Настоящее изобретение касается нового и улучшенного способа получения производных 5-(гидроксиалкил)-1-фенил-1,2,4-триазола формулы (I)

в которой
R1A и R1B независимо друг от друга выбраны из группы, состоящей из водорода, фтора, хлора, метила, монофторметила, дифторметила, трифторметила, этила, метокси, дифторметокси и трифторметокси,
нового предшественника для его получения, а также получения и применения кристаллической модификации I (5-(4-хлорфенил)-2-({1-(3-хлорфенил)-5-[(1S)-1-гидроксиэтил]-1Н-1,2,4-триазол-3-ил}метил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-она формулы (IA-1).
Соединения формулы (I) действуют как эффективные двойные антагонисты V1a/V2-рецептора и могут быть использованы в качестве средства для профилактики и/или лечения сердечно-сосудистых и почечных заболеваний, таких как острая и хроническая сердечная недостаточность (worsening chronic heart failure), кардиоренальный синдром, гиперволемическая и эуволемическая гипонатриемия, цирроз печени, асциты, отеки и синдром неадекватной секреции антидиуретического гормона (SIADH), как описано в международной заявке WO 2016/071212.
Общий способ получения 5-фенилзамещенных производных 1,2,4-триазола описан в международной заявке WO 2011/104322 (см. схему 8, примеры 21, 25, 54, 56-61, 68-70). Однако описанным там способом на одной стадии процесса не может быть реализовано 1,3,5-замещение 1,2,4-триазольного кольца и, в частности, 1-фенилзамещение 1,2,4-триазольного кольца.
Далее следующая схема 1 показывает способ получения 5-фенилзамещенных производных 1,2,4-триазола согласно международной заявке WO 2011/104322.
Схема 1: Синтез производных 1,2,4-триазола согласно международной заявке WO 2011/104322.

[WO 2011/104322: Схема 8, стр. 32; L2 = помимо прочего, химическая связь; Ar2 = помимо прочего, замещенный фенил; Alk = алкил].
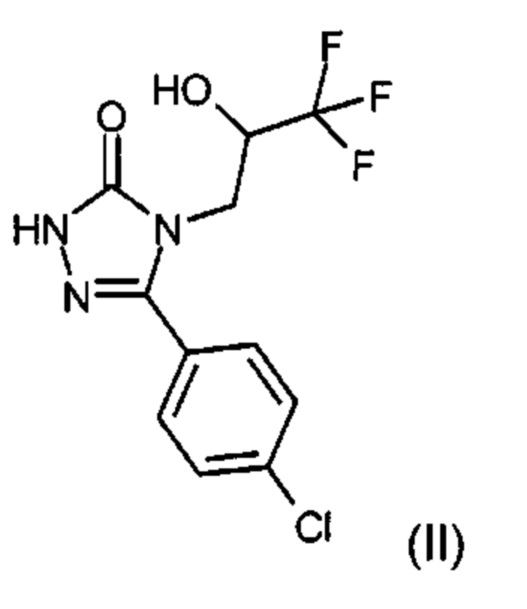
Соединения формулы (I) и их получение описаны в международной заявке WO 2016/071212. Описанный в ней исследовательский синтез рассматривается как ближайший уровень техники. Исходя из 5-(4-хлорфенил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-она (II) целевые соединения формулы (I) получают в 4 стадии с общим выходом не более ~12% от теории. На следующей стадии диастереомеры формулы (I-A) и (I-B) получают в лабораторном масштабе из смеси диастереомеров (I) посредством хирального разделения диастереомеров.
Соединение формулы (I) в международной заявке WO 2016/071212 получали в частности в виде твердого вещества, до сих пор не было описано конкретного способа кристаллизации на конечной стадии для получения фармацевтически приемлемых кристаллических форм.
Далее следующая схема 2 показывает способ получения соединений формулы (I).
Схема 2: Синтез соединений формулы (I) согласно международной заявке WO 2016/071212.

[R1A, R1B = водород, фтор, хлор, циано, метил, монофторметил, дифторметил, трифторметил, этил, метокси, дифторметокси и трифторметокси].
Синтез, раскрытый в WO 2016/071212, до стадии (V) включительно, а также на стадии (VII), аналогичен способу, раскрытому в WO 2011/104322.
Для получения соединения формулы (IV) 5-(4-хлорфенил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-он (II) подвергают взаимодействию с метил иловым эфиром бромуксусной кислоты (III) с образованием метил{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}ацетата (IV). Затем соединение (IV) преобразуют с гидразингидратом в 2-{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}ацетогидразид (V). Соединение (V) затем подвергают взаимодействию с имидным соединением (VI) с образованием смеси диастереомеров 5-(4-хлорфенил)-2-({5-[(1RS)-1-гидроксиэтил]-1Н-1,2,4-триазол-3-ил}метил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-она (VII). Катализируемое медью арильное сочетание («сочетание Чан-Лама») соединения (VII) с замещенной фенилбороновой кислотой (VIII) приводит к замещенным производным 5-(1-гидроксиэтил)-1-арил-1,2,4-триазола (I). Разделение с помощью хиральной хроматографии дает отдельные диастереомеры 5-(4-хлорфенил)-2-({5-[(1S)-1-гидроксиэтил]-1-(R1A,R1B)-фенил-1Н-1,2,4-триазол-3-ил}метил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-он (IA) и 5-(4-хлорфенил)-2-({5-[(1R)-1-гидроксиэтил]-1-(R1A,R1B)-фенил-1Н-1,2,4-триазол-3-ил}метил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-он (IB).
Упомянутая ранее схема реакции описана в WO 2016/071212 следующим образом: Реакционная последовательность от соединения формулы (II) через соединения формул (IV), (V) и (VII) к соединениям формулы (I), а также разделение на диастереомеры (IA) и (IB) представлены на схеме 2 и в примерах 1А, 2А, 4А и 10-83.
Однако данный способ, известный из WO 2016/071212, имеет различные недостатки в процедуре реакции, которые оказывают особенно неблагоприятное воздействие при получении соединений формулы (I) в промышленном масштабе. Общий выход после четырех стадий от (II) к (I) является очень низким, менее чем 15% от теории (от 1,3% до 13,1%). Многие стадии проводят при очень сильном разбавлении и с очень высоким избытком реагентов.
Синтез, описанный в WO 2016/071212, является особенно невыгодным на стадиях синтеза от соединения (VII) к (I) (промотируемое медью арильное сочетание, «сочетание Чан-Лама»), которое протекает лишь с 30% (между 3,0% и 30,1%) максимальным изолированным выходом и, следовательно, неблагоприятно с точки зрения атомной эффективности. Другим недостатком является то, что при реакции могут образовываться региоизомерные производные фенилтриазола, в результате реакции сочетания у другого кольцевого атома азота (кольцевая таутомерия производного 1,2,4-триазола (VII)). Это также отрицательно влияет на выход на этой стадии, и, кроме того, региоизомерные продукты затем должны быть с существенными затратами отделены на дополнительной стадии очистки. Кроме того, эта реакция является особенно невыгодной для синтеза в промышленном масштабе, поскольку в этой реакции используются стехиометрические количества ацетата меди. Это невыгодно, поскольку оставшиеся количества соли меди должны быть удалены ниже максимального предела, разрешенного в продукте по нормативным соображениям, что означает дополнительные расходы. Кроме того, реагенты должны быть легкодоступными и иметь невысокую цену.
В синтезе согласно международной заявке WO 2016/071212 стереоизомерную смесь формулы (I) в лабораторном масштабе разделяли на диастереомеры с помощью хиральной хроматографии. Такое хроматографическое разделение является очень затратным и трудоемким и, следовательно, неподходящим для синтеза в промышленном масштабе. Кроме того, в результате дополнительной стадии еще более снижается общий выход. Еще одним недостатком является то, что целевое соединение в соответствии с методикой, описанной в WO 2016/071212, не получают в определенной фармацевтически приемлемой кристаллической форме.
Поэтому существовала потребность в реализуемом на практике, промышленном синтезе, который воспроизводимо давал бы соединения формулы (I) с высоким общим выходом, низкими производственными расходами и высокой чистотой. Кроме того, существовала необходимость в крупномасштабном практическом синтезе, который отвечал бы всем нормативным требованиям, которые должны соблюдаться для того, чтобы активное вещество могло быть использовано в клинических испытаниях и для государственной регистрации.
Неожиданным образом был обнаружен очень эффективный способ получения соединения формулы (I), который соответствует вышеуказанным требованиям. Указанный новый способ делает возможным эффективный синтез 5-гидроксиалкилзамещенных производных 1-фенил-1,2,4-триазола.
Важным преимуществом способа согласно изобретению является значительно повышенный выход после всех стадий. Новый способ согласно изобретению по варианту проведения (А) обеспечивает целевые соединения (I) в четыре стадии с общим выходом более чем 20% от теории (от 23,8% до 53,2%). Хроматографическая очистка промежуточных продуктов не требуется. Таким образом, в альтернативном варианте проведения (В) последние два этапа и в другом альтернативном варианте проведения (С) - даже три последних этапа могут быть выполнены в виде однореакторного процесса. За счет этого можно добиться дальнейшего увеличения общего выхода после четырех стадий (до 63,2%).
Схемы и стадии способа, описанные ниже, представляют собой пути синтеза к соединениям общей формулы (I) согласно изобретению, и не должны пониматься как ограничивающие. Специалисту в данной области техники будет понятно, что последовательность превращений, как проиллюстрировано в качестве примера на схемах 3 и 4, может варьироваться различным образом, и поэтому показанную последовательность не следует рассматривать как ограничение. Кроме того, преобразование функциональных групп отдельных радикалов и заместителей, в частности тех, которые приведены для R1 и R2, может происходить до и/или после описанных для примера превращений, причем исходя из других, полученных вышеуказанным способом, соединений формулы (I) или их предшественников. Эти превращения осуществляют обычными методами, известными специалисту в данной области, и включают, например, реакции, такие как реакции нуклеофильного или электрофильного замещения, реакции сочетания, промотируемые переходными металлами, реакции получения и присоединения металлоорганических соединений (например, соединений Гриньяра или литийорганических соединений), реакции окисления и восстановления, гидрирование, галогенирование (например, фторирование, бромирование), дегалогенирование, аминирование, алкилирование и ацилирование, образование эфиров карбоновых кислот, амидов карбоновых кислот и сульфонамидов, расщепление и гидролиз сложных эфиров, введение и удаление временных защитных групп или других реакций, известных специалисту в данной области. Указанные превращения также включают те, которые подразумевают введение функциональной группы, которая делает возможным дальнейшее преобразование заместителей. Подходящие защитные группы, а также реагенты и условия реакции для их введения или соответственно отщепления, известны специалисту в данной области (см., например, T.W. Greene, P.G.M. Wuts; "Protective Groups in Organic Synthesis", 3. Издание; Wiley 1999). Конкретные примеры приведены в следующих разделах текста.
Ниже следующая схема 3 иллюстрирует новый способ согласно изобретению для получения соединений формулы (I).
Схема 3: Способ получения соединения формулы (I) согласно изобретению.

[R1A,R1B = водород, фтор, хлор, метил, монофторметил, дифторметил, трифторметил, этил, метокси, дифторметокси и трифторметокси]; а) Na2CO3, метилизобутилкетон; b) Метилат натрия, МеОН, с) 1. (XII-А), DIPEA, Толуол/ТГФ, 2. (XIII), DIPEA, ТГФ; d) NaOH, МеОН].
Причем в соединениях схемы синтеза 3
R1A и R1B независимо друг от друга выбраны из группы, состоящей из водорода, фтора, хлора, метила, монофторметила, дифторметила, трифторметила, этила, метокси, дифторметокси и трифторметокси,
Причем предпочтительно в соединениях схемы синтеза 3
R1A и R1B независимо друг от друга выбраны из группы, состоящей из водорода, фтора и хлора, причем по меньшей мере один из заместителей отличается от водорода.
Причем особо предпочтительно в соединениях схемы синтеза 3
R1A означает водород, и
R1B означает хлор в положении 2 или в положении 3.
Причем в высшей степени предпочтительно в соединениях схемы синтеза 3
R1A означает водород, и
R1B означает хлор в положении 3.
Ниже рассматриваются отдельные стадии способа согласно изобретению для получения соединения формулы (I) согласно схеме 3. Обсуждаются также альтернативы, характеризующиеся отсутствием выделения соединений формулы (XI) и (XIV).
Для получения производных 5-(1-гидроксиэтил)-1-арил-1,2,4-триазола (I) взаимодействию подвергают 5-(4-хлорфенил)-4-((2S)-3,3,3-трифтор-2-гидроксипропил)-2,4-дигидро-3Н-1,2,4-триазол-3-он (II) с образованием {3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}ацетонитрила (X) (стадия 1). Затем нитрильное соединение (X) посредством реакции с метилатом натрия превращают в метил-2-{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}этанимидат (XI) (стадия 2). Затем осуществляют образование 1,2,4-триазольного кольца посредством трехкомпонентной реакции циклизации, в которой иминослож-ноэфирное соединение (XI) реагирует с хлорангидридом 2-ацетоксипропионовой кислоты (XII) и замещенным фенилгидразиновым соединением (XIII), с получением 1-{3-({3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}метил)-1-[(R1A,R1B)-фенил]-1Н-1,2,4-триазол-5-ил}этилацетата (XIV) (стадия 3). Последующее отщепление ацетильной группы обеспечивает целевые соединения формулы (I) (стадия 4). В варианте (В) способ можно проводить таким образом, чтобы не выделять защищенный ацетат (XIV), а далее подвергнуть реакции непосредственно в растворе (стадии 3+4). В следующем варианте (С) способ может быть осуществлен в виде однореакторного процесса для этапов (X)→(XI)→(XIV)→(I) (стадии 2+3+4), ив этом случае общий процесс состоит только из двух изолированных этапов вместо 4 из уровня техники.
Особенно предпочтительной является трехкомпонентная реакция циклизации согласно изобретению для образования 1,2,4-триазольного кольца (стадия 3), которая позволяет вводить два кольцевых заместителя в 1- и 5-положение на одной стадии способа, так что для этой стадии достигается высокий выход (от 37,0% до 83,0% для стадии 3, после снятия защитной группы: от 36,7% до 82,2% после 2 этапов для стадий 3+4). Синтез, описанный в предшествующем уровне техники (схема 2), обеспечивает для аналогичной последовательности (V)→(VII)→(I) значительно более низкий выход после двух стадий от 2,9% до 28,9%.
Далее также предпочтительна надежность способа согласно изобретению, ведь, как описано выше, последовательность также может быть выполнена в виде однореакторного процесса (варианты проведения В и С), таким образом нет необходимости выделять промежуточные продукты и проводить хроматографическую очистку промежуточных продуктов, а они непосредственно подвергаются следующей стадии в том же реакционном сосуде и/или реакционной среде. Такого рода однореакторный процесс особенно выгоден для крупномаштабного синтеза, поскольку в результате можно избежать дополнительных стадий обработки, и добиться высокого общего выхода для процесса.
Получение исходного соединения формулы (II), описанного в схеме синтеза 3, может быть осуществлено в соответствии со следующей схемой синтеза 4, исходя из коммерчески доступных или соответственно известных специалисту в данной области исходных соединений:
Схема 4: Способ получения соединений формулы (II).

[е) ТГФ; f) водн. NaOH, Δ; g) 1. (CF3CO)2O / Пиридин, 2. водн. HCl, Δ; h) хиральный Ru(II)-катализатор, НСООН / Et3N.]
Исходные вещества формулы (II) описаны в международной заявке WO 2010/105770 (см. схемы 4 и 5, примеры 1А, 2А, 3А, 4А и 158А) и WO 2011/104322 (см. схему 1, примеры 1А, 2А, 3А, 4А и 5А). Указанные соединения формулы (II) получают взаимодействием 4-хлорбензогидразида (XV) с этил-2-изоцианатоацетатом (XVI) с образованием этил-N-({2-[(4-хлорфенил)карбонил]гидразинил}карбонил)глицината (XVI). Затем его превращают в [3-(4-хлорфенил)-5-оксо-1,5-дигидро-4Н-1,2,4-триазол-4-ил] уксусную кислоту (XVIII) посредством инициируемой основанием реакции циклизации. 5-(4-Хлорфенил)-4-(3,3,3-трифтор-2-оксопропил)-2,4-дигидро-3Н-1,2,4-триазол-3-он (IX) затем получают посредством реакции с трифто-руксусным ангидридом и последующей обработки соляной кислотой. Затем кетон (IX) превращают в хиральный спирт (II) путем асимметричного гидрирования с переносом водорода в присутствии энантиоселективного катализатора на основе рутения (II).
При помощи нового синтеза согласно изобретению целевое соединение (I) удалось получить очень эффективным образом. Этот способ дает значительные преимущества по сравнению с уровнем техники с точки зрения масштабируемости и технической реализации. Общий выход значительно выше по сравнению с опубликованными данными и кроме того в основном достигается очень высокая чистота активного вещества. Вариант осуществления нового способа делает возможным воспроизводимое экономически эффективное получение определенной кристаллической формы, не описанной в предшествующем уровне техники. Посредством представленного здесь способа согласно изобретению уже было успешно получено несколько кг материала для клинических испытаний.
В качестве солей в рамках настоящего изобретения предпочтительными являются физиологически приемлемые соли соединений согласно изобретению (например, см. S.М. Berge и др., Pharmaceutical Salts, J. Pharm. Sci. 977, 66, 1-19). Однако также включены соли, которые сами по себе не подходят для фармацевтических применений, но могут быть использованы, например, для выделения или очистки соединений согласно изобретению.
Физиологически приемлемые соли соединений согласно изобретению включают аддитивные соли минеральных кислот, карбоновых и сульфоновых кислот, например соли хлороводородной, бромоводородной, серной, фосфорной, метансульфокислоты, этансульфокислоты, толуолсульфокислоты, бензолсульфокислоты, нафталиндисульфокислоты, уксусной, три-фторуксусной, пропионовой, молочной, винной, яблочной, лимонной, фумаровой, малеиновой и бензойной кислоты.
Физиологически приемлемые соли соединений согласно изобретению включают также соли обычных оснований, как, например и предпочтительно, соли щелочных металлов (например, соли натрия и калия), соли щелочно-земельных металлов (например, соли кальция и магния) и соли аммония, производные от аммиака или органических аминов с 1-16 атомами углерода, как, например и предпочтительно, этиламин, диэтиламин, три-этиламин, этилдиизопропиламин, моноэтаноламин, диэтаноламин, три-этаноламин, дициклогексиламин, диметиламиноэтанол, прокаин, дибензи-ламин, N-метилморфолин, аргинин, лизин, этилендиамин, N-метилпиперидин и холин.
Сольватами в рамках изобретения обозначают такие формы соединений согласно изобретению, которые в твердом или жидком состоянии в результате координации образуют комплекс с молекулами растворителя. Гидраты являются особой формой сольватов, в которой осуществляется координация с водой.
Соединения формулы (IA), (XII-A), (XIV-A) и (IB), (XII-B), (XIV-B) являются в каждом случае частью соединений формулы (I), (XII) и (XIV), и в каждом случае представляют собой энантиомеры или диастереомеры относительно стереоцентра спиртовой группы в 5-положении 1,2,4-триазольного кольца или его защищенной формы. Соединения формулы (IA), (XII-А) и (XIV-A) при этом имеют (S)-конфигурацию стереоцентра, а соединения формулы (IB), (XII-В) и (XIV-A) в каждом случае имеют (R)-конфигурацию стереоцентра.
Если соединения согласно изобретению могут встречаться в таутомерных формах, настоящее изобретение охватывает все таутомерные формы.
Следующие три таутомерных варианта изображения (а), (b) и (с) производного триазола эквивалентны друг другу и являются равнозначными и во всех случаях описывают 1,4-дизамещенное производное триазола.



Это относится, в частности, к следующим структурным элементам: Н-1,2,4-триазол-3-ил, 1Н-1,2,4-триазол-5-ил, 4Н-1,2,4-триазол-3-ил и 4Н-1,2,4-триазол-5-ил. Y1 и Y2 представляют собой при этом различные заместители.
Объектом настоящего изобретения является способ получения соединений общей формулы (I), или их солей, их сольватов или сольватов их солей, отличающийся тем, что он включает стадии [С] и [D], причем
[С] Соединение общей формулы (XI),

причем
R2 означает алкил с 1-4 атомами углерода, предпочтительно метил,
подвергают взаимодействию один за другим на первой стадии
[С-1] в присутствии основания с хлорангидридом кислоты общей формулы (XII)
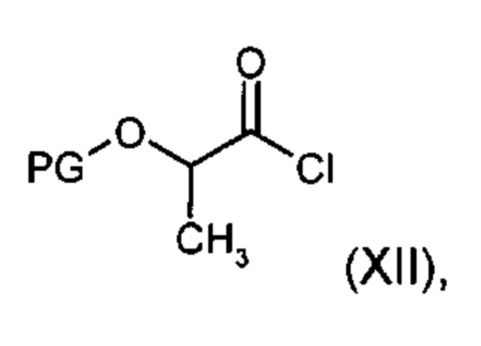
причем
PG означает защитную группу, предпочтительно ацетил,
и полученный при этом промежуточный продукт затем на следующей стадии подвергают взаимодействию
[С-2] в присутствии основания с фенилгидразинным соединением общей формулы (XIII)

причем R1A и R1B независимо друг от друга выбраны из группы, состоящей из водорода, фтора, хлора, метила, монофторметила, дифторметила, трифторметила, этила, метокси, дифторметокси и трифторметокси,
с получением 1,2,4-триазолильного соединения общей формулы (XIV)

причем остатки PG, R1A и R1B имеют вышеуказанные значения, и его на следующей стадии
[D] посредством удаления защитной группы PG превращают в соединение общей формулы (I)

причем остатки R1A и R1B имеют вышеуказанные значения.
В отличие от предшествующего уровня техники (WO 2016/071212) получение (I) протекает (через (XI)+(XII)+(XIII)→(XIV)→(I) см. также схему 3: стадии 3+4: от 36,7% до 82,2% после 2х стадий) со значительно более высоким выходом, чем для аналогичной последовательности из уровня техники (см. схему 2: (V)→(VII)→(I), от 2,9% до 28,9% после 2х стадий).
В предпочтительном варианте осуществления способ согласно изобретению осуществляют в виде однореакторной реакции по многоступенчатой процедуре, адаптированной к химическому механизму.
При этом способ осуществляют в присутствии подходящего растворителя и промежуточный продукт со стадии [С-1] без выделения, т.е. в растворе, затем подвергают взаимодействию на следующей стадии [С-2].
В еще одном предпочтительном варианте осуществления полученное на стадии [С-2] 1,2,4-триазолильное соединение общей формулы (XIV) без выделения, т.е. в растворе, превращают на следующей стадии [D] в соединение общей формулы (I).
В одном варианте осуществления способ согласно изобретению перед стадией [С] содержит дополнительную стадию [В], причем
[В] Соединение общей формулы (X)

подвергают взаимодействию с основным алкоксилатом с 1-4 атомами углерода, предпочтительно метилатом натрия, с получением иминосложно-эфирного соединения общей формулы (XI),
причем
R2 означает алкил с 1-4 атомами углерода, предпочтительно метил.
В предпочтительном варианте осуществления способ согласно изобретению осуществляют в виде однореакторной реакции по многоступенчатой процедуре, адаптированной к химическому механизму.
При этом реакцию осуществляют в присутствии подходящего растворителя и иминосложноэфирное соединение общей формулы (XI) со стадии [В] без выделения, т.е. в растворе, затем подвергают взаимодействию на следующей стадии [С].
В одном варианте осуществления способ согласно изобретению включает стадию [В] перед стадией [С] и стадию [В] перед стадией [А], причем
[А] Соединение общей формулы (II)

подвергают взаимодействию с нитрильным соединением (IX),

причем X означает уходящую группу, предпочтительно хлорид или бромид,
с получением соединения общей формулы (X)

В следующем варианте осуществления способ согласно изобретению включает указанные стадии [А], [В], [С] и [D], причем
[А] Соединение общей формулы (II)
подвергают взаимодействию с нитрильным соединением (IX),

причем X означает уходящую группу, предпочтительно хлорид или бромид,
с получением соединения общей формулы (X)

и его на следующей стадии
[В] подвергают взаимодействию с основным алкоксилатом с 1-4 атомами углерода, предпочтительно метилатом натрия, с получением имино-сложноэфирного соединения общей формулы (XI),

причем
R2 означает алкил с 1-4 атомами углерода, предпочтительно метил,
и его на следующей стадии
[С] один за другим на первой стадии
[С-1] подвергают взаимодействию в присутствии основания с хлорангидридом кислоты общей формулы (XII)

причем
PG означает защитную группу, предпочтительно ацетил,
и полученный при этом промежуточный продукт затем на следующей стадии
[С-2] в присутствии основания подвергают взаимодействию с фенилгидразинным соединением общей формулы (XIII)

причем R1A и R1B независимо друг от друга выбраны из группы, состоящей из водорода, фтора, хлора, метила, монофторметила, дифторметила, трифторметила, этила, метокси, дифторметокси и трифторметокси,
с получением 1,2,4-триазолильного соединения общей формулы (XIV)

причем остатки R1A и R1B имеют вышеуказанные значения, и
PG означает защитную группу, предпочтительно ацетил,
и его на следующей стадии
[D] посредством удаления защитной группы PG превращают в соединение общей формулы (I)

причем остатки R1A и R1B имеют вышеуказанные значения.
Предпочтительным является способ получения соединение формулы (I), отличающийся тем, что R1A и R1B независимо друг от друга выбраны из группы, состоящей из водорода, фтора и хлора, причем по меньшей мере один из заместителей отличается от водорода.
Особенно предпочтительным является способ получения соединений формулы (I), отличающийся тем, что R1A представляет собой водород и R1B представляет собой хлор в положении 2 или в положении 3.
В высшей степени предпочтительным является способ получения соединений формулы (I), отличающийся тем, что R1A представляет собой водород и R1B представляет собой хлор в положении 3.
В высшей степени предпочтительным является способ получения соединений формулы (I-A-1)

Другим объектом настоящего изобретения является способ получения соединений общей формулы (X), или их солей, их сольватов или сольватов их солей, отличающийся тем, что он включает стадию [А], причем
[А] Соединение общей формулы (II)

подвергают взаимодействию с нитрильным соединением (IX),
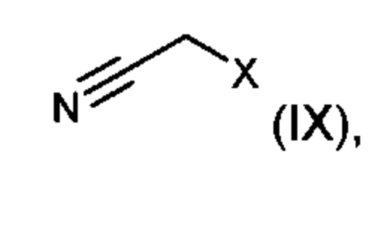
причем X означает уходящую группу, предпочтительно хлорид или бромид,
с получением соединения общей формулы (X)

Другим объектом настоящего изобретения является способ получения соединений общей формулы (XI), или их солей, их сольватов или сольватов их солей, отличающийся тем, что он включает стадию [В], причем
[В] Соединение общей формулы (X)

подвергают взаимодействию с основным алкоксилатом с 1-4 атомами углерода, предпочтительно метилатом натрия, с получением иминосложно-эфирного соединения общей формулы (XI),

причем
R2 означает алкил с 1-4 атомами углерода, предпочтительно метил,
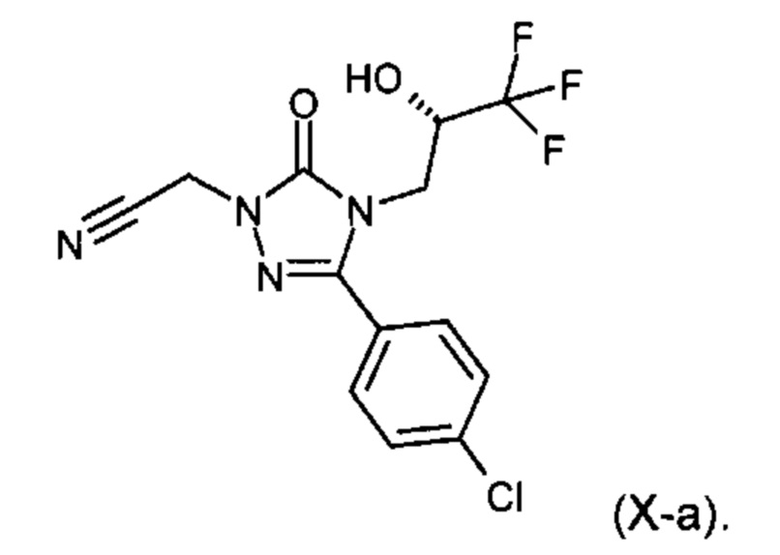
Другим объектом настоящего изобретения является соединение формулы (X)

В предпочтительном варианте осуществления настоящего изобретения соединение представляет собой {3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}ацетонитрил (Х-а)
Другим объектом настоящего изобретения является применение соединения общей формулы (X) для получения соединения общей формулы (I).
Другим объектом настоящего изобретения является соединение формулы (XI)

причем
R2 означает алкил с 1-4 атомами углерода, предпочтительно метил.
В предпочтительном варианте осуществления настоящего изобретения соединение представляет собой метил-2-{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}этанимидат (XI-a)

Другим объектом настоящего изобретения является применение соединения общей формулы (XI) для получения соединения общей формулы (I).
Другим объектом настоящего изобретения является применение соединения общей формулы (XIV) для получения соединения общей формулы (I).
Стадия 1: Подходящими основаниями для стадии способа [А]: (II)+(IX)→(X) являются обычные неорганические или органические основания, как например и предпочтительно карбонаты щелочных металлов, такие как карбонат натрия, карбонат калия или карбонат цезия, алкоголяты щелочных металлов, такой как трет-бутилат натрия или трет-бутилат калия, или органические амины, такие как N,N-диизопропилэтиламин (DIPEA) и три-этиламин. В качестве растворителя могут быть использованы инертные растворители, такие как ацетонитрил, метилизобутилкетон, диоксан, диметилформамид, диметилацетамид, N-метилпирролидинон, диметилсуль-фоксид или сульфолан. Предпочтительно используют карбонат калия в метилизобутилкетоне или ацетонитриле.
При необходимости указанные стадии способа преимущественно могут быть осуществлены с добавлением катализаторов алкилирования, таких как например бромид лития, иодид натрия, бромид тетра-н-бутиламмония или хлорид бензилтриэтиламмония. Кроме того, может оказаться выгодным в течение длительного периода времени медленно добавлять алкилирующий агент хлорацетонитрил или бромацетонитрил. Реакции обычно проводят в диапазоне температур от +40°С до +120°С, предпочтительно при температуре от +60°С до +80°С.
Взаимодействие можно проводить при нормальном, повышенном или пониженном давлении (например, от 0,5 до 5 бар); как правило работают при нормальном давлении.
Альтернативно, соединения формулы (X) также могут быть получены из соединений формулы (XX), известных в литературе (см. схему 5):
Схема 5:

[PvCl = хлорангидрид пивалиновой кислоты, TFAA = ангидрид трифторуксусной кислоты].
Реакция сочетания (XX)→(XXI) [образование амида] может быть осуществлена либо непосредственно с помощью конденсирующего или активирующего средства в присутствии основания, либо через промежуточное соединение получаемого из (XX) хлорангидрида карбоновой кислоты, сложного эфира карбоновой кислоты или имидазолида карбоновой кислоты.
В качестве таких конденсирующих или активирующих средств пригодными являются, например, карбодиимиды, такие как N,N'-диэтил-, N,N'-дипропил-, N,N'-диизопропил-, N,N'-дициклогексилкарбодиимид (DCC) или N-(3-диметиламинопропил)-N'-этилкарбодиимид гидрохлорид (EDC), производные фосгена, такие как N,N'-карбонилдиимидазол (CDI), изопропил-хлорформиат или изобутилхлорформиат, 1,2-оксазолиевые соединения, такие как 2-этил-5-фенил-1,2-оксазолия-3-сульфат или 2-трет-бутил-5-метилоксазолия перхлорат, ациламинные соединения, такие как 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин, α-хлоренамины, такие как 1-хлор-N,N,2-триметилпроп-1-ен-1-амин, производные 1,3,5-триазина, такие как хлорид 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния, фосфорные соединения, такие как ангидрид н-пропанфосфоновой кислоты (Т3Р, РРАСА), сложный диэтиловый эфир цианофосфоновой кислоты, дифенилфосфорилазид (DPPA), бис(2-оксо-3-оксазолидинил)-фосфорилхлорид, гексафторфосфат бензотриазол-1-илокси-трис(диметиламино)фосфония или гексафторфосфат бензотриазол-1-илокси-трис(пирролидино)фосфония (РуВОР) или урониевые соединения, такие как тетрафторборат O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (TBTU), гексафторфосфат O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HBTU), тетрафторборат O-(1Н-6-хлорбензотриазол-1-ил)-1,1,3,3-тетраметилурония (TCTU), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU), или тетрафторборат 2-(2-оксо-1-(2Н)-пиридил)-1,1,3,3-тетраметилурония (TPTU), при необходимости в комбинации с дополнительными вспомогательными веществами, такими как 1-гидроксибензотриазол (HOBt) или N-гидроксисукцинимид (HOSu), а также в качестве основания карбонаты щелочных металлов, такие как карбонат натрия или калия, или третичные аминные основания, такие как триэтиламин, N-метилморфолин (NMM), N-метил-пиперидин (NMP), N,N-диизопропилэтиламин (DIPEA), пиридин или 4-N,N-диметиламинопиридин (DMAP). Как правило, хлорангидриды кислот получают реакцией с тионилхлоридом или оксалилхлоридом в инертном растворителе, таком как дихлорметан или N,N-диметилформамид. Также возможно использовать смеси перечисленных растворителей.
Превращение в нитрил (XXI)→(X) может быть проведено в присутствии дегидратирующего агента. Типичными дегидратирующими агентами являются, например, трифторуксусный ангидрид (TFAA), пятиокись фосфора (Р4О10), фосфорилхлорид (POCl3), пентахлорид фосфора (PCl5), CCl4-PPh3 (реагент Аппеля), гексаметилфосфорамид (НМРА); метил-N-(триэтиламмонийсульфонил)карбамат (реагент Бурджесса), хлорид (хлор-метилен)диметилиминия (реагент Вилсмайера), оксалилхлорид/ДМСО и тионилхлорид (SOCl2).
Типичными растворителями для обеих стадий процесса (XX)→(XXI) и (XXI)→(X) являются, например, простые эфиры, такие как диэтиловый эфир, диоксан, тетрагидрофуран, диметиловый эфир этиленгликоля или диметиловый эфир диэтиленгликоля, углеводороды, такие как бензол, толуол, ксилол, гексан, циклогексан или нефтяные фракции, галогенированные углеводороды, такие как дихлорметан, трихлорметан, тетрахлорметан, 1,2-дихлорэтан, трихлорэтилен или хлорбензол или другие растворители, такие как ацетон, этилацетат, ацетонитрил, диметилсульфоксид, N,N-диметилформамид, N,N'-диметилпропиленмочевина (DMPU), N-метилпирролидон (NMP) или пиридин. Также возможно использовать смеси названных растворителей.
Обычно и предпочтительно карбоновую кислоту (XX) подвергают взаимодействию на первой стадии с пивалоил хлоридом в присутствии пиридина, причем получают промежуточное соединение, которое на следующей стадии подвергают взаимодействию с аммиаком. Как правило, образовавшееся промежуточное соединение не выделяют и проводят реакцию в два этапа в виде однореакторного процесса. Подходящими основаниями для первой стадии являются предпочтительно пиридин, 4-(N,N-диметиламино)пиридин или N,N-диизопропилзтиламин (DIPEA). Превращение карбоксамида (XX) в нитрил (X) затем обычно осуществляют путем взаимодействия с ангидридом трифторуксусной кислоты. Обе реакции проводят в инертном органическом растворителе, предпочтительно тетра-гидрофуране.
Соединения формулы (XX) известны из литературы (см. международную заявку WO 2010/105770, схема 2, примеры 8А и 9А и WO 2011/104322, схема 11).
Стадия 2: Основаниями, которые могут быть использованы для получения сложного иминоэфира (XI) на стадии способа [В], являются основные алкоголяты щелочных металлов с 1-4 атомами углерода, такие как, например, метилат натрия, этилат натрия, пропилат натрия, изопропилат натрия, трет-бутилат натрия или трет-бутилат калия. Подходящими спиртами являются спирты, такие как метанол, этанол, н-пропанол, 2-пропанол, н-бутанол, 2-бутанол, а также трет-бутанол. Предпочтительно используют метилат натрия в метаноле.
Реакции обычно проводят в интервале температур от +20 до +80°С, предпочтительно от +40 до +60°С. Сложный иминоэфир (XI) не обязательно промежуточно выделять, а можно использовать посредством передистилляции метанола на толуол или тетрагидрофуран непосредственно на следующей стадии.
Стадия 3: Реакцию многокомпонентной циклизации на стадии способа [С] проводят в виде двухступенчатого процесса. Сначала, на стадии способа [С-1] сложный иминоэфир (XI) подвергают взаимодействию с хлорангидридом кислоты (XII) в присутствии основания и затем полученное промежуточное соединение на стадии способа [С-2] в присутствии основания подвергают взаимодействию с фенилгидразинным соединением (XIII). Как правило, образовавшееся промежуточное соединение не выделяют и двухступенчатую реакцию проводят в виде однореакторного процесса.
При этом хлорангидрид кислоты (XII) на стадии [С-1] предпочтительно используют в количестве от 1,1 до 1,5 моль, предпочтительно в количестве 1,2 моль, в пересчете на 1 моль соединения формулы (XI). Основание на стадии [С-1] обычно используют в количестве от 1 до 2,5 моль, предпочтительно в количестве от 1,05 до 2,0 моль, особенно предпочтительно в количестве от 1,05 до 1,5 моль, в пересчете на 1 моль соединения формулы (XII).
Гидразин (XIII) на стадии [С-2] можно также использовать в форме соли, например, в виде гидрохлорида или в виде соли п-толуолсульфоновой кислоты (тозилата). В основных условиях реакции солевая форма затем превращается в свободный гидразин. В этом случае количество основания может быть подобрано соответствующим образом. В еще одном предпочтительном варианте осуществления перед добавлением проводят нейтрализацию соли гидразина в отдельном реакционном сосуде, и затем полученный раствор, при необходимости после отфильтровывания образовавшейся соли, в виде раствора добавляют к реакционной смеси.
Основание на стадии [С-2] обычно используют в количестве от 1,05 до 1,5 моль, предпочтительно в количестве от 1,2 до 1,5 моль, в пересчете на 1 моль соединения формулы (XIII).
Подходящими для обеих стадий основаниями обычно являются третичные аминные основания, такие как N,N-диизопропилэтиламин (DIPEA), триэти-ламин, триизопропиламин, N-метилимидазол, N-метилморфолин, пиридин, а также 4-(диметиламино)пиридин. Предпочтительным является триэти-ламин или N,N-диизопропилэтиламин. Особенно предпочтительным является диизопропилэтиламин.
Подходящими растворителями являются инертные органические растворители, как например дихлорметан, 1,2-дихлорэтан, метил-трет-бутиловый эфир, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, толуол, пиридин, этилацетат, ацетонитрил или N,N-диметилформамид или смеси указанных растворителей.
Предпочтительно применяют тетрагидрофуран (ТГФ) или смеси тетрагидрофурана и толуола.
Реакция с хлорангидридом кислоты (XII) на стадии [С-1] и с гидразином (XIII) на стадии [С-2] происходит в диапазоне температур от -20°С до +30°С, предпочтительно от 0°С до +10°С. Для образования триазола с удалением воды (циклизация) на стадии [С-2] реакционную смесь затем доводят до температуры от +20 до +150°С. Предпочтительно указанное взаимодействие проводят при температуре от +70 до +80°С.
Стадия 4: Введение и удаление защитной группы PG на стадии способа (D) осуществляют согласно обычным в литературе методам [см. например T.W. Greene und P.G.M. Wuts, Protective Groups in Organic Synthesis, Wiley, New York, 1999]. Таким образом, ацетильную группу предпочтительно удаляют с помощью основания, такого, например, как водный раствор гидроксида натрия.
Если на стадии способа [D] снятие защиты проводят при помощи водного раствора гидроксида натрия, обработку осуществляют, например, посредством экстракции подходящим растворителем с последующей многократной промывкой и сушкой. Предпочтительно экстрагируют метил-трет-бутиловым эфиром (MtBE).
Соединения формул (II), (IX), (XII), (XIII) и (XX) либо коммерчески доступны, либо как таковые описаны в литературе, либо они могут быть получены специалистом в данной области техники по аналогии с опубликованным в литературе методами. Многочисленные подробные методики и литературные ссылки для получения исходных материалов также находятся в экспериментальной части.
Поскольку соединение формулы (I-A-1) действует в форме таблетки, существует большая потребность в воспроизводимом выделении в определенной кристаллической форме изолированного соединения формулы (I-А-1), так чтобы можно было обеспечить воспроизводимую биологическую усвояемость.
Неожиданно было обнаружено, что соединение (5-(4-хлорфенил)-2-({1-(3-хлорфенил)-5-[(1S)-1-гидроксиэтил]-1Н-1,2,4-триазол-3-ил}метил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-он формулы (IA-1)

может кристаллизоваться из смеси метил-трет-бутилового эфира и диизопропилового эфира или из смеси метил-трет-бутилового эфира и н-гептана, причем воспроизводимо образуется кристаллическая модификация I (стадия 5).
Стадия 5: При этом раствор соединения формулы (I-A-1) в 5-10-кратном избытке метил-трет-бутилового эфира (MtBE) перемешивают при 20-80°С, предпочтительно при 50-60°С и более предпочтительно при температуре кипения с возвратом флегмы метил-трет-бутилового эфира (около 54°С), и смешивают при этой температуре с диизопропиловым эфиром. При непрерывном добавлении диизопропилового эфира отгоняют MtBE. При этом выкристаллизовывается соединение формулы (I-A-1). Охлаждают до температуры от 0 до 30°С, предпочтительно от 10 до 20°С, отделяют кристаллы и сушат их в вакууме при температуре от 40 до 60°С, предпочтительно при 40-50°С.
Альтернативно может быть использована смесь метил-трет-бутилового эфира и н-гептана. При этом раствор соединения формулы (I-A-1) в 5-10-кратном избытке метил-трет-бутилового эфира (MtBE) перемешивают при 20-80°С, предпочтительно при 50-60°С и более предпочтительно при температуре кипения с возвратом флегмы метил-трет-бутилового эфира (около 54°С), и смешивают при этой температуре с 1,5-2,5-кратным объемом н-гептана, причем выкристаллизовывается соединение формулы (I-A-1). Охлаждают до температуры от 0 до 30°С, предпочтительно от 10 до 20°С, отделяют кристаллы и сушат их в вакууме при температуре от 40 до 80°С, предпочтительно при 40-50°С.
Опираясь на технические требования стандарта GMP, может быть целесообразным перед нагреванием сначала подвергнуть раствор продукта в MtBE фильтрованию частиц.
Обработку обычно проводят посредством фильтрации, многократной промывки диизопропиловым эфиром или соответственно н-гептаном и последующей сушки.
Достигнутая химическая чистота >99% и содержание около 100% соответствуют критериям для коммерческих продуктов в соответствии с директивой ICH. Оптическая чистота составляет >>99% ее.
Способ кристаллизации является очень надежным и обеспечивает воспроизводимым образом желаемую кристаллическую форму. Соединение формулы (I) как правило микронизируют и в фармацевтике формуют в таблетки. Оказывается, что указанная кристаллическая форма обладает очень хорошими характеристиками стабильности (даже при высокой влажности воздуха) и может храниться в течение нескольких месяцев без потери стабильности.
Другим объектом настоящего изобретения является соединение (5-(4-хлорфенил)-2-({1-(3-хлорфенил)-5-[(1S)-1-гидроксиэтил]-1Н-1,2,4-триазол-3-ил}метил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-он в кристаллической форме модификации I.
Объектом настоящего изобретения является соединение формулы (I-A-1) в кристаллической форме модификации I, отличающееся тем, что рентгеновская дифрактограмма указанного соединения имеет максимумы пиков 2 тета-угла при 7,0, 8,9, 16,8, 17,7, 17,9, 18,1, 21,6, 21,8, 22,4 и 24,6.
Другим объектом настоящего изобретения является способ получения соединения формулы (I-A-1) в кристаллической форме модификации I, отличающийся тем, что соединение формулы (I-A-1), находящееся в одной или нескольких модификациях или в виде сольвата, перемешивают в смеси метил-трет-бутилового эфира и диизопропилового эфира или в смеси метил-трет-бутилового эфира и н-гептана при температуре от 20°С до 80°С, затем фильтруют, промывают и сушат в вакууме.
Предпочтительным растворителем для способа получения соединения формулы (I-A-1) в кристаллической форме модификации I является смесь метил-трет-бутилового эфира и диизопропилового эфира или смеси метил-трет-бутилового эфира и н-гептана.
Предпочтительный температурный диапазон для способа получения соединения формулы (I-A-1) в кристаллической форме модификации I находится при температуре кипения с возвратом флегмы метил-трет-бутилового эфира (около 54°С).
Другим объектом настоящего изобретения является соединение формулы (I-A-1) в кристаллической форме модификации I, как описано выше, для лечения заболеваний.
Другим объектом настоящего изобретения является лекарственное средство, содержащее соединение формулы (I-A-1) в кристаллической форме модификации I, как описано выше, и не содержащее значительной доли другой формы соединения формулы (I-A-1), чем кристаллическая форма модификации I, как описано выше. Еще одним объектом настоящего изобретения является лекарственное средство, содержащее соединение формулы (I-A-1) в кристаллической форме модификации I, как описано выше, в количестве более 90% масс, в пересчете на общее количество содержащегося соединения формулы (I-A-1).
Другим объектом настоящего изобретения является применение соединения формулы (I-A-1) в кристаллической форме модификации I, как описано выше, для получения лекарственного средства для лечения сердечнососудистых заболеваний и заболеваний почек.
Еще одним объектом настоящего изобретения является способ лечения сердечно-сосудистых заболеваний и заболеваний почек путем введения эффективного количества соединения формулы (I-A-1) в кристаллической форме модификации I, как описано выше.
Соединения формулы (I-A-1) по изобретению действуют как мощные двойные антагонисты V1a/V2-рецепторов и показывают непредсказуемый ценный фармакологический спектр действия. Поэтому они пригодны для использования в качестве лекарственных средств для лечения и/или профилактики заболеваний у людей и животных.
Соединения согласно изобретению пригодны по отдельности или в комбинации с одним или несколькими другими активными веществами для профилактики и/или лечения различных заболеваний, например заболеваний сердечно-сосудистой системы (сердечно-сосудистые заболевания), для кардиозащиты после повреждения сердца, а также при заболеваниях, вызванных нарушением обмена веществ, и почечных заболеваниях.
Соединения согласно изобретению пригодны по отдельности или в комбинации с одним или несколькими другими активными веществами для профилактики и/или лечения различных заболеваний, например заболеваний сердечно-сосудистой системы (сердечно-сосудистые заболевания), а также при почечных заболеваниях.
Соединения согласно изобретению обладают ценными фармакологическими свойствами и могут быть использованы для профилактики и/или лечения различных заболеваний и состояний, обусловленных заболеванием, у людей и животных.
Возможные целевые показания приведены в качестве примера и предпочтительного варианта в международной заявке WO 2016/071212, стр. 16-19.
Подходящие активные вещества для комбинаций и лекарственные формы приведены в качестве примера и предпочтительного варианта в международной заявке WO 2016/071212, стр. 19-27.
Следующие примеры осуществления иллюстрируют изобретение. Изобретение не ограничивается указанными примерами.
Указание процентов в следующих тестах и примерах соответствует массовым процентам, если не указано иное; части представляют собой массовые части. Соотношение растворителей, коэффициенты разбавления и данные по концентрации растворов жидкость/жидкость рассчитывают в каждом случае на объем.
А. Примеры
Сокращения:


мин минута(ы)
МС массспектрометрия
MtBE метил-трет-бутиловый эфир
ЯМР ядерный магнитный резонанс
Ph фенил
колич. количественно (при указании выхода)
rac рацемический, рацемат
RT комнатная температура
Rt время удерживания (в случае ВЭЖХ)
ТГФ тетрагидрофуран
УФ ультрафиолетовая оптическая спектроскопия
об/об соотношение объем к объему (раствора)
ЖХ/МС и ВЭЖХ методы:
Метод 1 (ЖХ-МС): MCW-SQ-HSST3
Прибор: Waters Acquity SQD UPLC System; колонка: Waters Acquity UPLC HSS T3 1.8 мкм 50 мм × 1 мм; элюент A: 1 л воды + 0,25 мл 99%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0,25 мл 99%-ной муравьиной кислоты, градиент: 0,0 мин 90% А→1,2 мин 5% А→2,0 мин 5% А; печь: 50°С; скорость потока: 0,40 мл/мин; УФ детектирование: 208-400 нм.
Метод 2 (ЖХ-МС): MCW-FT-MS-M1
Прибор: Thermo Scientific FT-MS; прибор УВЭЖХ+: Thermo Scientific Ulti-Mate 3000; колонка: Waters, HSST3, 2,1×75 мм, C18 1,8 мкм; элюент A: 1 л воды + 0,01 мл 0,01%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0,01 мл муравьиной кислоты, градиент: 0,0 мин 10% В → 2,5 мин 95% В → 3,5 мин 95% В; печь: 50°С; скорость потока: 0,90 мл/мин; УФ детектирование: 210 нм/ Optimum Integration Path 210-300 нм
Дополнительные данные:
Указание процентов в следующих описаниях примеров и экспериментов соответствует массовым процентам, если не указано иное; части представляют собой массовые части. Соотношение растворителей, коэффициенты разбавления и данные по концентрации растворов жидкость/жидкость рассчитывают в каждом случае на объем.
При очистке соединений согласно изобретению с помощью препаративной ВЭЖХ способами, описанными выше, в которых элюенты содержат добавки, такие как трифторуксусная кислота, муравьиная кислота или аммиак, соединения согласно изобретению могут находиться в форме соли, например, в виде трифторацетата, формиата или аммониевой соли, в случае, если соединения согласно изобретению содержат достаточно основные или соответственно кислотные функциональные группы. Такая соль может быть превращена в соответствующее свободное основание или кислоту различными способами, известными специалисту в данной области техники.
Данные по чистоте обычно относятся к соответствующим интегралам пиков на ЖХ/МС-хроматограмме, но могут быть дополнительно определены с помощью 1Н-ЯМР-спектра. Если чистота не указана, то, как правило, речь идет о 100% чистоте в соответствии с автоматической интеграцией пиков на ЖХ/МС-хроматограмме или чистоту не определяли.
Данные по выходу в % от теор., как правило, скорректированы в соответствии с чистотой, при условии, что приведенная чистота составляет <100%. Для содержащих растворитель или загрязненных партий выход может быть формально «>100%»; в этих случаях выход не скорректирован в соответствии с чистотой или содержанием растворителя.
Далее следующие описания образцов спин-спинового взаимодействия 1Н-ЯМР-сигналов были взяты частично непосредственно из расчетных спектров ACD SpecManager (ACD / Labs Release 12.00, версия продукта 12.5) и не требуют обязательного уточнения. Частично расчетные спектры SpecManager были адаптированы вручную. Адаптированые вручную или соответственно предоставленные описания, как правило, ориентированы на визуальное представление соответствующих сигналов и не обязательно соответствуют строгой, физически корректной интерпретации. Как правило, данные по химическому сдвигу относятся к центру соответствующего сигнала. Для широких мультиплетов данные приводят в виде интервала.
Сигналы, скрытые растворителем или водой, были либо назначены условно, либо не приведены. Сильно расширенные сигналы - например, обусловленные быстрым вращением фрагментов молекулы или из-за обмена протонами, также были назначены условно (часто называемые как широкий мультиплет или широкий синглет) или не приведены.
1Н-ЯМР-Данные выбранных примеров отмечают в виде списков 1Н-ЯМР-пиков. Для каждого пика сигнала сначала приводят δ-величину в м.д. и затем указывают интенсивность сигнала в круглых скобках. Числовые пары δ-величины и интенсивности сигнала различных сигнальных пиков отделены друг от друга запятой. Поэтому список пиков примера имеет следующую форму: δ1 (интенсивность1), δ2 (интенсивность2), …, δi (интенсивностьi), …, δn (интенсивностьn).
Интенсивность четких сигналов находится в корреляционной зависимости с высотой сигналов в печатном примере ЯМР-спектра в см и показывает в сравнении с другими сигналами действительное соотношение интенсивностей сигналов. В случае широких сигналов могут быть показаны несколько пиков или середина сигнала и ее относительная интенсивность в сравнении с самым интенсивным сигналом в спектре. Списки 1Н-ЯМР-пиков схожи с классическими печатными 1Н-ЯМР-спектрами и при этом обычно содержат все пики, которые приводятся при классической ЯМР- интерпретации. Кроме того, они могут, как и классические печатные 1Н-ЯМР-спектры, показывать сигналы растворителей, сигналы стереоизомеров целевых соединений, которые также являются объектом изобретения, и/или пики примесей. Пики стереоизомеров целевых соединений и/или пики примесей обычно имеют более низкую интенсивность, чем пики целевых соединений (например, с чистотой >90%). Такие стереоизомеры и/или примеси могут быть типичными для соответствующих способов получения. Таким образом их пики могут помогать при распознавании воспроизведения нашего способа получения при помощи "отпечатков пальцев побочных продуктов". Эксперт, который анализирует пики целевых соединений с помощью известных способов (MestreC, ACD-моделирование, или с применением эмпирически обработанных ожидаемых значений), по мере необходимости может выделить пики целевых соединений, причем, при необходимости, применять дополнительный фильтр интенсивности. Такое выделение было бы похоже на соответствующее выявление пиков в классической 1Н-ЯМР-интерпретации. Детальное описание представления ЯМР-данных в виде списков пиков можно найти в публикации "Citation of NMR Peaklist Data within Patent Applications" (см. номер в базе данных Research Disclosure 605005, 2014, 1. Август 2014 или http://www.researchdisclosure.com/searching-disclosures). В режиме выбора пиков, описанном в базе данных Research Disclosure под номером 605400, параметр "MinimumHeight" может быть установлен между 1% и 4%. В зависимости от типа химической структуры и/или в зависимости от концентрации анализируемого соединения может оказаться полезным установить параметр «MinimumHeight» на значение <1%.
Точки плавления и диапазоны плавления, если они указаны, не корректируются.
Для всех реактантов или реагентов, получение которых явно не описано ниже, действительным является, что они были получены коммерчески из общедоступных источников. Для всех других реактантов или реагентов, получение которых также не описано ниже и которые не были коммерчески доступны или получены из источников, которые обычно недоступны, описана ссылка на опубликованную литературу, в которой описывается их получение.
Примеры осуществления изобретения
Пример 1
{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}ацетонитрил (Х-а)

100 г (0,325 моль) 5-(4-хлорфенил)-4-((2S)-3,3,3-трифтор-2-гидроксипропил)-2,4-дигидро-3Н-1,2,4-триазола-3-on (II-А) (синтез, описан в примере 5А в международной заявке WO 2010/105770-А1) в виде раствора в 1,0 л метилизобутилкетона, смешивали с 135 г (0,975 моль) карбоната натрия и затем смесь нагревали до 60°С. Затем при этой температуре по каплям в течение 6 часов равномерно добавляли 27 г (0,358 моль) хлорацетонитрила, растворенного в 270 мл метилизобутилкетона (IX). Смесь дополнительно перемешивали в течение 15 ч при 60°С и затем охлаждали до 20°С, смешивали с 500 мл воды, дополнительно перемешивали и отделяли органическую фазу. Органическую фазу еще раз дополнительно промывали 500 мл воды и затем концентрировали в вакууме при температуре обогревательного контура 60°С до объема около 250 мл. Затем добавляли 250 мл н-гептана, в результате чего выкристаллизовался продукт. Для полноценной кристаллизации отгоняли в вакууме 500 мл смеси растворителей с одновременным добавлением 500 мл н-гептана при температуре обогревательного контура 60°С. Охлаждали до 20°С и перемешивали в течение одного часа при этой температуре. Продукт отфильтровывали и дополнительно промывали н-гептаном (2 раза по 150 мл). Затем сушили в вакууме при 40°С. Выход: 1 г (72% от теории) твердого вещества.
МС (Elpos): m/z=347.1 [М+Н]+
1Н-ЯМР (400 МГц, DMSO-d6): δ=3.81 (дд, 1Н), 3.98 (дд, 1Н), 4.23-4.34 (м, 1 Н), 5.17 (с, 2 Н), 6.91 (д, 1Н), 7.55 (д, 2Н), 7.78 (д, 2Н).
Пример 2
Метил-2-{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}этанимидат (XI-a)

Загрузили 200 г (576,9 ммоль) {3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}ацетонитрила (Х-а) в виде раствора в 1,6 л метанола и добавили 5,2 г (28 ммоль) метилата натрия (30%-ный раствор в метаноле). Перемешивали в течение 2 часов при 50°С и затем концентрировали при температуре обогревательного контура 50°С до маслянистого остатка. Смешивали с 2 л MtBE и концентрировали до объема около 0,8 л. Затем раствор медленно добавляли при перемешивании к 4 л н-гексана. При этом продукт кристаллизуется в виде густой кристаллической пульпы. Позволяли охладиться до 20°С и перемешивали в течение одного часа при комнатной температуре. Продукт отфильтровывали и дополнительно промывали н-гексаном (2 раза по 0,25 л). Затем сушили в вакууме при 40°С. Выход: 75 г (80% от теории) твердого вещества.
1Н-ЯМР (400 МГц, DMSO-d6): δ=3.67 (с, 3 Н), 3.81 (дд, 1Н), 3.96 (дд, 1Н), 4.23-4.35 (м, 1Н), 4.50 (с, 2 Н), 6.93 (уш. с, 1Н), 7.62 (д, 2Н), 7.78 (д, 2Н), 8.01 (с, 1Н).
Пример 3
(5-(4-хлорфенил)-2-({1-(3-хлорфенил)-5-[(1S)-1-гидроксиэтил]-1Н-1,2,4-триазол-3-ил}метил)-4-[(2S)-3,3,3-трифтор-2-гидроксипроп ил]-2,4-дигидро-3Н-1,2,4-триазол-3-он (I-A-1)

Вариант проведения способа В:
Метил-2-{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}этанимидат (XI-a) (164 г, 433 ммоль) растворяли в смеси ТГФ (1,0 л) и толуола (0,5 л). Смесь смешивали с N-этилдиизопропиламином (97,8 г, 757 ммоль) при 20°С и затем дополнительно перемешивали в течение 15 мин при 20°С. Затем при 0°С дозировали хлорангидрид (S)-2-ацетоксипропионовой кислоты (XII-А) (78,2 г, 519 ммоль) и дополнительно перемешивали при 0°С в течение 1 часа. Затем при 0°С добавляли раствор гидрохлорида 4-хлорфенилгидразина (XIII-1) (85,2 г, 476 ммоль) и N-этилдиизопропиламина (67,11 г, 519 ммоль) в ТГФ (0,5 л), причем осажденный N-этилдиизопропиламин-гидрохлорид отфильтровывали перед добавлением, затем перемешивали в течение 1 часа при 20°С и еще 2 ч при температуре кипения с возвратом флегмы (около 75°С). Позволяли охладиться до 20°С и к загрузке добавляли 5,7 л воды. После разделения фаз органическую фазу дважды промывали в каждом случае 0,5 л 1 н. раствора соляной кислоты, затем концентрировали в вакууме при температуре обогревательного контура 80°С до маслянистого остатка и дважды дистиллировали совместно с 1,0 л метанола соответственно. Маслянистый остаток затем растворяли в 0,6 л метанола, смешивали с 0,5 л 1 н. раствора гидроксида натрия при 0°С и дополнительно перемешивали в течение 1 часа при 20°С. После добавления 0,75 л воды и 0,75 л MtBE отделяли органическую фазу, дважды промывали соответственно 0,3 л полунасыщенного водного раствора хлорида натрия и затем концентрировали в вакууме при температуре обогревательного контура 80°С до объема около 0,3 л. После добавления 1,5 л диизопропилового эфира снова концентрировали в вакууме при температуре обогревательного контура 80°С до объема около 0,3 л, в результате чего продукт выпадал в осадок. Охлаждали до 10°С и перемешивали в течение одного часа при этой температуре. Продукт отфильтровывали и дополнительно промывали 0,3 л диизопропилового эфира. Затем сушили в вакууме при 50°С. Выход: 200 г (72% от теории).
МС (ESIpos): m/z=543.1 (100) [М+Н]+
1Н-ЯМР (400 МГц, DMSO-d6): δ=1.47 (д, 3 Н), 3.85 (дд, 1Н), 4.00 (дд, 1Н), 4.24-4.36 (м, 1Н), 4.81 (квин, 1Н), 5.07 (с, 2 Н), 5.75 (д, 1Н), 6.89 (д, 1Н), 7.54-7.66 (м, 5Н), 7.72-7.79 (м, 3H).
Вариант проведения способа С (с последующей кристаллизацией из метил-трет-бутилового эфира/диизопропилового эфира):
Загрузили 1,373 кг (3,96 моль) {3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}ацетонитрила (Х-а) в виде раствора в 6,9 л метанола и добавили 36 г (0,198 моль) метилата натрия (30%-ный раствор в метаноле). Перемешивали в течение 1,5 часов при 50°С и затем концентрировали при температуре обогревательного контура 50°С до еще перемешиваемого пастообразного остатка. Трижды смешивали соответственно с 3,0 л толуола и в каждом случае концентрировали до объема 5 л. К остатку добавляли ТГФ (9,5 л) и толуол (2,5 л), при 20°С смешивали с N-этилдиизопропиламином (0,896 кг, 6,93 моль) и перемешивали в течение 15 мин при 20°С. Затем при 0°С дозировали хлорангидрид (S)-2-ацетоксипропионовой кислоты (XII-А) (0,715 кг, 4,752 моль) и дополнительно перемешивали при 0°С в течение 1 часа. Затем при 0°С добавляли раствор гидрохлорида 4-хлорфенилгидразина (XIII-1) (0,78 кг, 4,356 моль) и N-этилдиизопропиламина (0,614 кг, 4,752 моль) в ТГФ (4,5 л), причем осажденный N-этилдиизопропиламин-гидрохлорид отфильтровывали перед добавлением, затем перемешивали в течение 1 часа при 20°С и еще 2 ч при температуре кипения с возвратом флегмы (около 75°С). Позволяли охладиться до 20°С и к загрузке добавляли 7,0 л воды. После разделения фаз органическую фазу дважды промывали в каждом случае 3,5 л 1 н. раствора соляной кислоты, концентрировали в вакууме при температуре обогревательного контура 80°С до маслянистого остатка и дважды дистиллировали совместно с 13,5 л метанола соответственно. Маслянистый остаток растворяли в 5,5 л метанола, смешивали с 4,0 л 1 н. раствора гидроксида натрия при 0°С и дополнительно перемешивали в течение 1 часа при 20°С. После добавления 7,0 л воды и 7,0 л MtBE отделяли органическую фазу, дважды промывали соответственно 2,75 л полунасыщенного водного раствора хлорида натрия и затем концентрировали в вакууме при температуре обогревательного контура 80°С до объема около 3,0 л. После добавления 16,0 л диизопропилового эфира снова концентрировали в вакууме при температуре обогревательного контура 80°С до объема около 6,0 л, в результате чего продукт выпадал в осадок. Затем охлаждали до 10°С и перемешивали в течение одного часа при этой температуре. Продукт отфильтровывали и дополнительно дважды промывали соответственно 1,0 л диизопропилового эфира. Затем сушили в вакууме при 50°С. Выход: 680 кг (78% от теории).
Чистота >99%; Оптическая чистота составляет >>99% ее.
Вариант проведения способа С (с последующей кристаллизацией из метил-трет-бутилового эфира/н-гептана):
Загрузили 50 г (144 ммоль) {3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}ацетонитрила (Х-а) в виде раствора в 250 мл метанола и добавили 1,58 г (7,3 моль) метилата натрия (25%-ный раствор в метаноле). Перемешивали в течение 1,5 часов при 50°С и затем концентрировали при температуре обогревательного контура 50°С до еще перемешиваемого пастообразного остатка. Трижды смешивали соответственно с 200 мл ТГФ и в каждом случае концентрировали в вакууме до сухого состояния. К остатку добавляли 325 мл ТГФ, при 20°С смешивали с N-этилдиизопропиламином (44 мл, 253 ммоль) и дополнительно перемешивали в течение 15 мин при 20°С. Затем при 0°С дозировали хлорангидрид (S)-2-ацетоксипропионовой кислоты (XII-А) (26 г, 173 ммоль) и дополнительно перемешивали при 0°С в течение 1 часа. Затем при 0°С добавляли раствор гидрохлорида 4-хлорфенилгидразина (XIII-1) (28,5 г, 159 ммоль) и N-этилдиизопропиламина (30 мл, 172 ммоль) в 150 мл ТГФ, причем осажденный N-этилдиизопропиламин-гидрохлорид отфильтровывали перед добавлением. Затем смесь перемешивали при 20°С в течение 30 мин и в течение еще 2,5 ч при температуре кипения с возвратом флегмы (около 75°С). Позволяли охладиться до 20°С и к загрузке добавляли 125 мл MtBE и 250 мл воды. После разделения фаз органическую фазу дважды промывали в каждом случае 125 г 1 н. раствора соляной кислоты, концентрировали в вакууме при температуре обогревательного контура 60°С до маслянистого остатка и дважды дистиллировали совместно с 500 мл метанола соответственно. Маслянистый остаток растворяли в 200 л метанола, смешивали с 35 мл 1 н. раствора гидроксида натрия при 0°С и дополнительно перемешивали в течение 1 часа при 20°С. После добавления 125 мл воды и 375 мл MtBE отделяли органическую фазу, дважды промывали соответственно 62 мл полунасыщенного водного раствора хлорида натрия и затем концентрировали в вакууме при температуре обогревательного контура 80°С до маслянистого остатка. После добавления 300 мл диизопропилового эфира снова концентрировали в вакууме при температуре обогревательного контура 80°С до объема около 150 мл, в результате чего продукт выпадал в осадок. Затем охлаждали до 10°С и перемешивали в течение одного часа при этой температуре. Продукт отфильтровывали и дополнительно дважды промывали соответственно 100 мл диизопропилового эфира. Затем сушили в вакууме при 50°С. Кристаллизат растворяли в 420 мл MtBE при кипячении с возвратом флегмы. После добавления 900 мл н-гептана при 50°С выкристаллизовывался продукт. Затем охлаждали до 20°С и перемешивали в течение одного часа при этой температуре. Отфильтровывали продукт, дополнительно промывали 100 мл воды и сушили в вакууме при 70°С. Выход: 8 г (74% от теории).
Чистота >99%; оптическая чистота составляет >>99% ее.
Пример 4
а) (1R)-1-[1-(3-хлорфенил)-3-({3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}метил)-1Н-1,2,4-триазол-5-ил]этилацетат (XIV-B-1)
При охлаждении льдом к смеси 200 мг (0,53 ммоль) метил-2-{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}этанимидата (XI) и 262 мкл (1,5 ммоль) DIPEA в 2 мл ТГФ добавляли по каплям 87 мг (0,58 ммоль) хлорангидрида (R)-(-)-2-ацетоксипропионовой кислоты (XII-В). Через 1 ч при 0°С добавляли 104 мг (0,58 ммоль) 3-хлорфенилгидразина (XIII) и смесь затем перемешивали при комнатной температуре в течение ночи. Реакционную смесь очищали хроматографией (препаративная ВЭЖХ, элюент: градиент ацетонитрил/вода, 0,1%-ная муравьиная кислота). После лиофилизации фракций, содержащих продукт, получали 208 мг (64% от теории) указанного в заголовке соединения.
ЖХ-МС (Метод 2): Rt=2,04 мин; МС (ESIpos): m/z=585.1 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.90-7.37 (м, 8Н), 6.89 (д, 1Н), 5.91 (д, 1Н), 5.09 (с, 2Н), 4.40-4.20 (м, 1Н), 4.09-3.71 (м, 2Н), 1.81(с, 3H), 1.56 (д, 3H)
b) 2-({1-(3-Хлорфенил)-5-[(1R)-1-гидроксиэтил]-1Н-1,2,4-триазол-3-ил}метил)-5-(4-хлорфенил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-он (I-B-1)

Смесь 200 мг (0,34 ммоль) соединения со стадии а) и 341мкл (0,34 ммоль) 1 М гидроксида натрия в 2,6 мл метанола перемешивали при комнатной температуре в течение 30 мин. Добавляли 1 г активированного ионообменного материала (Dowex 50WX8, 200-400 меш) и перемешивали при комнатной температуре в течение 5 мин. Затем ионообменный материал отфильтровывали и дополнительно промывали метанолом. Фильтрат упаривали. Получали 168 мг (90% от теории) указанного в заголовке соединения.
ЖХ-МС (Метод 2): Rt=1,85 мин; МС (ESIpos): m/z=543.1 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.98-7.48 (м, 8Н), 6.90 (д, 1Н), 5.76 (д, 1Н), 5.07 (с, 2Н), 4.81 (т, 1Н), 4.46-3.68 (м, 3H), 1.47 (д, 3H).
Пример 5
а) (1S)-1-[3-({3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}метил)-1-(2,4-дихлорфенил)-1Н-1,2,4-триазол-5-ил]этилацетат (XIV-A-2)
При охлаждении льдом к смеси 200 мг (0,53 ммоль) метил-2-{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}этанимидата (XI) и 262 мкл (1,51ммоль) DIPEA в 2 мл ТГФ добавляли по каплям 73 мкл (0,58 ммоль) хлорангидрида (S)-(-)-2-ацетоксипропионовой кислоты (ХII-А). Через 1 ч при 0°С добавляли 124 мг (0,58 ммоль) гидрохлорида 2,4-дихлорфенилгидразина (XIII) и смесь затем перемешивали при комнатной температуре в течение ночи. Реакционную смесь кипятили с возвратом флегмы в течение 2 часов и грели в микроволновой печи при 100°С в течение 5 часов. Растворитель удаляли в вакууме и сырой продукт очищали хроматографией (препаративная ВЭЖХ, элюент: градиент ацетонитрил/вода, 0,1%-ная муравьиная кислота). После лиофилизации фракций, содержащих продукт, получали 163 мг (48% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,13 мин; МС (ESIpos): m/z=619.0 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=8.04-7.49 (м, 7Н), 6.89 (д, 1Н), 5.90-5.44 (м, 1Н), 5.10 (д, 2Н), 4.45-4.16 (м, 1Н), 4.11-3.73 (м, 2Н), 1.81(с, 3H), 1.53 (д, 3H)
b) 5-(4-Хлорфенил)-2-({1-(2,4-дихлорфенил)-5-[(1S)-1-гидроксиэтил]-1Н-1,2,4-триазол-3-ил}метил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-он (I-A-2)

Смесь 160 мг (0,26 ммоль) соединения со стадии а) и 258 мкл (0,26 ммоль) 1 М гидроксида натрия в 2 мл метанола перемешивали в течение 2 мин при 0°С и 90 мин при комнатной температуре. Добавляли 1г активированного ионообменного материала (Dowex 50WX8, 200-400 меш) и перемешивали при комнатной температуре в течение 30 мин. Затем ионообменный материал отфильтровывали и дополнительно промывали метанолом.
Упаривали фильтрат и остаток сушили в вакууме. Получали 148 мг (колич.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,02 мин; МС (ESIpos): m/z=577.1 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.92 (д, 1Н), 7.78-7.71 (м, 2Н), 7.68-7.58 (м, 4Н), 6.89 (д, 1Н), 5.52 (д, 1Н), 5.06 (д, 2Н), 4.64 (с, 1Н), 4.43-4.21 (м, 1Н), 4.08-3.72 (м, 2Н), 1.39 (д, 3H)
Пример 6
а) (1S)-1-{3-({3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}метил)-1-[2-(дифторметокси)фенил)-1Н-1,2,4-триазол-5-ил]этилацетат (XIV-A-3)
При охлаждении льдом к смеси 150 мг (0,40 ммоль) метил-2-{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}этанимидата (XI) и 207 мкл (1,19 ммоль) DIPEA в 1,5 мл ТГФ добавляли по каплям 55 мкл (0,44 ммоль) хлорангидрида (S)-(-)-2-ацетоксипропионовой кислоты (XII-А). Через 30 мин при 0°С добавляли 76 мг (0,44 ммоль) 2-дифторметоксифенилгидразина (XIII) и смесь затем перемешивали при комнатной температуре в течение ночи. Реакционную смесь затем грели в микроволновой печи при 100°С в течение 3 часов. Реакционную смесь смешивали с несколькими каплями воды и очищали хроматографией (препаративная ВЭЖХ, элюент: градиент ацетонитрил/вода, 0,1%-ная муравьиная кислота). После лиофилизации фракций, содержащих продукт, получали 142 мг (58% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,09 мин; МС (ESIpos): m/z=617.1 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.83-6.82 (м, 10Н), 5.69 (д, 1Н), 5.09 (д, 2Н), 4.30 (д, 1Н), 4.07-3.77 (м, 2Н), 1.77 (с, 3H), 1.53 (д, 3H)
b) 5-(4-Хлорфенил)-2-({1-[2-(дифторметокси)фенил]-5-[(1S)-1-гидроксиэтил]-1Н-1,2,4-триазол-3-ил}метил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3H-1,2,4-триазол-3-он (I-A-3)

Смесь 132 мг (0,21 ммоль) соединения со стадии а) и 214 мкл (0,21 ммоль) 1 М гидроксида натрия в 1,3 мл метанола перемешивали в течение 2 мин при 0°С и 90 мин при комнатной температуре. Добавляли 0,5 г активированного ионообменного материала (Dowex 50WX8, 200-400 меш) и перемешивали при комнатной температуре в течение 30 мин. Затем ионообменный материал отфильтровывали и дополнительно промывали метанолом. Упаривали фильтрат и остаток сушили в вакууме. Получали 117 мг (95% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=0,99 мин; МС (ESIpos): m/z=575.3 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.75 (д, 2Н), 7.67-7.54 (м, 4Н), 7.45-6.97 (м, 3H), 6.89 (д, 1Н), 5.48 (д, 1Н), 5.19-4.94 (м, 2Н), 4.61 (квин, 1Н), 4.30 (д, 1Н), 4.09-3.76 (м, 2Н), 1.39 (д, 3H)
Пример 7
а) (1R)-1-{3-({3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}метил)-1-[2-(дифторметокси)фенил)-1Н-1,2,4-триазол-5-ил]этилацетат (XIV-B-3)
При охлаждении льдом к смеси 200 мг (0,53 ммоль) метил-2-{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}этанимидата (XI) и 276 мкл (1,58 ммоль) DIPEA в 2 мл ТГФ добавляли по каплям 73 мкл (0,58 ммоль) хлорангидрида (R)-(-)-2-ацетоксипропионовой кислоты (XII-В). Через 30 мин при 0°С добавляли 101 мг (0,58 ммоль) 2-дифторметоксифенилгидразина (XIII) и смесь затем перемешивали при комнатной температуре в течение ночи. Реакционную смесь затем грели в микроволновой печи при 150°С в течение 3 часов. Реакционную смесь смешивали с несколькими каплями воды и очищали хроматографией (препаративная ВЭЖХ, элюент: градиент ацетонитрил/вода, 0,1%-ная муравьиная кислота). После лиофилизации фракций, содержащих продукт, получали 202 мг (62% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,09 мин; МС (ESIpos): m/z=617.3 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.89-6.81 (м, 10Н), 5.79-5.59 (м, 1Н), 5.09 (д, 2Н), 4.35-4.22 (м, 1Н), 4.09-3.78 (м, 2Н), 1.76 (с, 3H), 1.53 (д, 3H)
b) 5-(4-Хлорфенил)-2-({1-[2-(дифторметокси)фенил]-5-[(1R)-1-гидроксиэтил]-1Н-1,2,4-триазол-3-ил}метил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3H-1,2,4-триазол-3-он (I-B-3)

Смесь 192 мг (0,31 ммоль) соединения со стадии а) и 310 мкл (0,31 ммоль) 1 М гидроксида натрия в 1,9 мл метанола перемешивали в течение 2 мин при 0°С и 90 мин при комнатной температуре. Добавляли 0,5 г активированного ионообменного материала (Dowex 50WX8, 200-400 меш) и перемешивали при комнатной температуре в течение 30 мин. Затем ионообменный материал отфильтровывали и дополнительно промывали метанолом. Упаривали фильтрат и остаток сушили в вакууме. Получали 172 мг (96% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=0,99 мин; МС (ESIpos): m/z=575.3 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.75 (д, 2Н), 7.68-7.53 (м, 4Н), 7.46-6.96 (м, 3H), 6.91 (д, 1Н), 5.48 (д, 1Н), 5.06 (с, 2Н), 4.61 (т, 1Н), 4.30 (д, 1Н), 4.07-3.75 (м, 1Н), 1.39 (д, 1Н)
Пример 8
а) (1S)-1-{3-({3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}метил)-1-[2-хлор-4-(трифторметил)фенил)-1Н-1,2,4-триазол-5-ил]этилацетат (XIV-A-4)
При охлаждении льдом к смеси 150 мг (0,40 ммоль) метил-2-{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}этанимидата (XI) и 207 мкл (1,19 ммоль) DIPEA в 1,5 мл ТГФ добавляли по каплям 55 мкл (0,44 ммоль) хлорангидрида (S)-(-)-2-ацетоксипропионовой кислоты (ХII-А). Через 30 мин при 0°С добавляли 91 мг (0,44 ммоль) 2-хлор-4-(трифторметил)фенилгидразина (XIII) и перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь затем грели в микроволновой печи при 100°С в течение 3 часов. Реакционную смесь смешивали с несколькими каплями воды и очищали хроматографией (препаративная ВЭЖХ, элюент: градиент ацетонитрил/вода, 0,1%-ная муравьиная кислота). После лиофилизации фракций, содержащих продукт, получали 111 мг (43% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод А): Rt=1,214 мин; МС (ESIpos): m/z=653.1 [M+H]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=8.25 (с, 1Н), 8.06-7.87 (м, 2Н), 7.81-7.54 (м, 4Н), 6.89 (д, 1Н), 5.75 (с, 1Н), 5.12 (д, 2Н), 4.29 (д, 1Н), 4.07-3.77 (м, 2Н), 1.76 (с, 3H), 1.55 (д, 3H)
b) 5-(4-Хлорфенил)-2-({1-[2-хлор-4-(трифторметил)фенил]-5-[(1S)-1-гидроксиэтил]-1Н-1,2,4-триазол-3-ил}метил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3H-1,2,4-триазол-3-он (I-A-4)
Смесь 104 мг (0,16 ммоль) соединения со стадии а) и 160 мкл (0,16 ммоль) 1 М гидроксида натрия в 1 мл метанола перемешивали в течение 2 мин при 0°С и 90 мин при комнатной температуре. Добавляли 0,5 г активированного ионообменного материала (Dowex 50WX8, 200-400 меш) и перемешивали при комнатной температуре в течение 30 мин. Затем ионообменный материал отфильтровывали и дополнительно промывали метанолом. Упаривали фильтрат и остаток сушили в вакууме. Получали 94 мг (96% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,06 мин; МС (ESIpos): m/z=611.1[М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=8.18 (с, 1Н), 7.99-7.82 (м, 2Н), 7.75 (д, 2Н), 7.62 (д, 2Н), 6.89 (д, 1Н), 5.54 (д, 1Н), 5.08 (д, 2Н), 4.71 (т, 1Н), 4.29 (д, 1Н), 4.11-3.77 (м, 2Н), 1.41 (д, 3H)
Пример 9
а) (1S)-1-[1-(2-хлор-6-фторфенил)-3-({3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}метил)-1Н-1,2,4-триазол-5-ил]этилацетат (XIV-A-5)
При охлаждении льдом к смеси 150 мг (0,40 ммоль) метил-2-{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}этанимидата (XI) и 207 мкл (1,19 ммоль) DIPEA в 1,5 мл ТГФ добавляли по каплям 55 мкл (0,44 ммоль) хлорангидрида (S)-(-)-2-ацетоксипропионовой кислоты (XII-А). Через 30 мин при 0°С добавляли 70 мг (0,44 ммоль) 2-хлор-6-фторфенилгидразина (XIII) и перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь затем грели в микроволновой печи при 100°С в течение 3 часов. Реакционную смесь смешивали с несколькими каплями воды и очищали хроматографией (препаративная ВЭЖХ, элюент: градиент ацетонитрил/вода, 0,1%-ная муравьиная кислота). После лиофилизации фракций, содержащих продукт, получали 139 мг (58% от теор.) указанного в заголовке соединения в виде атропоизомерной смеси.
ЖХ-МС (Метод А): Rt=1,10 мин; МС (ESIpos): m/z=603.1 [M+H]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.82-7.50 (м, 7Н), 6.89 (д, 1Н), 5.73 (д, 1Н), 5.25-5.04 (м, 2Н), 4.43-4.19 (м, 1Н), 4.10-3.78 (м, 2Н), 1.79 (с, 3H), 1.54 (м, 3H)
b) 2-({1-(2-Хлор-6-фторфенил)-5-[(1S)-1-гидроксиэтил]-1Н-1,2,4-триазол-3-ил}метил)-5-(4-хлорфенил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3H-1,2,4-триазол-3-он (I-A-5)

Смесь 129 мг (0,21 ммоль) соединения со стадии а) и 214 мкл (0,21 ммоль) 1 М гидроксида натрия в 1,3 мл метанола перемешивали в течение 2 мин при 0°С и 90 мин при комнатной температуре. Добавляли 0,5 г активированного ионообменного материала (Dowex 50WX8, 200-400 меш) и перемешивали при комнатной температуре в течение 30 мин. Затем ионообменный материал отфильтровывали и дополнительно промывали метанолом. Упаривали фильтрат и остаток сушили в вакууме. Получали 114 мг (95% от теор.) указанного в заголовке соединения в виде атропоизомерной смеси.
ЖХ-МС (Метод A): Rt=0,99 мин; МС (ESIpos): m/z=561.3 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.79-7.46 (м, 7Н), 6.89 (д, 1Н), 5.60 (дд, 1Н), 5.22-4.97 (м, 2Н), 4.84-4.55 (м, 1Н), 4.29 (д, 1Н), 4.08-3.73 (м, 2Н), 1.44-1.33 (м, 3H)
Пример 10
а) (1R)-1-[1-(2-хлор-6-фторфенил)-3-({3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}метил)-1Н-1,2,4-триазол-5-ил]этилацетат (XIV-B-5)
При охлаждении льдом к смеси 150 мг (0,40 ммоль) метил-2-{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}этанимидата (XI) и 207 мкл (1,19 ммоль) DIPEA в 1,5 мл ТГФ добавляли по каплям 55 мкл (0,44 ммоль) хлорангидрида (R)-(-)-2-ацетоксипропионовой кислоты (XII-В). Через 30 мин при 0°С добавляли 70 мг (0,44 ммоль) 2-хлор-6-фторфенилгидразина (XIII) и смесь затем перемешивали при комнатной температуре в течение ночи. Реакционную смесь затем грели в микроволновой печи при 150°С в течение 1 часа. Реакционную смесь смешивали с несколькими каплями воды и очищали хроматографией (препаративная ВЭЖХ, элюент: градиент ацетонитрил/вода, 0,1%-ная муравьиная кислота). После лиофилизации фракций, содержащих продукт, получали 162 мг (68% от теор.) указанного в заголовке соединения в виде смеси атропоизомеров.
ЖХ-МС (Метод A): Rt=1,10 мин; МС (ESIpos): m/z=603.3 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.83-7.49 (м, 7Н), 6.96-6.84 (м, 1Н), 5.73 (д, 1Н), 5.13 (д, 2Н), 4.29 (уш. с, 1Н), 4.09-3.76 (м, 2Н), 1.79 (д, 3H), 1.54 (дд, 3H)
b) 2-({1-(2-Хлор-6-фторфенил)-5-[(1R)-1-гидроксиэтил]-1Н-1,2,4-триазол-3-ил}метил)-5-(4-хлорфенил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3H-1,2,4-триазол-3-он (I-B-5)

Смесь 152 мг (0,25 ммоль) соединения со стадии а) и 250 мкл (0,25 ммоль) 1 М гидроксида натрия в 1,5 мл метанола перемешивали в течение 2 мин при 0°С и 90 мин при комнатной температуре. Добавляли 0,5 г активированного ионообменного материала (Dowex 50WX8, 200-400 меш) и перемешивали при комнатной температуре в течение 30 мин. Затем ионообменный материал отфильтровывали и дополнительно промывали метанолом. Упаривали фильтрат и остаток сушили в вакууме. Получали 137 мг (95% от теор.) указанного в заголовке соединения в виде атропоизомерной смеси.
ЖХ-МС (Метод A): Rt=0,99 мин; МС (ESIpos): m/z=561.2 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.83-7.43 (м, 7Н), 6.90 (д, 1Н), 5.60 (дд, 1Н), 5.26-4.92 (м, 2Н), 4.84-4.54 (м, 1Н), 4.29 (д, 1Н), 4.11-3.72 (м, 2Н), 1.44-1.33 (м, 3H)
Пример 11
а) (1S)-1-{3-({3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}метил)-1-[4-фтор-2-(трифторметил)фенил)-1Н-1,2,4-триазол-5-ил]этилацетат (XIV-A-6)
При охлаждении льдом к смеси 150 мг (0,40 ммоль) метил-2-{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}этанимидата (XI) и 207 мкл (1,19 ммоль) DIPEA в 1,5 мл ТГФ добавляли по каплям 55 мкл (0,44 ммоль) хлорангидрида (S)-(-)-2-ацетоксипропионовой кислоты (XII-А). Через 30 мин при 0°С добавляли 100 мг (0,44 ммоль) гидрохлорида 4-фтор-2-(трифторметил)фенилгидразина (XIII) и перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь затем грели в микроволновой печи при 150°С в течение 3 часов. Реакционную смесь смешивали с несколькими каплями воды и очищали хроматографией (препаративная ВЭЖХ, элюент: градиент ацетонитрил/вода, 0,1%-ная муравьиная кислота). После лиофилизации фракций, содержащих продукт, получали 129 мг (51% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,14 мин; МС (ESIpos): m/z=637.3 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=8.05-7.54 (м, 7Н), 6.88 (д, 1Н), 5.75 (с, 1Н), 5.09 (д, 2Н), 4.38-4.18 (м, 1Н), 4.08-3.74 (м, 2Н), 1.78 (уш. с, 3H), 1.51(д, 3H)
b) 5-(4-Хлорфенил)-2-({1-[4-фтор-2-(трифторметил)фенил]-5-[(1S)-1-гидроксиэтил]-1Н-1,2,4-триазол-3-ил}метил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3H-1,2,4-триазол-3-он (I-A-6)

Смесь 119 мг (0,19 ммоль) соединения со стадии а) и 187 мкл (0,19 ммоль) 1 М гидроксида натрия в 1,1 мл метанола перемешивали в течение 2 мин при 0°С и 90 мин при комнатной температуре. Добавляли 0,5 г активированного ионообменного материала (Dowex 50WX8, 200-400 меш) и перемешивали при комнатной температуре в течение 30 мин. Затем ионообменный материал отфильтровывали и дополнительно промывали метанолом. Упаривали фильтрат и остаток сушили в вакууме. Получали 110 мг (колич.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,03 мин; МС (ESIpos): m/z=595.1[М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.96-7.87 (м, 1Н), 7.83-7.55 (м, 6Н), 6.89 (д, 1Н), 5.50 (д, 1Н), 5.16-4.94 (м, 2Н), 4.69-4.50 (м, 1Н), 4.28 (уш. с, 1Н), 4.07-3.75 (м, 2Н), 1.37 (д, 3H)
Пример 12
а) (1S)-1-[1-(2-хлор-4-фторфенил)-3-({3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}метил)-1Н-1,2,4-триазол-5-ил]этилацетат (XIV-A-7)
При охлаждении льдом к смеси 150 мг (0,40 ммоль) метил-2-{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}этанимидата (XI) и 207 мкл (1,19 ммоль) DIPEA в 1,5 мл ТГФ добавляли по каплям 55 мкл (0,44 ммоль) хлорангидрида (S)-(-)-2-ацетоксипропионовой кислоты (XII-А). Через 30 мин при 0°С добавляли 85 мг (0,44 ммоль) гидрохлорида 2-хлор-4-фторфенилгидразина (XIII) и перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь затем грели в микроволновой печи при 150°С в течение 3 часов. Реакционную смесь смешивали с несколькими каплями воды и очищали хроматографией (препаративная ВЭЖХ, элюент: градиент ацетонитрил/вода, 0,1%-ная муравьиная кислота). После лиофилизации фракций, содержащих продукт, получали 157 мг (66% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,07 мин; МС (ESIpos): m/z=603.0 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.90-7.33 (м, 7Н), 6.88 (д, 1Н), 5.96-5.47 (м, 1Н), 5.10 (д, 2Н), 4.29 (д, 1Н), 4.11-3.74 (м, 2Н), 1.82 (уш. с, 3H), 1.53 (д, 3H)
b) 2-({1-(2-Хлор-4-фторфенил)-5-[(1S)-1-гидроксиэтил]-1Н-1,2,4-триазол-3-ил}метил)-5-(4-хлорфенил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3H-1,2,4-триазол-3-он (I-A-7)

Смесь 152 мг (0,25 ммоль) соединения со стадии А) и 252 мкл (0,25 ммоль) 1 М гидроксида натрия в 1,5 мл метанола перемешивали в течение 2 мин при 0°С и 90 мин при комнатной температуре. Добавляли 0,5 г активированного ионообменного материала (Dowex 50WX8, 200-400 меш) и перемешивали при комнатной температуре в течение 30 мин. Затем ионообменный материал отфильтровывали и дополнительно промывали метанолом. Упаривали фильтрат и остаток сушили в вакууме. Получали 140 мг (колич.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,00 мин; МС (ESIpos): m/z=561.2 [M+H]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.84-7.54 (м, 6Н), 7.42 (тд, 1Н), 6.89 (д, 1Н), 5.51 (д, 1Н), 5.06 (д, 2Н), 4.72-4.51(м, 1Н), 4.40-4.19 (м, 1Н), 4.10-3.74 (м, 2Н),1.38 (д, 3H)
Пример 13
а) (1R)-1-[1-(2-хлор-4-фторфенил)-3-({3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}метил)-1Н-1,2,4-триазол-5-ил]этилацетат (XIV-B-7)
При охлаждении льдом к смеси 250 мг (0,66 ммоль) метил-2-{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}этанимидата (XI) и 345 мкл (1,98 ммоль) DIPEA в 5 мл ТГФ добавляли по каплям 109 мг (0,73 ммоль) хлорангидрида (R)-(-)-2-ацетоксипропионовой кислоты (XII-В). Через 30 мин при 0°С добавляли 143 мг (0,73 ммоль) гидрохлорида 2-хлор-4-фторфенилгидразина (XIII) и смесь затем перемешивали при комнатной температуре в течение ночи. Реакционную смесь затем грели в микроволновой печи при 120°С в течение 3 часов и очищали хроматографией (препаративная ВЭЖХ, элюент: градиент ацетонитрил/вода, 0,1%-ная муравьиная кислота). После лиофилизации фракций, содержащих продукт, получали 177 мг (44% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,10 мин; МС (ESIpos): m/z=603.2 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.96-7.3 1 (м, 7Н), 6.89 (д, 1Н), 5.75 (с, 1Н), 5.26-4.96 (м, 2Н), 4.29 (уш. с, 1Н), 4.10-3.76 (м, 2Н), 1.82 (уш. с, 3H), 1.53 (д, 3H)
b) 2-({1-(2-Хлор-4-фторфенил)-5-[(1R)-1-гидроксиэтил]-1Н-1,2,4-триазол-3-ил}метил)-5-(4-хлорфенил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3H-1,2,4-триазол-3-он (I-B-7)

Смесь 165 мг (0,27 ммоль) соединения со стадии а) и 275 мкл (0,27 ммоль) 1 М гидроксида натрия в 3,3 мл метанола перемешивали при комнатной температуре в течение 30 мин. Реакционную смесь смешивали с несколькими каплями 50%-ной водной муравьиной кислоты и очищали посредством препаративной ВЭЖХ (препаративная ВЭЖХ, элюент: градиент ацетонитрил/вода, 0,1%-ная муравьиная кислота). После лиофилизации фракций, содержащих продукт, получали 144 мг (93% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=0,96 мин; МС (ESIpos): m/z=561.00 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.89-7.54 (м, 6Н), 7.42 (тд, 1Н), 6.90 (д, 1Н), 5.51 (д, 1Н), 5.06 (с, 2Н), 4.72-4.51 (м, 1Н), 4.30 (д, 1Н), 4.12-3.75 (м, 2Н), 1.38 (д, 3H)
Пример 14
а) (1S)-1-{3-({3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}метил)-1-[4-хлор-2-(трифторметил)фенил)-1Н-1,2,4-триазол-5-ил]этилацетат (XIV-A-8)
При охлаждении льдом к смеси 150 мг (0,40 ммоль) метил-2-{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}этанимидата (XI) и 207 мкл (1,19 ммоль) DIPEA в 1,5 мл ТГФ добавляли по каплям 55 мкл (0,44 ммоль) хлорангидрида (S)-(-)-2-ацетоксипропионовой кислоты (XII-А). Через 30 мин при 0°С добавляли 107 мг (0,44 ммоль) гидрохлорида 4-хлор-2-(трифторметил)фенилгидразина (XIII) и перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь затем грели в микроволновой печи при 150°С в течение 3 часов. Реакционную смесь смешивали с несколькими каплями воды и очищали хроматографией (препаративная ВЭЖХ, элюент: градиент ацетонитрил/вода, 0,1%-ная муравьиная кислота). После лиофилизации фракций, содержащих продукт, получали 125 мг (48% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,19 мин; МС (ESIpos): m/z=653.3 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=8.23-7.54 (м, 7Н), 6.88 (д, 1Н), 5.75 (с, 1Н), 5.22-4.96 (м, 2Н), 4.28 (д, 1Н), 4.08-3.72 (м, 2Н), 1.78 (уш. с, 3H), 1.51 (д, 3H)
b) 5-(4-Хлорфенил)-2-({1-[4-хлор-2-(трифторметил)фенил]-5-[(1S)-1-гидроксиэтил]-1Н-1,2,4-триазол-3-ил}метил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3H-1,2,4-триазол-3-он (I-A-8)

Смесь 129 мг (0,2 ммоль) соединения со стадии а) и 200 мкл (0,2 ммоль) 1 М гидроксида натрия в 1,2 мл метанола перемешивали в течение 2 мин при 0°С и 90 мин при комнатной температуре. Добавляли 0,5 г активированного ионообменного материала (Dowex 50WX8, 200-400 меш) и перемешивали при комнатной температуре в течение 30 мин. Затем ионообменный материал отфильтровывали и дополнительно промывали метанолом. Упаривали фильтрат и остаток сушили в вакууме. Получали 109 мг (90% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,08 мин; МС (ESIpos): m/z=611.0 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=8.15-7.92 (м, 2Н), 7.82-7.56 (м, 5Н), 6.89 (д, 1Н), 5.51 (д, 1Н), 5.18-4.96 (м, 2Н), 4.64 (т, 1Н), 4.29 (д, 1Н), 4.10-3.73 (м, 2Н), 1.37 (д, 3H)
Пример 15
а) (1S)-1-{3-({3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}метил)-1-[2-хлор-4-(трифторметокси)фенил)-1Н-1,2,4-триазол-5-ил]этилацетат (XIV-A-9)
При охлаждении льдом к смеси 150 мг (0,40 ммоль) метил-2-{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}этанимидата (XI) и 207 мкл (1,19 ммоль) DIPEA в 1,5 мл ТГФ добавляли по каплям 55 мкл (0,44 ммоль) хлорангидрида (S)-(-)-2-ацетоксипропионовой кислоты (XII-А). Через 30 мин при 0°С добавляли 107 мг (0,44 ммоль) 2-хлор-4-(трифторметокси)фенилгидразина (XIII) и смесь затем перемешивали при комнатной температуре в течение ночи. Реакционную смесь затем грели в микроволновой печи при 100°С в течение 3 часов. Реакционную смесь смешивали с несколькими каплями воды и очищали хроматографией (препаративная ВЭЖХ, элюент: градиент ацетонитрил/вода, 0,1%-ная муравьиная кислота). После лиофилизации фракций, содержащих продукт, получали 152 мг (57% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,23 мин; МС (ESIpos): m/z=669.1 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=8.01-7.53 (м, 7Н), 6.89 (д, 1Н), 5.92-5.60 (м, 1Н), 5.11 (д, 2Н), 4.39-4.19 (м, 1Н), 4.09-3.77 (м, 2Н), 1.77 (уш. с, 3H), 1.54 (д, 3H)
b) 5-(4-Хлорфенил)-2-({1-[2-хлор-4-(трифторметокси)фенил]-5-[(1S)-1-гидроксиэтил]-1Н-1,2,4-триазол-3-ил}метил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3H-1,2,4-триазол-3-он (I-A-9)
Смесь 141 мг (0,21 ммоль) соединения со стадии а) и 210 мкл (0,21 ммоль) 1 М гидроксида натрия в 1,3 мл метанола перемешивали в течение 2 мин при 0°С и 90 мин при комнатной температуре. Добавляли 0,5 г активированного ионообменного материала (Dowex 50WX8, 200-400 меш) и перемешивали при комнатной температуре в течение 30 мин. Затем ионообменный материал отфильтровывали и дополнительно промывали метанолом. Упаривали фильтрат и остаток сушили в вакууме. Получали 125 мг (94% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,11 мин; МС (ESIpos): m/z=627.3 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.87 (д, 1Н), 7.82-7.71 (м, 4Н), 7.67-7.51 (м, 4Н), 6.89 (д, 1Н), 5.54 (д, 1Н), 5.07 (д, 2Н), 4.75-4.58 (м, 1Н), 4.39-4.17 (м, 1Н), 4.08-3.74 (м, 2Н), 1.40 (д, 3H)
Пример 16
а) (1S)-1-{3-({3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}метил)-1-[2,6-дихлорфенил]-1Н-1,2,4-триазол-5-ил}этилацетат (XIV-А-10)
При охлаждении льдом к смеси 200 мг (0,53 ммоль) метил-2-{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}этанимидата (XI) и 276 мкл (1,58 ммоль) DIPEA в 2 мл ТГФ добавляли по каплям 73 мкл (0,58 ммоль) хлорангидрида (S)-(-)-2-ацетоксипропионовой кислоты (XII-А). Через 1 ч при 0°С добавляли 124 мг (0,58 ммоль) гидрохлорида 2,6-дихлорфенилгидразина (XIII) и смесь затем перемешивали при комнатной температуре в течение ночи. Реакционную смесь затем грели в микроволновой печи при 100°С в течение 3 часов. Реакционную смесь смешивали с несколькими каплями воды и очищали хроматографией (препаративная ВЭЖХ, элюент: градиент ацетонитрил/вода, 0,1%-ная муравьиная кислота). После лиофилизации фракций, содержащих продукт, получали 124 мг (37% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,08 мин; МС (ESIpos): m/z=619.0 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.84-7.52 (м, 1Н), 6.89 (д, 1Н), 5.78 (д, 1Н), 5.13 (д, 2Н), 4.41-4.20 (м, 1Н), 4.11-3.70 (м, 2Н), 1.78 (с, 3H), 1.55 (д, 3H)
b) 5-(4-Хлорфенил)-2-({1-(2,6-дихлорфенил)-5-[(1S)-1-гидроксиэтил]-1Н-1,2,4-триазол-3-ил}метил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3H-1,2,4-триазол-3-он (I-A-10)

Смесь 119 мг (0,19 ммоль) соединения со стадии а) и 190 мкл (0,19 ммоль) 1 М водного гидроксида натрия в 2 мл метанола перемешивали в течение 2 мин при 0°С и 90 мин при комнатной температуре. Добавляли 0,5 г активированного ионообменного материала (Dowex 50WX8, 200-400 меш) и перемешивали при комнатной температуре в течение 30 мин. Затем ионообменный материал отфильтровывали и дополнительно промывали метанолом. Упаривали фильтрат и остаток сушили в вакууме. Получали 110 мг (колич.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,00 мин; МС (ESIpos): m/z=576.9 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.81-7.54 (м, 7Н), 6.90 (д, 1Н), 5.55 (д, 1Н), 5.19-4.96 (м, 2Н), 4.63 (т, 1Н), 4.30 (д, 1Н), 4.08-3.77 (м, 2Н), 1.41 (д, 3H)
Пример 17
а) (1R)-1-[3-({3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}метил)-1-(2,6-дихлорфенил)-1Н-1,2,4-триазол-5-ил}этилацетат (XIV-B-10)
При охлаждении льдом к смеси 200 мг (0,53 ммоль) метил-2-{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}этанимидата (XI) и 276 мкл (1,58 ммоль) DIPEA в 2 мл ТГФ добавляли по каплям 73 мкл (0,58 ммоль) хлорангидрида (R)-(-)-2-ацетоксипропионовой кислоты (XII-В). Через 30 мин при 0°С добавляли 124 мг (0,58 ммоль) гидрохлорида 2,6-дихлорфенилгидразина (XIII) и смесь затем перемешивали при комнатной температуре в течение ночи. Реакционную смесь затем грели в микроволновой печи при 150°С в течение 3 часов. Реакционную смесь смешивали с несколькими каплями воды и очищали хроматографией (препаративная ВЭЖХ, элюент: градиент ацетонитрил/вода, 0,1%-ная муравьиная кислота). После лиофилизации фракций, содержащих продукт, получали 151 мг (46% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,12 мин; МС (ESIpos): m/z=619.2 [M+H]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.83-7.56 (м, 8Н), 6.90 (д, 1Н), 5.78 (д, 1Н), 5.13 (д, 2Н), 4.42-4.12 (м, 1Н), 4.06-3.74 (м, 2Н), 1.78 (с, 3H), 1.55 (д, 3H)
б) 5-(4-Хлорфенил)-2-({1-(2,6-дихлорфенил)-5-[(1R)-1-гидроксиэтил]-1Н-1,2,4-триазол-3-ил}метил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3H-1,2,4-триазол-3-он (I-B-10)

Смесь 141 мг (0,23 ммоль) соединения со стадии а) и 230 мкл (0,23 ммоль) 1 М гидроксида натрия в 2,4 мл метанола перемешивали в течение 2 мин при 0°С и 90 мин при комнатной температуре. Добавляли 0,5 г активированного ионообменного материала (Dowex 50WX8, 200-400 меш) и перемешивали при комнатной температуре в течение 30 мин. Затем ионообменный материал отфильтровывали и дополнительно промывали метанолом. Упаривали фильтрат и остаток сушили в вакууме. Получали 128 мг (97% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,01мин; МС (ESIpos): m/z=577.2 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.78-7.57 (м, 7Н), 6.90 (д, 1Н), 5.55 (д, 1Н), 5.08 (д, 2Н), 4.63 (т, 1Н), 4.40-4.19 (м, 1Н), 4.12-3.76 (м, 2Н), 1.41 (д, 3H)
Пример 18
a) (1S)-1-{3-({3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}метил)-1-[2-(трифторметокси)фенил)-1Н-1,2,4-триазол-5-ил]этилацетат (XIV-A-11)
При охлаждении льдом к смеси 150 мг (0,40 ммоль) метил-2-{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}этанимидата (XI) и 207 мкл (1,19 ммоль) DIPEA в 3 мл ТГФ добавляли по каплям 55 мкл (0,44 ммоль) хлорангидрида (S)-(-)-2-ацетоксипропионовой кислоты (XII-А). Через 30 мин при 0°С добавляли 143 мг (0,73 ммоль) гидрохлорида 2-трифторметоксифенилгидразина (XIII) и смесь затем перемешивали при комнатной температуре в течение ночи. Реакционную смесь затем грели в микроволновой печи при 120°С в течение 3 часов и очищали хроматографией (препаративная ВЭЖХ, элюент: градиент ацетонитрил/вода, 0,1%-ная муравьиная кислота). После лиофилизации фракций, содержащих продукт, получали 141 мг (56% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,14 мин; МС (ESIpos): m/z=635.3 [М+Н]+
b) 5-(4-Хлорфенил)-2-({5-[(1S)-1-гидроксиэтил]-1-[2-(трифторметокси)фенил]-1Н-1,2,4-триазол-3-ил}метил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3H-1,2,4-триазол-3-он (I-A-11)

Смесь 140 мг (0,22 ммоль) соединения со стадии а) и 220 мкл (0,22 ммоль) 1 М гидроксида натрия в 2 мл метанола перемешивали при комнатной температуре в течение 60 мин. Затем добавляли 17 мкл 50%-ной муравьиной кислоты и очищали хроматографией (препаративная ВЭЖХ, элюент: градиент ацетонитрил/вода, 0,1%-ная муравьиная кислота). После лиофилизации фракций, содержащих продукт, получали 123 мг (94% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,04 мин; МС (ESIpos): m/z=593.3 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.77-7.51 (м, 8Н), 6.89 (д, 1Н), 5.54 (д, 1Н), 5.06 (д, 2Н), 4.63 (т, 1Н), 4.41-4.18 (м, 1Н), 4.07-3.77 (м, 2Н), 1.40 (д, 3H)
Пример 19
а) (1R)-1-{3-({3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}метил)-1-[2-(трифторметокси)фенил)-1Н-1,2,4-триазол-5-ил]этилацетат (XIV-B-11)
При охлаждении льдом к смеси 250 мг (0,66 ммоль) метил-2-{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}этанимидата (XI) и 345 мкл (1,98 ммоль) DIPEA в 5 мл ТГФ добавляли по каплям 109 мг (0,73 ммоль) хлорангидрида (R)-(-)-2-ацетоксипропионовой кислоты (XII-В). Через 30 мин при 0°С добавляли 166 мг (0,73 ммоль) гидрохлорида 2-трифторметоксифенилгидразина (XIII) и смесь затем перемешивали при комнатной температуре в течение ночи. Реакционную смесь затем грели в микроволновой печи при 120°С в течение 3 часов и очищали хроматографией (препаративная ВЭЖХ, элюент: градиент ацетонитрил/вода, 0,1%-ная муравьиная кислота). После лиофилизации фракций, содержащих продукт, получали 167 мг (39% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,13 мин; МС (ESIpos): m/z=635.3 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.83-7.48 (м, 8Н), 6.89 (д, 1Н), 5.75 (д, 1Н), 5.10 (д, 2Н), 4.41-4.20 (м, 1Н), 4.11-3.76 (м, 2Н), 1.75 (с, 3H), 1.53 (д, 3H)
b) 5-(4-Хлорфенил)-2-({5-[(1R)-1-гидроксиэтил]-1-[2-(трифторметокси)фенил]-1Н-1,2,4-триазол-3-ил}метил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3H-1,2,4-триазол-3-он (I-B-11)

Смесь 158 мг (0,25 ммоль) соединения со стадии а) и 250 мкл (0,25 ммоль) 1 М гидроксида натрия в 3 мл метанола перемешивали в течение 2 мин при 0°С и 90 мин при комнатной температуре. Затем добавляли 19 мкл 50%-ной муравьиной кислоты и очищали хроматографией (препаративная ВЭЖХ, элюент: градиент ацетонитрил/вода, 0,1%-ная муравьиная кислота). После лиофилизации фракций, содержащих продукт, получали 139 мг (94% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,00 мин; МС (ESIpos): m/z=593.00 [M+H]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.80-7.51(м, 8Н), 6.90 (д, 1Н), 5.54 (д, 1Н), 5.06 (с, 2Н), 4.63 (т, 1Н), 4.39-4.21 (м, 1Н), 4.08-3.76 (м, 1Н), 1.40 (д, 1Н)
Пример 20
а) (1S)-1-[1-(2,6-дифторфенил)-3-({3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}метил)-1Н-1,2,4-триазол-5-ил]этилацетат (XIV-A-12)
При охлаждении льдом к смеси 100 мг (0,26 ммоль) метил-2-{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}этанимидата (XI) и 138 мкл (0,79 ммоль) DIPEA в 2 мл ТГФ добавляли по каплям 37 мкл (0,29 ммоль) хлорангидрида (S)-(-)-2-ацетоксипропионовой кислоты (XII-А). Через 30 мин при 0°С добавляли 53 мг (0,29 ммоль) 2,6-дифторфенилгидразина (XIII) и смесь затем перемешивали при комнатной температуре в течение ночи. Реакционную смесь затем грели в микроволновой печи при 120°С в течение 3 часов и очищали хроматографией (препаративная ВЭЖХ, элюент: градиент ацетонитрил/вода, 0,1%-ная муравьиная кислота). После лиофилизации фракций, содержащих продукт, получали 130 мг (83% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,07 мин; МС (ESIpos): m/z=587.1 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7:91-7,31 (м, 7Н), 6.89 (д, 1Н), 5.74 (д, 1Н), 5.12 (д, 2Н), 4.40-4.18 (м, 1Н), 4.09-3.74 (м, 2Н), 1.80 (с, 3H), 1.53 (д, 3H)
b) 5-(4-Хлорфенил)-2-({1-(2,6-дифторфенил)-5-[(1S)-1-гидроксиэтил]-1Н-1,2,4-триазол-3-ил}метил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3H-1,2,4-триазол-3-он (I-A-12)

Смесь 120 мг (0,20 ммоль) соединения со стадии а) и 200 мкл (0,20 ммоль) 1 М гидроксида натрия в 2 мл метанола перемешивали при комнатной температуре в течение 60 мин. Затем добавляли 16 мкл 50%-ной муравьиной кислоты и очищали хроматографией (препаративная ВЭЖХ, элюент: градиент ацетонитрил/вода, 0,1%-ная муравьиная кислота). После лиофилизации фракций, содержащих продукт, получали 110 мг (99% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=0,97 мин; МС (ESIpos): m/z=545.2 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.86-7.57 (м, 5Н), 7.38 (с, 2Н), 6.89 (д, 1Н), 5.63 (д, 1Н), 5.08 (с, 2Н), 4.74 (т, 1Н), 4.30 (д, 1Н), 4.11-3.71(м, 2Н), 1.39 (д, 3H)
Пример 21
а) (1S)-1-{3-({3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}метил)-1-[4-хлор-2-(трифторметокси)фенил)-1Н-1,2,4-триазол-5-ил]этилацетат (XIV-A-13)
При охлаждении льдом к смеси 100 мг (0,26 ммоль) метил-2-{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}этанимидата (XI) и 138 мкл (0,79 ммоль) DIPEA в 2 мл ТГФ добавляли по каплям 37 мкл (0,29 ммоль) хлорангидрида (S)-(-)-2-ацетоксипропионовой кислоты (XII-А). Через 30 мин при 0°С добавляли 53 мг (0,29 ммоль) 4-хлор-2-(трифторметокси)фенилгидразина (XIII) и смесь затем перемешивали при комнатной температуре в течение ночи. Реакционную смесь затем грели в микроволновой печи при 120°С в течение 3 часов и очищали хроматографией (препаративная ВЭЖХ, элюент: градиент ацетонитрил/вода, 0,1%-ная муравьиная кислота). После лиофилизации фракций, содержащих продукт, получали 76 мг (43% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,17 мин; МС (ESIpos): m/z=669.1 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=8.01-7.53 (м, 7Н), 6.88 (д, 1Н), 5.74 (д, 1Н), 5.10 (д, 2Н), 4.29 (д, 1Н), 4.10-3.75 (м, 2Н), 1.79 (с, 3H), 1.53 (д, 3H)
b) 5-(4-Хлорфенил)-2-({1-[4-хлор-2-(трифторметокси)фенил]-5-[(1S)-1-гидроксиэтил]-1Н-1,2,4-триазол-3-ил}метил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3H-1,2,4-триазол-3-он (I-A-13)

Смесь 70 мг (0,10 ммоль) соединения со стадии а) и 105 мкл (0,10 ммоль) 1 М гидроксида натрия в 1 мл метанола перемешивали при комнатной температуре в течение 30 мин. Затем добавляли 8 мкл 50%-ной муравьиной кислоты и очищали хроматографией (препаративная ВЭЖХ, элюент: градиент ацетонитрил/вода, 0,1%-ная муравьиная кислота). После лиофилизации фракций, содержащих продукт, получали 59 мг (90% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,12 мин; МС (ESIpos): m/z=627.3 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.87-7.56 (м, 7Н), 6.92-6.85 (м, 1Н), 5.55 (д, 1Н), 5.20-4.96 (м, 2Н), 4.68 (т, 1Н), 4.29 (д, 1Н), 4.10-3.73 (м, 1Н), 1.40 (д, 1Н)
Пример 22
а) (1S)-1-[1-(2-хлорфенил)-3-({3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}метил)-1Н-1,2,4-триазол-5-ил]этилацетат (XIV-A-14)
При охлаждении льдом к смеси 14,1г (37,3 ммоль) метил-2-{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}этанимидата (XI) и 32,5 мл (41,0 ммоль) DIPEA в 303 мл ТГФ добавляли по каплям 5,2 мл (41,0 ммоль) хлорангидрида (S)-(-)-2-ацетоксипропионовой кислоты (XII-А). Через 1,5 часа при 0°С добавляли 7,35 г (0,44 ммоль) гидрохлорида 2-хлорфенилгидразина (XIII) и перемешивали при комнатной температуре в течение 1,5 часов. Реакционную смесь затем грели в микроволновой печи при 100°С в течение 10 часов. Затем реакционную смесь смешивали с водой и этилацетатом и энергично перемешивали. Разделяли фазы. Водную фазу экстрагировали этилацетатом. Органическую фазу промывали насыщенным водным раствором хлорида аммония, сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали хроматографией (силикагель, элюент: градиент циклогексан/этилацетат). После упаривания фракций, содержащих продукт, в роторном испарителе получали 10,4 г (47% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,09 мин; МС (ESIpos): m/z=585.2 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.84-7.49 (м, 8Н), 6.89 (д, 1Н), 5.75 (уш. с, 1Н), 5.22-5.00 (м, 2Н), 4.4-3.70 (м, 3H), 1.77 (уш. с, 3H), 1.58-1.44 (м, 3H)
b) 2-({1-(2-Хлорфенил)-5-[(1S)-1-гидроксиэтил]-1Н-1,2,4-триазол-3-ил}метил)-5-(4-хлорфенил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3H-1,2,4-триазол-3-он (I-A-14)

Смесь 10,4 г (17,7 ммоль) соединения со стадии а) и 2,84 г (35,5 ммоль) 50%-ного водного раствора гидроксида натрия в 110 мл смеси метанол/вода (10:1) перемешивали в течение 2 мин при 0°С и 1 час при комнатной температуре. Реакционную смесь добавляли к воде и доводили рН до 7 раствором 1 н. соляной кислоты. Водную фазу экстрагировали MtBE. Органическую фазу промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния и концентрировали в вакууме. Получали 10,6 г (колич.) указанного в заголовке соединения.
ЖХ-МС (Метод 2): Rt=1,75 мин; МС (ESIpos): m/z=543.1 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.94-7.35 (м, 8Н), 6.89 (д, 1Н), 5.50 (д, 1Н), 5.07 (д, 2Н), 4.69-3.70 (м, 4Н), 1.38 (д, 3H)
Пример 23
а) (1R)-1-[1-(2-хлорфенил)-3-({3-(4-хлорфенил)-5-оксо-4-[(2R)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}метил)-1Н-1,2,4-триазол-5-ил]этилацетат (XIV-B-14)
К смеси 2,5 г (6,6 ммоль) метил-2-{3-(4-хлорфенил)-5-оксо-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-4,5-дигидро-1Н-1,2,4-триазол-1-ил}этанимидата (XI) и 4,6 мл (26,4 ммоль) DIPEA в 65 мл диоксана добавляли по каплям 1,09 г (7,3 ммоль) хлорангидрида (R)-(-)-2-ацетоксипропионовой кислоты (XII-В). Через 30 мин при комнатной температуре добавляли 1,3 г (7,3 ммоль) 2-хлорфенилгидразина (XIII). Реакционную смесь перемешивали в течение 2 ч при комнатной температуре и нагревали в течение ночи до кипения с возвратом флегмы. Реакционную смесь концентрировали и очищали хроматографией (силикагель, элюент: градиент циклогексан/этилацетат). После упаривания фракций, содержащих продукт, в роторном испарителе получали 2,87 г (74% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод A): Rt=1,12 мин; МС (ESIpos): m/z=585.2 [М+Н]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.90-7.45 (м, 8Н), 6.89 (д, 1Н), 5.86-5.53 (м, 1Н), 5.11 (д, 2Н), 4.37-4.22 (м, 1Н), 4.11-3.78 (м, 2Н), 1.77 (уш. с, 3H), 1.53 (д, 3H)
b) 2-({1-(2-Хлорфенил)-5-[(1R)-1-гидроксиэтил]-1Н-1,2,4-триазол-3-ил}метил)-5-(4-хлорфенил)-4-[(2S)-3,3,3-трифтор-2-гидроксипропил]-2,4-дигидро-3H-1,2,4-триазол-3-он (I-B-14)

Смесь 2,94 г (5,0 ммоль) соединения со стадии а) и 30 мг (0,27 ммоль) карбоната цезия в 52 мл метанола перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали. Остаток растворяли в этилацетате, промывали 1 н. раствором соляной кислоты и затем насыщенным водным раствором хлорида натрия. Органическую фазу сушили над сульфатом магния и концентрировали в вакууме. Получали 2,63 г (96% от теор.) указанного в заголовке соединения.
ЖХ-МС (Метод 1): Rt=0,94 мин; МС (ESIpos): m/z=543.0 [M+H]+
1Н-ЯМР (ДМСО-d6, 400 МГц): δ=7.93-7.42 (м, 8Н), 6.90 (д, 1Н), 5.50 (уш. д, 1Н), 5.07 (с, 2Н), 4.59 (уш. с, 1Н), 4.30 (уш. д, 1Н), 4.13-3.76 (м, 2Н), 1.38 (д, 3H)
Параметры измерения рентгеновской дифрактометрии для анализа соединения формулы (I) в кристаллической Форме модификации I:
Ось сканирования Gonio
Стартовая позиция [°2θ] 2,0066
Конечная позиция [°2θ] 37,9906
Температура измерения [°С] 25
Материал анода Cu
K-Альфа1 [ ] 1,54060
] 1,54060
K-Альфа2 [] 1,54443
K-Бета [] 1,39225
K-Альфа 2/K-Альфа 10,50000
Регулировки генератора 40 мА, 40 кВ
Монохроматор падающего луча Фокусировочное рентгеновское зеркало
Вращение образца Да


Описание чертежей:
Фигура 1: Рентгеновская дифрактограмма соединения формулы (I-A-1) в кристаллической форме модификации I
| название | год | авторы | номер документа |
|---|---|---|---|
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3K И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2600927C2 |
| БИЦИКЛИЧЕСКИЕ КЕТОНЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2018 |
|
RU2797922C2 |
| ПРОИЗВОДНЫЕ 1-(4-ИЗОКСАЗОЛ-5-ИЛ)-1Н-ПИРАЗОЛ-1-ИЛ)-2-МЕТИЛПРОПАН-2-ОЛА И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИРОРОВ ИЛ-17 И ИФН-ГАММА ДЛЯ ЛЕЧЕНИЯ АУТОИММУННЫХ ЗАБОЛЕВАНИЙ И ХРОНИЧЕСКОГО ВОСПАЛЕНИЯ | 2018 |
|
RU2785342C2 |
| Соединения триазоло-пиримидина и их применение | 2019 |
|
RU2802866C2 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2654068C1 |
| ПРОИЗВОДНЫЕ ПИРИДАЗИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ RORc | 2017 |
|
RU2757571C2 |
| ЗАМЕЩЕННЫЕ ДИГИДРОПИРАЗОЛОНЫ ДЛЯ ЛЕЧЕНИЯ КАРДИОВАСКУЛЯРНЫХ И ГЕМАТОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ, ИХ ПРИМЕНЕНИЕ, ЛЕКАРСТВЕННОЕ СРЕДСТВО И СПОСОБ ЛЕЧЕНИЯ И/ИЛИ ПРОФИЛАКТИКИ | 2012 |
|
RU2611012C2 |
| ИЗОИНДОЛИНОНОВЫЕ ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ MDM2-P53, ОБЛАДАЮЩИЕ ПРОТИВОРАКОВОЙ АКТИВНОСТЬЮ | 2016 |
|
RU2797295C1 |
| ЗАМЕЩЕННЫЕ [1,2,4]ТРИАЗОЛО[1,5-a]ПИРИМИДИН-7-ИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE2 | 2015 |
|
RU2659070C9 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |

Изобретение относится к способу получения соединения формулы (I), включающему стадии [С] и [D], причем [С] соединение общей формулы (XI), в которой R2 означает алкил с 1-4 атомами углерода, подвергают взаимодействию один за другим на первой стадии [С-1] в присутствии основания с хлорангидридом кислоты общей формулы (XII), где PG означает защитную группу, и полученный при этом промежуточный продукт затем на следующей стадии [С-2] в присутствии основания подвергают взаимодействию с фенилгидразинным соединением общей формулы (XIII), причем R1A и R1B независимо друг от друга выбраны из группы, состоящей из водорода, фтора, хлора, метила, моно-фторметила, дифторметила, трифторметила, этила, метокси, дифторметокси и трифторметокси, с получением 1,2,4-триазолильного соединения общей формулы (XIV), в котором остатки PG, R1A и R1B имеют вышеуказанные значения, и его на следующей стадии [D] посредством удаления защитной группы PG превращают в соединение общей формулы (I). Изобретение также относится к соединению общей формулы (X), к соединению общей формулы (XI). Технический результат: предложен новый способ получения соединения формулы (I) с высоким выходом, подходящий для синтеза в промышленном масштабе. 3 н. и 9 з.п. ф-лы, 1 ил., 1 табл., 23 пр.
 ,
, 

 ,
,
1. Способ получения соединения формулы (I), включающий стадии [С] и [D], причем
[С] Соединение общей формулы (XI),

причем
R2 означает алкил с 1-4 атомами углерода,
подвергают взаимодействию один за другим на первой стадии [С-1] в присутствии основания с хлорангидридом кислоты общей формулы (XII)

причем
PG означает защитную группу,
и полученный при этом промежуточный продукт затем на следующей стадии
[С-2] в присутствии основания подвергают взаимодействию с фенилгидразинным соединением общей формулы (XIII)

причем R1A и R1B независимо друг от друга выбраны из группы, состоящей из водорода, фтора, хлора, метила, моно-фторметила, дифторметила, трифторметила, этила, метокси, дифторметокси и трифторметокси,
с получением 1,2,4-триазолильного соединения общей формулы (XIV)

причем остатки PG, R1A и R1B имеют вышеуказанные значения,
и его на следующей стадии
[D] посредством удаления защитной группы PG превращают в соединение общей формулы (I)

причем остатки R1A и R1B имеют вышеуказанные значения.
2. Способ по п. 1, отличающийся тем, что R2 означает метил.
3. Способ по п. 1, отличающийся тем, что защитная группа PG представляет собой ацетил.
4. Способ по п. 1, отличающийся тем, что его осуществляют в виде однореакторного процесса в присутствии подходящего растворителя и промежуточный продукт со стадии [С-1] без выделения, т.е. в растворе, затем подвергают взаимодействию на следующей стадии [С-2].
5. Способ по п. 1, отличающийся тем, что его осуществляют в виде однореакторного процесса в присутствии подходящего растворителя и полученное на стадии [С-2] 1,2,4-триазолильное соединение общей формулы (XIV) без выделения, т.е. в растворе, превращают на следующей стадии [D] в соединение общей формулы (I).
6. Способ по п. 1, отличающийся тем, что R1A и R1B независимо друг от друга выбраны из группы, состоящей из водорода, фтора и хлора, причем по меньшей мере один из заместителей отличается от водорода.
7. Способ по п. 1, отличающийся тем, что перед стадией [С] включает дополнительную стадию [В], причем
[В] Соединение общей формулы (X)

подвергают взаимодействию с основным алкоксилатом с 1-4 атомами углерода, предпочтительно метилатом натрия, с получением иминосложноэфирного соединения общей формулы (XI),

причем
R2 означает алкил с 1-4 атомами углерода, предпочтительно метил.
8. Способ по п. 1, отличающийся тем, что его осуществляют в виде однореакторного процесса в присутствии подходящего растворителя и иминосложноэфирное соединение общей формулы (XI) со стадии [В] без выделения, т.е. в растворе, затем подвергают взаимодействию на следующей стадии [С].
9. Способ по одному из пп. 1-8, отличающийся тем, что перед стадией [В] включает дополнительную стадию [А], причем
[А] Соединение общей формулы (II)

подвергают взаимодействию с нитрильным соединением (IX),

причем X означает уходящую группу, предпочтительно хлорид или бромид,
с получением соединения общей формулы (X)

10. Соединение общей формулы (X)

11. Соединение общей формулы (XI)

причем
R2 означает алкил с 1-4 атомами углерода.
12. Соединение по п. 11, причем R2 означает метил.
| EA 201290836 A1, 29.03.2013 | |||
| WO 2010105770 A1, 23.09.2010 | |||
| Сверлильный станок | 1932 |
|
SU32137A1 |

