Изобретение относится к производным имидазо [1,5а] пиридина - ингибиторам серинпротеаз, способу их получения, фармацевтической композиции, содержащей указанные соединения, а также к применению указанных производных имидазо [1,5а] пиридина как ингибиторов серинпротеаз в лекарственной терапии и, в частности, для лечения и предупреждения тромбоза или других обусловленных тромбином заболеваний.
Производные имидазо [1,5а] пиридина известны, например 3-амино-6,7,8,8a- тетрагидро-8а-гидроксиимидазо[1, 5a] пиридин-1(5H)-он описан Klein с сотр. (Leibigs Ann. Chem., 1623-1637, 1983). О фармакологической активности этого соединения не упоминается.
Производные 3,8-диаминоимидазо [1,5a]пиридин-1(5H)-она настоящего изобретения, замещенные в положении 8, представляют собой новые соединения, которые являются селективными обратимо действующими ингибиторами серинпротеаз, которые требуют основного аминокислотного остатка в положении P1 их субстратов.
Настоящее изобретение относится к ингибиторам серинпротеаз - производным имидазо [1,5а] пиридина, содержащим структурную единицу (звено) с общей формулой I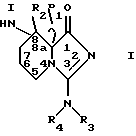
где R1 представляет собой водород, низшую алкильную или ацильную группу; R2 является водородом или низшим алкилом; R3 и R4, независимо, представляют водород, низший алкил или вместе образуют =CH-NR5R6, причем R5 и R6 представляют низший алкил; или к их фармацевтически приемлемым солям.
При определении соединений формулы I термин "низший алкил" обозначает разветвленную или линейную алкильную группу с 1-6 атомами углерода, подобную гексилу, изобутилу, пропилу, изопропилу, этилу и наиболее предпочтительно метилу.
Термин "ацильная группа" относится к 1-оксоалкильной группе, образованной от карбоновой кислоты с 1-6 атомами углерода, подобно гексаноилу, трет-бутаноилу, пропионилу, ацетилу и формилу. Предпочтительной ацильной группой является ацетильная группа.
Серинпротеазы составляют класс протеолитических ферментов, которые катализируют гидролиз специфических пептидных связей в белковых субстратах, Schechter Berger (Biochem. Biophys. Res. Commun., 27, 157-162, 1967) предложили номенклатуру, которую теперь часто используют для идентификации аминокислотных остатков в субстратах серинпротеаз:
Субстрат: ~ = расщепляющаяся связь
..Pn...P4-P3-P2-P1~ P'1-P'2-P'3-P'4...P'n
Фермент
..Sn...S4-S3-S2-S1-S'1-S'2-S'3-S'4....Sn..
Аминокислотные остатки субсайтов субстрата на N -конце расщепляющейся связи P1 - P'1 обозначаются P1, P2 и т.д., и P'1, P'2 и т.д. на С-конце. Эти субсайты субстрата соответствуют возможным субсайтам (S1, S2, и т.д.) на ферменте, с которым происходят взаимодействия связывания.
Соединения настоящего изобретения являются ингибиторами серинпротеаз, которые требуют присутствия основного аминокислотного остатка, подобного аргинину или лизину, в положении P1 своих субстратов. Характерными примерами таких серинпротеаз являются трипсин, плазмин, активатор плазминогенаурокиназа, калликреины, кальпаин, акрозин и тромбин.
Настоящее изобретение относится к аналогам пептидных субстратов, которые охватывают остатки P-области субстратов только протеаз, в которых терминальный P1-остаток заменяется звеном 3,8-диаминоимидазо [1,5а] пиридин-1(5H)-она формулы I.
Основной целью настоящего изобретения являются селективные ингибиторы некоторых серинпротеаз, которые составляют часть системы свертывания крови. В этой ферментативной системе активированная форма одного фактора свертывающей системы катализирует активацию следующего фактора, что приводит в конце концов к быстрой генерации аргинин- направленной (остаток субстрата P1 является аргинином) серинпротеазы тромбина (фактор IIа) из его предшественника протромбина (фактор II). Последний процесс катализируется фактором Ха, который также является аргинин-направленной серинпротеазой. Тромбин, последний фермент в системе коагуляции, будет расщеплять растворимый белок плазмы фибриноген с образованием мономеров фибрина, которые перекрестно сшиваются с образованием нерастворимого геля. Кроме участия в регуляции своего собственного продуцирования и активности, тромбин является потенциальным агонистом тромбоцитов, посредством чего индуцируется агрегация тромбоцитов. Активированные тромбоциты вместе с фибриновой полимерной матрицей и захваченными эритроцитами образуют сгусток крови или тромб.
Тромбин играет ключевую роль в процессе гемостаза - физиологическом процессе, который останавливает кровотечение из поврежденного кровеносного сосуда. Он также играет некую роль при тромбозе, который является патологическим состоянием, в соответствии с которым несоответствующая (нарушенная) активность механизма гемостаза проявляется в образовании внутрисосудистых тромбов, что в свою очередь ведет к нарушению кровотока. Тромбоз может иметь место как в артериях, так и венах.
На сегодняшний день клиническое применение для предупреждения тромбоза находят два типа антикоагулянтов, т.е., гепарины и антагонисты витамина К. Те и другие действуют косвенно, снижая активность тромбина. Гепарин действует главным образом путем ускорения инактивации тромбина его физиологическими ингибиторами, подобными антитромбину III и гепариновому кофактору II. Гепарин действует только тогда, когда дается парентерально. Антагонисты витамина К, из которых хорошо известным является, например, производное кумарина варфарин, активны при пероральном введении и действуют путем ингибирования продуцирования в функциональной форме ряда витамин-К-зависимых факторов коагуляции (II, VII, IX и X). Из-за своего механизма действия эти последние агенты имеют слабую атаку и обратимость действия. Основными клиническими проблемами, вызываемыми применением гепаринов и кумаринов, являются кровотечение и их небольшой и непредсказуемый запас терапевтической безопасности.
Поэтому существует необходимость разработки улучшенных ингибиторов коагуляции, которые, например, непосредственно ингибируют тромбин или фактор Ха.
Обнаружено, что соединения, которые содержат звено 3,8-диамино-8a-гидроксиимидазо[1,5 a]пиридин-1 (5H)-она, по изобретению, являются ингибиторами серинпротеаз, которые требуют основного аминокислотного остатка (т.е. аргинина, лизина) в положении P1 своих субстратов. Эти соединения предположительно способны взаимодействовать специфически в первую очередь с сайтом S1 протеазы. Селективность такого рода действия также определяется заместителем в 8-аминогруппе звена 3,8-диамино-8a-гидроксиимидазо [1,5 а] пиридин-1 (5H)-она. Заместитель может являться любой группой, которая способна взаимодействовать с субсайтами Sn. ..S2 и предпочтительно пептидильной группой, которая гомологична субсайтам Pn...P2 субстрата подходящего фермента, или может являться любым производным или подражанием сайтов субстрата Pn...P2, которые связываются с предполагаемыми субсайтами Sn...S2 активного сайта фермента.
Предпочтительный вариант осуществления настоящего изобретения относится к ингибиторам серинпротеаз, подобных тромбину и фактору Ха, которые участвуют в процессе тромбоза и гемостаза. Ингибиторы по предпочтительному варианту настоящего изобретения содержат звено формулы I и заместитель в 8-аминогруппе, который является гомологом, производным или подражанием субсайтов P3-P2 субстрата соответствующей серинпротеазы. Ряд таких P2-P2 - производных уже известен из уровня техники, например производные, описанные в работах Hauptmann u Markwardt (Seminars in Thrombosis and Hemostasis, 18, 200-217, 1992), Jakubowski et al. (Annual Reports in Medicinal Chemistry, 27, 99-108, 1992), u Shuman et al. (J. Med.Chem., 36, 314-319, 1993), которые включены здесь в качестве ссылок.
Предпочтительные соединения по настоящему изобретению представляют собой производные 3,8-диамино-8a-гидроксимидазо- [1,5a] -пиридин-1 (5н] -она формулы 1, в которой R1, R2, R3 и R4 представляют водород.
В более предпочтительном варианте осуществления настоящее изобретение относится к ингибиторам серинпротеаз, имеющим формулу II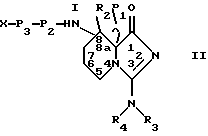
где R1, R2, R3 и R4 представляют собой водород; X является водородом, R7, R7-O-C(О)-, R7-C(О)-, R7-SO2-, - (CHR8)m COOR8 или N -защитной группой, где R7 представляет (1- 12C)алкил или (2-12C)алкенил, которые, необязательно, могут быть замещены (3-8C)циклоалкилом, (1-6C)алкокси, OH или галогеном, или R7 представляет собой (3-8C)циклоалкил, (4-10C)гетероциклил, (4- 14C)арил, (7-15C)аралкил и (8-16C)аралкенил, которые, необязательно, могут быть замещены (1-6C)алкилом, (3-8C)циклоалкилом, (1-6C)алкокси, OH или галогеном, и арильные группы которых могут содержать, необязательно, гетероатом; каждая группа R8, независимо, представляет водород или имеет то же значение, что R7; m равен 1, 2 или 3; P3 представляет связь, аминокислоту формулы -NH-CH [(CH2)pС(O)OH]-С(O)- или ее эфирное производное, а p равен 0, 1, 2 или 3, N - (бензил)-CH2-СО-,D-Tig, Atc, 3-Pig, 1-Pig или D- аминокислоту с гидрофобной боковой цепью; P2 представляет Pro или Pec, замещенный, необязательно, (1-4C)алкилом, галогеном, гидрокси или оксо, или аминокислоту, выбираемую среди Gly, Val, Tle, 2,4- MePro, 3,3-Dmp, Ilc, Thz, Hyp, 2,2-Dmt, 5.5-Dmt, Lac, Ару, AC5C, 1-Nal и 2-Nal, либо P2 является аминокислотой формулы - N[(3,8C)циклоалкил]-CH2-С(O)-, кольцо которое может быть, необязательно, замещено (1-6C)алкилом, галогеном, гидрокси или оксо; либо P2 является связью в случае, когда P3 также является связью, и Х является R7-SO2-; или P2 и P3 вместе представляют структуру, подобную дипептиду, имеющую формулу III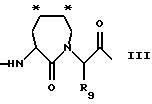
которая в положениях, отмеченных звездочками, может конденсироваться с бензольным кольцом, и в которой R9 является водородом или низшим алкилом.
N - Защитная группа, указанная при определении группы X, является любой N-защитной группой, которую обычно используют в химии пептидов для защиты  аминогруппы, подобной трет-бутоксикарбонильной (Boc) группе, бензилоксикарбонильной (Z) группе, 9-флуоренилметоксикарбонильной (Fmoc) группе или фталоильной (Phth) группе. Подходящие N-защитные группы можно также найти в T. W. Green and P.G.M. Wuts: Protective Groups in Organic Synthesis, Second Edition (Wiley, NY, 1991) и в The Peptides, Analysis, Synthesis, Biology", Vol. 3, E. Gross and J.Meienhofer, Eds., Academic Press, New York, 1981).
аминогруппы, подобной трет-бутоксикарбонильной (Boc) группе, бензилоксикарбонильной (Z) группе, 9-флуоренилметоксикарбонильной (Fmoc) группе или фталоильной (Phth) группе. Подходящие N-защитные группы можно также найти в T. W. Green and P.G.M. Wuts: Protective Groups in Organic Synthesis, Second Edition (Wiley, NY, 1991) и в The Peptides, Analysis, Synthesis, Biology", Vol. 3, E. Gross and J.Meienhofer, Eds., Academic Press, New York, 1981).
Термин "(1-12C)алкил" означает разветвленную или неразветвленную алкильную группу с 1-12 атомами углерода, такую как метил, этил, трет-бутил, изопентил, гептил, додецил и подобную группу. Предпочтительными алкильными группами являются (1-6C)алкильные группы с 1-6 атомами углерода. Наиболее предпочтительны (1- 4C)алкильные группы с 1-4 атомами углерода, такие как метил, этил, изопропил, н-бутил и трет-бутил.
(2-12C) Алкенильная группа представляет разветвленную или линейную ненасыщенную углеводородную группу с 2-12 атомами углерода. Примерами являются этенил, пропенил, аллил и подобные группы.
Термин "(1-6C)алкокси" означает алкоксигруппу с 1-6 атомами углерода, причем ее алкильная группа имеет ранее установленные значения.
Термин "(3- 8C)циклоалкил" означает циклоалкильную группу с 3-8 атомами углерода, представляя циклопропил, циклобутил, циклопентил, циклогексил, циклогептил или циклооктил. Циклопентил и циклогексил являются предпочтительными циклоалкильными группами.
Термин "(4-10C)гетероциклил" обозначает замещенную или незамещенную циклическую углеводородную группу с 4-10 атомами углерода, содержащую также один или два гетероатома, выбираемых среди N, О и S, подобную 3-метил-1,2,3,4-тетрагидро-8-хинолинилу. Заместители для гетероциклической группы можно выбрать среди таких групп, как (1-6C)алкокси, гидрокси, галоген, нитро, амино, диалкиламино или низший алкил. Термин "диалкиламино" обозначает диалкиламиногруппу, в которой алкил имеет значения, установленные ранее для низшего алкила.
(4-14C)Арильная группа является ароматической группой с 4-14 атомами углерода. Арильная группа также может содержать один или два гетероатома и может быть замещенной, например, (1-6C)алкилом, (3-8C)циклоалкилом, (1- 6C)алкокси, гидрокси, нитро, амино, диалкиламино или галогеном. Примерами арильных групп являются фенил, диметоксифенил, нафтил, 4-бифенил, имидазолил, тиенил, бензтиенил, (изо)хинолил, 3-метил- 8-хинолинил, инданил, индолил и подобные группы. Предпочтительными арильными группами являются фенил и нафтил.
(7-15C)Аралкильная и (8-16C)аралкенильная группы представляют алкильные и алкенильные группы, соответственно замещенные одной или несколькими арильными группами, причем общее число атомов углерода составляет от 7 до 15 и от 8 до 16 соответственно.
Термин "галоген" обозначает фтор, хлор, бром или иод.
Термин "сложноэфирное производное" обозначает любое соответствующее эфирное производное, предпочтительно (1-4C) алкильные эфиры, такие как метил-, этил- или бутилэфиры.
Термин "гидрофобная боковая цепь" обозначает (1- 12C)алкил, необязательно замещенный (3-8C)циклоалкильной группой или ароматической группой (которая может содержать гетероатом, например, азота), такой как циклогексил, циклооктил, фенил, пиридил, нафтил, тетрагидронафтил, и подобной группой, и такая гидрофобная боковая цепь может быть замещена, необязательно, такими заместителями, как галоген, нитро, трифторметил, низший алкил (например, метил или этил), (1- 6C)алкокси (например, метокси), фенилокси, бензилокси, и подобными группами. В соединениях по формуле III значение низшего алкила в определении R9 такое же, как указано ранее.
Особенно предпочтительными являются ингибиторы серинпротеаз формулы II, в которых R1, R2, R3 и R4 представляют собой водород; Х является водородом, низшим алкилом, ацильной группой, R7-SO2-, где R7 представляет (4-10C)гетероциклил, (6-14C)арил, причем арильные группы могут содержать гетероатом, или Х является N-защитной группой; P3 представляет связь в случае, когда Х является R7-SO2 -, либо P3 выбирают среди D-Phe, D-Nle, D-Dpa, D-MePhe, D-1-Tig, D-Cyk, D-Phg, D-Tic, D-Atc, D-2-Nal, D-2-Pal, D-Chg и D-2-Nag; P2 выбирают среди Pro, Pec, Gly, Val, Ile, 2,4-MePro, 3,3- Dmp, Ilc, Thz, Hyp, 2,2-Dmt, 5,5-Dmt, Lac, Ару и AC5C; либо P2 является связью в случае, когда P3 также является связью, и Х является R7-SO2-; P2 и P3 вместе представляют структуру, подобную дипептиду, имеющую формулу III, причем положения, указанные звездочками, конденсируются с бензолом. Ароматические аминокислотные остатки в определении P3 в формуле II в этих предпочтительных ингибиторах серинпротеаз, например, Phe, Dpa, Tig, Phg, Nal и Nag могут замещаться в соответствующем ароматическом кольце (кольцах) (1-6C)алкилом, (1-6C)алкокси, галогеном, гидрокси или нитро. Предпочтительные производные фенилаланина (Phe) или фенилглицина (Phg) имеют хлор- или нитрозаместитель в пара-положениях фенильной группы.
В наиболее предпочтительном варианте осуществления изобретения ингибитор серинпротеазы находится в форме ацетата.
Соединения, соответствующие общей формуле II, можно получить путем конденсации Х-P3-P2-OH с производным 3,8-диамино-8a- гидроксиимидазо [1,5a] пиридин-1(5H)-она формулы IY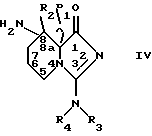
в которой R1, R2, R3, R4, P2, P3 и X имеют ранее указанные значения.
В тех случаях, когда X-P3-P2-OH представляет дипептидильную группу или R7-SO2-P2-OH или содержит дипептидподобную структуру формулы III, конденсацию можно осуществить путем активации функциональной группы карбоновой кислоты в защищенной тем или иным образом структуре, способами, применяемыми для конденсации пептидных фрагментов, например, азидным способом, способом со смешанным ангидридом, способом с активированным эфиром или предпочтительно карбодиимидным способом, особенно с добавлением каталитических и подавляющих рацемизацию соединений, подобных N-гидроксисук- цинимиду и N-гидроксибензотриазолу. Обзор таких способов конденсации, которые обычны в химии пептидов, дается в работе " The Peptides, Analysis, Synthesis, Biology", Vol. 3, цит. выше, которая включена здесь в качестве ссылки.
В тех случаях, когда Х представляет R7-SO2, а P2 и P3 представляют связи, конденсацию можно осуществить, используя активированное сульфонилгалоидное производное, такое как R7-SO2,Cl, где R7 имеет ранее установленные значения.
Соединения формулы IV можно получить из производных 3-амино-6-гуанидино-2-оксогексановой кислоты общей формулы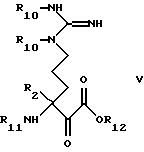
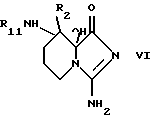
где R2 имеет значения, установленные при определении формулы I, и где R10 и R11 представляют N -защитную группу, обычную в химии пептидов, а R12 представляет низший алкил, определенный ранее, посредством удаления гуанидинозащитной группы R10, после чего полученное соединение формулы VI, где R10 и R11 имеют значения, указанные в случае формулы V, методами, известными в данной области, необязательно, алкилируют, ацилируют или конвертируют до 8- [(амино) метилен] аминопроизводного соединения VI, после чего удаляют R11.
Производные 3-амино-6-гуанидино-2-оксогексановой кислоты общей формулы V можно получить путем введения защитных групп в  аминогруппу и в гуанидиногруппу аминокислоты аргинина или 2-алкилзамещенного аргинина, и последующей конверсии функциональной группы карбоновой кислоты в функциональную группу
аминогруппу и в гуанидиногруппу аминокислоты аргинина или 2-алкилзамещенного аргинина, и последующей конверсии функциональной группы карбоновой кислоты в функциональную группу 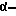 кетоэфира методами, известными в данной области.
кетоэфира методами, известными в данной области.
Соединения настоящего изобретения можно применять при производстве лекарственных препаратов для лечения и профилактики опосредованных тромбином и связанных с тромбином заболеваний. К таковым относятся легочная эмболия, тромбофлебит, артериальная окклюзия при тромбозе или эмболии, артериальная реокклюзия во время или после ангиопластики или тромболиза, тромбоз глубоких вен, рестеноз после повреждения артерии или инвазивных кардиологических процедур, постоперационный тромбоз вен или эмболия, острый или хронический атеросклероз, удар, инфаркт миокарда, рак и метастазы, и нейродегенеративные болезни.
Соединения настоящего изобретения можно применять в качестве антикоагулянтов также in vitro.
Новые соединения формул I или II, которые могут существовать в форме свободного основания, можно выделить из реакционной смеси в форме фармацевтически приемлемой соли. Фармацевтически приемлемые соли можно также получить путем обработки свободного основания формулы I или II органической кислотой, такой как уксусная кислота, пропионовая кислота, гликолевая кислота, малеиновая кислота, малоновая кислота, метансульфоновая кислота, фумаровая кислота, янтарная кислота, винная кислота, лимонная кислота, бензойная кислота и аскорбиновая кислота.
Соединения настоящего изобретения могут иметь один или несколько хиральных атомов углерода и, следовательно, могут быть получены в виде чистого энантиомера, или в виде смеси энантиомеров, или в виде смеси, содержащей диастереоизомеры. Способы получения чистых энантиомеров хорошо известны в данной области техники, например кристаллизация солей, которые получают из оптически активных кислот и рацемической смеси, или хроматография с использованием хиральных колонок.
Соединения настоящего изобретения можно вводить энтерально или парэнтерально, и для людей предпочтительна дневная доза 0,001-10 мг на кг массы тела. Смешанные с фармацевтически подходящими вспомогательными средствами, описанными, например, в работе Gennaro et al. Remington's Pharmaceutical Sciences (18 th ed, Mack Publishing Company, 1990, см. особенно часть 8: "Фармацевтические препараты и их производство"), соединения можно спрессовать в твердые стандартные дозы, такие как драже, таблетки, или переработать в капсулы или суппозитории. С помощью фармацевтически подходящих жидкостей соединения можно применять также в виде препарата для инъекции в форме раствора, суспензии, эмульсии или в виде спрея, например назального спрея.
При получении стандартных доз, например таблеток, рассматривают применение обычных наполнителей, красителей, полимерных связующих и подобных веществ. Вообще, можно использовать любую фармацевтически приемлемую добавку, которая не влияет на функцию активных соединений. Подходящими носителями, с которыми можно вводить композиции, являются лактоза, крахмал, производные целлюлозы и подобные вещества или их смеси, применяемые в соответствующем количестве.
Далее настоящее изобретение иллюстрируется примерами.
В описании и в формуле изобретения использованы следующие аббревиатуры названий аминокислот:
Aic = 2-аминоиндан-2-карбоновая кислота;
АС5С = аминоциклопентан-2-карбоновая кислота;
Ару = аминопирролидон;
Arg = аргинин;
Ate = 2-аминотетралин-2-карбоновая кислота;
Cha = циклогексилаланин;
Chg = циклогексилглицин;
Cyk = циклооктилаланин;
3,3-Dmp = 3,3-диметилпропин;
2,2-Dmt = 2,2-диметилтиазолидин-4-карбоновая кислота;
5,5-Dmt = 5,5-диметилтиазолидин-4-карбоновая кислота;
Dpa = 3,3-дифенилаланин;
Hyp = 4-оксипропин;
Ilc = (S)-индолин-2-карбоновая кислота;
Lac = 3-амино-2-оксо-1-пиперидин (" 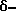 лактам");
лактам");
MePhe =  метилфенилаланин;
метилфенилаланин;
2-Nag = 2-нафтилглицин;
1-Nal = 1-нафталиналанин;
2-Nal = 2-нафтилаланин;
Nie = норлейцин;
2-Pа1 = 2-пиридилаланин;
Pec = пипеколиновая кислота;
Phg = фенилглицин;
1-Pig = 1-карбоксипергидроизохинолин;
3-Pig = 3-карбоксипергидроизохинолин;
Pro = пролин;
Thz = тиазолидин-4-карбоновая кислота;
Tic = 1,2,3,4- тетрагидроизохинолин-3-карбоновая кислота;
1-Tig = 1-карбокси- 1,2,3,4-тетрагидроизохинолин.
Используются и другие аббревиатуры:
АС = ацетил;
Pmc = 2,2,5,7,8-пентаметилхроман-6-сульфонил.
Все пептидные последовательности, упомянутые в заявке, написаны в соответствии с общепринятым соглашением, по которому N-концевая аминокислота располагается слева, а С-концевая аминокислота - справа. Если конфигурация аминокислоты не указана, все аминокислоты, как встречающиеся в природе, так и "небелковые" аминокислоты, упомянутые в настоящей заявке, находятся в L-форме.
Восходящую тонкослойную хроматографию (ТСХ) осуществляют с использованием пластин, предварительно покрытых диоксидом кремния (Merck, F254), в следующих системах растворителей:
Система А: дихлорметан:этилацетат = 9:1 (о/о);
Система В: н-бутанол: пиридин: уксусная кислота: вода = 10: 1:1:2 (о/о/о/о);
система С: этилацетат: пиридин: уксусная кислота: вода = 63:20:6:11 (о/о/о/о);
система D: н-бутанол: пиридин:уксусная кислота:вода = 6:1:1:2 (о/о/о/о);
Система E: толуол:этанол = 8:2 (о/о);
система F: этилацетат: пиридин: уксусная кислота: вода = 63:10:3:5,5 (о/о/о/о);
система G: дихлорметан: этилацетат = 95:5 (о/о);
система H: этилацетат: пиридин: уксусная кислота: вода = 6: 2:2:1 (о/о/о/о).
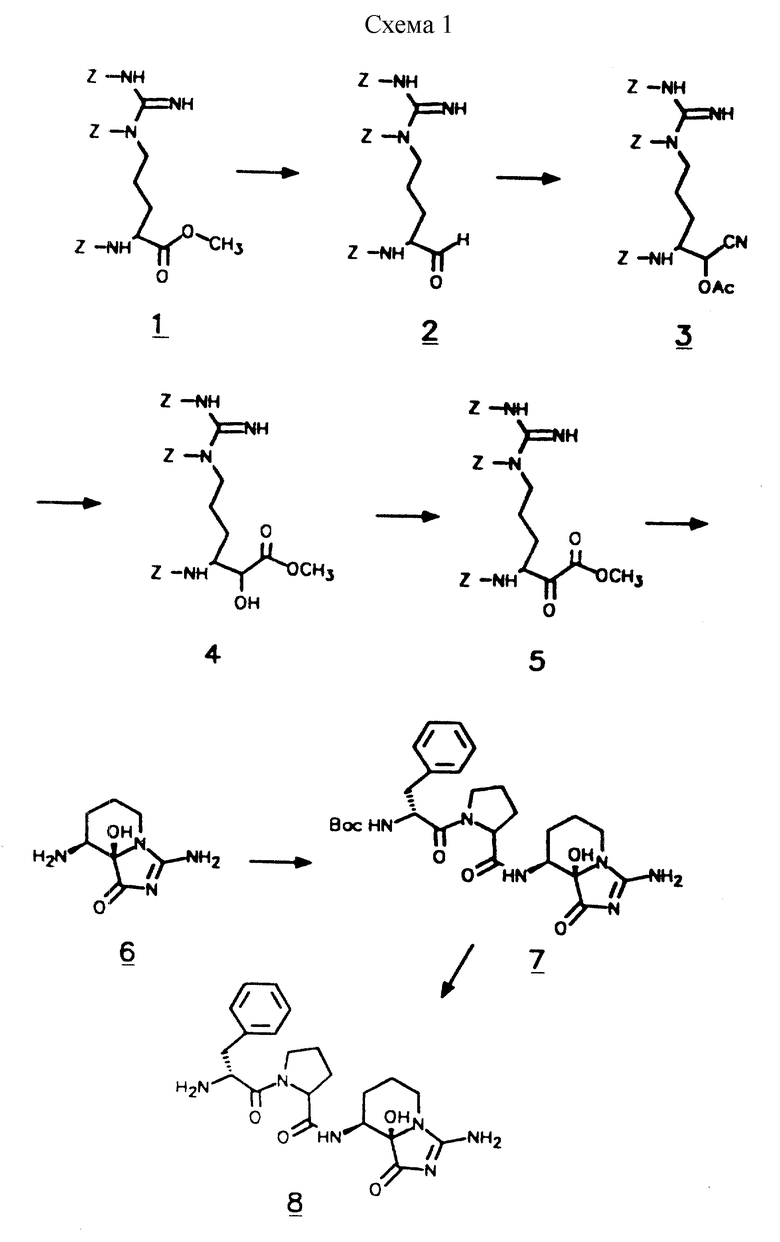
Пример 1 схема 1)
3,8-Диамино-6,7,8,8а-тетрагидро-8а- гидроксиимидазо [1.5a] -пиридин-[1(5H)]-он (6)
Метиловый эфир Nα,Nδ, , N -трибензилоксикарбонил-L - аргинина (Z-Arg(Z2)-OMe; (1)
Nα,Nδ, ,N - Трибензилоксикарбонил-L-аргинин (40 г), полученный, как описано (Jetten et al. Tetrahedron Lett., 1991, 32, 6025-6028), растворяют в смеси дихлорметана (1080 мл) и метанола (120 мл). К раствору добавляют 2-(1Н-бензотриазол-1-ил) -1,1,3,3- тетраметилурония тетрафторборат (TBTU; 22,4 г), после чего к раствору добавляют триэтиламин до pH 8. Смесь перемешивают при комнатной температуре в течение 1 часа, после чего раствор последовательно промывают водой, раствором бикарбоната натрия и водой, сушат и упаривают. Получают твердый остаток, который кристаллизуют из метанола. Выход 35 г. Rf 0,60 (система Ж).
Б. Nα,Nδ, , N -Трибензилоксикарбонил-L-аргиналь (Z-Arg(Z2)-H; (2)
Раствор диизобутилалюмогидрида в гексане (180 мл; 1 М) добавляют при -78oC, по каплям и при перемешивании, к раствору Z-Arg (Z2)- ОМе (90 г) в сухом дихлорметане (700 мл). Смесь перемешивают в течение 1 часа при -78oC, после чего добавляют 20% (о/о) раствор концентрированной хлористоводородной кислоты в этаноле до pH 2. Смесь экстрагируют дихлорметаном. Экстракты промывают водой, раствором бикарбоната натрия и водой, сушат (сульфат натрия), и упаривают, и получают сырой продукт (25 г), который используют без дополнительной очистки. Rf 0.48 (система А).
В. 2-Ацетокси-3- (бензилоксикарбониламино)-6-(дибензилоксикарбонилгуанидино) гексаннитрил (3)
Раствор цианида натрия (28 г) и триэтилбензиламмонийхлорида (35 г) в воде (700 мл) и уксусный ангидрид (14 мл) добавляют одновременно, при перемешивании, к предварительно охлажденному раствору Z-Arq(Z2)-H(30 г) в дихлорметане (700 мл). Смесь перемешивают в течение 30 минут при 0-5oC. Органический слой отделяют и последовательно промывают водой и рассолом, сушат (сульфат натрия), и упаривают, и получают остаток, который хроматографируют на диоксиде кремния. Элюирование смесью дихлорметана с этилацетатом (95:5, о/о) дает твердый продукт (17 г). Rf 0,76 (система А).
Г. Метиловый эфир 3- (бензилоксикарбониламино)-6-(дибензилоксикарбонилгуанидино)-2- оксигексановой кислоты (4)
2-Ацетокси-3-(бензилоксикарбониламино)-6-(дибензилоксикарбонилгуанидино) гексаннитрил (6,0 г) растворяют в смеси диэтилового эфира и метанола (3:1; о/о; 140 мл). Через раствор при -78 С пропускают газообразный хлористый водород до получения 3 М раствора. Смесь перемешивают в течение 16 часов при 5oC, после чего смесь экстрагируют дихлорметаном. Объединенные экстракты промывают водой, раствором бикарбоната натрия и водой, сушат (сульфат натрия) и упаривают. Получают смолу (6,1 г). Rf 0,48 (система А).
Д. Метиловый эфир 3- (бензилоксикарбониламино)-6-(дибензилоксикарбонилгуанидино)-2- оксогексановой кислоты (5)
Добавляют постепенно хромовую кислоту (1,3 мл 8 N раствора в водной серной кислоте) к предварительно охлажденному раствору метилового эфира 3- (бензилоксикарбониламино)-6-(дибензилоксикарбонилгуанидино) -2- оксигексановой кислоты (1,3 г) в ацетоне (130 мл). Смесь перемешивают в течение 1 часа при 0oC и затем выливают в смесь воды со льдом. Выпавший осадок отфильтровывают, промывают водой, и сушат в вакууме, и получают белое твердое вещество (1,15 г). Rf 0,80 (система А).
Е. 3,8-диамино-6,7,8,9а-тетрагидро-8а- гидроксиимидазо[1,5а] пиридин-1(5H)-он (6)
Хлористоводородную кислоту (1,04 мл 1 М водного раствора) и палладий на активированном угле (Pd/С 10%; 64 мг) добавляют к раствору метилового эфира 3-(бензилоксикарбониламино)-6- (дибензилоксикарбонилгуанидино) -2-оксогексановой кислоты (644 мг) в диметилформамиде (20 мл). Через раствор пропускают водород до завершения реакции, что контролируют с помощью тонкослойной хроматографии. Реакционную смесь фильтруют для удаления катализатора. Фильтрат упаривают в вакууме и получают масло (320 мг). Rf 0,50 (система В).
Пример 2 (схема 1)
N8 (D -фенилаланинпропил)-3,8-диамино- 6,7,8,8а-тетрагидро-8а-гидроксимидазо [1,5а]пиридин-1(5H)-он (8)
Добавляют последовательно 1-гидроксибензотриазол (233 мг) и дициклогексилкарбодиимид (261 мг) к раствору Boc-D-Phe -Pro-OH (0,41 г) в диметилформамиде (10 мл), поддерживая температуру 0-5oC. Реакционную смесь перемешивают в течение 15 минут, после чего добавляют раствор 3,8-диамино-6,7,8,8а- тетрагидро-8а-гидроксиимидазо [1,5а] пиридин-1(5H)-она (192 мг) в диметилформамиде (10 мл), pH которого предварительно доведена до 7 посредством добавления триэтиламина. Раствор перемешивают в течение 16 часов при комнатной температуре, после чего отфильтровывают выпавшую в осадок дициклогексилмочевину. Фильтрат упаривают до небольшого объема. Добавляют н-бутанол, после чего раствор промывают раствором бикарбоната натрия и водой, сушат (сульфат натрия) и упаривают, получают соединение 7 с 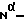 Вос- защитной группой (0,89 г). Rf 0,50 (система С).
Вос- защитной группой (0,89 г). Rf 0,50 (система С).
Сырой продукт растворяют при 0oC в 90% водном растворе трифторуксусной кислоты (15 мл), содержащем также анизол (0,43 мл). Смесь перемешивают в течение 2 часов при комнатной температуре и затем упаривают в вакууме. Остаток растворяют в смеси трет-бутанола с водой (1:1, о/о) и добавляют порциями Dowex -2 (Х-8, ацетатная форма) до тех пор, пока pH раствора не поднимется до 5-6. Отфильтровывают ионообменную смолу, после чего фильтрат лиофилизуют. Продукт хроматографируют на диоксиде кремния. Элюирование смесью н- бутанола с пиридином, уксусной кислотой и водой (8:1:1:2, о/о) дает названное в заголовке соединение 8 (120 мг) (см. в конце описания)
Rf 0,70 (система D). Данные спектров ЯМP соответствуют структуре с конфигурацией 8S, 8aR.
Пример 3
N8 Nα (Метил)-D-фенилаланилпролил)-3,8-диамино-6,7,8, 8а-тетрагидро-8а-гидроксиимидазо [1,5а]пиридин-1(5H)-он
A. Z -N(Me)-D-Phe-OH
Добавляют карбобензоксихлорид (6,4 ммоль) к раствору H-N(Me)-D-Phe-OH (4,0 ммоль) и гидроксида натрия (4,0 ммоль) в смеси диоксана с водой (1:1, о/о). Раствор перемешивают в течение 24 часов, поддерживая pH 12 посредством добавления гидроксида натрия (4 N раствор в воде). Реакционную смесь экстрагируют диэтиловым эфиром для удаления избытка карбобензоксихлорида. К раствору добавляют соляную кислоту до pH 2. Выпавший в осадок продукт экстрагируют этилацетатом. Органически слой промывают рассолом, сушат (сульфат натрия) и упаривают в вакууме. Получают сироп (76%). Rf 0,45 (система Е).
Б. Z-N(Me)-D-Phe-Pro-OMe
Z- N(Me)-D-Phe-OH (3 ммоль), H-Pro -OMe.HCI (3 ммоль) и N -гидроксибензотриазол (6 ммоль) растворяют в диметилформамиде (20 мл). К раствору добавляют 4-этилморфолин до pH 6,5, после чего раствор охлаждают до 0oС. К охлажденному раствору добавляют постепенно раствор дициклогексилкарбодиимида (3,3 ммоль) в диметилформамиде (5 мл). Смесь перемешивают в течение 1 часа при 0oC и затем в течение 16 часов при комнатной температуре. Выпавшую в осадок дициклогексилмочевину отфильтровывают, а фильтрат упаривают в вакууме. Получают сироп, который растворяют в этилацетате. Раствор последовательно промывают раствором бикарбоната натрия, раствором бисульфата натрия и рассолом, сушат (сульфат натрия), и упаривают в вакууме, и получают пену (96%). Rf 0,47 (система Е).
B.Z - (Me)-D-Phe-Pro-OH
Гидроксид натрия (6 ммоль; 4N водный раствор) добавляют при перемешивании к раствору Z - N(Me)-D-Phe-Pro-ОМе (3 ммоль) в смеси диоксана с водой (1: 1: о/о). Раствор выдерживают при комнатной температуре в течение 16 часов. Разбавляют раствор водой и экстрагируют диэтиловым эфиром. К водному раствору добавляют хлористоводородную кислоту до pH 2. Выпавший в осадок продукт экстрагируют этилацетатом. Объединенные экстракты промывают рассолом, сушат (сульфат натрия) и упаривают, получают пену (0,94 г; 77%). Rf 0,22 (система Е).
Г. Названное в заголовке соединение получают путем сочетания 3,8-диамино-6,7,8,8а-тетрагидро-8а- гидроксимидазо[1,5а] пиридин-1(5H)-она с Z -N(Me)-D-Phe-Pro - OH с использованием способа сочетания, описанного в примере 2, с последующим удалением 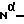 бензилоксикарбонильной защитной группы посредством каталитического дегидрирования. Rf 0,60 (система С).
бензилоксикарбонильной защитной группы посредством каталитического дегидрирования. Rf 0,60 (система С).
Пример 4
N8 (D-Дифенилаланилпролил)-3,8-диамино-6,7,8, 8а-тетрагидро-8а-гидроксимидазо [1,5а]пиридин-1(5H)-он
A. Z -D-Dpa-OH
Раствор N-(бензилоксикарбонилокси)сукцинимида (Z -ONSu; 2,0 ммоль) в диоксане (15 мл) добавляют, постепенно и при перемешивании, к раствору D-фенилаланина (H-D-Dpa-OH; 2,0 ммоль) в 10% (м/о) водном растворе бикарбоната натрия. Смесь перемешивают в течение 2 суток, после чего смесь промывают диэтиловым эфиром. Водный раствор подкисляют до pH 1-2, добавляя хлористоводородную кислоту. Выпавший в осадок продукт экстрагируют этилацетатом. Объединенные экстракты промывают рассолом, сушат (сульфат натрия), и упаривают, и получают масло (0,74 г; 100%). Rf 0,77 (система Е).
B. Z -D-Dpa-Pro-O-TpeT-Bu
Z-D-Dpa-OH (2,0 ммоль), H-Pro-О- трет-Bu.HCI (2,0 ммоль) и N-гидроксибензотриазол (4 ммоль) растворяют в диметилформамиде (15 мл). К раствору при перемешивании добавляют 4-этилморфолин до pH 6,5, после чего раствор охлаждают до 0oC. К охлажденной реакционной смеси добавляют постепенно раствор дициклогексилкарбодиимида (2,2 ммоль) в диметилформамиде (4 мл) и затем перемешивают ее в течение 1 часа при 0oC и затем в течение 16 часов при комнатной температуре. Выпавшую в осадок дициклогексилмочевину отфильтровывают, а фильтрат упаривают в вакууме. Получают сироп, который растворяют в этилацетате. Раствор последовательно промывают раствором бикарбоната натрия, раствором бисульфата натрия и рассолом, сушат (сульфат натрия), и упаривают в вакууме, и получают масло (0,88 г; 83%). Rf 0,69 (система Е).
В. Z -D-Dpa-Pro-OH
Z-D-Dpa-Pro-O-трет-Bu (1,67 ммоль) растворяют при 0oC в 90% водном растворе трифторуксусной кислоты (15 мл), содержащем также анизол (0,43 мл). Смесь перемешивают в течение 30 минут при комнатной температуре и затем упаривают в вакууме. Остаток обрабатывают диэтиловым эфиром и получают твердое вещество (0,33 г; 42%).
0,532 (система D) M.-c. FAB: мм 472.
Г. Названное в заголовке соединение получают путем сочетания 3, 8-диамино-6,7, 8,8а-тетрагидро-8а-гидроксиимидазо [1,5а]пиридин-1(5Н)-она c Z-D- Dpa-Pro-OH с использованием способа сочетания, описанного в примере 2, с последующим удалением  бензилоксикарбонильной защитной группы посредством каталитического дегидрирования. Rf 0,40 (система С).
бензилоксикарбонильной защитной группы посредством каталитического дегидрирования. Rf 0,40 (система С).
Пример 5
N8(H-D-Phe-Ilc)-3,8-диамино-6,7,8,8а- тетрагидро-8а-гидроксиимидазо [1, 5а] пиридин-1 (5Н)-он
A. Z-D-Phe-Ilc-OH
К раствору (8)-индолин-2-карбоновой кислоты (1 ммоль; 163 мг) и N-карбоксиангидрида Z-D -Phe-OH (4 ммоль; 1,3 г) в сухом тетрагидрофуране (10 мл) добавляют 4-этилморфолин (1 ммоль). Смесь перемешивают при комнатной температуре в течение 16 часов, после чего испаряют растворитель в вакууме. Сырой продукт очищают путем распределения в противотоке в системе растворителей дихлорметан-метанол-толуол-вода (5: 8: 5: 3; о/о/о/о) и получают Z-D-Phe-llc-OH с количественным выходом (0,45 г). Rf 0,60 (система F). M.-c. FAB:.мм 444.
Б. N8 (Z-Phe-Ilc)-3,8-диамино-6,7,8, 8а-тетрагидро-8а-гидроксиимидазо [1, 5а] пиридин-1 (5Н)-он
К раствору Z-D-Phe-Ilc-OH (0,67 ммоль; 300 мг) и 3,8 диамино-6,7, 8, 8а-тетрагидро-8а-гидроксиимидазо [1, 5а] пиридин-1 (5Н)-она (0,71 ммоль; 320 мг) в диметилформамиде (12 мл) добавляют 4- этилморфолин (0,67 ммоль). К раствору добавляют последовательно N- гидроксибензотриазол (1,1 ммоль; 150 мг) и дициклогексилкарбодиимид (0,71 ммоль; 147 мг), поддерживая при этом температуру 0-2oC. Смесь перемешивают при этой температуре в течение 1 часа, а затем в течение 16 часов при комнатной температуре. Отфильтровывают дициклогексилмочевину, после чего упаривают фильтрат в вакууме. Остаток растворяют в н-бутаноле. Раствор промывают раствором бикарбоната натрия и рассолом, сушат (сульфат натрия) и упаривают в вакууме. Получают пену (322 мг; 70%). Rf 0,37 (система С).
В. К раствору продукта примера 5Б (0,43 ммоль; 300 мг) в метаноле (10 мл) добавляют палладий на активированном угле (Pd/C 10%; 30 мг). Через раствор в течение 16 часов при перемешивании пропускают водород. Удаляют катализатор фильтрованием, после чего фильтрат упаривают в вакууме. Остаток хроматографируют на оксиде алюминия (Lichroprep AloxT; 25-30 мкм). Элюирование смесью этилацетата с пиридином, уксусной кислотой и водой (63:20:6:11; о/о/о/о) дает названное в заголовке соединение (135 мг; 45%). Rf 0,35 (система С).
Пример 6
Способами, подобными способам, описанным в примерах 1-5, получают перечисленные далее соединения.
N8(H- D-MePhe - Pro)-3,8-диамино-6,7,8,8а-тетрагидро-8а-гидроксиимидазо[1,5а] пиридин-1 (5H) -он;
N8(H-D-1-Tig-Pro)-3,8-диамино-6,7,8,8а- тетрагидро-8а-гидроксиимидазо[1,5а] пиридин-1 (5H-он;
N8(H-D - Nle-Pro)-3,8-диамино-6,7,8,8а-тетрагидро-8а-гидроксиимидазо [1,5a] пиридин-1 (5H)-он;
N8 (Pmc - Gly) -3, 8-диамино-6,7,8,8a-тетрагидро-8a- гидроксиимидазо[1,5а] пиридин-1 (5H)-он;
N8 (Phth - Gly) - 3,8-диамино-6,7,8,8a-тетрагидро-8a-гидроксиимидазо[1,5а] пиридин-1 (5H) -он;
N8(H-D - Atс- Pro) -3, 8-диамино-6,7,8, 8a- тетрагидро-8a-гидроксиимидазо[1,5а]пиридин-1(5H)-он;
N8 (Ac-D- Phe- Pro)-3,8-диамино-6,7,8,8a-тетрагидро-8a-гидроксиимидазо [1,5а]пиридин-1(5H)-он;
N8(H-D-2-Nag-Pro)-3,8-диамино- 6,7,8,8a-тетрагидро-8a-гидроксиимидазо [1, 5 а] пиридин-1 (5H) - он;
N8(H-D -Phe -3,3- Dmp)-3, 8-диамино-6,7,8,8a-тетрагидро- 8a-гидроксиимидазо [1,5а] пиридин-1 (5H)-он;
N8 (H-D-Phe -2,4- MePro) -3,8-диамино-6,7,8,8a-тетрагидро-8a-гидроксиимидазо [1, 5a]пиридин-1 (5H)-он;
N8(H- D -Phe -2,2-Dmt-3,8- диамино-6,7,8,8a-тетрагидро-8a-гидроксиимидазо [1,5а] пиридин-1 (5H)- он;
N8 (H- D-Phe-5,5- Dmt)-3, 8-диамино-6,7,8,8a-тетрагидро-8a- гидроксиимидазо [1, 5a]пиридин-1(5H)-он;
N8 (H-D- Phe- Thz)-3,8-диамино-6,7,8,8a-тетрагидро-8a-гидроксиимидазо [1, 5a]пиридин-1(5H)-он;
N8(H-D-Phe-Нyp)-3,8-диамино-6,7,8,8a- тетрагидро-8a-гидроксиимидазо [1, 5a] пиридин-1 (5H)-он.
Пример 7
N8 2-(S) [4(R)-(1,3-Дигидро-1,3-диоксо-2Н- изоиндол-2-ил) -1,3,4, 5-тетрагидро-3-оксо-2Н-2-бензазепин-2-ил] - 1-оксопропил] -3,8-диамино-6,7,8,8a-тетрагидро-8a- гидроксимидазо [1, 5a]пиридин-1(5H)-он
А. Метиловый эфир N-фталоил-D-фенилаланил-L-аланина
К раствору гидрохлорида метилового эфира L-аланина (1,15 г, 8,3 ммоль) в дихлорметане (20 мл) добавляют триэтиламин (1,15 мл, 8,3 ммоль), а затем добавляют N-фталоил-D-фенилаланин (2,44 г, 8,3 ммоль) и N-гидроксибензотриазол (1,27 г, 9,1 ммоль). Смесь перемешивают до образования прозрачного желтого раствора. Указанный раствор охлаждают до ОoC и добавляют 1- [3- (диметиламино)пропил-3-этил] карбодиимид (1,74 г, 9,1 ммоль). После перемешивания при комнатной температуре в течение 64 час раствор разбавляют дихлорметаном (50 мл). Добавляют соляную кислоту (1N; 50 мл) и фильтруют образовавшуюся в результате суспензию. Слои разделяют и органическую фазу последовательно промывают соляной кислотой (1N; 15 мл), насыщенным водным раствором бикарбоната натрия (50 мл), водой (50 мл) и рассолом (50 мл). Органический экстракт сушат (сульфат натрия), и упаривают, и получают 2,50 г (80%) кристаллического вещества. Образец для анализа кристаллизуют из этилацетата и гептана; т.пл. 118-120oC.
Б. N-фталоил-D-фенилаланил-L-аланин
К раствору метилового эфира N-фталоил-D-фенилаланил-L-аланина (1,46 г, 3,8 ммоль) в ацетоне (20 мл) добавляют воду (11 мл) и концентрированную HCl (6 мл). Смесь кипятят с обратным холодильником в течение 3,5 час. После охлаждения до комнатной температуры с помощью фильтрации выделяют 0,80 г (2,1 ммоль) названного в заголовке соединения. Маточный раствор концентрируют для удаления ацетона и водный раствор экстрагируют этилацетатом (3 х 20 мл). Органические слои экстрагируют насыщенным водным раствором бикарбоната натрия (3х25 мл). Объединенные водные экстракты промывают этил- ацетатом (25 мл) и доводят pH до 1 концентрированной соляной кислотой. Добавляют этилацетат (50 мл), слои разделяют и водный слой экстрагируют этилацетатом (2х25 мл). Объединенные этилацетатные экстракты промывают рассолом (2х50 мл), сушат (сульфат натрия), и упаривают, и получают 0,50 г названной в заголовке кислоты. Общий выход 1,30 г (3,6 ммоль, 92%). Кристаллизуют из метанола; т. пл. 241-242oC.
В. 3-[2(R)-(1,3- Дигидро-1,3-диоксо-2Н-изоиндол-2-ил)-4 (S) -метил-1-оксо-3- фенилпропил)-5-оксазолидинон
К раствору N-фталоил-D-фенилаланил- L-аланина (0,50 г, 1,4-ммоль) в сухом дихлорметане добавляют избыток параформальдегида (0,50 г) и молекулярное сито  (2,5 г). Суспензию перемешивают в течение 30 мин при комнатной температуре. Добавляют трифторметансульфокислоту (120 мкл, 1,4 ммоль) и продолжают перемешивание в течение 24 час. Раствор фильтруют, промывают насыщенным водным раствором бикарбоната натрия (2х25 мл) и рассолом (25 мл). Органическую фазу сушат (сульфат натрия) и упаривают досуха. Остаток очищают колоночной хроматографией (диоксид кремния, этилацетат:гептан=1:2) и получают 400 мг (1,1 ммоль, 80%) названного в заголовке соединения. Rf (этилацетат:гептан = 2:1) 0,45.
(2,5 г). Суспензию перемешивают в течение 30 мин при комнатной температуре. Добавляют трифторметансульфокислоту (120 мкл, 1,4 ммоль) и продолжают перемешивание в течение 24 час. Раствор фильтруют, промывают насыщенным водным раствором бикарбоната натрия (2х25 мл) и рассолом (25 мл). Органическую фазу сушат (сульфат натрия) и упаривают досуха. Остаток очищают колоночной хроматографией (диоксид кремния, этилацетат:гептан=1:2) и получают 400 мг (1,1 ммоль, 80%) названного в заголовке соединения. Rf (этилацетат:гептан = 2:1) 0,45.
Г. 4(R)-(1,3-Дигидро-1,3-диоксо-2Н-изоиндол-2-ил)- α (S) -метил-1,3,4,5-тетрагидро-3-оксо-2Н- 2-бензазепин-2-уксусная кислота
Оксазолидинон (250 мг, 0,7 ммоль), полученный, как описано выше, растворяют в сухом дихлорметане (1 мл) и добавляют к трифторметансульфокислоте (1 мл). Смесь энергично перемешивают
в течение 2 час. После разбавления реакционной смеси дихлорметаном (10 мл) осторожно добавляют воду (15 мл), продолжая энергичное перемешивание. Слои разделяют и водную фазу экстрагируют дихлорметаном (2 х 10 мл). Объединенные органические слои промывают рассолом (25 мл), сушат (сульфат натрия) и упаривают. Остаток кристаллизуют из этанола и диэтилового эфира и получают 150 мг (0,4 ммоль, 60%) названного в заголовке соединения; т.пл. 209-210oC.
Д. N8 [2-(S)-[4(R)-(1,3-Дигидро-1,3-диоксо-2Н-изоиндол- 2- ил)-1,3,4,5-тетрагидро-3-оксо-2Н-2-бензазепин-2-ил] -1- оксопропил]-3,8-диамино-6,7,8,8a-тетрагидро-8a-гидроксимидазо[1, 5а] пиридин-1 (5Н)-он
К раствору дигидрохлорида 3,8- диамино-6,7,8,8a-тетрагидро-8a-гидроксиимидазо [1, 5а] пиридин-1 (5Н)-она (90 мг, чистота 57%, 0,19 ммоль) в сухом диметилформамиде (15 мл) добавляют кислоту, описанную в n.Г (75 мг, 0,19 ммоль). С помощью 4-этилморфолина доводят pH до 7,4 и добавляют N- гидроксибензотриазол (45 мг, 0,3 ммоль). После охлаждения до 0oC добавляют дициклогексилкарбодиимид (42 мг, 0,2 ммоль) и получающийся в результате раствор перемешивают в течение 3 час при ОoC, а затем в течение 33 час при комнатной температуре. Растворяют, частично упаривают и добавляют несколько капель воды. Раствор перемешивают в течение 30 мин, фильтруют и упаривают досуха. Очистка колоночной хроматографией (оксид алюминия; элюирование смесью этилацетат: пиридин:уксусная кислота: вода = 6:2:2:1, о/о/о/о) дает 75 г названного в заголовке соединения. Rf 0,65 (система И).
Пример 8
3-[[(Диметиламино)метилен] амино] -N8(2- нaфтилcульфoнил)-8-амино-6,7,8,8a-тетрагидро-8a-гидроксиимидазо [1,5а]пиридин-1 (5H)-он
К раствору 3,8-диамино-6,7,8,8a-тетрагидро-8a- гидроксиимидазо [1,5а] пиридин-1(5H)-она (100 мг) в диметилформамиде (10 мл) добавляют триэтиламин до кажущегося pH 8. К раствору при перемешивании последовательно добавляют 2-нафтилсульфонилхлорид (135,5 мг) и эквимолярное количество триэтиламина. Смесь перемешивают в течение 16 часов при комнатной температуре, после чего удаляют летучие. Остаток очищают колоночной хроматографией (диоксид кремния, этилацетат: пиридин: уксусная кислота: вода = 5:2:2:1; о/о/о/о) и получают названное в заголовке соединение (12,6 мг). Rf 0,45 (система С).
Пример 9
N8 (2-Нафтилсульфонил)-3,8-диамино-6,7,8, 8a- тетрагидро-8a-гидроксимидазо [1, 5а] пиридин-1 (5H)-он
К раствору 3,8-диамино-6,7,8,8a-тетрагидро-8a-гидроксиимидазо [1,5а]пиридин-1(5H)-она (257 мг) в диметилформамиде (10 мл) добавляют триэтиламин до кажущегося pH 8. Добавляют последовательно при перемешивании 2-нафтилсульфонилхлорид (227 мг) и эквимолярное количество триэтиламина. Смесь перемешивают в течение 16 часов при комнатной температуре, после чего удаляют летучие. Остаток очищают колоночной хроматографией (диоксид кремния, этилацетат: пиридин: уксусная кислота: вода = 5:2:2:1; о/о/о/о) и получают названное в заголовке соединение (26 мг). Rf 0,30 (система С).
Пример 10
N8 Nα (2-Нафтилсульфонил)глицил]-3,8-диамино- 6,7,8,8a тетрагидро-8a-гидроксимидазо [1, 5а] пиридин-1 (5Н)-он
Nα (2-Haфтилcульфoнил) глицин (2-Nas-Gly-OH; 1,0 ммоль), полученный конденсацией 2-нафтилсульфонилхлорида и метилглицината с последующим омылением метилового эфира в водном растворе гидроксида натрия, дигидрохлорид 3,8-диамино-6, 7, 8, 8a- тетрагидро-8a-гидроксиимидазо[1, 5а] пиридин-1 (5Н) -она (1,0 ммоль) и N-гидроксибензотриазол (2,0 ммоль) растворяют в диметилформамиде (15 мл). Доводят pH раствора до 6,5, добавляя 4- этилморфолин, после чего раствор охлаждают до 0oC и добавляют дициклогексилкарбодиимид (1,1 ммоль). Смесь перемешивают в течение 1 часа при 0oC, а затем в течение 17 часов при комнатной температуре. Выпавшую в осадок дициклогексилмочевину отфильтровывают и фильтрат упаривают. Остается остаток, который растворяют в бутаноле. Органический раствор промывают 5% (м/о) раствором бикарбоната натрия и рассолом, после чего удаляют бутанол в вакууме. Затем сырой продукт очищают хроматографией на оксиде алюминия (Alox Т; 25-40 мкм) и получают названное в заголовке соединение (70 мг). Rf 0,40 (система И).
Антитромбиновый анализ
Тромбин (Фактор IIа), сериновая протеаза, является фактором в каскаде коагуляции. Активность соединения Примера 2 настоящего изобретения по ингибированию тромбина исследовали спектрофотометрическим измерением скорости гидролиза хромогенного субстрата S-2238, обработанного тромбином. Это испытание на антитромбиновую активность в буферной системе использовали для оценки величины IC50 исследуемого соединения.
Среда для испытания. Трометамин-NaCl-полиэтиленгликоль 6000 (TNP) буфер. Контрольное соединение: 12581 (Kabi). Растворитель: TNP буфер. Солюбилизация может быть осуществлена с помощью диметилсульфоксида, метанола, этанола, ацетонитрила или трет. - бутилового спирта, которые не оказывают неблагоприятного воздействия при концентрациях до 2,5% в конечной реакционной смеси.
Реагенты, используемые в методе*.
1. Трометамин-NaCI (TN) буфер. Состав буфера: Трометамин (Трис) 6,057 г (50 ммол); NaCI 5,844 г (100 ммол); вода до 1 л. pH раствора доводят до 7,4 при 37oC с помощью HCl (10 ммол. л-1).
2. TNP буфер. Полиэтиленгликоль 6000 растворяли в TN буфере до концентрации 3 г. л-1.
3. Раствор S-2238: Один флакон S-2238 (25 мг; от Kabi Diagnostica, Швеция) растворяли в 20 мл TN буфера с получением концентрации 1,25 мг. мл-1 (2 ммол.л-1).
4. Раствор тромбина: тромбин человека (16000 нКат.флак-1); от Centraal Laboratorium voor Bloedtransfusie, Амстердам, Нидерланды) растворяли в TNP буфере с получением маточного раствора 835 нКат.флак-1. Непосредственно перед использованием этот раствор разбавляют TNP буфером до концентрации 3,34 нКат.флак-1. *- Использовали все ингредиенты аналитической чистоты;
- Для водных растворов использовали ультрачистую воду (Milli-Q чистота).
Приготовление растворов исследуемого и контрольного соединения.
Исследуемое и контрольное соединение растворяли в ультрачистой воде для получения маточного раствора до концентрации 10-2 мол.л1.
Каждый образец пошагово разбавляли растворителем до концентраций 10-3, 10-4 и 10-5 мол.л-1. Разведения, включая маточный раствор, использовали в анализе (конечные концентрации в реакционной смеси: 3•10-3, 10-3, 3•10-4, 10-4, 3•10-5, 10-5, 3•10-6 и 10-1 мол.л соответственно).
Исследование
0,075 мл и 0,025 мл растворов исследуемого соединения или контрольного соединения или растворителя альтернативно дозировали с помощью пипетки в лунки микротитрационного планшета и эти растворы разбавляли 0,115 мл и 0,0165 мл TNP буфера соответственно. Добавляли аликвоту 0,030 мл раствора S-2238 в каждую лунку, и планшет подогревали, и преинкубировали со встряхиванием в инкубаторе (Amersham) в течение 10 мин при 37oС. Следующий после преинкубации гидролиз S-2238 начинается добавлением 0,030 мл раствора тромбина в каждую лунку. Планшет инкубируют (со встряхиванием в течение 30 сек) при 37oC. Начиная после 1 минуты инкубации, абсорбцию каждого образца при 405 нм измеряют каждые 2 минуты в течение 90 минут, используя кинетический микропланшетный ридер (Twinreader plus. Flow Laboratories).
Все данные были обработаны на IBM ПК с использованием программы LOTUS-MEASURE. Для каждой концентрации соединения (выраженной в мол. л-1 реакционной смеси) и для контролей строили графики зависимости абсорбции от времени реакции в минутах.
Вычисление результатов: Для каждой конечной концентрации максимальная абсорбция была вычислена из экспериментального графика. Величину IC50 (конечная концентрация, выраженная в мкмол.л-1, вызывающая 50% ингибирование максимальной абсорбции контрольных проб) вычисляли с использованием логнормального трансформационного анализа согласно Hafner et al.(Arzneim.-Forsch./ Drug. Res. 1977, 27(1): 1971-3).
Величина IC50 соединения Примера 2 (соединение 8) составляет 3•10-6М.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ПРОТЕАЗЫ СЕРИНА | 1997 |
|
RU2178419C2 |
| ИНГИБИТОРЫ СЕРИНПРОТЕАЗЫ | 1997 |
|
RU2191193C2 |
| ИНГИБИТОРЫ СЕРИНОВЫХ ПРОТЕАЗ | 1997 |
|
RU2172321C2 |
| ИНГИБИТОРЫ ТРОМБИНА | 1996 |
|
RU2178796C2 |
| ПРОИЗВОДНЫЕ БЕНЗОКСАЗЕПИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СТИМУЛЯТОРОВ РЕЦЕПТОРА АМРА | 2002 |
|
RU2279434C2 |
| СТЕРОИДНОЕ СОЕДИНЕНИЕ, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2182153C2 |
| ИНГИБИТОРЫ СЕРИНОВЫХ ПРОТЕАЗ | 1998 |
|
RU2183642C2 |
| ИНГИБИТОР СЕРИНОВЫХ ПРОТЕАЗ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 1999 |
|
RU2232760C2 |
| СИНТЕТИЧЕСКИЕ ПОЛИСАХАРИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2167163C2 |
| ПРОИЗВОДНЫЕ 11,21-БИСФЕНИЛ-19-НОРПРЕГНАНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1995 |
|
RU2152952C2 |
Изобретение относится к ингибитору серинпротеазы - производному имидазо [1,5a] пиридина, содержащему включающую структурную единицу (звено) общей формулы (I), где R1 и R2 представляют собой водород, R3 и R4, независимо, представляют водород, (1-6С) алкил или вместе образуют =СН-NR5R6, причем R5 и R6 представляют (1-6С) алкил; Х является водородом, R7, ацетилом, Рmс, R7-SO2, N-защитной группой, где R7 представляет (1-12С) алкил или (1-14С) арил; Р3 представляет связь, D-Tig, Atc, D - аминокислоту с гидрофобной боковой цепью; Р2 представляет Pro, Gly, Val, Jle, 2,4-Me P2O, 3,3-Dmp, Jlc, Thr, Hyp, 2,2-Dmt или 5,5-Dmt; или Р2 является связью в случае, когда Р3 также является связью, и Х является R7-SO2-; или Р2 и Р3 вместе представляют структуру, подобную дипептиду, имеющую формулу (II), которая в положениях, отмеченных звездочками, может конденсироваться с бензольным кольцом и в которой R9 является водородом; или его фармацевтически приемлемая соль. Описывается также фармацевтическая композиция. 3 с. и 2 з.п.ф-лы.
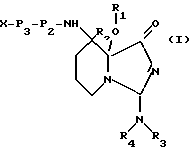
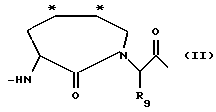
где R1 и R2 представляют собой водород;
R3 и R4, независимо, представляют водород, (1-6С)алкил или вместе образуют =CH-NR5-R6, причем R5 и R6 представляют (1-6С)алкил;
Х является водородом;
R7, ацетилом, Pmc, R7-SO2, N-защитной группой, где R7 представляет (1-12С)акил или (1-14С)арил;
P3 представляет связь, D-Tig, Atc, D-аминокислоту с гидрофобной боковой цепью;
Р2 представляет Pro, Gly, Val, Jle, 2,4-MePro, 3,3-Dmp, Jlc, Thz, Hyp, 2,2-Dmt или 5,5-Dmt; или Р2 является связью в случае, когда Р3 также является связью, и Х является R7-SO2-; или Р2 и Р3 вместе представляют структуру, подобную дипептиду, имеющую формулу II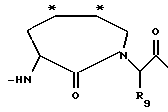
которая в положениях, отмеченных звездочками, может конденсироваться с бензольным кольцом и в которой R9 является водородом;
или его фармацевтически приемлемая соль.
| Способ получения производных борсодержащих пептидов | 1988 |
|
SU1807988A3 |
| RU 94034734 A1, 20.07.1996 | |||
| ВСЕСОЮЗНДЯ I РЯ?||ТЧч..Г::^/К7-'с-лг*t.. ^ ч Д,>&5 i «1. V ! :-,.;i;jii :_.' .;':j.;;;-;; | 0 |
|
SU335483A1 |
| C.KLE IN et al | |||
| Umwandlung von Omega - Guanidino - und Omega - Ureido - Alpha - aminosauren in alpha - Ket osauren und cleren hetorocyclische Folgeproducte | |||
| hiebigs aunalen des Chemic, 1988, № 9, p.1623-1637 | |||
| HANS LILYA A kallikrein like Serine Protease in Prostatic Fluid Cleaves the Predominant Seminal Vesicle Protein | |||
| Journal Clinic | |||
| Приспособление для установки двигателя в топках с получающими возвратно-поступательное перемещение колосниками | 1917 |
|
SU1985A1 |
