Область техники
Настоящее изобретение относится к новому длительно циркулирующему носителю лекарственного средства.
Предшествующий уровень техники
В последнее время внимание было сосредоточено на лекарственных средствах на основе нуклеиновых кислот, таких как синтетическая двухцепочечная РНК (такая как поли(I)-поли(C)), короткая интерферирующая РНК (миРНК), использующие РНК-интерференцию (РНКи), микроРНК (мкРНК), короткая шпилечная РНК (кшРНК), антисмысловая ДНК и антисмысловая РНК, которые интенсивно изучены. Такое лекарственное средство на основе нуклеиновой кислоты с трудом доставляется в ткань с участком повреждения, даже при системном введении лекарственного средства независимо в организм через, например, вену. Таким образом, для лекарственного средства на основе нуклеиновой кислоты требуется, например, введение после его включения в подходящий носитель или местное введение в ткань с участком поражения.
Примеры носителя лекарственного средства для доставки лекарственного средства на основе нуклеиновой кислоты в ткань с участком повреждения включают катионные липосомы, такие как ЛИПОФЕКТИН (зарегистрированный товарный знак), ЛИПОФЕКТАМИН 2000 (зарегистрированный товарный знак) и ОЛИГОФЕКТАМИН (зарегистрированный товарный знак), и катионная липосома (далее в настоящем документе обозначаемая как «липосома с соединением A»), содержащие в качестве обязательных компонентов 2-O-(2-диэтиламиноэтил)карбамоил-1,3-O-диолеоилглицерин (далее в настоящем документе обозначаемый как «соединение A») и фосфолипид (см., например, патентный документ 1). Так как подобные катионные липосомы имеют тенденцию легко накапливаться в печени и селезенке, при системном введении через, например, вену предполагается применение катионных липосом в качестве терапевтического средства для лечения злокачественной опухоли печени или гепатита посредством включения лекарственного средства на основе нуклеиновой кислоты в катионные липосомы. В настоящее время описывают, что комплекс «липосомы с соединением A» и синтетической двухцепочечной РНК, такой как поли (I)-поли (C), является эффективным в лечении злокачественной опухоли печени или гепатита (см., например, патентный документ 2, патентный документ 3, непатентный документ 1 и непатентный документ 2).
Для накапливания в печени или т.п. в качестве носителя лекарственного средства на основе нуклеиновой кислоты пригодны катионные липосомы. Однако они не являются достаточными в качестве носителя для применения в доставке лекарственного средства на основе нуклеиновой кислоты в ткань, отличную от печени или селезенки (такую как легкие, почки, поджелудочная железа или сердце), а также для обеспечения продолжительной циркуляции в крови.
Описывают, что посредством модифицирования липида, составляющего липосому, модифицированную полиэтиленгликолем, снижают поглощение в ретикулоэндотелиальной системе и, таким образом, увеличивают способность к циркулированию в крови (см., например, непатентный документ 3).
Однако модифицированный полиэтиленгликолем липид с увеличением содержания липида может снижать эффективность лекарственного средства, включенного в липосому. Таким образом, важно добавлять минимальное количество липида, позволяющее обеспечивать способность к длительному циркулированию (см., например, патентный документ 4).
Описывают, что модифицированный полиэтиленгликолем дистеароилфосфатидилэтаноламин, в качестве компонента липосомы, обеспечивает наиболее продолжительное время циркуляции в количестве приблизительно 4 мол.% в общем количестве липидов, составляющих липосому (см., например, непатентный документ 3).
Между тем также описано, что модифицированный полиэтиленгликолем липид, дозированный в количестве приблизительно 10 или 15 мол.%, обеспечивает продолжительное время циркуляции (см., например, патентные документы 5 до 9). По патентным документам от 5 до 9, в зависимости от различия в структуре модифицированного полиэтиленгликолем липида, или катионного липида, подлежащих использованию в качестве компонента липосомы, изменяется полученный эффект удлинения времени циркуляции или степень проявления эффективности лекарственного средства.
Патентный документ 1: WO 94/19314 A1
Патентный документ 2: WO 99/20283 A1
Патентный документ 3: WO 99/48531 A1
Патентный документ 4: WO 2005/051351 A2
Патентный документ 5: WO 2006/074546 A1
Патентный документ 6: WO 2006/007712 A1
Патентный документ 7: WO 2005/120152 A2
Патентный документ 8: CA 2271582 A1
Патентный документ 9: US 2004/0166150 A1
Непатентный документ 1: Kazuko Hirabayashi, et al., Cancer Research, 1999, Vol. 59, pp. 4325-4333
Непатентный документ 2: Kazuko Hirabayashi, et al., Oncology Research, 1999, Vol. 11, pp.497-504
Непатентный документ 3: Tatsuhiro Ishida, et al., Riposomu Oyo no Shintenkai, 2005, June, pp. 528-538
Описание изобретения
Задачи, подлежащие решению посредством изобретения
Основной целью настоящего изобретения является получение носителя лекарственного средства, который представляет собой длительно циркулирующий носитель лекарственного средства, в основном содержащий модифицированный полиэтиленгликолем фосфолипид и соединение A, где модифицированный полиэтиленгликолем фосфолипид содержится в концентрации в пределах определенного диапазона, и получение фармацевтической композиции, содержащей носитель лекарственного средства, включающей лекарственное средство.
Средства решения задач
После проведения авторами изобретения интенсивных исследований они обнаружили, что в носителе лекарственного средства, который в качестве существенных компонентов содержит модифицированный полиэтиленгликолем фосфолипид с определенной структурой и соединение A, модифицированный полиэтиленгликолем фосфолипид в высокой концентрации от 30 до 50% мас. от общей массы липидов в носителе лекарственного средства придает носителю лекарственного средства способность к длительному циркулированию, и также позволяет лекарственному средству, включенному в носитель лекарственного средства, проявлять эффективность in vivo. Таким образом, настоящее изобретение завершено.
Настоящее изобретение включает, например, следующие изобретения, описанные в патентных документах 1 и 2.
1. Долго циркулирующий носитель лекарственного средства, включающий модифицированный полиэтиленгликолем фосфолипид, представленный следующей общей формулой (I) (далее, для краткости, обозначаемый в настоящем документе как «модифицированный PEG фосфолипид») или его фармацевтически приемлемую соль:
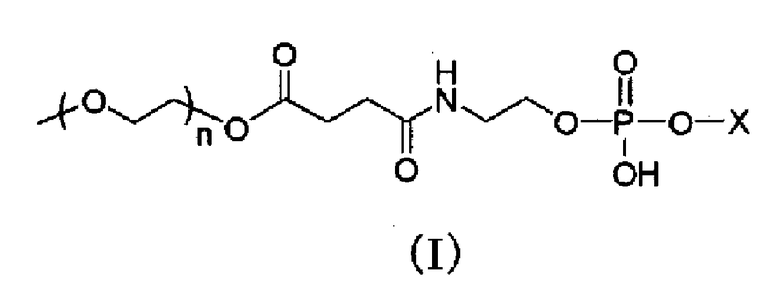
где X представляет собой следующие (II) или (III); и n представляет собой целое число от 30 до 150,

где R1 представляет собой остаток насыщенной неразветвленной жирной кислоты, имеющий от 17 до 22 атомов углерода, и соединение A, где модифицированный PEG фосфолипид, представленный общей формулой выше (I), содержится в количестве в пределах от 30 до 50% мас. от общей массы липидов в носителе лекарственного средства (далее в настоящем документе обозначаемый как «носитель по настоящему изобретению»).
2. Фармацевтическую композицию, содержащую носитель по настоящему изобретению, который включает лекарственное средство (далее в настоящем документе обозначаемую как «композиция по настоящему изобретению»).
Примеры остатка насыщенной неразветвленной жирной кислоты, имеющего от 17 до 22 атомов углерода, представляющего собой R1, включают стеароил, арахидоил и бегеноил. Среди них предпочтительным является остаток насыщенной неразветвленной жирной кислоты, имеющий от 17 до 20 атомов углерода, и более предпочтительным является стеароил.
n представляет собой целое число в пределах от 30 до 150, предпочтительно целое число в пределах от 30 до 100, более предпочтительно целое число в пределах от 30 до 65.
Фосфолипид, модифицированный PEG, можно использовать в виде свободной кислоты, как таковой, однако ее можно перевести традиционным способом в форму фармацевтически приемлемой соли и использовать.
Фармацевтически приемлемая соль конкретно не ограничена, но ее примеры включают натриевую соль и калиевую соль. Среди них особенно предпочтительной является натриевая соль.
Предпочтительные примеры модифицированного PEG фосфолипида включают 1,3-дистеароилглицеро-2-фосфатидил-N-(метоксиполиэтиленгликоль-сукцинил)этаноламин и N-(метоксиполиэтиленгликоль-сукцинил)дистеароилфосфатидилэтаноламин.
Краткое описание чертежей
[Фиг.1] На фиг.1 показан массовый спектр модифицированного PEG фосфолипида, синтезированного в примере получения 3.
[Фиг.2] На фиг.2 показан массовый спектр модифицированного PEG фосфолипида, использованного в примерах получения от 12 до 15.
[Фиг.3] На фиг.3 показано зависящее от времени изменение в способности к циркулированию в плазме. На вертикальной оси представлен коэффициент распределения (% дозы), а на горизонтальной оси представлен период времени после введения (час).
[Фиг.4] На фиг.4 показана способность к циркулированию в плазме, определенная через 24 часа после введения. На вертикальной оси показан коэффициент распределения (% дозы), а на горизонтальной оси представлено содержание использованного модифицированного PEG фосфолипида (% мас.).
[Фиг.5] На фиг.5 показана способность к циркулированию в плазме, определенная через 24 часа после введения. На вертикальной оси показан коэффициент распределения (% дозы), а на горизонтальной оси представлено содержание использованного модифицированного PEG фосфолипида (% мас.).
[Фиг.6] На фиг.6 показана способность к циркулированию в плазме. На вертикальной оси представлен коэффициент распределения (% от дозы), а на горизонтальной оси представлен период времени после введения фармацевтической композиции (час).
[Фиг.7] На фиг.7 показана способность к циркулированию в плазме. На вертикальной оси представлен коэффициент распределения (% дозы), а на горизонтальной оси представлен период времени после введения фармацевтической композиции (час).
[Фиг.8] На фиг.8 показана способность к доставке в печень. На вертикальной оси представлен коэффициент распределения (% дозы), а на горизонтальной оси представлен период времени после введения фармацевтической композиции (час).
[Фиг.9] На фиг.9 показана способность к доставке в селезенку. На вертикальной оси представлен коэффициент распределения (% дозы), а на горизонтальной оси представлен период времени после введения фармацевтической композиции (час).
[Фиг.10] На фиг.10 показана способность к доставке в легкие. На вертикальной оси представлен коэффициент распределения (% дозы), а на горизонтальной оси представлен период времени после введения фармацевтической композиции (час).
[Фиг.11] На фиг.11 показана способность к доставке в почки. На вертикальной оси представлен коэффициент распределения (% дозы), а на горизонтальной оси представлен период времени после введения фармацевтической композиции (час).
[Фиг.12] На фиг.12 показана способность к доставке в периферические ткани злокачественной опухоли. На вертикальной оси представлена концентрация носителя лекарственного средства (мкг/г), а на горизонтальной оси представлен период времени после введения фармацевтической композиции (час).
[Фиг.13] На фиг.13 показана способность к доставке в злокачественные узлы. На вертикальной оси представлена концентрация носителя лекарственного средства (мкг/г), а на горизонтальной оси представлен период времени после введения фармацевтической композиции (час).
[Фиг.14] На фиг.14 показана способность к доставке в плазму. На вертикальной оси представлена концентрация носителя лекарственного средства (мкг/г), а на горизонтальной оси представлен период времени после введения фармацевтической композиции (час).
[Фиг.15] На фиг.15 показано противоопухолевое действие. На вертикальной оси представлен объем опухоли (мм3), а на горизонтальной оси представлен период времени после имплантации злокачественных клеток.
[Фиг.16] На фиг.16 показано противоопухолевое действие в случае постоянного введения композиции по настоящему изобретению. На вертикальной оси представлен уровень выживаемости (%), а на горизонтальной оси представлен период выживаемости после имплантации злокачественных клеток (сутки).
[Фиг.17] На фиг.17 показано противоопухолевое действие в случае периодического введения композиции по настоящему изобретению. На вертикальной оси представлен уровень выживаемости в (%), а на горизонтальной оси представлен период выживаемости после имплантации злокачественных клеток (сутки).
[Фиг.18] На фиг.18 показано противоопухолевое действие. На вертикальной оси представлен уровень выживаемости (%), а на горизонтальной оси представлен период выживаемости после имплантации злокачественных клеток (сутки).
[Фиг.19] На фиг.19 показано противоопухолевое действие. На вертикальной оси показана масса поджелудочной железы (г).
Лучший способ осуществления изобретения
I. Способ получения модифицированного PEG фосфолипида
Модифицированный PEG фосфолипид (Ia), в котором X представлен формулой выше (II), можно получать, в присутствии подходящего основания, посредством взаимодействия аминового производного, представленного следующей общей формулой (1a), с производным PEG, представленным следующей общей формулой (2).
Растворитель, подлежащий использованию в реакции с производным PEG, представленным следующей общей формулой (2), конкретно не ограничивают при условии, что растворитель не участвует в реакции, и его примеры включают дихлорметан, диметоксиэтан и их жидкую смесь. Примеры основания включают триэтиламин, пиридин и водный раствор бикарбоната натрия. Температура реакции представлена, соответствующим образом, в пределах от 0°C до 50°C. Время реакции изменяется в зависимости от вида используемого исходного материала и температуры реакции. В основном, время реакции располагается, соответствующим образом, в пределах от 1 до 30 часов.
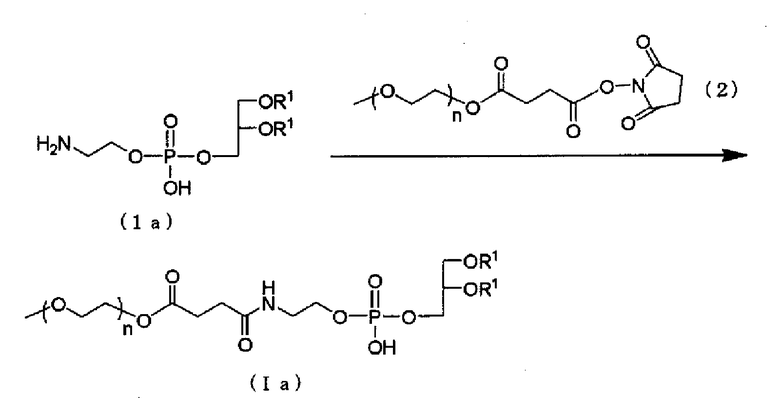
(В формуле n и R1 представляют собой то же, что определено выше).
Модифицированный PEG фосфолипид (Ib), в котором X представлен формулой выше (III), можно получать, в присутствии подходящего основания, посредством удаления, общепринятым способом защитной группы (R2) для аминогруппы и защитной группы (R3) для фосфорной кислоты аминового производного, представленного следующей общей формулой (1b), и затем посредством взаимодействия аминового производного с производным PEG, представленным выше общей формулой (2).
Удаление защитных групп R2 и R3 можно выполнять одновременно или последовательно. Примеры реагента для удаления защитной группы R2 включают кислоты, такие как трифторуксусная кислота, уксусная кислота и соляная кислота. Примеры реагента для удаления защитной группы R3 включают жидкую смесь пиридина, триэтиламина и воды (3:1:1); раствор триэтиламина в ацетонитриле; 50% водно-диоксановый раствор пиридин-2-карбоксальдоксима и N1,N1,N3,N3-тетраметилгуанидина; и кислот, таких как трифторуксусная кислота, уксусная кислота и соляная кислота.
Растворитель, используемый в реакции с производным PEG, представленным общей формулой выше (2), конкретно не ограничен при условии, что он не участвует в реакции, и его примеры включают дихлорметан, диметоксиэтан и их жидкую смесь. Примеры основания включают триэтиламин, пиридин и жидкий раствор бикарбоната натрия. Температура реакции представлена, соответствующим образом, в пределах от 0°C до 50°C. Время реакции изменяется в зависимости от вида используемого исходного материала и температуры реакции. В основном, время реакции представлено, соответствующим образом, в пределах от 1 до 30 часов.

В формуле n и R1 представляют собой то же, что определено выше. R2 обозначает защитную группу для аминогруппы. Защитная группа конкретно не ограничена и ее примеры включают трет-бутилоксикарбонил и бензилоксикарбонил. R3 обозначает защитную группу для фосфорной кислоты. Защитная группа конкретно не ограничена и ее примеры включают метил, цианоэтил и трет-бутил.
Аминовое производное, представленное выше общей формулой (1a), можно получать по способу, описанному в документе (J. Am. Chem. Soc., 1993, 115, pp. 10487-10491), используя фосфатидилхолин, представленный следующей общей формулой (3), аминоэтанол, представленный следующей общей формулой (4), и фосфолипазу D.

(В формуле R1 представляет собой то же, что определено выше).
Аминовое производное, представленное выше общей формулой (1b), можно получать посредством взаимодействия соединения амидита, представленного общей формулой (5), с аминоэтанолом, представленным следующей общей формулой (6), в присутствии подходящего активатора, а затем окислением полученного продукта подходящим окислителем.
Примеры активатора включают тетразол и 5-фенил-1H-тетразол. Примеры окислителя включают йодный раствор (0,1 M йод / тетрагидрофуран:пиридин:вода = 7:1:2) и раствор трет-бутил гидропероксида. Температура реакции, соответствующим образом, представлена в пределах от 0°C до 50°C. Растворитель, подлежащий использованию, конкретно не ограничен, при условии, что растворитель не участвует в реакции, и его примеры включают ацетонитрил и дихлорметан. Время реакции изменяется в зависимости от вида исходного материала, подлежащего использованию, и температуры реакции. В основном, время реакции соответствующим образом представлено в пределах от 1 до 30 часов.

(В формуле R1, R2 и R3 представляют собой то же, что определено выше. R4 обозначает алкил. Алкил конкретно не ограничен, и его примеры включают метил, этил, н-пропил и изопропил).
Амидит, представленный выше общей формулой (5), можно получать преобразованием спирта, представленного следующей общей формулой (7), в амидит в присутствии подходящего активатора.
Примеры активатора включают диизопропиламмония тетразолид, тетразол, 5-фенил-1H-тетразол и диизопропилэтиламин. Примеры реагента, подлежащего использованию в преобразовании в амидит, включают бис(N,N-диизопропиламино)цианоэтилфосфит, 2-цианоэтил-N,N-диизопропилхлорфосфорамидит и трет-бутил тетраизопропилфосфорамидит. Растворитель, подлежащий использованию, конкретно не ограничен, при условии, что растворитель не участвует в реакции, и его примеры включают ацетонитрил и дихлорметан. Температура реакции, соответствующим образом, представлена в пределах от 0°C до 50°C. Время реакции изменяется в зависимости от вида исходного материала, подлежащего использованию, и температуры реакции. В основном, время реакции соответствующим образом представлено в пределах от 1 до 30 часов.

(В формуле R1, R2 и R3 представляют собой то же, что определено выше).
Спирт, представленный выше общей формулой (7), можно получать по способу, описанному в документе (например, The Journal of Organic Chemistry, 1970, vol. 35, pp. 2082-2083), используя димер дигидроксиацетона (8). Примеры конденсирующего средства включают N,N'-дициклогексилкарбодиимид, 1-этил-3-(3-диметиламинопропил)карбодиимид и 1-гидроксибензотриазол. Примеры восстановителя включают натрия борогидрид.

(В формуле R1 представляет собой то же, что определено выше.)
II. Носитель по настоящему изобретению
Носитель по настоящему изобретению содержит модифицированный PEG фосфолипид и соединение A в качестве существенных компонентов. Конкретно, носитель по настоящему изобретению может приобретать форму липосомы, жировой эмульсии или им подобных. Примеры в форме липосомы включают многослойную и однослойную везикулу.
Соединение A можно синтезировать способом, описанным в WO 94/19314.
Количество в смеси модифицированного PEG фосфолипида в носителе по настоящему изобретению представлено, соответствующим образом, в пределах от 30% мас. до 50% мас., предпочтительно в пределах от 40% мас. до 50% мас. от общей массы липидов в носителе по настоящему изобретению.
Касаемо соотношения в смеси между модифицированным PEG фосфолипидом и соединением A в носителе по настоящему изобретению доля соединения A представлена, соответствующим образом, в пределах от 0,2 до 20 массовых частей на 1 массовую часть модифицированного PEG фосфолипида, предпочтительно в пределах от 0,5 до 10 массовых частей, более предпочтительно в пределах от 0,7 до 1,3 массовой части.
К носителю по настоящему изобретению, кроме модифицированного PEG фосфолипида и соединения A, которые являются существенными компонентами, дополнительно можно добавлять фосфолипид. Фосфолипид конкретно не ограничен, при условии, что он является фармацевтически приемлемым фосфолипидом, и его примеры включают фосфатидилхолин, фосфотидилэтаноламин, фосфатидилинозитол, фосфатидилсерин, сфингомиелин, лецитин, дипальмитоилфосфатидилхолин, дистеароилфосфатидилхолин и дипальмитоилфосфатидилглицерин. Их можно использовать отдельно или в комбинации из двух или более. Среди них особенно предпочтительными являются 1-пальмитоил-2-олеоил-sn-глицеро-3-фосфохолин, фосфатидилхолин и лецитин из бобов сои.
Что касается, в случае добавления такого фосфолипида, соотношения в смеси между модифицированным PEG фосфолипидом и фосфолипидом в носителе по настоящему изобретению, то фосфолипид соответствующим образом представлен в пределах от 0,03 до 100 массовых частей на 1 массовую часть модифицированного PEG фосфолипида, предпочтительно в пределах от 0,05 до 20 массовых частей, более предпочтительно в пределах от 0,2 до 1,1 массовой части.
К носителю по настоящему изобретению, кроме модифицированного PEG фосфолипида и соединения A, которые являются существенными компонентами, можно дополнительно добавлять холестерин. Что касается, в случае добавления холестерина, соотношения в смеси между модифицированным PEG фосфолипида и холестерином в носителе по настоящему изобретению, то холестерин соответствующим образом представлен в пределах от 0,01 до 200 массовых частей на 1 массовую часть модифицированного PEG фосфолипида, предпочтительно в пределах от 0,02 до 100 массовых частей.
Дисперсию носителя по настоящему изобретению можно получать смешиванием, например, модифицированного PEG фосфолипида и соединения A; модифицированного PEG фосфолипида, соединения A и фосфолипида; или модифицированного PEG фосфолипида, соединения A и холестерина и диспергированием компонентов в водном растворе по общепринятому способу. Процедуру диспергирования можно выполнять посредством подходящего прибора, такого как ультразвуковой диспергатор или диспергатор для эмульгирования.
III. Композиция по настоящему изобретению
Размер частицы носителя по настоящему изобретению, содержащей лекарственное средство, где носитель содержится в композиции по настоящему изобретению, конкретно не ограничен и соответствующим образом представлен в пределах от, например, 50 нм до 200 нм, предпочтительно в пределах от 60 нм до 150 нм.
Примеры “лекарственного средства”, которое можно использовать в композиции по настоящему изобретению, включают водорастворимые анионные соединения, противоопухолевые средства, противовирусные средства и антибиотики. Их конкретные примеры включают нуклеиновые кислоты, такие как одноцепочечные или двухцепочечные РНК, одноцепочечные или двухцепочечные ДНК и олигонуклеиновые кислоты, кислые сахара, такие как гепаран-сульфат и декстран сульфат, цитокины, вторичные мессенджеры, такие как циклические АМФ, АТФ и IP3, пенициллины и цефалоспорины, витамины, такие как витамин C и ретинолы, и другие, имеющиеся лекарственные средства с кислотной группой, такие как интерфероны (α, β, γ), интерлейкины (IL-1, IL-2), колониестимулирующие факторы (CSF), фактор некроза опухолей (TNF), левамизол, пестатин, ретиноевая кислота, 5-фторурацил (5-FU), цитозинарабинозид (Ara-C), аденинарабинозид (Ara-A), цисплатин (CDDP), циклофосфамид и азидотимидин (AZT).
Примеры синтетической двухцепочечной РНК включают те, которые описаны ниже.
1. Комплексы гомополимер-гомополимер
Полиинозиновая кислота-полицитидиловая кислота
Полиинозиновая кислота-поли(5-бромцитидиловая кислота)
Полиинозиновая кислота-поли(2-тиоцитидиловая кислота)
Поли(7-деазаинозиновая кислота)-полицитидиловая кислота
Поли(7-деазаинозиновая кислота)-поли(5-бромцитидиловая кислота)
Поли(2'-азидоинозиновая кислота)-полицитидиловая кислота
Полиинозиновая кислота-поли(цитидин-5'-тиофосфорная кислота)
2. Комплексы гомополимер-сополимер
Полиинозиновая кислота-поли(цитидиловая кислота, уридиловая кислота)
Полиинозиновая кислота-поли(цитидиловая кислота, 4-тиоуридиловая кислота)
3. Комплексы синтетическая нуклеиновая кислота и поликатион
Полиинозиновая кислота-полицитидиловая кислота-поли-L-лизин
4. Другие
Полиинозиновая кислота-поли(1-винилцитидиловая кислота)
Примеры олигонуклеиновой кислоты включают РНК, ДНК и их соединения, которые содержат от 10 до 3000 нуклеиновых оснований, предпочтительно от 15 до 2000 нуклеиновых оснований, более предпочтительно от 18 до 1000 нуклеиновых оснований на молекулу, например, миРНК, мкРНК, кшРНК, некодирующая РНК, антисмысловая ДНК, антисмысловая РНК, ДНК - ферменты, рибозимы и аптамеры.
Олигонуклеиновая кислота не ограничена встречающимися в природе видами, и по меньшей мере некоторую часть сахарного, фосфатного остова или т.п., которые образованы ее нуклеотидами, можно модифицировать для увеличения стабильности in vivo, такой как устойчивость к нуклеазам. Примеры такой модификации включают модификацию рибозы в 2'-положении, модификацию рибозы в других положениях и модификации фосфатного остова. Примеры модификации рибозы в 2'-позиции включают модификации посредством замещения гидроксильной группы в 2'-положении рибозы на H, OR5, R5, R6OR5, SH, SR5, NH2, NHR5, N(R5)2, N3, CN, F, Cl, Br и I. В настоящей работе R5 представлен алкилом или арилом, и R6 представлен алкиленом.
Алкил R5 конкретно не ограничен по образованию линейной или разветвленной цепи, и его примеры включают алкил, имеющий от 1 до 6 атомов углерода. Его конкретные примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, неопентил, трет-пентил, н-гексил и изогексил. Алкил можно замещать 1-3 заместителями, включая, например, галоген, алкил, алкокси, циано и нитро. Примеры галогена включают фтор, хлор, бром и йод. Примеры алкила включают те же группы, что и описанные выше для алкила. Алкокси конкретно не ограничен по образованию линейной или разветвленной цепи, и его примеры включают алкокси, имеющий от 1 до 6 атомов углерода. Его конкретные примеры включают метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси, н-пентокси, изопентокси, н-гексилокси и изогексилокси. Среди них особенно предпочтительным является алкокси, имеющий 1-3 атома углерода.
Примеры арила в R5 включают арил, имеющий от 6 до 10 атомов углерода. Конкретные примеры арил включают фенил, α-нафтил и β-нафтил. Среди них особенно предпочтительным является фенил.
Алкилен в R6 конкретно не ограничен по образованию линейной или разветвленной цепей, и его примеры включают алкилен, имеющий от 1 до 6 атомов углерода. Конкретные примеры алкилена включают метилен, этилен, триметилен, тетраметилен, пентаметилен, гексаметилен, 2-(этил)триметилен и 1-(метил)тетраметилен.
Примеры модификаций рибозы в остальных положениях включают 4'-тио-модификации. Примеры модификаций фосфатного остова включают фосфотиоатные, фосфодитиоатные, алкилфосфонатные и фосфорамидатные модификации.
Массовое отношение (носитель по настоящему изобретению / лекарственное средство) носителя по настоящему изобретению к лекарственному средству, включенному в композицию по настоящему изобретению, изменяется в зависимости от вида лекарственного средства, соотношения в смеси модифицированного PEG фосфолипида или соединения A в носителе по настоящему изобретению и т.д. Массовое отношение соответствующим образом представлено в пределах от 0,01 до 1000, предпочтительно в пределах от 10 до 300, более предпочтительно в пределах от 100 до 200. В случае, если лекарственное средство, включенное в композицию, представляет собой олигонуклеиновую кислоту, массовое отношение соответствующим образом представлено в пределах от 0,01 до 100, предпочтительно в пределах от 1 до 50, более предпочтительно в пределах от 5 до 30.
В композицию по настоящему изобретению, кроме указанного выше носителя по настоящему изобретению и лекарственного средства, можно, по мере необходимости, примешивать фармацевтически приемлемую добавку. Примеры добавки включают эмульгирующие вспомогательные средства (такие, как жирные кислоты, имеющие от 6 до 22 атомов углерода и их фармацевтически приемлемые соли, альбумин и декстран), стабилизаторы (такие, как холестерин и фосфатидная кислота), средства, регулирующие тоничность (такие, как хлорид натрия, глюкоза, мальтоза, лактоза, сахароза и трегалоза), и средства, регулирующие pH (такие, как соляная кислота, серная кислота, фосфорная кислота, уксусная кислота, гидроксид натрия, гидроксид калия и триэтаноламин). Их можно использовать индивидуально или в комбинации из двух или более.
Композицию по настоящему изобретению можно получать, добавляя лекарственное средство к дисперсии носителя по настоящему изобретению и посредством размешивания, соответствующим образом, полученной смеси. Композицию по настоящему изобретению также можно получать, добавляя лекарственное средство в процессе получения носителя настоящего изобретения. Указанную выше добавку можно добавлять в надлежащее время процесса или до, или после процедуры диспергирования.
Композицию по настоящему изобретению можно получать, например, в виде жидкого препарата или лиофилизированного препарата. В случае жидкого препарата концентрация носителя по настоящему изобретению, содержащегося в композиции по настоящему изобретению, соответствующим образом представлена в пределах от 0,001 мас./об.% до 50 мас./об.%, предпочтительно в пределах от 0,01 мас./об.% до 25 мас./об.%, более предпочтительно в пределах от 0,1 мас./об.% до 10 мас./об.%.
Лиофилизированный препарат можно получать, подвергая процедуре лиофилизации, посредством общепринятого способа, композицию по настоящему изобретению в форме жидкого препарата. Например, процедуру лиофилизации можно осуществлять так, как указано далее. После того как композицию по настоящему изобретению в форме жидкого препарата соответствующим образом стерилизуют, заданный объем переносят в пробирку с последующим предварительным замораживанием в условиях приблизительно от -40°C до -20°C в течение приблизительно 2 часов. Затем композицию подвергают первичному высушиванию при пониженном давлении при приблизительно от 0°C до 10°C и затем вторичному высушиванию при пониженном давлении при приблизительно от 15°C до 25°C. В основном, внутрь пробирки помещают газообразный азот, пробирку запечатывают, таким образом можно получать лиофилизированный препарат композиции по настоящему изобретению.
Лиофилизированный препарат композиции по настоящему изобретению, как правило, можно получать, добавляя подходящий раствор (раствор для повторного растворения) для повторного растворения препарата. Примеры раствора для повторного растворения включают воду для инъекций, физиологический раствор и другие общепринятые инфузионные растворы. Объем жидкости для раствора, для повторного растворения, изменяется в зависимости от его применения и т.д. и конкретно не ограничен, и соответствующим образом объем жидкости в растворе от 0,5 до 2 раз превышает объем жидкости в композиции по настоящему изобретению до лиофилизации или 500 мл или менее.
Заболевание, для которого можно применять композицию по настоящему изобретению, конкретно не ограничено и примеры заболевания включают злокачественную опухоль, вирусные, воспалительные, метаболические и неврологические заболевания.
Путь введения композиции по настоящему изобретению конкретно не ограничен, при условии, что он является фармацевтически приемлемым путем введения и может быть выбран в соответствии со способом лечения. Примеры пути введения включают внутривенное введение, внутриартериальное введение, пероральное введение, транспульмональное введение, введение внутрь ткани, трансдермальное введение, введение внутрь слизистой оболочки, ректальное введение, введение внутрь мочевого пузыря, интраперитонеальное введение, внутриглазное введение, внутрицеребральное введение и внутригрудное введение. Среди них внутривенное введение, трансдермальное введение и введение внутрь слизистой оболочки являются особенно предпочтительными. Лекарственная форма композиции по настоящему изобретению конкретно не ограничена, и примеры лекарственной формы включают различные формы для инъекций, оральные средства, формы для инфузий, ингаляций, глазные капли, мази, примочки и суппозитории.
Дозу композиции по настоящему изобретению в виде лекарственного средства устанавливают, предпочтительно, учитывая вид и лекарственную форму лекарственного средства, состояние пациента, такое как возраст и масса тела, путь введения и природу и тяжесть заболевания. Как правило, доза находится в пределах от 0,01 мг до 10 г/человека/сутки, предпочтительно в пределах от 0,1 мг до 5 г/человека/сутки в качестве дозы лекарственного средства для взрослого. В случае, если лекарственное средство, содержащееся в композиции по настоящему изобретению, представляет собой олигонуклеиновую кислоту, то доза олигонуклеиновой кислоты для взрослого находится, как правило, в пределах от 0,1 мг до 10 г/человека/сутки, предпочтительно в пределах от 1 мг до 5 г/человека/сутки. Численные значения иногда изменяются в зависимости от вида целевого заболевания, пути введения и целевой молекулы. Таким образом, в некоторых случаях, доза олигонуклеиновой кислоты может быть достаточной, когда она ниже диапазона, описанного выше. В некоторых случаях может требоваться доза выше диапазона, описанного выше. Дозу можно вводить один раз в сутки или несколько раз в сутки, или можно вводить в интервалах от одних до нескольких суток.
Примеры
Далее в настоящем документе настоящее изобретение будет более подробно проиллюстрировано со ссылкой на примеры получения, сравнительные примеры и примеры тестирования. Однако настоящее изобретение не ограничено объемом, описанным ниже.
Пример получения 1: Синтез олиго РНК
Используя автоматический синтезатор нуклеиновых кислот (Expedite 8909, произведено Applied BioSystems, Inc.), олиго РНК с нуклеотидной последовательностью, представленной SEQ ID NO: 1, олиго РНК с нуклеотидной последовательностью, представленной SEQ ID NO: 2, олиго РНК с нуклеотидной последовательностью, представленной SEQ ID NO: 3, и олиго РНК с нуклеотидной последовательностью, представленной SEQ ID NO: 4, синтезировали амидитным способом, описанным в документе (Nucleic Acid Research, 1984, Vol. 12, pp. 4539-4557).
Защитные группы основных молекул удалили отщеплением от CPG с использованием жидкости, смешанной из концентрированного гидроксида аммония и этанола (3/1) и, дополнительно, реакцией в том же растворе при 55°C в течение 18 часов. Затем, удалили защитную силильную группу в 2'-положении реакцией при комнатной температуре в течение 20 часов, используя 1 М раствор фторида тетрабутиламмония в тетрагидрофуране. Полученную, в результате, олиго РНК очистили обратно-фазовой хроматографией. Далее удалили защитную диметокситритильную группу в 5'-положении реакцией при комнатной температуре в течение 30 минут, используя 80% водный раствор уксусной кислоты, и затем полученную, в результате, олиго РНК снова очистили ионообменной хроматографией. Концентрации полученных олиго РНК с нуклеотидной последовательностью, представленной SEQ ID NO: 1, олиго РНК с нуклеотидной последовательностью, представленной SEQ ID NO: 2, олиго РНК с нуклеотидной последовательностью, представленной SEQ ID NO: 3, и олиго РНК с нуклеотидной последовательностью, представленной SEQ ID NO: 4, составили 3,37 мг/мл, 3,45 мг/мл, 10,00 мг/мл и 10,00 мг/мл, соответственно.
В данном случае капиллярным электрофорезом подтвердили, что 90% или более полученных олиго РНК являлись полноразмерными РНК.
Пример получения 2: Синтез меченной тритием двухцепочечной олиго РНК
Меченные тритием двухцепочечные олиго РНК, состоящие из олиго РНК с нуклеотидной последовательностью, представленной SEQ ID NO: 1, и олиго РНК с нуклеотидной последовательностью, представленной SEQ ID NO: 2, синтезировали включением меченной тритием [2,5',8-3H] аденозин-5'-трифосфата аммониевой соли (произведено Amersham BioSciences, Inc.), используя набор для in vitro транскрипции с Т7 (произведено Takara-Bio Co., Ltd.).
Затем, белки удалили из двухцепочечной олиго РНК, используя смешанный раствор фенол/хлороформа, и дополнительно мономеры, не вступившие в реакцию, удалили, используя спин-колонки G-25 (произведено Pharmacia, Inc.). Концентрация полученной двухцепочечной олиго РНК составила 4,07 мг/мл. Дополнительно, ее удельная радиоактивность составила 5,3×105 распадов в минуту/мкг.
В данном случае посредством электрофореза в 15% полиакриламиде подтвердили, что длина цепи полученной двухцепочечной олиго РНК составила приблизительно 21 пару оснований.
Пример получения 3: Синтез 1,3-дистеарилглицеро-2-фосфатидил-N-(метокси-полиэтиленгликоль-сукцинил)этаноламина
Стадия 1: Синтез 1,3-дистеарилглицерина
3 г димера дигидроксиацетона, 22,7 г стеариновой кислоты, 16,8 г 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорида и 10,8 г 4-диметиламинопиридина перемешивали в 100 мл дихлорметана в течение ночи при комнатной температуре. К реакционному раствору добавили 0,5 л метанола и полученный, в результате, порошок выделили фильтрацией, промыли метанолом и высушили. 6 г полученного порошка суспендировали в жидкости, смешанной из 400 мл тетрагидрофурана и 20 мл 10% водного раствора уксусной кислоты. К полученной, в результате, суспензии маленькими порциями при 0°C добавили 1,1 г борогидрида натрия. После того как полученную, в результате, смесь перемешивали при комнатной температуре в течение 6 часов, реакционный раствор перелили в концентрированный водный раствор бикарбоната натрия и выполнили экстракцию этилацетатом. Органический слой высушили, а затем сконцентрировали при пониженном давлении. Полученный, в результате, осадок очистили хроматографией на колонках с силикагелем, посредством чего получили 3,5 г целевого продукта.
Стадия 2: Синтез тетразолида диизопропиламина
365 мг тетразола растворили в 8 мл ацетонитрила и к ним по каплям добавили 1,20 г диизопропиламина. Полученную, в результате, смесь перемешивали при комнатной температуре в течение 20 минут, и реакционный раствор сконцентрировали при пониженном давлении, с последующей сушкой, посредством чего получили 852 мг целевого продукта.
Стадия 3: Синтез 1,3-дистеарилглицерин 2-О-(2-цианоэтил-N,N-диизопропилфосфорамидита)
1,39 г 1,3-дистеарилглицерина, полученного выше в стадии 1, суспендировали в жидкости, смешанной из 30 мл ацетонитрила и 10 мл дихлорметана, и к ним добавили 456 мг тетразолида диизопропиламина, полученного выше в стадии 2, и 1 г bis(N, N-диизопропиламин)цианоэтилфосфита, и полученную, в результате, смесь перемешивали при 40°C в течение 1,5 часов. Реакционный раствор подвергли фильтрации, и фильтрат сконцентрировали. Полученный, в результате, осадок очистили хроматографией на колонках с силикагелем, посредством чего получили 800 мг целевого продукта.
31P ЯМР (202 МГц, CDCl3, δ): 152,283
Стадия 4: Синтез 1,3-дистеарилглицерин 2-О-(2-цианоэтил 2- трет-бутоксикарбониламиноэтилфосфата)
700 мг 1,3-дистеарилглицерин 2-О-(2-цианоэтил-N,N-диизопропилфосфорамидита), полученного выше в стадии 3, 114 мг трет-бутил N-(2-гидроксиэтил)карбамата и 119 мг тетразола растворили в жидкости, смешанной из 5 мл ацетонитрила и 5 мл дихлорметана вместе с 0,5 г молекулярных сит 4 A, и полученную, в результате, смесь перемешивали при комнатной температуре в течение 30 минут. К реакционному раствору добавили 20 мл раствора йода (0,1 М йод / тетрагидрофуран:пиридин:вода = 7:1:2), и далее полученную, в результате, смесь дополнительно перемешивали при комнатной температуре в течение 20 минут. Реакционный раствор подвергли фильтрации, и к фильтрату добавляли насыщенный водный раствор тиосульфата натрия до тех пор, пока не исчез цвет йода. Затем выполнили экстракцию этилацетатом. Органический слой высушили и затем сконцентрировали при пониженном давлении. Полученный, в результате, осадок очистили хроматографией на колонках с силикагелем, посредством чего получили 700 мг целевого продукта.
31P ЯМР (202 МГц, CDCl3, δ): 0,12453
Масса MALDI-TOF (m/z) = 923,564 ([M+Na]+)
Стадия 5: Синтез 1,3-дистеарилглицерин 2-О-(2-аминоэтилфосфата)
15 мл жидкости, смешанной из пиридина, триэтиламина и воды (3:1:1), добавили к 660 мг 1,3-дистеарилглицерин 2-О-(2-цианоэтил 2-трет-бутоксикарбониламиноэтилфосфата), полученного выше в стадии 4, и полученную, в результате, смесь перемешивали при комнатной температуре в течение 2 часов. После того как реакционный раствор сконцентрировали при пониженном давлении, три раза выполнили азеотропную дистилляцию с пиридином. После этого дополнительно 3 раза выполнили азеотропную дистилляцию с дихлорметаном. Полученный, в результате, осадок растворили в 5 мл дихлорметана, к нему при 0°C добавили 5 мл трифторуксусной кислоты, и полученную, в результате, смесь перемешивали при комнатной температуре в течение 30 минут. После того как реакционный раствор сконцентрировали при пониженном давлении, 3 раза выполнили азеотропную дистилляцию с дихлорметаном, посредством чего получили 410 мг целевого продукта.
31P ЯМР (202 МГц, CDCl3, δ):-0,0833
Стадия 6: Синтез 1,3-дистеарилглицеро-2-фосфатидил-N-(метокси-полиэтиленгликоль-сукцинил)этаноламина
40 мл диметоксиэтана, 40 мл дихлорметана и 10 мл насыщенного водного раствора бикарбоната натрия добавили к 400 мг 1,3-дистеарилглицерин 2-О-(2-аминоэтил фосфата), полученного выше в стадии 5, и 1,24 г α-сукцинимидилоксисукцинил-ω-метоксиполиоксиэтилена [SUNBRIGHT (зарегистрированный товарный знак) ME-020CS, произведено NOF Corporation], и полученную, в результате, смесь перемешивали в течение ночи при комнатной температуре. После того как к реакционному раствору добавили воду, 3 раза выполнили экстракцию дихлорметаном. Органический слой высушили и затем сконцентрировали при пониженном давлении. Полученный, в результате, осадок очистили хроматографией на колонках с силикагелем, посредством чего получили 950 мг целевого продукта.
Молекулярную массу полученного продукта определили масс-спектрометрией, используя способ ионизации электрораспылением. В результате, как показано на фиг.1, у полученного продукта распределение молекулярной массы располагалось в пределах от 2000 до 3800.
Пример получения 4: Получение дисперсии носителя лекарственного средства
60 мг соединения A, 8 мг модифицированного ПЭГ фосфолипида, синтезированного в примере получения 3, и 92 мг 1-пальмитоил-2-олеоил-sn-глицеро-3-фосфохолина (произведено NOF Corporation, далее в настоящем документе применяют то же самое) растворили в 2 мл хлороформа в пробирке, которую продули азотным газом для того, чтобы удалить хлороформ, с формированием, таким образом, тонкой пленки на стенке пробирки. После того как пробирку оставили стоять в течение ночи при пониженном давлении, в пробирку добавили 1000 мг мальтозы (произведено Otsuka Pharmaceutical Co., Ltd.), 4,0 мл воды для инъекций (произведено Otsuka Pharmaceutical Co., Ltd., далее в настоящем документе применяют то же самое) и 81 мл 1 Н соляной кислоты, и тонкую пленку диспергировали, используя вихревую мешалку. После того как дисперсию оставили стоять при 4°C в течение 3 часов, осуществили обработку ультразвуком, используя микрозонд, в течение 10 минут, получая, таким образом, 32 мг/мл дисперсии носителя лекарственного средства. Затем, объем дисперсии увеличили до 5,0 мл водой для инъекций.
Пример получения 5: Получение дисперсии носителя лекарственного средства
Дисперсию носителя лекарственного средства получили тем же способом, что и в примере получения 4, используя 60 мг соединения A, 16 мг модифицированного ПЭГ фосфолипида, синтезированного в примере получения 3, и 84 мг 1-пальмитоил-2-олеоил-sn-глицеро-3-фосфохолина.
Пример получения 6: Получение дисперсии носителя лекарственного средства
Дисперсию носителя лекарственного средства получили тем же способом, что и в примере получения 4, используя 60 мг соединения A, 32 мг модифицированного ПЭГ фосфолипида, синтезированного в примере получения 3, и 68 мг 1-пальмитоил-2-олеоил-sn-глицеро-3-фосфохолина.
Пример получения 7: Получение дисперсии носителя лекарственного средства
Дисперсию носителя лекарственного средства получили тем же способом, что и в примере получения 4, используя 60 мг соединения A, 48 мг модифицированного ПЭГ фосфолипида, синтезированного в примере получения 3, и 52 мг 1-пальмитоил-2-олеоил-sn-глицеро-3-фосфохолина.
Пример получения 8: Получение дисперсии носителя лекарственного средства
Дисперсию носителя лекарственного средства получили тем же способом, что и в примере получения 4, используя 60 мг соединения A, 64 мг модифицированного ПЭГ фосфолипида, синтезированного в примере получения 3, и 36 мг 1-пальмитоил-2-олеоил-sn-глицеро-3-фосфохолина.
Пример получения 9: Получение дисперсии носителя лекарственного средства
Дисперсию носителя лекарственного средства получили тем же способом, что и в примере получения 4, используя 60 мг соединения A, 80 мг модифицированного ПЭГ фосфолипида, синтезированного в примере получения 3, и 20 мг 1-пальмитоил-2-олеоил-sn-глицеро-3-фосфохолина.
Пример получения 10: Получение дисперсии носителя лекарственного средства
Дисперсию носителя лекарственного средства получили тем же способом, что и в примере получения 4, используя 60 мг соединения A, 88 мг модифицированного ПЭГ фосфолипида, синтезированного в примере получения 3, и 12 мг 1-пальмитоил-2-олеоил-sn-глицеро-3-фосфохолина.
Пример получения 11: Получение дисперсии носителя лекарственного средства
Дисперсию носителя лекарственного средства получили тем же способом, что и в примере получения 4, используя 60 мг соединения A, и 100 мг модифицированного ПЭГ фосфолипида, синтезированного в примере получения 3.
Пример получения 12: Получение дисперсии носителя лекарственного средства
Дисперсию носителя лекарственного средства получили тем же способом, что и в примере получения 4, используя 60 мг соединения A, 32 мг N-(метокси-полиэтиленгликоль-сукцинил) дистеароилфосфатидилэтаноламина [SUNBRIGHT (зарегистрированный товарный знак) DSPE-020C, произведено NOF Corporation, далее в настоящем документе применяют то же самое] (далее в настоящем документе обозначаемый как “соединение В”) и 20 мг 1-пальмитоил-2-олеоил-sn-глицеро-3-фосфохолина.
В данном случае молекулярную массу используемого соединения B определили масс-спектрометрией, используя способ ионизации электрораспылением. В результате, как показано на фиг.2, у используемого соединения B распределение молекулярной массы лежит в пределах от 2200 до 3600.
Пример получения 13: Получение дисперсии носителя лекарственного средства
Дисперсию носителя лекарственного средства получили тем же способом, что и в примере получения 4, используя 60 мг соединения A, 48 мг соединения B и 52 мг 1-пальмитоил-2-олеоил-sn-глицеро-3-фосфохолина.
Пример получения 14: Получение дисперсии носителя лекарственного средства
Дисперсию носителя лекарственного средства получили тем же способом, что и в примере получения 4, используя 60 мг соединения A, 80 мг соединения B и 20 мг 1-пальмитоил-2-олеоил-sn-глицеро-3-фосфохолина.
Пример получения 15: Получение дисперсии носителя лекарственного средства
Дисперсию носителя лекарственного средства получили тем же способом, что и в примере получения 4, используя 60 мг соединения A, 88 мг соединения B и 12 мг 1-пальмитоил-2-олеоил-sn-глицеро-3-фосфохолина.
Пример получения 16: Получение дисперсии носителя лекарственного средства
Дисперсию носителя лекарственного средства получили тем же способом, что и в примере получения 4, используя 60 мг соединения A, 80 мг модифицированного ПЭГ фосфолипида, синтезированного в примере получения 3, и 20 мг лецитина яичного желтка (произведено QP Corporation, далее в настоящем документе применяют то же самое).
Пример получения 17: Получение фармацевтической композиции
(1) Получение раствора нуклеиновой кислоты
Раствор нуклеиновой кислоты получили смешиванием 36 мл меченной тритием двухцепочечной олиго РНК, синтезированной в примере получения 2, 1,50 мл олиго РНК с нуклеотидной последовательностью, представленной SEQ ID NO: 1, синтезированной в примере получения 1, 1,43 мл олиго РНК с нуклеотидной последовательностью, представленной SEQ ID NO: 2, синтезированной в примере получения 1, и 2,07 мл воды для инъекций.
(2) Получение фармацевтической композиции
5 мл дисперсии носителя лекарственного средства, полученного в примере получения 4, добавили к общему количеству раствора нуклеиновой кислоты, полученного выше (1), и выполнили разрушение ультразвуком в течение 5 минут. Затем, полученный, в результате, раствор центрифугировали при 5000 об/мин в течение 20 минут и фильтровали через 0,22 мкм фильтр, посредством чего получили 1,0 мг/мл фармацевтической композиции.
(3) Измерение средней величины частиц носителя лекарственного средства
Среднюю величину частиц (средний объем) носителя лекарственного средства в фармацевтической композиции измерили разведением водой для инъекций композиции настоящего изобретения, полученной выше (2), до 0,02 мг/мл. Конкретно, среднюю величину частиц носителя лекарственного средства измерили трижды, используя устройство для измерения величины частиц [Nicomp C380 (зарегистрированный товарный знак), произведено Particle Sizing Systems, Inc., далее в настоящем документе применяют то же самое], устанавливая показатель преломления 0,993, вязкость 1,333 и интервал времени измерения 5 минут. В результате, средняя величина частиц носителя лекарственного средства в фармацевтической композиции составила 102,7 нм.
Пример получения 18: Получение фармацевтической композиции
(1) Получение раствора нуклеиновой кислоты
Раствор нуклеиновой кислоты получили тем же способом, что и в примере получения 17 (1).
(2) Получение фармацевтической композиции
1,0 мг/мл фармацевтической композиции получили тем же способом, что и в примере получения 17 (2), используя общее количество раствора нуклеиновой кислоты, полученной выше (1), и 5 мл дисперсии носителя лекарственного средства, полученной в примере получения 5.
В данном случае среднюю величину частиц носителя лекарственного средства в фармацевтической композиции измерили тем же способом, что и в примере получения 17 (3), и она составила 104,0 нм.
Пример получения 19: Получение фармацевтической композиции
(1) Получение раствора нуклеиновой кислоты
Раствор нуклеиновой кислоты получили тем же способом, что и в примере получения 17 (1).
(2) Получение фармацевтической композиции
1,0 мг/мл фармацевтической композиции получили тем же способом, что и в примере получения 17 (2), используя общее количество раствора нуклеиновой кислоты, полученного выше (1), и 5 мл дисперсии носителя лекарственного средства, полученной в примере получения 6.
В данном случае среднюю величину частиц носителя лекарственного средства в фармацевтической композиции измерили тем же способом, что и в примере получения 17 (3), и она составила 103,4 нм.
Пример получения 20: Получение фармацевтической композиции
(1) Получение раствора нуклеиновой кислоты
Раствор нуклеиновой кислоты получили тем же способом, что и в примере получения 17 (1).
(2) Получение фармацевтической композиции
1,0 мг/мл фармацевтической композиции получили тем же способом, что и в примере получения 17 (2), используя общее количество раствора нуклеиновой кислоты, полученного выше (1), и 5 мл дисперсии носителя лекарственного средства, полученной в примере получения 7.
В данном случае среднюю величину частиц носителя лекарственного средства в фармацевтической композиции измерили тем же способом, что и в примере получения 17 (3), и она составила 102,5 нм.
Пример получения 21: Получение фармацевтической композиции
(1) Получение раствора нуклеиновой кислоты
Раствор нуклеиновой кислоты получили тем же способом, что и в примере получения 17 (1).
(2) Получение фармацевтической композиции
1,0 мг/мл фармацевтической композиции получили тем же способом, что и в примере получения 17 (2), используя общее количество раствора нуклеиновой кислоты, полученного выше (1), и 5 мл дисперсии носителя лекарственного средства, полученной в примере получения 8.
В данном случае среднюю величину частиц носителя лекарственного средства в фармацевтической композиции измерили тем же способом, что и в примере получения 17 (3), и она составила 97,9 нм.
Пример получения 22: Получение фармацевтической композиции
(1) Получение раствора нуклеиновой кислоты
Раствор нуклеиновой кислоты получили тем же способом, что и в примере получения 17 (1).
(2) Получение фармацевтической композиции
1,0 мг/мл фармацевтической композиции получили тем же способом, что и в примере получения 17 (2), используя общее количество раствора нуклеиновой кислоты, полученного выше (1), и 5 мл дисперсии носителя лекарственного средства, полученной в примере получения 9.
В данном случае среднюю величину частиц носителя лекарственного средства в фармацевтической композиции измерили тем же способом, что и в примере получения 17 (3), и она составила 91,6 нм.
Пример получения 23: Получение фармацевтической композиции
(1) Получение раствора нуклеиновой кислоты
Раствор нуклеиновой кислоты получили тем же способом, что и в примере получения 17 (1).
(2) Получение фармацевтической композиции
1,0 мг/мл фармацевтической композиции получили тем же способом, что и в примере получения 17 (2), используя общее количество раствора нуклеиновой кислоты, полученного выше (1), и 5 мл дисперсии носителя лекарственного средства, полученной в примере получения 10.
В данном случае среднюю величину частиц носителя лекарственного средства в фармацевтической композиции измерили тем же способом, что и в примере получения 17 (3), и она составила 65,1 нм.
Пример получения 24: Получение фармацевтической композиции
(1) Получение раствора нуклеиновой кислоты
Раствор нуклеиновой кислоты получили тем же способом, что и в примере получения 17 (1).
(2) Получение фармацевтической композиции
1,0 мг/мл фармацевтической композиции получили тем же способом, что и в примере получения 17 (2), используя общее количество раствора нуклеиновой кислоты, полученного выше (1), и 5 мл дисперсии носителя лекарственного средства, полученной в примере получения 11.
В данном случае среднюю величину частиц носителя лекарственного средства в фармацевтической композиции измерили тем же способом, что и в примере получения 17 (3), и она составила 63,5 нм.
Пример получения 25: Получение фармацевтической композиции
(1) Получение раствора нуклеиновой кислоты
Раствор нуклеиновой кислоты получили тем же способом, что и в примере получения 17 (1).
(2) Получение фармацевтической композиции
Фармацевтическую композицию в 1,0 мг/мл получили тем же способом, что и в примере получения 17 (2), используя общее количество раствора нуклеиновой кислоты, полученного выше (1), и 5 мл дисперсии носителя лекарственного средства, полученной в примере получения 12.
В данном случае среднюю величину частиц носителя лекарственного средства в фармацевтической композиции измерили тем же способом, что и в примере получения 17 (3), и она составила 97,3 нм.
Пример получения 26: Получение фармацевтической композиции
(1) Получение раствора нуклеиновой кислоты
Раствор нуклеиновой кислоты получили тем же способом, что и в примере получения 17 (1).
(2) Получение фармацевтической композиции
1,0 мг/мл фармацевтической композиции получили тем же способом, что и в примере получения 17 (2), используя общее количество раствора нуклеиновой кислоты, полученного выше (1), и 5 мл дисперсии носителя лекарственного средства, полученной в примере получения 13.
В данном случае среднюю величину частиц носителя лекарственного средства в фармацевтической композиции измерили тем же способом, что и в примере получения 17 (3), и она составила 97,7 нм.
Пример получения 27: Получение фармацевтической композиции
(1) Получение раствора нуклеиновой кислоты
Раствор нуклеиновой кислоты получили тем же способом, что и в примере получения 17 (1).
(2) Получение фармацевтической композиции
Фармацевтическую композицию в 1,0 мг/мл получили тем же способом, что и в примере получения 17 (2), используя общее количество раствора нуклеиновой кислоты, полученного выше (1), и 5 мл дисперсии носителя лекарственного средства, полученной в примере получения 14.
В данном случае среднюю величину частиц носителя лекарственного средства в фармацевтической композиции измерили тем же способом, что и в примере получения 17 (3), и она составила 98,0 нм.
Пример получения 28: Получение фармацевтической композиции
(1) Получение раствора нуклеиновой кислоты
Раствор нуклеиновой кислоты получили тем же способом, что и в примере получения 17 (1).
(2) Получение фармацевтической композиции
1,0 мг/мл фармацевтической композиции получили тем же способом, что и в примере получения 17 (2), используя общее количество раствора нуклеиновой кислоты, полученного выше (1), и 5 мл дисперсии носителя лекарственного средства, полученной в примере получения 15.
В данном случае среднюю величину частиц носителя лекарственного средства в фармацевтической композиции измерили тем же способом, что и в примере получения 17 (3), и она составила 78,2 нм.
Пример получения 29: Получение фармацевтической композиции
(1) Получение раствора нуклеиновой кислоты
Раствор нуклеиновой кислоты получили тем же способом, что и в примере получения 17 (1).
(2) Получение фармацевтической композиции
1,0 мг/мл фармацевтической композиции получили тем же способом, что и в примере получения 17 (2), используя общее количество раствора нуклеиновой кислоты, полученного выше (1), и 5 мл дисперсии носителя лекарственного средства, полученной в примере получения 16.
В данном случае среднюю величину частиц носителя лекарственного средства в фармацевтической композиции измерили тем же способом, что и в примере получения 17 (3), и она составила 92,7 нм.
Пример получения 30: Получение фармацевтической композиции
(1) Получение раствора нуклеиновой кислоты
Раствор нуклеиновой кислоты получили тем же способом, что и в примере получения 17 (1).
(2) Получение фармацевтической композиции
1,0 мг/мл фармацевтической композиции получили тем же способом, что и в примере получения 17 (2), используя общее количество раствора нуклеиновой кислоты, полученного выше (1), и 5 мл дисперсии носителя лекарственного средства, полученной в примере получения 9.
В данном случае среднюю величину частиц носителя лекарственного средства в фармацевтической композиции измерили тем же способом, что и в примере получения 17 (3), и она составила 84,9 нм.
Пример получения 31: Получение фармацевтической композиции
(1) Получение раствора нуклеиновой кислоты
Раствор нуклеиновой кислоты получили смешиванием 0,50 мл олиго РНК с нуклеотидной последовательностью, представленной SEQ ID NO: 3, синтезированной в примере получения 1, 0,50 мл олиго РНК с нуклеотидной последовательностью, представленной SEQ ID NO: 4, синтезированной в примере получения 1, и 4,0 мл воды для инъекций.
(2) Получение композиции по настоящему изобретению
1,0 мг/мл фармацевтической композиции получили тем же способом, что и в примере получения 17 (2), используя общее количество раствора нуклеиновой кислоты, полученного выше (1), и 5 мл дисперсии носителя лекарственного средства, полученной в примере получения 9.
В данном случае среднюю величину частиц носителя лекарственного средства в фармацевтической композиции измерили тем же способом, что и в примере получения 17 (3), и она составила 95,7 нм.
Кроме того, двухцепочечная олиго РНК, состоявшая из олиго РНК с нуклеотидной последовательностью, представленной SEQ ID NO: 3, и олиго РНК с нуклеотидной последовательностью, представленной SEQ ID NO: 4, представляла собой двухцепочечную олиго РНК, с ингибиторной активностью относительно экспрессии Bcl-2 (см. WO 2004/106511).
Сравнительный пример 1: Получение, в качестве контроля сравнения, дисперсии носителя лекарственного средства
Дисперсию носителя лекарственного средства в качестве контроля сравнения получили тем же способом, что и в примере получения 4, используя 60 мг соединения A и 100 мг лецитина яичного желтка.
Сравнительный пример 2: Получение, в качестве контроля сравнения, дисперсии носителя лекарственного средства
Дисперсию носителя лекарственного средства в качестве контроля сравнения получили, используя LIPOFECTIN (зарегистрированный товарный знак) (произведено Invitrogen, Inc.), способом получения по инструкции поставляющей компании.
Сравнительный пример 3: Получение, в качестве контроля сравнения, дисперсии носителя лекарственного средства
Дисперсию носителя лекарственного средства в качестве контроля сравнения получили, используя OLIGOFECTAMINE (зарегистрированный товарный знак) (произведено Invitrogen, Inc.), способом получения по инструкции поставляющей компании.
Сравнительный пример 4: Получение, в качестве контроля сравнения, фармацевтической композиции
(1) Получение раствора нуклеиновой кислоты
Раствор нуклеиновой кислоты получили тем же способом, что и в примере получения 17 (1).
(2) Получение, в качестве контроля сравнения, композиции
1,0 мг/мл фармацевтической композиции, в качестве контроля сравнения, получили тем же способом, что и в примере получения 17 (2), используя общее количество раствора нуклеиновой кислоты, полученного выше (1), и 5 мл дисперсии носителя лекарственного средства, полученной в сравнительном примере 1.
В данном случае среднюю величину частиц носителя лекарственного средства в фармацевтической композиции в качестве контроля сравнения измерили тем же способом, что и в примере получения 17 (3), и она составила 148,1 нм.
Сравнительный пример 5: Получение, в качестве контроля сравнения, фармацевтической композиции
(1) Получение раствора нуклеиновой кислоты
Раствор нуклеиновой кислоты получили тем же способом, что и в примере получения 17 (1).
(2) Получение, в качестве контроля сравнения, фармацевтической композиции
1,0 мг/мл фармацевтической композиции, в качестве контроля сравнения, получили тем же способом, что и в примере получения 17 (2), используя общее количество раствора нуклеиновой кислоты, полученного выше (1), и 5 мл дисперсии носителя лекарственного средства, полученной в сравнительном примере 1.
В данном случае среднюю величину частиц носителя лекарственного средства в фармацевтической композиции в качестве контроля сравнения измерили тем же способом, что и в примере получения 17 (3), и она составила 168,9 нм.
Сравнительный пример 6: Получение, в качестве контроля сравнения, фармацевтической композиции
(1) Получение раствора нуклеиновой кислоты
Раствор нуклеиновой кислоты получили тем же способом, что и в примере получения 31 (1).
(2) Получение, в качестве контроля сравнения, композиции
1,0 мг/мл фармацевтической композиции, в качестве контроля сравнения, получили тем же способом, что и в примере получения 31 (2), используя общее количество раствора нуклеиновой кислоты, полученного выше (1), и 5 мл дисперсии носителя лекарственного средства, полученной в сравнительном примере 1.
В данном случае среднюю величину частиц носителя лекарственного средства в фармацевтической композиции, в качестве контроля сравнения, измерили тем же способом, что и в примере получения 31 (3), и она составила 138,6 нм.
Пример тестирования 1: Оценка способности к циркулированию в крови
Способность к циркулированию в крови носителя лекарственного средства, содержащего модифицированный PEG фосфолипид, синтезированный в примере получения 3, оценивали c использованием, в качестве показателя, радиоактивности нуклеиновой кислоты, содержащейся в носителе лекарственного средства.
(1) Экспериментальный способ
Каждую из фармацевтических композиций, полученных в примерах получения от 17 до 24, внутривенно вводили самцу мыши (C57BL/6J, возраст 6 недель, получены CLEA Japan, Inc.) в хвостовую вену из расчета 2,5 мг/кг (содержание нуклеиновой кислоты). Через 2 часа, 8 часов и 24 часа после введения собирали цельную кровь из брюшной аорты мыши, находящейся под анестезией с Этраном и получали плазму. При получении плазмы в качестве противосвертывающего средства использовали гепарин. Во всех случаях введение осуществляли из расчета дозы 10 мл/кг. На группу использовали четыре мыши.
10 мл средства для сцинтиграфии (Hionic-Fluor, произведено PerkinElmer, Inc. далее использовали его же) добавляли к 50 мкл плазмы и перемешивали друг с другом, и затем измеряли радиоактивность каждого образца, используя жидкий сцинтилляционный счетчик. Исходя из результатов, рассчитывали коэффициент распределения (% дозы) носителя лекарственного средства.
(2) Экспериментальные результаты
Как показано на фиг.3 и 4, носитель лекарственного средства обладал наиболее продолжительным временем циркуляции в случае, когда содержание модифицированного PEG фосфолипида примера получения 3 располагалось в пределах от 30% мас. до 50% мас.
Пример тестирования 2: Оценка способности к циркулированию в крови
Способность к циркулированию в крови носителя лекарственного средства, который содержал соединение B, оценивали c использованием, в качестве показателя, радиоактивности нуклеиновой кислоты, содержащейся в носителе лекарственного средства.
(1) Экспериментальный способ
Каждую из фармацевтических композиций, полученных в примере получения от 25 до 28, внутривенно вводили самцу мыши (C57BL/6J, возраст 6 недель, получены CLEA Japan, Inc.) в хвостовую вену из расчета 2,5 мг/кг (содержание нуклеиновой кислоты). Через 24 часа после введения из брюшной аорты мыши, находящейся под анестезией с Этраном, собирали цельную кровь и получали плазму. При получении плазмы в качестве противосвертывающего средства использовали гепарин. Во всех случаях введение проводили из расчета дозы 10 мл/кг. На группу использовали четырех мышей.
10 мл средства для сцинтиграфии добавляли к 50 мкл плазмы и перемешивали друг с другом, а затем измеряли радиоактивность каждого образца, используя жидкий сцинтилляционный счетчик. Исходя из результатов, рассчитывали коэффициент распределения (% дозы) носителя лекарственного средства.
(2) Экспериментальные результаты
Как показано на фиг.5, носитель лекарственного средства обладал наиболее продолжительным временем циркуляции в случае, когда содержание соединения B составляло 30% мас. или более.
Пример тестирования 3: Оценка биораспределения носителя по настоящему изобретению
Биораспределение носителя по настоящему изобретению оценивали c использованием, в качестве показателя, радиоактивности нуклеиновой кислоты, содержащейся в носителе лекарственного средства.
Экспериментальный способ
Фармацевтическую композицию, полученную в примере получения 29 или сравнительном примере 4, внутривенно вводили самцу мыши (C57BL/6J, возраст 6 недель, получено CLEA Japan, Inc.) в хвостовую вену из расчета 2,5 мг/кг (содержание нуклеиновой кислоты). Через 5 минут, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 8 часов, 24 часа, 48 часов и 72 часа после введения из брюшной аорты мыши, находящейся под анестезией с Этраном, отбирали цельную кровь и получали плазму. При получении плазмы в качестве противосвертывающего средства использовали гепарин. Во всех случаях введение проводили из расчета дозы 10 мл/кг. На группу использовали трех или четырех мышей.
10 мл средства для сцинтиграфии добавляли к 50 мкл плазмы и перемешивали друг с другом, а затем измеряли радиоактивность каждого образца, используя жидкий сцинтилляционный счетчик. Исходя из результатов, рассчитывали коэффициент распределения (% дозы), объем распределения (л/кг), время полувыведения (часы), площадь под кривой концентрации в плазме (мкг·час/мл) и клиренс (л/час·кг).
(2) Экспериментальные результаты
(i) Концентрация в плазме
Как показано на фиг.6, носитель по настоящему изобретению в композиции по настоящему изобретению из примера получения 29 обладал более длительным временем циркуляции в плазме, чем носитель лекарственного средства в качестве сравнительного контроля в фармацевтической композиции в качестве сравнительного контроля из сравнительного примера 4.
(ii) Фармакокинетический параметр
Как показано в таблице 1, площадь под кривой концентрации в плазме (AUC0-8) носителя по настоящему изобретению в композиции по настоящему изобретению из примера получения 29 приблизительно в 5 раз превышала площадь под кривой носителя лекарственного средства, в качестве сравнительного контроля, в фармацевтической композиции, в качестве сравнительного контроля, из сравнительного примера 4. Дополнительно, объем распределения носителя по настоящему изобретению составлял приблизительно одну восьмую от объема распределения носителя лекарственного средства в качестве сравнительного контроля.
Пример тестирования 4: Оценка биораспределения носителя по настоящему изобретению
Биораспределение носителя по настоящему изобретению оценили, используя, в качестве показателя, радиоактивность нуклеиновой кислоты, содержащейся в носителе лекарственного средства.
(1) Экспериментальный способ
Фармацевтическую композицию, полученную в примере получения 29 или сравнительном примере 4, внутривенно ввели самцу мыши (C57BL/6J, возраст 6 недель, получено CLEA Japan, Inc.) в хвостовую вену из расчета 2,5 мг/кг (содержание нуклеиновой кислоты). Через 30 минут, 2 часа, 8 часов и 24 часа после введения из брюшной аорты мыши, находящейся под анестезией с Этраном, собирали цельную кровь и получали плазму. Для получения плазмы в качестве противосвертывающего средства использовали гепарин. Дополнительно, одновременно со взятием крови произвели резекцию печени, легких, селезенки и почек и измерили их массу, соответственно. Во всех случаях введение осуществляли в дозе 10 мл/кг. На группу использовали три или четыре мыши.
Каждый из органов растворили в пробирке посредством добавления 1 мл солюбилизатора ткани (SOLVABLE, произведено PerkinElmer, Inc.) и встряхивания полученной смеси при 40°C в течение двух ночей.
10 мл средства для сцинтиграфии добавляли к образцу, в котором растворили от 50 до 200 мг каждого органа и перемешали друг с другом, а затем измерили радиоактивность каждого образца, используя жидкий сцинтилляционный счетчик. Исходя из результатов, рассчитали коэффициент распределения (% дозы) носителя лекарственного средства в каждом органе. В данном случае коэффициент распределения (% дозы) носителя лекарственного средства в плазме рассчитали тем же способом, что и в примере тестирования 1.
(2) Экспериментальные результаты
Как показано на фиг.7, коэффициент распределения в плазме носителя по настоящему изобретению в композиции по настоящему изобретению из примера получения 29 был выше, чем коэффициент распределения носителя лекарственного средства в качестве сравнительного контроля в фармацевтической композиции в качестве сравнительного контроля из сравнительного примера 4. Как показано на фиг.8 и 9, коэффициент распределения в печени и селезенке носителя по настоящему изобретению в композиции по настоящему изобретению из примера получения 29 был ниже, чем коэффициент распределения в печени и селезенке носителя лекарственного средства в качестве сравнительного контроля в фармацевтической композиции в качестве сравнительного контроля из сравнительного примера 4. Как показано на фиг.10 и 11, не существовало различия в коэффициенте распределения в легких и почках между носителем лекарственного средства в фармацевтической композиции из примера получения 29 и сравнительного примера 4.
Пример тестирования 5: Оценка биораспределения носителя по настоящему изобретению в организме мыши, которой имплантировали злокачественные клетки
Биораспределение носителя по настоящему изобретению в организме мыши, которой имплантировали злокачественные клетки, оценили, используя, в качестве показателя, радиоактивность нуклеиновой кислоты, содержащейся в носителе лекарственного средства.
(1) Имплантация злокачественных клеток на модели мыши
Мышь с имплантированными злокачественными клетками получили посредством имплантации, подкожно, 1×106 клеток A431 (клетки плоского эпителия человека) самцу мыши nude (BALB/cA Jcl-nu, возраст 9 недель, получено CLEA Japan, Inc.) и выращиванием мыши в течение 9 суток после имплантации.
(2) Экспериментальный способ
Фармацевтическую композицию, полученную в примере получения 30 или сравнительном примере 5, внутривенно вводили в хвостовую вену мыши, полученной выше (1), из расчета 10 мг/кг (содержание нуклеиновой кислоты). Через 8 часов, 24 часа и 72 часа после введения из брюшной аорты мыши, находящейся под анестезией с Этраном, отбирали цельную кровь и получали плазму. При получении плазмы в качестве противосвертывающего средства использовали гепарин. Дополнительно, одновременно со взятием крови подвергали резекции периферическую ткань злокачественной опухоли и узлы злокачественной опухоли и взвешивали их, соответственно. Во всех случаях введение осуществляли в дозе 10 мл/кг. На группу использовали трех мышей.
Количество носителя лекарственного средства (мкг/г или мкг/мл), доставленного в периферические ткани злокачественной опухоли, узлы злокачественной опухоли и в плазму, измеряли тем же способом, что и в примере тестирования 1 или 4.
(3) Экспериментальные результаты
Как показано на фиг.12, количество носителя по настоящему изобретению в композиции по настоящему изобретению из примера получения 30, доставленного в периферические ткани злокачественной опухоли, было выше, чем количество носителя лекарственного средства в качестве сравнительного контроля в фармацевтической композиции в качестве сравнительного контроля из сравнительного примера 5. С другой стороны, как показано на фиг.13, не выявлено различия в коэффициенте распределения в узлах злокачественной опухоли между носителем лекарственного средства в фармацевтической композиции из примера получения 30 и сравнительного примера 5. В данном случае, как показано на фиг.14, коэффициент распределения носителя по настоящему изобретению в композиции по настоящему изобретению из примера получения 30 в плазме был выше, чем коэффициент распределения носителя лекарственного средства в качестве сравнительного контроля в фармацевтической композиции в качестве сравнительного контроля из сравнительного примера 5.
Пример тестирования 6: Оценка гемолитических свойств носителя по настоящему изобретению
(1) Экспериментальный способ
Кровь, взятую от самца крысы (Slc:SD, возраст 7 недель, получено Japan SLC, Inc.) центрифугировали при 3,000 об/мин в течение 10 минут, и затем удалили верхний слой, получив, тем самым, суспензию эритроцитов. К полученной суспензии эритроцитов добавили физиологический раствор для инъекций (произведено Otsuka Pharmaceutical Factory, Inc., далее в настоящем документе применяли то же самое) в количестве, вдвое превышающем количество суспензии эритроцитов, и перемешали друг с другом. Затем, полученную в результате смесь центрифугировали при 3,000 об/мин в течение 5 минут. Эту процедуру повторили еще два раза. Полученную суспензию эритроцитов разбавляли физиологическим раствором для инъекций до 1×109 клеток/мл.
Каждую дисперсию носителя лекарственного средства, полученную в примерах получения 16 и сравнительных примерах от 1 до 3, разводили 10% мальтозой до желаемой концентрации в пределах от 0,3 мкг/мл до 30 мкг/мл. Затем 285 мкл разведенной дисперсии носителя лекарственного средства подвергали предварительной инкубации при 37°C в течение 10 минут, к ней добавили 15 мкл суспензии эритроцитов и перемешали, и смесь, полученную, в результате, инкубировали при 37°C в течение 30 минут. Реакционный раствор центрифугировали при 3,000 об/мин в течение 3 минут и удалили супернатант. Оптическую плотность супернатанта измерили при 405 нм.
Степень гемолиза рассчитывали посредством учета оптической плотности, полученной в случае, когда не добавлен носитель лекарственного средства в качестве 0% гемолиза, и оптической плотности, полученной в случае, если добавлен 0,02% тритон X100 в качестве 100% гемолиза. Дополнительно, из рассчитанной степени гемолиза рассчитали концентрацию, вызывающую 50% гемолиза.
(2) Экспериментальные результаты
Как показано в таблице 2, концентрация носителя по настоящему изобретению из примера получения 16, приводящая к 50% гемолизу, была выше, чем та же концентрация носителя лекарственного средства у сравнительных контролей из сравнительных примеров от 1 до 3.
Пример тестирования 7: Оценка цитотоксичности носителя по настоящему изобретению
(1) Экспериментальный способ
Эндотелиальные клетки из пупочной вены человека (произведено Sanko Junyaku Co., Ltd.) высевали в 96-луночный планшет из расчета 3,000 клеток/лунку и культивировали в течение ночи. Каждую из дисперсий носителя лекарственного средства, полученных в примере получения 16 и сравнительных примерах от 1 до 3, разводили 10% мальтозой до желаемой концентрации в пределах от 3 мкг/мл до 10 мкг/мл. Разведенную дисперсию носителя лекарственного средства добавили в каждую лунку в количестве одной десятой объема среды для культивирования. После культивирования в течение 72 часов подсчитали количество жизнеспособных клеток, используя Cell Counting Kit-8 (WST-8, произведено Dojin Chemical Co., Ltd.), и из полученного значения рассчитали 50% ингибиторную концентрацию клеточного роста. В данном случае при культивировании эндотелиальных клеток из пупочной вены человека использовали среду для культивирования M199 (произведено Nissui Фармацевтическ Co., Ltd.).
(2) Экспериментальные результаты
Как показано в таблице 3, 50% ингибиторная концентрация клеточного роста у носителя по настоящему изобретению из примера получения 16 была выше, чем та же концентрация у носителей лекарственного средства в качестве сравнительных контролей из сравнительных примеров от 1 до 3.
Пример тестирования 8: Оценка способности композиции по настоящему изобретению к индуцированию цитокинов
(1) Экспериментальный способ
Брали свежую донорскую кровь человека и для предотвращения свертывания смешивали с ней HEPARIN SODIUM INJECTION (произведено Ajinomoto Co., Ltd.) в количестве 1 мл на 10 мл крови. Добавляли равный объем фосфатно-солевого буфера (далее в настоящем документе обозначаемый как “PBS”) и кровь в количестве 10 мл осторожно наслаивали на 3 мл Ficoll-Paque PLUS (произведено GE Healthcare BioSciences, Inc.), так чтобы не разрушить границу фаз. Затем проводили центрифугирование при 400×g в течение 30 минут при комнатной температуре, получая, тем самым, мононуклеарные клетки периферической крови. Полученные мононуклеарные клетки периферической крови дважды промывали PBS и затем суспендировали в среде для культивирования RPMI 1640 (произведено Nissui Pharmaceutical Co., Ltd.), содержащей 10% эмбриональную бычью сыворотку (произведено JRH BioSciences, Inc.), 100 Ед/мл пенициллина (произведено Nacalai Tesque, Inc.) и 100 мкг/мл стрептомицина (произведено Nacalai Tesque, Inc.). Затем, подсчитывали клетки и получали клеточную суспензию из расчета 2×106 клеток/мл.
Полученную клеточную суспензию засевали в 48-луночный планшет из расчета 300 мкл (6×105 клеток)/лунку, и клетки культивировали в условиях 37°C и 5% CO2 в течение 3 часов. Затем каждую из фармацевтических композиций, полученных в примере получения от 17 до 24 и 31 и сравнительном примере 6, добавляли к среде для культивирования до желаемой концентрации (примеры получения от 17 до 24: 100 нМ, пример получения 31 и сравнительный пример 6: 30, 100 и 300 нМ). После добавления фармацевтической композиции клетки культивировали в течение 24 часов, и затем удаляли супернатант культуры и выполняли ELISA. Уровень IFN-α измеряли, используя набор ELISA для определения интерферона-α человека (произведено Biosource, Inc.) по приложенному к набору протоколу.
(2) Экспериментальные результаты
Как показано в таблице 4, способность к индуцированию ИНФ-α фармацевтической композицией из примеров получения от 17 до 19 была выше, чем фармацевтической композицией из примеров получения от 20 до 24.
Как показано в таблице 5, уровень ИНФ-α, индуцированного лечением композицией по настоящему изобретению из примера получения 31 был ниже, чем уровень ИНФ-α, индуцированного лечением фармацевтической композицией в качестве сравнительного контроля из сравнительного примера 6.
В данном случае n.d. в таблицах 4 и 5 означает уровень, располагающийся ниже предела детекции.
Пример тестирования 9: Оценка эффективности композиции по настоящему изобретению в качестве лекарственного средства
(1) Имплантация злокачественных клеток на модели мыши
Мышь с имплантированными злокачественными клетками получили посредством подкожной имплантации 3×105 клеток PC-3 (злокачественные клетки предстательной железы человека) самцу мыши nude (BALB/cA Jcl-nu, возраст 6 недель, получено CLEA Japan, Inc.).
(2) Экспериментальный способ
Композицию по настоящему изобретению, полученную в примере получения 31, внутривенно вводили в хвостовую вену мыши, полученной выше (1), из расчета 10 мг/кг (содержание нуклеиновой кислоты) один раз в сутки в течение следующих подряд 5 суток, начиная с 10 суток после имплантации. Дополнительно, композицию по настоящему изобретению вводили тем же способом в течение 5 следующих подряд суток, начиная с 17 суток после имплантации. Во всех случаях введение осуществляли в дозе 10 мл/кг. На группу использовали 6 мышей. В данном случае мышь, которой вводили 10% мальтозу, использовали в качестве отрицательного контроля.
Объем опухоли определили посредством измерения большой и малой осей опухоли после 10, 13, 17, 20 и 24 суток после имплантации и проведения расчета по формуле: [(малая ось)2 × (большая ось) / 2].
(3) Экспериментальные результаты
Как показано на фиг.15, введение композиции по настоящему изобретению, полученной в примере получения 31, подавляет увеличение опухоли в объеме.
Пример тестирования 10: Оценка эффективности композиции по настоящему изобретению в качестве лекарственного средства
(1) Имплантация злокачественных клеток на модели мыши
Мышь с имплантированными злокачественными клетками получали посредством интраперитонеальной имплантации 7×105 клеток MCAS (злокачественные клетки яичников человека) самке мыши nude (BALB/cA Jcl-nu, возраст 6 недель, получено CLEA Japan, Inc.).
(2) Экспериментальный способ
Оценку проводили в случае, если композицию по настоящему изобретению вводили постоянно, и в случае, если композицию по настоящему изобретению вводили периодически. В схеме с постоянным введением композицию по настоящему изобретению, полученную в примере получения 31, вводили один раз в сутки, в сутки с 3 до 7 и с 10 до 14 после имплантации. В случае периодической схемы введения композицию по настоящему изобретению, полученную в примере получения 31, вводили один раз в сутки, в сутки 4, 7, 11, 14, 18, 21, 25, 28, 32 и 35 после введения. Композицию по настоящему изобретению вводили внутривенно в хвостовую вену из расчета 10 мг/кг (содержание нуклеиновой кислоты). Во всех случаях введение осуществляли в дозе 10 мл/кг. Использовали шесть мышей на группу. В данном случае мышь, которой вводили 10% мальтозу, использовали в качестве отрицательного контроля.
(3) Экспериментальные результаты
Как показано на фиг.16 и 17, посредством введения композиции по настоящему изобретению, полученной в примере получения, увеличили уровень выживаемости.
Пример тестирования 11: Оценка эффективности композиции по настоящему изобретению в качестве лекарственного средства
(1) Получение мыши, имеющей метастазы злокачественных клеток в печени
Мышь, имеющую метастазы злокачественных клеток в печени, получали посредством имплантации 1×106 клеток A549 (немелкоклеточный рак легких человека) в селезенку самца мыши nude (BALB/cA Jcl-nu, возраст 6 недель, получено CLEA Japan, Inc.) и удалили селезенку через 10 минут после имплантации.
(2) Экспериментальный способ
Композицию по настоящему изобретению, полученную в примере получения 31, внутривенно вводили в хвостовую вену мыши, полученной выше (1), из расчета 10 мг/кг (концентрация нуклеиновой кислоты) один раз в сутки, в течение следующих подряд 5 суток, начиная с 7 суток после имплантации. Дополнительно, композицию по настоящему изобретению вводили тем же способом, в течение 5 следующих подряд суток, начиная с 14 суток после имплантации. Во всех случаях введение осуществляли в дозе 10 мл/кг. Использовали 6 мышей на группу. В данном случае мышь, которой вводили 10% мальтозу, использовали в качестве отрицательного контроля.
(3) Экспериментальные результаты
Как показано на фиг.18, посредством введения композиции по настоящему изобретению, полученной в примере получения 31, увеличили уровень выживаемости.
Пример тестирования 12: Оценка эффективности композиции по настоящему изобретению в качестве лекарственного средства
(1) Имплантация злокачественных клеток на модели мыши
Мышь с имплантированными злокачественными клетками получали посредством имплантации 1×106 клеток HPAC (клетки злокачественной опухоли поджелудочной железы человека) в поджелудочную железу самца мыши nude (BALB/cA Jcl-nu, возраст 7 недель, получено CLEA Japan, Inc.). Мышь, подвергнутую воздействию тем же способом, с использованием среды для культивирования вместо клеток HPAC, использовали в качестве ложно оперированной группы.
(2) Экспериментальный способ
Композицию по настоящему изобретению, полученную в примере получения 31, внутривенно вводили из расчета 10 мг/кг (содержание нуклеиновой кислоты) в хвостовую вену мыши, полученной выше (1), один раз в сутки в период от 6 до 10 суток и от 13 до 17 после имплантации. Через 29 суток после имплантации мышь вскрыли, измерили массу поджелудочной железы и оценили противоопухолевый эффект. В данном случае использовали 8 мышей на группу. Дополнительно, в качестве отрицательного контроля, использовали мышь, которой ввели 10% мальтозу.
(3) Экспериментальные результаты
Как показано на фиг.19, посредством введения композиции по настоящему изобретению, полученной в примере получения 31, увеличение массы поджелудочной железы подавлено.

| название | год | авторы | номер документа |
|---|---|---|---|
| АКТИВАТОР ВНУТРИКЛЕТОЧНОЙ НУКЛЕАЗЫ РАКОВОЙ КЛЕТКИ | 1998 |
|
RU2183460C2 |
| СПОСОБ АКТИВАЦИИ/ПРОЛИФЕРАЦИИ T-КЛЕТОК | 2019 |
|
RU2806549C2 |
| НОВЫЕ КОНЪЮГАТЫ ОЛИГОНУКЛЕОТИДОВ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2599449C1 |
| ОДНОРАЗОВАЯ СИСТЕМА ДЛЯ СТЕРИЛЬНОГО ПОЛУЧЕНИЯ ЧАСТИЦ ИЗ ЛИПИДОВ И НУКЛЕИНОВЫХ КИСЛОТ | 2012 |
|
RU2642640C2 |
| КОМПЛЕКСЫ НУКЛЕИНОВЫХ КИСЛОТ - ЛИГАНДОВ СОСУДИСТОГО ЭНДОТЕЛИАЛЬНОГО ФАКТОРА РОСТА (VEGF) | 1997 |
|
RU2177950C2 |
| СТАБИЛЬНЫЕ СОСТАВЫ ЛИПИДОВ И ЛИПОСОМ | 2015 |
|
RU2738060C2 |
| СРЕДСТВА ИНГИБИРОВАНИЯ ЭКСПРЕССИИ ПРОТЕИНКИНАЗЫ 3 | 2007 |
|
RU2553561C2 |
| КАТИОННЫЕ ЛИПИДЫ | 2018 |
|
RU2782218C2 |
| ЛИПИДНЫЕ НАНОЧАСТИЦЫ ДЛЯ ДОСТАВКИ ЛЕКАРСТВЕННОГО СРЕДСТВА IN VIVO И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2799045C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ АНИОННОЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО, И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2016 |
|
RU2721558C2 |




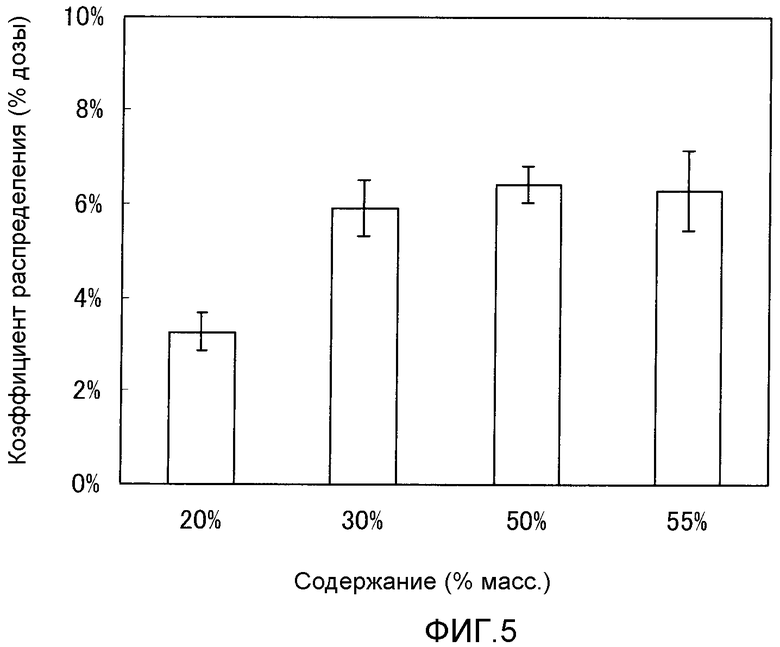

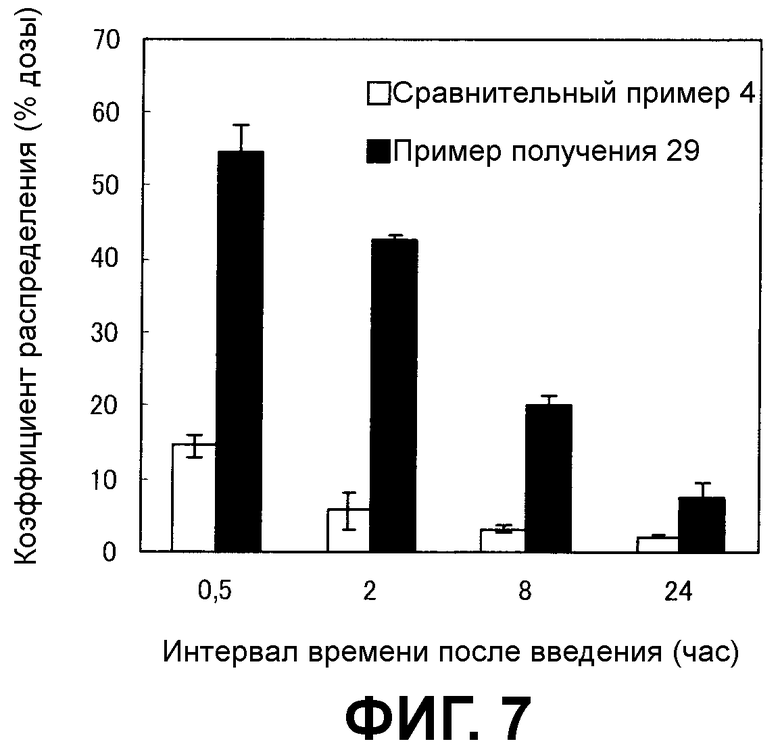





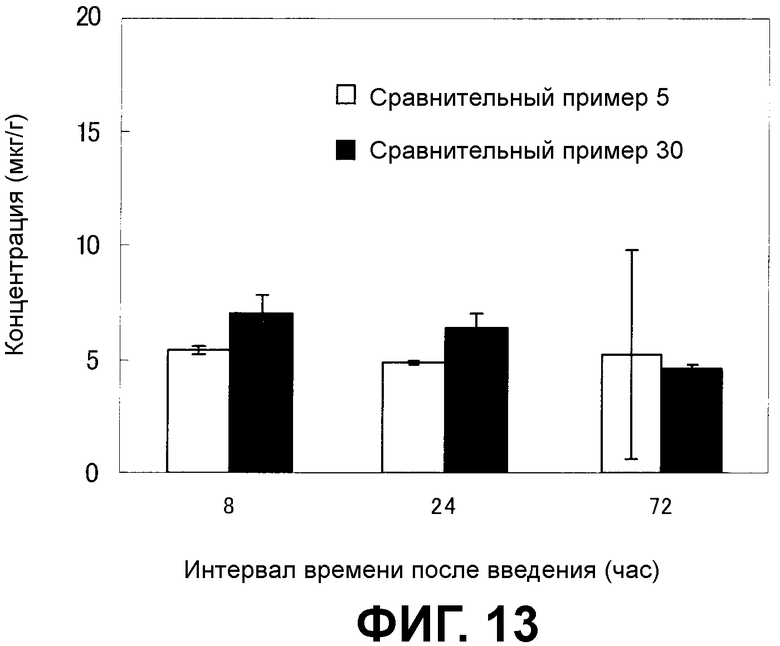

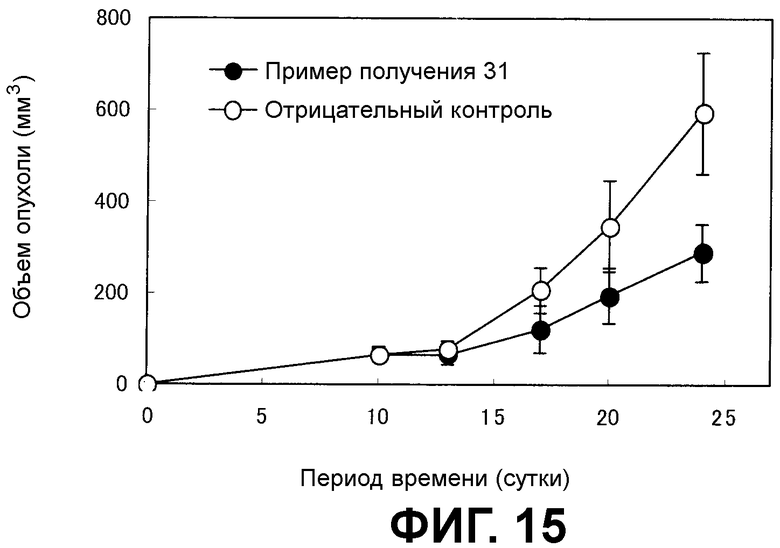
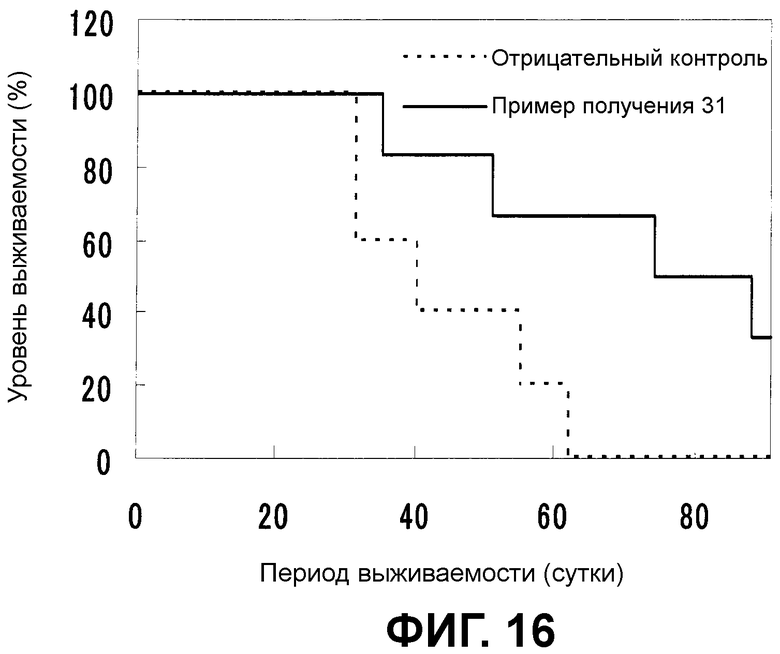



Группа изобретений относится к фармакологии и касается циркулирующего носителя для лекарственного средства. Носитель содержит модифицированный полиэтиленгликолем фосфолипид, представленный общей формулой (I) или его фармацевтически приемлемой солью:
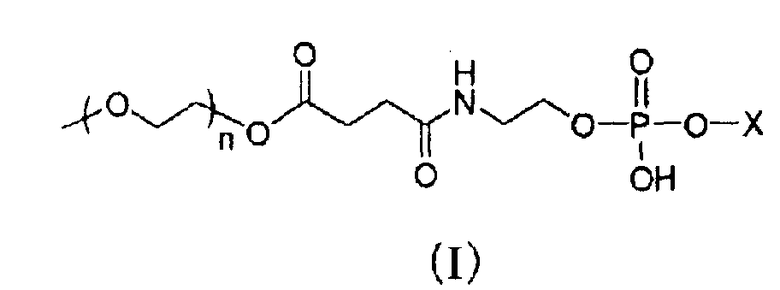
где Х представляет собой следующие (II) или (III); и n представляет собой целое число от 30 до 150,

где R1 представляет собой остаток насыщенной неразветвленной жирной кислоты, имеющей от 17 до 22 атомов углерода, и 2-O-(2-диэтиламиноэтил)карбамоил-1,3-O-диолеоилглицерин. При этом модифицированный полиэтиленгликолем фосфолипид содержится в количестве 30 - 50% мас. от общей массы липидов в носителе. Предложена также фармацевтическая композиция, содержащая вышеописанный носитель и лекарственное средство. Группа изобретений обеспечивает увеличение времени циркуляции и соответственно повышение эффективности лекарственного средства. 2 н. и 4 з.п. ф-лы, 19 ил., 5 табл., 49 пр.
1. Циркулирующий носитель для лекарственного средства, содержащий модифицированный полиэтиленгликолем фосфолипид, представленный следующей общей формулой (I) или его фармацевтически приемлемой солью
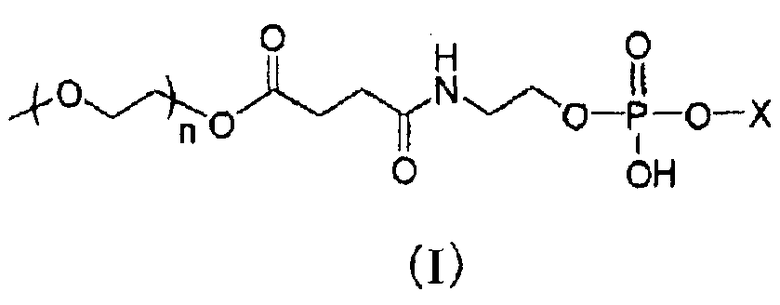
где Х представляет собой следующие (II) или (III); и n представляет собой целое число от 30 до 150

где R1 представляет собой остаток насыщенной неразветвленной жирной кислоты, имеющей от 17 до 22 атомов углерода, и 2-O-(2-дизтиламипоэтил)карбамоил-1,3-O-диолеоилглицерин, где модифицированный полиэтиленгликолем фосфолипид, представленный общей формулой (I), содержится в количестве в пределах от 30 мас.% до 50 мас.% от общей массы липидов в носителе лекарственного средства.
2. Циркулирующий носитель для лекарственного средства по п.1, где модифицированный полиэтиленгликолем фосфолипид представляет собой 1,3-дистеароилглицеро-2-фосфатидил-N-(метокси-полиэтиленгликоль-сукцинил)этаноламин или N-(метокси-полиэтилен гликоль-сукцинил)дистеароилфосфатидилэтаноламин.
3. Циркулирующий носитель для лекарственного средства по любому из пп.1 и 2, дополнительно содержащий фосфолипид.
4. Фармацевтическая композиция, содержащая циркулирующий носитель для лекарственного средства по любому из пп.1-3, который включает лекарственное средство.
5. Фармацевтическая композиция по п.4, где лекарственное средство представляет собой одноцепочечную или двухцепочечную РНК, одноцепочечную или двухцепочечную ДНК, олигонуклеиновую кислоту или водорастворимое анионное соединение.
6. Фармацевтическая композиция по п.5, где олигонуклеиновая кислота представляет собой короткую интерферирующую РНК, микроРНК, короткую шпилечную РНК, антисмысловую ДНК, антисмысловую РНК, ДНК-фермент, рибозим, аптамер или некодирующую РНК.
| ПРОИЗВОДНЫЕ ГЛИЦЕРИНА, СРЕДСТВО ДЛЯ ДОСТАВКИ ФИЗИОЛОГИЧЕСКИ АКТИВНОГО ВЕЩЕСТВА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2123492C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ СОПОЛИМЕР НУКЛЕИНОВОЙ КИСЛОТЫ | 1994 |
|
RU2143903C1 |
| АКТИВАТОР ВНУТРИКЛЕТОЧНОЙ НУКЛЕАЗЫ РАКОВОЙ КЛЕТКИ | 1998 |
|
RU2183460C2 |
| ЛЕКАРСТВЕННЫЕ СРЕДСТВА ПРОТИВ ГЕПАТИТА | 1999 |
|
RU2226102C2 |
| ЕР 1637144 А1, 22.03.2006 | |||
| WATANABE T | |||
| et al | |||
| Liver target delivery of small interfering RNA to the HCV gene by lactosylated cationic liposome | |||
| J Hepatol | |||
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |

