Предпосылки к созданию изобретения
Данное изобретение относится к способу и промежуточным соединениям для получения ингибиторов 5-липоксигеназы. Ингибиторы 5-липоксигеназы, полученные в соответствии с настоящим изобретением, раскрыты в патентной заявке США серийный номер 09/0200140, которая представляет собой продолжение 08/809901, поданной 13 июня 1997, в настоящее время отозвана. Данная находящаяся на рассмотрении заявка озаглавлена "ингибиторы 5-липоксигеназы" и включена ссылкой в описание в полном объеме.
Ингибиторы 5-липоксигеназы, полученные в соответствии с настоящим изобретением, представляют собой селективные ингибиторы действия фермента липоксигеназы и полезны при лечении или облегчении воспалительных заболеваний, аллергии и сердечно-сосудистых заболеваний у млекопитающих.
Краткое описание изобретения
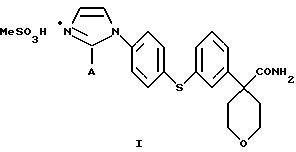
Настоящее изобретение относится к способу получения соединения формулы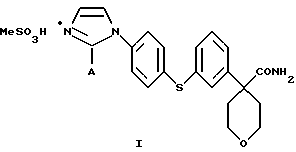
в которой А представляет собой C1-С6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, который включает взаимодействие соединения формулы II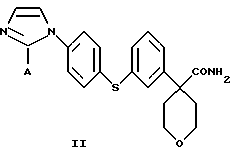
в которой А представляет собой C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, с сульфокислотой в C1-C5 алкиловом спирте; и осаждение соединения формулы I путем добавления органического растворителя, менее полярного, чем спирт.
Кислота представляет собой метансульфокислоту, и органический растворитель является диизопропиловым эфиром или этилацетатом.
В следующем аспекте настоящего изобретения соединение формулы II получают взаимодействием соединения формулы III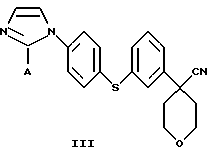
в которой А представляет собой C1-С6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, с гидроксидом в спиртовом растворителе.
Гидроксид представляет собой гидроксид калия, и спирт является трет-бутиловым спиртом.
В следующем аспекте настоящего изобретения соединение формулы III получают взаимодействием соединения формулы IV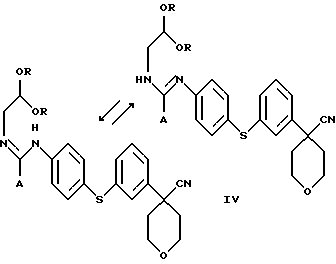
в которой А представляет собой C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, с органической или минеральной кислотой.
Кислота представляет собой уксусную кислоту, серную кислоту, муравьиную кислоту или п-толуолсульфокислоту. Предпочтительной кислотой является муравьиная кислота.
В следующем аспекте настоящего изобретения соединение формулы IV получают при взаимодействии соединения формулы V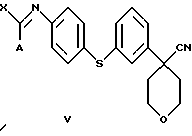
в которой А представляет собой C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, и в которой Х является Cl, Br, I или ОСН3, с избытком ацеталя аминоацетальдегида.
В следующем аспекте настоящего изобретения соединение формулы V, в которой Х является Cl, Br или I, получают взаимодействием соединения формулы VI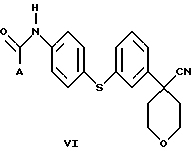
в которой А представляет собой C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, с пентагалогенидом фосфора в инертном растворителе. Соединение формулы V можно также получить взаимодействием соединения формулы VI с (CH3)3O+BF4 - с образованием промежуточного соединения, в котором Х представляет собой ОСН3.
Пентагалогенид представляет собой пятихлористый фосфор(III), пятииодистый фосфор (III) или пятибромистый фосфор (III), а растворитель представляет собой толуол. Предпочтительным А является СН3.
В дополнительном аспекте настоящего изобретения соединение формулы VI получают взаимодействием соединения формулы VIII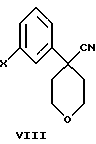
в которой Х является Сl, Br или I, с избытком 4-аминотиофенола с основанием в инертном растворителе с образованием соединения формулы VII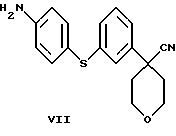
и последующей обработкой соединения формулы VII путем ацилирования галогенангидридом или ангидридом кислоты.
Другой, наиболее предпочтительный путь получения соединения формулы VI состоит во взаимодействии соединения формулы VIII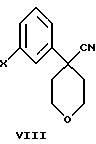
в которой Х является Сl, Br или I, с избытком 4-амидотиофенола с основанием в инертном растворителе.
4-амидотиофенол представляет собой 4-ацетамидотиофенол. Растворитель представляет собой N-метилпирролидон (NMP) или диметисульфоксид (DMSO).
Основание представляет собой карбонат натрия/карбонат цезия.
В следующем аспекте настоящего изобретения соединение формулы VIII можно получить взаимодействием соединения формулы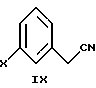
в которой Х является Сl, Br или I, с бис 2-хлорэтиловым эфиром, щелочным основанием и межфазным катализатором в инертном растворителе.
Межфазным катализатором является кислый сульфат тетрабутиламмония. Основание представляет собой гидроксид натрия.
Инертный растворитель представляет собой смесь тетрагидрофурана с водой.
Изобретение относится также к новому соединению формулы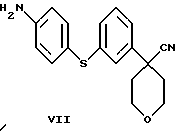
Изобретение также относится к новому соединению формулы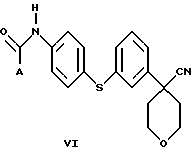
в которой А представляет собой C1-С6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом.
Изобретение относится также к новому соединению формулы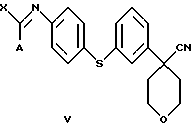
в которой А представляет собой C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, и в которой Х является Cl, Br, I или ОСН3.
Изобретение относится также к новому соединению формулы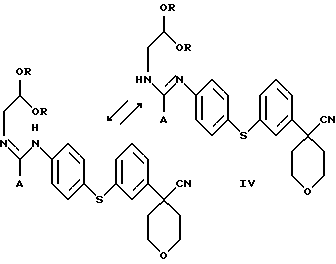
в которой А представляет собой C1-С6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, и в которой R является C1-С6 алкилом.
Изобретение относится также к новому соединению формулы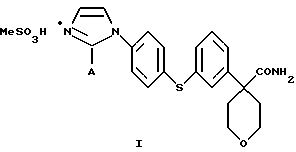
в которой А представляет собой C1-C6-алкил, арил, который моно- или дизамещен F, Сl, Br, ОСН3, C1-С3-алкилом или бензилом.
Предпочтительным соединением является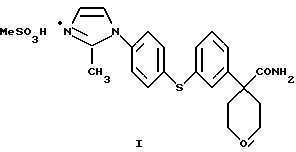
Эти новые соединения используются при получении ингибиторов 5-липоксигеназы и их фармацевтической композиции, полезных при лечении или облегчении воспалительных заболеваний, аллергии и сердечно-сосудистых заболеваний у млекопитающих.
Подробное описание изобретения
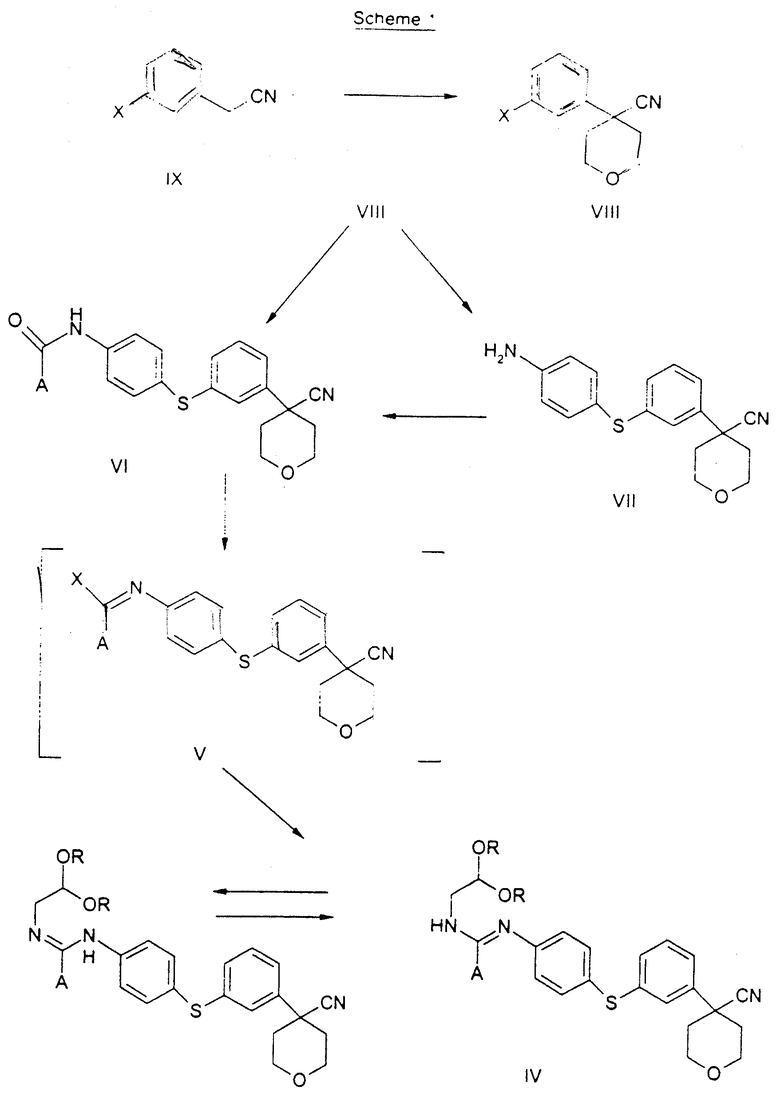
Новый способ синтеза представлен на реакционной схеме 1.
3-Бромфенилацетонитрил в тетрагидрофуране обрабатывают водным NaOH, кислым сульфатом тетрабутиламмония и бис 2-хлорэтиловым эфиром, получая арилбромидное соединение формулы VIII.
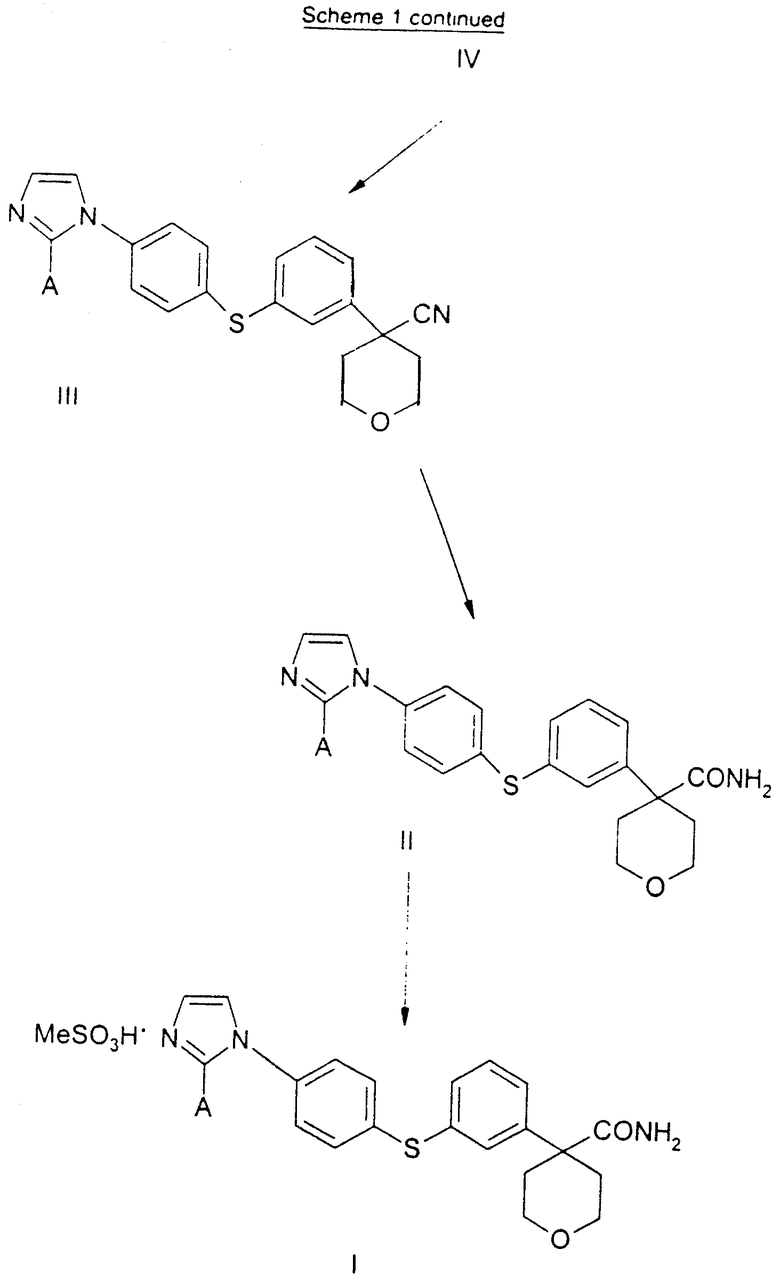
Арилбромидное соединение формулы VIII обрабатывают либо 4-аминотиофенолом, получая анилиновое соединение VII, с последующим ацилированием, либо 4-амидотиофенолом, получая амидное соединение VI. Имидазольную функцию вводят при превращении амидогруппы формулы VI путем нагревания соединения VI с пентагалогенидом фосфора (III), получая соединение V, которое обрабатывают алкилацеталем аминоацетальдегида, что приводит к амидиновому соединению IV. Амидиновое соединение IV существует в виде смеси таутомеров, которые не выделяют и немедленно подвергают кислотно-индуцированной циклизации, получая имидазольное соединение III. Последующий гидролиз нитрильной функции имидазольного соединения III приводит к соединению II - ингибитору 5-липоксигеназы. Найдена предпочтительная солевая форма путем обработки соединения II метансульфокислотой, что приводит к соединению I.
В новом способе настоящего изобретения исключены две предыдущие дорогостоящие реакции сочетания на палладии (0) для введения в молекулу сульфидной связи, как описано в патентной заявке США 09/0200140, которая включена ссылкой. Кроме того, упомянутый предпочтительный атом серы ранее вводили с использованием ТИПС-тиольного реагента (ТИПС представляет собой триизопропилсилил), получаемого из токсичного сероводорода и дорогостоящего ТИПС-хлорида.
Соединение I, в котором А представляет собой СН3, является предпочтительной солевой формой ингибитора 5-липоксигеназы, полезного при лечении или облегчении воспалительных заболеваний, аллергии и сердечно-сосудистых заболеваний у млекопитающих. В особенности соединение I полезно при лечении или облегчении воспалительных заболеваний.
Эти полезные ингибиторы 5-липоксигеназы можно вводить в виде самых разнообразных лекарственных форм.
Для лечения различных описанных выше состояний эти соединения и их фармацевтически приемлемые соли можно вводить человеку либо сами по себе, либо предпочтительно в сочетании с фармацевтически приемлемыми носителями или разбавителями в фармацевтической композиции в соответствии со стандартной фармацевтической практикой. Соединения можно вводить перорально или парентерально обычным образом.
Когда соединения вводят человеку для предотвращения или лечения воспалительного заболевания, пероральная дозировка составит от примерно 0,1 до 10 мг/кг на вес тела субъекта, подлежащего лечению, в день, предпочтительно от примерно 0,1 до 4 мг/кг в день в виде одной или разделенной доз. Если желательно парентеральное введение, тогда эффективная доза составит от примерно 0,05 до 5 мг/кг на вес тела субъекта, подлежащего лечению, в день. В некоторых случаях может быть необходимым применять дозировки, выходящие за эти пределы, поскольку дозировки будут неизбежно меняться в соответствии с возрастом, весом и реакцией индивидуального пациента, а также с серьезностью симптомов заболевания и эффективностью определенного вводимого соединения.
Для перорального введения соединения данного изобретения и их фармацевтически приемлемые соли можно вводить, например, в виде таблеток, порошков, лепешек, сиропов, или капсул, или в виде водного раствора или суспензии. В случае таблеток для перорального применения обычно используемые носители включают лактозу и кукурузный крахмал. Кроме этого, обычно добавляют смазывающие агенты, такие как стеарат магния. В случае капсул полезными разбавителями являются лактоза и высушенный кукурузный крахмал. Когда для перорального применения требуются водные суспензии, активный ингредиент объединяют с эмульгирующими и суспендирующими агентами. При желании можно добавить определенные подслащивающие и/или ароматизирующие агенты. Для внутримышечного, внутрибрюшинного, подкожного и внутривенного применения обычно готовят стерильные растворы активного ингредиента и рН этих растворов должен быть соответствующим образом установлен и забуферен. При внутривенном применении следует контролировать общую концентрацию растворенного вещества для того, чтобы получить препарат изотоническим.
Кроме того, особенно для лечения астмы, соединения формулы I данного изобретения можно вводить человеку путем ингаляции. Для этой цели их вводят в виде спрея или аэрозоля в соответствии со стандартной практикой.
Настоящее изобретение иллюстрируется следующими примерами, но не ограничивается их деталями.
Пример 1
Нитрил 4-(3-бромфенил)-тетрагидропиран-4-карбоновой кислоты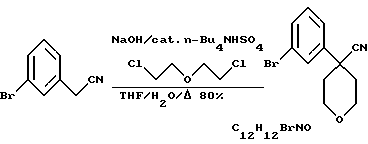
3-Бромфенилацетонитрил (51 г) в ТГФ (300 мл) обработали 40%-ным водным NaOH (470 мл), кислым сульфатом тетрабутиламмония (9 г) и добавили по каплям бис 2-хлорэтиловый эфир (32 мл). Реакционную смесь нагревали при температуре образования флегмы 4 часа и затем охладили. Смесь разбавили EtOAc (400 мл), промыли 5%-ной НСl (200 мл), водой (200 мл) и насыщенным раствором NаНСО3. После высушивания над Mg2SO4 растворитель удалили, получив сырой СР-399,554 в виде воскообразного твердого вещества (75,4 г). Это твердое вещество суспендировали в смеси 1:1 изопропилового эфира и гексана (100 мл), получив нитрил 4-(3-бромфенил)-тетрагидропиран-4-карбоновой кислоты (55,3 г, выход 80%).
Пример 2
Нитрил 4-[3-(4-аминофенилсульфанил)-фенил]-тетрагидропиран-4-карбоновой кислоты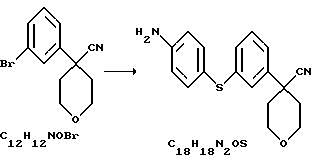
Нитрил 4-(3-бромфенил)-тетрагидропиран-4-карбоновой кислоты (133,4 г), Na2СО3 (363,6 г), Сs2СО3 (223,1 г) и аминотиофенол (62,8 г) нагревали в N-метилпирролидиноне (2,3 л) при 130oС в течение 24 часов. Добавили еще аминотиофенол (35,6 г) и продолжали нагревание в течение еще 8 часов. Смесь охладили до комнатной температуры, вылили в ледяную воду (6,8 л) и отфильтровали. Продукт суспендировали в воде (2,5 л), снова отфильтровали и промыли водой (1,5 л). После этого продукт суспендировали в EtOH (0,5 л), отфильтровали и высушили при 40oС/20 мбар, получив в результате нитрил 4-[3-(4-аминофенилсульфанил)-фенил] -тетрагидропиран-4-карбоновой кислоты (134,3 г, 86%).
Пример 3
N-{4-[3-(4-циано-тетрагидропиран-4-ил)-фенилсульфанил]-фенил}ацетамид
Нитрил 4-(3-бромфенил)-тетрагидропиран-4-карбоновой кислоты (1,33 г) смешали с Na2СО3 (1,59 г), Сs2СО3 (0,651 г) и 4-ацетамидотиофенолом (1 г) в N-метилпирролидиноне (15 мл). Реакционную смесь нагревали при 130oС в течение ночи. После охлаждения смесь вылили в ледяную воду. Продукт, выпавший в виде твердого вещества, собрали при помощи фильтрования с отсасыванием. Твердое вещество перекристаллизовали из смеси EtOAc и гексана, получив при этом N-{ 4-[3-(4-циано-тетрагидропиран-4-ил)-фенилсульфанил]-фенил}ацетамид (1,4 г, выход 80%).
Объединенные примеры 2 и 3
N-{4-[3-(4-циано-тетрагидропиран-4-ил)-фенилсульфанил]фенил}ацетамид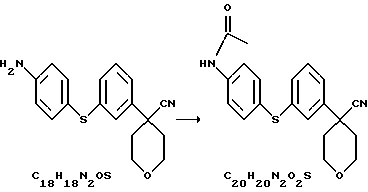
Нитрил 4-[3-(4-аминофенилсульфанил)фенил] -тетрагидропиранкарбоновой кислоты (93,57 г) и Et3N (53,1 мл) растворили в EtOAc (1,23 л) и нагрели до 50-60oС. К этому раствору в течение 30 минут добавили хлористый ацетил (27,7 мл) в EtOAc (73 мл). Полученную суспензию отфильтровали и промыли фильтровальную лепешку EtOAc (3•150 мл). Объединенные растворы EtOAc промыли водой (0,5 л), наполовину насыщенным водным раствором Na2СО3 (2•0,5 л), водой (0,5 л) и насыщенным водным раствором NaCl (0,25 л). Органические слои высушили над Na2SO4 и выпарили при 40oС. Сырой продукт перекристаллизовали из кипящего EtOH (0,52 л), получив после охлаждения, фильтрования и высушивания при 40oС/20 мбар N-{4-[3-(4-циано-тетрагидропиран-4-ил)-фенилсульфанил]-фенил} ацетамид (55,27 г, выход 52%).
Пример 4
Нитрил 4-{ 3-[4-(2-метилимидазол-1-ил)-фенилсульфанил] фенил}-тетрагидропиран-4-карбоновой кислоты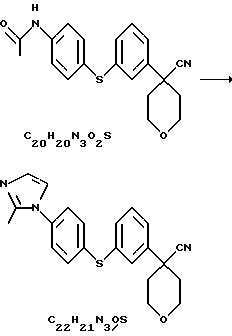
Нитрил N-4-[3-(4-аминофенилсульфанил)фенил] -тетрагидропиранкарбоновой кислоты (49,77 г) растворили в толуоле (545 мл) и нагрели до 60oС в условиях азеотропа. Из этого раствора изеотронно отогнали 20 мл растворителя для удаления оставшейся воды. К этому раствору в виде нескольких порций добавили PCl5 (35,0 г). После перемешивания в течение 1 часа при 60oС растворитель отогнали. Остаток охладили до 10oС и добавили смесь Еt3N (19,8 мл) и диметилацеталя аминоацетальдегида (15,2 мл) в EtOAc (500 мл). Полученную суспензию перемешивали в течение 30 минут при 10oС и после этого добавили еще EtOAc (150 мл). Смесь промыли водой (360 мл), а затем насыщенным водным раствором NaCl (150 мл). Органические слои высушили над Na2SO4 (52 г) и выпарили при 50oС. Остаток растворили в муравьиной кислоте (250 мл) и нагревали до температуры образования флегмы в течение 1 часа. Реакционную смесь сконцентрировали при 50oС/100 мбар до состояния масла. Это масло растворили в 10%-ной лимонной кислоте (400 мл) и EtOAc (200 мл). Водный слой экстрагировали EtOAc (350 мл). рН водного слоя довели до 9-10 при помощи наполовину насыщенного раствора К2СО3 (175 мл) и экстрагировали этот раствор EtOAc (200 мл). Экстракт высушили над Na2SO4 (48 г) и выпарили при 50oС/100 мбар, получив после фильтрования через слой двуокиси кремния с использованием в качестве элюента СН2Сl2/ 10% МеОН, нитрил 4-{3-[4-(2-метилимидазол-1-ил)-фенилсульфанил] фенил} -тетрагидропиран-4-карбоновой кислоты (27,6 г, общий выход 55%).
Пример 5
Амид 4-{3-[4-(2-метилимидазол-1-ил)-фенилсульфанил]фенил}-тетрагидропиран-4-карбоновой кислоты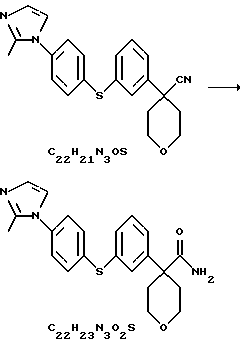
Нитрил 4-{ 3-[4-(2-метилимидазол-1-ил)-фенилсульфанил] фенил}-тетрагидропиран-4-карбоновой кислоты (27,35 г) растворили в трет-BuOH (280 мл) при 50oС. К этому раствору добавили КОН (12,28 г) и перемешивали смесь в течение ночи. Суспензию охладили до комнатной температуры и добавили воду (180 мл). Полученную суспензию отфильтровали, а фильтровальную лепешку высушили при 50oС, получив амид 4-{3-[4-(2-метилимидазол-1-ил)-фенилсульфанил]фенил}-тетрагидропиран-4-карбоновой кислоты (17,52 г, выход 55%).
Пример 6
Метилсульфонат амида 4-{3-[4-(2-метилимидазол-1-ил)-фенилсульфанил]фенил}-тетрагидропиран-4-карбоновой кислоты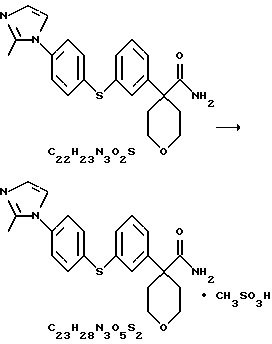
Амид 4-{ 3-[4-(2-метилимидазол-1-ил)-фенилсульфанил] фенил}-тетрагидропиран-4-карбоновой кислоты (5,05 г) суспендировали в МеОН (39 мл) при комнатной температуре. К этой суспензии по каплям добавляли метансульфокислоту до тех пор, пока все вещество не растворилось. Полученный раствор отфильтровали и промыли фильтрат МеОН (20 мл). Объединенные растворы МеОН обработали диизопропиловым эфиром (280 мл) при комнатной температуре. После перемешивания в течение ночи образовались кристаллы, которые собрали при фильтровании и высушили при 40oС/19 мбар, получив при этом метилсульфонат амида 4-{ 3-[4-(2-метилимидазол-1-ил)-фенилсульфанил] фенил} -тетрагидропиран-4-карбоновой кислоты (4,85 г, выход 77%).
Изобретение относится к новому способу получения соединений формулы I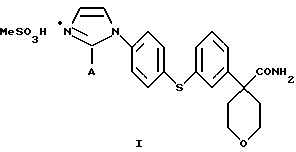
где А представляет собой C1-С6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, - ингибиторов 5-липоксигеназы, полезных для лечения или облегчения воспалительных заболеваний, аллергии и сердечно-сосудистых заболеваний. Изобретение также относится к новым соединениям формулы I, где А - С1-C6, и промежуточным соединениям формулы VIII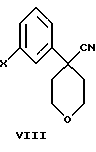
где Х - Br или Cl, необходимым для получения соединений формулы I. Технический результат состоит в разработке нового более простого и дешевого способа получения ингибиторов 5-липоксигеназы. 3 с. и 23 з.п. ф-лы.
в которой А представляет C1-С6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом,
а) включающий взаимодействие соединения формулы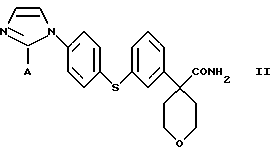
в которой А представляет C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3-алкилом или бензилом, с сульфокислотой в C1-C5-алкиловом спирте;
в) осаждение соединения формулы I путем добавления органического растворителя, полярность которого меньше, чем у упомянутого спирта.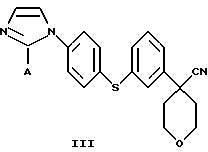
в которой А представляет C1-С6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3 алкилом или бензилом,
с гидроксидом в спиртовом растворителе.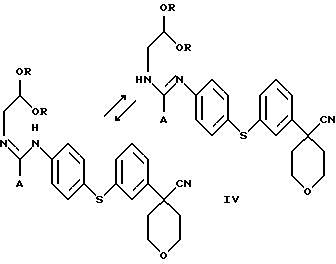
в которой А представляет C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3 алкилом или бензилом,
с органической или минеральной кислотой.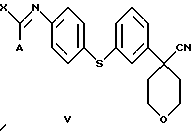
в которой А представляет C1-C6-алкил, арил, который моно- или дизамещен F, Сl, Br, ОСН3, C1-С3 алкилом или бензилом;
Х представляет Сl, Br, I или ОСН3, с избытком ацеталя аминоацетальдегида.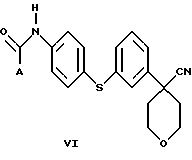
в которой А представляет C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3 алкилом или бензилом, с пентагалогенидом фосфора в инертном растворителе; или в которой Х представляет ОСН3 в формуле V, с (CH3)3O+BF4 - с образованием промежуточного соединения.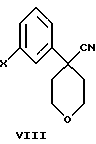
в которой Х представляет Cl, Br, или I, с избытком 4-аминотиофенола с основанием в инертном растворителе, с образованием соединения формулы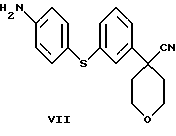
и последующей обработкой соединения формулы VII ацилированием с использованием галогенангидрида или ангидрида кислоты.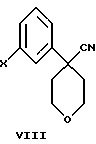
в которой Х представляет Сl, Br, или I, с избытком 4-амидотиофенола с основанием в инертном растворителе.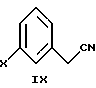
в которой Х представляет Сl, Br, или I, с бис 2-хлорэтиловым эфиром, щелочным основанием и межфазным катализатором в инертном растворителе.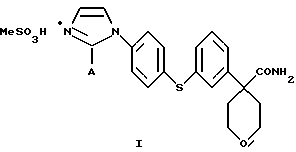
в которой А представляет C1-C6-алкил, арил, который моно- или дизамещен F, Cl, Br, ОСН3, C1-С3 алкилом или бензилом.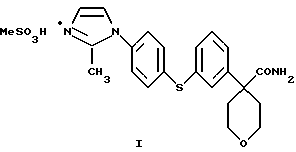
26. Соединение формулы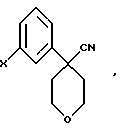
в которой Х представляет Br или Cl.
| ВЕТРОЭЛЕКТРИЧЕСКАЯ СИЛОВАЯ УСТАНОВКА | 1935 |
|
SU46830A1 |
| US 5446165 А, 29.08.1995 | |||
| US 5212198 А, 18.05.1993 | |||
| ПРОИЗВОДНЫЕ N-(3-ГИДРОКСИ-4-ПИПЕРИДИНИЛ)-(ДИГИДРОБЕНЗОФУРАН, ДИГИДРО-2Н-БЕНЗОПИРАН ИЛИ ДИГИДРОБЕНЗОДИОКСИН)-КАРБОКСАМИДА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2108332C1 |

