ССЫЛКА НА НАХОДЯЩИЕСЯ В ПРОЦЕССЕ ОДНОВРЕМЕННОГО РАССМОТРЕНИЯ ЗАЯВКИ
Дается ссылка на находящуюся в процессе одновременного рассмотрения заявку с регистрационным номером 09/207342, поданную 8 декабря 1998 года (номер в реестре поверенного PC8708C), которая является выделенной из заявки с регистрационным номером 09/020014, поданной 6 февраля 1998 года (номер в реестре поверенного PC8708B), теперь патент США N 5883106, которая является продолжением заявки с регистрационным номером 08/809901, поданной 29 мая 1995 года (номер в реестре поверенного PC8708A), в настоящее время отозванной; требуемый приоритет по заявке с регистрационным номером PCT/JP94/01747, поданной 18 октября 1994 года (номер в реестре поверенного PC8708), в настоящее время отозванной; и параграфа 371 заявки с регистрационным номером PCT/IB95/00408, поданной 29 мая 1995 года (с номером в реестре поверенного PC8708A), в настоящее время прекратившей действие и опубликованной как WO 96/11911 25 апреля 1996 года, в которых описаны ингибиторы 5-липоксигеназы, используемые при лечении воспалительных заболеваний и аллергии. В них описаны некоторые способы получения ингибиторов 5-липоксигеназы, но ничто из того, что описано, не позволило бы обычному специалисту в данной области предположить улучшенный способ по данному изобретению.
Делается также ссылка на находящуюся в процессе одновременного рассмотрения заявку с регистрационным номером 60/113221, поданную 22 декабря 1998 года (номер в реестре поверенного PC10097), в которой описан новый способ получения метилсульфоната амида 4-{3-[4-(2-метилимидазол-1-ил)фенилсульфанил] фенил} тетрагидропиран-4- карбоновой кислоты. Однако описанный способ не идентичен способу данного изобретения.
Далее дается ссылка на находящиеся в процессе одновременного рассмотрения заявки, поданные одновременно с данной заявкой, с номерами реестра поверенного PC10530 и PC10683, которые также включают в себя способы получения ингибиторов 5-липоксигеназы, имеющих различающиеся гетероциклические циклические системы, и которые имеют некоторые элементы способов, являющиеся общими со способом данного изобретения.
В WO 96/11911 описан класс новых соединений, активных в качестве ингибиторов активности фермента 5-липоксигеназы, характеризующихся следующей структурной формулой (1.1.0)
где Ar1 обозначает гетероциклическую часть молекулы, выбранную из группы, включающей имидазолил; пирролил; пиразолил; 1,2,3-триазолил; 1,2,4-триазолил; индолил; индазолил и бензимидазолил; связанную с X1 через атом азота кольца и замещенную 0-2 заместителями, выбранными из группы, включающей галоген; гидрокси; циано; амино; (C1-C4) алкил, (C1-C4) алкокси; (C1-C4) алкилтио; (C1-C4) галогензамещенный алкил; (C1-C4) галогензамещенный алкокси; (C1-C4) алкиламино и ди(C1-C4) алкиламино;
X1 обозначает прямую связь или (C1-C4) алкилен;
Ar2 обозначает фенилен, замещенный 0-2 заместителями, выбранными из группы, включающей галоген; гидрокси; циано; амино; (C1-C4) алкил; (C1-C4) алкокси; (C1-C4) алкилтио; (C1-C4) галогензамещенный алкил и (C1-C4) галогензамещенный алкокси;
X2 обозначает -A-X- или -X-A-, где A обозначает прямую связь или (C1-C4) алкилен и X обозначает окси; тио; сульфинил или сульфонил;
Ar3 обозначает радикал, выбранный из группы, включающей фенилен; пиридилен; тиенилен; фурилен; оксазолилен и тиазолилен; замещенный 0-2 заместителями, выбранными из галогена; гидрокси; циано; амино; (C1-C4) алкила; (C1-C4) алкокси; (C1-C4) алкилтио; (C1-C4) галогензамещенного алкила; (C1-C4) галогензамещенного алкокси; (C1-C4) алкиламино и ди(C1-C4) алкиламино;
R1 и R2 обозначают каждый (C1-C4) алкил; или вместе они образуют группу формулы: -D1-Z-D2-, которая вместе с атомом углерода, к которому она присоединена, образует циклическую структуру, имеющую 3-8 атомов углерода, где D1 и D2 представляют собой (C1-C4) алкилен и Z обозначает прямую связь или окси; тио; сульфонил; или винилен; и D1 и D2 могут быть замещены (C1-C3) алкилом; и
Y обозначает CONR3R4; CN; C(R3)=N-OR4; COOR3; COR3 или CSNR3R4; где R3 и R4 каждый обозначают H или (C1-C4) алкил.
Что касается вышеуказанных соединений, предпочтительным значением для (C1-C4) галогензамещенного алкила является трифторметил и предпочтительным значением для (C1-C4) галогензамещенного алкокси является трифторметокси. Предпочтительная группа вышеуказанных соединений состоит из соединений, в которых Ar2 представляет собой 1,4-фенилен и Ar3 представляет собой 1,3-фенилен или 5-фтор-1,3-фенилен. В этой предпочтительной группе более предпочтительными соединениями являются соединения, в которых Ar1 представляет собой 2-алкилимидазолил; X1 представляет собой прямую связь и Y представляет собой CONH2; и соединения, в которых Ar1 представляет собой пирролил; X1 представляет собой CH2 и Y представляет собой CONH2.
Особенно предпочтительным вариантом вышеописанного класса ингибиторных соединений является соединение формулы (1.0.0)
Соединения, которые ингибируют действие фермента липоксигеназы, могут использоваться при лечении или облегчении воспалительных заболеваний, аллергии и сердечно-сосудистых заболеваний у млекопитающих, в том числе человека. Активность фермента липоксигеназы проявляется как часть метаболизма арахидоновой кислоты. Арахидоновая кислота является биологическим предшественником нескольких групп биологически активных эндогенных метаболитов. Арахидоновая кислота сначала высвобождается из фосфолипидов мембран под действием фосфолипазы A2. Затем арахидоновая кислота метаболизируется (i) циклооксигеназой с образованием простагландинов, в том числе простациклина, и тромбоксанов; или (ii) липоксигеназой с образованием гидропероксижирных кислот, которые могут быть затем превращены в лейкотриены.
Лейкотриены, в свою очередь, являются чрезвычайно сильнодействующими и индуцируют большое разнообразие биологических эффектов, например, пептидолейкотриены, LTC4, LTD4 и LTE4, являются важными бронхоконстрикторами и вазоконстрикторами и вызывают экстравазацию плазмы путем увеличения проницаемости капилляров. LTB4 является сильным хемотаксическим агентом, который усиливает инфильтрацию и дегрануляцию лейкоцитов в месте воспаления. Полагают, что лейкотриены задействованы в ряде патологических состояний человека, в том числе астме, хронической обструктивной болезни легких, аллергическом рините, ревматоидном артрите, подагре, псориазе, атоническом дерматите, респираторном дистресс-синдроме взрослых (ARDS) и воспалительных заболеваниях пищеварительного тракта, в том числе болезни Крона. Средство, которое активно ингибирует липоксигеназы и вследствие этого продуцирование лейкотриенов, будет иметь существенную терапевтическую ценность при лечении острых и хронических воспалительных состояний. См. Masamune and Melvin, Annual Reports in Medicinal Chemistry 24, 71-80 (1989). Конкретные ингибиторы липоксигеназы были описаны в EP 0462830; EP 0505122 и EP 0540165.
Несколько способов получения ингибиторов липоксигеназы, описанных в вышеупомянутой опубликованной заявке WO 96/39408, приведены здесь. Примером такого способа получения является присоединение соединения формулы (1.2.0) и соединения формулы (1.2.1), которое может быть представлено приведенной ниже схемой реакции
где X1 обозначает тио и Q обозначает замещаемую группу, в присутствии тиомочевины и подходящего катализатора, например тетракис(трифенилфосфин) палладия. Приводится ссылка на Chem. Lett., 1379-1380 (1986). Указывается, что подходящие замещаемые группы Q включают в себя галоген или сульфонилоксигруппу.
Данное изобретение относится к области способов, используемых для синтетического получения соединений типа формулы (1.0.0), некоторые из которых являются известными соединениями, некоторые являются новыми соединениями и некоторые не относятся к общедоступной области, поскольку они не могут быть получены с использованием способов получения, известных до настоящего времени в данной области. Однако все эти соединения обладают биологической активностью в качестве ингибиторов 5-липоксигеназы.
Как уже отмечалось выше, в данной области известно, что соединения типа, представленного в формуле (1.0.0), могут быть получены по способу, в котором первоначально используют катализируемое палладием нуклеофильное замещение арилгалогенидов тиолатными анионами или самими тиолами. Как в реакции Вильямсона, которая является наилучшим общим способом получения несимметричных, а также симметричных простых эфиров, выходы улучшаются при помощи межфазного катализа. В отношении подробных действий при использования межфазного катализа в получении серусодержащих соединений, см., например, Weber; Gokel, Phase Transfer Catalysis in Organic Synthesis, Springer; New York, 221-233 (1977). Дальнейшие подробности, касающиеся начальной стадии способа данного изобретения, могут быть найдены в Migita et al., Bull. Chem. Soc. Japan 52, 1385-1389 (1980). Указанная начальная стадия может быть представлена следующей схемой реакции:
где X обозначает I или Br и R обозначает фенил или (C1-C4) алкил.
В литературе имеется ряд описаний, относящихся к катализируемому палладием синтезу. См., например, Brocato et al., Tetrahedron Lett., 33, 7433 (1992), где описана реакция замыкания цикла на основе катализируемой палладием, в частности Pd(PPh3)4, реакции бифункциональных ароматических соединений с концевыми алкинами и моноксидом углерода, требующей катализаторов, содержащих как палладий (0), так и палладий (II).
Arcadi et al. , Tetrahedron Lett. 34, 2813 (1993) описывают синтез 2,3,5-тризамещенных фуранов из арилгалогенидов и 2-пропаргил-1,3-дикарбонил-соединений в присутствии тетракис(трифенилфосфин) палладия (0) и K2CO3. Авторы наблюдают, что характер основания сильно влияет на ход реакции.
В McClure and Danishefsky, L. Am. Chem. Soc. 115, 6094-6100 (1993) описан синтез соединений типа 1,5-эпоксибензазоцина с 90% выходом с использованием каталитического тетракис(трифенилфосфин) палладия (0) в ацетонитриле, содержащем триэтиламин.
В Nuss et al., J. Am. Chem. Soc. 115, 6991-6992 (1993) описан синтез аналогов хромофора неокарциностатина с использованием каталитического тетракис(трифенилфосфин) палладия (0) в ТГФ и реагентов алкинилстаннанов.
В Paquette and Astles, J. Org. Chem. 58, 165-169 (1993) описан синтез фураноцембранолидов с удлинением боковой цепи, медиируемый катализируемым палладием (0) связыванием с винилстаннаном, проводимый в нагреваемом при температуре кипячения с обратным холодильником бензоле или диметоксиэтане. Авторы отмечают, что эта реакция зависит от растворителя и, в частности, предпочтительным является замена указанных растворителей на хлороформ.
Техническая литература содержит также ряд описаний, относящихся к применению других металлов переходной группы, кроме палладия, для катализа реакций. См., например, Takagi, Chemistry Letters, 2221-2224 (1987), где описано применение комплексов никеля (0) и палладия (0) в качестве катализаторов в синтезе диарилсульфидов из арилгалогенидов и ароматических тиолов.
Однако ни одна из этих ссылок не описывает и не предполагает конкретные способы получения по данному изобретению, которые являются как легкими, так и эффективными, дающими вместе с тем приемлемые выходы, не достигаемые прежде.
Данное изобретение относится к способам получения, в которых ряд конечных продуктов этих способов являются известными соединениями, используемыми в качестве ингибиторов 5-липоксигеназы. Данное изобретение относится также к ряду других конечных продуктов этих способов, которые не были известны до сих пор, поскольку они не были доступны синтетически до того, как стали доступными вышеупомянутые способы данного изобретения. Эти новые конечные продукты также могут использоваться в качестве ингибиторов 5-липоксигеназы, как описано более подробно далее в этом описании. Все из указанных способов получения данного изобретения описываются в общем виде в приводимых непосредственно ниже абзацах.
Ключевым промежуточным продуктом, используемым в способах получения по данному изобретению является тетрагидро-4-[3-(4-фторфенил)тио]фенил-2H-пиран-4-карбоксамид формулы (2.0.0)
Таким образом, данное изобретение относится также к способу получения соединения формулы (2.0.0), который может иллюстрироваться схемой синтеза (10.0.0) (см. в конце описания), предусматривающему (a) образование реакционной смеси, состоящей из (1) тетрагидро-4-(3-бром- или йодфенил)-2H-пиран-4-нитрила формулы (3.0.0)
где X обозначает бром или йод, и (2) 4-фтортиофенола формулы (4.0.0)
(3) в растворителе, состоящем из алифатического спирта с прямой или разветвленной цепью, имеющего в целом 2-7 атомов углерода, необязательно в виде его водной смеси; и более предпочтительно, если этот спирт является вторичным спиртом, выбранным из группы, включающей изопропиловый спирт, втор-бутиловый спирт, изопентилововый спирт и 2-гептанол, необязательно в виде водной смеси указанного вторичного спирта, (4) в присутствии сильного основания формулы (5.0.0)
M-O-R5,
где M обозначает щелочной металл, элемент группы 1/Ia, выбранный из группы, включающей литий, Li; натрий, Na; калий, K; рубидий, Rb; и цезий, Cs; и R5 обозначает водород, H; или (C1-C4) алкил с прямой или разветвленной цепью; предпочтительно представитель, выбранный из группы, включающей гидроксид лития, LiOH; гидроксид натрия, NaOH; гидроксид калия, KOH; гидроксид рубидия, RbOH; гидроксид цезия, CsOH; метоксид лития, LiOCH3; метоксид натрия, NaOCH3; метоксид калия, KOCH3; метоксид рубидия, RbOCH3; метоксид цезия, CsOCH3; этоксид лития, LiOCH2CH3; этоксид натрия, NaOCH2CH3; этоксид калия, KOCH2CH3; этоксид рубидия, RbOCH2CH3; этоксид цезия, CsOCH2CH3; трет-бутоксид лития, LiOC(CH3)3; трет-бутоксид натрия NaOC(CH3)3; трет-бутоксид калия KOC(CH3)3; трет-бутоксид рубидия RbOC(CH3)3 и трет-бутоксид цезия CsOC(CH3)3, в том числе их смеси; и, кроме того, (5) в присутствии катализатора, являющегося металлом переходной группы, включающего комплекс металла палладия, который предпочтительно является представителем, выбранным из группы, включающей
тетракис(трифенилфосфин)палладий (0), [(C6H5)3P]4Pd (0);
тетракис(метилдифенилфосфин)палладий (0), [(C6H5)2PCH3]4Pd (0);
транс-дихлорбис(метилдифенилфосфин)палладий (II), [(C6H5)2PCH3]2PdCl2; аддукт дихлорбис[метиленбис(дифенилфосфин)]дипалладий-дихлорметан;
дихлорбис(трифенилфосфин)палладий (II), [(C6H5)3P]2PdCl2;
аддукт трис(дибензилиденацетон)дипалладий (0) - хлороформ, (C6H5CH=CHCOCH=CHC6H5)3Pd2• CHCl3;
бис(дибензилиденацетон)палладий (0), (C6H5CH=CHCOCH=CHC6H5)2Pd;
[1,1'-бис(дифенилфосфино)ферроцен] дихлорпалладий (II), комплекс с дихлорметаном;
бис [1,2-бис(дифенилфосфино)этан]палладий (0) и
димер (π-аллил)палладий (II) хлорида; с последующим (b) нагреванием указанной реакционной смеси, предпочтительно при кипячении с обратным холодильником, предпочтительно в течение периода от 12 до 36 часов, более предпочтительно от 18 до 24 часов; в результате чего образуется вышеуказанное соединение формулы (2.0.0), которое необязательно выделяют путем общепринятых способов разделения.
Вышеописанный способ получения, в котором 4-карбоксамидная часть пирановой части молекулы образуется во время стадии добавления тиогруппы, является предпочтительным способом проведения этой части способа по данному изобретению. Применимый альтернативный вариант предусматривает образование 4-карбоксамидной части пирановой части молекулы перед стадией добавления тиогруппы. Этот альтернативный вариант данной части способа данного изобретения включает способ получения соединения формулы (2.0.0), который может иллюстрироваться схемой реакции (10.1.0) (см. в конце описания), предусматривающий (a) образование реакционной смеси, состоящей из (1) тетрагидро-4-(3-бром- или йодфенил)-2H-пиран-4-нитрила формулы (3.0.0)
где X обозначает бром или йод, (2) в растворителе, состоящем из алифатического спирта, как определено выше, необязательно в виде его водной смеси; предпочтительно вторичного спирта, как определено выше; более предпочтительно изопропилового спирта, необязательно в виде водной смеси указанного вторичного спирта; (3) в присутствии сильного основания формулы (5.0.0)
M-O-R5,
где M и R5 имеют указанные выше значения; причем предпочтительно указанным сильным основанием является гидроксид натрия, NaOH; гидроксид калия, KOH; этоксид натрия, NaOCH2CH3 или трет-бутоксид калия, KOC(CH3)3; с последующим (b) нагреванием указанной реакционной смеси предпочтительно при кипячении с обратным холодильником, предпочтительно в течение периода от 3 до 8 часов, более предпочтительно от 5 до 6 часов; в результате чего образуется вышеуказанное соединение формулы (3.1.0)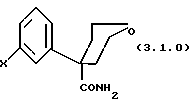
где X обозначает бром или йод; с последующим (c) образованием реакционной смеси, состоящей из соединения формулы (3.1.0) и 4-фтортиофенола формулы (4.0.0)
(1) в растворителе, состоящем из спирта, как описано выше, необязательно в виде его водной смеси; предпочтительно вторичного спирта, как определено выше; более предпочтительно изопропилового спирта, необязательно в виде водной смеси указанного вторичного спирта; (2) в присутствии сильного основания формулы (5.0.0)
M-O-R5,
где M и R5 имеют указанные выше значения; причем предпочтительно указанное сильное основание является гидроксидом натрия, NaOH; гидроксидом калия, KOH; этоксидом натрия, NaOCH2CH3 или трет-бутоксидом калия, KOC(CH3)3; и, кроме того, (3) в присутствии катализатора, являющегося металлом переходной группы, включающего в себя комплекс металла палладия, который предпочтительно выбран из группы, включающей
тетракис(трифенилфосфин)палладий (0), [(C6H5)3P]4Pd (0);
тетракис(метилдифенилфосфин)палладий (0), [(C6H5)2PCH3]4Pd (0);
транс-дихлорбис(метилдифенилфосфин)палладий (II), [(C6H5)2PCH3]2PdCl2; аддукт дихлорбис[метиленбис(дифенилфосфин)]дипалладий-дихлорметан;
дихлорбис(трифенилфосфин)палладий (II), [(C6H5)3P]2PdCl2;
аддукт трис(дибензилиденацетон)дипалладий (0) - хлороформ, (C6H5CH=CHCOCH=CHC6H5)3Pd2• CHCl3;
бис(дибензилиденацетон)палладий (0), (C6H5CH=CHCOCH=CHC6H5)2Pd;
[1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном;
бис [1,2-бис(дифенилфосфино)этан]палладий (0) и
димер (π-аллил) палладий (II) хлорида; с последующим (d) нагреванием указанной реакционной смеси предпочтительно при кипячении с обратным холодильником, предпочтительно в течение периода от 5 до 15 часов, более предпочтительно от 8 до 10 часов; в результате чего образуется вышеуказанное соединение формулы (2.0.0).
Далее, данное изобретение относится к способу получения соединения формулы (1.3.0)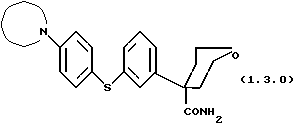
который может иллюстрироваться схемой синтеза (10.2.0) (см. в конце описания), где часть молекулы следующей формулы (1.3.1)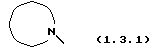
является электрондефицитной моноциклической или бензоконденсированной бициклической N-гетероциклической группой, содержащей два атома азота, формулы (1.3.2), (1.3.3), (1.3.4) или (1.3.5)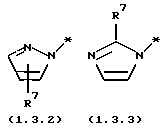

где * - символ, который представляет точку присоединения части молекулы формулы (1.3.2), (1.3.3), (1.3.4) или (1.3.5);
R7 и R8 независимо выбраны из группы, включающей H; (C1-C4) алкил с прямой или разветвленной цепью и (C6-C10) арил; где указанные группы арил и алкил замещены 0-2 заместителями, выбранными из группы, включающей галоген; гидрокси; циано; амино; (C1-C4) алкил; (C1-C4) алкокси; (C1-C4) алкилтио; (C1-C4) галогензамещенный алкил; (C1-C4) галогензамещенный алкокси; (C1-C4) алкиламино и ди(C1-C4) алкиламино; включающему (a) образование реакционной смеси, состоящей из (1) тетрагидро-4-[3-(4-фторфенил)тио]фенил-2H-пиран-4-карбоксамида формулы (2.0.0)
и (2) электрондефицитного моноциклического или бензоконденсированного бициклического N-гетероцикла, содержащего два атома азота, формулы (1.3.6), (1.3.7), (1.3.8) или (1.3.9)

где R7 и R8 имеют указанные выше значения;
(3) в апротонном растворителе, предпочтительно диметилсульфоксиде (ДМСО);
(4) в присутствии сильного основания в твердой форме, выбранного из группы, включающей гидроксид натрия, NaOH, и гидроксид калия, KOH; и (необязательно) (5) в присутствии каталитического количества карбоната цезия, Cs2CO3, или катализатора межфазного переноса, предпочтительно выбранного из группы, включающей цетилтриметиламмонийбромид (CTMAB); дибензо-18-краун-6 (DB-18-c-6); дициклогексано-18-краун-6 (DC-18-c-6); 18-краун-6 (18-c-6); (-)-N-додецил-N-метилэфедринийбромид (DMCOH); гексаметилтриамид фосфорной кислоты (HMPT); цетилпиридинийбромид (NCPB); N-бензилхининийхлорид (QUIBEC); тетра-н-бутиламмонийбромид (TBAB); тетра-н-бутиламмонийхлорид (TBAC); тетра-н-бутиламмонийгидроксид (TBAH); тетра-н-бутиламмонийгидросульфат (TBAHS); тетра-н-бутиламмониййодид (TBAI); тетраэтиламмонийхлорид, гидрат (TEAC); три-н-бутиламин (TBA); бензилтрибутиламмонийбромид (TBBAB); гексадецилтрибутилфосфонийбромид (TBHDPB); бензилтриэтиламмонийбромид (TEBAB); бензилтриэтиламмонийхлорид (TEBA); гексадецилтриэтиламмонийхлорид (TEHDAC); тетраметиламмонийхлорид (TMAC); гексадецилтриметиламмонийхлорид (TMHDAC) и октилтриметиламмонийхлорид (TMOAC) и более предпочтительно соли четвертичного аммония или соли фосфония, содержащей представитель вышеописанных групп; с последующим (b) нагреванием указанной реакционной смеси, предпочтительно при кипячении с обратным холодильником, в атмосфере азота; в результате чего образуется соединение формулы (1.3.0).
Далее данное изобретение относится к вышеуказанному способу получения соединения формулы (1.3.0), в котором указанное соединение формулы (1.3.0.) выбрано из группы, включающей:
тетрагидро-4-{ 3-[4-(2-метил-1H-имидазол-1-ил)фенил] тио} фенил-2H- пиран-4-карбоксамид;
тетрагидро-4-{ 3-[4-(1H-имидазол-1-ил)фенил)тио} фенил-2H-пиран-4- карбоксамид;
тетрагидро-4-{ 3-[4-(1H-бензоимидазол-1-ил)фенил)тио}фенил-2H- пиран-4-карбоксамид;
тетрагидро-4-{ 3-[4-(1H-пиразол-1-ил)фенил] тио} фенил-2H- пиран-4-карбоксамид и
тетрагидро-4-{ 3-[4-(4-метил-1H-пиразол-1-ил)фенил)тио} - фенил-2H-пиран-4-карбоксамид.
Вышеописанные конечные продукты не были известны до настоящего времени, так как они были синтетически недоступными прежде, до доступности способов данного изобретения. Эти новые конечные продукты также могут использоваться в качестве ингибиторов 5-липоксигеназы и выбраны из группы, включающей:
тетрагидро-4-{ 3-[4-(1H-имидазол-1-ил)фенил] тио} фенил-2H-пиран-4- карбоксамид;
тетрагидро-4-{ 3-[4-(1H-бензоимидазол-1-ил)фенил]тио}фенил-2H- пиран-4-карбоксамид;
тетрагидро-4-{ 3-[4-(1H-пиразол-1-ил)фенил] тио} фенил-2H-пиран-4- карбоксамид и
тетрагидро-4-{ 3-[4-(4-метил-1H-пиразол-1-ил)фенил]тио}фенил-2H- пиран-4-карбоксамид.
Данное изобретение относится также к способу получения соединения формулы (1.0.0)
включающему (a) образование реакционной смеси, состоящей из
(1) тетрагидро-4-[3-(4-фторфенил)тио] фенил-2H-пиран-4-карбоксамида формулы (2.0.0)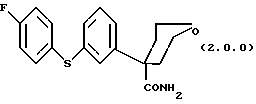
и (2) 2-метилимидазола;
(3) в апротонном растворителе, предпочтительно диметилсульфоксиде (ДМСО);
(4) в присутствии сильного основания в твердой форме, выбранного из группы, состоящей из гидроксида натрия, NaOH, и гидроксида калия, KOH; и (необязательно) (5) в присутствии каталитического количества карбоната цезия, Cs2CO3, или катализатора межфазного переноса, предпочтительно выбранного из группы, включающей цетилтриметиламмонийбромид (CTMAB); дибензо-18-краун-6 (DB-18-c-6); дициклогексано-18-краун-6 (DC-18-c-6); 18-краун-6 (18-c-6); (-)-N-додецил-N-метилэфедринийбромид (DMCOH); гексаметилтриамид фосфорной кислоты (HMPT); цетилпиридинийбромид (NCPB); N-бензилхининийхлорид (QUIBEC); тетра-н-бутиламмонийбромид (TBAB); тетра-н-бутиламмонийхлорид (TBAC); тетра-н-бутиламмонийгидроксид (TBAH); тетра-н-бутиламмонийгидросульфат (TBAHS); тетра-н-бутиламмониййодид (TBAI); тетраэтиламмонийхлорид, гидрат (TEAC); три-н-бутиламин (TBA); бензилтрибутиламмонийбромид (TBBAB); гексадецилтрибутилфосфонийбромид (TBHDPB); бензилтриэтиламмонийбромид (TEBAB); бензилтриэтиламмонийхлорид (TEBA); гексадецилтриэтиламмонийхлорид (TEHDAC); тетраметиламмонийхлорид (TMAC); гексадецилтриметиламмонийхлорид (TMHDAC) и октилтриметиламмонийхлорид (TMOAC) и более предпочтительно соли четвертичного аммония или соли фосфония, содержащей представитель вышеописанной группы; с последующим (b) нагреванием указанной реакционной смеси, предпочтительно при кипячении с обратным холодильником, предпочтительно при 115-145oC, более предпочтительно при 125-130oC, в атмосфере азота, предпочтительно в течение 12-30 часов, более предпочтительно в течение 17-24 часов; в результате чего образуется указанное соединение формулы (1.3.0).
Далее, данное изобретение относится к способу получения, по существу, чистой мезилатной соли формулы (1.0.1)
который может иллюстрироваться схемой синтеза (10.3.0) (см. в конце описания), включающей (a) получение соединения формулы (2.0.0)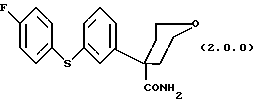
предусматривающее (1) образование реакционной смеси, состоящей из (i) тетрагидро-4-(3-бромфенил)-2H-пиран-4-нитрила формулы (3.2.0)
и (ii) 4-фтортиофенола формулы (4.0.0)
(iii) в растворителе, выбранном из группы, состоящей из изопропилового спирта, втор-бутилового спирта, изопентилового спирта и 2-гептанола, предпочтительно изопропилового спирта, необязательно, в виде его водной смеси; (iv) в присутствии сильного основания, выбранного из группы, состоящей из гидроксида натрия, NaOH, и гидроксида калия, KOH; и, кроме того, (v) в присутствии катализатора, содержащего металл переходной группы, в том числе независимо выбранного из группы, состоящей из комплексов металла палладия; предпочтительно такой комплекс металла палладия является членом, выбранным из группы, включающей
тетракис(трифенилфосфин) палладий (0), [(C6H5)3P]4Pd (0);
тетракис(метилдифенилфосфин)палладий (0), [(C6H5)2PCH3]4Pd (0);
транс-дихлорбис(метилдифенилфосфин) палладий (II), [(C6H5)2PCH3]2PdCl2; аддукт дихлорбис[метиленбис(дифенилфосфин)]дипалладий-дихлорметан;
дихлорбис(трифенилфосфин)палладий (II), [(C6H5)3P]2PdCl2;
аддукт трис(дибензилиденацетон)дипалладий (0) - хлороформ, (C6H5CH=CHCOCH=CHC6H5)3Pd2• CHCl3;
бис(дибензилиденацетон)палладий (0), (C6H5CH=CHCOCH=CHC6H5)2Pd;
[1,1'-бис(дифенилфосфино)ферроцен] дихлорпалладий (II), комплекс с дихлорметаном;
бис[1,2-бис(дифенилфосфино)этан]палладий (II) и
димер (π-аллил)палладий (II) хлорида; с последующим (2) нагреванием указанной смеси при кипячении с обратным холодильником при 80-84o в течение периода от 18 до 30 часов, предпочтительно 24 часов; в результате чего образуется вышеуказанное соединение формулы (2.0.0);
(b) образование реакционной смеси, состоящей из соединения формулы (2.0.0) и соединения формулы (1.3.10)
(1) в апротонном растворителе, предпочтительно диметилсульфоксиде (ДМСО);
(2) в присутствии сильного основания в твердой форме, выбранного из группы, состоящей из гидроксида натрия, NaOH, и гидроксида калия, KOH; и необязательно (3) в присутствии каталитического количества карбоната цезия, Cs2CO3, или катализатора межфазного переноса, предпочтительно выбранного из группы, включающей цетилтриметиламмонийбромид (CTMAB); дибензо-18-краун-6 (DB-18-c-6); дициклогексано-18-краун-6 (DC-18-c-6); 18-краун-6 (18-c-6); (-)-N-додецил-N-метилэфедринийбромид (DMCOH); гексаметилтриамид фосфорной кислоты (HMPT); цетилпиридинийбромид (NCPB); N-бензилхининийхлорид (QUIBEC); тетра-н-бутиламмонийбромид (TBAB); тетра-н-бутиламмонийхлорид (TBAC); тетра-н-бутиламмонийгидроксид (TBAH); тетра-н-бутиламмонийгидросульфат (TBAHS); тетра-н-бутиламмониййодид (TBAI); тетраэтиламмонийхлорид, гидрат (TEAC); три-н-бутиламин (TBA); бензилтрибутиламмонийбромид (TBBAB); гексадецилтрибутилфосфонийбромид (TBHDPB); бензилтриэтиламмонийбромид (TEBAB); бензилтриэтиламмонийхлорид (TEBA); гексадецилтриэтиламмонийхлорид (TEHDAC); тетраметиламмонийхлорид (TMAC); гексадецилтриметиламмонийхлорид (TMHDAC) и октилтриметиламмонийхлорид (TMOAC) и более предпочтительно соли четвертичного аммония или соли фосфония, содержащей представитель вышеописанной группы; с последующим (c) нагреванием указанной реакционной смеси при кипячении с обратным холодильником в атмосфере азота; в результате чего образуется указанное соединение формулы (1.0.0)
с последующим (d) образованием концентрированного раствора в метаноле указанного соединения формулы (1.0.0), который затем фильтруют предпочтительно через активированный уголь, после чего добавляют к фильтрату метансульфоновую кислоту, MeSO3H; с последующим дополнительным концентрированием и добавлением этилацетата ad seriatim до выделения кристаллического продукта, содержащего, по существу, чистую мезилатную соль формулы (1.0.1)
или альтернативно с последующим (e) образованием концентрированного раствора в метаноле указанного соединения формулы (1.0.0), к которому затем добавляют метансульфоновую кислоту, MeSO3H; с последующим фильтрованием этой смеси предпочтительно через активированный уголь, после чего следует дополнительное концентрирование и добавление этилацетата ad seriatim до выделения кристаллического продукта, содержащего по существу чистую мезилатную соль формулы (1.0.1)
Данное изобретение относится к улучшенному способу получения известных соединений, показавших возможность использования их в качестве ингибиторов 5-липоксигеназы, и, в частности, соединения формулы (1.0.0)
Далее, данное изобретение относится к получению ряда других соединений, которые не были известны до настоящего времени, так как они были синтетически недоступны прежде, до доступности улучшенного способа данного изобретения. Эти новые соединения также могут использоваться в качестве ингибиторов 5-липоксигеназы и включают в себя, среди других соединений, следующие соединения формул (1.1.1); (1.1.2); (1.1.3) и (1.1.4):
Тетрагидро-4-{ 3-[4-(1H-имидазол-1-ил)фенил] тио} фенил-2H-пиран-4- карбоксамид
Тетрагидро-4-{ 3-[4-(1H-бензоимидазол-1-ил)фенил]тио}фенил-2H- пиран-4-карбоксамид
Тетрагидро-4-{ 3-[4-(1H-пиразол-1-ил)фенил] тио} фенил-2H-пиран-4- карбоксамид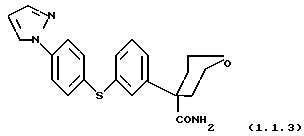
Тетрагидро-4-{ 3-[4-(4-метил-1H-пиразол-1-ил)фенил]тио}фенил-2H- пиран-4-карбоксамид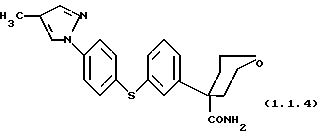
Для получения вышеуказанных соединений формул (1.1.1)-(1.1.4) и подобных соединений этого типа предпочтительно использовать следующий способ данного изобретения для получения соединения формулы (1.3.0)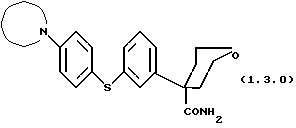
где часть молекулы формулы (1.3.1)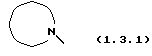
является электронодефицитной моноциклической бензоконденсированной бициклической N-гетероциклической группой, содержащей два атома азота, формулы (1.3.2), (1.3.3), (1.3.4) или (1.3.5)
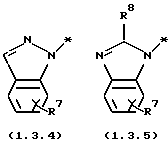
где * - обозначает символ, который представляет точку присоединения части молекулы формулы (1.3.2), (1.3.3), (1.3.4) или (1.3.5);
R7 и R8 независимо выбраны из группы, включающей H; (C1-C4)
алкил с прямой или разветвленной цепью и (C6-C10) арил; где указанные группы арил и алкил замещены 0-2 заместителями, выбранными из группы, включающей галоген; гидрокси; циано; амино; (C1-C4) алкил; (C1-C4) алкокси; (C1-C4) алкилтио; (C1-C4) галогензамещенный алкил; (C1-C4) галогензамещенный алкокси; (C1-C4) алкиламино и ди (C1-C4) алкиламино.
Вышеупомянутый вариант способа получения данного изобретения может иллюстрироваться схемой синтеза (10.2.0) (см. в конце описания), где реагирующее вещество формулы (1.4.0)
является электронодефицитным моноциклическим бензоконденсированным бициклическим N-гетероциклом, содержащим два атома азота, формулы (1.3.6), (1.3.7), (1.3.8) или (1.3.9), определенных дополнительно выше.
Таким образом, вышеупомянутый способ данного изобретения, иллюстрированный в схеме синтеза (10.2.0), может проводиться путем:
(a) образования реакционной смеси, состоящей из
(1) тетрагидро-4-[3-(4-фторфенил)тио] фенил-2H-пиран-4-карбоксамида формулы (2.0.0)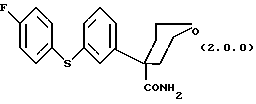
и (2) электронодефицитного моноциклического или бензоконденсированного бициклического N-гетероцикла, содержащего два атома азота, формулы (1.3.6), (1.3.7), (1.3.8) или (1.3.9)

где R7 и R8 имеют указанные выше значения;
(3) в апротонном растворителе, предпочтительно выбранном из группы, включающей, в основном, гексан; 1,4-диоксан; тетрахлорид углерода; бензол; толуол; ксилолы; диэтиловый эфир; хлороформ; этилацетат; тетрагидрофуран (ТГФ); метиленхлорид; гексаметилтриамид фосфорной кислоты (HMPT); нитрометан; N, N-диметилформамид (ДМФ); ацетонитрил; сульфолан и диметилсульфоксид (ДМСО); более предпочтительно диметилсульфоксид (ДМСО);
(4) в присутствии сильного основания в твердой форме, выбранного из группы, состоящей из гидроксида натрия, NaOH, и гидроксида калия, KOH; и (необязательно) (5) в присутствии каталитического количества карбоната цезия, Cs2CO3, или катализатора межфазного переноса, предпочтительно выбранного из группы, включающей цетилтриметиламмонийбромид (CTMAB); дибензо-18-краун-6 (DB-18-c-6); дициклогексано-18-краун-6 (DC-18-c-6); 18-краун-6 (18-c-6); (-)-N-додецил-N-метилэфедринийбромид (DMCOH); гексаметилтриамид фосфорной кислоты (HMPT); цетилпиридинийбромид (NCPB); N-бензилхининийхлорид (QUIBEC); тетра-н-бутиламмонийбромид (TBAB); тетра-н-бутиламмонийхлорид (TBAC); тетра-н-бутиламмонийгидроксид (TBAH); тетра-н-бутиламмонийгидросульфат (TBAHS); тетра-н-бутиламмониййодид (TBAI); тетраэтиламмонийхлорид, гидрат (TEAC); три-н-бутиламин (TBA); бензилтрибутиламонийбромид (TBBAB); гексадецилтрибутилфосфонийбромид (TBHDPB); бензилтриэтиламмонийбромид (TEBAB); бензилтриэтиламмонийхлорид (TEBA); гексадецилтриэтиламмонийхлорид (TEHDAC); тетраметиламмонийхлорид (TMAC); гексадецилтриметиламмонийхлорид (TMHDAC) и октилтриметиламмонийхлорид (TMOAC), и более предпочтительно соли четвертичного аммония или соли фосфония, содержащей представитель вышеописанной группы; с последующим (b) нагреванием указанной реакционной смеси предпочтительно при кипячении с обратным холодильником, в атмосфере азота; в результате чего образуется вышеуказанное соединение формулы (1.3.0).
Как понятно среднему специалисту в области получения органических соединений того типа, с которым имеет дело данное изобретение, замещение арилфторида в присутствии основания азотсодержащим гетероциклом с недостатком электронов является относительно неизвестным способом образования связей углерод-азот и явно является способом, который не предполагался до настоящего времени в качестве способа, который можно использовать при получении типов рассматриваемых соединений. Обычно сильная оттягивающая электрон группа, например нитро, расположенная в пара- или ортоположении относительно атома фтора, требуется для достижения приемлемого уровня замещения азотным нуклеофилом в присутствии основания. Такие реакции замещения обычно дают лишь низкие выходы, часто требуют повышенных температур и продолжительных периодов реакции и приводят к продуктам, требующим дополнительной очистки. См., например, Morgan et al. , J. Med. Chem., 33, 1091-1097 (1990), где описан способ получения, в котором метиловый или этиловый эфир 4-фторбензойной кислоты взаимодействует с подходящим имидазолом в ДМСО с использованием основания, такого как K2CO3, NaOH или NaH. Соединение этиловый эфир 4-(2-метил-1H-имидазол-1-ил) бензойной кислоты получали лишь с 33% выходом неперекристаллизованного продукта. В противоположность этому, способы получения данного изобретения дают высокие выходы, результат, который является полностью неожиданным, поскольку реагент арилфторид в способах данного изобретения не имеет оттягивающих электроны заместителей, присоединенных к арильному кольцу.
Наиболее предпочтительным растворителем для использования в вышеописанном способе по данному изобретению является диметилсульфоксид (ДМСО), хотя пригоден любой апротонный растворитель, и растворители, перечисленные выше, являются предпочтительными. В другом предпочтительном варианте способа твердый гидроксид натрия, NaOH, используют в реакционной смеси, для которой растворителем является ДМСО. Сильное основание в твердой форме, которое используют в этой стадии способа получения данного изобретения, выбирают из гидроксида натрия, NaOH, и гидроксида калия, KOH. Термин "твердый", используемый в этой связи, относится к фазе, в которой сильное основание, которое присутствует, должно быть обнаружено в этой реакционной смеси. Предпочтительно указанное твердое вещество применяют раздробленным, а не целиком, обеспечивая тем самым более обширную площадь поверхности, на которой другие реагирующие вещества способны контактировать с сильным основанием во время этой стадии способа. Так, сильное основание в твердой форме может быть использовано в виде порошка или в виде шариков (гранул). С другой стороны, не является обязательным, чтобы эта твердая форма сильного основания была мелкоизмельченной. Твердые формы сильного основания, предпочтительно используемые в способах данного изобретения, являются коммерчески легко доступными.
Необязательно, но предпочтительно на этой стадии замещения арилфторида по способам данного изобретения использовать также каталитическое количество карбоната цезия, Cs2CO3, или катализатора межфазного переноса ("PTC"). Используемое количество может варьироваться между 0,5% моля и 10% моля, т.е. мол. %, но предпочтительно между 1 мол.% и 5 мол.%. Количества катализатора, которые пригодны для применения в способах данного изобретения, могут быть выражены как находящиеся в диапазоне от 0,005 до 0,5 эквивалентов, предпочтительно от 0,01 до 0,1 эквивалентов и более предпочтительно приблизительно 0,05 эквивалентов относительно других соединений-участников в этой реакции.
Было обнаружено, что карбонат цезия, Cs2CO3, вещество, которое используют в качестве катализатора в полимеризации этиленоксида и других проводимых с катализатором реакций, может быть использован в качестве альтернативного катализатора относительно катализатора межфазного переноса, как описано здесь.
Концентрации реагирующих веществ в одной и той же фазе во время этой стадии могут быть более низкими, чем оптимальные, для достижения удобных скоростей реакции, и, следовательно, применение катализатора межфазного переноса часто может быть выгодным для снижения температур и времени реакции. Например, при использовании катализатора межфазного переноса эту реакцию можно проводить при 100oC со временем реакции 28 часов. Соответственно, при температуре реакции 130oC и использовании катализатора межфазного переноса время реакции уменьшается до 2-4 часов, в то время как требуется 3-4 часа в отсутствие катализатора межфазного переноса. Тем не менее должно быть понятно, что данное изобретение предполагает, что твердый NaOH или KOH могут быть использованы одни, т.е. без применения катализатора межфазного переноса.
Существуют два принципиальных типа катализаторов межфазного переноса на основе их способа действия. Первый тип включает в себя соли четвертичного аммония или фосфония, тогда как второй тип включает в себя краун-эфиры и другие криптанды. Соли четвертичного аммония могут включать в себя, кроме более типичных алифатических конфигураций, соединения, в которых кватернизованный атом азота является частью гетероциклической циклической системы, например соли пиридиния или соли хининия. Первый тип катализатора межфазного переноса, т. е. соли четвертичного аммония или соли фосфония, является предпочтительным для использования в качестве катализатора межфазного переноса в способах получения данного изобретения. Среди катализаторов этого типа более предпочтительными являются соли четвертичного аммония, а среди них наиболее предпочтительные катализаторы межфазного переноса включают выбранный из группы, включающей тетра-н-бутиламмонийбромид (TBAB); тетра-н-бутиламмонийхлорид (TBAC); тетра-н-бутиламмонийгидроксид (TBAH); тетра-н-бутиламмониййодид (TBAI) и гидрат тетраэтиламмонийхлорида.
Должно быть понятно, что, несмотря на вышеуказанные предпочтения в отношении конкретных катализаторов межфазного переноса, которые выбирают для использования в способах получения по данному изобретению, имеется значительное количество катализаторов межфазного переноса, известных в данной области и пригодных для использования в данном изобретении. Специалисту хорошо известны идентичность таких катализаторов межфазного переноса, а также подходящие стадии, при помощи которых может быть продемонстрирована их эффективность в способах получения данного изобретения. Например, среди катализаторов межфазного переноса, известных в данной области, следующие катализаторы пригодны для использования в способах получения данного изобретения: цетилтриметиламмонийбромид (CTMAB); дибензо-18-краун-6 (DB-18-c-6); дициклогексано-18-краун-6 (DC-18-c-6); 18-краун-6 (18-c-6); (- )-N-додецил-N-метилэфедринийбромид (DMCOH); гексаметилтриамид фосфорной кислоты (HMPT); цетилпиридинийбромид (NCPB); N-бензилхининийхлорид (QUIBEC); тетра-н-бутиламмонийбромид (TBAB); тетра-н-бутиламмонийхлорил (TBAC); тетра-н-бутиламмонийгидроксид (TBAH); тетра-н-бутиламмонийгидросульфат (TBAHS); тетра-н-бутиламмониййодид (TBA1); гидрат тетраэтиламмонийхлорида (TEAC); три-н-бутиламин (TBA); бензилтрибутиламмонийбромид (TBBAB); гексадецилтрибутилфосфонийбромид (TBHDPB); бензилтриэтиламмонийбромид (TEBAB); бензилтриэтиламмонийхлорид (TEBA); гексадецилтриэтиламмонийхлорид (TEHDAC); тетраметиламмонийхлорид (TMAC); гексадецилтриметиламмонийхлорид (TMHDAC) и октилтриметиламмонийхлорид (TMOAC).
Основным механизмом действия систем катализаторов межфазного переноса является непрерывное образование липофильных ионных пар желаемых анионов с липофильными катионами, обеспечиваемыми этими катализаторами. В результате эти анионы способны входить в неполярные органические среды, в которых может иметь место желательная реакция. Типичными источниками липофильных катионов, которые могут действовать как катализаторы в таких системах, являются тетраалкиламмониевые или другие -ониевые соли, краун-эфиры, криптанды, простые эфиры полиэтиленгликоля и т.д. Фундаментальной природой межфазного катализа является образование липофильных ионных пар, которое может быть обнаружено в неполярных средах. С высоколипофильными катионами даже небольшие неорганические анионы образуют такие ионные пары. Межфазный катализ может действовать только в гетерогенных, большей частью двухфазных системах. В таких системах органическая фаза содержит органические реагирующие вещества и катализатор, например липофильный тетраалкиламмонийхлорид, тогда как водная, или в общем неорганическая, фаза содержит соли желательных анионов или основание, которое может образовывать органические анионы из соответствующих предшественников, локализованных в органической фазе.
В этих системах катализ состоит из переноса анионов из неорганической фазы или альтернативно органических анионов, образующихся на поверхности раздела, в органическую фазу, где они вступают в желательную реакцию, тогда как высвободившийся катализатор может приносить другой анион в органическую фазу. Путем непрерывного повторения этого действия 1 моль катализатора может промотировать превращение ≥ 100 моль реагирующих веществ. В зависимости от состояния агрегации, типов анионов и некоторых других факторов можно дифференцировать несколько вариантов вышеописанного межфазного каталитического процесса. Несмотря на это, специалист в данной области сможет легко приспособить основные требования для проведения межфазного катализа к способам получения данного изобретения.
Теперь вернемся к описанию способов получения данного изобретения. После образования вышеописанной смеси ее нагревают до температуры кипячения с обратным холодильником в атмосфере азота. В большинстве условий окружающей среды температура кипячения с обратным холодильником данной реакционной смеси будет находиться в диапазоне от 120 до 140oC, обычно от 125oC до 135oC и наиболее часто 130oC.
Реакционную смесь необходимо нагревать при более низких перечисленных температурах в течение значительного периода времени, от 12 до 30 часов, предпочтительно от 16 до 24 часов, наиболее предпочтительно от 18 до 20 часов. Однако при более высоких указанных температурах реакция протекает более быстро, и необходимо нагревать реакционную смесь в течение более коротких периодов времени, от 1/2 до 4 часов, обычно от 3/4 до 3 часов и наиболее типично от 1 до 2 часов.
Выбор подходящих температуры и времени проведения реакции до завершения находится в пределах квалификации специалиста, знакомого со способами органического синтеза. Выделение продукта из вышеописанного процесса, например вакуум-фильтрацию, промывание водой и сушку в вакуумном термостате, осуществляют с использованием общепринятых процедур, которые также находятся в пределах обычной квалификации в данной области. В качестве дополнительного руководства для специалиста здесь дается табл. 1 величин, показывающая различные результаты выхода способа, которые могут быть получены с опосредованными твердым гидроксидом натрия замещениями арилфторида 2-метилимидазолом.
Следует отметить, что в вышеописанном способе по данному изобретению одним их ключевых реагирующих веществ является соединение формулы (2.0.0)
Это соединение является также новым промежуточным продуктом данного изобретения, тетрагидро-4-[3-(4-фторфенил)тио] фенил-2H-пиран-4-карбоксамидом. Для проведения вышеописанного способа по данному изобретению необходимо, следовательно, разработать способ, при помощи которого может быть получено само это новое реагирующее вещество/промежуточный продукт. Таким образом, далее следует описание другого способа данного изобретения, посредством которого может быть получено соединение формулы (2.0.0).
Данное изобретение относится далее к способу получения соединения формулы (2.0.0)
Один из предпочтительных способов данного изобретения для получения нового промежуточного продукта формулы (2.0.0) может иллюстрироваться следующей схемой синтеза (10.0.1) (см. в конце описания), где X, M и R5 имеют указанные в другом месте описания значения.
Таким образом, вышеуказанный способ по данному изобретению, иллюстрируемый в схеме синтеза (10.0.1), может проводиться путем (a) образования реакционной смеси, состоящей из
(1) тетрагидро-4-(3-бром- или йодфенил)-2H-пиран-4-нитрила формулы (3.0.0)
где X обозначает бром или йод; и (2) 4-фтортиофенола формулы (4.0.0)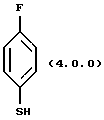
(3) в растворителе, состоящем из алифатического спирта с прямой или разветвленной цепью, имеющего в целом 2-7 атомов углерода, необязательно в виде его водной смеси; и, более предпочтительно, если этот спирт является вторичным спиртом, выбранным из группы, состоящей из изопропилового спирта, втор-бутилового спирта, изопентилового спирта и 2-гептанола, необязательно, в виде водной смеси указанного вторичного спирта;
(4) в присутствии сильного основания формулы (5.0.0):
M-O-R5,
где M обозначает щелочной металл, элемент группы 1/Ia, выбранный из группы, состоящей из лития, Li; натрия, Na; калия. K; рубидия, Rb; и цезия, Cs; и
R5 обозначает водород, H; или (C1-C4) алкил с прямой или разветвленной цепью; предпочтительно представитель, выбранный из группы, включающей гидроксид лития, LiOH; гидроксид натрия, NaOH; гидроксид калия, KOH; гидроксид рубидия, RbOH; гидроксид цезия, CsOH; метоксид лития, LiOCH3; метоксид натрия, NaOCH3; метоксид калия, KOCH3; метоксид рубидия, RbOCH3; метоксид цезия, CsOCH3; этоксид лития, LiOCH2CH3; этоксид натрия, NaOCH2CH3; этоксид калия, KOCH2CH3; этоксид рубидия, RbOCH2CH3; этоксид цезия, CsOCH2CH3; трет-бутоксид лития, LiOC(CH3)3; трет-бутоксид натрия NaOC(CH3)3; трет-бутоксид калия KOC(CH3)3; трет-бутоксид рубидия RbOC(CH3)3 и трет-бутоксид цезия CsOC(CH3)3, в том числе их смеси; и, кроме того, (5) в присутствии катализатора, являющегося металлом переходной группы, включающего в себя комплекс металла палладия, который предпочтительно выбран из группы, включающей
тетракис(трифенилфосфин)палладий (0), [(C6H5)3P]4Pd (0);
тетракис(метилдифенилфосфин)палладий (0), [(C6H5)2PCH3]4Pd (0);
транс-дихлорбис(метилдифенилфосфин)палладий (II), [(C6H5)2PCH3]2PdCl2; аддукт(дихлорбис[метиленбис(дифенилфосфин)]дипалладий-дихлорметан;
дихлорбис(трифенилфосфин)палладий (II), [(C6H5)3P]2PdCl2;
аддукт трис(дибензилиденацетон)дипалладий (0) - хлороформ, (C6H5CH=CHCOCH=CHC6H5)3Pd2• CHCl3;
бис(дибензилиденацетон)палладий (0), (C6H5CH=CHCOCH=CHC6H5)2Pd;
[1,1'-бис(дифенилфосфино)ферроцен] дихлорпалладий (II), комплекс с дихлорметаном;
[1,2-бис(дифенилфосфино)этан]палладий (II) и
димер (π-аллил)палладий (II) хлорида; с последующим (b) нагреванием указанной реакционной смеси, предпочтительно при кипячении с обратным холодильником, предпочтительно в течение периода от 12 до 36 часов, более предпочтительно от 18 до 24 часов; в результате чего образуется вышеуказанное соединение формулы (2.0.0), которое необязательно выделяют при помощи общепринятых способов разделения.
Вышеописанный способ является способом, который дает асимметрично замещенный диариловый простой эфир. В то же самое время реакция, которая происходит, приводит также к гидролизу нитрильного заместителя до соответствующего карбоксамидного заместителя. Было обнаружено, что несколько факторов являются важными в гарантии завершения вышеописанного процесса с приемлемыми выходами нового промежуточного продукта формулы (2.0.0).
Одним из таких факторов является растворитель, в котором проводят рассматриваемую реакцию. Растворитель состоит из алифатического спирта с прямой или разветвленной цепью, имеющего в целом от 2 до 7 атомов углерода. Спиртовой растворитель может также использоваться в смеси с водой, т.е. в виде водной смеси спирта в подходящих соотношениях. Хотя спирт и вода являются смешивающимися почти во всех соотношениях, было обнаружено, что желательно поддерживать отношение объем:объем спирта к воде соответственно в диапазоне от 25:1 до 3:1, предпочтительно в диапазоне от 10:1 до 5:1.
Было обнаружено, что наиболее подходящим алифатическим спиртом с прямой или разветвленной цепью, имеющим в целом от 2 до 7 атомов углерода, для использования в качестве растворителя в способе данного изобретения является вторичный спирт, выбранный из группы, включающей изопропиловый спирт, втор-бутиловый спирт, изопентиловый спирт и 2-гептанол. Из этих предпочтительных вторичных спиртов наиболее предпочтительным является изопропиловый спирт. Вышеуказанные вторичные спирты используют также необязательно в виде водной смеси, как описано подробно выше.
Должно быть понятно, что температура реакции, используемая в вышеописанном способе данного изобретения, может регулироваться выбором спиртового растворителя в зависимости, в свою очередь, от степени реакционной способности субстрата. Например, для реагирующего вещества формулы (3.0.0), где X обозначает йод, было обнаружено, что эта реакция может проводиться в мягких условиях в изопропиловом спирте при кипячении с обратным холодильником. Для реагирующего вещества формулы (3.0.0), где X обозначает бром, было обнаружено, что эта реакция может проводиться в мягких условиях во втор-бутиловом спирте при кипячении с обратным холодильником. Должно быть также понятно, что реакция с участием арилйодида в вышеописанном способе данного изобретения, т. е. когда X обозначает йод в реагирующем веществе формулы (3.0.0), протекает быстро и может быть завершена в пределах периода нескольких часов. С другой стороны, реакция с участием арилбромида, т.е. когда X обозначает бром в реагирующем веществе формулы (3.0.0), протекает более медленно, чем реакция с участием арилйодида, и для завершения реакции требуется нагревание реакционной смеси в течение значительно более продолжительного периода времени, более 10 часов. Однако длительное нагревание реакционной смеси в случае любой реакции не оказывает неблагоприятного действия на выход полученного диарилтиоэфира, т.е. диарилсульфида.
Другим подобным фактором является использование сильного основания формулы (5.0.0)
M-O-R5,
где M обозначает щелочной металл, элемент группы 1/Ia, выбранный из группы, включающей литий, Li; натрий, Na; калий, K; рубидий, Rb; и цезий, Cs; и R5 обозначает водород, H; или (C1-C4)алкил с прямой или разветвленной цепью. Предпочтительные сильные основания включают представитель, выбранный из группы, включающей гидроксид лития, LiOH; гидроксид натрия, NaOH; гидроксид калия, KOH; гидроксид рубидия, RbOH; гидроксид цезия, CsOH; метоксид лития, LiOCH3; метоксид натрия, NaOCH3; метоксид калия, KOCH3; метоксид рубидия, RbOCH3; метоксид цезия, CsOCH3; этоксид лития, LiOCH2CH3; этоксид натрия, NaOCH2CH3; этоксид калия, KOCH2CH3; этоксид рубидия, RbOCH2CH3; этоксид цезия, CsOCH2CH3; трет-бутоксид лития, LiOC(CH3)3; трет-бутоксид натрия NaOC(CH3)3; трет-бутоксид калия KOC(CH3)3; трет-бутоксид рубидия RbOC(CH3)3 и трет-бутоксид цезия CsOC(CH3)3.
Вышеуказанные сильные основания могут быть использованы в виде их смесей, но предпочтительно использовать только одно сильное основание. Более предпочтительными среди вышеуказанных сильных оснований являются гидроксид натрия, NaOH; гидроксид калия, KOH; этоксид натрия, NaOCH2CH3 и трет-бутоксид калия KOC(CH3)3.
Еще одним фактором в достижении удовлетворительного завершения вышеописанного способа по данному изобретению является использование в качестве катализатора металла переходной группы, включающего комплексы металла палладия. Среди комплексов металла палладия, которые являются предпочтительными для применения в способе данного изобретения, находятся более предпочтительные разновидности таких катализаторов, которые используют в вышеописанном процессе. Эти более предпочтительные разновидности выбраны из группы, включающей тетракис(трифенилфосфин)палладий (0), [(C6H5)3P]4Pd (0);
тетракис (метилдифенилфосфин) палладий (0), [(C6H5)2PCH3]4Pd (0);
транс-дихлорбис(метилдифенилфосфин) палладий (II), [(C6H5)2PCH3]2PdCl2; аддукт (дихлорбис[метиленбис(дифенилфосфин)] дипалладий-дихлорметан формулы (6.0.0)
дихлорбис(трифенилфосфин)палладий (II), [(C6H5)3P]2PdCl2;
аддукт трис(дибензилиденацетон)дипалладий (0) - хлороформ: (C6H5CH= CHCOCH=CHC6H5)3Pd2• CHCl3;
бис(дибензилиденацетон)палладий (0): (C6H5CH=CHCOCH=CHC6H5)2Pd;
[1,1'-бис(дифенилфосфино)ферроцен] дихлорпалладий (II), комплекс с дихлорметаном, формулы (6.1.0)
бис[1,2-бис(дифенилфосфино)этан]палладий (II) формулы (6.2.0)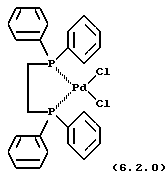
димер (π-аллил) палладий (II) хлорида формулы (6.3.0)
Из комплексов металла палладия, описанных выше, наиболее предпочтительным является тетракис(трифенилфосфин)палладий (0), [(C6H5)3P]4Pd (0). Этот предпочтительный катализатор может быть использован с лигандом или без лиганда. При использовании лиганда с [(C6H5)3P]4Pd (0) предпочтительными лигандами являются трифенилфосфин (TPP), этиленбис (дифенилфосфин) и три(2-толил)фосфин. Предпочтительное отношение катализатора к лиганду равно приблизительно 1: 2 мол. экв. , но специалисту понятно, что использование избыточных количеств лиганда может привести к уменьшению общего выхода реакции, в которой используется такой лиганд. Таким же образом, другие комплексы металла палладия, используемые в качестве катализаторов в способах данного изобретения, используют как с лигандом, так и без лиганда. Использование лиганда может влиять на выход конечного продукта, т.е. соединения формулы (2.0.0), как иллюстрируется табл. 2 величин, непосредственно после указания выходов из вышеописанного процесса данного изобретения, где используют различные комплексы металла палладия без лиганда или с одним из множества лигандов.
Вышеописанные лиганды, а также другие лиганды, хорошо известные в данной области, могут быть использованы с комплексами металла палладия, используемыми в качестве катализаторов в способе данного изобретения.
Как указано дополнительно выше, особое преимущество вышеописанного способа заключается в том, что в ходе проведения реакции в описанных выше условиях, которые являются подходящими или предпочтительными, нитрильная группа молекулы соединения формулы (3.0.0) гидролизуется до соответствующей карбоксамидной группы, которая образуется в конечном продукте, соединении формулы (1.0.0). Тем не менее данное изобретение обеспечивает также альтернативный способ получения нового промежуточного продукта, соединения формулы (2.0.0), в котором указанная нитрильная часть молекулы сначала гидролизуется до соответствующего карбоксамида, образуя тем самым соединение формулы (3.1.0). После проведения этой стадии синтеза карбоксамидное соединение формулы (3.1.0) реагирует с фтортиофенольным соединением формулы (4.0.0) с образованием вышеуказанного нового промежуточного продукта формулы (2.0.0).
Дополнительно следует отметить, что вторую стадию вышеупомянутого альтернативного способа проводят по существу таким же образом, как это иллюстрировано в схеме 2 выше.
Следовательно, данное изобретение относится также к альтернативному способу получения соединения формулы (2.0.0)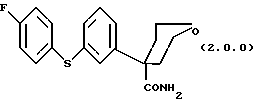
который может иллюстрироваться схемой синтеза (10.1.0) (см. в конце описания), где X, M и R5, все, имеют определенные в другом месте описания значения.
Альтернативный способ данного изобретения, показанный в схеме синтеза (10.1.0), может проводиться путем (a) образования реакционной смеси, состоящей из (1) тетрагидро-4-(3-бром- или йодфенил)-2H-пиран-4-нитрила формулы (3.0.0)
где X обозначает бром или йод; (2) в растворителе, состоящем из алифатического спирта с прямой или разветвленной цепью, имеющего в целом 2-7 атомов углерода, необязательно в виде его водной смеси; и более предпочтительно, если этот спирт является вторичным спиртом, выбранным из группы, состоящей из изопропилового спирта, втор-бутилового спирта, изопентилового спирта и 2-гептанола, необязательно в виде водной смеси указанного вторичного спирта; (3) в присутствии сильного основания формулы (5.0.0)
M-O-R5,
где M обозначает щелочной металл, элемент группы 1/Ia, выбранный из группы, состоящей из лития, Li; натрия, Na; калия, K; рубидия, Rb; и цезия, Cs; и R5 обозначает водород, H; или (C1-C4)алкил с прямой или разветвленной цепью; предпочтительно представитель, выбранный из группы, включающей гидроксид лития, LiOH; гидроксид натрия, NaOH; гидроксид калия, KOH; гидроксид рубидия, RbOH; гидроксид цезия, CsOH; метоксид лития, LiOCH3; метоксид натрия, NaOCH3; метоксид калия, KOCH3; метоксид рубидия, RbOCH3; метоксид цезия, CsOCH3; этоксид лития, LiOCH2CH3; этоксид натрия, NaOCH2CH3; этоксид калия, KOCH2CH3; этоксид рубидия, RbOCH2CH3; этоксид цезия, CsOCH2CH3; трет-бутоксид лития, LiOC(CH3)3; трет-бутоксид натрия NaOC(CH3)3; трет-бутоксид калия KOC(CH3)3; трет-бутоксид рубидия RbOC(CH3)3 и трет-бутоксид цезия CsOC(CH3)3, в том числе их смеси; с последующим (b) нагреванием указанной реакционной смеси, предпочтительно при кипячении с обратным холодильником, предпочтительно в течение периода от 3 до 8 часов, более предпочтительно от 5 до 6 часов; в результате чего образуется вышеуказанное соединение формулы (3.1.0)
где X обозначает бром или йод; с последующим (c) образованием реакционной смеси, состоящей из соединения формулы (3.1.0) и 4-фтортиофенола формулы (4.0.0)
(1) в растворителе, состоящем из алифатического спирта с прямой или разветвленной цепью, имеющего в целом 2-7 атомов углерода, необязательно в виде его водной смеси; и более предпочтительно, если этот спирт является вторичным спиртом, выбранным из группы, состоящей из изопропилового спирта, втор-бутилового спирта, изопентилового спирта и 2-гептанола, необязательно в виде водной смеси указанного вторичного спирта; (2) в присутствии сильного основания формулы (5.0.0)
M-O-R5,
где M обозначает щелочной металл, элемент группы 1/Ia, выбранный из группы, состоящей из лития, Li; натрия, Na; калия, K; рубидия, Rb; и цезия, Cs; и R5 обозначает водород, H; или (C1-C4) алкил с прямой или разветвленной цепью; предпочтительно представитель, выбранный из группы, включающей гидроксид лития, LiOH; гидроксид натрия, NaOH; гидроксид калия, KOH; гидроксид рубидия, RbOH; гидроксид цезия, CsOH; метоксид лития, LiOCH3; метоксид натрия, NaOCH3; метоксид калия, KOCH3; метоксид рубидия, RbOCH3; метоксид цезия, CsOCH3; этоксид лития, LiOCH2CH3; этоксид натрия, NaOCH2CH3; этоксид калия, KOCH2CH3; этоксид рубидия, RbOCH2CH3; этоксид цезия, CsOCH2CH3; трет-бутоксид лития, LiOC(CH3)3; трет-бутоксид натрия NaOC(CH3)3; трет-бутоксид калия KOC(CH3)3; трет-бутоксид рубидия RbOC(CH3)3 и трет-бутоксид цезия CsOC(CH3)3, в том числе их смеси; и, кроме того, (3) в присутствии катализатора, являющегося металлом переходной группы, включающего в себя представитель, независимо выбранный из группы, включающей комплекс металла палладия, который предпочтительно выбран из группы, включающей тетракис(трифенилфосфин) палладий (0), [(C6H5)3P]4Pd (0); тетракис(метилдифенилфосфин)палладий (0), [(C6H5)2PCH3] 4Pd (0); транс-дихлорбис(метилдифенилфосфин)палладий (II), [(C6H5)2PCH3]2PdCl2; аддукт дихлорбис[метиленбис(дифенилфосфин)]дипалладий-дихлорметан; дихлорбис(трифенилфосфин)палладий (II), [(C6H5)3P]2PdCl2; аддукт трис(дибензилиденацетон)дипалладий (0) - хлороформ, (C6H5CH= CHCOCH= CHC6H5)3Pd2• CHCl3; бис(дибензилиденацетон) палладий (0), (C6H5CH=CHCOCH= CHC6H5)2Pd; [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном; бис[1,2-бис(дифенилфосфино)этан] палладий (II) и димер (π-аллил)палладий (II) хлорида; с последующим (d) нагреванием указанной реакционной смеси, предпочтительно при кипячении с обратным холодильником, предпочтительно в течение периода от 5 до 15 часов, более предпочтительно от 8 до 10 часов; в результате чего образуется вышеуказанное соединение формулы (2.0.0).
Одним из ключевых аспектов способов получения данного изобретения является усовершенствованный способ получения известного ингибирующего 5-липоксигеназу соединения формулы (1.0.0)
Этот усовершенствованный способ включает в себя большинство предпочтительных вариантов данного изобретения и может иллюстрироваться схемой синтеза (10.3.1) (см. в конце описания).
Предполагается, что этот усовершенствованный способ данного изобретения, показанный в схеме синтеза (10.3.0), включает в себя в целом шесть вариантов данного изобретения. Первым вариантом является стадия a, которая является первой стадией, показанной в схеме синтеза (10.3.0), и представляет собой способ получения нового промежуточного продукта данного изобретения формулы (2.0.0). Вторым вариантом является стадия b, которая является второй или средней стадией, показанной на схеме синтезе (10.3.0), и представляет собой способ получения известного ингибирующего 5-липоксигеназу соединения формулы (1.0.0) в виде соединения per se. Третьим вариантом является стадия c или стадия d, которая является последней стадией в схеме синтеза (10.3.0) и представляет собой способ получения мезилатной соли указанного известного соединения формулы (1.0.0). Четвертым вариантом является стадия b + стадия c или d. Пятым вариантом является стадия a + стадия b. Шестым вариантом является стадия а + стадия b + стадия c или d.
Для краткости только второй и шестой варианты описаны подробно ниже. Таким образом, второй вышеупомянутый вариант, стадию b в схеме синтеза (10.3.1), проводят следующим образом: (a) образование реакционной смеси, состоящей из (1) тетрагидро-4-[3-(4-фторфенил)тио]фенил-2H-пиран-4-карбоксамида формулы (2.0.0)
и (2) 2-метилимидазола;
(3) в апротонном растворителе, предпочтительно выбранном из группы, включающей в основном гексан; 1,4-диоксан; тетрахлорид углерода; бензол; толуол; ксилолы; диэтиловый эфир; хлороформ; этилацетат; тетрагидрофуран (ТГФ); метиленхлорид; гексаметилтриамид фосфорной кислоты (HMPТ); нитрометан; N, N-диметилформамид (ДМФ); ацетонитрил; сульфолан и диметилсульфоксид (ДМСО); более предпочтительно диметилсульфоксид (ДМСО);
(4) в присутствии сильного основания в твердом виде, выбранного из группы, состоящей из гидроксида натрия, NaOH, и гидроксида калия, KOH; и (необязательно)
(5) в присутствии каталитического количества карбоната цезия, Cs2CO3, или катализатора межфазного переноса, предпочтительно выбранного из группы, включающей цетилтриметиламмонийбромид (CTMAB); дибензо-18-краун-6 (DB-18-c-6); дициклогексано-18-краун-6 (DC-18-c-6); 18-краун-6 (18-c-6); (-)-N-додецил-N-метилэфедринийбромид (DMCOH); гексаметилтриамид фосфорной кислоты (HMPT); цетилпиридинийбромид (NCPB); N-бензилхининийхлорид (QUIBEC);
бутиламмонийхлорид (TBAC); тетра-н-бутиламмонийгидроксид (TBAH); тетра-н-бутиламмонийгидросульфат (TBAHS); тетра-н-бутиламмониййодид (TBAI); гидрат тетраэтиламмонийхлорида, (TEAC); три-н-бутиламин (TBA); бензилтрибутиламмонийбромид (TBBAB); гексадецилтрибутилфосфонийбромид (TBHDPB); бензилтриэтиламмонийбромид (TEBAB); бензилтриэтиламмонийхлорид (TEBA); гексадецилтриэтиламмонийхлорид (TEHDAC); тетраметиламмонийхлорид (TMAC); гексадецилтриметиламмонийхлорид (TMHDAC) и октилтриметиламмонийхлорид (TMOAC) и более предпочтительно соли четвертичного аммония или соли фосфония, содержащей представитель вышеописанной группы; с последующим (b) нагреванием указанной реакционной смеси, предпочтительно при кипячении с обратным холодильником, предпочтительно при 115-145oC, более предпочтительно при 125-130oC, в атмосфере азота, предпочтительно в течение 12-30 часов, более предпочтительно в течение 17-24 часов; в результате чего образуется указанное соединение формулы (1.3.0).
Вышеупомянутый шестой вариант, стадия a + стадия b + стадия c схемы синтеза (10.3.0) по данному изобретению представляет собой способ получения по существу чистой мезилатной соли формулы (1.0.1)
предусматривающий (a) получение соединения формулы (2.0.0)
предусматривающее (1) образование реакционной смеси, состоящей из
(i) тетрагидро-4-(3-бромфенил)-2H-пиран-4-нитрила формулы (3.2.0)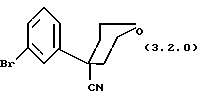
и (ii) 4-фтортиофенола формулы (4.0.0)
(iii) в растворителе, выбранном из группы, состоящей из изопропилового спирта, втор-бутилового спирта, изопентилового спирта и 2-гептанола, необязательно в виде его водной смеси;
(iv) в присутствии сильного основания, выбранного из группы, состоящей из гидроксида натрия, NaOH, и гидроксида калия, KOH; и, кроме того, (v) в присутствии катализатора, содержащего представитель, независимо выбранный из группы, включающей следующие комплексы металла палладия:
тетракис(трифенилфосфин)палладий (0), [(C6H5)3P]4Pd (0);
тетракис(метилдифенилфосфин)палладий (0), [(C6H5)2PCH3]4Pd (0);
транс-дихлорбис(метилдифенилфосфин)палладий (II), [(C6H5)2PCH3]2PdCl2; аддукт дихлорбис[метиленбис(дифенилфосфин)]дипалладий-дихлорметан;
дихлорбис(трифенилфосфин)палладий (II), [(C6H5)3P]2PdCl2;
аддукт трис(дибензилиденацетон)дипалладий (0) - хлороформ, (C6H5CH=CHCOCH-CHC6H5)3Pd2• CHCl3;
бис(дибензилиденацетон)палладий (0), (C6H5CH=CHCOCH=CHC6H5)2Pd;
[1,1'-бис(дифенилфосфино)ферроцен] дихлорпалладий (II), комплекс с дихлорметаном;
бис[1,2-бис(дифенилфосфино)этан]палладий (II) и
димер (π-аллил)палладий (II) хлорида; с последующим (2) нагреванием указанной реакционной смеси при температуре кипячения с обратным холодильником от 80 до 84oC в течение периода от 18 до 30 часов, предпочтительно 24 часов; в результате чего образуется вышеуказанное соединение формулы (2.0.0),
(b) образование реакционной смеси, состоящей из соединения формулы (2.0.0) и соединения формулы (1.3.10)
(1) в апротонном растворителе, выбранном из группы, состоящей в основном из тетрагидрофурана (ТГФ); метиленхлорида; N, N-диметилформамида (ДМФ) и диметилсульфоксида (ДМСО); более предпочтительно в диметилсульфоксиде (ДМСО);
(2) в присутствии сильного основания в твердой форме, выбранного из группы, состоящей из гидроксида натрия, NaOH, и гидроксида калия, KOH; и (необязательно) (3) в присутствии каталитического количества карбоната сезия, Cs2CO3, или катализатора межфазного переноса, предпочтительно члена, выбранного из группы, включающей цетилтриметиламмонийбромид (CTMAB); дибензо-18-краун-6 (DB-18-c-6); дициклогексано-18-краун-6 (DC-18-c-6); 18-краун-6 (18-c-6); (-)-N-додецил-N-метилэфедринийбромид (DMCOH); гексаметилтриамид фосфорной кислоты (HMPT); цетилпиридинийбромид (NCPB); N-бензилхининийхлорид (QUIBEC); тетра-н-бутиламмонийбромид (TBAB); тетра-н-бутиламмонийхлорид (TBAC); тетра-н-бутиламмонийгидроксид (TBAH); тетра-н-бутиламмонийгидросульфат (TBAHS); тетра-н-бутиламмониййодид (TBAI); тетраэтиламмонийхлорид, гидрат (TEAC); три-н-бутиламин (TBA); бензилтрибутиламмонийбромид (TBBAB); гексадецилтрибутилфосфонийбромид (TBHDPB); бензилтриэтиламмонийбромид (TEBAB); бензилтриэтиламмонийхлорид (TEBA); гексадецилтриэтиламмонийхлорид (TEHDAC); тетраметиламмонийхлорид (TMAC); гексадецилтриметиламмонийхлорид (TMHDAC) и октилтриметиламмонийхлорид (TMOAC) и более предпочтительно соли четвертичного аммония или соли фосфония, содержащей представитель вышеописанной группы; с последующим (c) нагреванием указанной реакционной смеси при кипячении с обратным холодильником, в атмосфере азота; с получением в результате соединения формулы (1.0.0)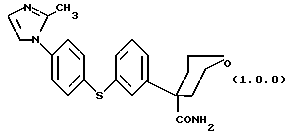
с последующим (d) образованием концентрированного раствора в метаноле указанного соединения формулы (1.0.0), который затем фильтруют предпочтительно через активированный уголь, после чего добавляют к фильтрату метансульфоновую кислоту, MeSO3H; с последующим дополнительным концентрированием и добавлением этилацетата ad seriatim до выделения кристаллического продукта, содержащего по существу чистую мезилатную соль формулы (1.0.1)
или альтернативно с последующим (e) образованием концентрированного раствора в метаноле указанного соединения формулы (1.0.0), к которому затем добавляют метансульфоновую кислоту, MeSO3H; с последующим фильтрованием этой смеси, предпочтительно через активированный уголь, после чего следует дополнительное концентрирование и добавление этилацетата ad seriatim до выделения кристаллического продукта, содержащего по существу чистую мезилатную соль формулы (1.0.1).
Предпочтительным способом образования мезилатной соли является способ образования концентрированного раствора в метаноле указанного соединения формулы (1.0.0), который также содержит метансульфоновую кислоту, MeSO3H, с последующим фильтрованием. Было обнаружено, что этот способ приводит к значительному уменьшению объемов по способу и уменьшению количества остаточного палладия в конечном продукте. Первичной целью вышеописанной перекристаллизации в метаноле в процессе образования мезилатной соли формулы (1.0.1) является удаление всего остаточного палладия из конечного продукта, который не был удален во время стадии фильтрования, проводимой предпочтительно с использованием активированного угля.
Следует учесть, что вышеописанный способ получения мезилатной соли соединения формулы (1.0.0) может быть легко приспособлен с использованием квалификации и знаний, доступных в данной области, для получения других, аналогичных сульфонатных солей соединения формулы (1.0.0), в частности тозилатной соли.
ПРИМЕРЫ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ
Способы, новые промежуточные продукты и новые конечные продукты данного изобретения будут более понятны из их иллюстрации в виде рабочих примеров, иллюстрирующих детали их проведения. Однако примеры предпочтительных вариантов данного изобретения, которые следуют ниже, предназначены только для целей иллюстрации и не должны восприниматься как ограничивающие каким-либо образом объем данного изобретения, и для цели ограничения служит только прилагаемая формула изобретения.
Пример 1. Синтез тетрагидро-4-(3-бромфенил)-2H-пиран-4-нитрила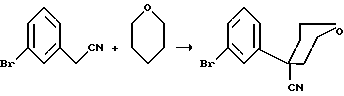
3-Бромфенилацетонитрил (20,0 г, 102 ммоль, 1 экв.), от фирмы Aldrich Chemical Co. Milwaukee, WI, тетрагидрофуран (120 мл), 40% водный раствор гидроксида натрия (180 мл, ммоль, экв.), гидросульфат тетрабутиламмония (3,46 г, ммоль, 0,1 экв.) перемешивали в реакционной установке с колбой для кипения при температуре кипения с обратным холодильником. После этого добавляли 2,2'-дихлордиэтиловый эфир (13,75 мл, 117, ммоль, 0,1 экв.) с перемешиванием при комнатной температуре, 20-25oC. Полученную реакционную смесь кипятили с обратным холодильником в течение 5-8 ч при приблизительно 64oC. Реакционную смесь охлаждали до температуры окружающей среды и добавляли этилацетат (154 мл). Нижний водный слой отделяли и органический слой упаривали до красного масла. К этому маслу добавляли изопропанол (100 мл) и воду (10 мл) и перемешивали при 0oC в течение ночи с получением кристаллической суспензии. Кристаллическую суспензию фильтровали в вакууме, промывали изопропанолом (2 х 20 мл). Белое кристаллическое твердое вещество сушили в вакууме при 40-45oC. Выход 18,57 г (68,4%): т.пл. 82-85oC; m/z 267 (m+1);
1H-ЯМР (300 МГц, ДМСО) δ 7,75 (с, 1H), 7,6 (м, 2H), 7,44 (т, 1H), 4,02 (м, 2H), 3,66 (м, 2H), 2,14 (м, 4H).
Пример 2. Синтез тетрагидро-4-[3-(4-фторфенил)тио] фенил-2H-пиран-4-карбоксамида
Пропан-2-ол (311 мл), тетрагидро-4-(3-бромфенил)-2H-пиран-4-нитрил (51,91 г, 0,195 моль, 1 экв.), гидроксид калия (25,16 г, 0,39 моль, 2 экв.), воду (4 мл, 0,39 моль, 2 экв.), тетракис(трифенилфосфин)палладий (0) (2,26 г, 0,00195 моль, 0,01 экв. ) и 4-фтортиофенол (25 г, 0,195 моль, 1 экв.) добавляли в реакционную установку с колбой для кипения с обратным холодильником в атмосфере азота. Полученную реакционную смесь кипятили с обратным холодильником в течение 20-24 ч при приблизительно 82oC. Реакционную смесь охлаждали до температуры окружающей среды, 20-25oC, и добавляли воду (315 мл) с получением суспензии. Сырой продукт выделяли фильтрованием и промывали смесью 1:1 вода:пропан-2-ол (125 мл) и отсасывали досуха. Неочищенный сухой продукт растворяли в метаноле (1900 мл), обрабатывали активированным углем, Darco KB-B (2,5 г) и ускорителем фильтрования Celite (10 г) при температуре кипения с обратным холодильником приблизительно 60oC в течение 20 минут, и отфильтровывали продукт, не содержащий активированный уголь и ускоритель фильтрования. Осадок на фильтре промывали горячим метанолом (200 мл) и промывной раствор соединяли с основным фильтратом. Продукт, содержащий объединенные фильтрат и промывной раствор, концентрировали перегонкой до объема приблизительно 700 мл. Концентрат охлаждали до 10-0o, гранулировали в этом диапазоне температур в течение 1-3 ч с получением кристаллов. Кристаллический продукт выделяли фильтрованием, промывали холодным метанолом (125 мл) и сушили под вакуумом при 40-45oC. Выход 40,2 г (62,2%): т.пл. 175-178oC; m/z 332 (m+1); 1H-ЯМР (300 МГц, ДМСО) δ 7,37 (м, 8H), 7,11 (м, 2H), 3,60 (м, 2H), 2,30 (м, 2H), 2,40 (м, 2H), 1,77 (м, 2H); ИК (смещения) vmax 3394, 3198, 3078, 3014, 2970, 2931, 2880, 2824, 1681, 1664, 1664, 1623, 1588, 1569.
Пример 3. Синтез тетрагидро-4-{3-[4-(2-метил-1H-имидазол-1- ил)фенил] тио}фенил-2H-пиран-4-карбоксамида с использованием твердого NaOH и Cs2CO3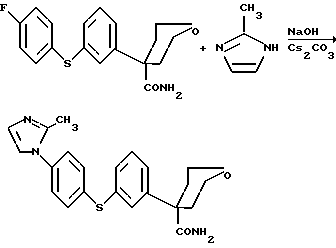
Тетрагидро-4-[3-(4-фторфенил)тио] фенил-2H-пиран-4-карбоксамид (25,0 г, 75,4 ммоль, 1 экв. ), диметилсульфоксид (250 мл, 10 об.), 2-метилимидазол (12,39 г, 150,9 ммоль, 2,0 экв.), гидроксид натрия (6,03 г, 150,9 ммоль, 2,0 экв. ) и карбонат цезия (1,23 г, 0,38 ммоль, 0,005 экв.) добавляли в реакционную установку с колбой для кипячения с обратным холодильником в атмосфере азота и реакционную смесь нагревали при 125-130oC в течение 17-24 ч в атмосфере азота. По завершении реакции ее охлаждали (<30oC) и гасили водой (250 мл, 10 об.), что приводило к образованию осадка. Во время добавления воды наблюдали экзотерму 10-15oC. Образовавшуюся таким образом суспензию охлаждали до комнатной температуры (15-25oC) и затем гранулировали в течение 1 часа. Продукт выделяли фильтрацией в вакууме и промывали водой (140 мл, 5,6 об. ). Продукт сушили в течение ночи в вакуумном термостате при 40-45oC. Количество полученного продукта было 29,4 г, что соответствует выходу 99%. Аналитические данные для продукта следующие: т.пл. 198-200oC; m/z 396 (m+1); 1H-ЯМР (300 МГц, ДМСО) δ 7,41 (м, 10H), 7,12 (с, 1H), 6,93 (д, 1H), 3,75 (м, 2H), 3,48 (т, 2H), 2,48 (д, 2H), 2,3 (с, 3H), 1,75 (м, 2H); ИК (смещения) vmax 3402, 3301, 3123, 3096, 2971, 2930, 2880, 1680, 1663, 1622, 1593, 1569, 1528.
Пример 4. Синтез тетрагидро-4-{3-[4-(2-метил-1H-имидазол-1- ил)фенил] тио} фенил-2H-пиран-4-карбоксамида с использованием твердого NaOH и катализатора межфазного переноса
Тетрагидро-4-[3-(4-фторфенил)тио] фенил-2H-пиран-4-карбоксамид (25,0 г, 75,4 ммоль, 1 экв. ), диметилсульфоксид (250 мл, 10 об.), 2-метилимидазол (12,39 г, 150,9 ммоль, 2,0 экв.), гидроксид натрия (6,03 г, 150,9 ммоль, 2,0 экв. ) и тетра-н-бутиламмонийхлорид (TBAC) (0,210 г, 0,75 ммоль, 0,05 экв.) добавляли в реакционную установку с колбой для кипячения с обратным холодильником в атмосфере азота и реакционную смесь нагревали при 125-130oC в течение 17-24 ч в атмосфере азота. По завершении реакции ее охлаждали (<30oC) и гасили водой (250 мл, 10 об.), что приводило к образованию осадка. В процессе добавления воды наблюдали экзотерму 10-15oC. Образовавшуюся таким образом суспензию охлаждали до комнатной температуры (15-25oC) и затем гранулировали в течение 1 часа. Продукт выделяли фильтрованием в вакууме и промывали водой (140 мл, 5,6 об.). Продукт сушили в течение ночи в вакуумном термостате при 40-45oC. Количество полученного продукта было 27,6 г, что соответствует выходу 93,0%. Аналитические данные для продукта были следующими: т.пл. 198-200oC; m/z 396 (m+1); 1H-ЯМР (300 МГц, ДМСО) δ 7,41 (м, 10H), 7,12 (с, 1H), 6,93 (д, 1H), 3,75 (м, 2H), 3,48 (т, 2H), 2,48 (д, 2H), 2,3 (с, 3H), 1,75 (м, 2H); ИК (смещения) vmax 3402, 3301, 3123, 3096, 2971, 2930, 2880, 1680, 1663, 1622, 1593, 1569, 1528.
Пример 5. Синтез тетрагидро-4-{3- [4-(2-метил-1H-имидазол-1- ил)фенил] тио}фенил-2H-пиран-4-карбоксамида с использованием только твердого NaOH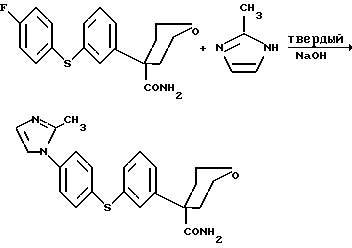
Тетрагидро-4-[3-(4-фторфенил)тио] фенил-2H-пиран-4-карбоксамид (6,5 г, 19,6 ммоль, 1 экв. ), диметилсульфоксид (65 мл, 10 об.), 2-метилимидазол (3,22 г, 39,23 ммоль, 2,0 экв.) и гидроксид натрия (1,57 г, 39,23 ммоль, 2,0 экв. ) добавляли в реакционную установку с колбой для кипячения с обратным холодильником в атмосфере азота и реакционную смесь нагревали при 125-130oC в течение 4-6 ч в атмосфере азота. По завершении реакции ее охлаждали (<30oC) и гасили водой (65 мл, 10 об.), что приводило к образованию осадка. В процессе добавления воды наблюдали экзотерму 10-15oC. Образовавшуюся таким образом суспензию охлаждали до комнатной температуры (15-25oC) и затем гранулировали в течение 1 часа. Продукт выделяли фильтрованием в вакууме и промывали водой (80 мл, 12,3 об.). Продукт сушили в течение ночи в вакуумном термостате при 40-45oC. Количество полученного продукта было 6,98 г, что соответствовало выходу 90,4%. Аналитические данные для продукта следующие: т. пл. 198-200oC; m/z 396 (m+1); 1H-ЯМР (300 МГц, ДМСО) δ 7,41 (м, 10H), 7,12 (с, 1H), 6,93 (д, 1H), 3,75 (м, 2H), 3,48 (т, 2H), 2,48 (д, 2H), 2,3 (с, 3H), 1,75 (м, 2H); ИК (смещения) vmax 3402, 3301, 3123, 3096, 2971, 2930, 2880, 1680, 1663, 1622, 1593, 1569, 1528.
Пример 6. Образование мезилатной соли тетрагидро-4-{3-[4-(2-метил-1H-имидазол-1-ил)фенил]тио}фенил-2H- пиран-4-карбоксамида
Метанол (640 мл, 40 об.), тетрагидро-4-{3-[4-(2-метил-1H-имидазол-1-ил)фенил] тио}фенил-2H- пиран-4-карбоксамид, полученный по способу примера 3 (16,0 г, 40,7 ммоль, 1,0 экв.), активированный уголь, DARCO KB-B (0,80 г), и ускоритель фильтрования, целит (2,4 г), добавляли в реакционную установку с колбой для кипячения с обратным холодильником. Смесь нагревали до кипения, приблизительно 66oC, для растворения органического субстрата. Содержимое колбы охлаждали до температуры в диапазоне 55-60oC и уголь и ускоритель фильтрования удаляли фильтрованием в диапазоне температур 55-60oC. Остаток промывали метанолом (50 мл) и промывные растворы соединяли с первоначальным фильтратом. Полученные таким образом прозрачные объединенные фильтрат и промывные растворы концентрировали перегонкой при атмосферном давлении до объема приблизительно 700 мл. К концентрированному раствору в метаноле добавляли метансульфоновую кислоту (4,1 г, 42,7 ммоль, 1,05 экв.). Полученный раствор дополнительно концентрировали перегонкой при атмосферном давлении до объема приблизительно 250 мл и добавляли этилацетат (500 мл) в виде двух аликвот, общий объем уменьшали перегонкой до 250 мл после каждого добавления этилацетата. Полученную кристаллическую суспензию охлаждали до комнатной температуры 15-25oC и гранулировали 4-16 ч в диапазоне температур 15-25oC. Белый кристаллический продукт выделяли фильтрованием и промывали этилацетатом (135 мл) и сушили в вакууме при 45-50oC. Выход 18,39 г, 92,3%. Полученную таким образом соль характеризовали картиной дифракции рентгеновских лучей порошка с главными пиками, представленными в таблице 3.
Пример 7. Перекристаллизация тетрагидро-4-{3-[4-(2-метил-1H-имидазол-1-ил)фенил]тио}фенил-2H- пиран-4-карбоксамида.
Метанол (3200 мл, 40 об.), тетрагидро-4-{3-[4-(2-метил-1H-имидазол-1-ил)фенил] тио}фенил-2H- пиран-4-карбоксамид, полученный по способу примера 3 (80,2 г), активированный уголь, DARCO KB-B (4,0 г), и ускоритель фильтрования, целит, (10 г) добавляли в реакционную установку с колбой для кипячения с обратным холодильником. Смесь нагревали до кипения, приблизительно 66oC, для растворения органического субстрата. Содержимое колбы охлаждали до температуры в диапазоне 55-60oC и уголь и ускоритель фильтрования удаляли фильтрованием в диапазоне температур 55-60oC. Остаток промывали метанолом (300 мл) и промывные растворы соединяли с первоначальным фильтратом. Полученные таким образом прозрачные объединенные фильтрат и промывные растворы концентрировали перегонкой при атмосферном давлении до объема приблизительно 1000 мл. Полученный таким образом метанольный концентрат охлаждали до диапазона температур 3-7oC для получения кристаллизации продукта и гранулировали в течение 6-24 часов в этом диапазоне температур. Белые кристаллы продукта выделяли фильтрованием и стушили в вакууме при 40-45oC. Выход 70,3 г, 87,7%, т.пл. 198-200oC; m/z 396 (m+1); спектральные данные идентичны данным примера 3.
Пример 8. Синтез тетрагидро-4-(3-бромфенил)-2H-пиран-4-карбоксамида
Пропан-2-ол (100 мл), тетрагидро-4-(3-бромфенил)-2H-пиран-4-нитрил (20,0 г, 0,075 моль, 1 экв.), гидроксид калия (13,74 г, 0,245 моль, 3,26 экв.) добавляли в реакционную установку с колбой для кипячения с обратным холодильником в атмосфере азота, и реакционную смесь нагревали при перемешивании при кипячении с обратным холодильником, приблизительно 82oC, в течение 5-6 часов в атмосфере азота. По завершении реакции смесь охлаждали (до <30oC) и гасили водой (100 мл). Полученную суспензию фильтровали и остаток-продукт промывали водой (30 мл) и сушили в вакууме при 45-50oC с получением белого твердого вещества. Выход 19,05 г, 89,2%, т.пл. 245-247oC; m/z 285 (m+1); 1H-ЯМР (300 МГц, ДМСО) δ 7,43 (м, 5H), 7,14 (с, 1H), 3,76 (д, 2H), 3,47 (т, 2H), 2,44 (д, 2H), 1,79 (м, 2H); ИК (смещения) vmax 3363, 3174, 3062, 2973, 2935, 2879, 2828, 1685, 1631, 1588.
Пример 9. Синтез тетрагидро-4-[3-(4-фторфенил)тио] фенил-2H-пиран-4-карбоксамида
Бутан-1-ол (8 мл), тетрагидро-4-(3-бромфенил)-2H-пиран-4-карбоксамид (2,97 г, 10,45 ммоль, 1 экв.) трет-бутоксид калия (2,34 г, 20,9 ммоль, 2 экв. ), воду (4 мл, 0,39 моль, 1 экв.), тетракис(трифенилфосфин)палладий (0) (0,242 г, 0,209 ммоль, 0,02 экв.) и 4-фтортиофенол (1,34 г, 10,45 ммоль, 1 экв. ) добавляли в реакционную установку с колбой для кипячения с обратным холодильником в атмосфере азота. Полученную реакционную смесь нагревали при приблизительно 100oC в течение 8-10 ч до завершения реакции. Реакционную смесь охлаждали до температуры окружающей среды, 20-25oC, и добавляли бутан-1-ол (10 мл) для получения суспензии. Неочищенный продукт выделяли фильтрованием и промывали бутан-1-олом (3 мл) и отсасывали досуха. Неочищенный сухой продукт перемешивали в метаноле (15 мл), полученную суспензию фильтровали и фильтровальный осадок (продукт) промывали метанолом (5 мл) и сушили в вакууме при 40-45oC. Частично очищенный продукт нагревали до кипячения с обратным холодильником в пропан-2-оле (45 мл) в течение 30 минут, охлаждали и полученную суспензию фильтровали и фильтровальный осадок (продукт) промывали пропан-2-олом (5 мл) и сушили в вакууме при 40-45oC. Полученное твердое вещество (3,22 г) дополнительно очищали перемешиванием в тетрагидрофуране (240 мл) при 20-25oC. Нерастворимые твердые примеси удаляли фильтрованием и содержащий продукт фильтрат концентрировали до 20 мл и обрабатывали гептаном (20 мл). Полученную суспензию продукта фильтровали и осадок на фильтре промывали гептаном (8 мл) и сушили в вакууме при 40-45oC. Выход 1,62 г (46,8%): т.пл. 175-178oC; m/z 332 (m+l); спектральные данные одинаковы с данными примера 2.
Пример 10. Синтез тетрагидро-4-{3-[4-(1H-пиразол-1-ил)фенил]тио}фенил-2H-пиран-4-карбоксамида с использованием твердого NaOH и катализатора межфазного переноса
Тетрагидро-4-[3-(4-фторфенил)тио] фенил-2H-пиран-4-карбоксамид (5,0 г, 15,09 ммоль, 1 экв.), ДМСО (50 мл, 10 об.), пиразол (2,05 г, 30,17 ммоль, 2,0 экв. ), гидроксид натрия (1,21 г, 30,17 ммоль, 2,0 экв.) и тетрабутиламмонийхлорид (0,042 г, 0,151 ммоль, 0,01 экв.) перемешивали при 140oC в атмосфере азота в течение 2-6 часов. По завершении реакции ее охлаждали (<30oC) и гасили водой (50 мл, 10 об.). Это давало осадок и экзотерму 20-25oC. Погашенную реакционную смесь охлаждали до комнатной температуры и гранулировали в течение 1 часа, фильтровали в вакууме и промывали водой (28 мл, 5,6 об. ) с получением продукта. Затем продукт вновь суспендировали в воде (55 мл, 11 об.) при 60oC в течение 1 часа. Суспензии давали охладиться до комнатной температуры и перемешивали в течение 1 часа, затем фильтровали в вакууме и промывали водой (28 мл, 5,6 об.). Белые твердые вещества помещали в вакуумный термостат при 40oC на 24-48 ч. Количество полученного продукта было 5,42 г, что давало выход 95%. Анализ продукта подтвердил его предполагаемую структуру.
Пример 11. Синтез тетрагидро-4-{3-[4-(1H-имидазол-1- ил)фенил]тио}фенил-2H-пиран-4-карбоксамида с использованием твердого NaOH и катализатора межфазного переноса
Тетрагидро-4-[3-(4-фторфенил)тио] фенил-2H-пиран-4-карбоксамид (5,0 г, 15,09 ммоль, 1 экв.), ДМСО (50 мл, 10 об.), имидазол (2,05 г, 30,17 ммоль, 2,0 экв. ), гидроксид натрия (1,21 г, 30,17 ммоль, 2,0 экв.) и тетрабутиламмонийхлорид (0,042 г, 0,151 ммоль, 0,01 экв.) перемешивали при 140oC в атмосфере азота в течение 2-6 часов. По завершении реакции ее охлаждали (<30oC) и гасили водой (50 мл, 10 об.). Это давало осадок и экзотерму 20-25oC. Погашенную реакционную смесь охлаждали до комнатной температуры и гранулировали в течение 1 часа, фильтровали в вакууме и промывали водой (28 мл, 5,6 об. ) с получением продукта. Затем продукт вновь суспендировали в воде (55 мл, 11 об.) при 60oC в течение 1 часа. Суспензии давали охладиться до комнатной температуры и перемешивали в течение 1 часа, затем фильтровали в вакууме и промывали водой (28 мл, 5,6 об.). Белые твердые вещества помещали в вакуумный термостат при 40oC на 24-48 ч. Количество полученного продукта было 5,35 г, что давало выход 93%. Анализ продукта соответствовал его предполагаемой структуре.
Пример 12. Синтез тетрагидро-4-{3-[4-(1H-бензоимидазол-1- ил)фенил]тио} фенил-2H-пиран-4-карбоксамида с использованием твердого KOH и катализатора межфазного переноса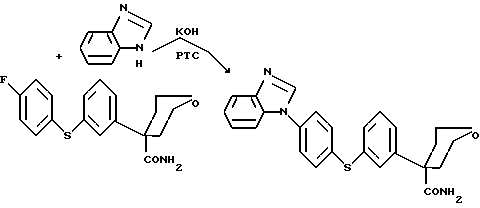
Тетрагидро-4-[3-(4-фторфенил)тио] фенил-2H-пиран-4-карбоксамид (5,0 г, 15,09 ммоль, 1 экв. ), ДМСО (50 мл, 10 об.), бензоимидазол (3,56 г, 30,17 ммоль, 2,0 экв. ), гидроксид калия (KOH содержал 12% воды) (1,96 г, 30,17 ммоль, 2,0 экв.) и тетрабутиламмонийхлорид (0,042 г, 0,151 ммоль, 0,01 экв.) перемешивали при 140oC в атмосфере азота в течение 2-6 часов. По завершении реакции смесь охлаждали (<30oC) и гасили водой (50 мл, 10 об.). Это давало осадок и экзотерму 20-25oC. Гашеную реакционную смесь охлаждали до комнатной температуры и гранулировали в течение 1 часа, фильтровали в вакууме и промывали водой (28 мл, 5,6 об.) с получением продукта. Затем продукт вновь суспендировали в воде (55 мл, 11 об.) при 60oC в течение 1 часа. Суспензии давали охладиться до комнатной температуры и перемешивали в течение 1 часа, затем фильтровали в вакууме и промывали водой (28 мл, 5,6 об.). Белые твердые вещества помещали в вакуумный термостат при 40oC на 24-48 ч. Количество полученного продукта было 6,34 г, что давало выход 98%. Анализ продукта согласовался с его предполагаемой структурой.
Пример 13. Синтез тетрагидро-4-{3-[4-(1H-пиразол-1-ил)фенил]тио}фенил-2H-пиран-4-карбоксамида с использованием только твердого KOH
Тетрагидро-4-[3-(4-фторфенил)тио] фенил-2H-пиран-4-карбоксамид (5,0 г, 15,09 ммоль, 1 экв.), ДМСО (50 мл, 10 об.), пиразол (2,05 г, 30,17 ммоль, 2,0 экв. ) и гидроксид калия (KOH содержал 12% воды) (1,96 г, 30,17 ммоль, 2,0 экв.) перемешивали при 140oC в атмосфере азота в течение 22-24 часов. По завершении реакции смесь охлаждали (<30oC) и гасили водой (50 мл, 10 об.). Это давало осадок и экзотерму 20-25oC. Гашеную реакционную смесь охлаждали до комнатной температуры и гранулировали в течение 1 часа, фильтровали в вакууме и промывали водой (28 мл, 5,6 об.) с получением продукта. Затем продукт вновь суспендировали в воде (55 мл, 11 об.) при 60oC в течение 1 часа. Суспензии давали охладиться до комнатной температуры и перемешивали в течение 1 часа, затем фильтровали в вакууме и промывали водой (28 мл, 5,6 об. ). Белые твердые вещества помещали в вакуумный термостат при 40oC на 24-48 ч. Количество полученного продукта было 5,53 г, что давало выход 97%. Анализ продукта подтвердил его предполагаемую структуру.
Пример 14. Синтез тетрагидро-4-{ 3-[4-(1H-имидазол-1-ил)фенил]тио}фенил-2H-пиран-4-карбоксамида с использованием только твердого NaOH
Тетрагидро-4-[3-(4-фторфенил)тио] фенил-2H-пиран-4-карбоксамид (5,0 г, 15,09 ммоль, 1 экв.), ДМСО (50 мл, 10 об.), имидазол (2,05 г, 30,17 ммоль, 2,0 экв. ) и гидроксид натрия (1,21 г, 30,17 ммоль, 2,0 экв.) перемешивали при 140oC в атмосфере азота в течение 22-24 часов. По завершении реакции смесь охлаждали (<30oC) и гасили водой (50 мл, 10 об.). Это давало осадок и экзотерму 20-25oC. Гашеную реакционную смесь охлаждали до комнатной температуры и гранулировали в течение 1 часа, фильтровали под вакуумом и промывали водой (28 мл, 5,6 об.) с получением продукта. Затем продукт вновь суспендировали в воде (55 мл, 11 об.) при 60oC в течение 1 часа. Суспензии давали охладиться до комнатной температуры и перемешивали в течение 1 часа, затем фильтровали в вакууме и промывали водой (28 мл, 5,6 об.). Белые твердые вещества помещали в вакуумный термостат при 40oC на 24-48 ч. Количество полученного продукта было 4,87 г, что давало выход 85%. Анализ продукта согласовался с его предполагаемой структурой.
Пример 15. Синтез тетрагидро-4-{3-[4-(1H-бензоимидазол-1- ил)фенил]тио} фенил-2H-пиран-4-карбоксамида с использованием только твердого NaOH
Тетрагидро-4-[3-(4-фторфенил)тио] фенил-2H-пиран-4-карбоксамид (5,0 г, 15,09 ммоль, 1 экв. ), ДМСО (50 мл, 10 об.), бензоимидазол (3,56 г, 30,17 ммоль, 2,0 экв. ) и гидроксид натрия (1,21 г, 30,17 ммоль, 2,0 экв.) перемешивали при 140oC в атмосфере азота в течение 22-24 часов. По завершении реакции ее охлаждали (<30oC) и гасили водой (50 мл, 10 об.). Это давало осадок и экзотерму 20-25oC. Гашенную реакционную смесь охлаждали до комнатной температуры и гранулировали в течение 1 часа, фильтровали под вакуумом и промывали водой (28 мл, 5,6 об.) с получением продукта. Затем продукт вновь суспендировали в воде (55 мл, 11 об.) при 60oC в течение 1 часа. Суспензии давали охладиться до комнатной температуры и перемешивали в течение 1 часа, затем фильтровали в вакууме и промывали водой (28 мл, 5,6 об.). Светлокоричневые твердые вещества помещали в вакуумный термостат при 40oC на 24-48 ч. Количество полученного продукта было 6,27 г, что давало выход 97%. Было показано, что анализ продукта согласовался с его предполагаемой структурой.
Пример 16. Синтез тетрагидро-4-{3-[4-(1H-пиразол-1-ил)фенил]тио}фенил-2H-пиран-4-карбоксамида с использованием твердого NaOH и катализатора Cs2CO3
Тетрагидро-4-[3-(4-фторфенил)тио] фенил-2H-пиран-4-карбоксамид (5,0 г, 15,09 ммоль, 1 экв.), ДМСО (50 мл, 10 об.), пиразол (2,05 г, 30,17 ммоль, 2,0 экв.), гидроксид натрия (1,21 г, 30,17 ммоль, 2,0 экв.) и карбонат цезия (катализатор) (0,246 г, 0,754 ммоль, 0,05 экв.) перемешивали при 140oC в атмосфере азота в течение 4-6 часов. По завершении реакции ее охлаждали (<30oC) и гасили водой (50 мл, 10 об.). Это давало осадок и экзотерму 20-25oC. Гашеную реакционную смесь охлаждали до комнатной температуры и гранулировали в течение 1 часа, фильтровали под вакуумом и промывали водой (28 мл, 5,6 об.) с получением продукта. Затем продукт вновь суспендировали в воде (55 мл, 11 об.) при 60oC в течение 1 часа. Суспензии давали охладиться до комнатной температуры и перемешивали в течение 1 часа, затем фильтровали в вакууме и промывали водой (28 мл, 5,6 об.). Белые твердые вещества помещали в вакуумный термостат при 40oC на 24-48 ч. Количество полученного продукта было 5,53 г, что давало выход 97%. Анализ продукта подтверждал его предполагаемую структуру.
Пример 17. Синтез тетрагидро-4-{ 3-[4-(1H-имидазол-1-ил)фенил]тио}фенил-2H-пиран-4-карбоксамида с использованием твердого KOH и катализатора Cs2CO3
Тетрагидро-4-[3-(4-фторфенил)тио] фенил-2H-пиран-4-карбоксамид (5,0 г, 15,09 ммоль, 1 экв.), ДМСО (50 мл, 10 об.), имидазол (2,05 г, 30,17 ммоль, 2,0 экв.), гидроксид калия (KOH содержал 12% воды) (1,96 г, 30,17 ммоль, 2,0 экв. ) и карбонат цезия (катализатор) (0,246 г, 0,754 ммоль, 0,05 экв.) перемешивали при 140oC в атмосфере азота в течение 4-6 часов. По завершении реакции смесь охлаждали (<30oC) и гасили водой (50 мл, 10 об.). Это давало осадок и экзотерму 20-25oC. Гашеную реакционную смесь охлаждали до комнатной температуры и гранулировали в течение 1 часа, фильтровали в вакууме и промывали водой (28 мл, 5,6 об.) с получением продукта. Затем продукт вновь суспендировали в воде (55 мл, 11 об.) при 60oC в течение 1 часа. Суспензии давали охладиться до комнатной температуры и перемешивали в течение 1 часа, затем фильтровали под вакуумом и промывали водой (28 мл, 5,6 об.). Белые твердые вещества помещали в вакуумный термостат при 40oC на 24-48 ч. Количество полученного продукта было 5,29 г, что давало выход 92%. Анализ продукта соответствовал его предполагаемой структуре.
Пример 18. Синтез тетрагидро-4-{3-[4-(1H-бензоимидазол-1- ил)фенил]тио} фенил-2H-пиран-4-карбоксамида с использованием твердого NaOH и катализатора Cs2CO3
Тетрагидро-4-[3-(4-фторфенил)тио] фенил-2H-пиран-4-карбоксамид (5,0 г, 15,09 ммоль, 1 экв. ), ДМСО (50 мл, 10 об.), бензоимидазол (3,56 г, 30,17 ммоль, 2,0 экв. ), гидроксид натрия (1,21 г, 30,17 ммоль, 2,0 экв.) и карбонат цезия (катализатор) (0,246 г, 0,754 ммоль, 0,05 экв.) перемешивали при 140oC в атмосфере азота в течение 4-6 часов. По завершении реакции смесь охлаждали (<30oC) и гасили водой (50 мл, 10 об.). Это давало осадок и экзотерму 20-25oC. Гашеную реакционную смесь охлаждали до комнатной температуры и гранулировали в течение 1 часа, фильтровали под вакуумом и промывали водой (28 мл, 5,6 об.) с получением продукта. Затем продукт вновь суспендировали в воде (55 мл, 11 об.) при 60oC в течение 1 часа. Суспензии давали охлаждаться до комнатной температуры и перемешивали в течение 1 часа, затем фильтровали в вакууме и промывали водой (28 мл, 5,6 об.). Белые твердые вещества помещали в вакуумный термостат при 40oC на 24-48 ч. Количество полученного продукта было 6,27 г, что давало выход 97%. Анализ продукта соответствовал его предполагаемой структуре.












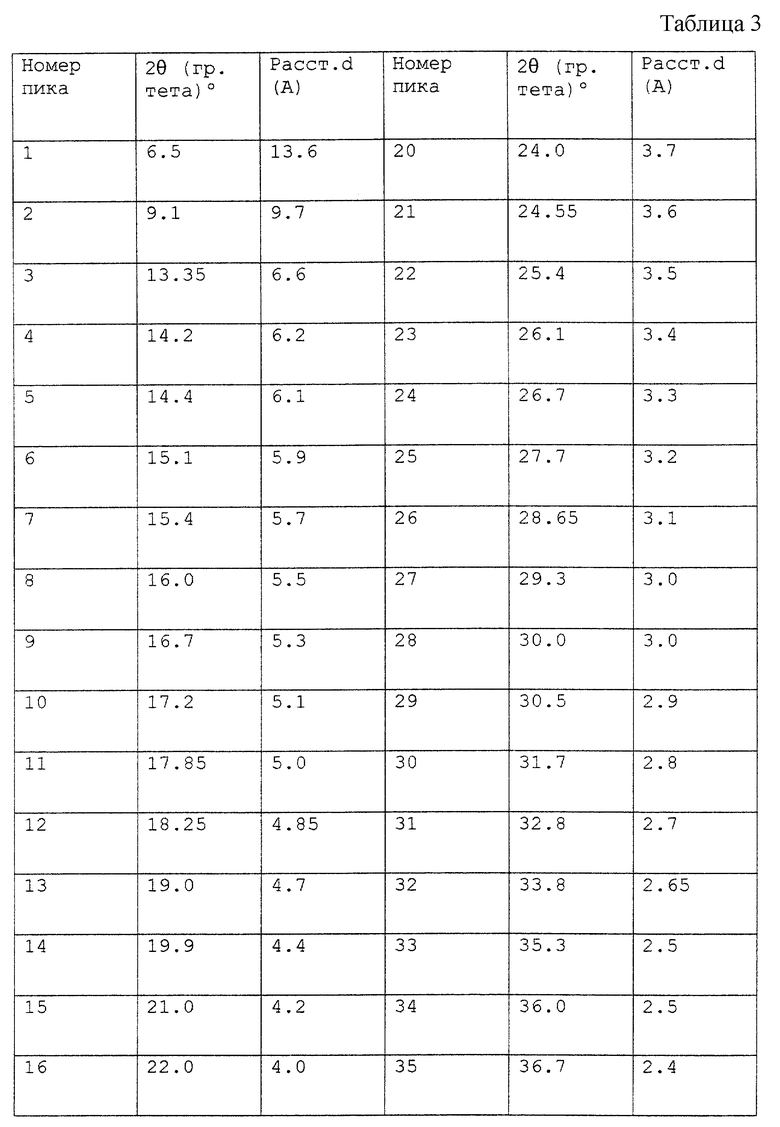

Описан способ получения соединения формулы (1.3.0), где 
является группой, выбранной из формул (1.3.2), (1.3.3), (1.3.4) или (1.3.5), включающий образование реакционной смеси, состоящей из карбоксамида формулы (2.0.0) и соответствующего бициклического N-гетероцикла в апротонном растворителе в присутствии сильного основания в твердой форме, выбранного из группы, включающей гидроксид натрия и гидроксид калия, и (необязательно) в присутствии каталитического количества карбоната цезия, Cs2СО3 или катализатора межфазного переноса с последующим нагреванием указанной реакционной смеси в атмосфере азота, в результате чего образуется целевое соединение. Также раскрыты способ получения одного из конкретных соединений и способ получения мезилатной соли соединения. Изобретение представляет собой улучшенный способ получения известных и новых ингибиторов 5-липоксигеназы, полезных при лечении воспалительных заболеваний и аллергии. 4 с. и 4 з.п. ф-лы, 3 табл.
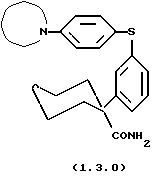


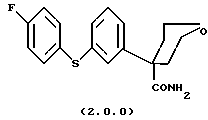

где часть молекулы формулы (1.3.1)
является электронодефицитной моноциклической или бензоконденсированной бициклической N-гетероциклической группой, содержащей два атома азота, формулы (1.3.2), (1.3.3), (1.3.4) или (1.3.5):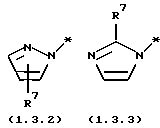

где * - символ, который представляет точку присоединения части молекулы формулы (1.3.2), (1.3.3), (1.3.4) или (1.3.5);
R7 и R8 независимо выбраны из группы, включающей Н; (C1-C4)алкил с прямой или разветвленной цепью, возможно замещенный галогеном,
включающий (а) создание реакционной смеси, состоящей из (1) тетрагидро-4-[3-(4-фторфенил)тио]фенил-2Н-пиран-4-карбоксамида формулы (2.0.0)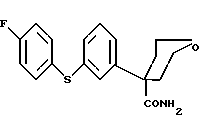
и (2) имеющего недостаток электронов моноциклического или бензоконденсированного бициклического N-гетероцикла, содержащего два атома азота, формулы (1.3.6), (1.3.7), (1.3.8) или (1.3.9):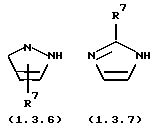

где R7 и R8 имеют указанные выше значения;
(3) в апротонном растворителе (4) в присутствии сильного основания в твердой форме, выбранного из группы, включающей гидроксид натрия и гидроксид калия, и (необязательно) (5) в присутствии каталитического количества карбоната цезия, Сs2СО3 или катализатора межфазного переноса с последующим (b) нагреванием указанной реакционной смеси в атмосфере азота, в результате чего образуется соединение формулы (1.3.0).
тетрагидро-4-{ 3-[4-(1H-имидазол-1-ил)фенил] тио} фенил-2Н-пиран-4-карбоксамид;
тетрагидро-4-{ 3-[4-(1Н-бензоимидазол-1-ил)фенил] тио}фенил-2Н-пиран-4-карбоксамид;
тетрагидро-4-{ 3-[4-(1Н-пиразол-1-ил)фенил] тио}фенил-2Н-пиран-4-карбоксамид и
тетрагидро-4-{3-[4-(4-метил-1Н-пиразол-1-ил)фенил]тио}
фенил-2Н-пиран-4-карбоксамид.
включающий (а) создание реакционной смеси, состоящей из (1) тетрагидро-4-[3-(4-фторфенил)тио]фенил-2Н-пиран-4-карбоксамида формулы (2.0.0)
и (2) 2-метилимидазола, (3) в апротонном растворителе, (4) в присутствии сильного основания в твердой форме, выбранного из группы, включающей гидроксид натрия и гидроксид калия, и (необязательно) (5) в присутствии каталитического количества карбоната цезия, Сs2СО3 или катализатора межфазного переноса с последующим (b) нагреванием указанной реакционной смеси в атмосфере азота, в результате чего образуется указанное соединение формулы (1.3.0).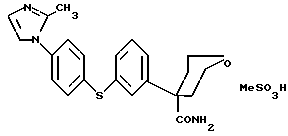
включающий (а) получение соединения формулы (2.0.0)
предусматривающий (1) образование реакционной смеси, состоящей из (i) тетрагидро-4-(3-бромфенил)-2Н-пиран-4-нитрила формулы (3.2.0)
(ii) 4-фтортиофенола формулы (4.0.0)
(iii) в растворителе, выбранном из группы, включающей изопропиловый спирт, втор-бутиловый спирт, изопентиловый спирт и 2-гептанол, необязательно в виде его водной смеси, (iv) в присутствии сильного основания, выбранного из группы, включающей гидроксид натрия и гидроксид калия, и, кроме того, (v) в присутствии катализатора, содержащего металл переходного ряда, в том числе независимо выбранного из группы, состоящей из комплексов металла палладия, с последующим (2) нагреванием указанной реакционной смеси с получением соединения формулы (2.0.0) (b) образование реакционной смеси, состоящей из соединения формулы (2.0.0) и соединения формулы (1.3.10)
(1) в апротонном растворителе, (2) в присутствии сильного основания в твердой форме, выбранного из группы, включающей гидроксид натрия и гидроксид калия, и (необязательно) (3) в присутствии каталитического количества карбоната цезия или катализатора межфазного переноса с последующим (с) нагреванием указанной реакционной смеси в атмосфере азота с образованием соединения формулы (1.0.0)
с последующим (d) образованием концентрированного раствора в метаноле указанного соединения формулы (1.0.0), содержащегося в указанной нагретой реакционной смеси, (1) фильтрованием указанного раствора в метаноле все еще в нагретом состоянии и после этого концентрированием полученного раствора-фильтрата, (2) обработкой указанного раствора-фильтрата метансульфоновой кислотой, (3) индуцированием кристаллизации указанного соединения формулы (1.0.0) из раствора-фильтрата путем вытеснения остаточного метанола в указанном растворе-фильтрате этилацетатом и (4) извлечением после этого, по существу, чистой мезилатной соли формулы (1.0.1) в кристаллической форме.
тетракис(трифенилфосфин)палладий (0), [(C6H5)3Р]4Pd (0);
тетракис(метилдифенилфосфин)палладий (0), [(C6H5)2PСН3]4Pd (0);
транс-дихлорбис(метилдифенилфосфин)палладий (II), [(С6Н5)2РСН3]2РdСl2; аддукт дихлорбис[метиленбис(дифенилфосфин)]дипалладий-дихлорметан;
дихлорбис(трифенилфосфин)палладий (II), [(С6Н5)3Р]2PdCl2;
аддукт трис(дибензилиденацетон)дипалладий (0)-хлороформ, (С6Н5СН=СНСОСН= СНС6Н5)2Рd2•СНСl3;
бис(дибензилиденацетон)палладий (0), (С6Н5СН=СНСОСН=СНС6Н5)2Pd;
[1,1'-бис(дифенилфосфино)ферроцен] дихлорпалладий (II), комплекс с дихлорметаном;
бис[1,2-бис(дифенилфосфино)этан]палладий (II) и
димер (π-аллил)палладий (II) хлорид.
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1994 |
|
RU2126004C1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
