Изобретение относится к активатору нуклеазы раковой клетки.
Термин "активатор нуклеазы раковой клетки", используемый в этом описании, означает лекарственное средство, которое активирует нуклеазы в раковых клетках и таким образом вызывает апоптоз и, следовательно, гибель раковых клеток. Также используемый здесь код "I" означает инозиновую кислоту, "С" означает цитидиловую кислоту, "А" означает адениловую кислоту, и "U" означает уридиловую кислоту.
Термины "не спариваемая по основаниям поли(1) • поли(С) и не спариваемая по основаниям поли(А) • поли(U)" означают поли(I) поли (С) и поли (А) • поли(U), которые содержат некомплементарные основания нуклеиновых кислот среди тех, которые составляют двойную цепь, что хорошо известно в данной области.
Поли(I) • поли(С) является двухцепочечной РНК, включающей полирибонуклеотидный сополимер полиинозиновой кислоты и полицитидиловой кислоты, и, как известно, является активным лекарственным веществом, обладающим сильной интерферон-индуцирующей активностью и иммуностимулирующей активностью. Тот факт, что поли (I) • поли(С) обладает иммуностимулирующей активностью, предполагает, что вещество будет косвенно ингибировать рост раковых клеток с помощью иммунных реакций, таким образом, побуждая многих исследователей изучать их потенциальную применимость в качестве лекарственного средства против злокачественных опухолей. Однако опосредствованное через иммунитет непрямое действие поли(I) • поли(С) не является достаточно сильным для ингибирования роста раковых клеток, и противораковая терапия еще не осуществлялась с помощью поли(I) • поли(С). Также не были разработаны лечебные схемы, использующие поли(I) • поли(С) для других показаний, основывающиеся на ее индуцирующей интерферон активности и иммуностимулирующей активности.
Считают также, что поли(А) • поли(U), которая является полирибонуклеотидным сополимером полиадениловой кислоты и полиуридиловой кислоты, не спариваемая по основаниям поли(I) • поли(С) и не спариваемая по основаниям поли(А) • поли(U) обладают похожей активностью, хотя отличающейся до некоторой степени.
Между тем, в качестве эффективного носителя для доставки лекарств внутрь клетки известны носители, обычно называемые катионными липосомами, как, например, липофектин (зарегистрированная торговая марка), а также носитель, включающий производное глицерина, как, например, 2-О-(2-диэтиламиноэтил)-карбамоил-1,3-О-диолеоилглицерин следующей химической формулы [I], и фосфолипид в качестве неотъемлемых компонентов [например, международные заявки на патент РСТ WO 91/17424, РСТ WO 94/19314].
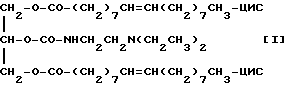
Катионная липосома рассматривается как маленькая везикула, имеющая липидную двухслойную структуру и приобретающая положительный заряд в водном растворе. Поскольку такая катионная липосома положительно заряжена, а двухцепочечная РНК, такая, как поли(I) • поли(С), заряжена отрицательно в водном растворе, катионная липосома и поли(I) • поли(С), например, могут легко образовать комплекс.
Однако совсем не было известно, будет ли в состоянии сама по себе двухцепочечная РНК, например поли(I) • поли(С), или ее комплекс с катионной липосомой активировать нуклеазы в раковых клетках, чтобы таким образом вызвать апоптоз и, следовательно, гибель раковых клеток.
Объектом настоящего изобретения является обеспечение эффективного лекарственного средства для терапии рака. Изобретение для этой цели должно также обеспечить новое лекарственное средство, содержащее двухцепочечную РНК, как, например, поли(I) • поли(С).
После большого числа исследований было обнаружено, что активирующее нуклеазу раковой клетки средство эффективно в терапии злокачественных опухолей, и в соответствии с этим было разработано настоящее изобретение.
Настоящее изобретение поэтому направлено на активатор нуклеазы раковой клетки. Является ли вещество активатором нуклеазы раковой клетки может быть легко определено экспериментально, например, наблюдая фрагментацию ДНК или РНК, как в примере тестирования 2, который приведен ниже. В общем случае изобретение охватывает активирующую нуклеазу раковой клетки композицию, включающую комплекс носителя, эффективного для внутриклеточной доставки лекарственного вещества, с веществом, выбранным из группы, состоящей из поли(I) • поли(С), не спариваемой по основаниям поли(I) • поли(С), поли(А) • поли(U) и не спариваемой по основаниям поли(А) • поли(U) (на такие двухцепочечные РНК далее будут ссылаться на каждую и вместе как на "поли(I) • поли(С) или эквивалент", и активирующую нуклеазу раковой клетки композицию, включающую комплекс катионной липосомы с поли(I) • поли(С) или эквивалентом.
Предпочтительный вариант осуществления изобретения включает активирующую нуклеазу раковой клетки композицию, включающую комплекс (кратко, комплекс) носителя (кратко, носитель), состоящего из 2-О-(2-диэтиламиноэтил)карбамоил-1,3-О-диолеоилглицерина (далее упоминается как производное глицерина) и фосфолипида в качестве основных компонентов, с поли(I) • поли(С) или эквивалентом (далее на композицию ссылаются как на активатор по изобретению).
Предпочтительный пример активатора по изобретению описывается далее подробно.
Носитель может обычно рассматриваться как катионная липосома, но необязательно находящаяся точно в виде катионной липосомы при условии, что носитель функционально пригоден для доставки лекарственного вещества в клетки.
Длина цепи поли (I) • поли(С) или эквивалента согласно настоящему изобретению особым образом не ограничивается, но, принимая поли(I) • поли(С) в качестве примера, считают подходящим использовать длину в пределах 50-2000 пар нуклеотидов (п.н.). Предпочтительна РНК длиной от 100 до 500 п.н., еще более предпочтительна РНК длиной от 200 до 400 п.н.. Если длина цепи менее 50 п.н., РНК не будет достаточно эффективной, в то время, как использование РНК с длиной цепи более 2000 п.н. может представлять проблему с точки зрения безопасности. Считают, что поли(I) • поли(С) с длиной цепи в пределах от 100 до 500 п. н. является РНК, сбалансированная с точки зрения эффективности и безопасности. Поскольку поли(I) • поли(С) или эквивалент обычно существует с заданным распределением различных длин цепи, вышеупомянутая длина цепи поли(I) • поли(С) является средней длиной цепи.
Фосфолипид для использования в настоящем изобретении особым образом не ограничивается при условии, что он является фармацевтически приемлемым фосфолипидом, таким образом включая, но не ограничиваясь этим, фосфатидилхолин, фосфатидилэтаноламин, фосфатидилинозит, фосфатидилсерин, сфингомиелины и лецитин. Гидрогенизированные фосфолипиды также могут использоваться. Предпочтительный фосфолипид включает фосфатидилхолин яичного желтка, лецитин яичного желтка, лецитин сои культурной и фосфатид яичного желтка. Могут также использоваться два или несколько различных фосфолипидов в комбинации. По сравнению с фосфатидилэтаноламином, который обычно используется в катионных липосомах, фосфатидилхолин и лецитин приводят к существенному ослаблению токсичности без ущерба активности.
Отношение носителя к поли(I) • поли(С) или эквиваленту в комплексе зависит от видов фосфолипида и поли(I) • поли(С) или эквивалента, от типа рака и других факторов, но рекомендуемое отношение поли (I) • поли(С) к 10 массовым (масс. ) частям носителя составляет 0,05-10 масс. частей, предпочтительно 0,1-4 масс. части и более предпочтительно 0,5-2 масс. части.
Отношение производного глицерина к фосфолипиду в носителе зависит от вида и количества поли(I) • поли(С) или эквивалента и вида фосфолипида, но рекомендуемое отношение фосфолипида по отношению к каждой масс. части производного глицерина составляет 0,1-10 масс. частей, предпочтительно 0,5-5 масс. частей и более предпочтительно 1-2 масс. части.
Активатор по изобретению может быть, например, представлен в виде жидкого препарата (инъекция или капельное вливание), в котором комплекс диспергирован в водном растворе, или в виде его лиофилизата. В случае жидкого препарата рекомендуемая концентрация комплекса составляет 0,001-25% (масса/объем (м/о)), предпочтительно 0,01-5% (м/о) и более предпочтительно 0,1-1% (м/о).
Активатор по изобретению может содержать фармацевтически приемлемые добавки, как, например, вспомогательный эмульгатор, стабилизатор, изотонирующее средство и/или средство для регулирования pH, в подходящих количествах. Конкретно могут быть упомянуты вспомогательные эмульгаторы, как, например, жирные кислоты с 6-22 атомами углерода (например, каприловая кислота, каприновая кислота, лауриновая кислота, миристиновая кислота, пальмитиновая кислота, стеариновая кислота, олеиновая кислота, линолевая кислота, арахидоновая кислота, докозагексеновая кислота), их фармацевтически приемлемые соли (например, натриевые соли, калиевые соли, кальциевые соли и т.д.), альбумин, декстран и т. д. , стабилизаторы, такие как холестерин, фосфатидин и т.д., агенты, обеспечивающие изотонические свойства, такие как хлористый натрий, глюкоза, мальтоза, лактоза, сахароза, трегалоза и т.д., и регулирующие рН средства, как, например, соляная кислота, серная кислота, фосфорная кислота, уксусная кислота, гидроокись натрия, гидроокись калия, триэтаноламин и т.д..
Активатор по настоящему изобретению может быть получен с применением обычной технологии для получения липосом. Типичный способ включает смешение заранее установленного количества воды (например, воды для инъекций, дистиллированной воды для инъекций или физиологического раствора) с заранее установленным количеством производного глицерина и фосфолипида при перемешивании, диспергирование смеси с помощью подходящего диспергирующего устройства, как, например, гомосмеситель, гомогенизатор, ультразвуковой диспергатор, ультразвуковой гомогенизатор, эмульгатор-диспергатор высокого давления, устройство Microfluidizer (торговое название), устройство Nanomizer (торговое название), устройство Ultimizer (торговое название) или гомогенизатор высокого давления Manton-Gaulin, затем прибавление заранее установленного количества поли(I) • поли(С) или эквивалента и редиспергирование смеси для получения активатора по изобретению для инъекций. Необязательные добавки, упомянутые выше, могут быть прибавлены на подходящем этапе до или после диспергирования. Альтернативно, активатор по изобретению может быть получен при прибавлении воды к трехкомпонентной смеси производного глицерина, фосфолипида и поли(I) • поли(С) или эквивалента и диспергировании всей смеси. Более того, может быть включена стадия получения неочищенной дисперсии.
Полученный таким образом активатор по вышеуказанной методике диспергирования может быть высушен при температуре ниже точки замерзания для получения лиофилизованного активатора по изобретению. Эта операция лиофильной сушки может быть осуществлена общепринятым способом. Например, активатор по изобретению, полученный согласно упомянутой методике диспергирования, стерилизуют и распределяют по пузырькам. Заполненные пузырьки подвергают предварительному замораживанию при примерно -40 - -20oС в течение примерно 2 часов и затем подвергают первичному высушиванию в вакууме при примерно 0 - 10oС и далее вторичному высушиванию в вакууме при примерно 15 - 25oС. Обычно пузырьки продувают газообразным азотом и укупоривают, получают целевой лиофилизованный активатор по изобретению.
Лиофилизованный стабилизатор по изобретению обычно может быть восстановлен путем прибавления подходящего растворителя (для восстановления) и взят для использования. Растворитель для восстановления включает воду для инъекций, физиологический раствор и другие обычные инфузионные растворы. Объем растворителя для восстановления меняется в зависимости от предназначения использования и, в частности, не ограничивается этим, но может предпочтительно быть 0,5-2-кратным объемом от объема до лиофильной сушки или не более 500 мл.
Активатор по изобретению активирует нуклеазы раковых клеток, чтобы индуцировать апоптоз и гибель клеток, и является лишь умеренно токсичным, так что он с высокой эффективностью находит применение в терапии рака, например, злокачественной гепатомы у млекопитающих, включая человека.
В особенности высоко эффективен и, кроме того, очень малотоксичен активатор, содержащий комплекс, образованный носителем и поли(I) • поли(С).
Активатор по изобретению может быть введен внутривенно, применен местно, введен через слизистую оболочку или другими способами при лечении опухолевых заболеваний. Когда активатор по изобретению применяют для терапии злокачественной гепатомы, его предпочтительно вводят внутривенно в печеночную артерию или в воротную вену.
Дозировка активатора по изобретению при терапии рака зависит среди прочих условий от типов поли(I) • поли(С) или эквивалента и фосфолипида, вида рака, стадии заболевания, возраста реципиента и видов, способа введения и способа лечебного воздействия. Что касается поли (I) • поли(С) или эквивалента, рекомендуемая дозировка обычно составляет 50 мкг - 50 мг/на человека на дозу и предпочтительно 100 мкг - 2 мг/на человека на дозу. Что касается поли(I) • поли(С) как таковой, рекомендуемая дозировка обычно составляет 50 мкг - 50 мг/на человека на дозу и предпочтительно 100 мкг - 2 мг/на человека на дозу. Активатор по изобретению может быть введен в один прием или путем капельной инъекции от одного до трех раз в день, каждый день, через день или раз в неделю, или раз в две недели.
Следующие рабочие примеры и примеры испытаний иллюстрируют настоящее изобретение более детально. Следует иметь в виду, что концентрация активатора по изобретению постоянно выражается в концентрации упомянутой поли(I) • поли(С) в активаторе.
Пример 1. Раствор 40 г мальтозы в 100 мл воды для инъекций смешивали с 2 г производного глицерина и 2 г очищенного лецитина из яичного желтка, и смесь обрабатывали с помощью гомогенизатора в течение 5 минут для приготовления неочищенной дисперсии носителя. Эта неочищенная дисперсия далее обрабатывалась с помощью настольного компактного эмульгатора-диспергатора в течение 1 часа и к ней добавляли воду для инъекций до 250 мл. В результате получали дисперсию носителя. К 250 мл этой дисперсии носителя прибавляли при перемешивании 150 мл водного раствора, содержащего 500 мг поли(I) • поли(С) [средняя длина цепи: около 200 п.н.], и, используя настольный компактный эмульгатор-диспергатор, смесь далее обрабатывали в течение 1 часа для получения активатора по изобретению. Этот активатор далее помещали в пузырьки, по 1 мл в пузырек, и подвергали лиофильной сушке общепринятым способом.
Пример 2. Раствор 4 кг сахарозы в 10 л воды для инъекций смешивали с 50 г производного глицерина и 30 мг фосфатида яичного желтка и смесь обрабатывали с помощью Manton-Gaulin-гомогенизатора высокого давления в течение 10 минут. Прибавляли воду для инъекций до объема в 25 л и в результате получали дисперсию. К 20 л этой дисперсии носителя прибавляли при перемешивании 12 л водного раствора, содержащего 10 г поли(I) • поли(С) [средняя длина цепи: около 200 п.н.], и в смеси устанавливали pH 5,5 с помощью соляной кислоты и далее обрабатывали с помощью Manton-Gaulin-гомогенизатора высокого давления в течение 30 минут для получения активатора по изобретению. Этот активатор затем заливали в пузырьки, по 20 мл в пузырек, и подвергали лиофильной сушке обычным способом для получения лиофилизата. Лиофилизованный стабилизатор восстанавливали путем прибавления 500 мл имеющегося в продаже 5%-ного раствора глюкозы для вливания.
Пример 3. Раствор 20 г глюкозы в 100 мл воды для инъекций смешивали с 2 г производного глицерина и 2 г лецитина сои, и смесь обрабатывали с помощью гомогенизатора в течение 5 минут для получения неочищенной дисперсии носителя. Неочищенную дисперсию далее обрабатывали с помощью настольного компактного эмульгатора-диспергатора в течение 1 часа и добавляли воду для инъекций до объема 250 мл. В результате получали дисперсию носителя. К 250 мл этой дисперсии носителя прибавляли при перемешивании 150 мл водного раствора, содержащего 50 мг поли(I) • поли(С) [средняя длина цепи: около 200 п. н. ], и, используя настольный компактный эмульгатор-диспергатор, смесь далее обрабатывали в течение 1 часа для получения активатора по изобретению.
Пример 4. Раствор 40 г мальтозы в 100 мл воды для инъекций смешивали с 1,2 г производного глицерина и 2,0 г очищенного лецитина яичного желтка, и эту неочищенную дисперсию далее обрабатывали с помощью настольного компактного эмульгатора-диспергатора в течение 30 минут и доводили объем до 250 мл водой для инъекций. В результате получали дисперсию носителя. К 250 мл этой дисперсии носителя прибавляли при перемешивании 150 мл водного раствора, содержащего 200 мг поли(I) • поли(С) [средняя длина цепи: около 200 п. н. ], и, используя настольный компактный эмульгатор-диспергатор, смесь далее обрабатывали в течение 2 часов для получения активатора по изобретению.
Пример 5. Раствор 40 г мальтозы в 100 мл воды для инъекций смешивали с 1,2 г производного глицерина и 2,0 г очищенного лецитина яичного желтка, и эту неочищенную дисперсию далее обрабатывали с помощью настольного компактного эмульгатора-диспергатора в течение 30 минут и доводили объем до 250 мл водой для инъекций. В результате получали дисперсию носителя. К 250 мл этой дисперсии носителя прибавляли при перемешивании 150 мл водного раствора, содержащего 100 мг поли(I) [средняя длина цепи: 360 оснований] и 100 мг поли (С) [средняя длина цепи: 318 оснований], и, используя настольный компактный эмульгатор-диспергатор, смесь далее обрабатывали 2 часа для получения активатора по изобретению.
Пример 6. Раствор 40 г мальтозы в 100 мл воды для инъекций смешивали с 2 г производного глицерина и 2 г очищенного лецитина яичного желтка, и смесь обрабатывали с помощью гомогенизатора в течение 5 минут для получения неочищенной дисперсии носителя. Эту неочищенную дисперсию далее обрабатывали с помощью настольного компактного эмульгатора-диспергатора в течение 1 часа и доводили объем до 250 мл водой для инъекций. В результате получали дисперсию носителя. К 250 мл этой дисперсии носителя прибавляли при перемешивании 150 мл водного раствора, содержащего 250 мг поли(I) [средняя длина цепи: 1419 оснований] и 250 мг • поли(С) [средняя длина цепи: 1491 основание], и, используя настольный компактный эмульгатор-диспергатор, смесь далее обрабатывали в течение 1 часа для получения активатора по изобретению. Этот активатор затем расфасовывали в пузырьки, 1 мл в пузырек, и подвергали лиофилизации общепринятым способом.
Пример 7. Раствор 40 г мальтозы в 100 мл воды для инъекций смешивали с 1,2 г производного глицерина и 2,0 г очищенного лецитина яичного желтка, и эту неочищенную дисперсию далее обрабатывали с помощью настольного компактного эмульгатора-диспергатора в течение 30 минут и доводили объем до 250 мл водой для инъекций. В результате получали дисперсию носителя. К 250 мл этой дисперсии носителя прибавляли при перемешивании 150 мл водного раствора, содержащего 100 мг поли(I) [средняя длина цепи: 84 основания] и 100 мг поли(С) [средняя длина цепи: 76 оснований], и, используя настольный компактный эмульгатор-диспергатор, смесь далее обрабатывали в течение 2 часов для получения активатора по изобретению.
Пример 8. Активатор по изобретению, содержащий поли(I) • поли(С) [средняя длина цепи: около 350 п.н.], получали таким же способом, как описано в примере 4.
Пример 9. Активатор по изобретению, содержащий поли(I) • поли(С) [средняя длина цепи: около 1450 п.н.], получали таким же способом, что и в примере 4.
Пример 10. Активатор по изобретению, содержащий поли(I) • поли(С) [средняя длина цепи: около 80 п.н.], получали таким же способом, что и в примере 4.
Пример тестирования 1: эффект ингибирования роста на различных линиях клеток (in vitro)
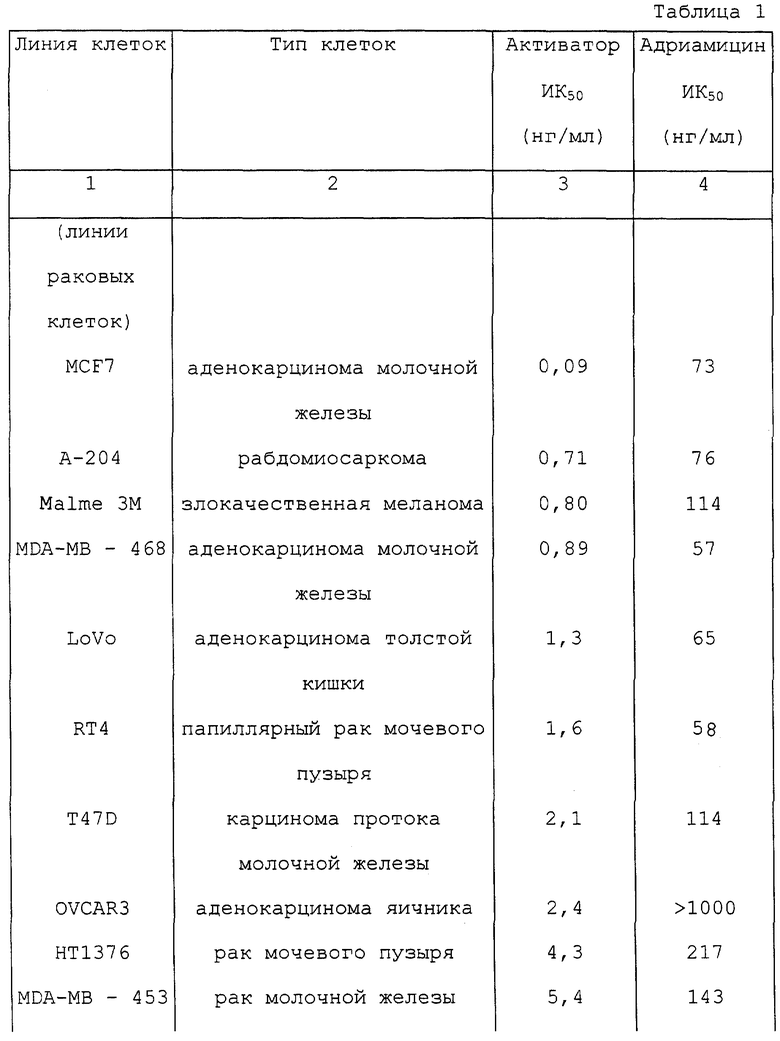
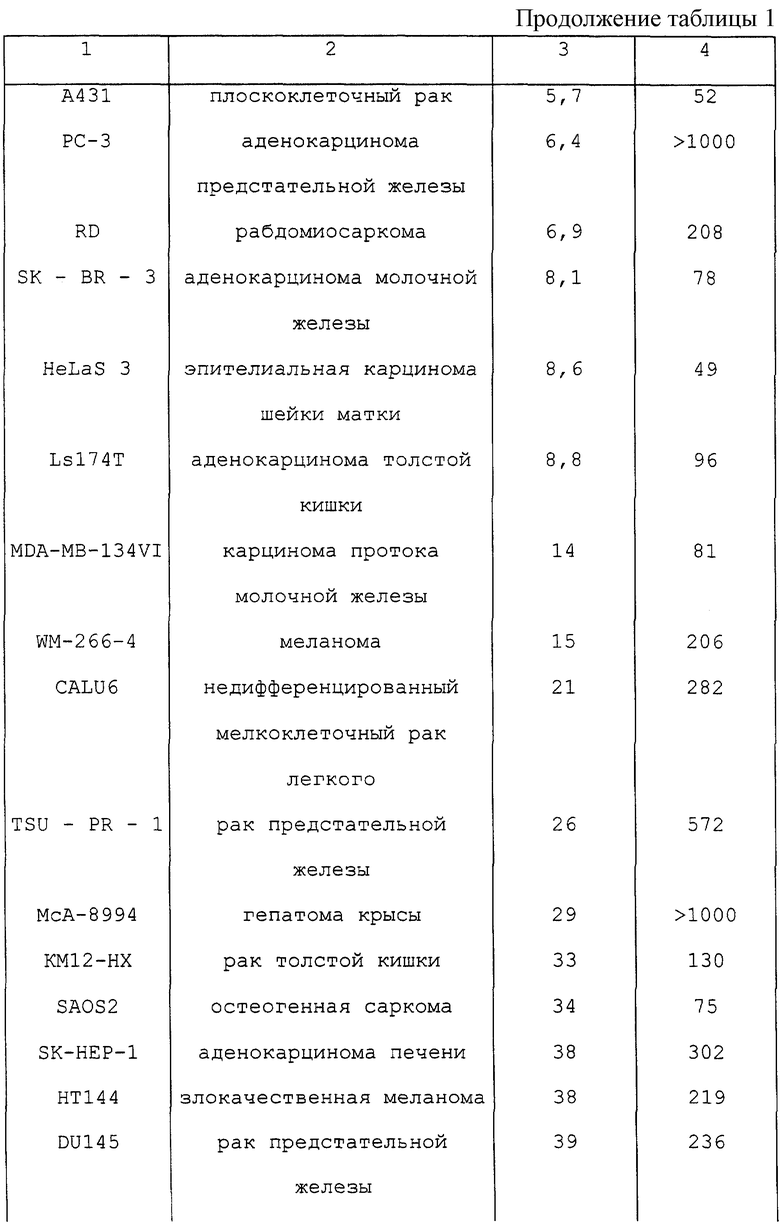
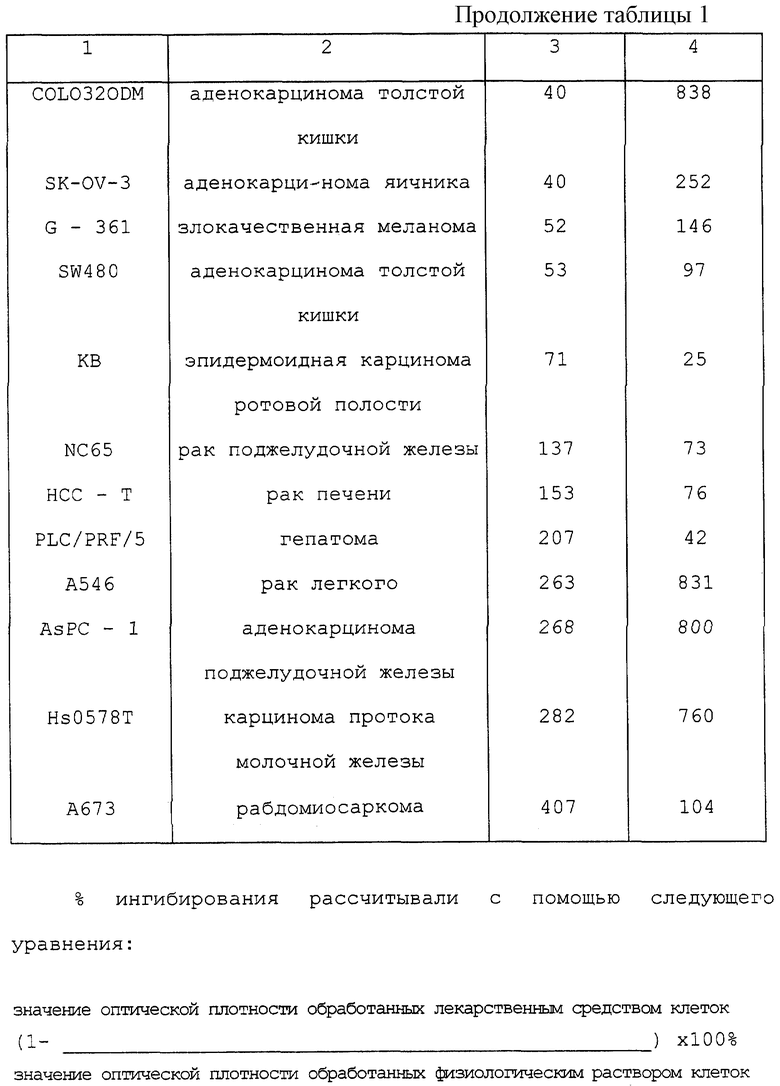
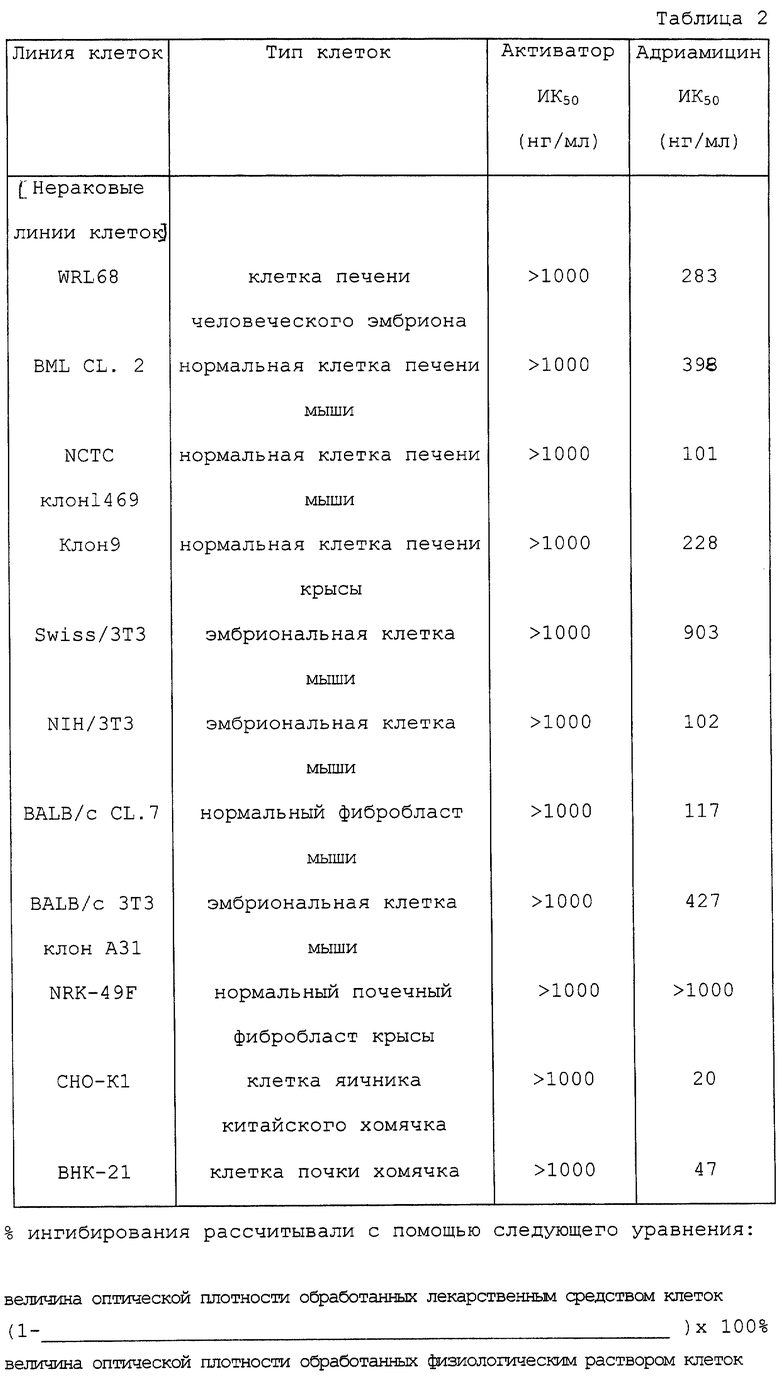
В 96 -ячеечный планшет засевали клетки с плотностью 104 клеток/в ячейке. На следующий день прибавляли активатор из примера 4 или адриамицин и культивирование продолжали. Через 3 дня подсчет жизнеспособных клеток проводили по способу с использованием бромида 3-[4,5-диметилтиазол-2-ил]-2,5-дифенилтетразолия (МТТ). Результаты представлены в таблицах 1 и 2.
Из таблиц 1 и 2 очевидно, что активатор оказывал сильные ингибирующие рост воздействия на многие эпителиальные клетки и фибробласты - воздействия, полностью сравнимые с таковыми для адриамицина, который проявляет противораковую активность путем ингибирования синтеза нуклеиновых кислот. Активатор оказывал воздействие на раковые клетки любого органа, тем самым не проявляя специфичности к органу. С другой стороны, активатор по изобретению не ингибировал рост 4 производных клеточных линий печени даже при концентрации 1000 нг/мл. Нужно отметить, что этот противораковый эффект in vitro совсем не наблюдался с одним поли(I) • поли(С) или только с одним носителем, и что это явление необъяснимо с точки зрения только простой транслокации поли(I) • поли(С) в клетки.
Пример тестирования 2: наблюдение апоптоза
(1) Фрагментация ДНК и РНК фрагментация ДНК
фрагментация ДНК
К каждой из линий клеток А431 и КМ12-НХ прибавляли 1 мкг/мл активатора из примера 4. Клетки А431 и клетки КМ12-НХ выделяли через 5 часов и 7,5 часов, соответственно. Клетки лизировали с 5 мМ трис-НСl (рН 8,0) - 10 мМ этилен-диаминтетрауксусной кислотой (ЭДТК) - 0,5% (о/о) тритоном X-100 и центрифугировали при 13000хg в течение 20 минут для выделения фрагментированной ДНК (надосадочная жидкость) и хроматиновой фракции (осадок в центрифужной пробирке). Затем действовали рибонуклеазой А 100 мкг/мл на надосадочную жидкость при 37oС в течение 1 часа и после этого прибавляли 200 мкг/мл протеиназы К и 1% (м/о) додецилсульфата натрия для проведения реакции при 50oС в течение 1,5 часов. Фрагментированную ДНК экстрагировали с фенолом-хлороформом и подвергали электрофорезу в 1,8% агарозном геле. В результате, фрагментацию ДНК наблюдали в обеих линиях клеток.
Время протекания фрагментации ДНК исследовали для линии клеток А431. Так, 6-ячеечный планшет засевали клетками А431 с плотностью 2,8•105 клеток/в ячейке, и на следующий день клеточную ДНК метили с помощью 2 мкКи (3Н] тимидина. Затем прибавляли активатор из примера 4 (1 мкг/мл) и клетки собирали через определенные временные интервалы. Клетки подвергали лизису с помощью 5 мМ трис-НСl (pH 8,0)-10 мМ ЭДТК - 0,5% (о/о) тритона Х-100 и центрифугировали при 13000xg в течение 20 минут для отделения фрагментированной ДНК (надосадочная жидкость) от хроматиновой фракции (осадок в центрифужной пробирке). На основании измеренной радиоактивности в надосадочной жидкости и в осадке после центрифугирования, соответственно, рассчитывали отношение фрагментированной ДНК к общей ДНК. Результаты представлены на фиг.1.
Степень фрагментации составляла примерно 30% от общей ДНК через 3 часа после прибавления и не менее 55% через 5 часов, указывая на то, что эта фрагментация происходит для выделения фрагментированной ДНК (надосадочная жидкость) и хроматиновой фракции (осадок в центрифужной пробирке). Затем действовали рибонуклеазой А 100 мкг/мл на надосадочную жидкость при 37 oС в течение 1 часа и после этого прибавляли 200 мкг/мл протеиназы К и 1% (м/о) додецилсульфата натрия для проведения реакции при 50oС в течение 1,5 часов. Фрагментированную ДНК экстрагировали с фенолом-хлороформом и подвергали электрофорезу в 1,8% агарозном геле. В результате, фрагментацию ДНК наблюдали в обеих линиях клеток.
Время протекания фрагментации ДНК исследовали для линии клеток А431. Так, 6-ячеечный планшет засевали клетками А431 с плотностью 2,8•105 клеток/в ячейке, и на следующий день клеточную ДНК метили с помощью 2 мкКи [3Н] тимидина. Затем прибавляли активатор из примера 4 (1 мкг/мл) и клетки собирали через определенные временные интервалы. Клетки подвергали лизису с помощью трис-НСl (рН 8,0)-10 мМ ЭДТК - 0,5% (о/о) тритона Х-100 и центрифугировали при 13000xg в течение 20 минут для отделения фрагментированной ДНК (надосадочная жидкость) от хроматиновой фракции (осадок в центрифужной пробирке). На основании измеренной радиоактивности в надосадочной жидкости и в осадке после центрифугирования, соответственно, рассчитывали отношение фрагментированной ДНК к общей ДНК. Результаты представлены на фиг.1.
Степень фрагментации составляла примерно 30% от общей ДНК через 3 часа после прибавления и не менее 55% через 5 часов, указывая на то, что эта фрагментация происходит незамедлительно после внутриклеточного поглощения активатора по изобретению.
 фрагментация РНК
фрагментация РНК
К каждой из линий клеток А431, MDA-MB-468, KB, HeLa S3 и MCF-7 прибавляли активатор из примера 4 в виде 1 мкг/мл поли(I) • поли(С) и обработанные клетки выделяли через 4 часа. Из выделенных клеток отделяли рибосомную фракцию и общую РНК экстрагировали по методу AGPC (кислота-гуанидиний-фенол-хлороформ). РНК подвергали электрофорезу в модифицированном формальдегидом геле (1,8% агарозный гель) и окрашиванию бромидом этидия. В результате фрагментацию 28S и 18S рибосомных РНК наблюдали во всех линиях клеток.
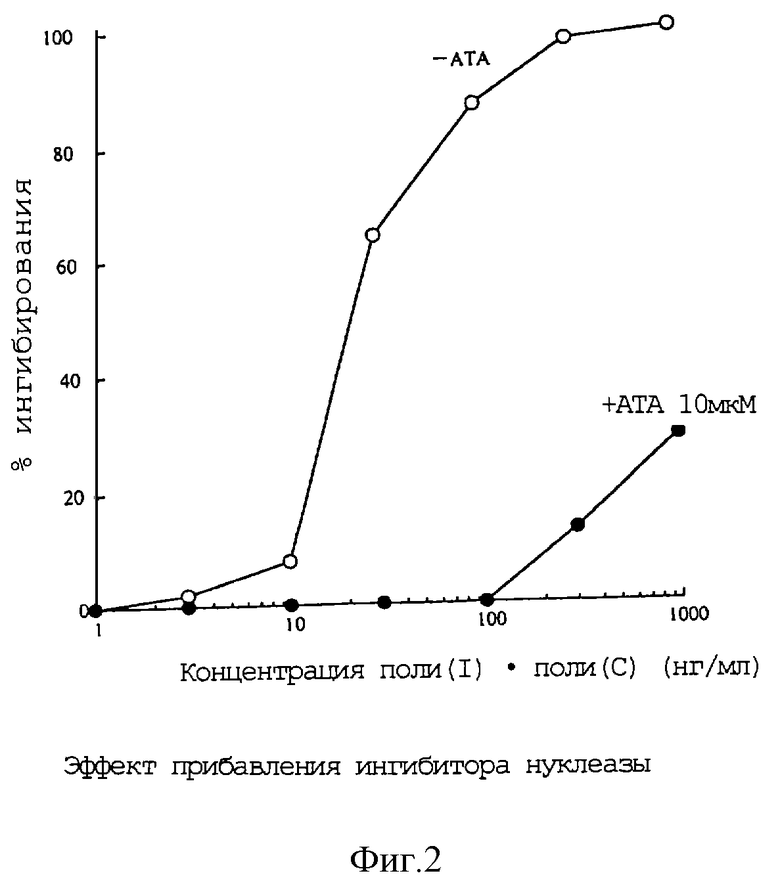
(2) Действие ингибитора нуклеазы
В 96-ячеечный планшет засевали клетки HeLaS3 с плотностью 104 клеток/в ячейке, и на следующий день одновременно прибавляли 10 мкМ ингибитора нуклеазы ауринтрикарбоновую кислоту (АТА) и активатор согласно примеру 4 настоящего изобретения. Клетки выращивали далее в течение 3 дней, и число жизнеспособных клеток определяли способом с применением МТТ. Результаты приведены на фиг.2.
Из фиг. 2 очевидно, что, когда активность внутриклеточной нуклеазы ингибировалась при прибавлении АТА, активатору по изобретению не удавалось тормозить рост раковых клеток. Более того, когда добавленную АТА удаляли из среды через 8 часов и перед добавлением активатора по изобретению, активатору не удавалось проявить свою активность. Поэтому считали, что действие АТА заключалось не в ингибировании внутриклеточного поглощения активатора, а в том, что она действовала как ингибитор нуклеазы.
(3) Результаты описываемого выше опыта показывают, что активатор по изобретению активирует внутриклеточные нуклеазы, чтобы таким образом вызвать апоптоз раковых клеток.
Пример тестирования 3: действие на модель мышиной метастатической злокачественной гепатомы (in vivo)
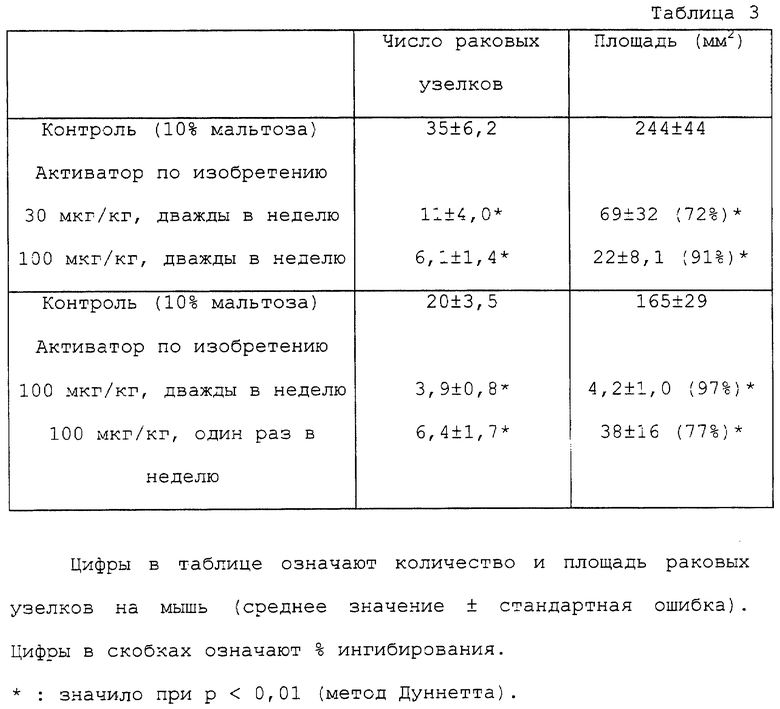
Используя безволосых мышей Balb/c, nu/nu (возраст 5 недель, самцы), инъецировали в селезенку 106 клеток/на мышь линии клеток КМ12-НХ (линия клеток рака толстой кишки человека, которая при трансплантировании в селезенку безволосых мышей метастазирует с высокой эффективностью в печень, вызывая опухолевые поражения) и через 10 минут селезенку энуклеировали. Через 3 дня после этого вводили внутривенно активатор согласно примеру 4 изобретения дважды в неделю в основном с постоянными интервалами в 5 последующих недель. Через два дня после последней дозы печень отделяли и определяли число и площадь раковых узелков, образовавшихся в печени. Результаты приведены в таблице 3.
По сравнению с контрольной группой (которой вводили 10% мальтозу) ингибирование роста раковых клеток печени составляло 72% в группе, которой вводили 30 мкг/кг, и 91% в группе, которой вводили 100 мкг/кг. В группе, которой вводили 100 мкг/кг, 77%-ное ингибирование наблюдали даже тогда, когда схема применения предусматривала введение один раз в неделю.
Кроме того, получали и изучали в отношении патологии образцы ткани печени. В результате, злокачественное новообразование печени в контрольной группе являлось эпителиальной низкодифференцированной аденокарциномой. Наблюдали обычную степень васкуляризации протока. Ничто не свидетельствовало о заметной инфильтрации иммунных клеток. Более того, опухолевая ткань обнаруживала местную кальцификацию. В группе, обработанной активатором по изобретению, не определяли явных раковых клеток, а только был отмечен кальцификат, оставшийся после лечения.
Таким образом, активатор по изобретению проявляет значительную эффективность на модели животного с злокачественной гепатомой в диапазоне доз 10 мкг/кг 100 мкг/кг по схеме, предусматривающей применение два раза в неделю.
Пример тестирования 4: изучение токсичности
(1) Экспрессия гепатотоксичности у крыс, получавших разовую дозу (изучение острой токсичности)
Использовали 8 самцов крыс SD в возрасте 6 недель, активатор согласно примеру 4 изобретения вводили в виде разовой внутривенной дозы и через 20 часов определяли активность аминоацилтрансферазы сыворотки. В результате, гибель не наблюдалась вплоть до 5 мг/кг и слабое повышение сывороточной аминоацилтрансферазы больше всего наблюдали при 5 мг/кг. При 1 мг/кг уровень аминоацилтрансферазы сыворотки был незначительно повышен.
(2) Двухнедельное изучение подострой токсичности у крыс
Активатор согласно примеру 4 изобретения вводили внутривенно 6 самцам крыс SD (возраст 6 недель) ежедневно в течение 14 дней подряд. В результате никаких заметных признаков токсичности не наблюдали в случае доз до 1 мг/кг.
(3) Изучение антигенности
Используя самцов морских свинок (штамм Hartley, возраст 5 недель), исследовали антигенность активатора согласно примеру 4 изобретения. В результате антигенность не была обнаружена при 50 мкг/на животное.
(4) Соответствующее изучение мутагенности
Активатор согласно примеру 4 изобретения подвергали соответствующему анализу на обратную мутацию и соответствующему анализу на хромосомную аберрацию. В результате, мутагенность не наблюдалась при 10 мкг/мл.
Краткое описание чертежей
Фиг.1 представляет кривую зависимости, показывающую степень фрагментации ДНК. Ордината представляет процент (%) фрагментации ДНК и абсцисса представляет время (час).
Фиг. 2 представляет кривую зависимости, показывающую эффект прибавления ингибитора нуклеазы АТА, где абсцисса показывает концентрацию (нг/мл) активатора согласно примеру 4 изобретения, и _•_ обозначает данные, полученные в не содержащей АТА системе, и -•- обозначает данные, полученные в системе с АТА.
Изобретение относится к медицине и представляет лекарственное средство, эффективное для терапии рака, и новое лекарственное средство, содержащее двухцепочечную РНК, как, например, поли (I) • поли (С), конкретно, и активатор внутриклеточной нуклеазы раковой клетки, содержащий 2-O-(2-диэтиламиноэтил)-карбамоил-1,3-O-диолеоилглицерин, и композицию, включающую носитель, полученный из фосфолипида как основного компонента, и поли (I) • поли (С) или не спариваемую по основаниям поли (I) • поли (С). Изобретение обеспечивает получение лекарственного средства, которое эффективно в терапии рака и малотоксично. 3 с. и 3 з.п. ф-лы, 2 ил., 3 табл.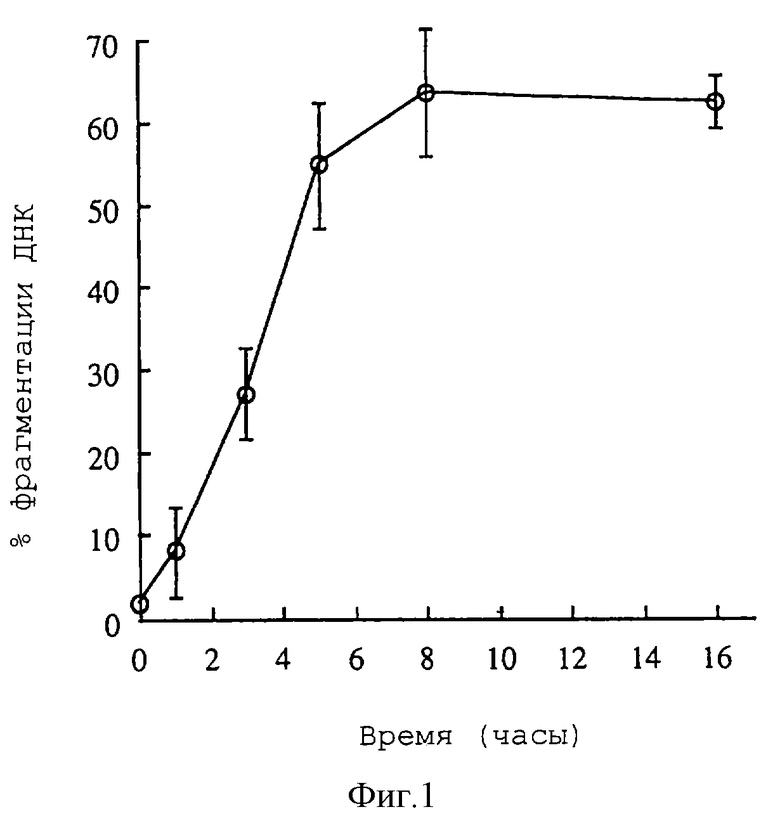
| Экономайзер | 0 |
|
SU94A1 |
| SU 774455 А1, 30.10.1980. | |||

