Данное изобретение относится к новым производным гидроксамовой кислоты и N-гидроксимочевины и к их использованию. Соединения настоящего изобретения ингибируют действие фермента липоксигеназы и являются полезными при лечении воспалительных заболеваний или состояний вообще, например аллергии, сердечнососудистых заболеваний млекопитающих, включая людей. Данное изобретение также относится к фармацевтическим композициям, включающим такие соединения, способам получения таких соединений и к способам использования их и к композициям для лечения и к композициям для лечения упомянутых выше заболеваний и состояний.
Известно, что арахидоновая кислота является биологическим предшественником нескольких групп эндогенных метаболитов, простагландинов, включающих простациклины, тромбоксаны и лейкотриены. Первой стадией метаболизма арахидоновой кислоты является высвобождение сложноэтерифицированной аразидиновой кислоты и родственных ненасыщенных жирных кислот из фосфолипидов мембраны с помощью действия фосфолипазы. Свободные жирные кислоты затем метаболизируются или циклогеназой, давая простагландины и тромбоксаны, или липоксигеназой, давая гидроперокси-жирные кислоты, которые могут далее превращаться в лейкотриены. Лейкотриены вовлечены в патофизиологию воспалительных заболеваний, включая ревматоидные артриты, подагру, астму, ишемические реперфузионные повреждения, псориаз и воспалительные заболевания пищеварительного тракта. Ожидается, что любой лекарственный препарат, который ингибирует липоксигеназу, дает важный новый метод лечения, как острых, так и хронических воспалительных состояний.
В недавнее время появился ряд обзорных статей об ингибиторах липоксигеназы. См. , например, работу H.Masamure, L.S. Melvin. - Annual Reports in Median Chemistry, 24, 71-80 (Академик Пресс, 1989) и В. J.Jitixsimmons, J. Rokach. - Lenkotrienes and Lipoxygenases, 424-502 (Elsevier, 1989).
Кроме того, ингибиторы липоксигеназы описываются в EP 279263, EP 196184, патенте Японии 63502179 и патенте США 4822809.
Настоящее изобретение проводили работы по получению соединений способных ингибировать действие липоксигеназы, и после обширных исследований достигли успеха в синтезе ряда соединений, раскрываемых здесь подробно ниже.
Настоящее изобретение представляет новые производные гидроксамовых кислот и N-гидроксимочевины общей формулы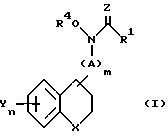
где
R1 - водород, C1-C4-алкил или -NH2;
R4 - водород;
X - химическая связь, кислород или NP5 , где R5 - водород, тиенил (низший) алкил, пиридил, пиридинил-C1-C3-алкил, C3-C6-алкил, циано, фенил, фенил-C1= C3-алкил, бензоил, причем фенил может быть необязательно замещен низшим алкилом, галоидом, трифторметилом, низшим алкокси, прямым или разветвленным низшим алкенилокси, нитрилом, низшим алкоксиалкилом, аминокарбонилом, галоид(низшим) алкокси или цилогексил(низший) алкилом;
m - 0 или 1;
n - 1 - 3;
A - C1 -C3-алкил или C2-C5-алкенилен;
Y каждый - водород, галоген, фенил, триазолилокси, C1-C2-алкокси, низший алкил, фенил - C1-C3-алкилокси, оксанилокси, дифенилокси, фенилокси, пиридилокси, C3-C12 - алкенилокси, 3,4-диметилендиоксифенокси, тиазолил или фенилалкенилокси, причем, указанное пиридильное или фенильное кольцо может быть необязательно замещено заместителем или заместителями, выбранными из группы, включающей галоид, низший алкил, низший алкокси, метилендиокси и трифторметил;
Z - кислород или сера.
Заместитель(ли) Y и связующая группа A могут быть присоединены в любом доступном положении у любого кольца.
В используемом здесь смысле "галоид" означает фтор, хлор, бром и йод.
Термин "алкил" означает необязательно прямую или разветвленную цепь.
Некоторые из соединений формулы I могут образовывать кислотно-аддитивные соли. Фармацевтически приемлемыми кислотно-аддитивными солями являются соли, образуемые из кислот, которые образуют нетоксичные кислотно-аддитивные соли, например хлоргидрат, бромгидрат, сульфат или бисульфат, фосфат или кислый фосфат, ацетат, цитрат, фумарат, глюконат, лактат, малеат, сукцинат, тартрат, метансульфонат, бензолсульфонат, толуолсульфонат и форматную соли.
Данное изобретение включает фармацевтические композиции для лечения воспалительных заболеваний, аллергий и сердечно-сосудистых заболеваний млекопитающих, которые включают фармацевтически приемлемый носитель или разбавитель и соединение формулы I или его фармацевтически приемлемую соль.
Данное изобретение включает также фармацевтические композиции для ингибирования действия фермента липоксигеназы у млекопитающих, которые включают фармацевтически приемлемый носитель и соединение формулы I или его фармацевтически приемлемую соль.
Данное изобретение далее включает способы синтеза соединений формулы I.
Соединения формулы I могут быть получены с помощью ряда способов синтеза. В приведенных ниже формулах II, III, IV и V Q представляет группу ,
,
и
X, Y, m и n имеют значения, определенные ранее. Хотя в реакционных схемах 1 и 2 ниже R1 представляет метил и NH2 соответственно и Z представляет кислород, аналогичным образом могут быть получены соединения формулы I, в которой R1 и Z имеют определенные ранее значения.
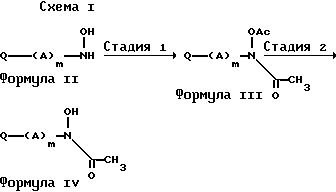
В одном из воплощений соединения формулы IV получаются согласно стадиям реакций, показанным на схеме I.
 .
.
На стадии I диацетильное соединение III получатся с помощью стандартных методов, известных в технике. Например, гидроксиламин (II) подвергается реакции с ацетилхлоридом или уксусным ангидридом в реакционно инертном растворителе и присутствии подходящего основания. Предпочтительными основаниями являются триэтиламин и пиридин. Подходящие реакционно инертные растворители включают метиленхлорид, хлороформ, тетрагидрофуран, бензол и толуол. Реакция обычно осуществляется в интервале температур от 0oC до температуры окружающей среды. Обычными являются периоды реакции от 30 мин до нескольких часов. Продукт может выделяться и очищаться с помощью общепринятых приемов, например, перекристаллизации или хроматографии.
Стадия 2 предусматривает селективный гидролиз диацетильного соединения (III) соответствующим основанием. Основания, применяемые подходящим образом в данной реакции, включают аммиак, гидроокись аммония, гидроокись натрия, гидроокись калия и гидроокись лития, предпочтительно в метаноле, этаноле, изопропиловом спирте или воде, хотя могут применяться бинарные системы растворителей, такие как спирт-вода, тетрагидрофуран-вода и аналогичные. Температуры реакции обычно составляют величины в интервале от -10oC до температуры окружающей среды, и реакция обычно завершается за период от нескольких минут до нескольких часов. Продукт, имеющий структуру, показанную с помощью формулы (IV), выделяется с помощью стандартных методов, а очистка может достигаться с помощью общепринятых средств, например перекристаллизации и хроматографии.
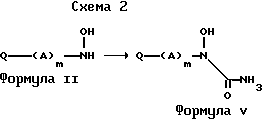
Согласно еще одному воплощению соединения формулы V получаются, как проиллюстрировано на схеме реакции 2.
 .
.
На данной стадии гидроксиламин (II) обрабатывается триметилсилилизоцианатом в реакционно инертном растворителе, обычно при температуре окружающей среды и до температуры дефлегмации. Подходящие растворители, которые не реагируют с реагентами и/или продуктами реакции, включают, например, тетрагидрофуран, диоксан, метиленхлорид и бензол. Согласно альтернативной процедуре применяют обработку гидроксиламина (II) газообразным хлористым водородом в реакционно инертном растворителе, таком как бензол или толуол, а затем последующую обработку фосгеном. Температуры реакции лежат обычно в пределах от температуры окружающей среды до точки кипения растворителя. Промежуточный карбамоилхлорид не выделяется, а подвергается реакции (т.е. ин ситу) с водным аммиаком. Полученный таким образом продукт, имеющий структуру, показанную формулой V, выделяется с помощью стандартных методов, и очистка может достигаться с помощью общепринятых средств, таких как перекристаллизация и хроматография.
Упомянутый выше гидроксиламин (II) легко получается с помощью стандартных приемов синтеза из легко доступных карбонильных соединений, например кетонов или альдегидов, или из спиртов или галогеновых соединений. Например, подходящее карбонильное соединение превращается в его оксим, а затем восстанавливается до требуемого гидроксиламина (II) подходящим восстанавливающим агентом (например, см. работу R.F. Borch и др., J. Am. Chem. Soc, 93, 2897 (1971). Предпочтительные восстанавливающие агенты включают цианоборгидрид натрия и борановые комплексы, такие как бор-пиридин, бор-триэтиламин, и бор-диметилсульфид. Может также применяться триэтилсилан в трифторуксусной кислоте.
Альтернативно, гидроксиламин II) может быть получен с помощью обработки соответствующего спирта N, O - бис(трет-бутилокси-карбонил)-гидроксиламином в условиях реакции типа Мицунобу, с последующим катализируемым кислотой гидролизом N, O - защищенного промежуточного продукта (см. FP 1045344). Примечательно также, что N, O - диацетильное соединение (III) может получаться с применением N, O - диацетил-гидроксиламина вместо N, O - бис(трет-бутилоксикарбонил)гидроксиламина, обеспечивая таким образом удобный путь к получению продукта формулы IV.
Упомянутый выше гидроксиламин (II) может также получаться из подходящего галогенидного соединения с помощью реакции с O-защищенным гидроксиламином и последующего снятия защиты (см. работу W.p. Jacks on, и др., J. Med. Chem., 31, 499 (1988). Предпочтительные O-защищенные гидроксиламины включают O-тетрагидропиринил-, O-триметилсилил- и O-бензилгидроксиламин.
Гидроксиламин формулы II, полученный таким образом с помощью представленных выше характерных приемов, выделяется с помощью стандартных методов, а очистка может достигаться с помощью общепринятых средств, таких как перекристаллизация и хроматография.
Фармацевтически приемлемые соли новых соединений настоящего изобретения свободно получаются с помощью контактирования указанных соединений со стехиометрическим количеством соответствующей минеральной или органической кислоты или в водном растворе, или подходящем органическом растворителе. Соль может затем получаться с помощью осаждения или путем выпаривания растворителя.
Соединения данного изобретения ингибируют активность фермента липоксигеназы. Данное ингибирование было продемонстрировано с помощью анализа с использованием резидентных клеток перитональной полости крыс, который определяет влияние указанных соединений на метаболизм арахидоновой кислоты.
В данном испытании некоторые предпочтительны соединения показывают низкие величины ИК50, в интервале от 0,1 до 30 мкМ, в отношении ингибирования липоксигеназы. В используемом здесь смысле величины ИК50 относится к концентрации испытываемого соединения, необходимой для достижения 50% ингибирования липоксигеназы.
Способность соединений настоящего изобретения ингибировать липоксигеназный фермент делает их полезными для регулирования симптомов, вызываемых эндогенными метаболитами, возникающими у млекопитающих от арахидоновой кислоты. Таким образом соединения являются ценными при профилактике и лечении таких состояний и заболеваний, при которых причинным фактором является накопление метаболитов арахидоновой кислоты. Примеры таких болезненных состояний включают аллергическую бронхиальную астму, кожные расстройства, ревматоидный артрит, остеоартрит и тромбоз.
Таким образом, соединения формулы I и их фармацевтически приемлемые соли представляют особую пользу при лечении или облегчении воспалительных заболеваний, аллергий, сердечно-сосудистых заболеваний у людей, а также при ингибировании фермента липоксигеназы.
Для лечения различных состояний, описанных выше, соединения формулы I и их фармацевтически приемлемые соли могут назначаться людям или по одному, или предпочтительно в сочетании с фармацевтически приемлемыми носителями или разбавителями в виде фармацевтической композиции, в соответствии со стандартной фармацевтической практикой. Соединение может назначаться для приема с помощью большого разнообразия обычных способов назначения, включающих оральное назначение, парентеральное и с помощью ингаляции. Когда соединения назначаются для приема орально, интервал доз обычно составляет в сутки примерно от 0,1 до 20 мг/кг веса тела субъекта, подвергаемого лечению, предпочтительно в день примерно от 0,1 до 1,0 мг/кг в виде единичной или разделительных доз. Если желательно парентеральное назначение, тогда эффективная доза обычно составляет в день примерно от 0,1 до 1,0 мг/кг веса тела подвергаемого лечению субъекта. В некоторых случаях может быть необходимо использовать дозы, находящиеся за указанными пределами, поскольку доза обязательно будет варьировать в зависимости от возраста, веса и ответной реакции индивидуального пациента, а также от тяжести симптомов у пациента, и силы или активности конкретного назначаемого соединения.
Для орального применения соединения формулы I и их фармацевтически приемлемые соли могут назначаться, например, в виде таблеток, порошков, пилюль, сиропов или капсул, или в виде водных растворов или суспензий. В случае таблеток для орального применения носителя, которые обычно используются, включают лактозу и кукурузный крахмал. В дополнение обычно добавляются смазочные агенты, такие как стеарат магния. В случае капсул полезными разбавителями являются лактоза и высушенный кукурузный крахмал. Когда требуются водные суспензии для орального применения, активный ингредиент смешивается с эмульгирующим и суспендирующим агентами. Если необходимо, могут добавляться некоторые подслащивающие и/или ароматизирующие или вкусовые агенты. Для внутримышечного, внутрибрюшинного, подкожного и внутривенного использования обычно приготавливаются стерильные инъецируемые растворы активного ингредиента, и pH растворов следует подходящим образом доводить до нужной величины и буферировать. Для внутривенного применения общая концентрация растворенного вещества должна соответствующим регулироваться чтобы сделать препарат изотоническим.
Настоящее изобретение иллюстрируется с помощью нижеследующих примеров. Однако, следует понимать, что изобретение не ограничивается конкретными подробностями этих примеров. Спектры протонного ядерного магнитного резонанса (ЯМР) измерялись при 270 МГц, если не указано иное, для растворов в пердейтеродиметилсульфоксиде (ДМСО-d6), и пиковые положения выражаются в частях на миллион /м,д, вниз поля от тетраметилсилана. Виды пиков обозначаются следующим образом: с. , синглет; д. , дублет; т., триплет; кв., квартет; квинт. квинтет; м, мультиплет; шир., широкий.
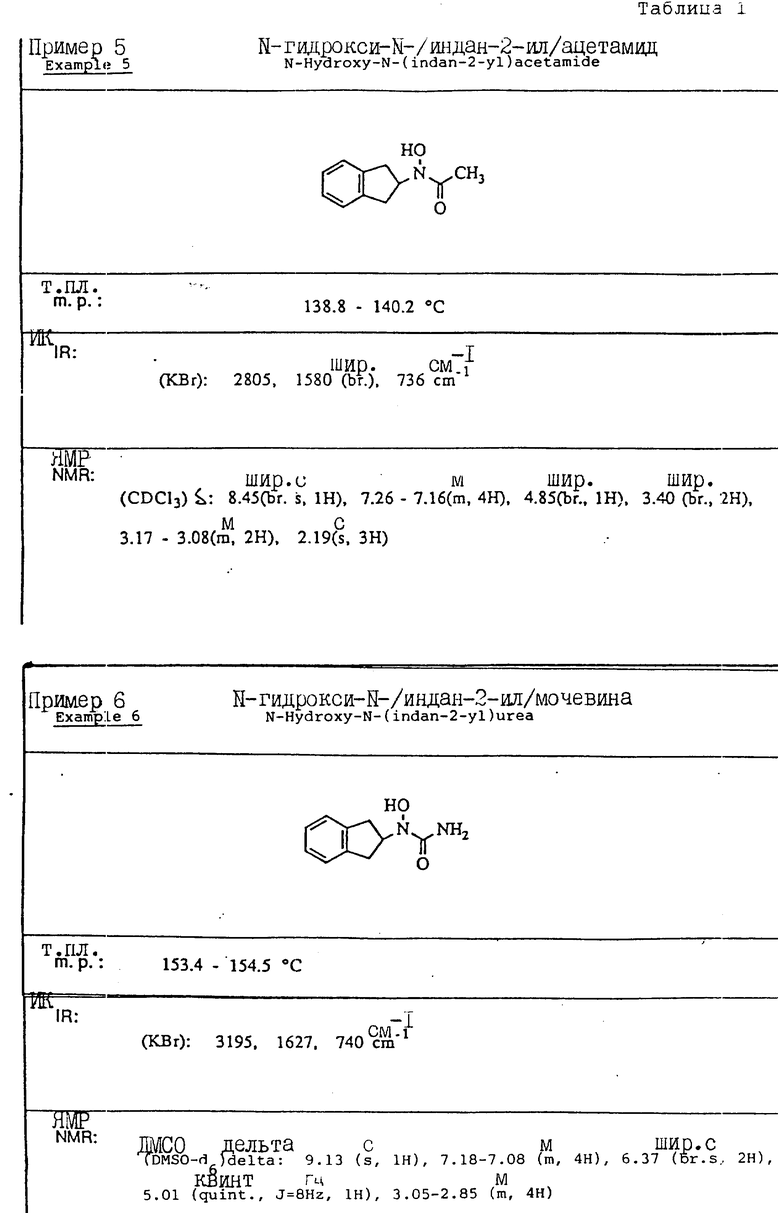
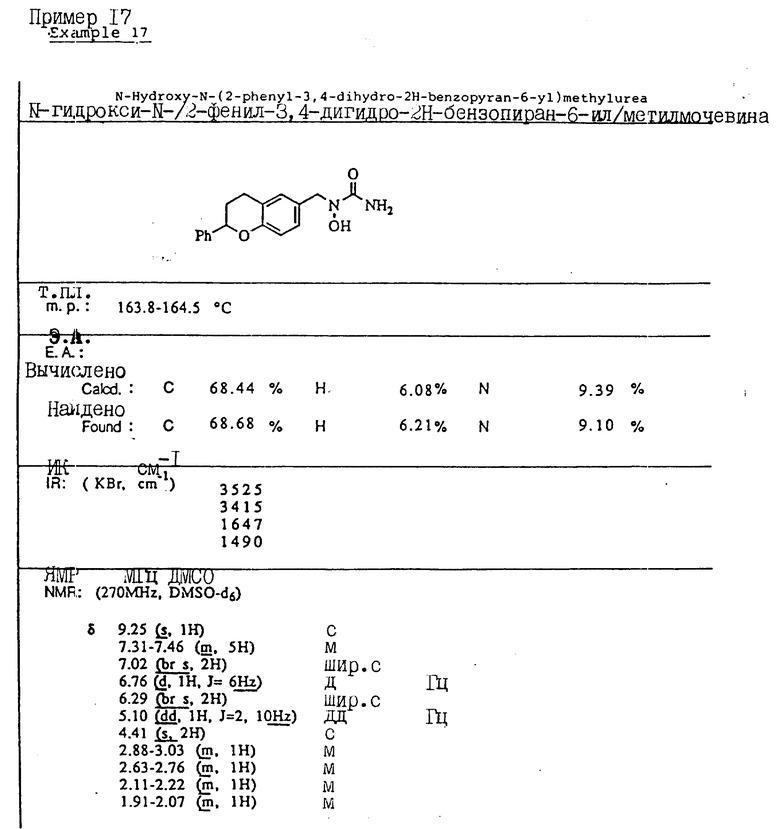
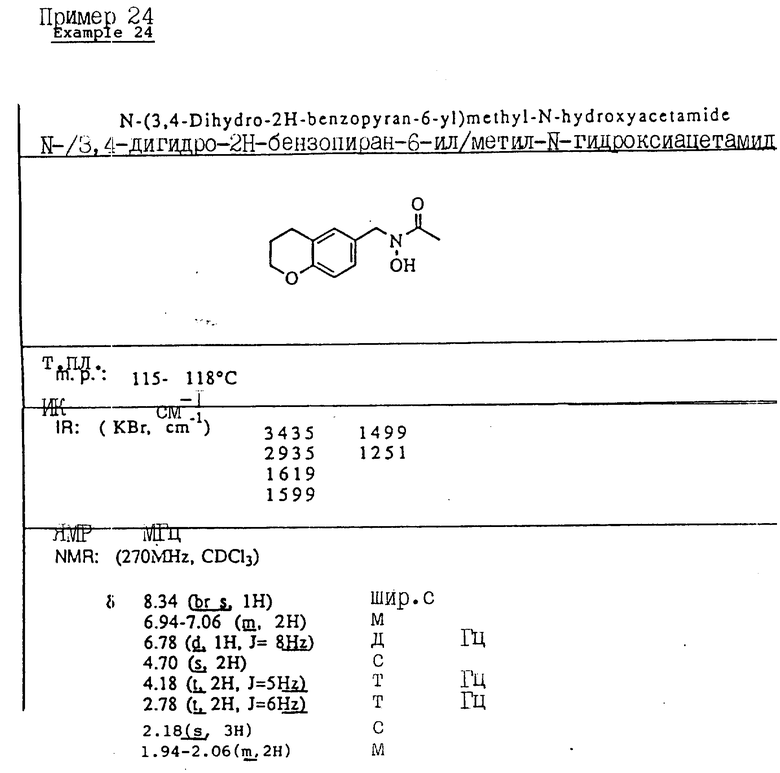
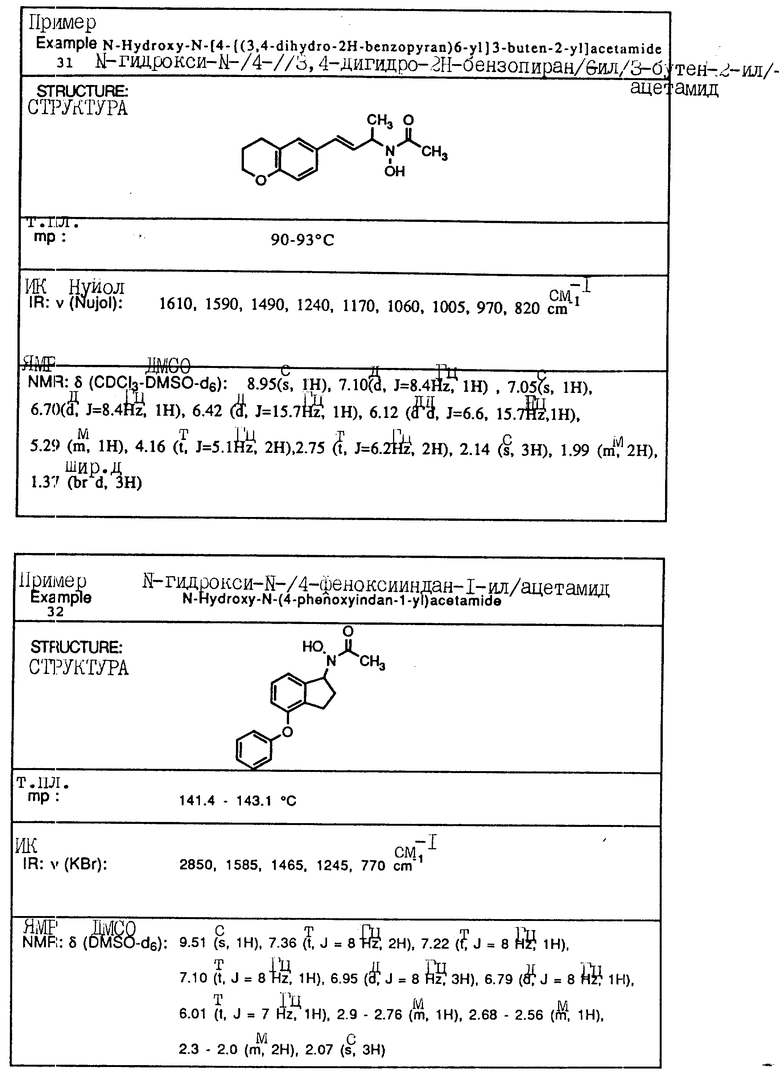
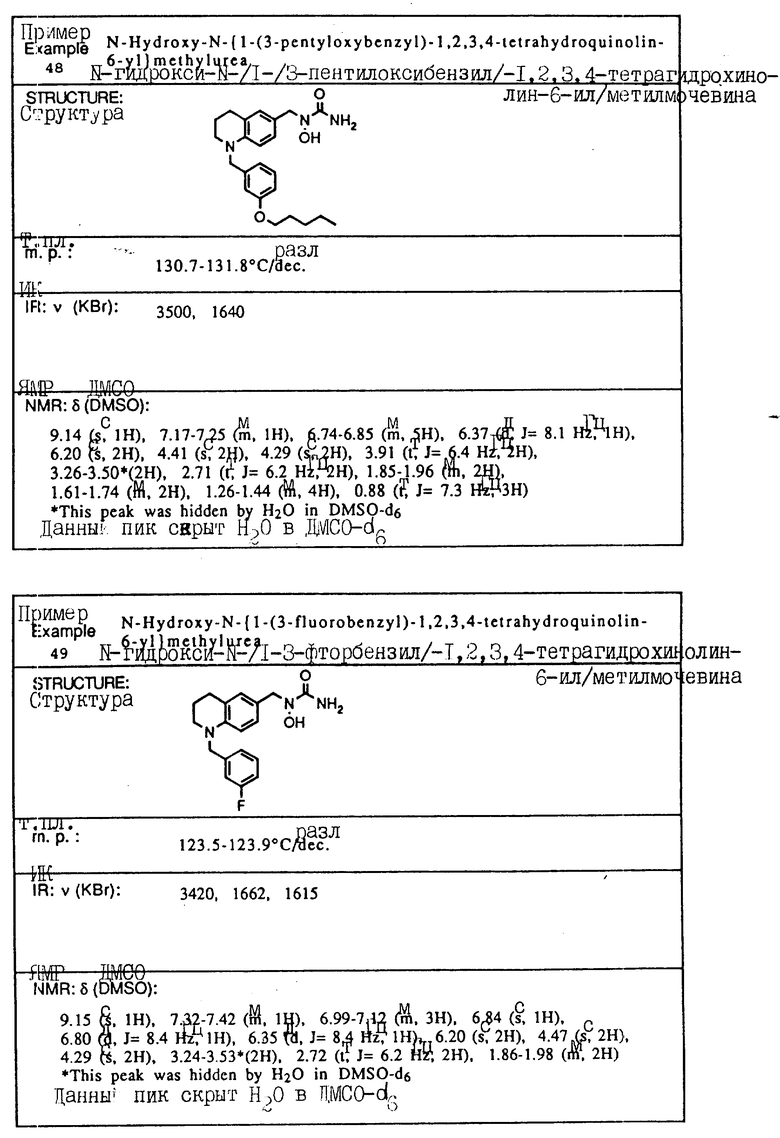
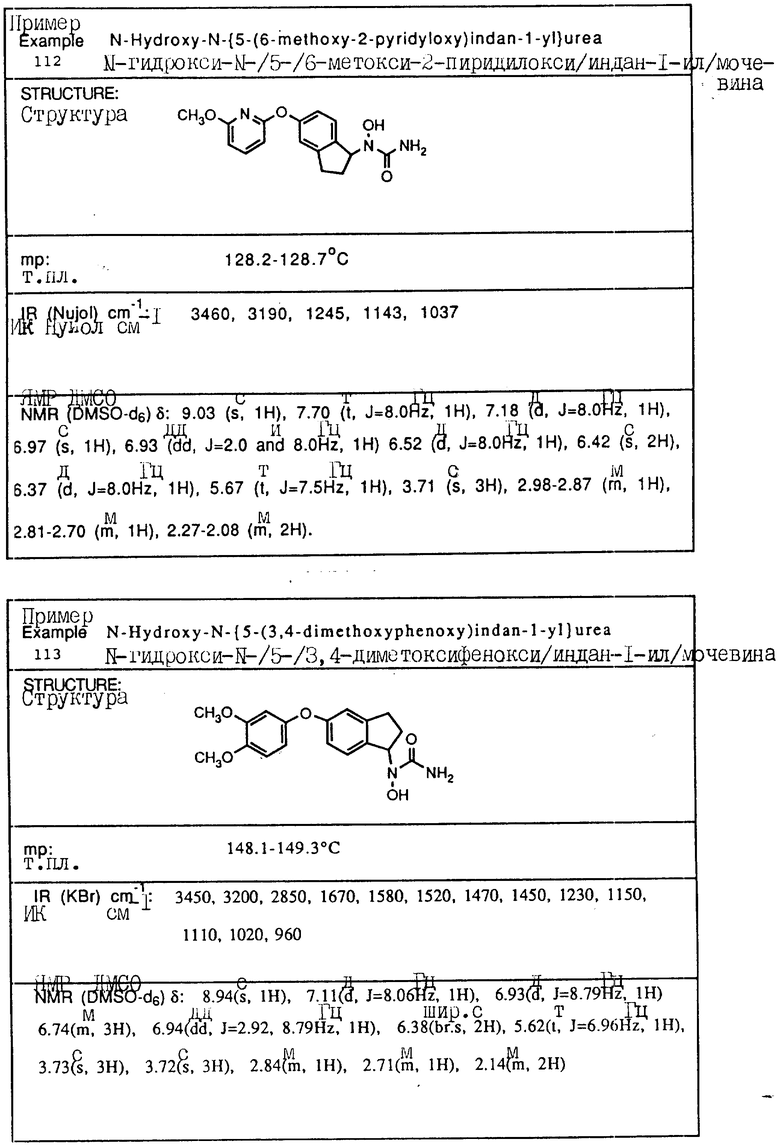
Пример 1. N-(Гидрокси)-N-(индан-1-ил)мочевина
1-Инданон (4,00 г, 303 ммоля) и хлоргидрат гидроксиламина (5,26 г, 75,7 ммоля) растворялись в смеси метанола (40 мл) и пиридина (10 мл) и перемешивались в течение 3 ч при температуре окружающей среды. Реакционная смесь концентрировалась в вакууме и получающийся остаток разбавлялся 1 норм. HCl (100 мл) и экстрагировался три раза метиленхлоридом. Органический слой сушился над сульфатом магния и концентрировался в вакууме, давая 4,13 г (923% выход) требуемого 1-инданон-оксима в виде белых игл.
Оксим (4,08 г, 27,7 ммоля), полученный на предыдущей стадии, растворялся в уксусной кислоте (50 мл), и порциями на протяжении 1 ч добавлялся цианоборгидрид натрия (9,40 г, 63 ммоля). После того, как реакция завершилась, реакционная смесь осторожно выливалась в охлажденный льдом водный карбонат натрия, так чтобы величина pH доводилась до 9. Смесь экстрагировалась метиленхлоридом, сушилась над сульфатом магния и концентрировалась в вакууме, давая 3,6 г 1-индангидроксиламина (87% выход) в виде рыжевато-коричневого порошка..
Гидроксиламин (1,26 г, 8,4 ммоля), полученный на описанной выше стадии, перемешивался в течение 1 ч с триметилсилилизоцианатом (1,65 г, 16,8 ммоля) в тетрагидрофуране. Реакционная смесь концентрировалась в вакууме, и остаток перекристаллизовывался из этилацетата, давая 0,78 г (48% выход) продукта в виде тонкодисперсного белого порошка.
Т.пл.: 158,7 - 159,4oC.
ИК (KBr): 3465, 3190, 1667, 1654, 1573, 759, 741 см-1
ЯМР (CDCl3) δ : 7,34-7,21 (м., 4H), 5,92 (дд., J = 5,8 и 8,1 Гц, 1H), 5,3 (шир. с. , 2H), 5,16 (с., 1H), 3,07-3,02 (м., 1H), 2,95-2,83 (м., 1H), 2,46-2,35 (м., 1H), 2,26-2,13 (м., 1H).
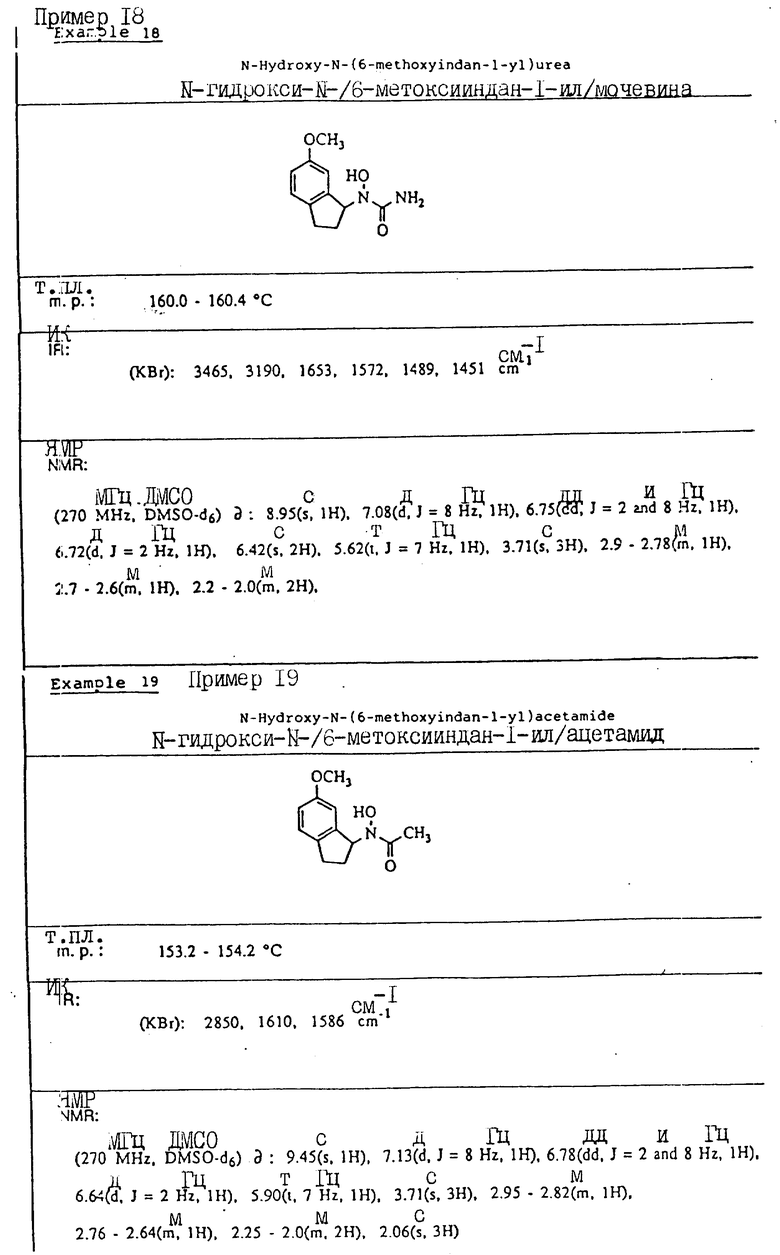
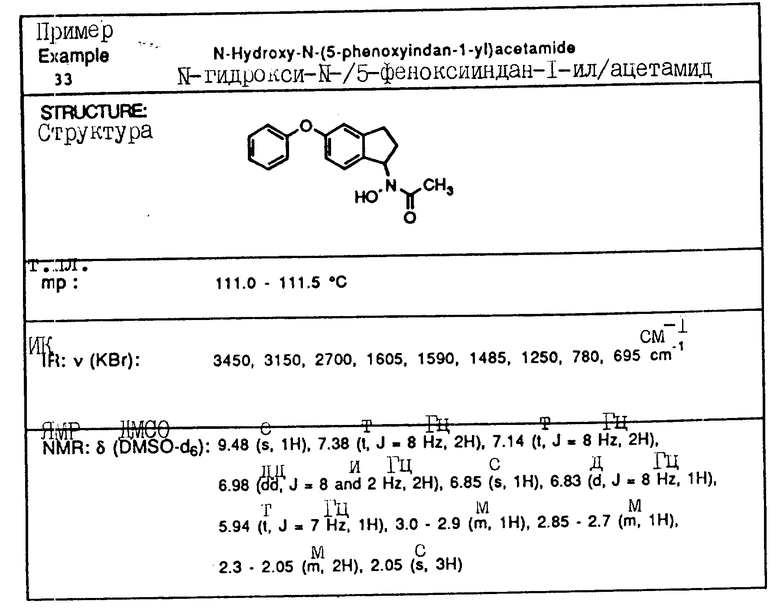
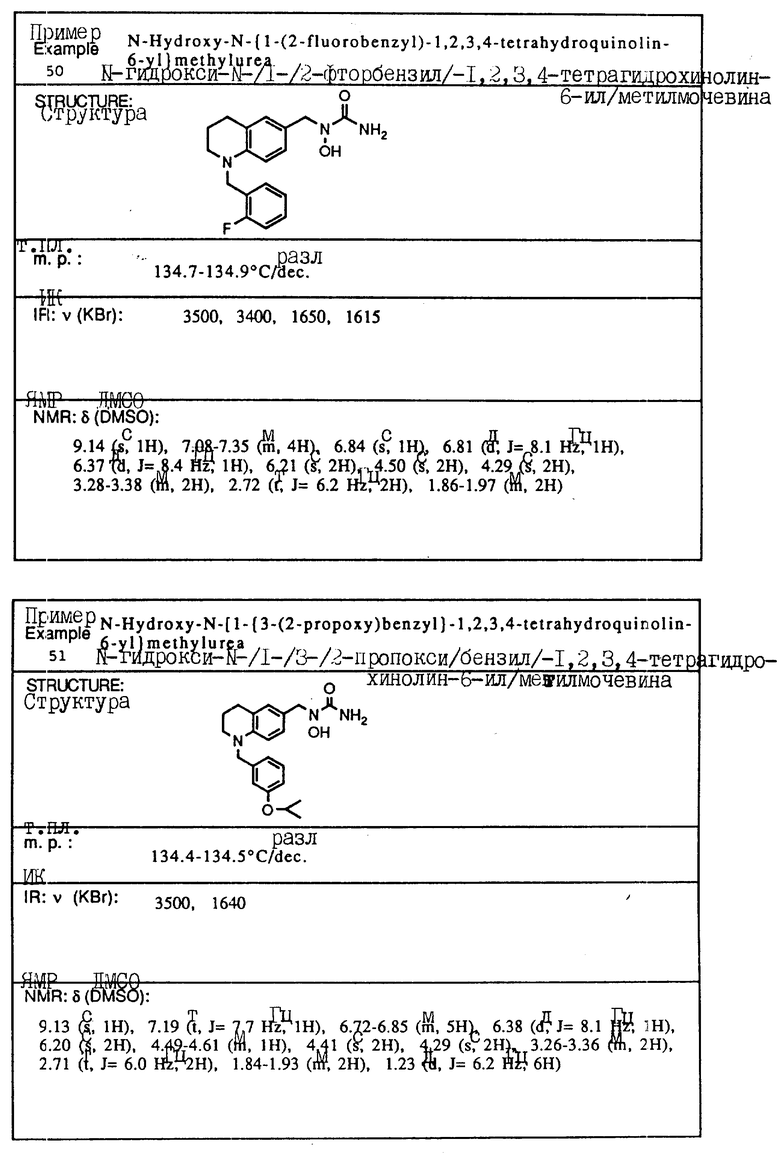
Пример 2. N-Гидрокси-N-(индан-1-ил)ацетамид
1-Индан-гидроксиламин (2,33 г, 15,6 ммоля), полученный как описано в примере 1, и триэтиламин (3,48 г, 34,3 ммоля) растворялись в метиленхлориде (40 мл), охлаждались до 0oC, и добавлялся ацетилхлорид (2,33 мл, 32,8 ммоля). Смесь перемешивалась в течение 30 мин и выливалась в 1 норм. HCl. Органический слой отделялся, сушился над сульфатом магния и концентрировался в вакууме, давая 3,58 г (98% выход) N-ацетокси -N-(индан-1-ил) ацетамида.
Ацетамид (3,65 г, 15,3 моля) растворяли в смеси метанола (20 мл) и водного аммиака (10 мл) при температуре окружающей среды. Через 30 мин смесь концентрировалась в вакууме, и остаток распределялся между водой и метиленхлоридом. Органическая фаза сушились над сульфатом магния и концентрировалась в вакууме. Полученный в результате остаток перекристаллизовывался из бензола, давая 2,06 г (70% выход) продукта в виде тонкодисперсного белого порошка.
Т.пл.,: 137,9 - 139,5oC
ИК (KBr): 3090, 2925, 1615 (шир.), 757 см-1
ЯМР (DMCO-d6) δ : 9,46 (с., 1H), 7,22-7,12 (м., 4H), 5,96 (шир.т J=8 Гц, 1H), 3,05-2,90 (м., 1H), 2,85-2,70 (м., 1H), 2,25-2,05 (м., 2H), 2,06 (с., 3H).
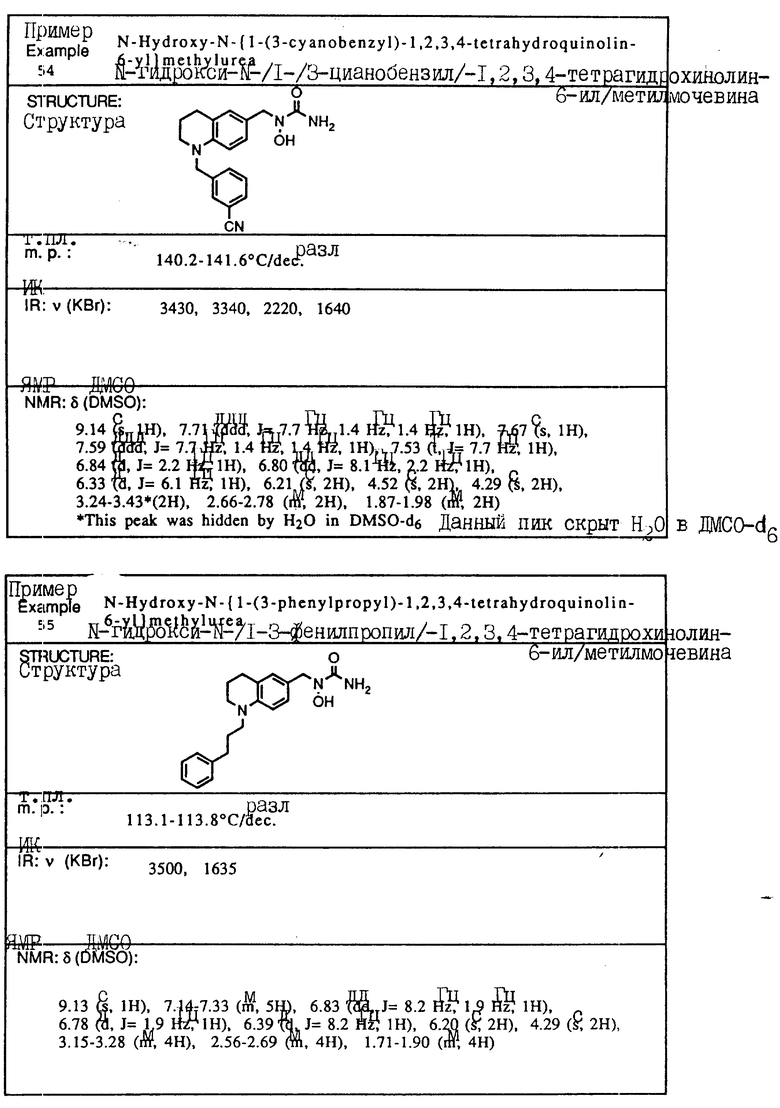
Пример 3. N-Гидрокси-N-[2-(2,3-дигидро-1H-инден-1-илиден)этил]ацетамид
Диэтил-азодикарбоксилат (3,94 г) в сухом толуоле (10 мл) добавлялся к перемешиваемому раствору 2-(2,3-дигидро-1H-инден-1-илиден)-этанола (2,41 г), N,O-диацетилгидроксиламина (1,85 г), и трифенилфосфина (5,94 г) в сухом толуоле (60 мл) при -78oC в атмосфере азота. Смесь перемешивалась при температуре окружающей среды в атмосфере азота в течение 30 мин. Смесь фильтровалась, и остаток тщательно промывался этилацетатом и гексаном (1:1). Объединенный фильтрат и промывные воды концентрировались при пониженном давлении. Хроматография на силикагеле при элюировании смесью гексана и этилацетата (3: 1) давала N-ацетокси-N-[2-(2,3-дигидро-1H-инден-1-илиден)этил]ацетамид (1,34 г). Диацетат растворялся в метаноле (10 мл), добавлялась концентрированная гидроокись аммония, смесь перемешивалась при температуре окружающей среды в течение 1 ч и концентрировалась при пониженном давлении. Получающееся в результате бледно-желтое масло экстрагировалась этилацетатом и промывалась солевым раствором. Раствор сушился над сульфатом магния и концентрировался, давая бледно-желтое масло. Хроматография на силикагеле, элюируемом смесью гексан-этилацетат (1:1) с последующей кристаллизацией из изопропилового эфира давала требуемое соединение, белое твердое вещество (0,46 г).
Т.пл.: 96,0-96,6oC
ИК- (KBr): 1650, 1610
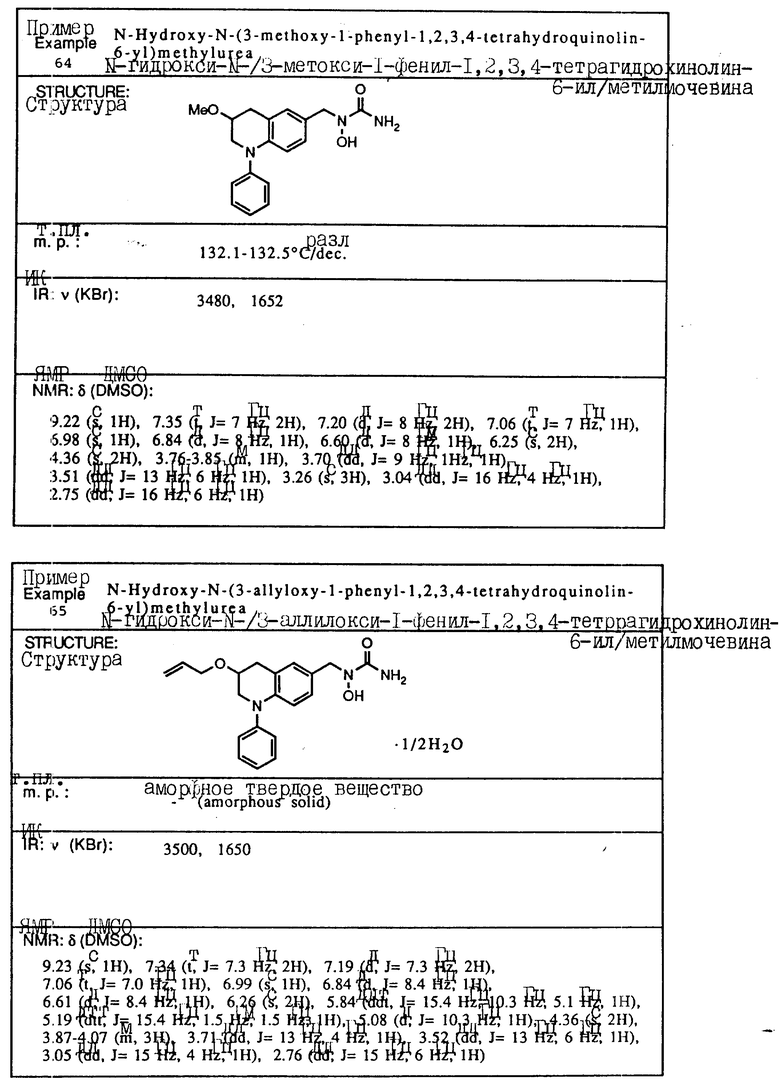
ЯМР (270 МГц, CDCl3) δ : 8,30 и 6,40 (шир., с., 1H), 7,44-7,51 (м., 1H), 7,16-7,31 (м., 3H), 6,08-6,18 (м., 1H), 4,40 (д., 2H, J= 6,2 Гц), 3,00-3,09 (м., 2H), 2,78-2,87 (м., 2H), 2,16 (с., 3H)
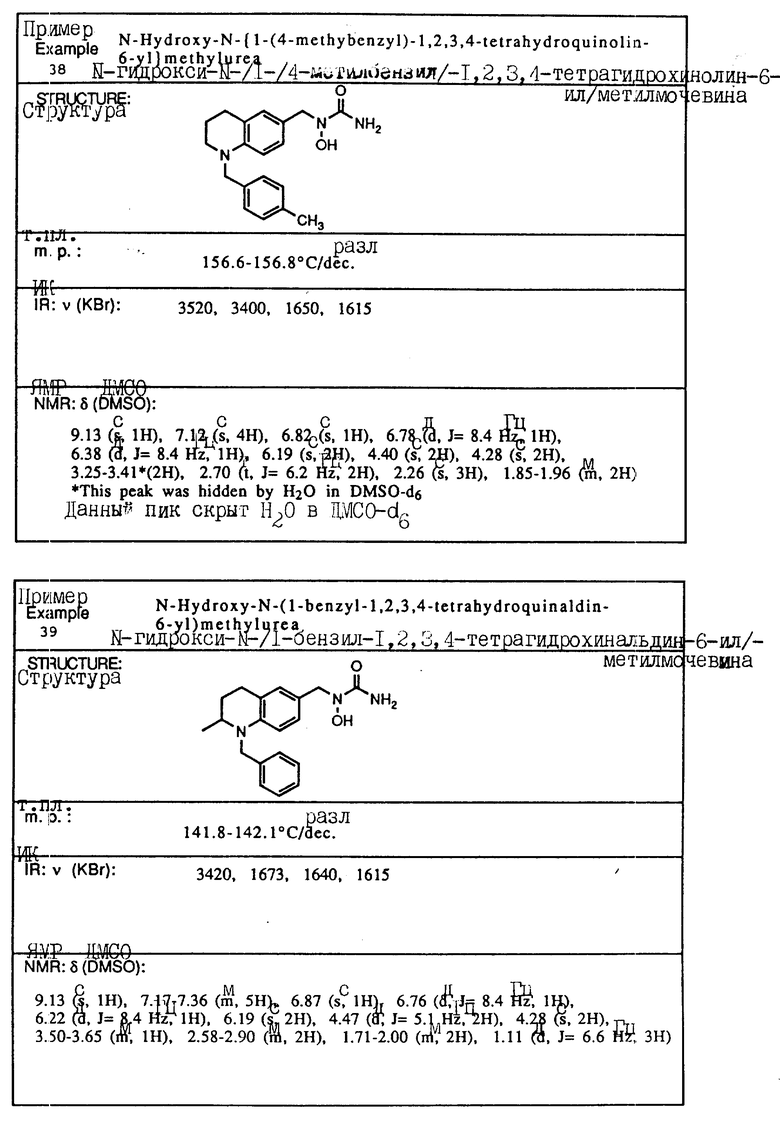
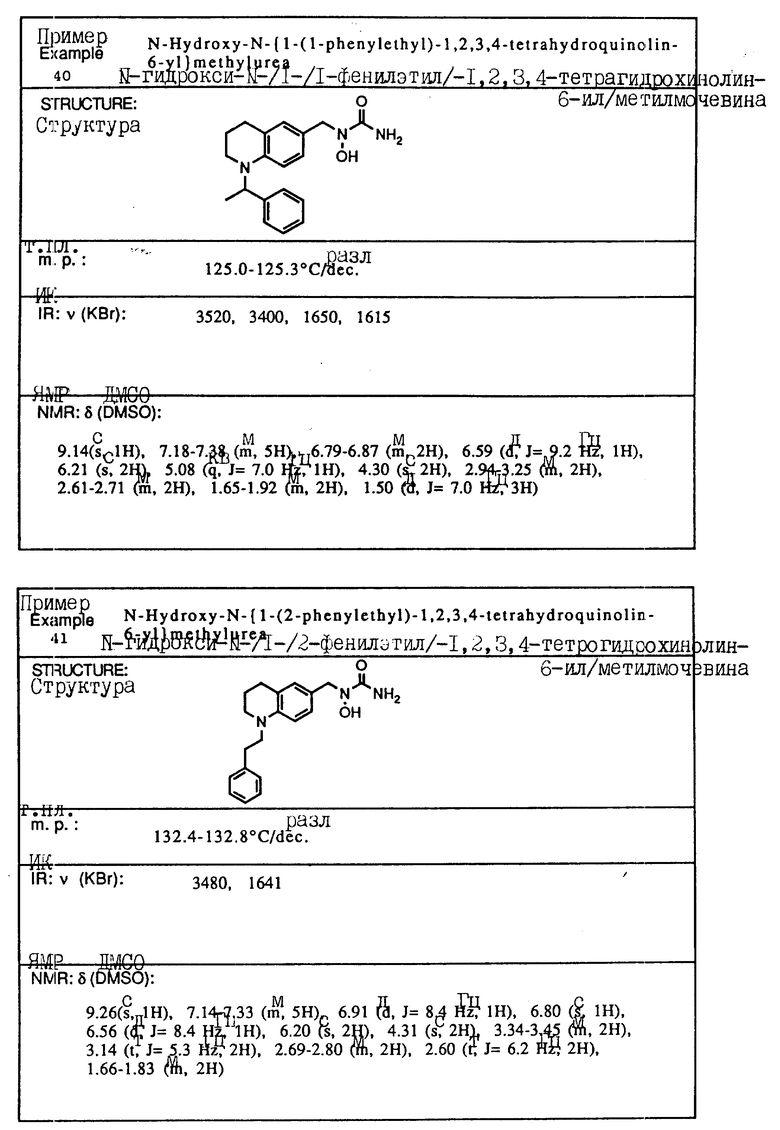
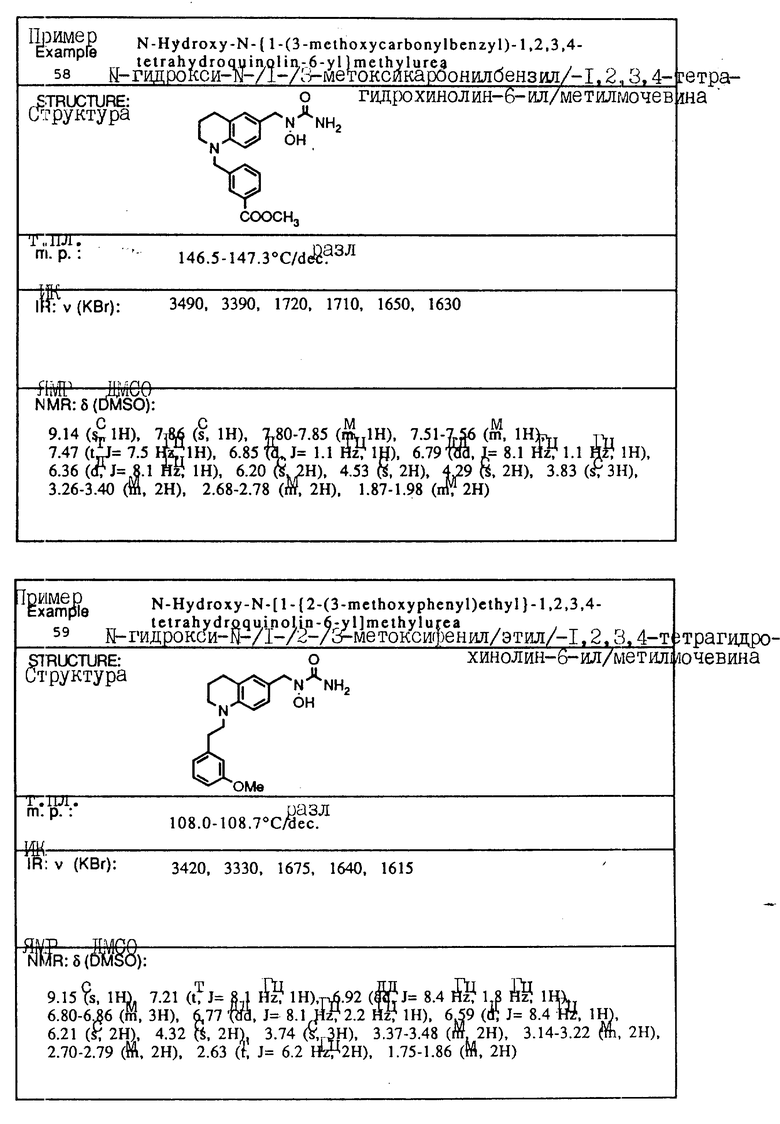
Пример 4. N-Гидрокси-N-[1-(1-бензил-1,2,3,4-тетрагидрохинолин-6-ил)-этил] мочевина
К смеси 1-бензил-1,2,3,4-тетрагидрохинолин-6-илэтан-1-ол/ (2,82 г, 10,6 ммоля), BocNh-OBoc (2,48 г, 11,1 ммоля) и трифенилфосфина (3,62 г, 13,8 ммоля) в толуоле (20 мл) добавлялся диэтил азодикарбоксилат (2,40 г, 13,8 ммоля) при -78oC в атмосфере азота. Смесь перемешивалась при -78oC до температуры окружающей среды в течение 30 мин. Смесь концентрировалась в вакууме, давая красновато-коричневое масло (11,87 г). Хроматография на силикагеле, элюируемом смесью гексан-этилацетат (15:1) давала N,O-дибутоксикарбонил-N-[1-(1-бензил-1,2,3,4-тетрагидрохинолин-6-ил)этил] гидроксиламин(2,57 г, 53,8% выход).
ЯМР (CDCl3) δ: 7,17-7,35 (м., 5H), 6,19-7,05 (м., 2H), 6,43 (д., J=8,1 Гц, 1H), 5,24 (кв., J=6,8 Гц, 1H), 4,45 (с., 2H), 3,34 (т., J=5,5 Гц, 2H), 2,79 (т., J=5,9 Гц, 2H), 1,92-2,05 (м., 2H), 1,21-1,63 (м., 21H)
К раствору N,O-дибутоксикарбонил-N-[1-(1бензил-1,2,3,4-тетрагидрохинолин-6-ил)этил] гидроксиламина (2,57 г, 5.70 ммоля) в метиленхлориде (30 мл) добавлялась трифторуксусная кислота (9 мл) при температуре окружающей среды. Смесь перемешивалась при температуре окружающей среды в течение 1 ч, концентрировалась в вакууме, давая вязкое масло, которое экстрагировалось этилацетатом и промывалось водой и солевым раствором. Раствор сушился над сульфатом магния и концентрировался, давая желтое масло. (1,38 г). Без очистки сырой продукт растворялся в тетрагидрофуране (5 мл) и обрабатывался 90% триметилсилилизоцианатом (1,1 мл, 7,33 ммоля) в течение 1 ч при температуре окружающей среды. К смеси добавлялась вода (1 мл), и смесь затем концентрировалась в вакууме. Остаток растворялся в этилацетате, и нерастворимый материал удалялся с помощью фильтрования. Фильтрат концентрировался в вакууме и кристаллизовался из смеси изопропилового эфира и этилацетата: давая белое твердое вещество. Перекристаллизация из смеси этилацетат-изопропиловый эфир (4: 1) давала целевое соединение в виде белого твердого вещества (0,233 г, 12% выход).
Т.пл.,: 127,8 - 128,2oC (разл.)
ИК (KBr): 3500, 3460, 1645
ЯМР (DMCO) δ : 8,84 (с., 1H), 7,18-7,37 (м., 5H), 6,87 (с., 1H), 6,84 (д., J=8,8 Гц, 1H), 6,36 (д., J=8,8 Гц, 1H), 6,15 (с., 2H), 5,11 (кв., J=7,0 Гц, 1H), 4,45 (с., 2H), 3,20-3,56х (2H), 2,70 (т., J=6,2 Гц, 2H), 1,80-1,97 (м., 2H), 1,30 (д., J=7,0 Гц, 3H)
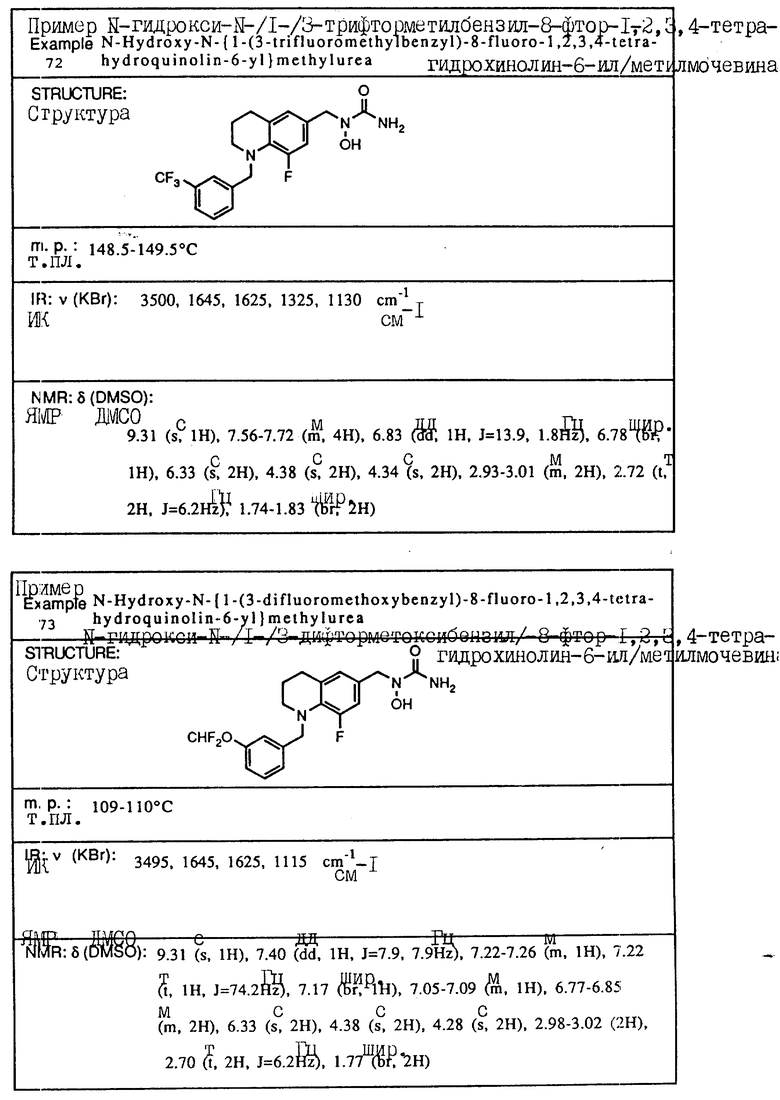
x Данный пик был скрыт водой в DMCO-d6.
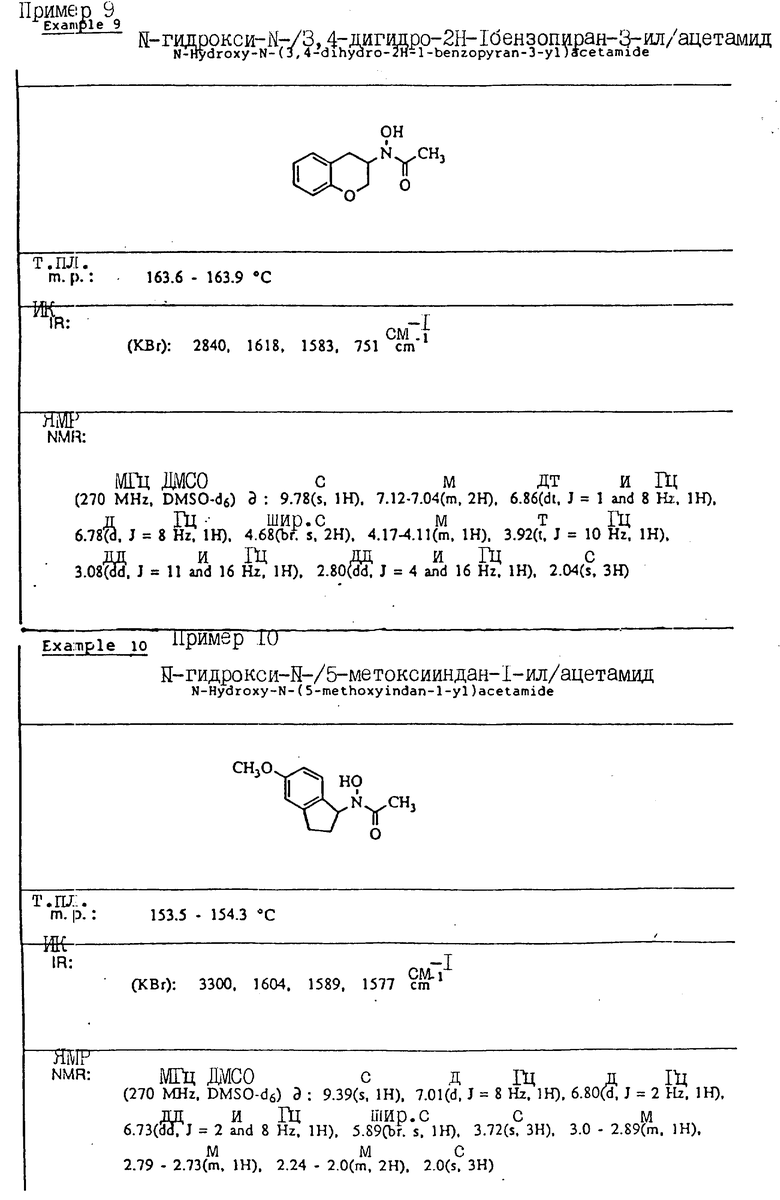
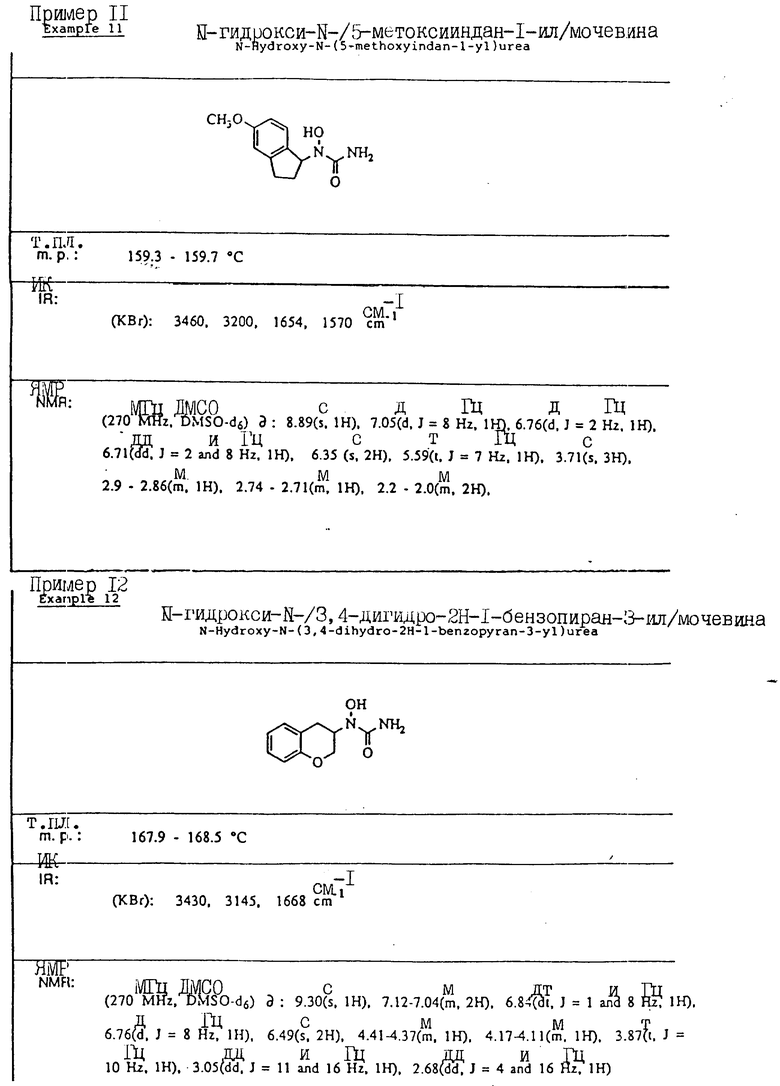
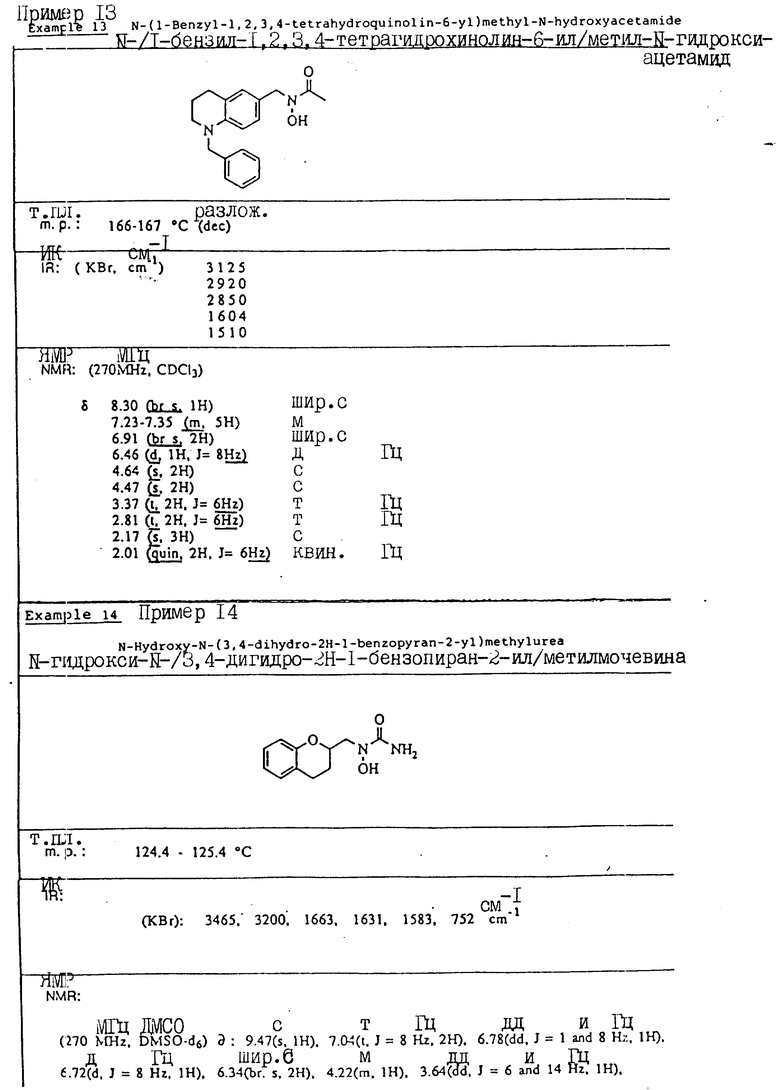
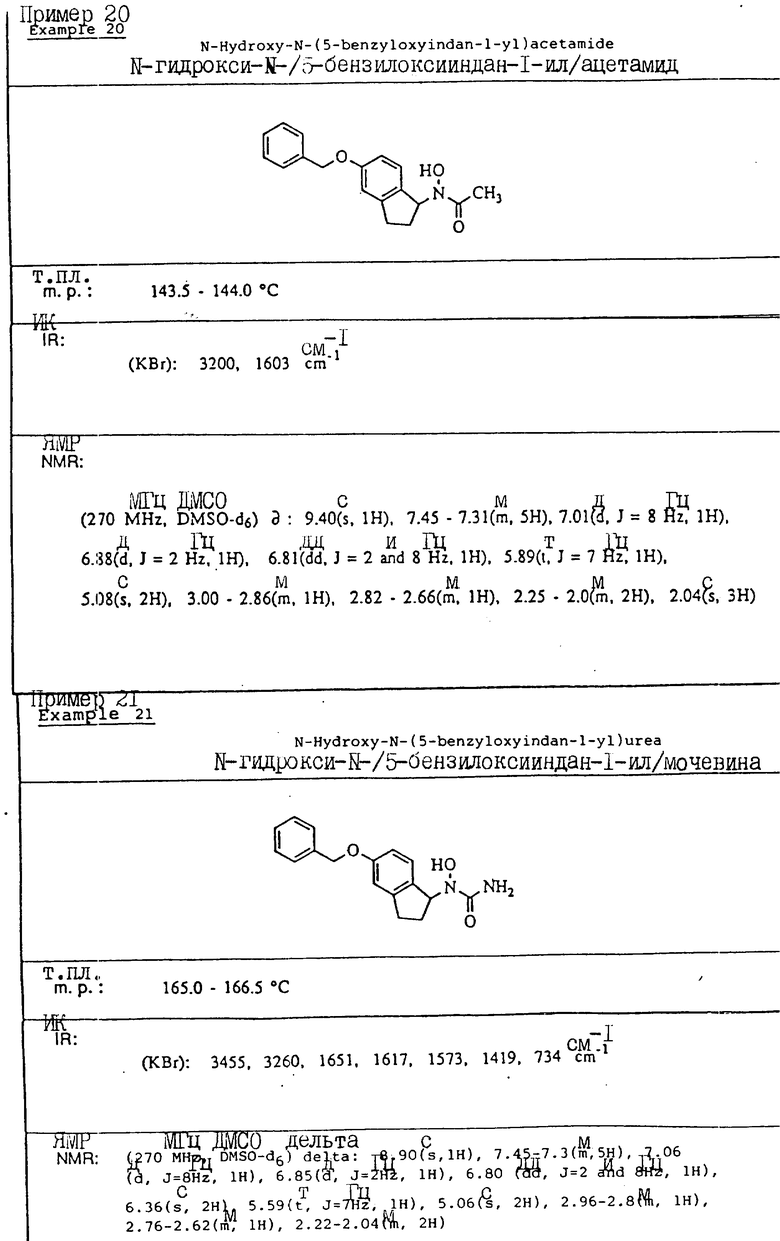
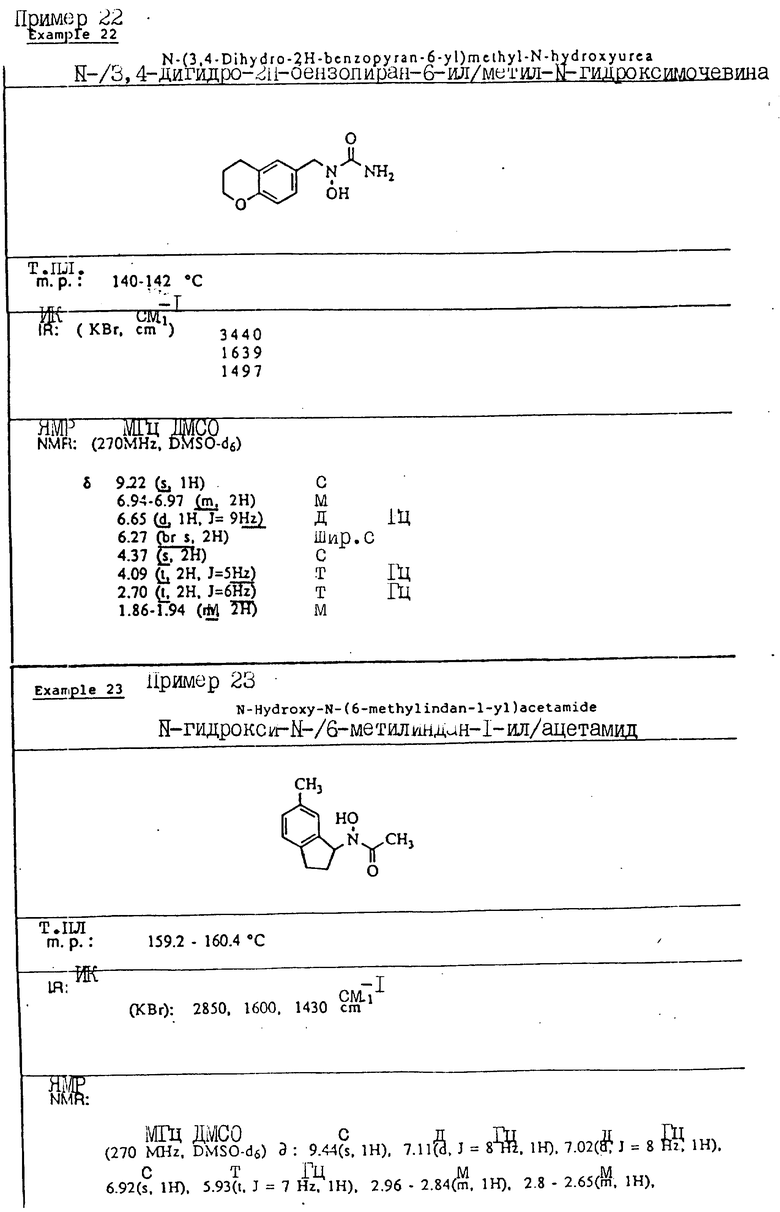
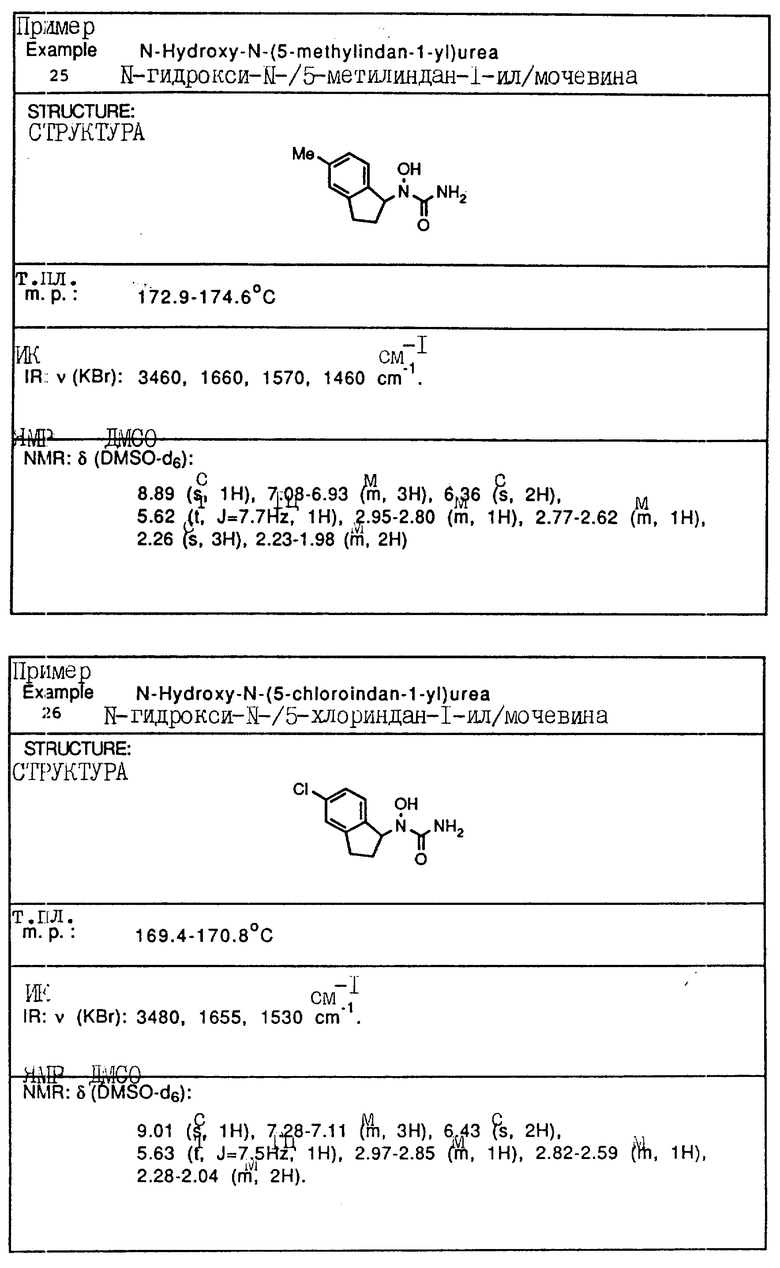
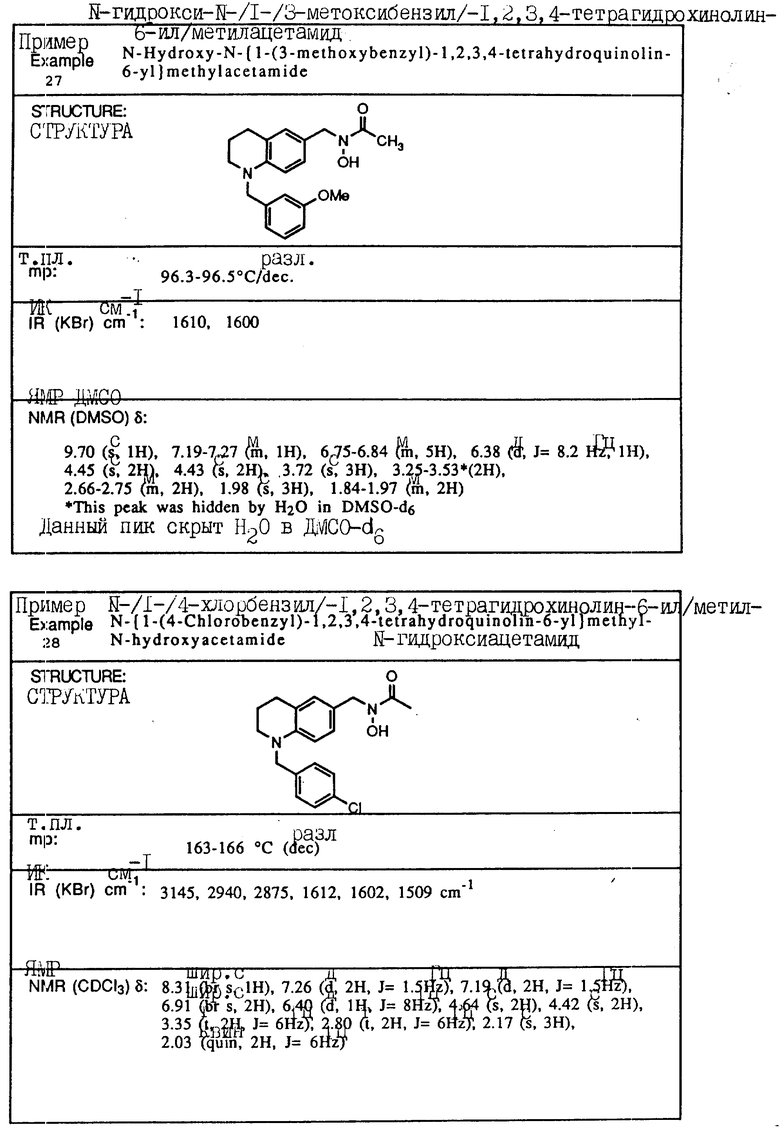
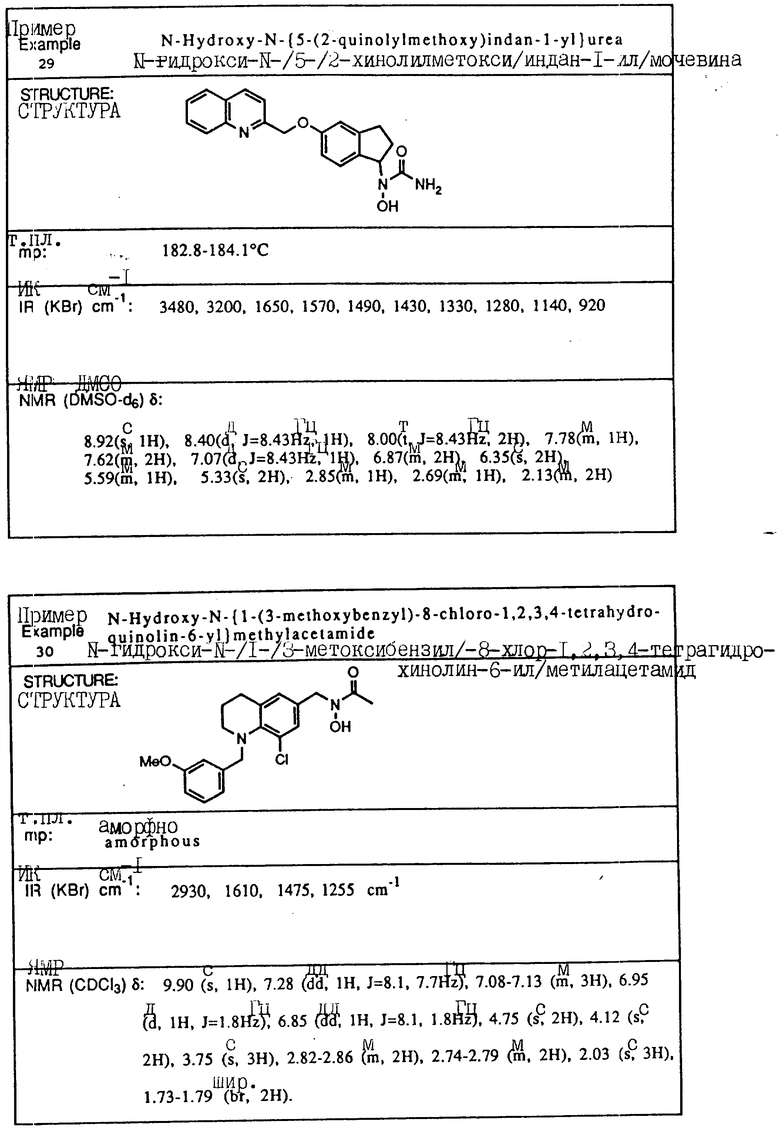
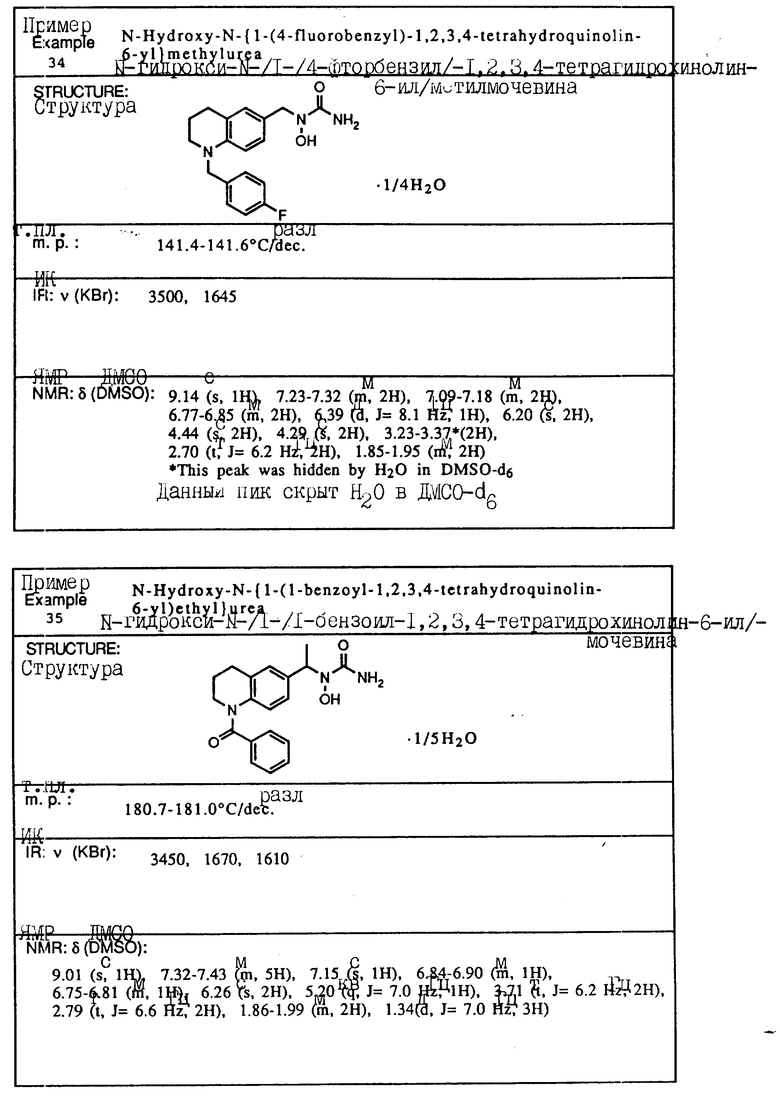
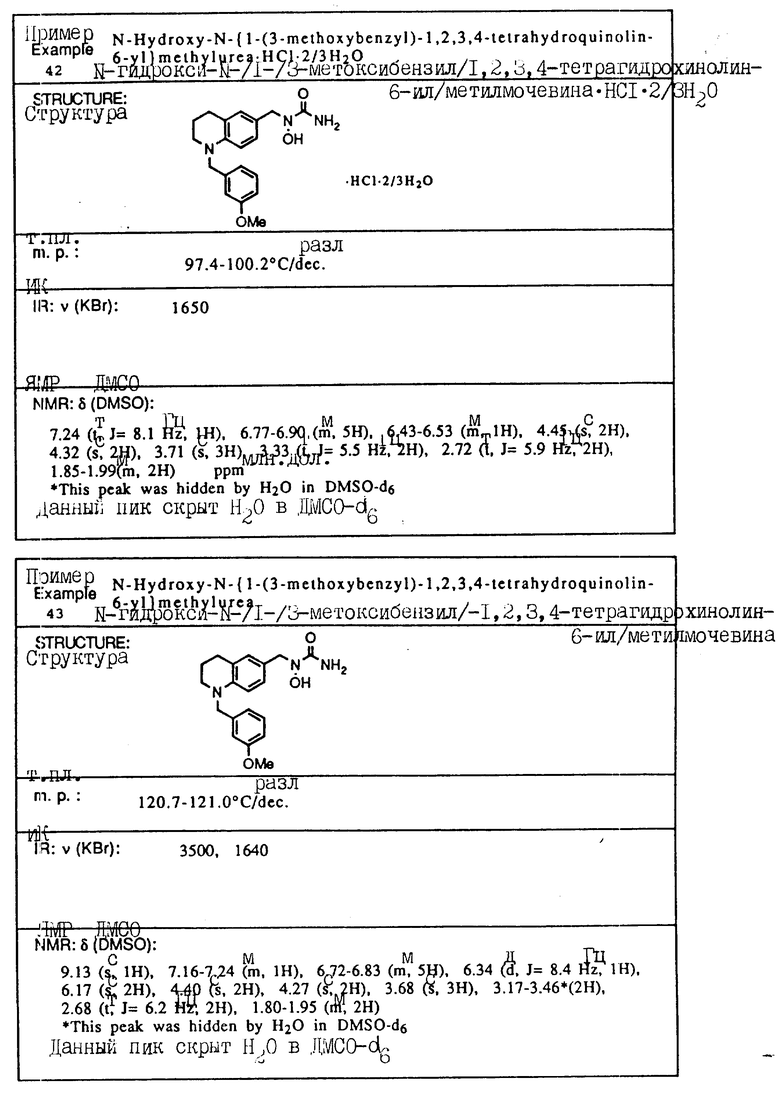
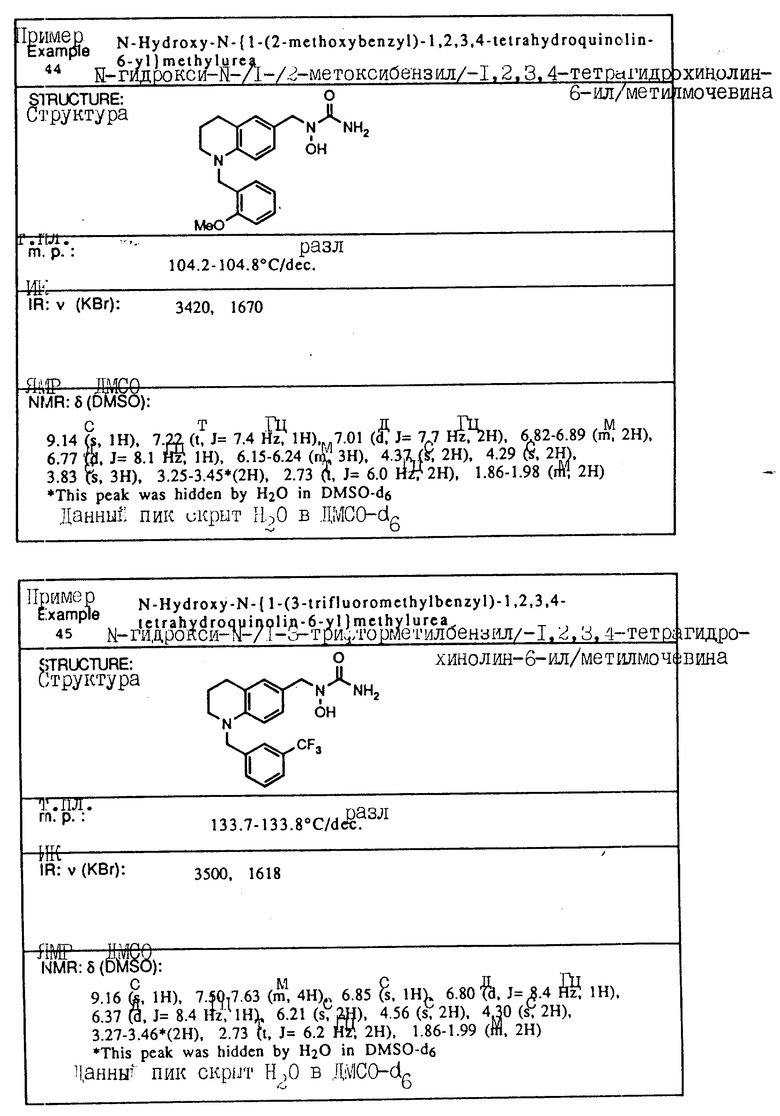
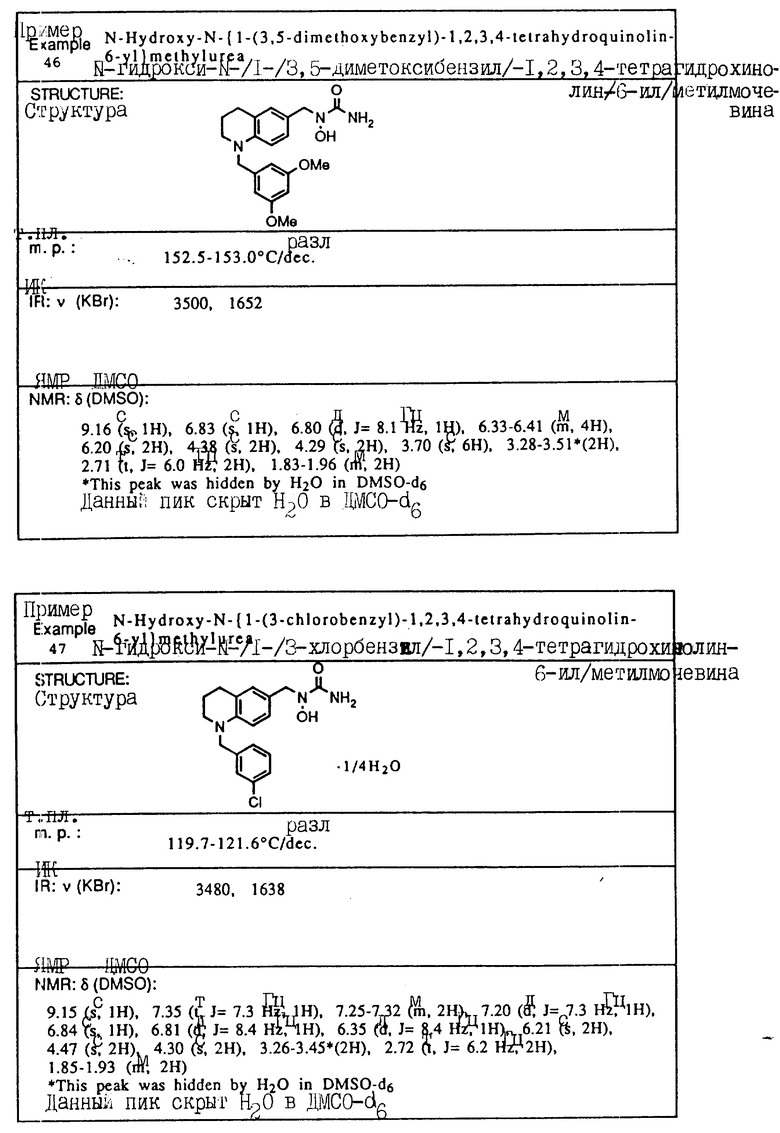
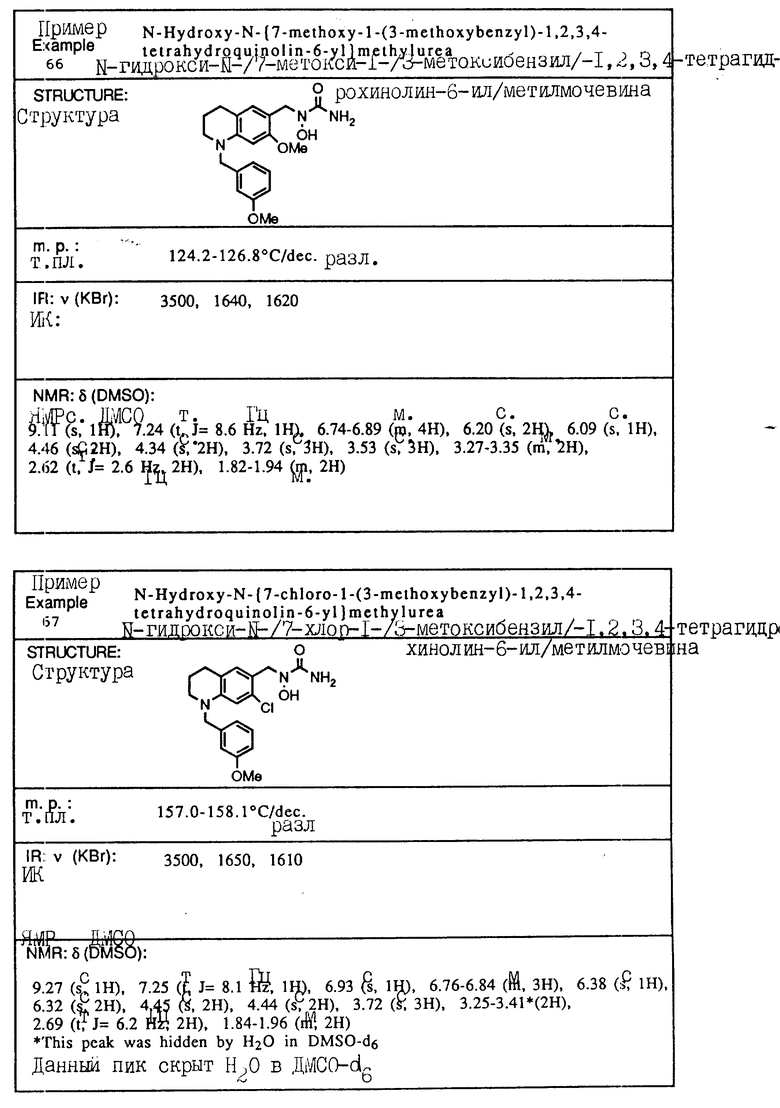
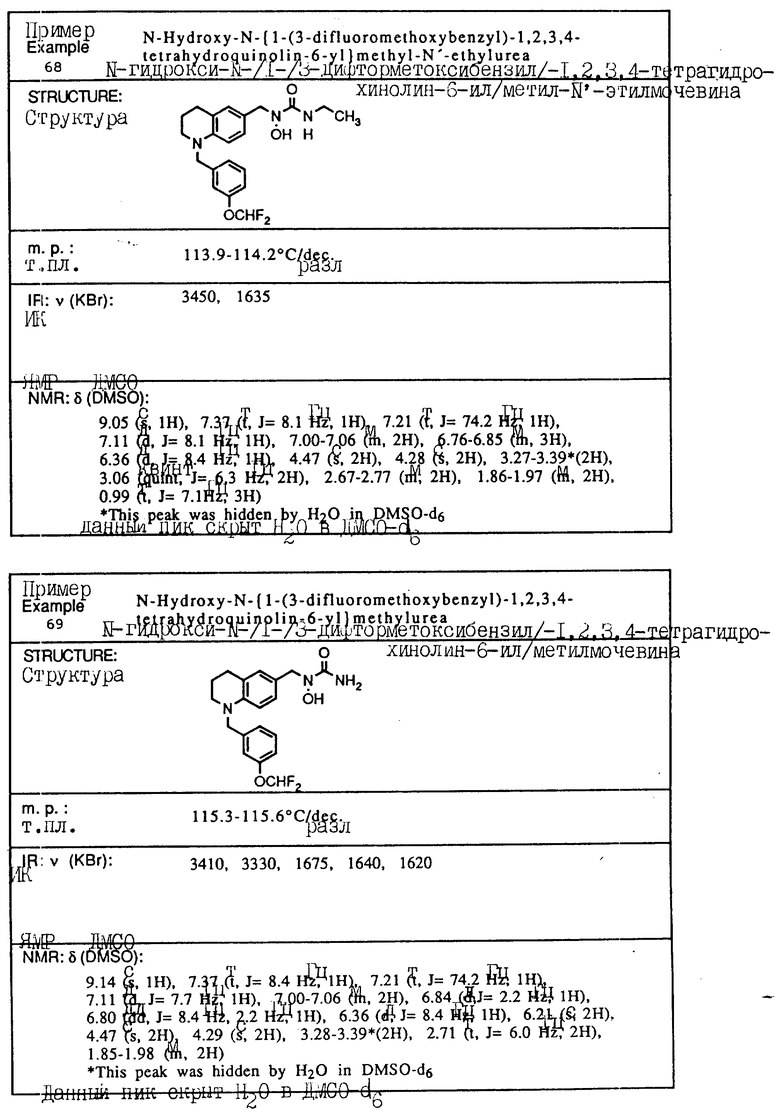
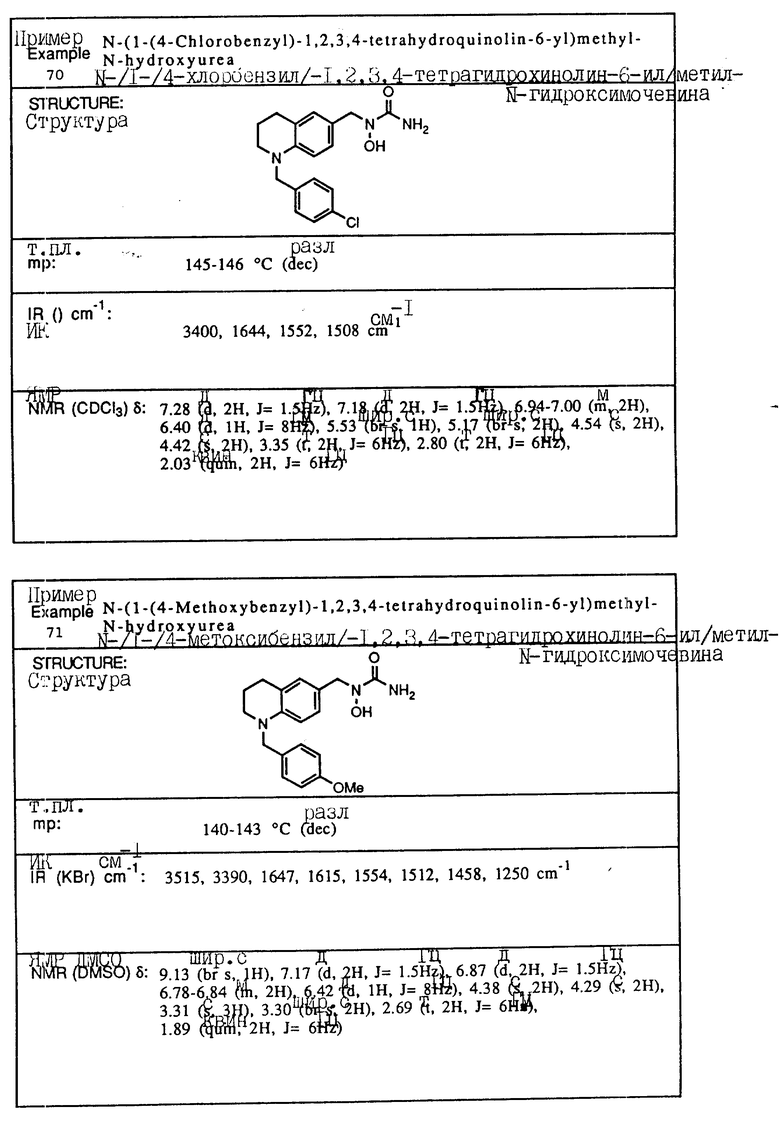
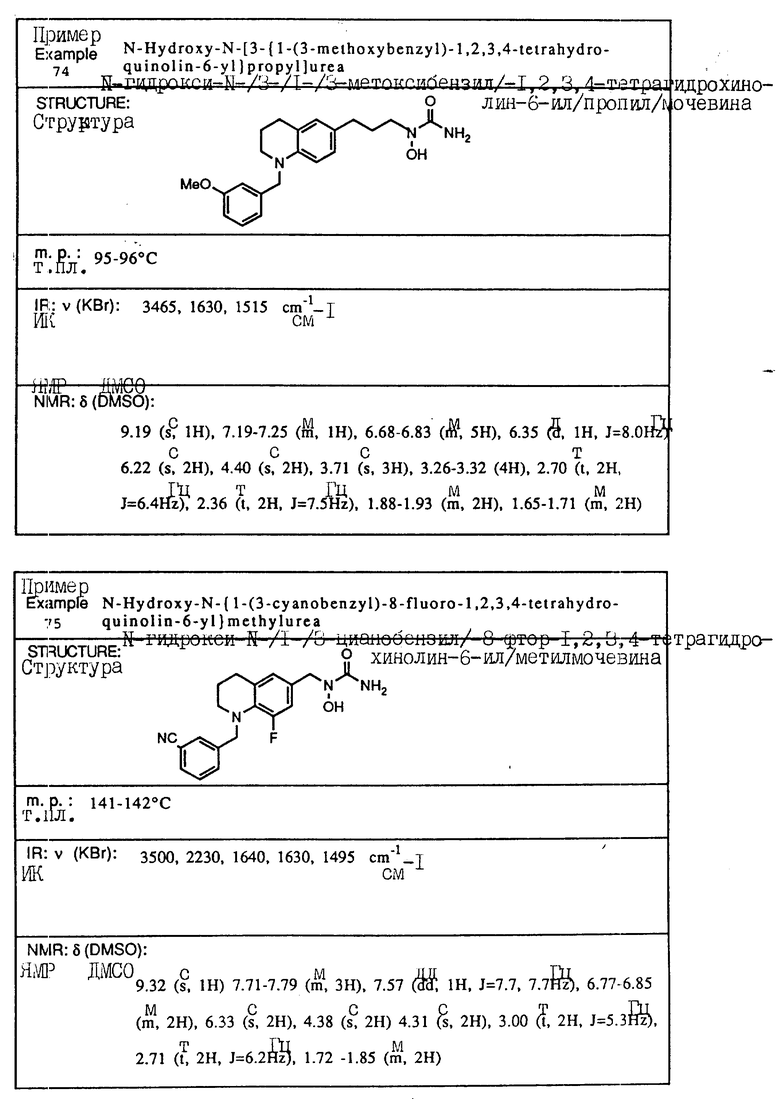
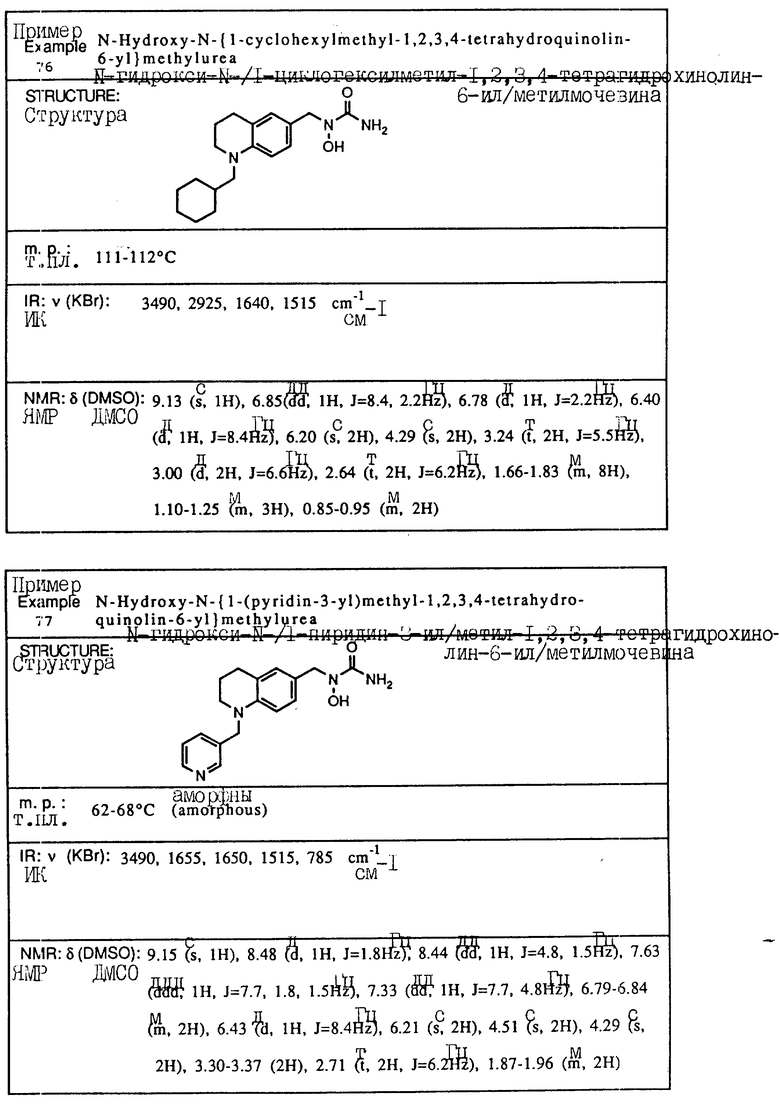
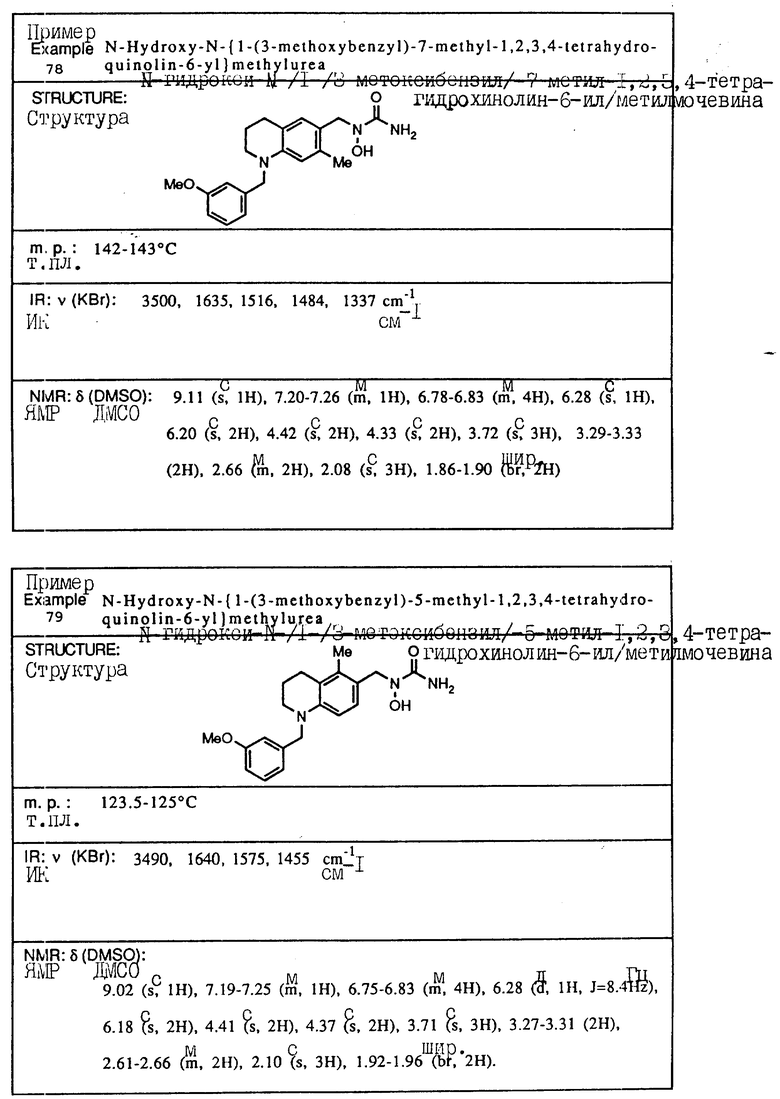
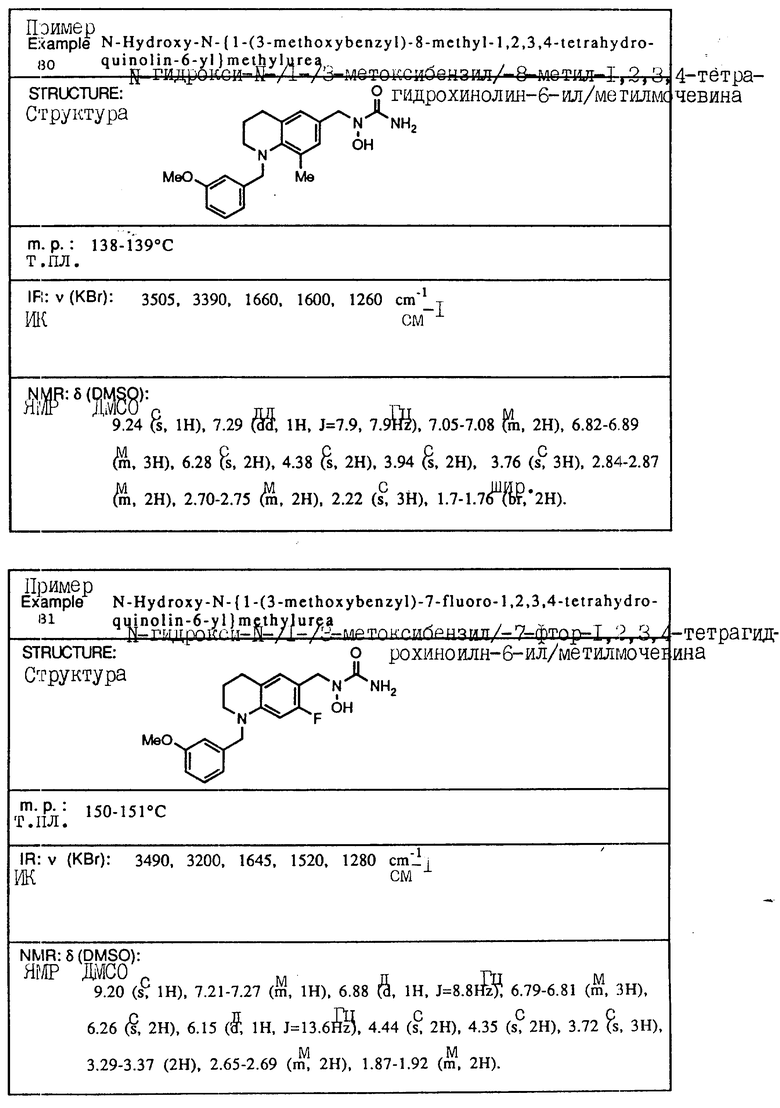
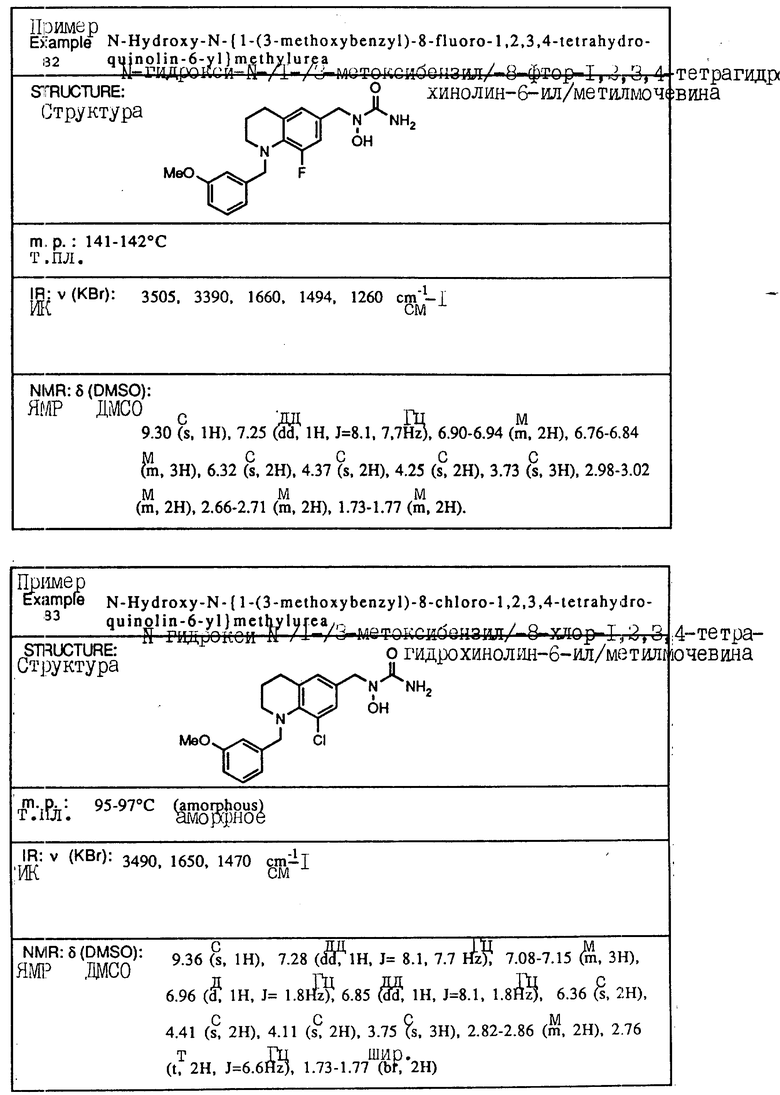
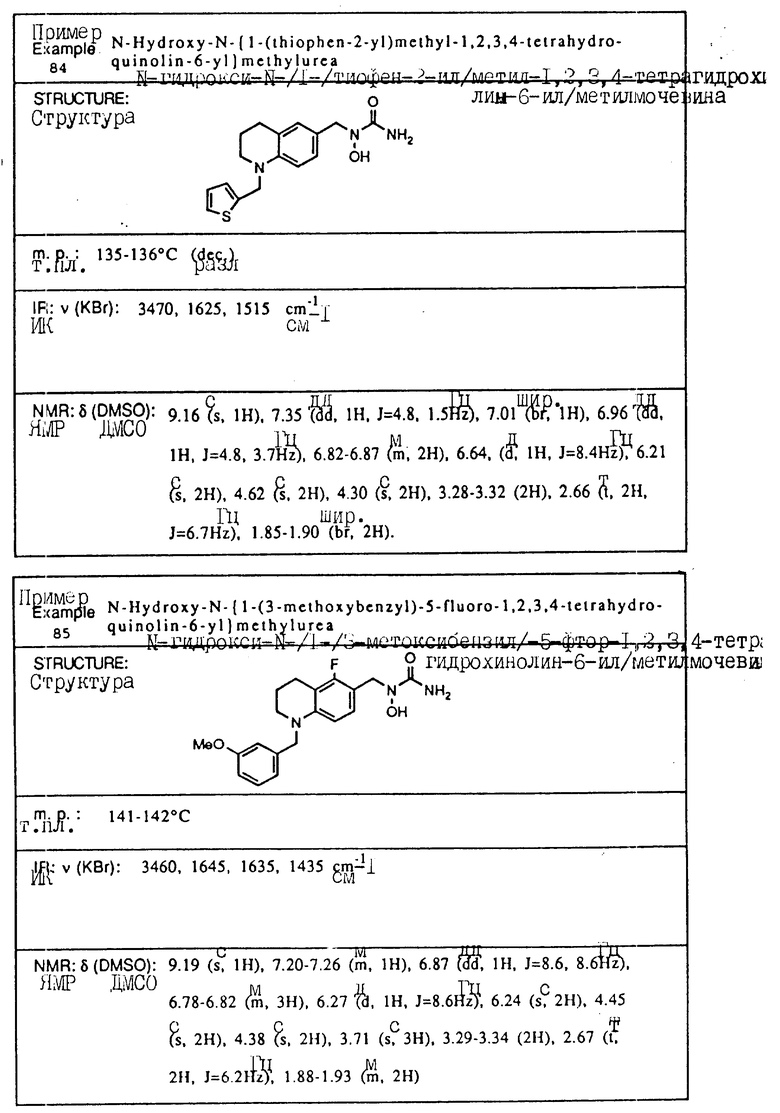
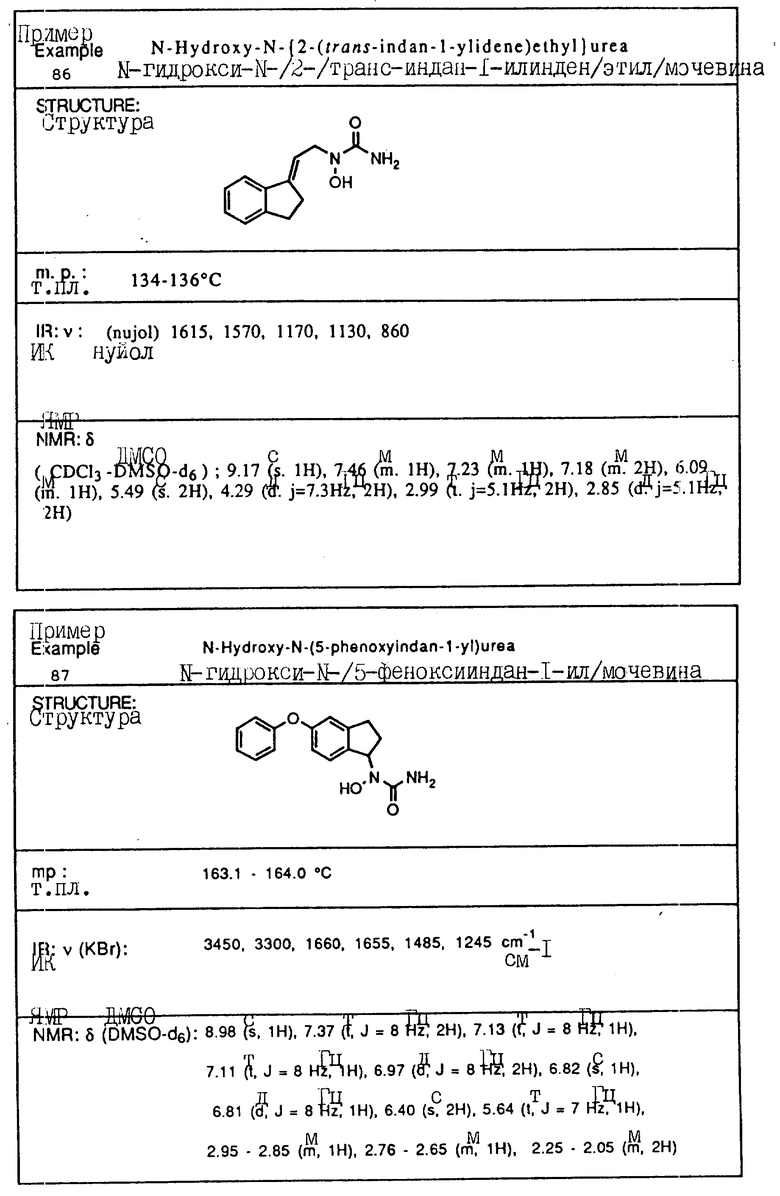
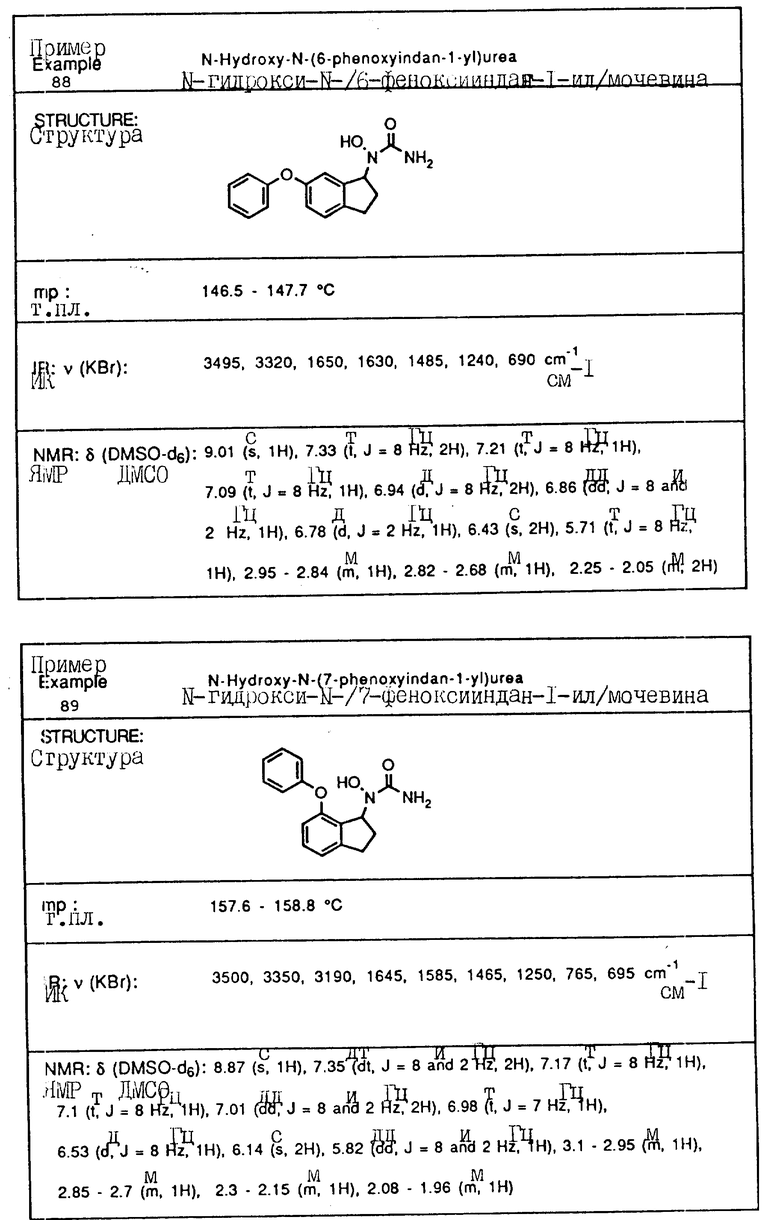
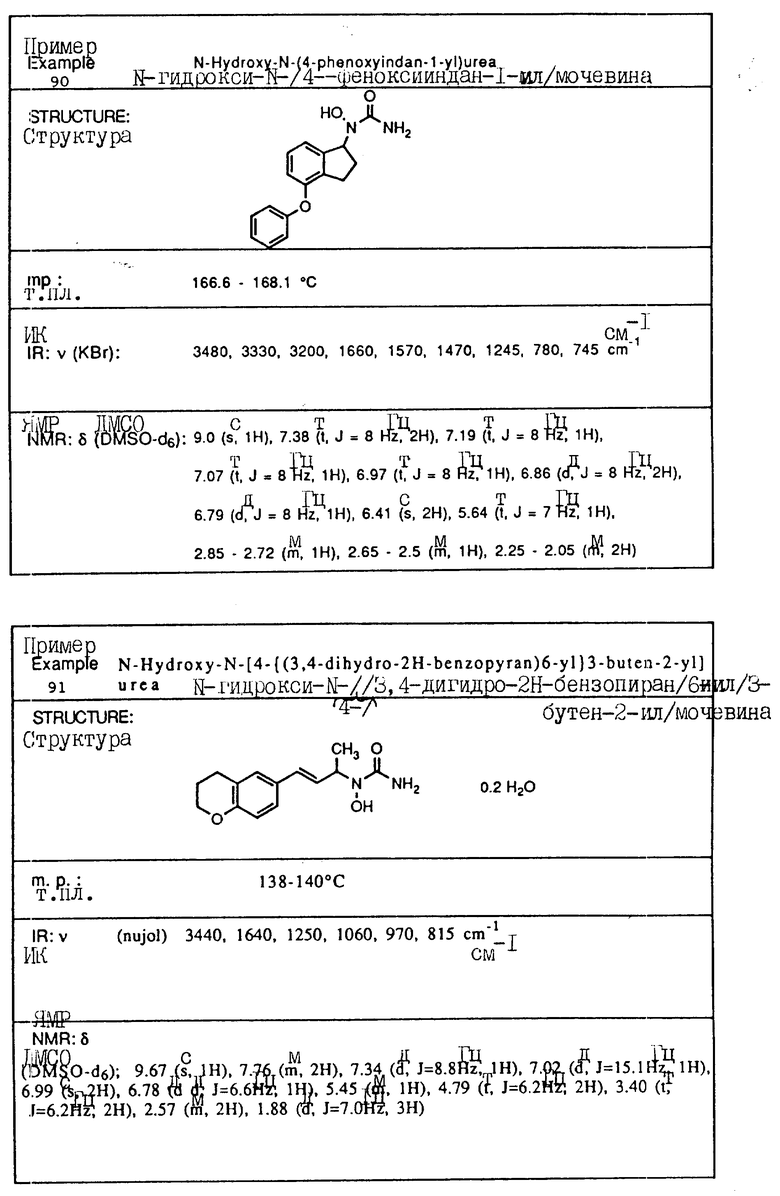
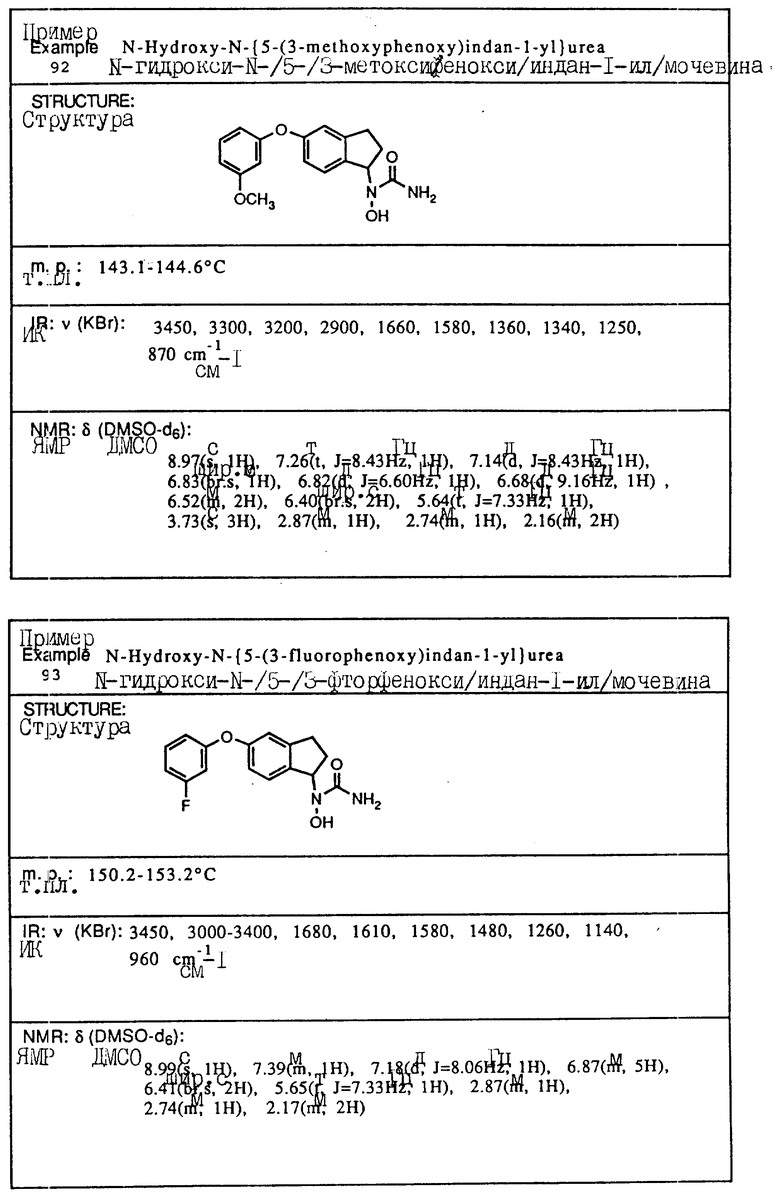
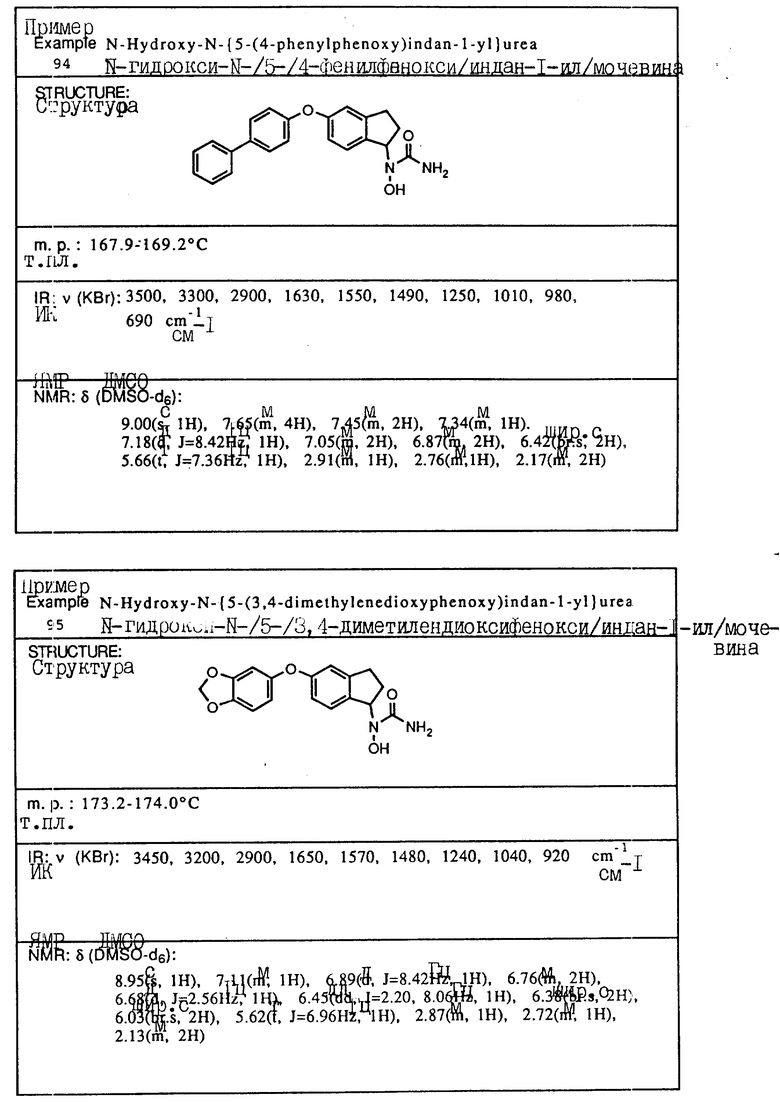
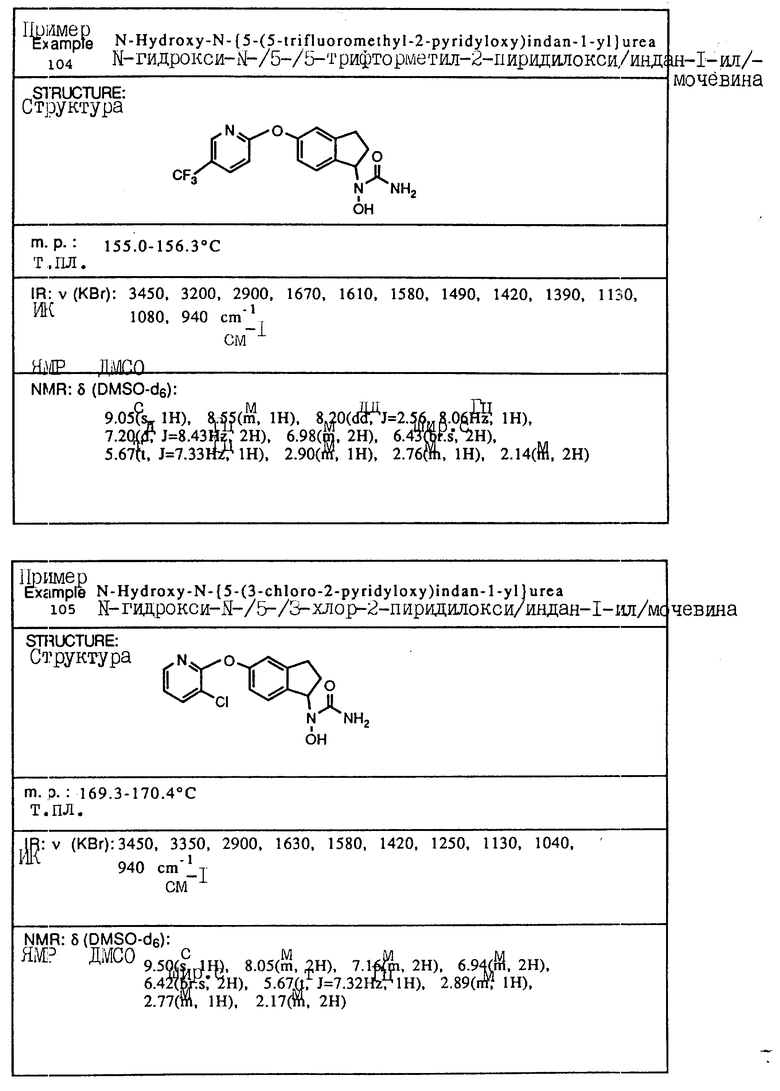
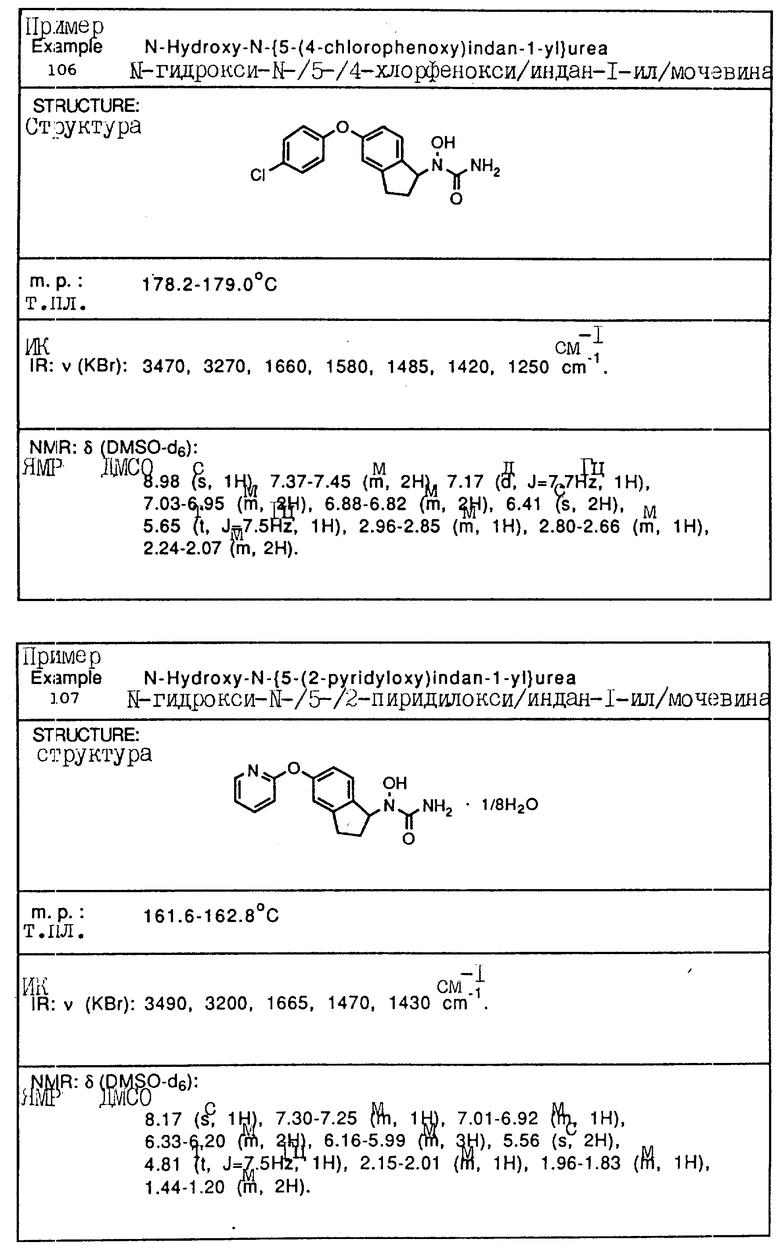
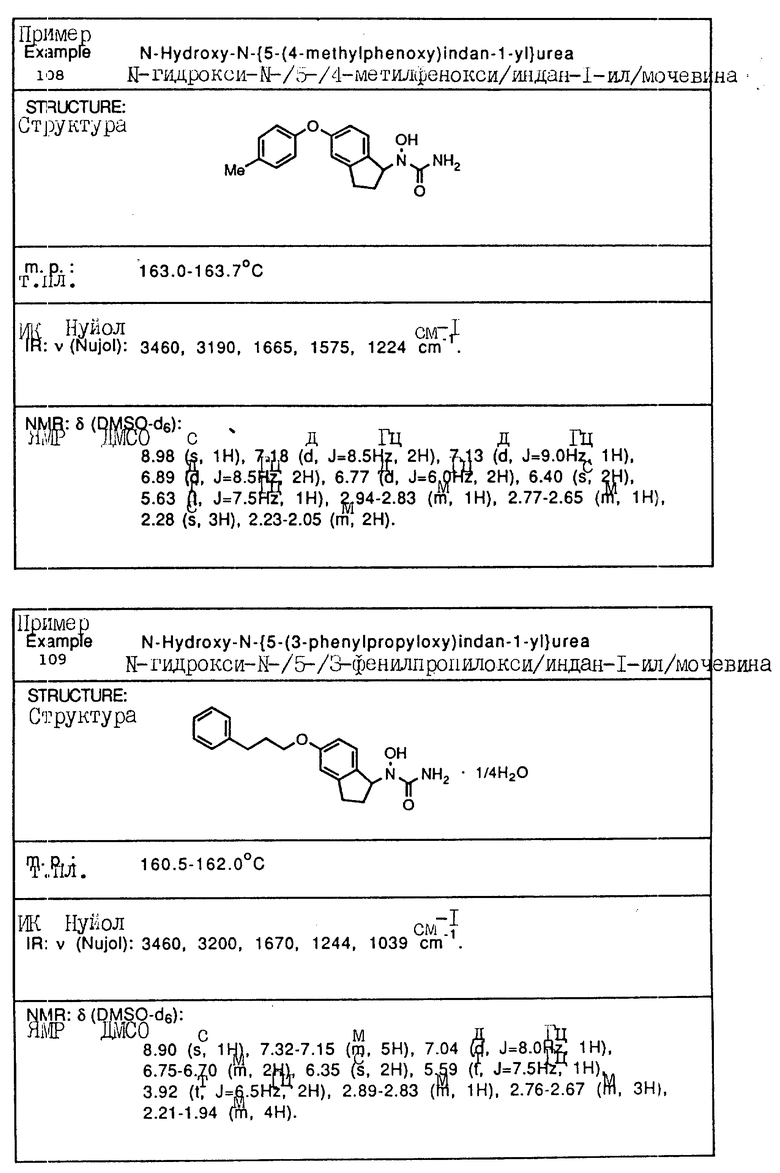
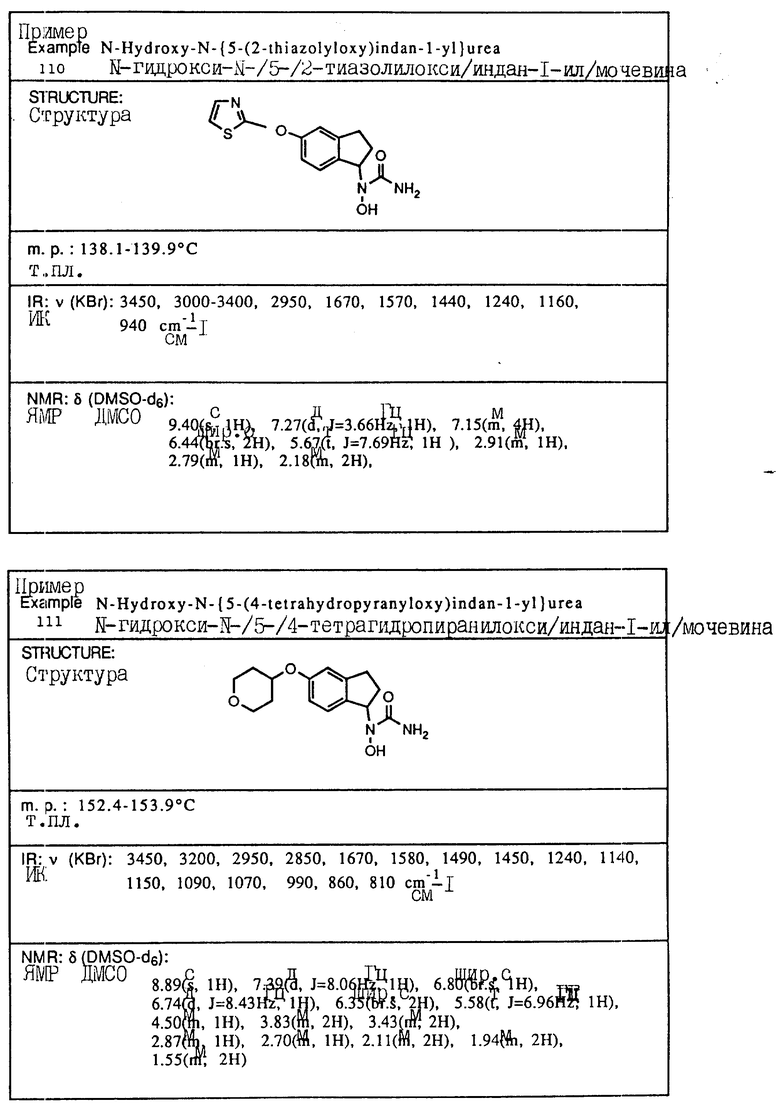
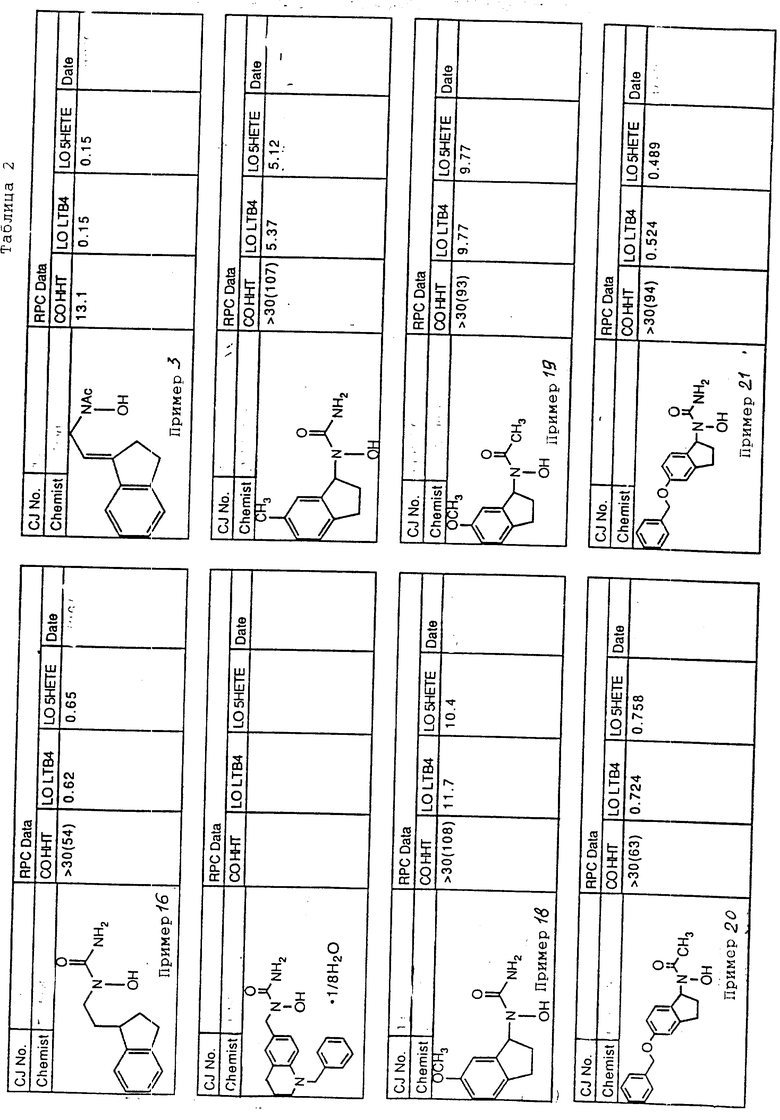
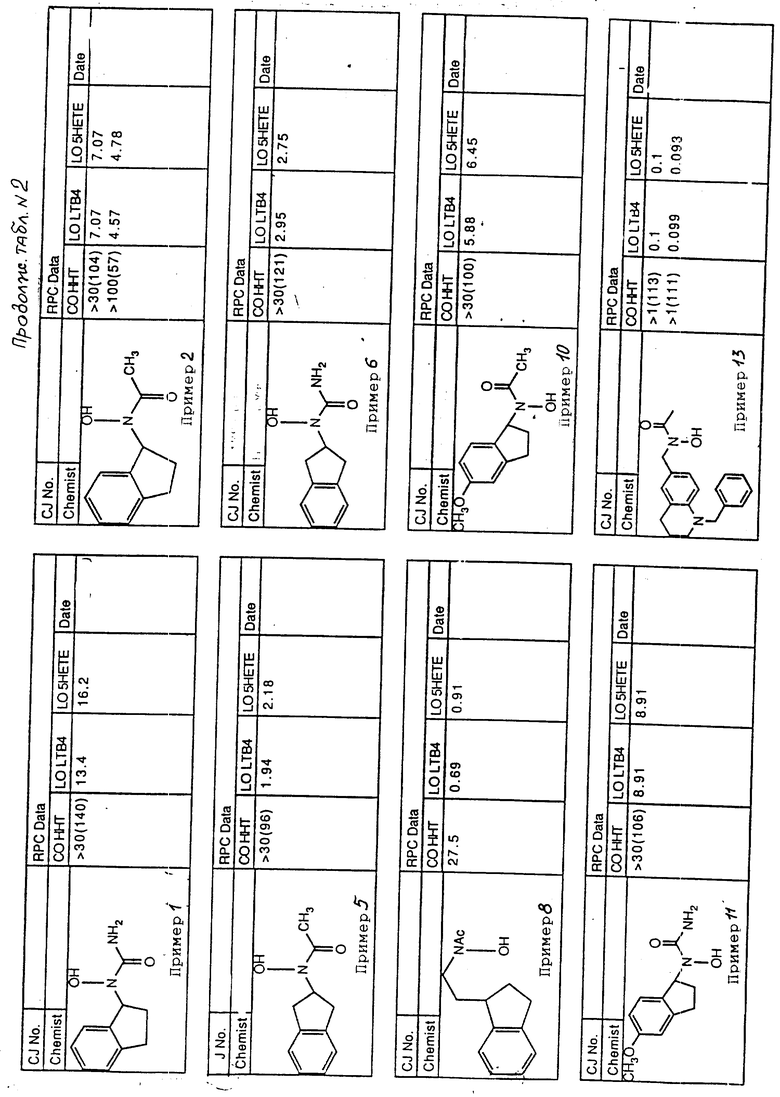
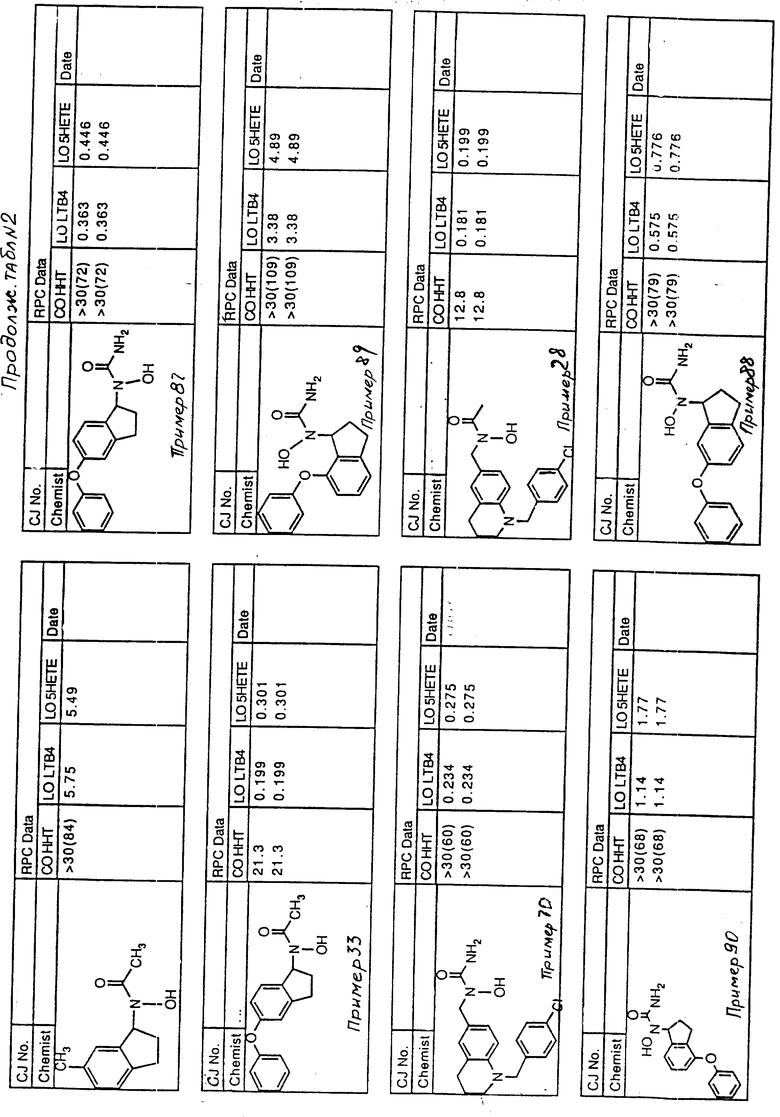
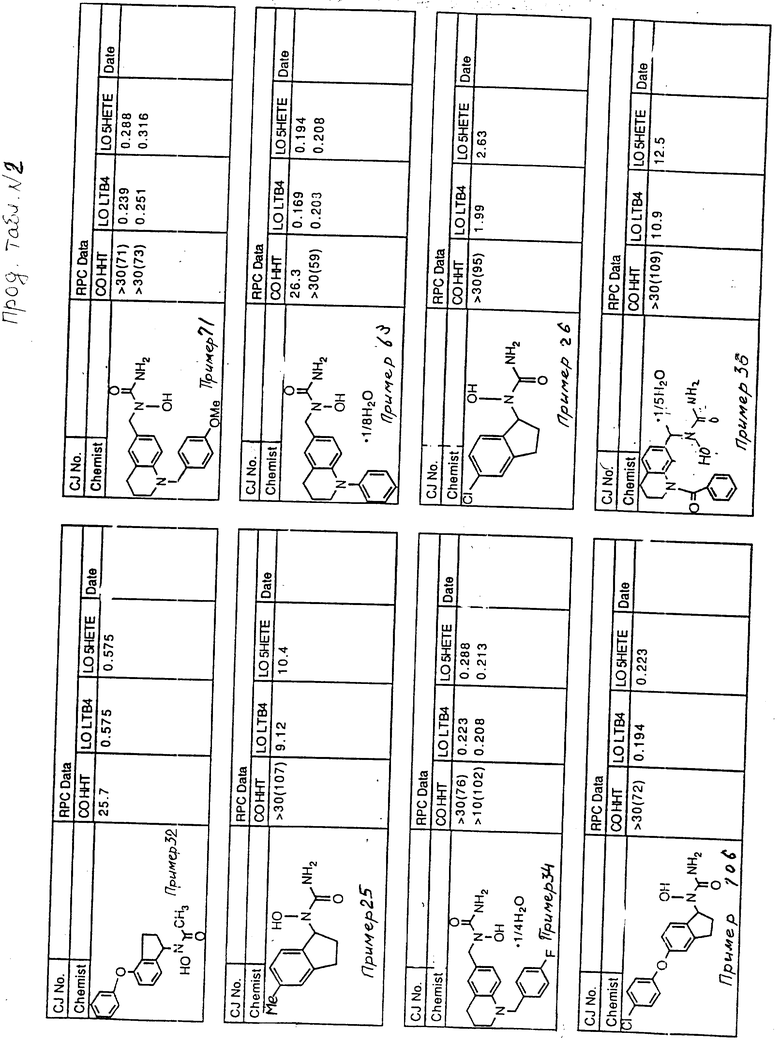
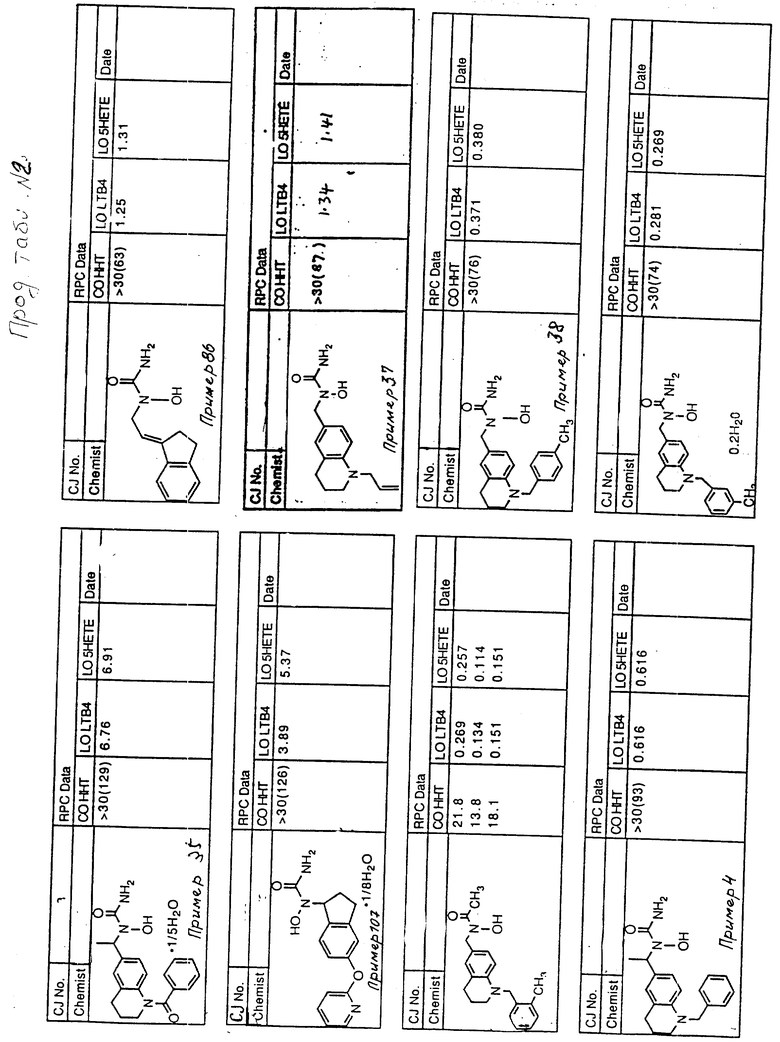
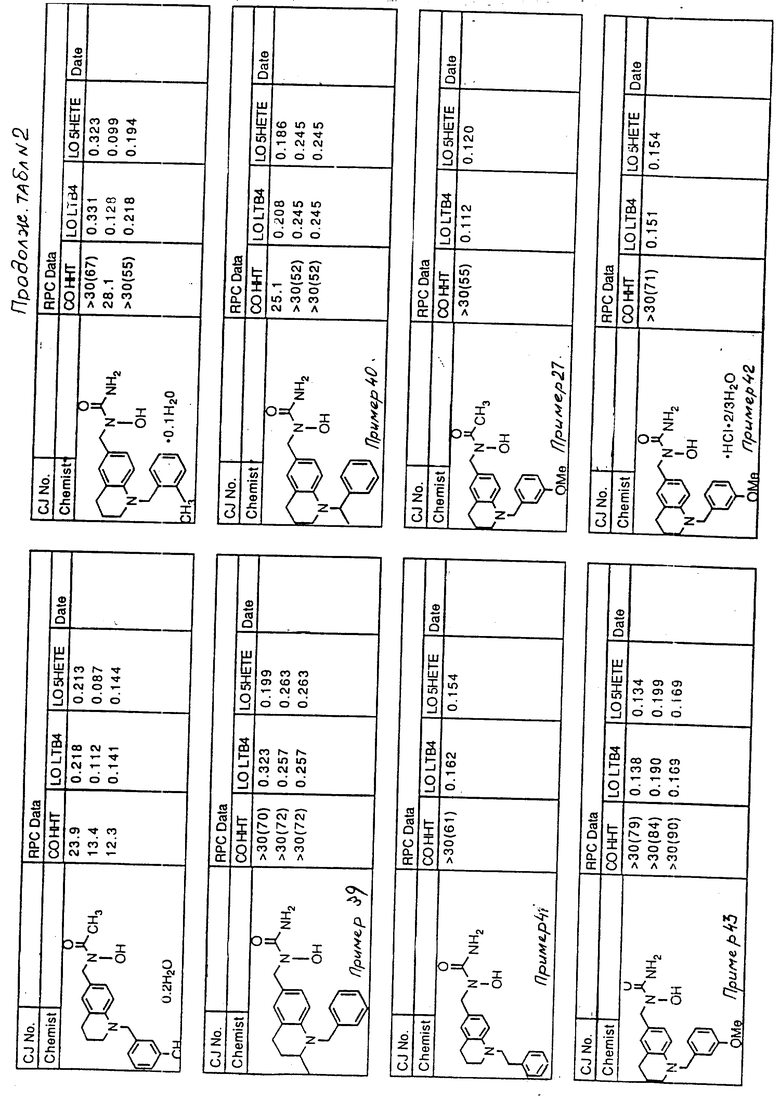
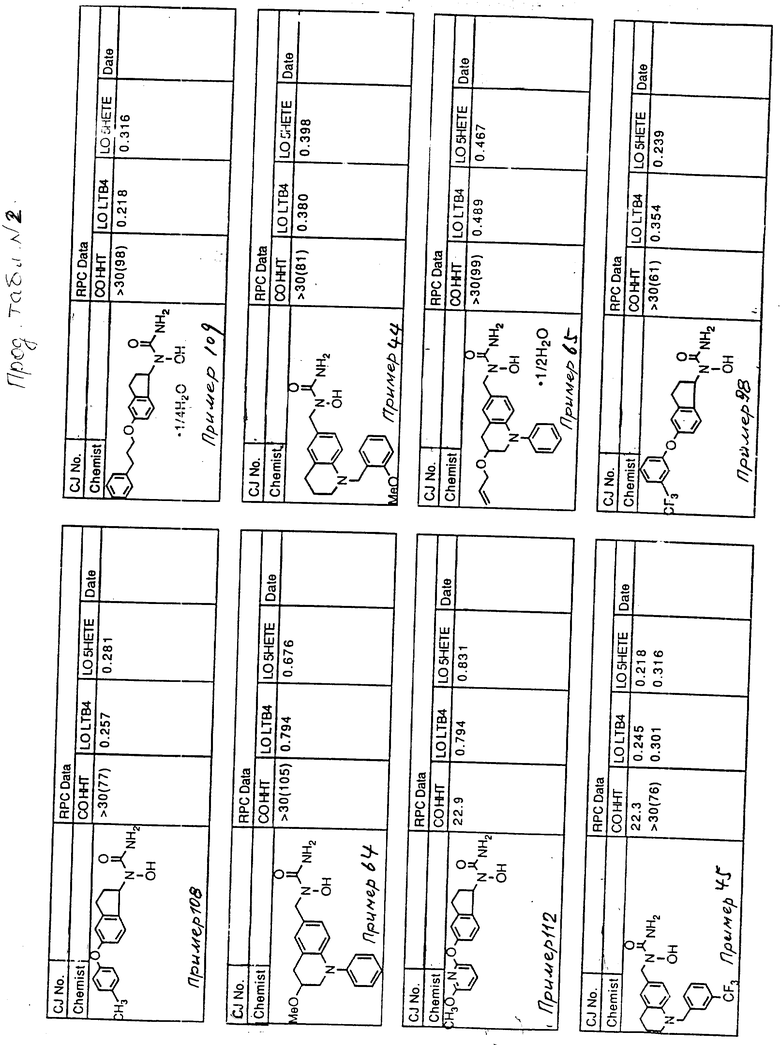
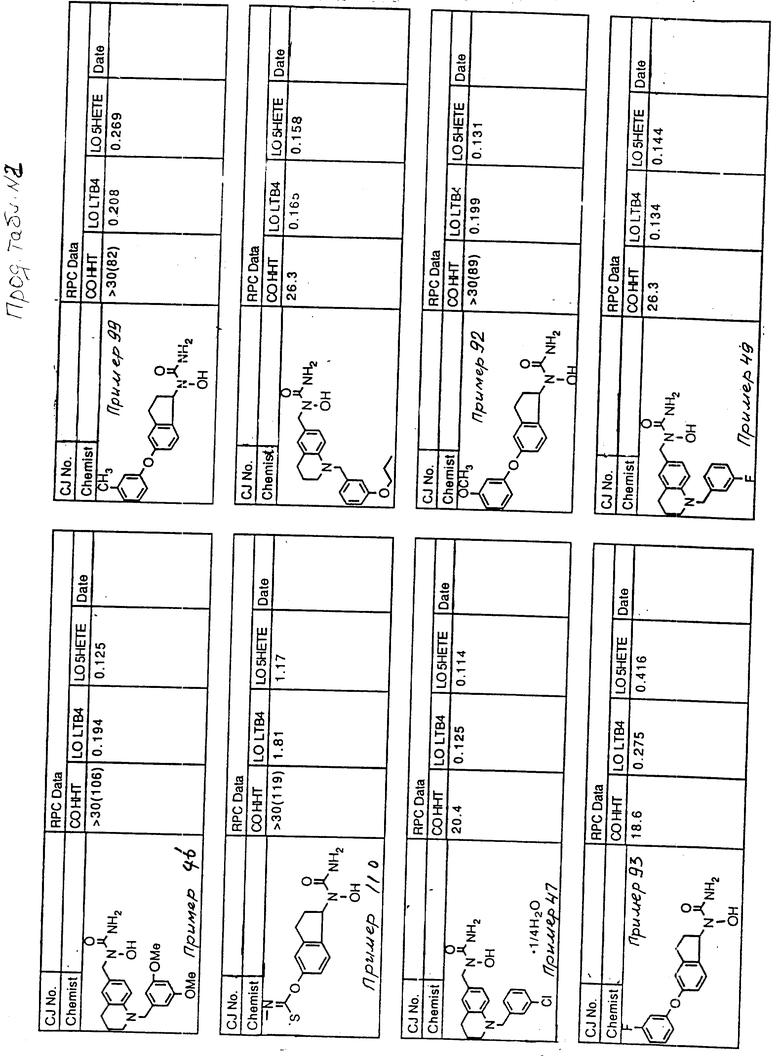
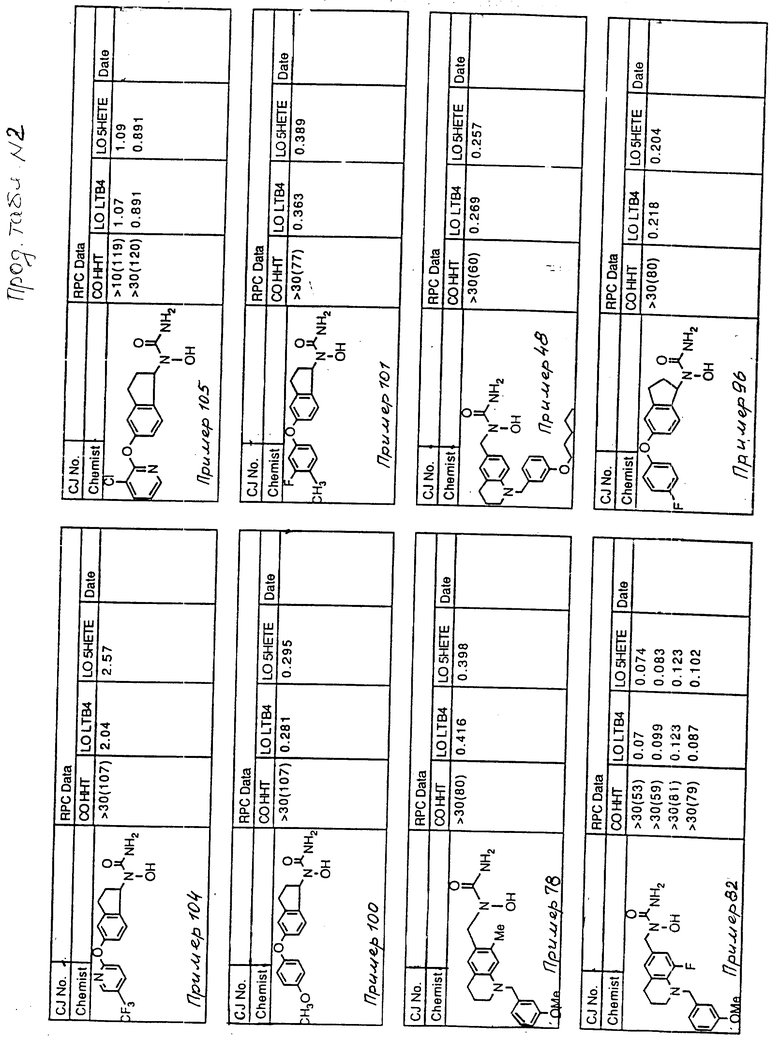
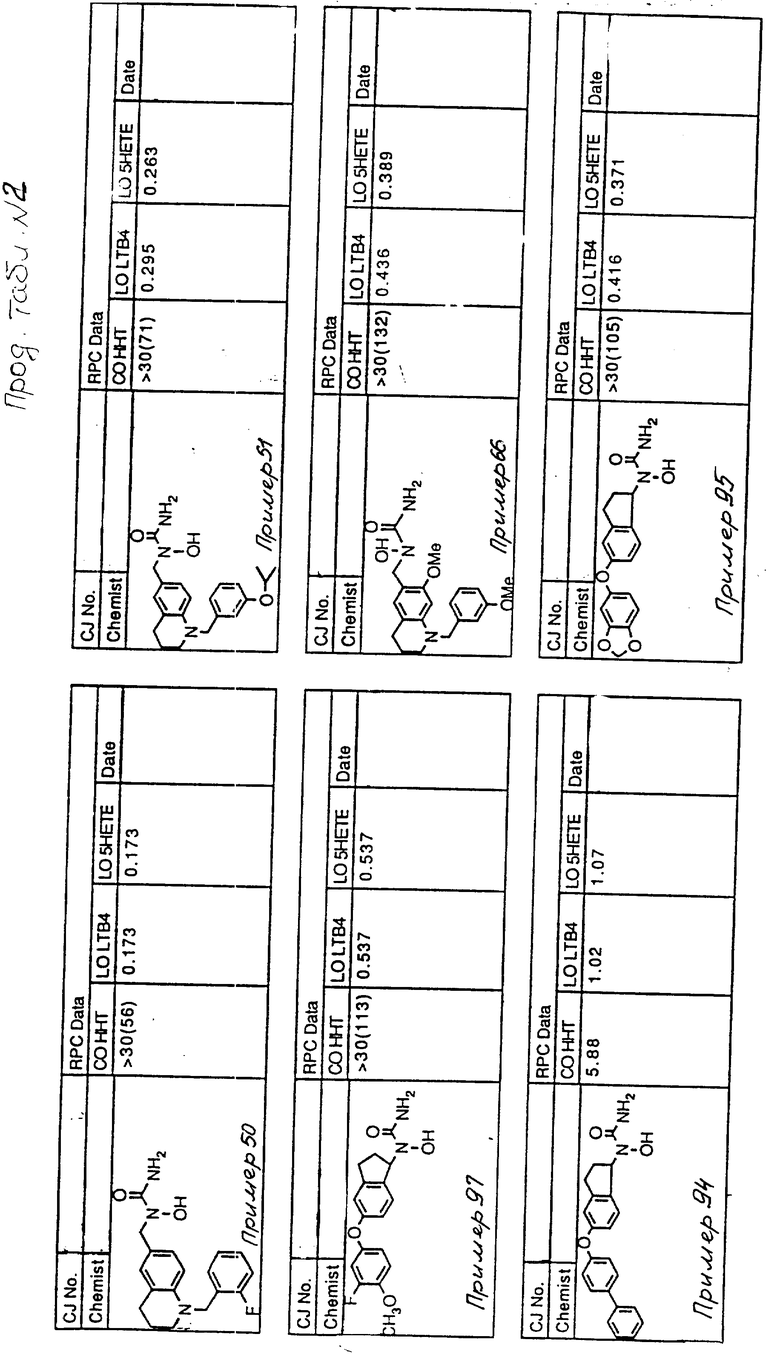
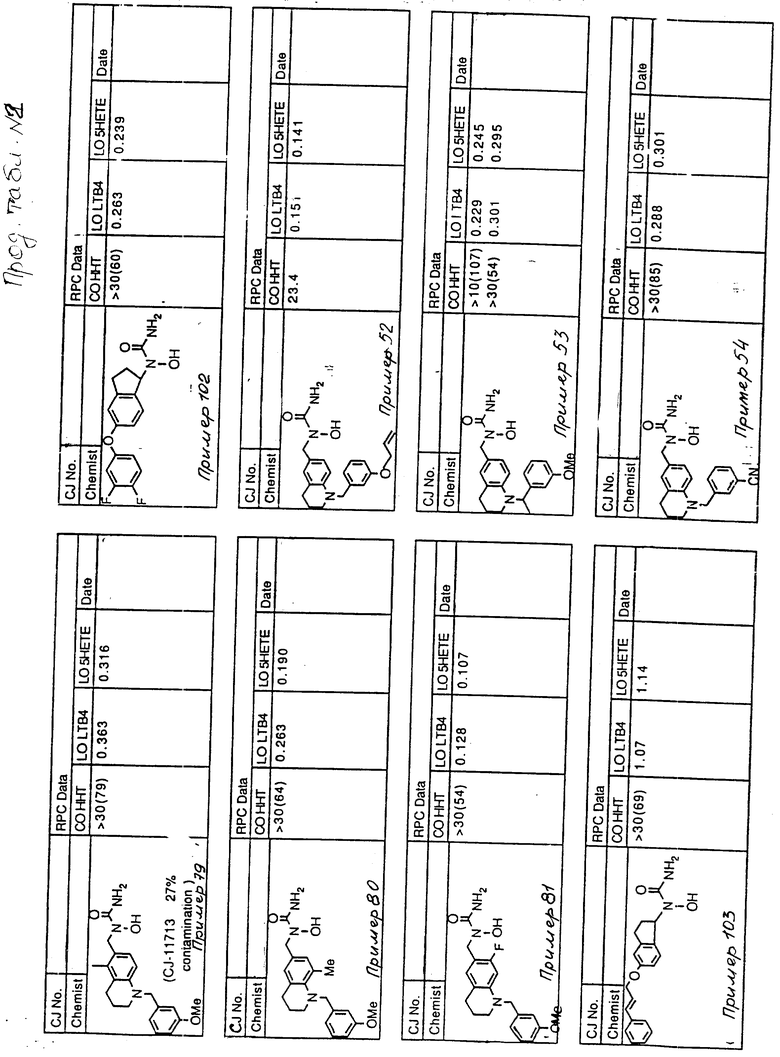
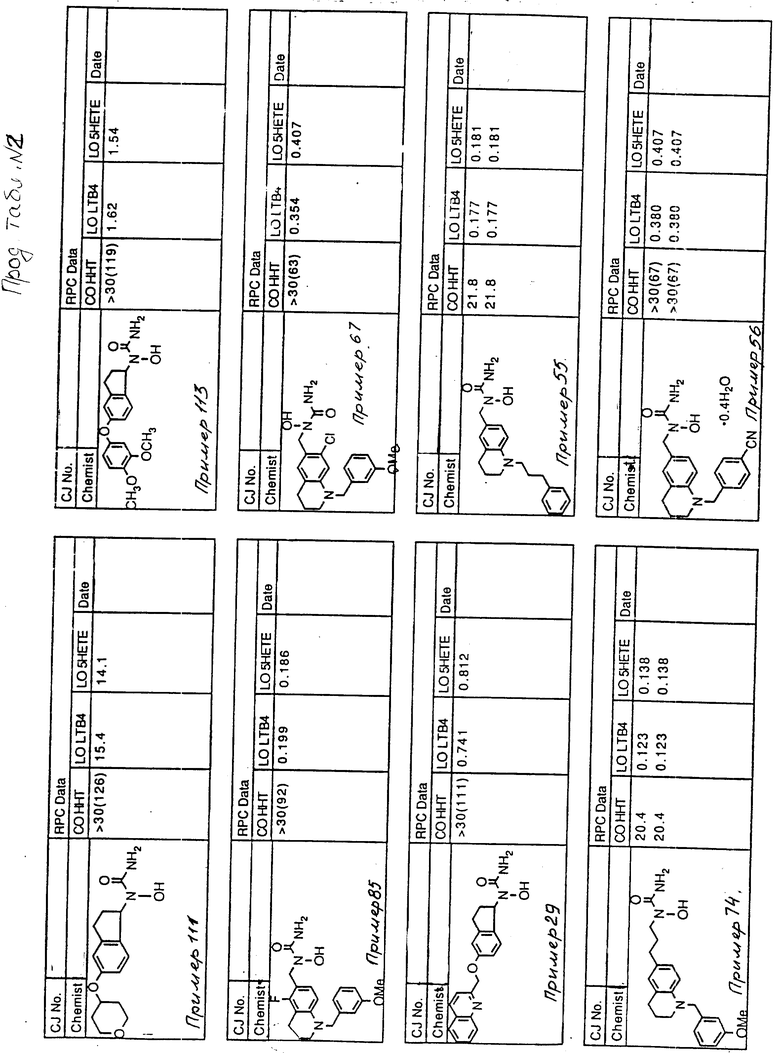
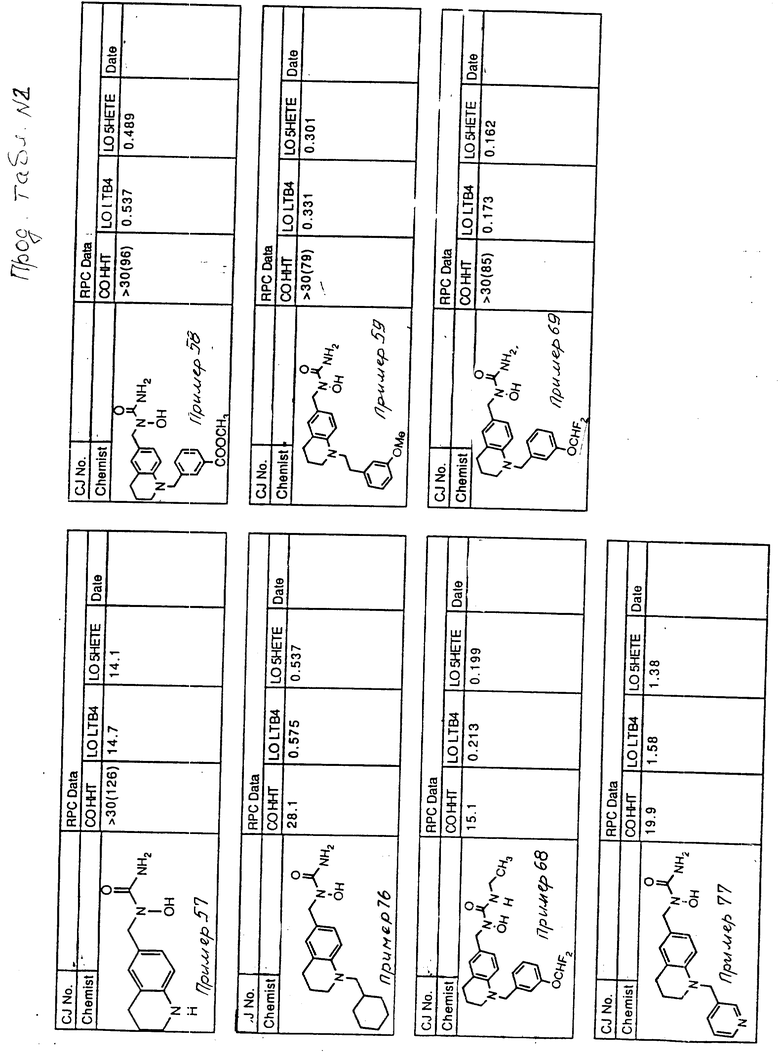
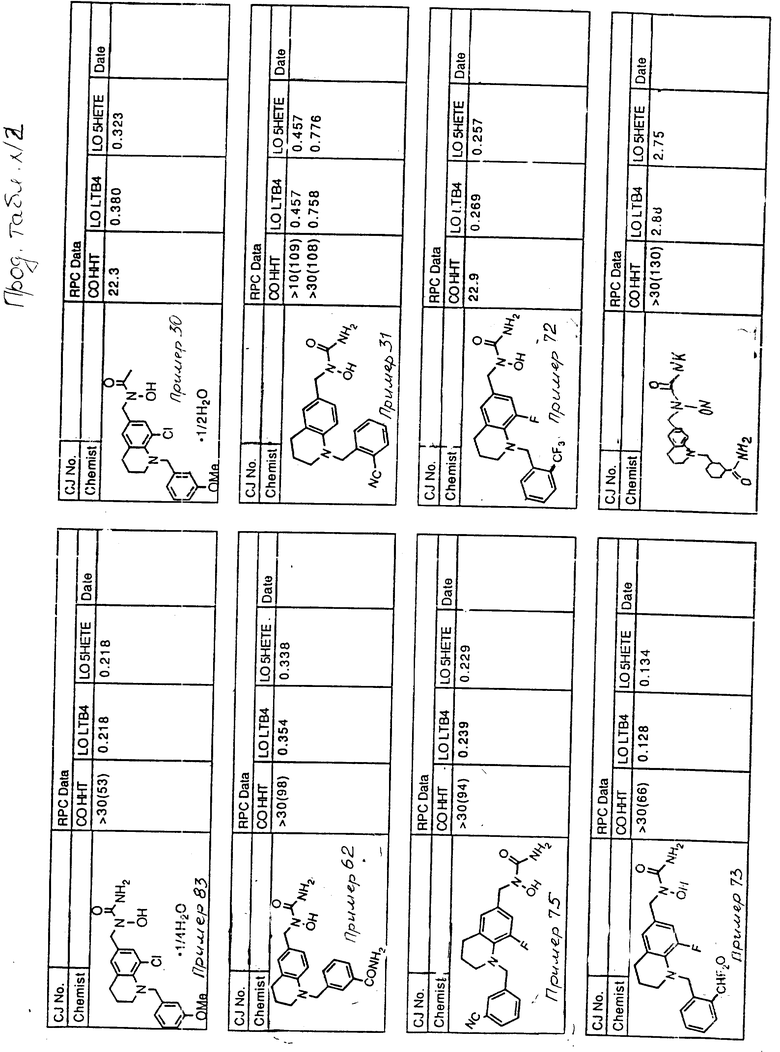
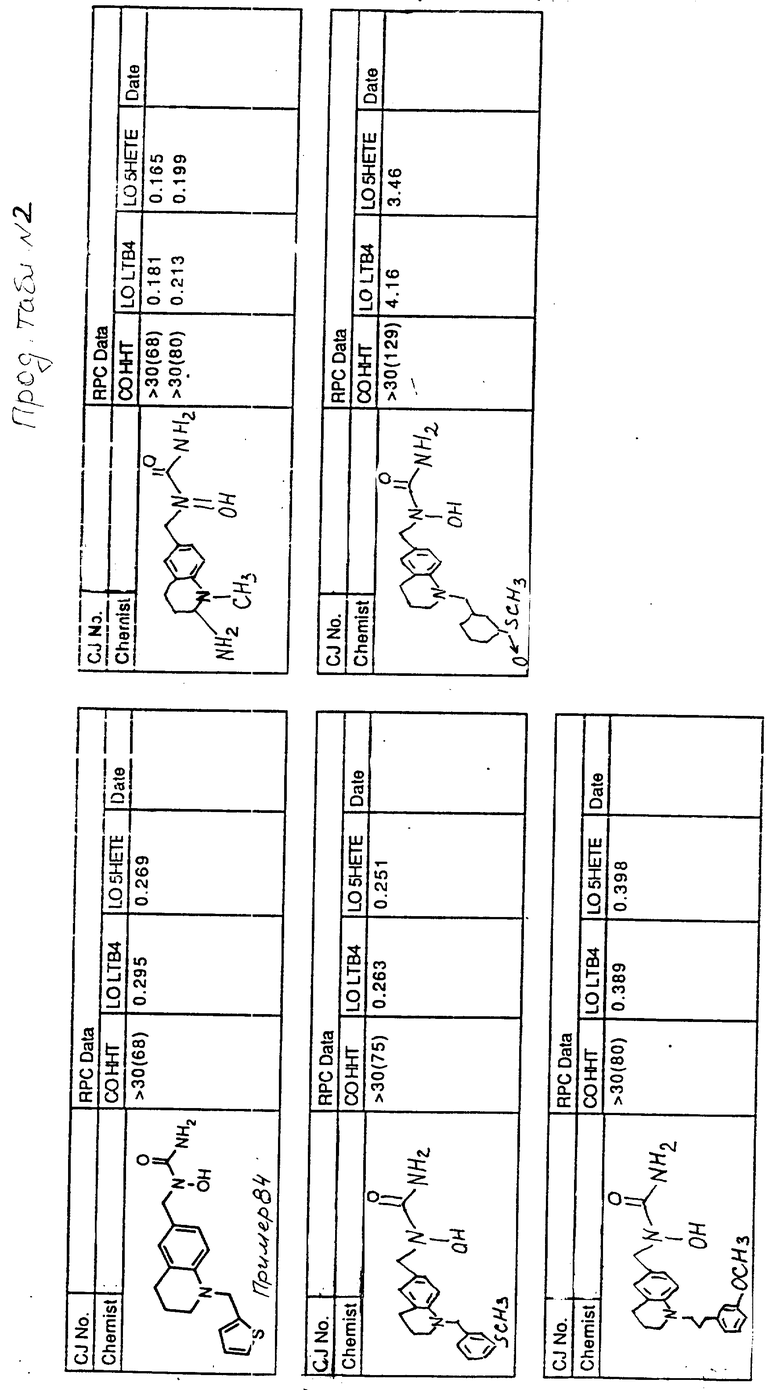
С помощью аналогичных способов были получены соединения примеров 5-113 (табл.1).
Результаты испытаний соединений изобретения на ингибирующую активность в отношении ряда химических веществ в виде показателя ИК50 (концентрации, дающей 50% ингибирования) представлены в табл.2.
Испытания проводились методом анализа резидентных клеток перитональной полости крыс с использованием каждого из соединений в соотношении следующих соединений, показанных в колонках 2, 3 и 4 соответственно табл.2.
Пояснения к табл.2:
COHHT - циклооксигеназа-гидроксигептадекатриеновая кислота;
LOLTB4 - липоксигеназа -лейкотриен B4;
LO5HETE - липоксигеназа-5-гидроксиэйкозатетраеновая кислота.







Описываются соединения формулы I, представленной в описании, где X - кислород, химическая связь или NR5; Z - кислород или сера. Эти соединения являются ингибиторами липоксигеназы и полезны в качестве активных агентов в фармацевтических композициях для лечения воспалительных состояний людей и других млекопитающих, причиной которых является активность липоксигеназа. 4 с. и 12 з.п.ф-лы, 1 табл.
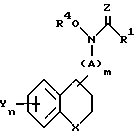
в которой R1 - С1 - С4-алкил или -NH2;
R4 - водород;
X - кислород, химическая связь или NR5, где R5 - водород, тиенил (низший)алкил, пиридил, пиридинил-С1 - С3-алкил, С3 - С6-алкенил, циано, фенил, фенил-С1 - С3-алкил, бензоил, причем указанный фенил является необязательно замещенным низшим алкилом, галоидом, трифторметилом, низшим алкокси, прямым или разветвленным низшим алкенилокси, нитрилом, низшим алкоксиалкилом, аминокарбонилом, галоид (низшим)алкокси или циклогексил (низшим)алкилом;
m = 0 или 1;
n = 1 - 3;
А - С1 - С3-алкилен или С2 - С5-алкенилен;
Y каждый - водород, галоген, фенил, триазолилокси, С1 - С2-алкокси, низший алкил, фенил-С1 - С3-алкилокси, оксанилокси, дифенилокси, фенилокси, пиридилокси, С3 - С1 2-алкенилокси, 3,4-диметилендиоксифенокси, тиазолил или фенилалкенилокси, причем указанное пиридильное или фенильное кольцо является необязательно замещенным галогеном, низшим алкилом, низшим алкокси, метилендиокси или трифторметилом;
Z - кислород или сера.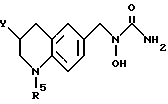
6. Соединение по п.5, в котором R5 - фенил или фенил - С1 - С3-алкил.
в которой Y - водород, галоген, фенил, триазолилокси, С1 - С2-алкокси, низший алкил, фенил-С1 - С3-алкилокси, оксинилокси, дифенилокси, фенилокси, пиридилокси, С3 - С1 2-алкенилокси, 3,4-диметилендиоксифенокси, тиазолил или фенил (низший)алкенилокси, причем, указанное пиридильное или фенильное кольцо является необязательно замещенным галогеном, низшим алкилом, низшим алкокси, метилендиокси или трифторметилом.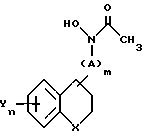
в которой A, X, Y, m и n имеют значения, определенные в п.1,
отличающийся тем, что гидроксиламин формулы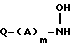
в которой Q - группа формулы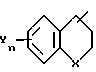
в которой Y и n имеют указанные значения,
подвергают взаимодействию с ацетилхлоридом или уксусным ангидридом в присутствии основания в нереакционноспособном растворителе с получением диацетильного соединения формулы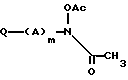
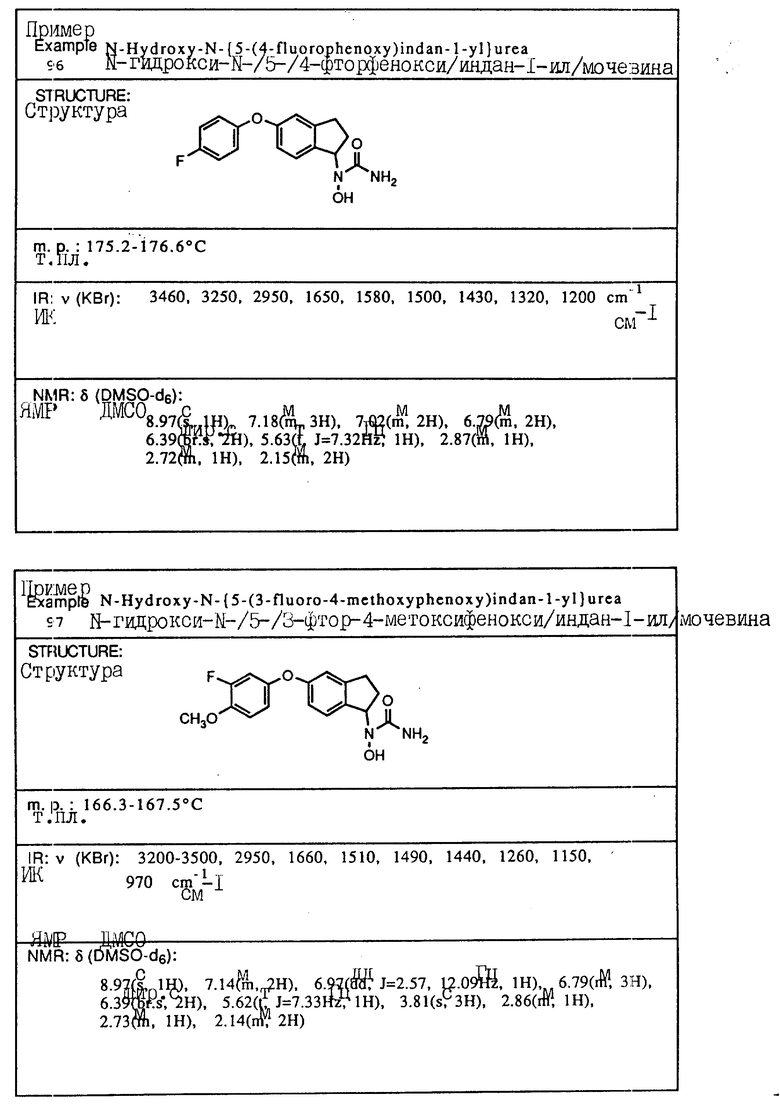
в которой A, Q и m имеют указанные значения,
которое подвергают селективному гидролизу с помощью реакции с основанием с последующим выделением полученного соединения.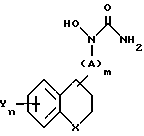
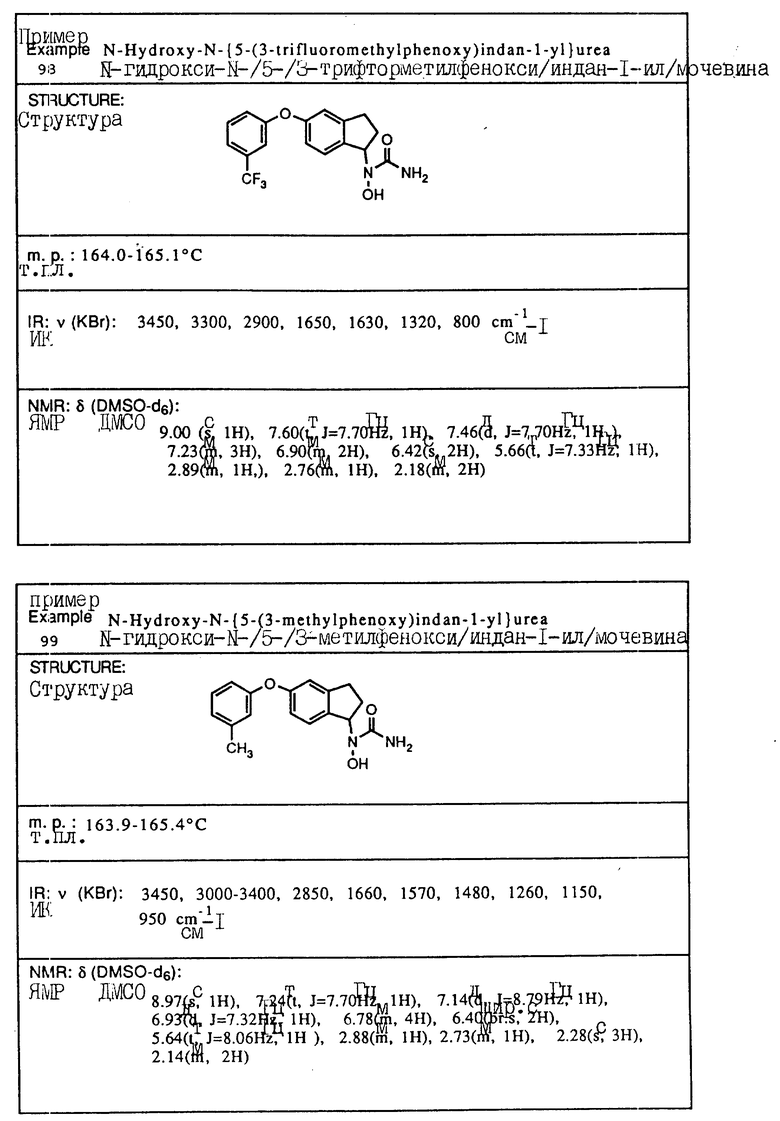
в которой A, Y, m и n имеют указанные значения,
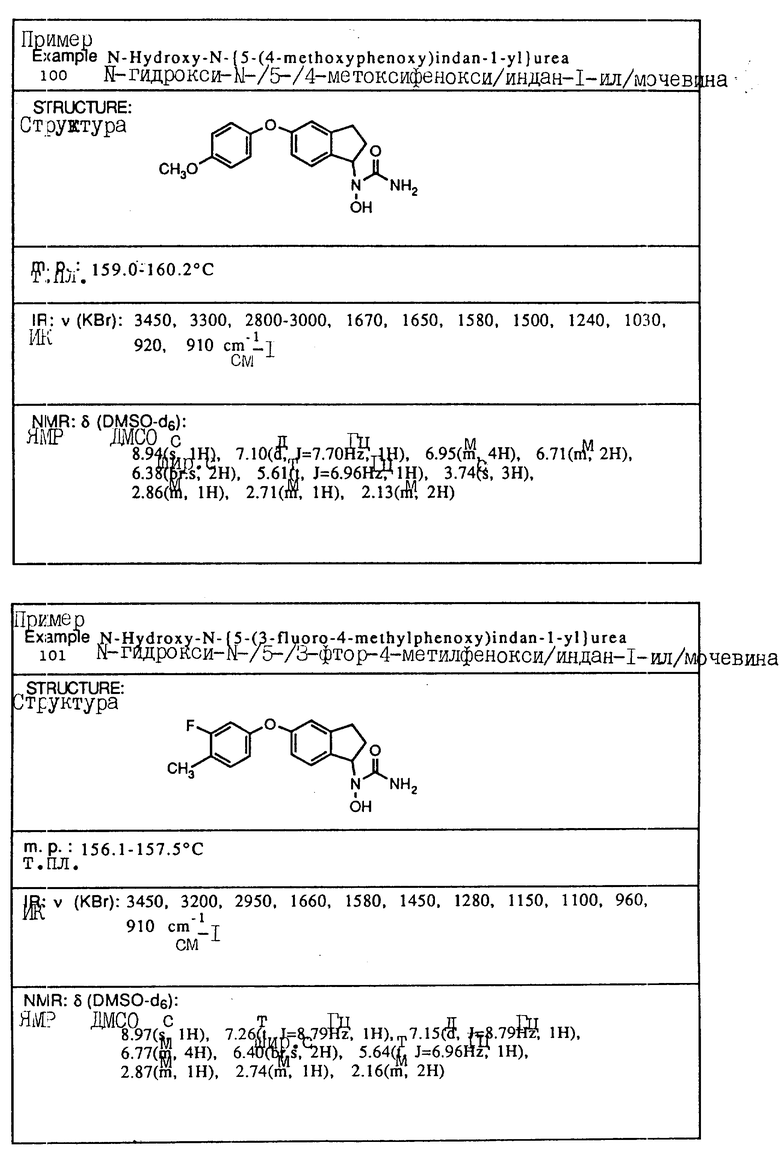
отличающийся тем, что гидроксиламин формулы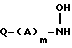
в которой Q - группа формулы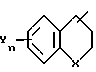
в которой X, Y и n имеют указанные значения,
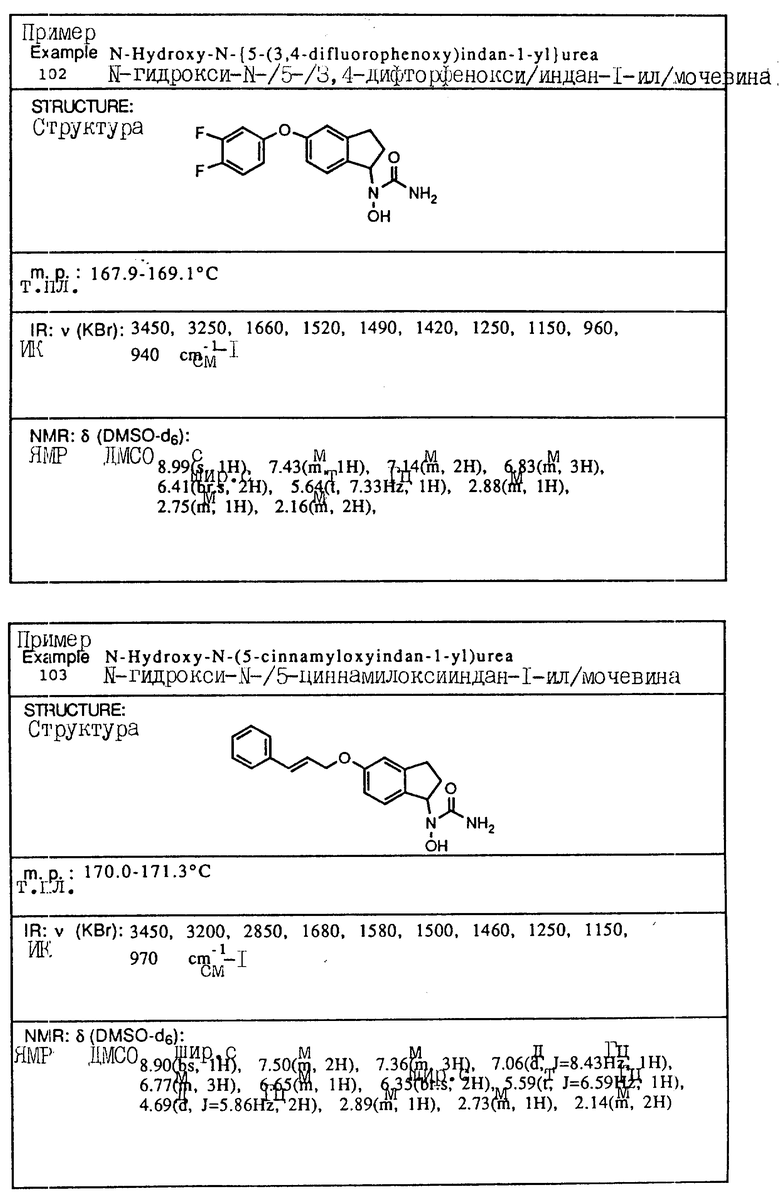
обрабатывают триметилсилилизоцианатом в нереакционноспособном растворителе с последующим выделением целевого продукта.
| EP, патент, 0196184, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, патент, 0279263, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| US, патент, 4822809, кл | |||
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |
