(Cbz)-(2-тиазолил). ТФУ был выделен с количественным выходом после удаления растворителя путем выпаривания и немедленно использован в следующей стадии. ТСХ: Rf = 0,25, силикагель, EPAW = 63/20/6/11 по объему.
3,3-Дифенилпропионил-Pro-Lys(Cbz)-(2-тиазолил)
К холодному (0oС) раствору 3,3-дифенилпропионил-пролил-ОН (385 мг) в диметилформамиде (5 мл) последовательно добавляли DCCI (270 мг), HOBt (176 мг), H-Lys(Cbz)-(2-тиазолил). TFA (515 мг) и N-этилморфолин (0,28 мл). Реакционную смесь перемешивали в течение 1 часа при 0oС, а затем выдерживали в течение ночи при комнатной температуре. Затем смесь охлаждали до -20oС и путем фильтрации удаляли DCU. Фильтрат выпаривали досуха. Осадок растворяли в этилацетате и последовательно промывали 5%-ным водным гидрокарбонатом натрия, водой, 5%-ным водным гидросульфатом калия и насыщенным водным хлоридом натрия, высушивали над сульфатом натрия и концентрировали в вакууме. Осадок хроматографически очищали на кремнеземе (элюент: этилацетат/гептан 4/1 по объему) с получением 3,3-дифенилпропионил-Pro-Lys(Cbz)-(2-тиазолил) (332 мг). ТСХ: Rf = 0,40, силикагель, этилацетат/гептан; 3/1 по объему.
3,3-Дифенилпропионил-Pro-Lys-(2-тиазолил)
3,3-Дифенилпропионил-Pro-Lys(Cbz)-(2-тиазолил) (320 мг) обрабатывали с помощью ТФУ/тиоанизола, 10/1 по объему (3,3 мл), в течение 3 часов при комнатной температуре. Реакционную смесь концентрировали в вакууме и осадок растворяли в воде. Водную фазу экстенсивно высушивали диэтилэфиром.
Водный слой, содержащий 3,3-дифенилпропионил-Pro-Lys(Cbz)-(2-тиазолил) заряжали прямо на препаративной колонке Supelcosil LC-18-DB высокоэффективной жидкостной хроматографии с помощью системы градиентной элюции от 20% А/60% В/20% С до 20% А/80% С через 45 минут при скорости элюции 20 мл/мин. (А: 0,5 М натрий-фосфатный буфер, рН 2,1, В: вода, С: ацетонитрил/вода; 3/2 по объему).
Выход: 47 мг 3,3-дифенилпропионил-Pro-Lys-(2-тиазолил). ТСХ: Rf = 0,57, силикагель, EPAW; 63/20/6/11 по объему, Rt(ЖХ): 32,9 мин; 20% А/60% В/20% С до 20% А/0% В/80% C через 40 минут.
Пример 2.
Подобным описанному в примере 1 способом было получено:
(a). H-D-Phe-Pro-Lys-(2-тиазолил) Rt(ЖХ): 25,67 мин; 20% А/80% В/60% С до 20% А/20% В/60% С через 40 мин.
(b). H-D-1-Tiq-Pro-Lys-(2-тиазолил)
Rt(ЖХ): 23,40 мин; 20% А/80% В/0% С до 20% А/20% В/60% С через 40 мин. (Tiq= тетрагидроизохинолин)
(c). H-D-(p-Cl)-Phe-Pro-Lys-(2-тиазолил)
Rt(ЖX): 30,47 мин; 20% А/80% В/0% С до 20% А/20% В/60% С через 40 мин.
(d). Инданеглицил-(N-циклопропил)-Gly-Lys-(2-тиазолил)
Rt(ЖX): 27,88 мин; 20% А/80% В/0% С до 20% А/20% В/60% С через 40 мин.
(e). H-D-Phe-(N-циклопентил)-Gly-Lys-(2-тиазолил)
Rt(ЖХ): 31,07 мин; 20% А/80% В/0% С до 20% А/20% В/60% С через 40 мин.
(f). Ацетил-D-Phe-(N-циклопропил)-Gly-(2-тиазолил)
Rt(ЖХ): 33,73 мин; 20% А/80% В/0% С до 20% А/20% В/60% С через 40 мин.
(g). H-D-Cha-Pro-Lys-(2-тиазолил)
Rt(ЖX): 30,59 мин; 20% А/80% В/0% С до 20% А/20% В/60% С через 40 мин. (Cha = циклогексилаланин).
(h). H-D-Phe-(N-циклопропил)-Gly-Lys-(2-тиазолил)
Rt(ЖХ): 5.1 мин; изократически; 55/45 MeOH/25 mM фосфат, рН 7.
(i). 3,3-Дифенилпропионил-(N-циклопропил)-Gly-Lys-(2-тиазолил)
Rt(ЖХ): 8,1 мин; изократически; 75/25 MeOH/25 mM фосфат, рН 7.
(j). H-D-Phe-(N-циклобутил)-Gly-Lys-(2-тиазолил)
Rt(ЖХ): 30,59 мин; 20% А/80% В/0% С до 20% А/20% В/60% С через 40 мин.
(k). H-Atc-Pro-Lys-(2-тиазолил)
Rt(ЖХ): 27,79+28,04 мин; 20% А/80% В/0% С до 20% А/20% В/60% С через 40 мин.
(Atc = 2-аминотетралин-2-карбоновая кислота)
(l). H-D-Phe-(N-бензил)-Gly-Lys-(2-тиазолил)
Rt(ЖХ): 16,99 мин; 20% А/80% В/20% С до 20% А/0% В/80% С через 40 мин.
(m). H-D-Cha-(N-циклопропил)-Gly-Lys-(2-тиазолил)
Rt(ЖХ): 30/84 мин; 20% А/80% В/0% С до 20% А/20% В/60% С через 40 мин.
(n). р-хлоро-3-фенилпропионил-(N-циклопентил)-Gly-Lys-(2-тиазолил)
Rt(ЖX): 36,15 мин; 20% А/60% В/20% С до 20% А/0% В/80% С через 40 мин.
(о). (N-бензил)-Gly-(N-циклопентил)-Gly-Lys-(2-тиазолил)
Rt(ЖХ): 28,14 мин; 20% А/80% В/0% С до 20% А/80% С через 40 мин.
Пример 3.
НООН-СН2-(N-циклопентил)-Gly-Lys-(2-тиазолил)
(N-циклопентил)-Gly-OMe
К раствору, содержащему 23,2 г H-Gly-OMe. HCl в 200 мл метанола добавляли 15,6 г циклопентанона. Смесь размешивали в течение 15 минут и добавляли 7 г цианоборгидрида. Устанавливали рН 6. Реакционную смесь выдерживали при комнатной температуре при постоянном перемешивании. Для завершения реакции добавляли 1 г циклопентанона и продолжали перемешивать. Регистрировали реакцию на ТСХ. Когда весь первоначальный материал исчезал, смесь подкисляли до рН 2 и перемешивали в течение 30 минут. Растворитель удаляли, а осадок разбавляли водой. Раствор промывали эфиром, рН доводили 6 N NaOH до 12 и экстрагировали дихлорметаном. Объединенные органические слои промывали насыщенным раствором хлористого натрия, высушивали на сульфате натрия и выпаривали в вакууме до получения 16 г масла. Rf = 0,46 в этилацетате/пиридине/уксусной кислоте/воде = 63/20/6/11 по объему на кремнеземе.
N-(t-бутилоксикарбонил-метил)-D-Cha-OMe
К перемешиваемому раствору, содержащему 26 г H-D-Cha-OМe. HCl в 300 мл ацетонитрила, добавляли 17 г t-бутилбромоацетата. рН реакции доводили диизопропилэтиламином до 8,5. Смесь перемешивали в течение 16 часов при комнатной температуре и выпаривали в вакууме. Осадок растворяли в дихлорметане, раствор промывали водой, сушили на сульфате натрия и выпаривали в вакууме. Хроматографировали на силикагеле в гексане: этилацетате, 9: 1 по объему, получали 20 г вышеназванного продукта. Rf = 0/46 в этилацетате/пиридине/уксусной кислоте/воде, 15,75/5/1,5/2,75 по объему, на кремнеземе.
N, N-Boc. (t-бутилоксикарбонил-метил)-D-Cha-OMe
Диизопропилэтиламином доводили рН раствора, содержащего 20 г N-(t-бутилоксиметил)-D-Cha-OMe и 17 г ди-t-бутилбикарбоната, до 8,5. Полученную смесь перемешивали в течение 16 часов при комнатной температуре. Растворитель удаляли под вакуумом. К осадку добавляли дихлорметан и воду. Органическую фракцию отделяли, промывали охлажденной 1 N соляной кислотой, водой, 5%-ным бикарбонатом натрия и водой. Органическую фракцию высушивали на сульфате натрия, и фильтрат выпаривали до аморфного твердого состояния с выходом, составляющим 28 г.
Rf = 0,60 в этилацетате/пиридине/уксусной кислоте/воде, 252/20/6/11 по объему на кремнеземе.
N, N-Boc. (t-бутилоксикарбонилметил)-D-Cha-ОН
pH раствора, содержащего 28 г N, N-Boc. (t-бутилкарбонилметил)-D-Cha-Оме в 420 мл диоксана с водой, 9/1, доводили 1 N гидроокисью натрия до 13 в течение 90 минут при комнатной температуре. После закисления смесь вливали в воду и экстрагировали дихлорметаном. Органическую фракцию промывали водой и сушили на сульфате натрия. Фильтрат выпаривали с получением 24 г целевого продукта.
Rf = 0,23 в смеси дихлорметан/метанол, 9/1 по объему, на кремнеземе.
N, N-Boc. (t-бутилоксикарбонил-метил)-D-Cha-(N-циклопентил)-Gly-OMe
К раствору, содержащему 24 г N, N-Boc. (t-бутилоксикарбонил-метил)-D-Cha-OH в 300 мл N, N-диметилформамида добавляли 10,2 г (N-циклопентил)-Gly-OMe и 21,2 г 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурониум-тетрафторбората (TBTU).
pH раствора доводили до 8,5. Смесь перемешивали в течение ночи при комнатной температуре и концентрировали путем выпаривания. К осадку добавляли воду и этилацетат. Отделяли органический слой и промывали его 1 N соляной кислотой, водой, 5%-ным бикарбонатом натрия и водой и сушили над сульфатом натрия. Фильтрат выпаривали, и осадок хроматографировали на силикагеле в смеси гексана и этилацетата, 8: 2, в качестве элюэнта. Фракции, содержащие целевой продукт, объединяли и выпаривали. Выход составлял 17 г.
Rf = 0,57 в гексане/этилацетате 7/3 по объему на кремнеземе.
N, N-Boc. (t-бутилоксикарбонилметил)-D-Cha-(N-циклопентил)-Gly-OH
Используя тот же прием, что и для N, N-Boc. (t-бутилоксикарбонилметил)-D-Cha-OH, омыляли 17 г N, N-Boc. (t-бутилоксикарбонилметил)-D-Cha-(N-циклопентил)-Gly-ОМе, и на выходе получали 15 г аморфного твердого вещества. Хроматография на силикагеле с дихлорметаном/метанолом, 95/5 по объему, в качестве элюэнта давала на выходе 13 г целевого продукта.
Rf = 0,30 в метилхлориде/метаноле, 9/1 по объему на кремнеземе.
HOOH-CH2-D-Cha-(N-циклопентил)-Gly-Lys-(2-тиазолил)
Это соединение получали с помощью той же процедуры, которая описана в примере 1, но с использованием N, N-Boc. (t-бутилоксикарбонилметил)-D-Cha-(N-циклопентил)-Gly-OH.
Rt(ЖХ): 23,57 мин; 20% А, 60% В, 20% С до 20% А, 80% С через 40 мин.
(а). Аналогичным описанному выше образом, используя Proline. HCl и HONSu (как в примере 11), было получено соединение HOOC-CH2-D-Cha-Pro-Lys-(2-тиазолил). Rt(ЖХ): 31,44 мин; 20% А, 80% В, 0% С до 20% А, 20% В, 60% С через 40 мин.
Пример 4.
3-Фенилпропионил-Pro-Lysψ[COCO] -ОН
Boc-Lys(Cbz)-OMe
Boc-Lys(Cbz)-OH (25 г) растворяли в дихлорметане/метаноле = 9/1 по объему (500 мл). Добавляли 21,1 г TBTU и триэтиламином доводили рН раствора до 8. Реакционную смесь перемешивали в течение часа при комнатной температуре. Затем смесь последовательно промывали охлажденным раствором 2 N соляной кислоты, водой, 5% бикарбонатом натрия и водой. Органический слой высушивали над сульфатом натрия, фильтровали и выпаривали. Осадок хроматографически очищали на кремнеземе (элюэнт: этилацетат/гептан = 1/4 по объему) и в результате получали 26,7 г Boc-Lys(Cbz)-Оме.
ТСХ: Rf = 0,79, силикагель, этилацетат/гептан = 3/1 по объему.
Boc-Lysψ[цианоацетат]
К холодному (-78oС) раствору Boc-Lys(Cbz)-OMe (20 г) в сухом дихлорметане (600 мл) добавляли по каплям диизобутилалюминиумгидрид (127 мл 1 М раствора в гексане) при такой скорости, чтобы температура реакции оставалась ниже -70oС. Полученный в результате раствор перемешивали в течение 30 минут при -78oС. 5% раствор лимонной кислоты (500 мл) добавляли к реакционной смеси. Двуфазный раствор перемешивали при комнатной температуре в течение 10 минут, слои разделяли, и водную фазу дважды экстрагировали в дихлорметане. Смесь дихлорметановых слоев промывали водой, сушили над сульфатом натрия и фильтровали. Раствор замораживали под азотом в водной ледяной бане. Добавляли водный (500 мл) раствор цианида натрия (24,85 г) и бензилтриэтилхлористого аммония (2,89 г). При интенсивном перемешивании порционно (6•6 мл) через каждые 30 мин добавляли уксусный ангидрид. Отделяли органический слой, а водный слой экстрагировали дважды дихлорметаном. Смесь дихлорметановых слоев промывали водой, сушили над сульфатом натрия, фильтровали и выпаривали под вакуумом. Осадок хроматографически очищали на кремнеземе (элюэнт: дихлорметан/этилацетат = 9/1 по объему) с выходом 17,2 г Boc-Lys(Cbz)ψ[цианоацетата] . ТСХ: Rf = 0,60, силикагель, дихлорметан/этилацетат = 7/3 по объему.
Boc-Lys(Cbz)ψ([СНОНСО] -ОМе
Раствор Boc-Lys(Cbz)ψ(цианоацетата (17,2 г) в смеси диэтилэфир/метанол = 3/1 по объему (500 мл) охлаждали до -20oС под азотом и затем, поддерживая температуру ниже -5oС, вводили 54,7 г газообразной соляной кислоты. Реакционную смесь выдерживали в течение ночи при 4oС. Поддерживая температуру ниже 5oС, к реакционной смеси по каплям добавляли воду (85 мл). После перемешивания в течение 4 часов при комнатной температуре отделяли органическую фазу и промывали ее водой. Водную фазу насыщали хлористым натрием и экстрагировали сухим бутанолом/дихлорметаном = 3/2 по объему. Органическую фазу промывали рассолом, высушивали над сульфатом натрия, фильтровали и выпаривали в вакууме с получением 17,4 г сырого амина. К осадку добавляли диметилформамид (DMF, 200 мл), bis-(трет-бутил)ангидрид (8,7 г) и триэтиламин до достижения рН 8. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали при пониженном давлении. Осадок растворяли в этилацетате, последовательно промывали водой и рассолом, высушивали над сульфатом натрия, фильтровали и сушили под вакуумом. Осадок хроматографически очищали на кремнеземе (элюэнт: этилацетат/гептан = 1/1 по объему) и получали 6,22 г продукта Boc-Lys(Cbz)ψ[СНОНСО] -Оме. ТСХ: Rf = 0,75, силикагель, этилацетат/гептан = 3/1 по объему.
3-Фенилпропионил-Pro-Lys(Cbz)ψ([СНОНСО] -ОМе
60 мг Boc-Lys (Cbz)ψ([СНОНСО] -ОМе растворяли в 50% трифторуксусной кислоте/дихлорметане (6 мл) и перемешивали в течения 1 часа при комнатной температуре. После удаления путем выпаривания растворителя получали с количественным выходом сырой амин и немедленно использавали его для получения 3-фенилпропионил-Pro-Lys(Cbz)ψ([СНОНСО] -ОМе.
3-Фенилпропионил-Pro-ОН растворяли в сухом DMF (5 мл). После добавления 196 мл триэтиламина реакционную смесь помещали под азот и охлаждали до -15oС. Затем добавляли 183 мл изобутилхлороформата, после чего в течение 15 мин смесь перемешивали при -15oС. Сырой амин растворяли в сухом DMF (5 мл), нейтрализовали триэтиламином и затем капельным способом добавляли на холоду к перемешиваемому раствору ангидрида. Реакцию проводили в течение 1 часа при -15oС, а затем реакционную смесь выдерживали при 0oС в течение ночи. Смесь затем выпаривали до сухого состояния. Осадок растворяли в этилацетате и последовательно промывали водой, 5% бикарбонатом натрия и рассолом, сушили над сульфатом натрия и концентрировали под вакуумом. Осадок очищали хроматографически на кремнеземе (элюэнт: дихлорметан/метанол = 95/5 по объему) и получали 246 мг 3-фенилпропионил-Pro-Lys(Cbz)ψ[СНОНСО] -Оме.
ТСХ: Rf = 0,92, силикагель/EPAW = 63/20/6/11 по объему.
3-Фенилпропионил-Pro-Lysψ[СНОНСО] -ОМе
К 240 мг растворенного в 5 мл метанола 3-фенилпропионил-Pro-Lys(Cbz)ψ[CHOHCO] -Оме добавляли 10% палладия-на-угле (50 мг) и 216 мл раствора 2 N соляной кислоты. Смесь гидрогенизировали при атмосферном давлении и при комнатной температуре в течение 1 часа. Палладиевый катализатор удаляли путем фильтрации, а растворитель удаляли выпариванием при пониженном давлении, с количественным выходом 3-фенилпропионил-Pro-Lysψ[СНОНСО] -Оме. ТСХ: Rf = 0,13, силикагель, дихлорметан/метанол = 9/1 по объему.
3-Фенилпропионил-Pro-Lys(Воc)ψ([СНОНСО] -ОМе
К раствору фенилпропионил-Pro-Lysψ[СНОНСО] -ОМе (196 мл) в сухом DMF (5 мл) добавляли бис(трет-бутил)ангидрид (102 мг) и рН раствора доводили триэтиламином до 8,5. Реакционную смесь размешивали при комнатной температуре в течение ночи. Далее смесь выпаривали в вакууме и полученный осадок хроматографически очищали на кремнеземе (элюэнт: дихлорметан/этанол = 98/2 по объему, выход 3-фенилпропионил-Pro-Lys(Boc)ψ([CHOHCO] -OMe составлял 189 мг. ТСХ: Rf = 0,43, силикагель, дихлорметан/метанол = 9/1 по объему.
3-Фенилпропионил-Pro-Lys(Воc)ψ([СНОНСО] -ОН
Фенилпропионил-Pro-Lys(Воc)ψ([СНОНСО] -ОМе (185 мг) растворяли в диоксане/воде = 7/3 (5 мл) и постепенно, малыми порциями добавляли раствор 2 М гидроокиси натрия (267 мл) в течение 30 минут при комнатной температуре, удерживая рН на уровне 10-10,5.
Через 30 минут реакционную смесь разбавляли водой (20 мл), 2 М раствором соляной кислоты доводили рН раствора до 2,0 и экстрагировали водную фракцию дихлорметаном. Объединенные органические фазы промывали водой (50 мл), рассолом (50 мл) и сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением 228 мг 3-фенилпропионил-Pro-Lys(Воc)ψ[СНОНСО] -ОН. ТСХ: Rf = 0,60, силикагель, EPAW = 63/20/6/11 по объему.
3-Фенилпропионил-Pro-Lys(Воc)ψ([COCO] -ОН
К раствору фенилпропионил-Pro-Lys(Воc)ψ([СНОНСО] -ОН (220 мг) в сухом дихлорметане (5 мл) добавляли 255 мг периодинана (реагент Dess-Martin). Через 1 час размешивания при комнатной температуре добавляли 15 мл 2% раствора тиосульфата натрия, после чего в течение 30 минут раствор перемешивали при комнатной температуре. Отделяли органический слой, промывали его водой, сушили на сульфате натрия, фильтровали и выпаривали в вакууме для получения сырой 3-фенилпропионил-Pro-Lys(Воc)ψ[COCO] -ОН кетокислоты (411 мг). ТСХ: Rf = 0,47, силикагель, EPAW = 63/20/6/11 по объему.
3-Фенилпропионил-Pro-Lysψ[COCO] -ОН
3-Фенилпропионил-Pro-Lys(Воc)ψ[COCO] -ОН (411 мг) обрабатывали в течение 1 часа при комнатной температуре 90%-ой трифторуксусной кислотой в воде. Реакционную смесь концентрировали в вакууме и осадок растворяли в воде и заряжали прямо на препаративной колонке Supelcosil LC-18-DB высокоэффективной жидкостной хроматографии с помощью системы градиентной элюции от 20% А/70% В/10% С до 20% А/0% В/80% С через 45 минут при скорости элюции 20 мл/мин. (А: 0,5 М фосфатный буфер, рН 2,1, В: вода. С: ацетонитрил/вода = 3/2). Выход: 71 мг 3-фенилпропионил-Pro-Lysψ[СОСО] -ОН. Rt(ЖХ): 24,9 мин; 20% А/80% В до 20% А/20% В/60% С через 40 минут.
Подобным вышеописаному способом были получены:
(а). 3,3-дифенилпропионил-Pro-Lysψ[СОСО] -ОН.
Rt (ЖХ): 36,42 мин; 20% А/80% В/0% С до 20% А/20% В/60% С через 40 минут.
(b). 3-фенилпропионил-(N-циклопентил)-Gly-Lysψ[СОСО] -ОН.
Rt(ЖХ): 34,29 мин; 20% А/80% В/0% С до 20% А/20% В/60% С через 40 минут.
(c). 3-[(р-Сl)-фенил] пропионил-(N-циклопентил)-Gly-Lysψ[СОСО] -ОН.
Rt(ЖХ): 39/52 мин; 20% А/80% В/0% С до 20% А/20% В/60% С через 40 минут.
(d) 3-[(p-Cl)-фенил] пропионил-Pro-Lysψ[COCO] -ОН.
Rt(ЖХ): 31,31 мин; 20% А/80% В/0% С до 20% А/20% В/60% С через 40 минут.
(e) Нафтилсульфонил-Asp-Pro-Lysψ[СОСО] -ОН.
Rt(ЖХ): 30,45 мин; 20% А/80% В/0% С до 20% А/20% В/60% С через 40 минут.
Пример 5.
Н-D-Cha-Pro-Lys-(2-бензотиазолил)
Вос-Lys(Cbz)-ψ[СНОН] -(2-бензотиазолил)
К холодному (-78oС) раствору Вос-Lys(Cbz)-ОМе (1 г) в дихлорметане (25 мл) добавляли по каплям диизобутилалюминиумгидрид (DiBAL -Н; 7,6 мл 1 М раствора в гексане), так, чтобы температура реакции поддерживалась ниже -70oС. Результирующий раствор перемешивали 30 минут при температуре -78oС. К реакционной смеси добавляли 5% раствор лимонной кислоты. Двуслойную смесь перемешивали при комнатной температуре в течение 10 минут, разделяли фазы, и водную фазу дважды экстрагировали в дихлорметане. Объединенные дихлорметановые слои промывали водой, 5% бикарбонатом натрия, водой, сушили над сульфатом натрия и фильтровали. Под азотам добавляли 0,79 г 2-(триметилсилил)бензотиазола, реакционную смесь перемешивали в течение 16 часов при комнатной температуре. После выпаривания досуха осадок растворяли в сухом тетрагидрофуране (15 мл) и добавляли тетрабутиламмониумфторид (3,8 мл 1 М раствора в THF). Смесь перемешивали в течение 2 часов при комнатной температуре, затем добавляли воду. Продукт экстрагировали в дихлорметане и очищали хроматографически на кремнеземе (элюэнт: дихлорметан/этилацетат; 9/1 по объему) с выходом 724 мг Вос-Lys(Cbz)-ψ[CHOH] -(2-бензотиазолил). ТСХ: Rf = 0,35, силикагель, дихлорметан/этилацетат = 7/3 по объему.
Boc-Lys(Cbz)-(2-бензотиазолил)
К раствору Boc-Lys(Cbz)-ψ[CHOH] -(2-бензотиазолил) (700 мл) в сухом дихлорметане (10 мл) добавляли 1 г периодинана (реагента Dess-Martin). После перемешивания в течение 1 часа при комнатной температуре добавляли 2% раствор тиосульфата натрия и перемешивали еще 30 минут при комнатной температуре. Органический слой отделяли, промывали водой, сушили над сульфатом натрия, фильтровали и выпаривали в вакууме. Осадок хроматографически очищали на кремнеземе (элюэнт: этилацетат/гептан = 3/1 по объему) с выходом 193 мг Вос-Lys(Cbz)-(2-бензотиазолил). ТСХ: Rf = 0,85, силикагель, этилацетат/гептан = 3/1 по объему.
Boc-D-Cha-Pro-Lys(Cbz)-(2-бензотиазолил)
Boc-Lys(Cbz)-(2-бензотиазолил) (193 мг) растворяли в 50% ТФУ/дихлорметане (2 мл) и размешивали в течение 1 часа, при комнатной температуре. Сырой амин выделяли с количественным выходом после удаления растворителя путем выпаривания и немедленно использовали для получения Boc-D-Cha-Pro-Lys(Cbz)-(2-бензотиазолила). Boc-D-Cha-Pro-OH растворяли в сухом диметилформамиде (4 мл). После добавления диизопропилэтиламина (DIPEA, 66 мл) реакционную смесь помещали под азот и охлаждали до -15oС. Затем добавляли изобутилхлорформат (50 мл) и оставляли смесь на мешалке в течение 15 минут при температуре -15oС. Сырой амин растворяли в сухом DMF (4 мл), нейтрализовали с помощью диизопропилэтиламина и по каплям добавляли к холодному перемешиваемому раствору ангидрида. Смесь перемешивали в течение 1 часа при -15oС, а затем держали в течение ночи при 0oС. Смесь выпаривали до сухого состояния. Осадок растворяли в этилацетате и последовательно промывали водой, 5% бикарбонатом натрия, водой и рассолом, сушили над сульфатом натрия и концентрировали в вакууме. Осадок хроматографически очищали на кремнеземе (элюэнт: этилацетат/гептан = 1/1 по объему) с выходом 191 мг Boc-D-Cha-Pro-Lys(Cbz)-(2-бензотиазолила). ТСХ: Rf = 0,66, силикагель, этилацетат/гептан = 3/1 по объему.
H-D-Cha-Pro-Lys-(2-бензотиазолил)
Boc-D-Cha-Pro-Lys(Cbz)-(2-бензотиазолил) обрабатывали трифторуксусной кислотой/тиоанизолом, 10/1 по объему (2,2 мл) в течение 3,5 часов при комнатной температуре. Реакционную смесь концентрировали под вакуумом и осадок растворяли в воде. Водную фазу экстенсивно промывали диэтиловым эфиром. Водный слой, содержащий H-D-Cha-Pro-Lys-(2-бензотиазолил), заряжали прямо на препаративной колонке Supelcosil LC-18-DB высокоэффективной жидкостной хроматографии с помощью системы градиентной элюции от 20% А/80% В до 20% А/20% В/60% С через 40 мин при скорости элюции 20 мл/мин. (А: 0,5 мМ натрийфосфатный буфер, рН 2,1, В: вода, С: ацетонитрил/вода, 3/2 по объему). Выход: 98 мг H-D-Cha-Pro-Lys-(2-бензотиазолила). Rt(ЖХ): 42 мин. 20% А/80% В до 20% А/20% В/60% С через 40 мин.
Пример 6.
H-D-Cha-Pro-Lys-(2-тетразолил)
Boc-Lys(Cbz)ψ(СНОАс)-(2-тетразолил)
К раствору Boc-Lys(Cbz)ψ[цианоацетата] (5,4 г) в 39 мл диметилфтормамида добавляли 801 мг хлорида аммония и 975 мг азида натрия. Реакционную смесь нагревали до 80oС и перемешивали под азотом в течение 48 часов. Преципитировавшие соли отфильтровывали, а раствор выпаривали при пониженном давлении досуха. Получали 4,9 г требуемого соединения. ТСХ: Rf= 0,42, силикагель, толуол/этанол = 6/4 по объему.
Boc-Lys(Сbz)ψ(СНООН)-(2-тетразолил)
Boc-Lys(Cbz)ψ(CHOAc)-(2-тетразолил) (1,25 г) растворяли в 60 мл смеси диоксана с водой = 7/3 и 2,65 мл 2 N раствора гидроокиси натрия. Раствор перемешивали в течение 2,5 часов при комнатной температуре, после чего рН доводили до 5, полученную смесь выпаривали досуха при пониженном давлении. Осадок растворяли в метаноле/дихлорметане, 1/1, а нерастворимую соль удаляли путем фильтрации. Это давало 1,27 г деацетилированного продукта ТСХ: Rf = 0,40, силикагель, толуол/этанол = 6/4 по объему.
Boc-Lys(Cbz)-(2-тетразолил)
К раствору, содержащему 0,56 г Boc-Lys(Cbz)ψ(СНООН)-(2-тетразолила) в 37 мл сухого дихлорметана добавляли 1,38 г периодана, реагента Dess-Martin. Смесь перемешивали при комнатной температуре в течение 30 минут, после чего реакцию останавливали с помощью 10%-го раствора тиосульфата натрия в воде. Органический слой экстрагировали водой и бикарбонатом натрия (5% в воде), водные слои объединяли и экстрагировали 1-бутанолом. Фракцию 1-бутанола выпаривали досуха при пониженном давлении. Осадок хроматографировали на силикагелевой колонке, используя элюэнт: этилацетат/пиридин/уксусная кислота/вода = 263/20/6/11 по объему. Выход: 0,22 г. ТСХ: Rf = 0,30, силикагель, этилацетат/пиридин/уксусная кислота/вода = 63/20/6/11 по объему.
H-Lys (Cbz)-(2-тетразолил)ТФУ
Boc-Lys(Cbz)-(2-тетразолил) (0,21 г) растворяли в 16 мл смеси трифторуксусной кислоты с водой, 9: 1. Смесь перемешивали в течений 1 часа при комнатной температуре, после чего раствор концентрировали в вакууме до получения масла. Выход: 0,34 г; использовали незамедлительно для получения трипептида Boc-D-Cha-Pro-Lys(Cbz)-(2-тетразолила).
Boc-D-Cha-Pro-Lys(Cbz)-(2-тетразолил)
Boc-D-Cha-Pro-OH (0,19 г) получали согласно процедуре, описанной в примере 1 для получения дипептидной части. Связывание с H-Lys(Cbz)-(2-тетразолилом) (0,17 г) было произведено наподобие того, как описано в примере 5. Очистка на силикагеле (элюэнт: этилацетат/пиридин/уксусная кислота/вода = 163/20/6/11 по объему) давала 0,21 г требуемого соединения. ТСХ: Rf = 0,17, силикагель, этилацетат/пиридин/уксусная кислота/вода = 63/20/6/11 по объему.
H-D-Cha-Pro-Lys-(2-тетразолил)
Удаление защитных групп и очистку методом высокоэффективной жидкостной хроматографии производили аналогично тому, как было описано для примера 5. Выход: 20 мг. Rt(ЖХ): 23,3 и 24,5 мин, 20% А, 80% В до 20% А, 20% В и 60% С через 40 мин.
Пример 7.
HOOC-CH2-CO-D-Cha-Pro-Lys-(2-тиазолил)
H-D-Cha-Pro-Lys(Cbz)-(2-тиазолил). ТФУ
Boc-D-Cha-Pro-Lys(Cbz)-(2-тиазолил) получали согласно процедуре, описанной в примере 5, 0,30 г этого трипептида растворяли в 3 мл трифторуксусной кислоты/дихлорметане, 1/1 по объему, и раствор перемешивали в течение 1 часа при комнатной температуре. Раствор выпаривали досуха при пониженном давлении и трижды совыпаривали с толуолом. Выход: количественный, полученное масло незамедлительно использовали для следующей стадии. ТСХ: Rf = 0,30, силикагель, этилацетат/пиридин/уксусная кислота/вода = 163/20/6/11 по объему.
(t-бутил-OOC-CH2-CO)-D-Cha-Pro-Lys(Cbz)-(2-тиазолил)
H-D-Cha-Pro-Lys(Cbz)-(2-тиазолил). ТФУ (0,33 г) растворяли в 3 мл сухого дихлорметана и добавляли 76 мг моно-третичного бутилмалоната, триэтиламином доводили рН до 8. Далее, добавляли бензотриазолилокситрис(диметиламино)-фосфониумгексафторфосфат (211 мг), и реакционную смесь перемешивали в течение 2 часов при комнатной температуре, а последующие 16 часов - при температуре 4oС. Раствор концентрировали в вакууме, растворяли в этилацетате и трижды промывали водой и рассолом. Органический слой снова концентрировали в вакууме после просушивания на сульфате магния. Осадок хроматографировали на кремнеземе с помощью гептана/этилацетата, 2/8 по объему, в качестве элюэнта. Получалось в результате 270 мг ацетилированного трипептида. ТСХ: Rf = 0,21, силикагель, гептан/этилацетат = 8/2 по объему.
HOOC-CH2-CO-D-Cha-Pro-Lys-(2-тиазолил)
Удаление защитных групп и очистку методом высокоэффективной хроматографии производили аналогично тому, как описано в примере 5. Выход: 124 мг. Rt(ЖХ): 38,23 мин, 20% А, 80% В до 20% А, 20% В и 60% С через 40 мин.
Пример 8.
НООС-(CH2)2-CO-D-Cha-Pro-Lys-(2-тиазолил)
НООС-(СН2)2-CO)-D-Cha-Pro(Cbz)-Lys-(2-тиазолил)
Н-D-Cha-Pro(Cbz)-Lys-(2-тиазолил). ТФУ (0,33 г) растворяли в 3 мл сухого дихлорметана и 0,335 мл пиридина. К этому раствору добавляли 48 мг янтарного ангидрида, и полученный раствор перемешивали при температуре под азотом. Через 4 часа реакция завершалась, и ее гасили несколькими каплями воды. Полученную смесь концентрировали под вакуумом, растворяли в этилацетате, промывали разбавленной кислотой, водой и рассолом и сушили на сульфате магния. После удаления соли органический слой концентрировали в вакууме, в результате чего получали 320 мг масла. ТСХ: Rf = 0,37, силикагель, дихлорметан/метанол = 9/1 по объему.
НООС-(СН2)2-CO-D-Cha-Pro-Lys-(2-тиазолил)
Удаление защитных групп и очистку методом высокоэффективной хроматографии производили аналогично тому, как описано в примере 5. Выход: 187 мг.
Rt(ЖХ): 38,31 мин, 20% А, 80% В до 20% А, 20% В и 60% С через 40 мин.
Пример 9.
НООС-СН(СН3)-D-Cha-Pro-Lys-(2-тиазолил)
(t-бутил-ООС-СН(СН3))-D-Cha-Pro-Lys(Cbz)-(2-тиазолил)
H-D-Cha-Pro-Lys(Cbz)-(2-тиазолил). ТФУ (0,33 г) растворяли в 2 мл ацетонитрила. Далее, добавляли 0,50 г 2-бромопропионовой кислоты трет-бутилэфира, а затем 25 мг иодида натрия. рН раствора доводили диизопропилэтиламином до 8 и поддерживали в течение 12 дней при комнатной температуре. Реакционную смесь концентрировали в вакууме, растворяли в этилацетате, промывали водой, сушили на сульфате магния и вновь концентрировали. Осадок хроматографировали на кремнеземе, используя этилацетат/толуол, 1/1 по объему, в качестве элюэнта. Выход: 279 мг. ТСХ: 0,75, силикагель, этилацетат.
НООС-СН(СН3)-D-Cha-Pro-Lys-(2-тиазолил)
Удаление защитных групп, очистка методом высокоэффективной жидкостной хроматографии были проделаны аналогично тому, как описано в примере 5. Выход: 40 мг и 29 мг (разделенные диастереомеры). Rt(ЖХ): 30,06 мин и 34,87 мин (разделенные диастереомеры), 20% А, 80% В до 20% А, 20% В и 60% С через 40 мин.
Пример 10.
НООС-(СH2)2-D-Cha-Pro-Lys-(2-тиазолил)
(t-Бутил-OОС-(СН2)2-D-Cha-Pro-Lys(Cbz)-(2-тиазолил)
H-D-Cha-Pro-Lys(Cbz)-(2-тиазолил). ТФУ (0,21 г) растворяли в 5 мл ацетонитрила. Далее, 1,84 мл сложный трет-бутиловый эфир акриловой кислоты добавляли в три этапа. рН раствора с помощью диизопропилэтиламина доводили до 8 и поддерживали на этом уровне в течение 13 дней при комнатной температуре. Реакционную смесь концентрировали в вакууме, растворяли в этилацетате, промывали водой, сушили на сульфате магния и вновь концентрировали. Осадок хроматографировали на кремнеземе с использованием этилацетата/толуола = 2/1 по объему в качестве элюэнта. Выход: 92 мг. ТСХ: Rf = 0,62, силикагель, этилацетат.
НООС-(СН2)2-D-Cha-Pro-Lys-(2-тиазолил)
Удаление защитных групп и очистка методом высокоэффективной жидкостной хроматографии были проделаны аналогично тому, как это описано в примере 5. Выход: 40 мг. Rt(ЖХ): 32,83 мин, 20% А, 80% В до 20% А, 20% В и 60% С через 40 мин.
Пример 11.
N-Me-D-norLeu-Pro-Lys-(2-тиазолил)
H-D-norLeu-OMe. HCl
К 270 мл метанола, охлажденного до -15oС добавляли 18,2 г тионилхлорида. Постепенно температуру повышали до -10oС и через 20 мин после установления такой температуры добавляли 10 г H-D-norLeu-OH. Температуру медленно повышали, а во время орошения поддерживали постоянной в течение 5 часов. Продукт кристаллизовали из метанола и диэтилового эфира при 4oС с выходом 12,9 г. ТСХ: Rf = 0,61, силикагель, n-бутанол/уксусная кислота/вода, 10/1/3 по объему.
Boc-D-norLeu-OMe
H-D-norLeu-OMe. HCl (12,9 г) растворяли в 200 мл сухого метанола, затем добавляли ди-трет-бутилбикарбонат (15,5 г) и триэтиламин (19,8 мл). Реакционную смесь перемешивали в течение 3 часов при комнатной температуре, после чего концентрировали в вакууме. Затем осадок растворяли в этилацетате и промывали водой. Продукт хроматографировали на кремнеземе с помощью смеси гептана с этилацетатом = 3/1 по объему. Выход: 16,9 г. ТСХ: Rf = 0,55, силикагель, гептан/этилацетат = 3/1 по объему.
N-Me-Boc-D-norLeu-OMe
Boc-D-norLeu-OMe (16,9 г) растворяли в 200 мл сухого диметилформамида под азотом. Затем добавляли метилиодид (24,9 мл) охлаждали до 0oС, добавляли гидрид натрия (3,31 г) и оставляли на 16 ч при комнатной температуре. Смесь концентрировали в вакууме, растворяли в этилацетате, промывали разведенной соляной кислотой (0,1 N), водой, 5% бикарбонатом натрия, опять водой, затем сушили и вновь концентрировали. Получали 18,8 г алкилированного продукта. ТСХ: Rf = 0,56, силикагель, гептан/этилацетат = 3/1 по объему.
N-Me-Boc-D-norLeu-OH
N-Me-Boc-D-norLeu-OMe (18 г) растворяли в 400 мл смеси диоксана с водой = 9/1 по объему и рН раствора доводили до 12 1 N гидроокисью натрия. Реакцию проводили в течение 2 часов, поддерживая рН постоянным, равным 12. Затем рН понижали до 2, раствор охлаждали на льду, добавляли воду (400 мл) и экстрагировали дихлорметаном. Органический слой промывали рассолом, сушили, фильтровали и концентрировали в вакууме. Это давало 18,9 г продукта, содержащего некоторое количество диоксана. ТСХ: Rf = 0,26, силикагель, дихлорметан/метанол = 9/1 по объему.
N-Me-Boc-D-norLeu-Pro-OH
Сначала сложный эфир N-сукцинимида получали из N-Me-Boc-D-norLeu-OH. 18 г этой производной растворяли в ацетонитриле (250 мл), затем добавляли EDCI (14,5 г) и N-гидроксисукцинимид (HONSu) (8,7 г), осадок растворяли в этилацетате, промывали водой и сушили. Это давало 24,3 г сырого сложного эфира ONSu. Следующий шаг - растворение пролин. НСl (20,9 г) в 300 мл диметилформамида и 300 мл воды, рН доводили до 8 с помощью 2 N гидроокиси натрия. Раствор сложного эфира ONSu (24,3 г в 300 мл диметилформамида) по каплям добавляли к этому раствору при постоянном значении рН. Реакция завершалась через 5 часов, после чего органический растворитель удаляли выпариванием при пониженном давлении. Затем добавляли воду (300 мл) и рН раствора доводили до 2. Продукт экстрагировали этилацетатом и промывали водой. После процессов сушки, фильтрации и концентрирования получали 22,2 г продукта в виде желтого масла. Сырой продукт хроматографировали на кремнеземе, используя этилацетат/метанол = 8/2 по объему в качестве элюэнта. Выход: 13,2 г. ТСХ: Rf = 0,65, силикагель, этилацетат/пиридин/уксусная кислота/вода = 163/20/6/11 по объему.
N-Me-D-norLeu-Pro-Lys-(2-тиазолил)
Связывание смешанным ангидридом N-Me-Boc-D-norLeu-Pro-OH и H-Lys(Cbz)-(2-тиазолил). ТФУ, защиту и очистку производили согласно процедурам, описываемым в примере 5. Выход: 107 мг. Rt(ЖХ): 23,22 мин, 20% А, 80% В до 20% А, 20% В и 60% С через 40 мин.
Пример 12.
N-Me-D-Cha-Pro-Lys-(2-тиазолил)
Все этапы, ведущие к получению этого трипептида, производили аналогично описанным в примере 11, и начинали с Boc-D-Cha-OH. Выход: 253 мг. Rt(ЖХ): 31,82 мин, 20% А, 80% В до 20% А, 20% В и 60% С через 40 мин.
Пример 13.
N-Ме-D-Phe-N-циклопентил-Gly-Lys-(2-тиазолил)
N-Ме-Вос-D-Phe-N-циклопентил-Gly-ОМе
N-Me-Boc-D-Phe-OH (полученный согласно примеру 11) (26 г) и N-циклопентил-Gly-ОМе (21 г, см. пример 3) растворяли в 300 мл диметилформамида. Затем добавляли 36 г TBTU и рН доводили диизопропилэтиламином (20 мл) до 8. Реакционную смесь размешивали в течение 16 часов, затем концентрировали в вакууме, растворяли в этилацетате, промывали бикарбонатом натрия (5%) и рассолом, сушили на сульфате магния и вновь концентрировали в вакууме. Выход: 24,8 г. ТСХ: Rf = 0,62, силикагель, дихлорметан/метанол = 95/5 по объему.
N-Ме-Вос-D-Phe-N-циклопентил-Gly-ОН
N-Ме-Вос-D-Phe-N-циклопентил-Gly-ОМе (17,3 г растворяли в 150 мл смеси тетрагидрофурана с водой = 135/15 по объему и добавляли 4 г гидроокиси натрия (в воде). Через 2 часа реакцию останавливали доведением рН до 2, и полученный продукт экстрагировали дихлорметаном. После промывания водой, сушки на сульфате магния, концентрировании в вакууме и кристаллизации из дихлорметана/диэтилэфира выход составлял 13,1 г. ТСХ: Rf = 0,52, силикагель, дихлорметан/метанол = 9/1 по объему.
N-Me-D-Phe-N-циклопентил-Gly-Lys-(2-тиазолил)
Последующие этапы проводили согласно процедуре, описанной в примере 11. Выход: 110 мг.
Rt(ЖX): 33,43 мин, 20% А, 80% В до 20% А, 20% В и 60% С через 40 мин.
Пример 14.
N-Me-D-Phe-Pro-Lys-(2-тиазолил)
N-Me-Boc-D-Phe-Pro-OH получали согласно описанию в примере 1. Привязывание смешанного ангидрида к H-Lys(Cbz)-(2-тиазолилу), депротекция и очистка были проделаны согласно приемам, описанным в примере 5. Выход: 148 мг.
Rt(ЖХ): 27,22 мин, 20% А, 80% В до 20% А, 20% В и 60% С через 40 мин.
Пример 15.
3,3-Дифенилпропионил-Pro-Lys(этоксикарбонил)-(2-тиазолил)
3,3-дифенилпропионил-Pro-Lys-(2-тиазолил) получали, как описано в примере 1. Раствор готовили из 20 мг этого дипептида в смеси диоксан/вода 4/1 (4 мл), и рН доводили 1 N гидроокисью натрия до 12. Затем добавляли 22 мг этилхлороформиата и 2 часа раствор перемешивали при комнатной температуре. Смесь разбавляли водой и экстрагировали дихлорметаном, промывали водой, сушили на сульфате магния, концентрировали в вакууме и, наконец, лиофилизировали из смеси трет-бутанол/вода, 1/1 по объему. Выход: 15 мг. ТСХ: Rf = 0,92, силикагель, этилацетат/пиридин/уксусная кислота/вода = 63/20/6/11 по объему.
Пример 16.
HOOC-CH2-D-Cha-Pro-Lys-(2-оксазолил)
Boc-Lys(Cbz)ψ[CHOH] -(2-оксазолил)
К раствору, содержащему 0,975 г Boc-Lys(Cbz)-OMe в 25 мл дихлорметана при -78oС в атмосфере азота добавляли 6 мл диизобутилалюминиумгидрида (1 М раствор в гексане). Через 15 мин реакция завершалась, реакционную смесь переливали в 150 мл 2%-го раствора лимонной кислоты и отфильтровывали. Органический слой отделяли, промывали водой и рассолом, сушили (на сульфате магния) и концентрировали. Осадок совыпаривали с толуолом для получения 0,92 г Boc-Lys(Cbz)-Н. Этот альдегид (0,89 г) растворяли в 1,4 мл толуена и добавляли 0,90 г 2-(триметилсилил)оксазола (полученного по методу Edwards, P. D. , Wolanin, D. J. , Andisik D. W. , and Davis W. , J. Med. Chem. 38. 76 (1995)), затем нагревали при 80oС. Через 60 часов реакционную смесь концентрировали, осадок растворяли в 5 мл тетрагидрофурана, обрабатывали 3 мл 3 М тетрабутиламмониумфторида в растворе тетрагидрофурана и перемешивали при комнатной температуре в течение 2 часов. Смесь концентрировали, растворяли в этилацетате, промывали 3% водным раствором бикарбоната натрия, сушили (на сульфате магния) и выпаривали. Очищали на хроматографической силикагелевой колонке, элюируя в градиенте этилацетата/дихлорметана = 2/1 (по объему) до этилацетата: получали масло, которое вновь хроматографировали на силикагеле, элюируя в градиенте этилацетата/гептана = 1/1 (по объему) до этилацетата/гептана = 1/3 (по объему) до получения 0,22 г названного соединения. ТСХ: Rf = 0,7, силикагель, этилацетат.
Boc-Lys(Cbz)-(2-оксазолил)
К раствору, содержащему 0,22 г Boc-Lys(Cbz)ψ[СНОН] -(2-оксазолила) в 10 мл дихлорметана, добавляли 0,22 г периодиана (реагент Dess-Martin). Через 1,5 часа перемешивания при комнатной температуре добавляли 10 мл водного раствора 5% тиосульфата натрия и перемешивали еще 15 минут при комнатной температуре. Органический слой отделяли, промывали водой, 5% водным раствором бикарбоната натрия, сушили над сульфатом магния и концентрировали. Очищали хроматографически на силикагеле, элюируя гептаном/этилацетатом 1/1 (по объему), получая 162 мг указанного продукта. ТСХ: Rf = 0,5, гептан/ацетат = 1/3 по объему.
(tBuOOCCH2)(Воc)-D-Cha-Pro-Lys(Cbz)-(2-оксазолил)
Использовали метод, описанный в примере 5. Снятие защиты производили, используя Boc-Lys(Cbz)-(2-оксазолил) (0,16 г), а соединение - с помощью (tBuOOCCH2) (Boc)-D-Cha-Pro-OH (0,19 г), получая 0,19 г (tBuOOCCH2) (Воc)-D-Cha-Pro-Lys(Cbz)-(2-оксазолила). ТСХ: Rf = 0,3, силикагель, гептан/этилацетат = 1/3 по объему.
HOOCCH2-D-Cha-Pro-Lys-(2-оксазолил)
Использовали описанную для примера 5 процедуру. 0,19 г (tBuOOCCH2) (Воc)-D-Cha-Pro-Lys(Cbz)-(2-оксазолила) давали 52 мг указанного соединения.
Rt(ЖХ): 28,46 мин, 20% А/80% В до 20% А/20% В/60% С через 40 мин.
Пример 17.
Этил-SO2-norLeu(цикло)(Gly-Lys-(2-тиазолил)
Вос-L-α-амино-ε-капролактам
К перемешиваемому раствору L-α-амино-ε-капролактама (10 г) в смеси диоксан/вода (2/1 по объему) (30 мл) добавляли 1 N раствор гидроокиси натрия (7,8 мл), а затем - ди-t-бутилкарбонат (18,8 г). Смесь перемешивали в течение 16 часов при комнатной температуре и концентрировали в вакууме. Осадок растворяли в этилацетате и промывала водой и рассолом, сушили над сульфатом натрия. Фильтровали и выпаривали под вакуумом. Сырой продукт превращали в порошок с помощью гексана, фильтровали и сушили в вакууме с получением 16 г Boc-L-α-амино-ε-капролактама. ТСХ: Rf = 0,85, этилацетат/гептан 1/1 по объему, на кремнеземе.
Boc-norLeu(цикло)Gly-OMe
Boc-L-α-амино-ε-капролактам (10 г) растворяли в дихлорметане (100 мл). При -20oС медленно добавляли 1 М раствор бис(триметилсилил)амида в THF/циклогексане (1/1 по объему) (1 эквив. ) и размешивали в течение 30 мин. Затем добавляли метилбромацетат (4 мл) и 2 часа смесь размешивали при комнатной температуре. Затем добавляли еще бис(триметилсилил)амид в THF/циклогексане (1/1 по объему) для завершения реакции. Смесь разбавляли дихлорметаном и промывали 0,1 N раствором соляной кислоты, водой, 5% раствором бикарбоната натрия и рассолом, сушили над сульфатом натрия, фильтровали и выпаривали в вакууме. Осадок очищали хроматографически на кремнеземе (элюэнт: гептан/этилацетат = 6/4 по объему, выход составлял 12 г Boc-norLeu(цикло)Gly-OMe. ТСХ: Rf = 0,55, этилацетат/гептан = 6/4 по объему на кремнеземе.
Этил SO2-norLeu(цикло)Gly-OMe
Boc-norLeu(цикло)Gly-OMe (3 г) растворяли в 50% ТФУ/дихлорметане (30 мл) и перемешивали при комнатной температура 1 час. Реакционную смесь выпаривали в вакууме. Сырой амин растворяли в дихлорметане (30 мл) и медленно при 0oС добавляли раствор этансульфонилхлорида (1,29 г) в дихлорметане (10 мл). Для поддержания рН 8 добавляли в процессе реакции триэтиламин. Смесь перемешивали в течение 1 часа при комнатной температуре, затем концентрировали в вакууме. Осадок растворяли в этилацетате и промывали 5% раствором бикарбоната натрия, водой и рассолом, сушили над сульфатом натрия, фильтровали и выпаривали под вакуумом. Осадок хроматографически очищали на кремнеземе (элюэнт: дихлорметан/этилацетат = 95/5 по объему %). Выход этил SO2-norLeu(цикло)Gly-OMe составлял 1,45 г. ТСХ: Rf= 0,30, дихлорметан/этилацетат = 9/1 по объему на кремнеземе.
Этил SO2-norLeu(цикло)Gly-OH
Этил SO2-norLeu(цикло)Gly-OMe (1,45 г) растворяли в 50 мл смеси диоксан/вода = 9/1 по объему, этот раствор обрабатывали 1 N гидроокисью натрия для достижения рН 13 и поддержания этого значения в течение 2 часов при комнатной температуре. После закисления раствор переливали в воду и экстрагировали дихлорметаном. Органический слой промывали водой и сушили на сульфате натрия. Фильтрат выпаривали и получали 600 мг указанного соединения. ТСХ: Rf = 0,45, этилацетат/пиридин/уксусная кислота/вода = 63/20/6/11 по объему на кремнеземе.
Этил SO2-norLeu(цикло)Gly-Lys(Cbz)-(2-тиазолил)
Этил SO2-norLeu(цикло)Gly-OH (482 мг) растворяли в сухом диметилформамиде (5 мл). После добавления этилдиизопропиламина (0,36 мл) реакционную смесь помещали под азот и охлаждали до -20oС. Затем добавляли изобутилхлороформат (140 мл) и размешивали в течение 15 минут при -20oС, H-Lys(Cbz)-(2-тиазолил). ТФУ растворяли в сухом диметилформамиде (3 мл) и по каплям добавляли к холодному раствору смешанного ангидрида, удерживая рН 8,5 добавлением этилдиизопропиламина. Реакционную смесь перемешивали в течение 15 мин при -20oС, затем выпаривали досуха. Осадок растворяли в этилацетате и последовательно промывали 5% водным раствором бикарбоната натрия, водой, рассолом, сушили над сульфатом натрия и концентрировали в вакууме. Осадок хроматографически очищали на кремнеземе (элюэнт: дихлорметан/метанол = 95/5 по объему) с выходом 607 мл этил SO2-norLeu(цикло)Gly-Lys(Cbz)-(2-тиазолила). ТСХ: Rf = 0,63, этилацетат/пиридин/уксусная кислота/вода = 60/3/1/2 по объему, кремнезем.
Этил SO2-norLeu(цикло)Gly-Lys-(2-тиазолил)
Этил SO2-norLeu(цикло)Gly-Lys(Cbz)-(2-тиазолил) (600 мг) обрабатывали в течение 4 часов при комнатной температуре трифторуксусной кислотой/тиоанизолом, 10/1 по объему (10 мл). Реакционную смесь концентрировали в вакууме и осадок растворяли в воде. Водную фазу экстенсивно промывали диэтиловым эфиром, затем концентрировали в вакууме, вновь совыпаривали с разведенной соляной кислотой и лиофилизировали из воды. Сырой продукт заряжали на препаративной колонке Deltapack C18 RP высокоэффективной жидкостной хроматографии, используя систему градиентной элюции 20% А/80% В до 40% А/40% В/40% С через 40 минут при скорости элюции 50 мл/мин. Выход: 233 мг этил SO2-norLeu(цикло)Gly-Lys-(2-тиазолила).
Rt(ЖХ): 26,73 мин. 20% А/80% В до 20% А/20% В/60% С через 40 минут.
Пример 18.
Бензил SO2-norLeu(цикло)Gly-Lys-(2-тиазолил)
Это соединение получали аналогично тому, как описано в примере 17.
Rt(ЖХ): 37,05 мин. 20% А/80% В до 20% А/20% В/60% С через 40 минут.
Пример 19.
7-Метокси-2-нафтилсульфонил-norLeu(цикло)Gly-Lys-(2-тиазолил)
Это соединение получали, аналогично тому, как описано в примере 17.
Rt(ЖХ): 26,40 мин. 20% А/60% В/20% С до 100% С через 40 минут.
Пример 20.
(4aR, 8aR)-пергидроизохинолин-1(R)-карбонил-Pro-Lys-(2-тиазолил)
2-Cbz-(4aR, 8aR)-пергидроизохинолин-1(R, S)-карбоновая кислота
2-Cbz-(4aR, 8aR)-пергидроизохинолин-1(R, S)-карбоновая кислота была синтезирована, как описано в ЕР-0643073, пример 1.
ТСХ: Rf= 0,85, этилацетат/пиридин/уксусная кислота/вода = 63/20/6/11 по объему на кремнеземе.
2-Cbz-(4aR, 8aR)-пергидроизохинолин-1(R, S)-карбонил-Pro-O-tBu
К холодному (0oС) 2-Cbz-(4aR, 8aR)-пергидроизохинолин-1(R, S)-карбоновой кислоты (500 мг) в диметилформамиде (5 мл) последовательно добавляли DCCI (1,3-дициклогексилкарбодиимид; 342 мг), НОВТ (1-гидроксибензотриазол-гидрат; 319 мг), Н-Pro-OtBu (270 мг) и триэтиламин (0,55 мл). Реакционную смесь перемешивали в течение 1 часа при 0oС, затем оставляли на ночь при комнатной температуре. Затем охлаждали до -20oС и путем фильтрации удаляли DCU (1,3-дициклогексилмочевина). Фильтрат концентрировали под вакуумом и осадок растворяли в этилацетате. Этот раствор последовательно промывали 5% водным раствором бикарбоната натрия, 3% водным раствором лимонной кислоты, водой и рассолом, сушили над сульфатом натрия и концентрировали в вакууме. Осадок очищали хроматографически на кремнеземе (элюэнт: гептан/этилацетат = 4/1 по объему) с выходом 2-Cbz-(4aR, 8aR)-пергидроизохинолин-1(R/S)-карбонил-Pro-O-tBu (634 мг). TCX: Rf = 0,90, этилацетат/пиридин/уксусная кислота/вода = 63/20/6/11 по объему на кремнеземе.
2-Cbz-(4aR, 8aR)-пергидроизохинолин-1(R, S)-карбонил-Pro-ОН
2-Cbz-(4aR, 8aR)-пергидроизохинолин-1(R, S)-карбонил-Pro-O-t-бутил-сложный эфир (600 мг) перемешивали в смеси с дихлорметаном (1 мл), трифторуксусной кислоты ТФУ (3 мл), анизола (0,15 мл) в течение 1 часа при комнатной температуре. Реакционную смесь концентрировали в вакууме при низкой температуре и осадок растворяли в воде при рН 9,5. Водную фазу промывали диэтиловым эфиром, потом подкисляли до рН 2,5 2 М раствором соляной кислоты. Водный слой экстрагировали этилацетатом, а органический - рассолом, высушивали над сульфатом натрия и концентрировали в вакууме с выходом 2-Cbz-(4aR, 8aR)-пергидроизохинолин-1(R, S)-карбонил-Pro-ОН (588 мг). TCX: Rf = 0,54, этилацетат/пиридин/уксусная кислота/вода = 60/3/1/2 по объему на кремнеземе.
2-Cbz-(4aR, 8aR)-пергидроизохинолин-1(R, S)-карбонил-Pro-Lys(Cbz)-(2-тиазолил)
2-Cbz-(4aR, 8aR)-пергидроизохинолин-1(R, S)-карбонил-Pro-ОН (500 мг) растворяли в сухом диметилформамиде (5 мл). После добавления этилдиизопропиламина (0,41 мл) реакционную смесь помещали под азотом и охлаждали до -20oС. Затем добавляли изобутилхлороформат (156 мл) и оставляли смесь на мешалке при -20oС. 594 мг H-Lyz(Cbz)-(2-тиазолил). ТФУ растворяли в 3 мл сухого диметилформамида и по каплям добавляли к холодному раствору смешанного ангидрида, удерживая с помощью этилдиизопропиламина рН 8,5. Реакционную смесь перемешивали в течение 15 мин при -20oС, затем досуха выпаривали. Осадок растворяли в этилацетате и последовательно промывали 5% водным раствором бикарбоната натрия, водой и рассолом, сушили над сульфатом натрия и концентрировали в вакууме. Осадок хроматографически очищали на кремнеземе (элюэнт: дихлорметан/метанол = 95/5 по объему - %) с выходом 880 мг 2-Cbz-(4aR, 8aR)-пергидроизохинолин-1(R, S)-карбонил-Pro-Lys(Cbz)-(2-тиазолила). TCX: Rf = 0,42, этилацетат/гептан = 3/1 по объему, на кремнеземе.
(4aR, 8aR)-пергидроизохинолин-1(R)-карбонил-Pro-Lys-(2-тиазолил)
(4aR, 8aR)-пергидроизохинолин-1(R, S)-карбонил-Pro-Lys-(2-тиазолил) (875 мг) обрабатывали в течение 4 часов при комнатной температуре трифторуксусной кислотой/тиоанизолом, 10/1 по объему (10 мл). Реакционную смесь концентрировали в вакууме, и осадок растворяли в воде. Водную фазу экстенсивно отмывали диэтиловым эфиром, концентрировали в вакууме, совыпаривали с разбавленной соляной кислотой и лиофилизировали из воды. Сырой продукт заряжали на препаративной колонке Deltapack C18RP высокоэффективной жидкостной хроматографии, используя систему градиентной элюции от 20% А/80% В до 20% А/53% В/27% С через 40 минут при скорости элюции 50 мл/мин. Выход: 211 мг (4aR, 8aR)-пергидроизохинолин-1(R)-карбонил-Pro-Lys-(2-тиазолила).
Rt(ЖХ): 28 мин, 20% А/80% В до 20% А/20% В/60% С через 40 минут.
Пример 21.
Этил SO2-D-Cha-Pro-Lys-(2-тиазолил)
Boc-D-Cha-Pro-OBzl (Bzl = бензил)
Boc-D-Cha-Pro-OBzl получали аналогично тому, как описано в примере 1, используя Boc-D-Cha и Pro-OBzl. ТСХ: Rf = 0,5, дихлорметан/метанол = 95/5 по объему на кремнеземе.
Этил SO2-D-Cha-Pro-OBzl
Boc-D-Cha-Pro-OBzl (3,6 г) растворяли в 50% ТФУ/дихлорметане (25 мл) и размешивали при комнатной температуре в течение 30 минут, затем выпаривали в вакууме. Сырой амин растворяли в дихлорметане (50 мл) и при -78oС добавляли этансульфонилхлорид (0,8 мл). Для поддержания рН 8 в процессе реакции добавляли триэтиламин. Смесь перемешивали в течение 3 часов при 0oС, после чего добавляли воду (25 мл). После дополнительного перемешивания в течение 30 минут при комнатной температуре реакционную смесь концентрировали в вакууме. Осадок растворяли в диэтилэфире и промывали 1 N раствором соляной кислоты, водой, 5% раствором бикарбоната натрия и рассолом, сушили на сульфате натрия, фильтровали и выпаривали в вакууме. Превращение сырого материала в порошок с помощью метанола давало на выходе 3,0 г этил SO2-D-Cha-Pro-OBzl. TCX: Rf = 0,6, дихлорметан/метанол = 95/5 по объему, на кремнеземе.
Этил SO2-D-Cha-Pro-OH
К раствору этил SO2-D-Cha-Pro-OBzl (10 г) в тетрагидрофуране (250 мл) добавляли 1 М раствор тетрабутиламмониумфторида в тетрагидрофуране (84 мл). Реакционную смесь перемешивали в течение 30 минут при комнатной температуре и выливали в воду (1 л). Водный раствор экстрагировали этилацетатом. Смешанные органические слои последовательно промывали 1 N раствором соляной кислоты и водой, сушили над сульфатом натрия и концентрировали в вакууме. Осадок очищали методом кристаллизации из этилацетата/диизопропилэфира с выходом этил SO2-D-Cha-Pro-OH (6,0 г). TCX: Rf = 0,2, этилацетат/пиридин/уксусная кислота/вода = 163/20/6/11 по объему на кремнеземе.
Этил SO2-D-Cha-Pro-Lys(Cbz)-(2-тиазолил)
Этил SO2-D-Cha-Pro-OH (397 мг) растворяли в сухом диметилформамиде (3 мл). После добавления этилдиизопропиламина (0,19 мл) реакционную смесь помещали под азот и охлаждали до -20oС. Затем добавляли изобутилхлороформат (130 мл), на мешалке, 15 минут при -20oС -H-Lys(Cbz)-(2-тиазолил). ТФУ растворяли в сухом диметилформамиде (3 мл) и по каплям добавляли в холодный раствор смешанного ангидрида, поддерживая с помощью этилдиизопропиламина рН 8,5. Реакционную смесь перемешивали 15 минут при -20oС, затем 1 час при комнатной температуре. После этого выпаривали досуха, осадок растворяли в этилацетате и последовательно промывали 5% водным раствором бикарбоната натрия, водой и рассолом, сушили над сульфатом натрия и концентрировали в вакууме. Осадок хроматографически очищали на кремнеземе (элюэнт: этилацетат/гептан = 2/1 по объему) с выходом 575 этил SO2-D-Cha-Pro-Lys(Cbz)-(2-тиазолила). ТСХ: Rf = 0,32, этилацетат/гептан = 2/1 по объему на кремнеземе.
Этил SO2-D-Cha-Pro-Lys-(2-тиазолил)
Этил SO2-D-Cha-Pro-Lys(Cbz)-(2-тиазолил) (570 мг) в течение 4 часов при комнатной температуре обрабатывали трифторуксусной кислотой/тиоанизолом, 10/1 по объему (44 мл). Реакционную смесь концентрировали в вакууме и осадок растворяли в воде. Водную фазу обильно промывали диэтиловым эфиром, затем концентрировали в вакууме, совыпаривали с разведенной соляной кислотой и лифилизировали из воды. Сырой продукт заряжали на препаративной колонке Deltapack C18RP высокоэффективной жидкостной хроматографии, используя систему градиентной элюции от 20% А/80% В до 20% А/30% В/50% С через 40 минут, при скорости элюции 80 мл/мин. Выход: 275 мг этил SO2-D-Cha-Pro-Lys-(2-тиазолила).
Rt(ЖХ): 26,06 мин, 20% А/60% В/20% С до 100% С через 40 минут.
Пример 22.
Этил SO2-D-Phe-Pro-Lys-(2-тиазолила)
Boc-D-Phe-Pro-OBzl
Это соединение получали аналогично тому, как описано в примере 1, используя Boc-D-Phe и Pro-OBzl. ТСХ: Rf = 0,9, этилацетат/пиридин/уксусная кислота/вода = 60/3/1/2 по объему, на кремнеземе.
Этил SO2-D-Phe-Pro-OH
Это соединение получали аналогично тому, как описано в примере 21, используя Boc-D-Phe-Pro-OBzl. ТСХ: Rf = 0,48, этилацетат/пиридин/уксусная кислота/вода = 163/20/6/11 по объему, на кремнеземе.
Этил SO2-D-Phe-Pro-Lys(Gbz)-(2-тиазолил)
Этил SO2-D-Phe-Pro-Lys(Gbz)-(2-тиазолил) получали аналогично способу, описанному в примере 21, используя этил SO2-D-Phe-Pro-OH и Lys(Gbz)-(2-тиазолил). ТСХ: Rf = 0,32, этилацетат/гептан = 6/2 по объему на кремнеземе.
Этил SO2-D-Phe-Pro-Lys-(2-тиазолил)
Этил SO2-D-Phe-Pro-Lys(Gbz)-(2-тиазолил) (336 мг) обрабатывали в течение 4 часов при комнатной температуре смесью трифторуксусной кислоты с тиоанизолом, 10/1 по объему (44 мл). Реакционную смесь концентрировали в вакууме, и осадок растворяли в воде. Водную фазу обильно промывали диэтиловым эфиром, затем концентрировали в вакууме, совыпаривали с разбавленной соляной кислотой и лиофилизировали из воды. Сырой продукт заряжали на препаративной колонке Deltapack C18RP высокоэффекивной жидкостной хроматографии, используя систему градиентной элюции от 20% А/65% В/15% С до 20% А/20% В/60% С через 40 минут при скорости элюции 50 мл/мин. Выход: 160 мг этил SO2-D-Phe-Pro-Lys-(2-тиазолила). Rt(ЖХ): 39,47 мин, 20% А/80% В до 20% А/20% В/60% С через 40 минут.
Пример 23.
D-Hpl-Pro-Lys-(2-тиазолил) (Hpl = 3-гексагидрофенилмолочная кислота)
H-D-HPl-ОМе
H-D-Cha-OH (1,0 г) растворяли в смеси 1 N соляной кислоты (4,8 мл), воды (19,4 мл) и уксусной кислоты (9,7 мл). При 0oС медленно добавляли раствор нитрита натрия (3,4 г) в воде (5,8 мл) и оставляли на мешалке в течение ночи при комнатной температуре. Затем добавляли 4,8 мл 37%-ной соляной кислоты, размешивая в течение 15 минут при комнатной температуре. Реакционную смесь выпаривали, а осадок растворяли в эфире/ацетоне. После фильтрации раствор концентрировали в вакууме, и сырой продукт перемешивали в метаноле (25 мл) в течение 18 часов, рН 1,5. Реакционную смесь выпаривали досуха, осадок хроматографически очищали на кремнеземе (элюэнт: толуол/метанол = 97/3 по объему), выход - 612 мг Н-D-Hpl-OMe. TCX: Rf = 0,9, этилацетат/пиридин/уксусная кислота/вода 163/20/6/11 по объему на кремнеземе.
THP-D-HPl-ОМе (ТНР = тетрагидропиран)
К перемешиваемому раствору H-D-Hpl-OMe (450 мг) в дихлорметане (2 мл) последовательно добавляли 3,4-дигидро-2Н-пиран (0,285 мл) пара-толуолсульфонат пиридиния (60 мг). Смесь перемешивали в течение 6 часов при комнатной температуре и разбавляли эфиром. Эту смесь промывали рассолом, сушили над сульфатом натрия, фильтровали и выпаривали в вакууме. Сырой продукт хроматографически очищали на кремнеземе (элюэнт: этилацетат/гептан = 1/4 по объему) с выходом THP-D-Hpl-OMe (498 мг). TCX: Rf = 0,64, этилацетат/гептан = 1/2 по объему на кремнеземе.
THP-D-Hpl-OH
Раствор THP-D-Hpl-OMe (10,3 г) в смеси диоксан/вода, 9/1 (200 мл), обрабатывали в течение 18 часов при комнатной температуре 1 N NaOH для установления рН 12. После подкисления смесь вливали в воду (500 мл) и экстрагировали дихлорметаном. Органическую фазу промывали водой и сушили на сульфате натрия. Фильтрат выпаривали, и получали 6,6 г целевого продукта. TCX: Rf = 0,76, этилацетат/пиридин/уксусная кислота/вода = 163/20/6/11 по объему на кремнеземе.
THP-D-Hpl-Pro-OH
К раствору THP-D-Hpl-OH (5,87 г) в ацетонитриле (75 мл) последовательно добавляли EDCI (1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид) (4,84 г) и N-гидроксисукцинимид (2,9 г). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, затем концентрировали, в вакууме. Осадок растворяли в этилацетате. Этот раствор промывали водой и рассолом, сушили над сульфатам натрия и концентрировали в вакууме. Сырой материал растворяли в диметилформамиде (100 мл) и добавляли к раствору пролин. НСl, (6,99 г) в смеси диметилформамида с водой, 1/1 по объему, (200 мл), рН которого с помощью NaOH устанавливали 8,5. После перемешивания в течение ночи реакционную смесь концентрировали в вакууме и осадок растворяли в воде. рН этого водного раствора доводили до 2,5 при 0oС, затем экстрагировали этилацетатом. Объединенные органические фазы последовательно промывали водой и рассолом, сушили над сульфатом натрия и концентрировали в вакууме. Сырой материал очищали хроматографически на кремнеземе (элюэнт: этилацетат/метанол, 8/2 6/4, по объему - %) с выходом 6,75 г THP-D-Hpl-Pro-OH. ТСХ: Rf = 0,52, этилацетат/пиридин/уксусная кислота/вода = 163/20/6/11 по объему на кремнеземе.
THP-D-Hpl-Pro-Lys(Cbz)-(2-тиазолил)
THP-D-Hpl-Pro-OH (390 мг) растворяли в сухом диметилформамиде (5 мл). После добавления этилдиизопропиламина (0,19 мл) реакционную смесь помещали под азот и охлаждали до -20oС. Затем добавляли изобутилхлорформат (130 мл), перемешивали 15 минут при -20oС. HP-Lys(Cbz)-(2-тиазолил). ТФУ (1,05 экв. ) растворяли в сухом диметилформамиде (5 мл) и по каплям добавляли к холодному раствору смешанного ангидрида, с помощью этилдиизопропиламина устанавливали рН 8,5. Реакционную смесь перемешивали 15 минут при -20oС и затем 2,5 часа при комнатной температуре. Затем упаривали досуха. Осадок растворяли в этилацетате и последовательно промывали в 5% растворе бикарбоната натрия в воде, водой и рассолом, сушили над сульфатом натрия и концентрировали в вакууме. Осадок хроматографически очищали на кремнеземе (элюэнт: этилацетат/гептан, 2/1 по объему), выход составил 497 мг THP-D-Hpl-Pro-Lys(Cbz)-(2-тиазолила). ТСХ: Rf = 0,42 в этилацетате/гептане = 2/1 по объему на кремнеземе.
D-Hpl-Pro-Lys-(2-тиазолил)
THP-D-Hpl-Pro-Lys(Cbz)-(2-тиазолил)(470 мг) в течение 4 часов при комнатной температуре обрабатывали трифторуксусной кислотой, 10/1 по объему (38,5 мл). Реакционную смесь концентрировали в вакууме и осадок растворяли в воде. Водную фазу обильно промывали диэтиловым эфиром. Водную фазу концентрировали в вакууме, совыпаривали с разбавленной соляной кислотой и лиофилизировали из воды. Сырой продукт заряжали на препаративной колонке Deltapack C18RP высокоэффективной жидкостной хроматографии, используя систему градиентной элюции от 20% А/65% В/15% С до 20% А/20% В/60% С через 40 минут при скорости элюции 50 мл/мин. Выход: 75 мг D-Hpl-Pro-Lys-(2-тиазолила).
Rt(ЖХ): 40,00 мин, 20% А/80% В до 20% А/20% В/60% С через 40 минут.
Пример 24.
HOOC-CH2-D-Phe-Pro-Lys-(2-тиазолил)
Н-D-Phe-ОМе-НСl
К холодному (-20oС) и сухому метанолу (1 л) по каплям добавляли тионилхлорид (130 мл). Затем добавляли 147,6 г H-D-Phe-OH. HCl, и реакционную смесь нагревали с обратным холодильником до комнатной температуры в течение 30 минут и оставляли на ночь. Затем смесь концентрировали в вакууме и совыпаривали с метанолом (3 раза). Осадок кристаллизовали из метанола/диэтилового эфира с выходом H-D-Phe-ОМе. НСl в виде белого кристаллического порошка (187,4 г). ТСХ: Rf = 0,54, силикагель, n-бутанол/уксусная кислота/вода = 10/1/3 по объему.
N-(t-бутилоксикарбонилметил)-D-Phe-OMe
t-Бутил-бромацетат (65 мл) добавляли на мешалке к раствору H-D-Phe-OMe. HCl (65,2 г) в 400 мл ацетонитрила. рН смеси доводили до 8,5 N, N-диизопропилэтиламином. Смесь перемешивали в течение 16 часов при комнатной температуре и выпаривали в вакууме. Осадок растворяли в дихлорметане, и раствор промывали водой, сушили на сульфате натрия и выпаривали в вакууме. Хроматографировали на силикагеле в гептане/этилацетате, 9/1 по объему; получали 96,4 г N-(t-бутилоксикарбонилметил)-D-Phe-OMe. ТСХ: Rf = 0,90, силикагель, этилацетат/пиридин/уксусная кислота/вода = 376/31/18/7 по объему.
N-(t-бутилоксикарбонилметил)-N-Boc-D-Phe-OMe
рН раствора N-(t-бутилоксикарбонилметил)-D-Phe-OMe (96,4 г) и ди-t-бутилбикарбоната (72,2 г) в N, N-диметилформамиде (400 мл) доводили до 8,5 N, N-диизопропилэтиламином. Смесь перемешивали в течение 48 часов при комнатной температуре. Под вакуумом удаляли растворитель. К осадку добавляли дихлорметан и воду. Органическую фракцию отделяли, промывали холодной 1 N соляной кислотой, водой, насыщенным раствором бикарбоната натрия и снова водой. Органическую фракцию сушили над сульфатом натрия и фильтрат выпаривали. Осадок хроматографировали на кремнеземе в толуоле/этилацетате, 9/1 по объему, в качестве элюэнта. Фракции, содержащие N-(t-бутилоксикарбонилметил)-N-Boc-D-Phe-OMe, объединяли и выпаривали. Выход: 115,3 г. ТСХ: Rf = 0,77, силикагель, толуол/этилацетат, 9/1 по объему.
N-(t-бутилоксикарбонилметил)-N-Boc-D-Phe-OH
Раствор N-(t-бутилоксикарбонилметил)-N-Boc-D-Phe-OMe (115,3 г) в 800 мл диоксана с водой = 9/1 (по объему) обрабатывали 2 N гидроокисью натрия для установления и поддержания рН 12 в течение 16 часов при комнатной температуре. После закисления смесь вливали в воду и экстрагировали дихлорметаном. Органическую фракцию промывали водой и сушили над сульфатом натрия. Фильтрат выпаривали и получали 104 г N-(t-бутилоксикарбонилметил)-N-Boc-D-Phe-OH. ТСХ: Rf = 0,10, силикагель, толуол/метилацетат, 7/3 по объему.
N-(t-бутилоксикарбонилметил)-N-Boc-D-Phe-Pro-OBzl
К холодному (0oС) раствору N-(t-бутилоксикарбонилметил)-N-Boc-D-Phe-OH (5,3 г) в N, N-диметилформамиде (40 мл) последовательно добавляли 1-гидроксибензотриазол (2,8 г), дициклогексилкарбодиимид (3,2 г), H-Pro-OBzl. HC1 (3,78 г) и триэтиламин (2,16 мл). Смесь перемешивали в течение 1 часа при 0oС, а затем - в течение ночи при комнатной температуре. Смесь охлаждали до -20oС и путем фильтрации освобождались от дициклогексилмочевины. Фильтрат упаривали досуха. Осадок растворяли в этилацетате и последовательно промывали 5% бикарбонатом натрия, водой, 2% лимонной кислотой и рассолом, высушивали над сульфатом натрия и концентрировали в вакууме. Осадок хроматографировали на силикагеле в гептане/этилацетате, 6/4 (по объему) в качестве элюэнта. Фракции, содержащие N-(t-бутилоксикарбонилметил)-N-Boc-D-Phe-Pro-OBzl объединяли и выпаривали. Выход: 4,35 г. ТСХ: Rf = 0,74, силикагель, гептан/этилацетат, 1/1 по объему.
N-(t-бутилоксикарбонилметил)-N-Boc-D-Phe-Pro-OH
К раствору N-(t-бутилоксикарбонилметил)-N-Boc-D-Phe-Pro-OBzl (4,35 г) в метаноле (50 мл) добавляли 10% палладий-на-угле (450 мг). Смесь гидрогенизировали при атмосферном давлении и при комнатной температуре в течение 45 минут. Палладиевый катализатор удаляли путем фильтрации, а растворитель - выпариванием при пониженном давлении, с выходом 3,48 г N-(t-бутилоксикарбонилметил)-N-Boc-D-Phe-Pro-ОН. ТСХ: Rf = 0,63, силикагель, этилацетат/пиридин/уксусная кислота/ вода = 664/31/18/7 по объему.
N-(t-бутилоксикарбонилметил)-N-Boc-D-Phe-Pro-Lys(Cbz)-(2-тиазолил)
К охлажденному (-20oС) раствору, содержащему 375 N-(t-бутилоксикарбонилметил)-N-Boc-D-Phe-Pro-OH и 276 мл N, N-диизопропилэтиламина в 10 мл N, N-диметилформамида, добавляли 100 мл изобутилхлороформата. Реакционную смесь перемешивали последующие 20 минут при -20oС. 362 мг H-Lys(Cbz)-(2-тиазолил). ТФУ растворяли в 5 N, N-диметилформамида и N, N-диизопропилэтиламином рН доводили до 8. Этот раствор медленно добавляли к реакционной смеси, которую перемешивали 15 минут при -20oС, а затем оставляли нагреваться до комнатной температуры. Затем ее выпаривали досуха, а осадок растворяли в этилацетате. Органическую фазу промывали 5% бикарбонатом натрия, водой и рассолом, сушили над сульфатом натрия и концентрировали до получения 622 мг сырого продукта. Очистка на силикагеле с использованием в качестве элюэнта дихлорметана/метанола, 97/3 по объему, давала выход в 394 мг N-(t-бутилоксикарбонилметил)-N-Boc-D-Phe-Pro-Lys(Cbz)-(2-тиазолила). ТСХ: Rf = 0,50, силикагель, дихлорметан/метанол = 95/5 по объему.
HOOC-CH2-D-Phe-Pro-Lys-(2-тиазолил)
Защищенный трипептид (394 мг) обрабатывали трифторуксусной кислотой и тиоанизолом согласно способу, описанному в примере 1, с получением после очистки методом высокоэффективной жидкостной хроматографии 206 мг HOOC-CH2-D-Phe-Pro-Lys-(2-тиазолила).
Rt(ЖХ): 27,9 мин, 20% А, 80% В до 20% А, 20% В и 60% С через 40 минут.
Пример 25.
НООС-СН2-D-р-ОСН3-Phe-Pro-Lys-(2-тиазолил)
НООС-СН2-D-р-ОСН3-Phe-Pro-Lys-(2-тиазолил) получали способом, аналогичным описанному в примере 24, начиная с H-D-р-ОСН3-Phe-OH. HCl. Снятие защиты (см. пример 1) с 345 мг N-(t-бутилоксикарбонилметил)-N-Boc-D-p-OCH3-Phe-Pro-Lys(Cbz)-(2-тиазолила) давало после очистки методом высокоэффективной жидкостной хроматографии 153 мг вышеуказанного продукта.
Rt(ЖХ): 28,9 мин, 20% А, 80% В до 20% А, 20% В и 60% С через 40 минут.
Пример 26.
HOOC-CH2-D/L-m-F-Phe-Pro-Lys-(2-тиазолил)
N-(t-бутилоксикарбонилметил)-N-Boc-D/L-m-F-Phe-OH
Аналогично способу, описанному в примере 24, H-D/L-m-F-Phe-OH. HCl (5 г) превращали в N-(t-бутилоксикарбонилметил)-N-Boc-D/L-m-F-Phe-OH. Выход: 8 г. TCX: Rf = 0,65, силикагель, этилацетат/метанол = 9/1 по объему.
N-(t-бутилоксикарбонилметил)-N-Boc-D/L-m-F-Phe-Pro-OMe
К холодному (0oС) раствору N-(t-бутилоксикарбонилметил)-N-Boc-D/L-m-F-Phe-OH (7,9 г) в диметилформамиде (80 мл) последовательно добавляли 1-гидроксибензотриазол (4,0 г), дициклогексилкарбодиимид (4,5 г), H-Pro-OMe. HCl (3,6 г) и триэтиламин (3,25 мл). Смесь перемешивали при 0oС в течение 1 часа, затем выдерживали в течение ночи при комнатной температуре. Затем смесь охлаждали до -20oС и путем фильтрации удаляли дициклогексилмочевину. Фильтрат выпаривали досуха, осадок растворяли в этилацетате и последовательно промывали 5% бикарбонатом натрия, водой, 2% лимонной кислотой и рассолом, сушили над сульфатом натрия и концентрировали в вакууме. Осадок очищали на силикагеле в гептане/этилацетате, 7/3 по объему, до получения 6,9 г целевого продукта. TCX: Rf = 0,65 гептан/этилацетат = 1/1 по объему.
N-(t-бутилоксикарбонилметил)-N-Boc-D/L-m-F-Phe-Pro-OH
6,9 г N-(t-бутилоксикарбонилметил)-N-Boc-D/L-m-F-Phe-Pro-OMe, растворенного в смеси диоксан/вода = 9/1 по объему, (60 мл), постепенно обрабатывали в течение 16 часов раствором 1 N гидроокиси натрия (13,8 мл) для достижения и поддержания рН на уровне 10-10,5. Реакционную смесь затем разбавляли ледяной водой и закисляли раствором 2 N соляной кислоты до рН 2. Водную фракцию экстрагировали дихлорметаном. Органическую фазу промывали холодной водой, сушили над сульфатом натрия и концентрировали до получения 14,7 г сырого продукта. Очистка на силикагеле с использованием этилацетата/метанола, 9/1 по объему, давала на выходе 5,22 г. ТСХ: Rf = 0,20, силикагель, этилацетат/метанол = 8/2 по объему.
N-(t-бутилоксикарбонилметил)-N-Boc-D/L-m-F-Phe-Pro-Lys(Cbz)-(2-тиазолил)
Связывание N-(t-бутилоксикарбонилметил)-N-Boc-D/L-m-F-Phe-OH (601,3 мг) с H-Lys(Cbz)-(2-тиазолилом) производили в тех же условиях, какие описаны в примере 24. Выход: 684,3 г. ТСХ: Rf = 0,74, силикагель, дихлорметан/метанол = 95/5 по объему.
HOOC-CH2-D/L-m-F-Phe-Pro-Lys-(2-тиазолил)
N-(t-бутилоксикарбонилметил)-N-Boc-D/L-m-F-Phe-Pro-Lys(Cbz)-(2-тиазолил) (673,5 мг) обрабатывали трифторуксусной кислотой и тиоанизолом в тех же условиях, что и в примере 1, и получали 259 мг чистого продукта после очистки методом высокоэффективной жидкостной хроматографии.
Rt(ЖХ): 28,4 мин и 29,0 мин, 20% А, 80% В до 20% А, 20% В и 60% С через 40 минут.
Пример 27
HOOC-CH2-D-p-CF3-Phe-Pro-Lys-(2-тиазолил)
N-(t-бутилоксикарбонилметил)-N-Boc-D/L-p-CF3-Phe-OH
Аналогично тому, как описано в примере 24, Н-D/L-р-CF3-Phe-OH. HCl (10/12 г) превращали в N-(t-бутилоксикарбонилметил)-N-Boc-D/L-p-CF3-Phe-OH. Выход: 12,23 г. ТСХ: Rf = 0,64, силикагель, этилацетат/метанол = 9/1 по объему.
N-(t-бутилоксикарбонилметил)-N-Boc-D-p-CF3-Phe-Pro-OBzl
Способом, описанным в примере 24, соединяли 6,10 г N-(t-бутилоксикарбонилметил)-N-Boc-D/L-p-CF3-Phe-OH с Н-Pro-OBzl. HCl. После соединения диастереоизомеры можно было разделить на силикагеле с использованием гептана/этилацетата, 75/25 по объему, для получения 0,63 г чистого N-(t-бутилоксикарбонилметил)-N-Вос-D-р-CF3-Phe-Pro-ОВzl. ТСХ: Rf = 0,35, силикагель, гептан/этилацетат = 7/3 по объему.
N-(t-бутилоксикарбонилметил)-N-Boc-D-p-CF3-Phe-Pro-Lys(Cbz)-(2-тиазолил)
N-(t-бутилоксикарбонилметил)-N-Boc-D-p-CF3-Phe-Pro-OBzl (630 мг) восстанавливали и затем связывали с H-Lys(Cbz)-(2-тиазолилом), используя способ, описанный в примере 24. Выход: 317,7 мг. ТСХ: Rf = 0,46, дихлорметан/метанол = 95/5 по объему.
HOOC-CH2-D-p-CF3-Phe-Pro-Lys-(2-тиазолил)
Удаление защищенных групп из N-(t-бутилоксикарбонилметил)-N-Boc-D-p-CF3-Phe-Pro-Lys(Cbz)-(2-тиазолил) (306,5 мг) производили по способу, описанному в примере 24. После очистки методом высокоэффективной жидкостной хроматографии было получено 157 мг продукта.
Rt(ЖХ): 36,7 мин, 20% А, 80% В до 20% А, 20% В и 60% С через 40 минут.
Пример 28.
HOOC-CH2-D-p-Cl-Phe-Pro-Lys-(2-тиазолил)
N-(t-бутилоксикарбонилметил)-N-Boc-D-p-Cl-Phe-OH
Аналогично способу, описанному в примере 24, H-D-p-Cl-Phe-OH. HCl (10 г) превратили N-(t-бутилоксикарбонилметил)-N-Boc-D-p-Cl-Phe-OH. Выход: 16,7 г. TCX: Rf = 0,27, силикагель, этилацетат/метанол = 9/1 по объему.
N-(t-бутилоксикарбонилметил)-N-Boc-D-p-Cl-Phe-ONSu
Раствор N-(t-бутилоксикарбонилметил)-N-Boc-D-p-Cl-Phe-OH (14,67 г) в 250 мл ацетонитрила обрабатывали в течение ночи при комнатной температуре N-гидроксисукцинимидом (4,11 г) и t-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлоридом (6,86 г). Реакционную смесь упаривали досуха и осадок растворяли в этилацетате. Органическую фазу промывали водой, сушили над сульфатом натрия и концентрировали с получением 19,11 г активного сложного эфира, который тут же использовали для следующей стадии.
N-(t-бутилоксикарбонилметил)-N-Boc-D-p-Cl-Phe-Pro-OH
H-Pro-OH. HCl (10,79 г) растворяли в 100 мл N, N-диметилформамида и 100 мл воды. рН реакционной смеси доводили до 8 с помощью 1 N раствора гидроокиси натрия, после чего в реакционную смесь по каплям добавляли N-(t-бутилоксикарбонилметил)-N-Boc-D-p-Cl-Phe-ONSu (19,11 г), растворенный в 120 мл N, N-диметилформамида. Реакцию проводили на мешалке при комнатной температуре в течение ночи при рН 8. Затем реакционную смесь охлаждали и подкисляли с помощью 1 N соляной кислоты до рН 2. Водную фракцию экстрагировали дихлорметаном. Органическую фракцию промывали водой, сушили над сульфатом натрия и выпаривали в вакууме. Затем очищали на силикагеле, используя градиент этилацетата/метанола = 9/1. ТСХ: Rf = 0,24, силикагель, этилацетат/метанол = 8/2 по объему.
N-(t-бутилоксикарбонилметил)-N-Boc-D-p-Cl-Phe-Pro-Lys(Cbz)-(2-тиазолил)
По способу, описанному в примере 24, N-(t-бутилоксикарбонилметил)-N-Boc-D-p-Cl-Phe-OH (369,4 мг) превращали в целевое соединение. Выход: 249,1 мг. ТСХ: Rf = 0,25, силикагель, дихлорметан/метанол = 97/3 по объему.
HOOC-CH2-D-p-Cl-Phe-Pro-Lys-(2-тиазолил)
Как описано в примере 1, 231,5 мг N-(t-бутилоксикарбонилметил)-N-Boc-D-p-Cl-Phe-Pro-Lys(Cbz)-(2-тиазолила) освобождали от защиты и очищали с получением 109,8 мг продукта.
Rt(ЖХ) = 33,8 мин, 20% А, 80% В до 20% А, 20% В и 60% С через 40 минут.
Пример 29.
HOOC-CH2-D-o-Cl-Phe-Pro-Lys-(2-тиазолил)
HOOC-CH2-D-o-Cl-Phe-Pro-Lys-(2-тиазолил) получали так, как описано в примере 26, начиная с H-D/L-o-Cl-Phe-OH. HCl. На защищенной трипептидной стадии были выделены два диастереоизомера. Снятие защиты с 230 мг N-(t-бутилкарбонилметил)-N-Boc-D-o-Cl-Phe-Pro-Lys(Cbz)-(2-тиазолила) по способу, описанному в примере 1, давало на выходе 116 мг продукта после проведения очистки методом высокоэффективной жидкостной хроматографии.
Rt(ЖХ): 30,0 мин, 20% А, 80% В до 20% А, 20% В и 60% С через 40 минут.
Пример 30.
HOOC-CH2-D/L-m, p-di-F-Phe-Pro-Lys-(2-тиазолил)
Это соединение получали по способу, сходному с описанным в примере 26, начиная с H-D/L-m, p-di-F-Phe-OH. HCl. Удаление блокирующих групп защищенных трипептидов (720 мг), а затем очистка методом высокоэффективной жидкостной хроматографии, как описано в примере 1, давали 170 мг целевого продукта.
Rt(ЖХ): 30,7 мин. и 31,1 мин, 20% А, 80% В до 20% А, 20% В и 60% С через 40 минут.
Пример 31.
HOOC-CH2-D/L-o, p-di-Cl-Phe-Pro-Lys-(2-тиазолил)
Это соединение получали способом, сходным с тем, который описан в примере 26, начиная с H-D/L-o, p-di-Cl-Phe-OH. HCl. Удаление блокирующих групп защищенного трипептида (1,07 г), за которым следовала очистка методом высокоэффективной жидкостной хроматографии, как описано в примере 1, на выходе давало 100 мг продукта.
Rt(ЖХ) = 35,4 мин и 36,1 мин, 20% А, 80% В до 20% А, 20% В и 60% С через 40 минут.
Пример 32.
HOOC-CH2-D-Tyr-Pro-Lys-(2-тиазолил)
Cbz-D-Tyr(tBu)-ОН
N-бензилоксикарбонилоксисукцинимид (5,75 г) добавляли к суспензии D-Tyr(tBu)-ОН (5,0 г) в N, N-диметилформамиде (40 мл). рН раствора доводили триэтиламином до 8. Реакционную смесь перемешивали при комнатной температуре в течение ночи, а затем упаривали в вакууме досуха. Осадок растворяли в дихлорметане и разбавляли ледяной водой. рН водной фракции доводили до 2,5 2 N соляной кислотой. Органическую фракцию отделяли, а водную фазу экстрагировали в дихлорметане. Органические слои объединяли и промывали водой, сушили над сульфатом натрия и концентрировали. Выход: 9,95 г. ТСХ: Rf = 0,31, гептан/этилацетат = 1/1.
Cbz-D-Tyr(tBu)-OМe
[2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурониум-тетрафторборат] (7,45 г) добавляли к раствору Cbz-D-Tyr(tBu)-ОН (9,95 г) в дихлорметане (45) и метаноле (5 мл). рН раствора доводили до 8 N, N-диизопропилэтиламином. Реакционную смесь перемешивали в течение 1 часа при комнатной температуре, а затем гасили 5% бикарбонатом натрия. Органическую фазу отделяли и промывали водой, 2% лимонной кислотой и рассолом, сушили над сульфатом натрия и концентрировали при пониженном давлении. Выход: 10,2 г. ТСХ: Rf = 0,74, гептан/этилацетат = 1/1.
H-D-Tyr(tBu)-OMe. HCl
10% палладиум-на-угле (1,2 г) добавляли к Cbz-D-Tyr(tBu)-OMe (10,2 г) в метаноле (100 мл) и 4 N соляной кислоте (5 мл). Смесь гидрогенизировали при атмосферном давлении и при комнатной температуре в течение 2 часов. Палладиевый катализатор удаляли путем фильтрации. Фильтрат концентрировали до малого объема, затем кристаллизовали из диэтилового эфира. Выход: 5,87 г. ТСХ: Rf = 0,10, гептан/этилацетат = 1/1.
HOOC-CH2-D-Tyr-Pro-Lys-(2-тиазолил)
Это соединение получали методом, аналогичным описанному в примере 24, начиная с H-D-Tyr(tBu)-OMe. HCl. В результате снятия защиты, из 586 мг N-(t-бутилоксикарбонилметил)-N-Boc-D-Tyr(tBu)-Pro-Lys(Cbz)-(2-тиазолила) согласно способу, описанному в примере 1, получали 283 мг продукта после очистки методом высокоэффективной жидкостной хроматографии.
Rt(ЖХ) = 20,9 мин, 20% А, 80% В до 20% А, 20% В и 60% С через 40 минут.
Пример 33.
HOOC-CH2-D/L-p-CH3-Phe-Pro-Lys-(2-тиазолил)
Н-D/L-р-СН3-Phe-ОН. НСl
Суспензию гидрида натрия (3,28 г, 60% дисперсия в минеральном масле) в этаноле (40 мл) добавляли к раствору хлор-пара-ксилола (10 г), диэтилацетамидомалоната (19,3 г) и йодида натрия (8,55 г) в диоксане (80 мл) и этаноле (20 мл). Реакционную смесь термостатировали при 80oС в течение 90 минут. В условиях пониженного давления удаляли растворитель, а осадок растворяли в этилацетате. Органическую фазу промывали 5% бисульфатом натрия, водой, 5% бикарбонатом натрия и рассолом, сушили над сульфатом натрия и концентрировали в вакууме. Продукт кристаллизовали из гептана с получением 19,8 г конденсированного продукта. После обработки 6 N соляной кислотой (420 мл) и уксусной кислотой (210 мл) в течение ночи при 95oС получали после процедуры выпаривания досуха 21,6 г продукта. ТСХ: Rf = 0,15, силикагель, этилацетат/пиридин/уксусная кислота/вода = 664/31/18/7 по объему.
НООС-СН2-D/L-р-СН3-Phe-Pro-Lys-(2-тиазолил)
Согласно методу, описанному в примере 24, HOOC-CH2-D/L-р-СН3-Phe-Pro-Lys-(2-тиазолил) получали, начиная с H-D/L-p-СН3-Phe-OH. HCl. Удаление блокирующих групп из защищенного трипептида (582 мг) и очистка методом высокоэффективной жидкостной хроматографии проводили в условиях, описанных в примере 1. Выход: 120 мг.
Rt(ЖХ) = 31,9 мин, 20% А, 80% В до 20% А, 20% В и 60% С через 40 минут.
Пример 34.
HOOC-CH2-D-m-Cl-Phe-Pro-Lys-(2-тиазолил)
Начиная с 3-хлоробензилбромида с H-D/L-m-Cl-Phe-OH. HCl получали, как описано в примере 33. Далее, полностью защищенный трипептид был получен согласно той же методике, что и в примере 26. На конечной стадии 1 г N-(t-бутилоксикарбонилметил)-N-Boc-D/L-m-Cl-Phe-Pro-Lys(Cbz)-(2-тиазолил) обрабатывали трифторуксусной кислотой и тиоанизолом (см. пример 1). После очистки методом высокоэффективной жидкостной хроматографии получали 195 мг HOOC-CH2-D-m-Cl-Phe-Pro-Lys-(2-тиазолила).
Rt(ЖХ) = 31,7 мин, 20% А, 80% В до 20% А, 20% В и 60% С через 40 минут.
Пример 35.
HOOC-CH2-D-DPA-Pro-Lys-(2-тиазолил) (DPA = дифениламин)
Это соединение получали аналогично тому, как описано в примере 24, начиная с H-D-DPA-OH. HCl. При снятии защиты согласно методам, описанным в примере 1, из 570 мг N-(t-бутилоксикарбонилметил)-N-Boc-D-DPA-Pro-Lys(Cbz)-(2-тиазолила) после очистки методом высокоэффективной жидкостной хроматографии получается 194 мг конечного продукта.
Rt(ЖX) = 35,6 мин, 20% А, 80% В до 20% А, 20% В и 60% С через 40 минут.
Пример 36.
HOOC-CH2-D-m-OH-Phe-Pro-Lys-(2-тиазолил)
Это соединение получали аналогично тому, как описано в примере 24, начиная с H-D/L-m-OH-Phe-OH. HCl. Фенольная гидроксильная функция также была защищена группой Воc в процессе введения ее на N-конец. Снятие защиты (см. пример 1) с 1,21 г N-(t-бутилоксикарбонилметил)-N-Boc-D/L-m-OBoc-Phe-Pro-Lys(Cbz)-(2-тиазолила) после очистки методом высокоэффективной жидкостной хроматографии давало на выходе 99 мг требуемого диастереоизомера.
Rt(ЖX) = 23,8 мин, 20% А, 80% В до 20% А, 20% В и 60% С через 40 минут.
Пример 37.
HOOC-CH2-D/L-m-OCH3-Phe-Pro-Lys-(2-тиазолил)
Boc-D/L-m-OH-Phe-OH
H-D/L-m-OH-Phe-OH. HCl (5,25 г) растворяли в диоксане (55 мл), воде (28 мл) и 1 N растворе гидроокиси натрия (29,0 мл). Затем добавляли Di-t-бутилбикарбонат (6,95 г) и реакционную смесь перемешивали в течение ночи при комнатной температуре при рН 9. Затем реакционную смесь разбавляли водой (200 мл) и экстрагировали гептаном. Водный слой разбавляли этилацетатом (150 мл) и 1 N соляной кислотой подкисляли до рН 2. Органическую фазу отделяли, а водный слой экстрагировали этилацетатом. Органические фракции объединяли и промывали водой и рассолом, сушили над сульфатом натрия и концентрировали в вакууме. Выход: 8,49 г. ТСХ: Rf = 0,67, силикагель, этилацетат/пиридин/уксусная кислота/вода = 126/20/6/11 по объему.
Вос-D/L-m-ОСН3-Phe-ОМе
Смесь, состоящую из Boc-D/L-m-OH-Phe-OH (8,49 г), карбоната натрия (23,9 г) и йодометана (20,3 мл) в N, N-диметилформамиде (60 мл), перемешивали при 60oС в течение 48 часов. Затем реакционную смесь вливали в ледяную воду и подкисляли 2 N соляной кислотой до рН 2,5, а потом экстрагировали этилацетатом. Органические слои объединяли и промывали водой и рассолом, сушили над сульфатом натрия и концентрировали в вакууме. Сырой продукт хроматографически очищали на силикагеле с использованием гептана/этилацетата = 7/3 по объему. Выход: 6,66 г. ТСХ: Rf = 0,56, силикагель, гептан/этилацетат = 3/2 по объему.
Н-D/L-m-ОСН3-Phe-ОМе. ТФУ
Boc-D/L-m-ОСН3-Phe-ОМе (6,66 г) растворяли в дихлорметане (20 мл) и трифторуксусной кислоте (20 мл) и перемешивали при комнатной температуре в течение 2 часов. Растворитель удаляли при пониженном давлении, а сырой продукт дважды совыпаривали из толуола. Выход: 9,56 г. TCX: Rf = 0,32, силикагель, этилацетат/пиридин/уксусная кислота/вода = 126/20/6/11 по объему.
HOOC-CH2-D/L-m-OCH3-Phe-Pro-Lys-(2-тиазолил)
Н-D/L-m-ОСН3-Phe-ОМе. ТФУ использовали для получения N-(t-бутилоксикарбонилметил)-N-Boc-D/L-m-OCH3-Phe-Pro-Lys(Cbz)-(2-тиазолила) согласно методике, описанной в примере 24. При обработке 624 мг защищенного трипептида трифторуксусной кислотой и тиоанизолом (см. пример 1), за которой следует очистка методом высокоэффективной жидкостной хроматографии, получается на выходе 114 мг продукта.
Rt(ЖX) = 29,3 мин и 29,8 мин, 20% А, 80% В до 20% А, 20% В и 60% С через 40 минут.
Пример 38.
HOOC-CH2-D/L-p-Br-Phe-Pro-Lys-(2-тиазолил)
Boc-D/L-p-Br-Phe-OH
рН суспензии H-D/L-p-Br-Phe-OH (2,44 г) в 25 мл смеси t-бутанола с водой = 1/1 по объему доводили до 9 1 N раствором гидроокиси натрия. Di-t-бутилбикарбонат (3,27 г) добавляли к этой суспензии, и реакционную смесь перемешивали в течение ночи, поддерживая рН постоянным и равным 9. Затем реакционную смесь разбавляли водой, затем экстрагировали гептаном. Водную фазу разбавляли этилацетатом и закисляли до рН 2,5 с помощью 2 N соляной кислоты. Органическую фазу отделяли, а водный слой экстрагировали этилацетатом. Органические слои объединяли и промывали водой и рассолом, сушили над сульфатом натрия и концентрировали в вакууме. Выход: 3,35 г. ТСХ: Rf = 0,32, силикагель, этилацетат/пиридин/уксусная кислота/вода = 126/20/6/11 по объему.
Boc-D/L-p-Br-Phe-Pro-OH
Boc-D/L-p-Br-Phe-OH (3,35 г) соединяли с H-Pro-OMe. HCl и затем омыляли гидроокисью натрия согласно методике, описанной в примере 26. Выход: 3,13 г. ТСХ: Rf = 0,45, силикагель, этилацетат/метанол = 9/1.
Boc-D/L-p-Br-Phe-Pro-Lys(Cbz)-(2-тиазолил)
Соединение Boc-D/L-p-Br-Phe-Pro-OH (750 мг) с Н-Lys(Cbz)-(2-тиазолилом) проводили в тех же условиях, которые описаны в примере 24. Выход: 1,01 г. ТСХ: Rf = 0,85, силикагель, дихлорметан/метанол = 9/1 по объему.
H-D/L-p-Br-Phe-Pro-Lys(Cbz)-(2-тиазолил). ТФУ
Boc-D/L-p-Br-Phe-Pro-Lys(Cbz)-(2-тиазолил) (1,01 г) растворяли в трифторуксусной кислоте (ТФУ, 10 мл) и перемешивали в течение 1 часа при комнатной температуре. Растворитель удаляли при пониженном давлении. Выход: 879 мг. ТСХ: Rf = 0,75 и 0,68, силикагель, этилацетат/пиридин/уксусная кислота/вода = 63/20/6/11 по объему.
N-(t-бутилоксикарбонилметил)-D/L-p-Br-Phe-Pro-Lys(Cbz)-(2-тиазолил)
t-Бутилбромацетат (264 мл) добавляли к раствору H-D/L-p-Br-Phe-Pro-Lys(Cbz)-(2-тиазолил). ТФУ (879 мл) в ацетонитриле (25 мл), рН реакционной смеси доводили до 8 N, N-диизопропилэтиламином, после чего реакционную смесь выдерживали в течение ночи при комнатной температуре. Растворитель удаляли путем выпаривания, а осадок растворяли в этилацетате. Органическую фазу промывали водой, 5% бикарбонатом натрия и рассолом, сушили над сульфатом натрия и концентрировали в вакууме. Сырой продукт очищали на силикагеле, используя дихлорметан/метанол = 95/5 по объему, и получали 850 мг защищенного трипептида. ТСХ: Rf = 0,91, силикагель, этилацетат/пиридин/уксусная кислота/вода = 63/20/6/11 по объему.
HOOC-CH2-D/L-p-Br-Phe-Pro-Lys-(2-тиазолил)
N-(t-бутилоксикарбонилметил)-D/L-p-Br-Phe-Pro-Lys(Cbz)-(2-тиазолил) (850 мг) обрабатывали трифторуксусной кислотой и тиоанизолом в тех же условиях, которые описаны в примере 1, с получением после очистки методом высокоэффективной жидкостной хроматографии 123 мг продукта.
Rt(ЖХ) = 33,9 мин и 34,4 мин, 20% А, 80% В до 20% А, 20% В и 60% С через 40 минут.
Пример 39.
HOOC-CH2-D-p-F-Phe-Pro-Lys-(2-тиазолил)
Это соединение получали аналогичным описанному в примере 38 способом, начиная с H-D-p-F-Phe-OH. Снятие защиты (см. пример 1) с 563 мг N-(t-бутилоксикарбонилметил)-D-p-F-Phe-Pro-Lys(Cbz)-(2-тиазолил) давало после очистки методом высокоэффективной жидкостной хроматографии 182 мг продукта.
Rt(ЖХ) = 29,7 мин, 20% А, 80% В до 20% А, 20% В и 60% С через 40 минут.
Пример 40.
HOOC-CH2-D/L-m, p-di-Cl-Phe-Pro-Lys-(2-тиазолил)
Это соединение получали аналогичным описанному в примере 38 способом, начиная с H-D/L-m, p-di-Cl-Phe-OH. Снятие защиты (см. пример 1) с 480 мг N-(t-бутилоксикарбонилметил)-D/L-m, p-di-Cl-Phe-Pro-Lys(Cbz)-(2-тиазолила) давало после очистки методом высокоэффективной жидкостной хроматографии 191 мг продукта.
Rt(ЖХ) = 36,8 мин и 37,8 мин, 20% А, 80% В до 20% А, 20% В и 60% С через 40 минут.
Пример 41.
BzlSO2-norLeu(цикло)Gly-Lysψ[COCO] -ОН (Bzl = бензил)
Cbz-Lys(Воc)-ОМе
Cbz-Lys(Воc)-ОН (28 г) растворяли в дихлорметане/метаноле = 9/1 по объему (500 мл), 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурониумтетрафторборат (23,6 г) добавляли к раствору и триэтиламином доводили рН раствора до 8. Реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Смесь последовательно промывали 1 N раствором соляной кислоты, 5% бикарбонатом натрия и водой и сушили над сульфатом натрия. Фильтрат выпаривали, а осадок хроматографировали на силикагеле в этилацетате/гептане = 1/4 по объему в качестве элюэнта. Фракции, содержащие Cbz-Lys (Воc)-ОМе, объединяли и упаривали. Выход: 29,1 г. ТСХ: Rf = 0,85, силикагель, этилацетат/гептан = 3/1 по объему.
Cbz-Lys(Воc)ψ[цианоацетат]
К холодному (-78oС) раствору Cbz-Lys(Воc)-ОМе (29,1 г) в сухом дихлорметане (800 мл) по каплям добавляли диизобутил алюминиумгидрид (222 мл 1 М раствора в гексане) с такой скоростью, чтобы поддерживать температуру реакции ниже -70oС. Результирующий раствор перемешивали при -78oС в течение 1 часа. Затем к реакционной смеси добавляли 5% раствор лимонной кислоты (600 мл). Двуслойную смесь перемешивали 10 минут при комнатной температуре, слои разделяли, а водный слой экстрагировали дихлорметаном. Объединенные дихлорметановые слои промывали водой, сушили над сульфатом натрия и фильтровали. Раствор помещали под азотом и охлаждали в ледяной бане. Добавляли раствор цианида натрия (36,3 г) и бензилтриэтиламмониумхлорида (4,2 г) в воде. При интенсивном перемешивании порционно (2•9 мл) в течение 30 минут добавляли уксусный ангидрид. Органический слой отделяли, а водный экстрагировали дихлорметаном. Объединенные дихлорметановые фазы промывали водой, сушили над сульфатом натрия, фильтровали и выпаривали в вакууме. Осадок очищали методом хроматографии на кремнеземе (элюэнт: гептан/этилацетат = 1/1 по объему) и на выходе получали 26,3 г Cbz-Lys(Воc)ψ[цианоацетата] . TCX: Rf = 0,60, силикагель, дихлорметан/этилацетат = 7/3 по объему.
Cbz-Lys(Воc)ψ[СНОНСО] -ОМе
Раствор Cbz-Lys(Воc)ψ[цианоацетата] (26,3 г) в смеси диэтилового эфира с метанолом = 3/1 по объему (600 мл) охлаждали до -20oС под азотом, затем вводили в него 66 г газообразной соляной кислоты, удерживая температуру ниже -5oС. Реакционную смесь выдерживали в течение ночи при 4oС. Затем к реакционной смеси по каплям добавляли воду (100 мл), соблюдая температурный режим ниже 5oС. После перемешивания в течение 16 часов при комнатной температуре органический слой отделяли и промывали водой. Водный слой насыщали хлоридом натрия и экстрагировали сухим бутанолом/дихлорметаном = 3/2 по объему. Органическую фазу промывали рассолом, сушили над сульфатом натрия, фильтровали и выпаривали в вакууме с получением 25,4 г сырого амина. Осадок растворяли в N, N-диметилформамиде (400 мл), затем добавляли бис-(трет-бутил)ангидрид (16 г) и триэтиламин до установления рН 8. Реакционную смесь перемешивали в течение ночи при комнатной температуре. Растворитель удаляли путем выпаривания при пониженном давлении. Осадок растворяли в этилацетате, промывали последовательно водой и рассолом, сушили над сульфатом натрия, фильтровали и выпаривали под вакуумом. Осадок очищали методом хроматографии на кремнеземе (элюэнт: этилацетат/гептан = 4/6 по объему), в результате получали 15,8 г Cbz-Lys(Boc)ψ[CHOHCO] -OMe. TCX: Rf = 0,75, силикагель, этилацетат/пиридин/уксусная кислота/вода = 63/20/6/11 по объему.
H-Lys(Воc)ψ[СНОНСО] -ОМе
10% палладий-на-угле (92 мг) и 2,18 мл 1 N раствора соляной кислоты добавляли к Cbz-Lys(Воc)ψ[СНОНСО] -ОМе (0,92 г) в N, N-диметилформамиде (20 мл). Смесь гидрогенизировали при атмосферном давлении при комнатной температуре в течение 3 часов. Палладиевый катализатор удаляли путем фильтрации, а растворитель - путем выпаривания при пониженном давлении, получая в результате H-Lys(Воc)ψ[СНОНСО] -OMe. HCl с количественным выходом. TCX: Rf = 0,47, силикагель, этилацетат/пиридин/уксусная кислота/вода = 88/31/18/7 по объему.
BzlSO2-norLeu(цикло)Gly-Lys(Воc)ψ[СНОНСО] -ОМе
(S)-3-бензилсульфониламидо-2-оксо-1-азепинуксусная кислота была получена согласно способу, описанному в примере 18. К холодному (0oС) (S)-3-бензилсульфониламидо-2-оксо-1-азепинуксусной кислоты (BzlSO2-norLeu(цикло)Gly) (400 мг) в N, N-диметилформамиде (20 мл) последовательно добавляли 1-гидроксибензотриазол (238 мг), дициклогексилкарбодиимид (267 мг), H-Lys(Воc)ψ[СНОНСО] -OMe. HCl (385 мг) и триэтиламин (0,32 мл). Смесь перемешивали в течение 1 часа при 0oС, а затем в течение ночи выдерживали при комнатной температуре. После этого смесь охлаждали до -20oС и путем фильтрации удаляли дициклогексилмочевину. Фильтрат выпаривали досуха. Осадок растворяли в этилацетате и последовательно промывали 5% бикарбонатом натрия, водой, 2% лимонной кислотой, насыщенным водным раствором хлористого натрия, сушили над сульфатом натрия и концентрировали в вакууме. Осадок хроматографировали на силикагеле, используя дихлорметан/метанол = 9/1 (по объему) в качестве элюэнта. Фракции, содержащие BzlSO2-norLeu(цикло)Gly-Lys(Boc)ψ[CHOHCO] -OMe, объединяли и упаривали. Выход: 663 мг. ТСХ: Rf = 0,91, силикагель, этилацетат/пиридин/уксусная кислота/вода = 63/20/6/11 по объему.
BzlSO2-norLeu(цикло)Gly-Lys(Воc)ψ[СНОНСО] -ОН
BzlSO2-norLeu(цикло)Gly-Lys(Воc)ψ[СНОНСО] -ОМе (650 мг) растворяли в смеси диоксана с водой = 7/3 по объему (20 мл) и обрабатывали малыми порциями в течение 1 часа при комнатной температуре 2 М раствором гидроокиси натрия (1,05 мл) до установления рН 12-13. Затем реакционную смесь разбавляли водой (20 мл), раствором 2 М соляной кислоты рН раствора доводили до 2,0, и водную фракцию экстрагировали дихлорметаном. Объединенные органические фазы промывали водой, рассолом и сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с выходом 740 мг BzlSO2-norLeu(цикло)Gly-Lys(Воc)ψ[СНОНСО] -ОН. ТСХ: Rf = 0,44, силикагель, этилацетат/пиридин/уксусная кислота/вода = 63/20/6/11 по объему.
BzlSO2-norLeu(цикло)Gly-Lys(Воc)ψ[COCO] -ОН
К раствору BnSO2-norLeu(цикло)Gly-Lys(Воc)ψ[СНОНСО] -ОН (740 мл) в сухом дихлорметане (20 мл) добавляли 450 мг периодината (реагент Dess-Martin). Через 1 час перемешивания при комнатной температуре добавляли 20 мл 2% раствора тиосульфата натрия, и смесь выдерживали в течение получаса при комнатной температуре. Органический слой отделяли, промывали водой, сушили над сульфатом натрия, фильтровали и выпаривали в вакууме для получения 497 мг сырого BzlSO2-norLeu(цикло)Gly-Lys(Воc)ψ[COCO] -ОН. ТСХ: Rf = 0,45, силикагель, этилацетат/пиридин/уксусная кислота/вода = 63/20/6/11 по объему.
BzlSO2-norLeu(цикло)Gly-Lysψ[COCO] -ОН
BzlSO2-norLeu(цикло)Gly-Lys(Воc)ψ[COCO] -ОН (497 мг, сырого) обрабатывали 90% трифторуксусной кислотой (10 мл) в течение 1 часа при комнатной температуре. Реакционную смесь концентрировали в вакууме, а осадок растворяли в воде и прямо заряжали на препаративной колонке Deltapack С18RP высокоэффективной жидкостной хроматографии с помощью системы градиентной элюции от 20% А/80% В до 20% А/45% В/35% С через 45 минут при скорости элюционного тока 80 мл/мин. Выход: 200 мг BzlSO2-norLeu(цикло)Gly-Lysψ[COCO] -ОН.
Rt(ЖХ) = 26,37 мин, 20% А/80% В до 20% А/20% В/60% С через 40 минут.
Пример 42.
Н-(N-СН3)-D-norLeu-Pro-Lysψ[COCO] -ОН
Воc-(N-СН3)-norLeu-Pro-ОН
Это соединение получали по способу, описанному в примере 11. Согласно тому, как изложено в примере 1, получали Н-(N-СН3)-D-norLeu-Pro-Lysψ[COCO] -ОН. Выход: 69 мг.
Rt(ЖХ)= 13,27 мин; 20% А/80% В до 20% А/20% В/60% С через 40 минут.
Пример 43.
H-D-Phe-Pro-Lysψ[COCO] -ОН
Boc-D-Phe-Pro-OMe
К холодному (0oС) раствору Boc-D-Phe-ОН (5 г) в N, N-диметилформамиде (200 мл) последовательно добавляли 1-гидроксибензотриазол (4,29 г), дихлоргексилкарбодиимид (4,29 г), H-Pro-OMe. HCl (3,1 г) и N-этилморфолин (3 мл). Смесь перемешивали при 0oС в течение 1 часа, а затем выдерживали в течение 2 дней при комнатной температуре. Затем смесь охлаждали до -20oС и путем фильтрации удаляли дициклогексилмочевину. Фильтрат выпаривали досуха. Осадок растворяли в этилацетате и последовательно промывали 5% бикарбонатом натрия, 0,1 М раствором соляной кислоты, насыщенным водным раствором хлористого натрия, сушили над сульфатом аммония и концентрировали в вакууме. Осадок хроматографировали на силикагеле в гептане/этилацетате = 6/4 (по объему) в качестве элюэнта. Фракции, содержащие Boc-D-Phe-Pro-OMe объединяли и выпаривали. Выход: 1,5 г. ТСХ: Rf = 0,90, силикагель, этилацетат/пиридин/уксусная кислота/вода = 163/20/6/11 по объему.
Boc-D-Phe-Pro-OH
Boc-D-Phe-Pro-OMe (8,3 г) растворяли в смеси диоксан/вода = 6/4 по объему (150 мл) и порционно обрабатывали раствором 2 М гидроокиси натрия в течение 1 часа при комнатной температуре, рН 12,5. Затем 2 М раствором соляной кислоты доводили рН раствора до 3,0 и водный слой экстрагировали этилацетатом. Комбинированные органические фазы промывали водой, рассолом и сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с выходом, равным 6,9 г Boc-D-Phe-Pro-OH. ТСХ: Rf = 0,30, силикагель, этилацетат/пиридин/уксусная кислота/вода = 213/20/6/11 по объему.
Аналогичным описанному в примере 1 способом получали: H-D-Phe-Pro-Lysψ[COCO] -OH. Выход: 417 мг.
Rt(ЖХ) = 16,22 мин, 20% А/80% В до 20% А/20% В/60% С через 40 минут.
Пример 44.
Н-(N-СН3)-D-Phe-(N-циклопентил)-Gly-Lysψ[COCO] -ОН
Воc-(N-СН3)-D-Phe-(N-циклопентил)-Gly-ОН
Это соединение получали, как описано в примере 3, с использованием Воc-(N-СН3)-D-Phe-OH и НСl. Н-(N-циклопентил)-Gly-OMe. ТСХ: Rf = 0,52, силикагель, дихлорметан/метанол = 9/1 по объему.
Аналогичным описанному в примере 1 способом получали: Н-(N-СН3)-D-Phe-(N-циклопентил)-Gly-Lysψ[COCO] -ОН. Выход: 87 мг.
Rt(ЖХ) = 23,92 мин, 20% А/80% В до 20% А/20% В/60% С через 40 минут.
Пример 45.
Этилсульфонил-D-Phe-Pro-Lysψ[COCO] -ОН
Этилсульфонил-D-Phe-Pro-OH
Это соединение получали согласно описанию к примеру 22.
Аналогичным описанному в примере 41 способу получали: этилсульфонил-D-Phe-Pro-Lysψ[COCO] -OH. Выход: 90 мг.
Rt(ЖХ) = 28,04 мин, 20% А/80% В до 20% А/20% В/60% С через 40 минут.
Пример 46.
(4aR, 8aR)-пергидроизохинолин-1(R)-карбонил-Pro-Lysψ[COCO] -ОН
2-Cbz-(4aR, 8aR)-пергидроизохинолин-1(R)-карбонил-Pro-OH
Это соединение получали согласно описанию к примеру 20.
Аналогично описанному в примере 41 способу получали: (4aR, 8aR)-пергидроизохинолин-1(R)-карбонил-Pro-Lysψ[COCO] -ОН. Выход: 170 мг.
Rt(ЖХ) = 18,95 мин, 20% А/80% В до 20% А/20% В/60% С через 40 минут.
Пример 47.
HOOC-CH2-D-Coa-Pro-Lys-(2-тиазолил) (Соа = циклоокталанин)
Циклооктилметилбромид
Циклооктилметанол (8,16 г) растворяли в 47%-ном растворе HBr (70 мл) и нагревали с обратным холодильником в течение 1 часа при 130oС. Реакционную смесь затем вливали в ледяную воду (500 мл) и добавляли насыщенный раствор бикарбоната натрия (500 мл). Водный раствор экстрагировали дихлорметаном. Объединенную органическую фазу промывали водой, рассолом и сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Фракции, содержащие цикло-октилметилбромид, объединяли и выпаривали. Выход: 9,85 г. ТСХ: Rf = 0,95, силикагель, толуол.
(R, S)-этил-2-ацетиламино-2-циано-3-циклооктилпропионат
Трет-бутилат калия (6,85 г) и этилацетамидоцианоацетат (8,1 г) растворяли в диметилсульфоксиде (100 мл) при комнатной температуре. Циклооктилметилбромид растворяли в диметилсульфоксиде (25 мл) и по каплям добавляли в реакционную смесь и перемешивали ее 44 часа при комнатной температуре. Затем эту смесь вливали в 500 мл воды, осадок фильтровали и сушили, получая на выходе 2,95 г (R, S)-этил-2-ацетиламино-2-циано-3-циклооктилпропионата. ТСХ: Rf = 0,50, силикагель, гептан/этилацетат = 3/7 по объему.
H-D, L-Циклооктилаланин-ОН. НСl
(R, S)-этил-2-ацетиламино-2-циано-3-циклооктилпропионат (2,95 г) суспензировали в 100 мл 20% раствора соляной кислоты и нагревали с обратным холодильником в течение 22 часов. Затем реакционную смесь охлаждали до 5oС и сформировавшийся осадок фильтровали, промывали диэтиловым эфиром и сушили. Выход: 2,69 г H-D, L-циклооктилаланин-ОН. НСl (H-D, L-Coa-ОН. НСl). ТСХ: Rf = 0,27, силикагель, этилацетат/пиридин/уксусная кислота/вода = 63/20/6/11 по объему.
Подобно тому, как описано в примере 24, получали с выходом в 162 мг HOOC-CH2-D-Coa-Pro-Lys-(2-тиазолил).
Rt(ЖХ) = 38,35 мин, 20% А/80% В до 20% А/20% В/ и 60% С через 40 минут.
Подобно тому, как описано в примере 24, получали:
Пример 48.
HOOC-CH2-D-2-Nal-Pro-Lys-(2-тиазолил) (Nal = нафтилаланин) Выход: 423 мг.
Rt(ЖX) = 35,78 мин, 20% А/80% В до 20% А/20% В/ и 60% С через 40 минут.
Пример 49.
HOOC-CH2-D-2-norLeu-Pro-Lys-(2-тиазолил) Выход: 344 мг.
Rt(ЖХ) = 24,84 мин, 20% А/80% В до 20% А/20% В/60% С через 40 минут.
Пример 50.
HOOC-CH2-D-Leu-Pro-Lys- (2-тиазолил) Выход: 138 мг.
Rt(ЖХ) = 24,50 мин, 20% А/80% В до 20% А/20% В/60% С через 40 минут.
Пример 51.
Анти-тромбиновая проба
Тромбин (фактор IIа) является одним из факторов каскада свертываемости крови.
Антитромбиновую активность соединений согласно изобретению устанавливали спектрофотометрически путем измерения скорости гидролиза хромогенного субстрата S-2238. Такую пробу антитромбиновой активности в буферной системе использовали для установления величины IС50 для тестируемого соединения.
Тест-среда: Буфер трометамин-NaCl-полиэтиленгликоль 6000 (TNP). Близкое из известных соединение: 12581 (Kabi).
Растворитель: буфер TNP. Растворимость можно улучшать с помощью диметилсульфоксида, метанола, этанола, ацетонитрила или трет. -бутилового спирта, которые до концентрации 2,5% в конечной реакционной смеси не обладают побочными эффектами.
Технические реагенты (все использованные реагенты имеют маркировку "Чистые для анализа", ЧДА; для приготовления водных растворов использовали сверхчистую воду (качества Milli-Q).
1. Буфер трометамин-NaCl (TN). Состав буфера: Трометамин (Трис) 6,057 г (50 ммоль), NaCl 5,844 г (100 ммоль), вода до 1 литра. рН раствора доводили до 7,4 с помощью HCl (10 ммоль/л) при 37oС.
2. Буфер TNP: полиэтиленгликоль 6000 растворяли в буфере TN для получения концентрации 3 г/л.
3. Раствор S-2238: один флакон S-2238 (25 мг; Kabi Диагностика, Швеция) растворяли в 20 мл буфера TN для достижения концентрации 1,25 мг/мл (2 ммоль/л).
4. Раствор тромбина: тромбин человека (16000 nKat/флакон; Центральная Лаборатория переливания крови, Амстердам, Нидерланды) растворяли в буфере TNP для получения концентрации 3,34 nKat/мл.
Приготовление растворов тестируемых соединений и соединений, близких по уровню техники
Для проверки тестируемых и близких по уровню техники соединений их растворяли в воде Milli-Q для получения исходных концентраций 10-2 моль/л. Каждую концентрацию получали последовательным разведением наполнителем для получения концентраций 10-3, 10-4 и 10-5 моль/л. Все разведения, включая и исходный раствор, использовали в опыте (конечные концентрации в реакционной смеси: 3•10-3, 10-3, 3•10-4, 10-4, 3•10-5, 10-5, 3•10-6, 10-6 моль/л, соответственно).
Способ
При комнатной температуре 0,075 мл и 0,025 мл раствора, содержащего тестируемое соединение или соединение, близкое по уровню техники, или наполнителя альтернативно вносили дозатором в лунки микротитрационного планшета, и эти растворы разбавляли, соответственно, 0,115 мл и 0,0165 мл буфера TNP. Затем в каждую лунку добавляли аликвоту в 0,030 мл раствора S-2238, и планшет проинкубировали в термостате (фирмы Amersham) на шейкере в течение 10 минут при 37oС. Продолжая преинкубацию, запускали процесс гидролиза S-2238 внесением в каждую лунку по 0,030 мл раствора тромбина. Планшет инкубировали (на шейкере в течение 30 секунд) при 37oС. Начиная с 1 минуты инкубации, каждые 2 минуты измеряли поглощение каждого образца в течение 90 минут при длине волны 405 нм, используя для этого кинетический аппарат для прочтения планшетов (планшет-ридер) (Twinreader plus, Flow Laboratories).
Все полученные данные фиксировались на персональном компьютере IBM с помощью системы LOTUS-MEASURE. Для каждой концентрации соединения (выраженной в моль/л реакционной среды) и для контрольного образца вычерчивались кривые зависимости поглощения от продолжительности реакции в минутах.
Оценка результатов.
Из экспериментальных кривых рассчитывали максимум поглощения для каждой конечной концентрации. Величина IС50 рассчитывалась на основании анализа преобразований (logit transformation analysis) согласно Hafner et al. , (Arzneim. -Forsch. /Drug Res. 1977; 27(II): 1871-3).
Значения IC50 для соединений согласно изобретению следующие:
Образец - Значения IC50 (мкМ)
2(е) - 4,5
4(b) - 4,34
39 - 19
41 - 0,135
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ПРОТЕАЗЫ СЕРИНА | 1997 |
|
RU2178419C2 |
| ИНГИБИТОРЫ СЕРИНОВЫХ ПРОТЕАЗ | 1997 |
|
RU2172321C2 |
| ИНГИБИТОРЫ СЕРИНПРОТЕАЗЫ | 1997 |
|
RU2191193C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗО[1,5-А]ПИРИДИНА КАК ИНГИБИТОРЫ СЕРИНПРОТЕАЗ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2175327C2 |
| ИНГИБИТОРЫ СЕРИНОВЫХ ПРОТЕАЗ | 1998 |
|
RU2183642C2 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМКОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ ТРОМБИНА У МЛЕКОПИТАЮЩЕГО | 1995 |
|
RU2148585C1 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОАРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ LH АГОНИСТОВ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2248979C2 |
| ПРОИЗВОДНЫЕ 5-АМИНО-4-ОКСИГЕКСАНОВОЙ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2067585C1 |
| Способ получения производных борсодержащих пептидов | 1988 |
|
SU1807988A3 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2087480C1 |
Описывается новый ингибитор тромбина формулы 1: А-В-С-Lys-D, в котором А представляет собой Н, 2-гидрокси-3-циклогексилпропионил-, R1, R1-CO-, R1-SO2-, -(СНR2)nСООR3-, где R1 выбран из -(1-6С)алкилен-СOOН, (1-12С)алкила, (6-14С)арила, (7-15С)аралкила и (8-16С)аралкенила, арильная группа которого может быть заменена (1-6С)алкилом, (2-12С)алкокси или галогеном; R2 представляет собой Н или (1-12С)алкил; R3 представляет собой Н или (1-12С)алкил; n является целым числом от 1 до 3; В представляет собой связь L-Asp, Leu, norLeu, -N(бензил)-СН2-СО-, -N(2-индан)-СН2-СО-, D-1-Piq, D-3-Piq, D-Tiq, Atc или D-аминокислоту, имеющую гидрофобную ароматическую боковую цепь; С является Pro, norLeu(цикло)Gly, аминокислотой одной из формул -N[(3-8С)циклоалкил] -СН2-СО- или -N(бензил)-СН2-СО-; D выбрано из СООН, тетразола, оксазола, тиазола и бензотиазола; или А и С имеют указанные выше значения, В является D-(3-8С)циклоалкилаланином, а D является тетразолом, оксазолом, тиазолом или бензотиазолом; или его пролекарство; или его фармацевтически приемлемая соль; за исключением соединения Me-D-Phe-Pro-Lys-COOH. Вышеуказанные соединения могут быть использованы в качестве антитромботических агентов. 2 с. и 7 з. п. ф-лы.
A-B-C-Lys-D,
где А представляет собой Н, 2-гидрокси-3-циклогексилпропионил; R1, R1-CO-, R1-SO2-, -(СНR2)nCOOR3, где R1 - выбран из -(1-6С)алкилен-СООН, (1-12С)алкила, (6-14С)арила, (7-15С)аралкила и (8-16С) аралкенила, арильная группа которого может быть заменена (1-6С) алкилом, (2-12С) алкокси или галогеном; R2 представляет собой Н или (1-12С)алкил; R3 представляет собой Н или (1-12С) алкил; n является целым числом от 1 до 3;
В представляет собой связь, L-Asp, Leu, norLeu, -N(бензил)-СН2-СО-, -N(2-индан)-СН2-СО-, D-1-Piq, D-3-Piq, D-Tiq Atc или D-аминокислоту, имеющую гидрофобную ароматическую боковую цепь;
С является Pro, norLeu(цикло)Gly, аминокислотой одной из формул -N[(3-8C) циклоалкил] -СН2-СО- или -N(бензил)-СН2-СО-;
D выбрано из СООН, тетразола, оксазола, тиазола и бензотиазола;
или
А и С имеют указанные выше значения, В является D-(3-8C) циклоалкилаланином, а D является тетразолом, оксазолом, тиазолом или бензотиазолом;
или его пролекарство, или его фармацевтически приемлемая соль; за исключением Me-D-Phe-Pro-Lys-COOH.
Приоритет по признакам:
03.11.1995 - B= L-Asp, -N(бензил)-СН2-СО-, D-Tiq, Atc; C= -N(бензил)-СН2-СО-.
| ПРОИЗВОДНЫЕ ПЕПТИДОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПРЕДУПРЕЖДЕНИЯ И УСТРАНЕНИЯ ТРОМБОЗОВ | 1992 |
|
RU2103276C1 |
| US 5523308 А, 04.06.1996 | |||
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| S.D | |||
| LEWIS et al | |||
| Inhibition of trombin by peptides containing lysyl-α-ketocarbonyl derivatives | |||
| Топка с качающимися колосниковыми элементами | 1921 |
|
SU1995A1 |
| D.M | |||
| JONES | |||
| Trombin inhibitors based on Ketone derivatives of Arg and Lys | |||
| Топка с качающимися колосниковыми элементами | 1921 |
|
SU1995A1 |

