Изобретение относится к некоторым новым производным пиперидина, к способам их получения, к содержащим их фармацевтическим препаратам и к их применению при медикаментозном лечении, в особенности при лечении психических расстройств.
В патенте США 2739968 описаны производные замещенного пиперидина, обладающие антигистаминной, антиспазматической, антиацетилхолиновой и болеутоляющей активностью.
В Британском патенте GB 1320481 описаны некоторые гем-диарилэтиленовые производные, обладающие антигистаминной активностью.
Эффективные антипсихотические (нейролептические) агенты включают трициклические фенотиазины, тиоксантены и дибензазепины, а также бензамиды и бутирофеноны. Эти соединения блокируют рецепторы допамина D2 и инактивируют перенос допамина. В результате эти соединения вызывают характерные неврологические побочные эффекты у человека, такие как экстрапирамидальные побочные эффекты, например дистонию и дискинезию (R.J. Baldessarini, 1996, Goodman, Gilman's, The Pharmacological Basis of Therapeutics, 9-th ed. eds. J.G. Hardman et. al). В тестах на животных такие побочные эффекты проявлялись в виде каталепсии. Таким образом, существует необходимость в разработке ряда антипсихотических агентов, которые не имеют таких отрицательных побочных эффектов.
Настоящее изобретение предлагает некоторые производные пиперидина, которые обладают сильной антипсихотической активностью, но не проявляют каталептического действия, и, следовательно, не могут вызывать экстрапирамидальные побочные эффекты при их применении в терапевтических дозах.
Таким образом, настоящее изобретение предлагает соединения формулы (I):
где заместитель R1 представляет собой бензотиенил, бензофуранил или нафтил (где бензотиенильный, бензофуранильный или нафтильный фрагмент может быть необязательно замещен одним или несколькими заместителями, выбираемыми из атома галогена, C1-6-алкоксигруппы, C1-6-алкила, С3-6-циклоалкила и C1-6-алкенила), замещенный тиенил или замещенный фуранил (где тиенильный или фуранильный фрагмент замещены одним или несколькими заместителями, выбираемыми из атома галогена, C1-6-алкила, С3-6-циклоалкила и C1-6-алкенила);
заместитель R2 представляет собой атом галогена и
заместитель R3 представляет собой C1-6-алкил или С3-6-циклоалкилметил;
или их фармацевтически приемлемые соли или сольваты.
Настоящее изобретение включает производные пиперидина формулы (I), где заместитель R1 представляет собой бензотиенил, бензофуранил, нафтил (где бензотиенильный, бензофуранильный или нафтильный фрагмент может быть необязательно замещен одним или несколькими заместителями, выбираемыми из атома галогена или C1-6-алкоксигруппы), замещенный тиенил или замещенный фуранил (где тиенильный или фуранильный фрагмент замещены одним или несколькими заместителями, выбираемыми из атома галогена или C1-6-алкила); заместитель R2 представляет собой атом галогена и заместитель R3 представляет собой C1-6-алкил.
Более предпочтительными являются производные пиперидина формулы (I), где заместитель R1 представляет собой бензотиенил, бензофуранил (где бензотиенильный или бензофуранильный фрагмент может быть необязательно замещен одним или несколькими заместителями, выбираемыми из атома галогена или C1-6-алкоксигруппы), замещенный тиенил или замещенный фуранил (где тиенильный или фуранильный фрагмент замещены одним или несколькими заместителями, выбираемыми из атома галогена или C1-6-алкила); заместитель R2 представляет собой атом галогена и заместитель R3 представляет собой C1-6-алкил.
Примеры приведенных выше соединений формулы (I) включают производные пиперидина, описанные в Примерах 1-7.
Используемое в данном описании понятие "алкил" означает линейную или разветвленную алкильную группу. К таким группам относятся метил, этил, изопропил, н-пропил, н-бутил, втор-бутил, трет-бутил, н-пентил, изопентил, неопентил, н-гексил, изогексил и неогексил. Такие алкильные группы предпочтительно представляют собой С1-4-алкильные группы. К циклоалкильным группам относятся циклопропил и циклопентил. С3-6-циклоалкилметильные группы включают циклопропилметил и циклопентилметил.
Примерами алкенильных групп являются группы, которые могут иметь Е- и Z-форму или их смесь и которые могут быть разветвленными, когда содержат, по меньшей мере, три атома углерода. Такие алкенильные группы предпочтительно представляют собой C1-4-алкенильные группы. Примерами таких алкенильных групп являются винил, аллил, изопропенил, бутенил, изобутенил, пентенил, изопентенил, гексенил, изогексенил и неогексенил.
Определение алкоксигруппа имеет понятное для специалиста в данной области значение и включает линейные и разветвленные группы. Примерами алкоксигрупп служат метокси- и этоксигруппы. Предпочтительными алкоксигруппами являются С1-4-алкоксигруппы.
Понятие "галоген" включает атомы хлора, брома, фтора и йода.
Бензотиенильный, бензофуранильный, нафтильный, замещенный тиенильный и замещенный фуранильный фрагменты включают 2- и 3-бензотиенил, 2- и 3-бензофуранил, 2- и 3-нафтил, замещенный 2-тиенил, замещенный 3-тиенил, замещенный 2-фуранил и замещенный 3-фуранил. Заместитель(и) в бензотиенильном, бензофуранильном, нафтильном, тиенильном и фуранильном кольцах могут находиться в любом из доступных положений. Конкретными примерами таких заместителей являются атомы фтора и хлора и метоксигруппа.
Предпочтительным примером заместителя R1 является замещенный тиенил, наиболее предпочтительно замещенный 2-тиенил, где тиенильный фрагмент замещен одним или несколькими заместителями, выбираемыми из атомов галогена, предпочтительно атомов хлора, и C1-6-алкила, предпочтительно метила и этила, наиболее предпочтительно метила.
Предпочтительным примером заместителя R2 является атом фтора, наиболее предпочтительно 4-фтор. Заместитель R3 предпочтительно является метилом.
Предпочтительными соединениями в соответствии с настоящим изобретением являются соединения формулы (I), где заместитель R1 представляет собой замещенный тиенил, где тиенильный остаток замещен одним или несколькими заместителями, выбираемыми из атома галогена и C1-6-алкила; заместитель R2 представляет собой атом галогена; и заместитель R3 представляет собой C1-6-алкил; или их фармацевтически приемлемые соли или сольваты.
Другими предпочтительными соединениями формулы (I) являются соединения, где заместитель R1 представляет собой замещенный 2-тиенил, где тиенильный остаток замещен одним или несколькими заместителями, выбираемыми из атома хлора и метильной группы; предпочтительно 4-хлор и 4-метила; заместитель R2 представляет собой атом фтора, предпочтительно 4-фтор; и заместитель R3 представляет собой метил; или их фармацевтически приемлемые соли или сольваты.
Особенно предпочтительными соединениями в соответствии с настоящим изобретением, которые, как установлено, могут быть использованы при лечении психических заболеваний, являются:
1-метил-4-[(4-хлор-2-тиенил)-(4-фторфенил)]метиленпиперидин и
1-метил-4-[(4-метил-2-тиенил)-(4-фторфенил)]метиленпиперидин, а также их фармацевтически приемлемые соли или сольваты.
Для терапевтического применения соли соединений формулы (I) представляют собой соли, в которых противоион является фармацевтически приемлемым. Однако соли и кислоты, которые не являются фармацевтически приемлемыми, также могут найти применение, например, при получении или очистке фармацевтически приемлемых соединений. Все соли, независимо от того, являются ли они фармацевтически приемлемыми или нет, включены в объем настоящего изобретения.
Примерами фармацевтически приемлемых кислотно-аддитивных солей являются соли, полученные из минеральных кислот, таких как соляная, бромистоводородная, йодистоводородная, фосфорная, метафосфорная, азотная и серная кислоты; и органических солей, таких как винная, уксусная, трифторуксусная, лимонная, малеиновая, молочная, малоновая, фумаровая, бензойная, аскорбиновая, пропионовая, гликолевая, глюконовая, янтарная кислоты, а также метансульфоновая и арилсульфоновые кислоты, например бензол- или п-толуолсульфоновая кислоты.
Предпочтительными солями в соответствии с настоящим изобретением являются кислотно-аддитивные соли соляной, малеиновой, янтарной и фумаровой кислот.
Сольваты настоящего изобретения представляют собой гидраты. В соответствии с еще одним аспектом настоящее изобретение предлагает соединения формулы (I) и их фармацевтически приемлемые соли и сольваты для терапевтического применения, более конкретно, для лечения или профилактики психических заболеваний, таких как шизофрения, мания, повышенная активность, злоупотребление химическими веществами, рвота и подобные шизофрении заболевания.
Настоящее изобретение также включает способ лечения животных, например, млекопитающих, включая человека, страдающих или подверженных психическим заболеваниям, в том числе перечисленным выше заболеваниям, и этот способ включает введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли или сольвата.
Еще одним объектом настоящего изобретения является применение соединения формулы (I) или его фармацевтически приемлемой соли или сольвата при производстве лекарственного средства для лечения или профилактики любого из названных выше заболеваний.
Количество соединения формулы (I) или его фармацевтически приемлемой соли или сольвата (называемых также активным ингредиентом), которое необходимо для достижения терапевтического эффекта, конечно, будет меняться в зависимости от конкретного соединения, способа его введения, возраста и состояния пациента, а также от конкретного расстройства или заболевания, которое подвергается лечению.
Приемлемая ежедневная доза для каждого из названных выше заболеваний будет лежать в интервале от 0,001 до 25 мг на кг веса тела пациента (например, человека) в день, предпочтительно в интервале от 0,1 до 10 мг/кг веса тела в день, и наиболее предпочтительно в интервале от 0,25 до 5 мг/кг веса тела в день. Требуемая доза может быть представлена в виде одной, двух, трех, четырех, пяти и более суб-доз, назначаемых через соответствующие промежутки времени в течение дня.
Хотя активный ингредиент можно вводить отдельно, предпочтительно готовить его в виде фармацевтического препарата. Таким образом, настоящее изобретение также предлагает фармацевтический препарат, содержащий соединение формулы (I) или его фармацевтически приемлемые соль или сольват и фармацевтически приемлемый носитель, а также необязательные другие терапевтические агенты. Носитель считается приемлемым, если он совмещается с другими ингредиентами препарата и не оказывает отрицательного воздействия на пациента.
Такие препараты включают препараты, пригодные для перорального, ректального, назального, локального (в том числе трансдермального, трансбуккального и подъязычного), вагинального или парентерального (в том числе подкожного, внутримышечного, внутривенного, внутрикожного и intravitreal) введения. Препараты могут быть получены любым из способов, известных в фармацевтической области, например, с использованием методов, описанных в работе Gennaro и др. Remington's Pharmaceutical Sciences (18-th ed. Mack Publishing Company, 1990; см. в особенности часть 8 - "Фармацевтические препараты и их получение"). Такие способы включают стадию объединения активного ингредиента с носителем, который состоит из одного или нескольких вспомогательных ингредиентов. К названным вспомогательным ингредиентам относятся обычные в данной области ингредиенты, такие как наполнители, связующие вещества, разбавители, дезинтегрирующие агенты, смазывающие вещества, красители, корригирующие вкус и запах добавки и смачиватели.
Препараты, приемлемые для перорального применения, могут быть приготовлены в виде отдельных стандартных лекарственных форм, таких как пилюли, таблетки или капсулы, которые содержат предопределенное количество активного ингредиента; в виде порошков или гранул; в виде растворов или суспензий. Активный ингредиент также может присутствовать в виде шариков (болусов) или пасты, или может находиться в липосомах.
Препараты для ректального применения могут быть получены в виде свечи или клизмы. Для парентерального применения приемлемыми рецептурами являются водные и неводные стерильные препараты для инъекций. Эти препараты могут быть представлены в виде контейнеров, содержащих единичную дозу или несколько доз, например в виде герметично закрытых пузырьков или ампул. Они могут храниться в виде высушенного вымораживанием (лиофилизацией) продукта, к которому перед применением необходимо только добавить жидкий стерильный носитель, например воду.
Препараты, приемлемые для ингаляции через нос, представляют собой тонко измельченные дусты или аэрозоли, которые можно получать с помощью находящейся под повышенным давлением отмеренной дозы аэрозоля, распылителя или порошковдувателя.
Соединения формулы (I) могут быть получены различными способами, которые в целом известны в области органической химии. Исходные материалы или известны, или легко доступны из различных источников, или могут быть получены по обычным методикам. Например, соединения могут быть синтезированы с использованием методов, описанных в книгах "The Chemistry of Heterocyclic Compounds", Vol. 44, "Thiophene and its derivatives", part 1-5, Ed. S. Gronowitz, J. Wiley and Son; A. R. Katritsky and C.W. Rees, "Comprehensive Heterocyclic Chemistry", Part 4, Ed. C.W. Bird and G.H. Cheesman, Pergamon Press.
Настоящее изобретение дополнительно включает следующие способы получения соединений формулы (I).
В последующем описании, если не оговорено особо, символы R1, r2 и r3 имеют значения, указанные для них при рассмотрении формулы (I).
Соединения формулы (I) могут быть получены при взаимодействии соединения формулы (II)
где заместитель R4 представляет собой группу R3, значения которой определены выше при описании формулы (I), с приемлемым дегидратирующим агентом, например с минеральной кислотой, такой как соляная кислота, или с оксихлоридом фосфора. Реакция может быть проведена при условиях, стандартных для дегидратации спирта. Например, при использовании оксихлорида фосфора в присутствии подходящего растворителя, такого как пиридин, при температуре в интервале от 80 до 120oС.
Для дегидратации могут быть использованы и другие способы, хорошо известные квалифицированным специалистам или описанные в химической литературе, в том числе с использованием серной кислоты, 4-метилбензолсульфокислоты, трифторуксусной кислоты, метансульфокислоты, триформетансульфокислоты, тионилхлорида, или при использовании сульфуранового дегидратирующего агента Мартина, и с использованием, при необходимости, подходящего растворителя.
С другой стороны, соединения формулы (II) (см. выше), где заместитель R4 представляет собой группу, защищающую атом азота, например тритильную группу, с помощью способов, известных квалифицированным специалистам или описанных в литературе, могут быть или одновременно или последовательно подвергнуты реакции дегидратации и снятия защиты с получением соединения формулы (III):
Алкилирование соединений формулы (III) дает соединения настоящего изобретения.
Подходящими алкилирующими агентами являются алкилгалогениды, такие как алкилйодиды, например этилйодид или н-бутил-бромид. Реакцию обычно проводят в присутствии основания, например ацетата калия и триэтиламина, в подходящем растворителе, таком как ацетон, при температуре в интервале от 0 до 50oС, или в инертном растворителе, таком как толуол или ксилол, при температуре в интервале 80-120oС.
Соединения формулы (III) могут быть также ацилированы с помощью соответствующего хлорангидрида кислоты в присутствии пиридина с последующим восстановлением до соединения формулы (I) с использованием способов, хорошо известных для восстановления амидов. Например, восстановление может быть проведено с помощью алюмогидрида лития.
Если необходимо или желательно, то после проведения одного из описанных выше процессов, в любом порядке может быть осуществлена одна или несколько из следующих дополнительных стадий:
(I) превращение фармацевтически приемлемой соли или сольвата соединения формулы (I) в соединение формулы (I).
(II) превращение фармацевтически приемлемой соли или сольвата соединения формулы (I) в другую фармацевтически приемлемую соль или сольват формулы (I).
(III) превращение соединения формулы (I) в фармацевтически приемлемую соль или сольват соединения формулы (I).
Соединения формулы (II), где заместитель R4 представляет собой группу R3, значения которой определены выше при рассмотрении формулы (I), могут быть получены при добавлении подходящего металлорганического реагента, такого как реактив Гриньяра, полученного, например, из 1-метил-4-хлор-пиперидина, к соединению формулы (IV)
Реакцию обычно проводят в присутствии неполярного апротонного растворителя, такого как эфир или тетрагидрофуран, при температуре от -30 до 67oС.
Соединение формулы (II) также может быть получено путем обработки соединений формулы (V)
где заместитель R4 представляет собой группу R3, значения которой определены выше при рассмотрении формулы (I), или группу, защищающую атом азота, подходящим металлорганическим реагентом, таким как реактив Гриньяра, или литиевым реагентом, полученным из R1-L, где L представляет собой соответствующий атом галогена, такой как атом брома или атом хлора, или литиевым реагентом, полученным из активированного арилом атома водорода. Реакцию обычно проводят в присутствии апротонного неполярного растворителя, такого как эфир или тетрагидрофуран, при температуре в интервале от -60 до 67oС.
С другой стороны, соединения формулы (II) могут быть получены путем обработки соединения формулы (VI)
где заместитель R4 представляет собой группу R3, значения которой определены выше при рассмотрении формулы (I), или группу, защищающую атом азота, подходящим металлорганическим реагентом, таким как реактив Гриньяра, или литиевым реагентом, полученным из замещенного атомом галогена бензола. Например, соединения формулы (II), где фенильная группа замещена атомом галогена, могут быть получены обработкой соединения формулы (VI) соответствующим замещенным атомом галогена фенилмагнийгалогенидом с использованием стандартных реакционных условий.
Соединения формулы (IV) могут быть получены методами, известными из химической литературы. Например, соединения, где заместитель R1 представляет собой 4-хлор- или 2,3-дихлортиенил, могут быть получены по методике Примера 1, путем хлорирования соответствующим образом замещенного галогенбензоилтиофена. Эти соединения являются коммерчески доступными или их получают по известным методикам, например, бензоилированием по Фриделю-Крафтцу тиофена или других групп, представленных заместителем R1.
В соответствии с другим способом соединения формулы (IV) могут быть получены путем ацилирования по Фриделю-Крафтцу или путем бензоилирования 3-хлор-2-бром-тиофена в присутствии катализатора, такого как хлорид железа или хлорид алюминия, в неполярном растворителе, таком как дихлорметан, при температуре в интервале 10-25oС. Восстановительное дебромирование полученного вначале продукта дает требуемое соединение. Такое восстановление может быть осуществлено путем каталитического гидрирования с использованием подходящего катализатора, такого как палладий на угле, в подходящем растворителе, таком как этанол или уксусная кислота, при температуре в интервале от 15 до 25oС и при давлении от 1 до 50 фунтов/кв. дюйм (3,51 кг/см2), или при использовании активированного цинка в названных выше растворителях при температуре 20-65oС.
Соединения формул (V) и (VI) могут быть получены, например, при добавлении подходящего реактива Гриньяра к этил-N-метил- или N-тритилизонипекотату. Последнее соединение является коммерчески доступным соединением или может быть получено из коммерчески доступных соединений по известным в данной области методикам.
С другой стороны, соединения формулы (V), где заместитель R4 представляет собой метил или атом водорода, а заместитель R2 представляет собой 4-фтор, могут быть получены по методикам, описанным в статье J. Med. Chem., 1970, 13, 1. Соединения формулы (V), где заместитель R4 представляет собой тритил, могут быть получены из соединений формулы (V), где заместитель R4 представляет собой атом водорода, например, при взаимодействии с тритил-бромидом по методике, описанной ниже в Примере 4.
Соединения формулы (III) (см. выше) могут быть получены путем превращения соединения формулы (I), где заместитель R3 представляет собой метил, в его уретановое производное. Реакцию можно проводить с хлорформиатом, таким как этил-, бензил- или трихлорэтил-хлорформиат (см. Baldwin S.W., Jeffs P.W. , Natarajan S., Gross P.M., Synthetic. Commun. 1977, 7, 79; Kraiss G., Nader K. Tetrahedron Letters, 1971, 57). Гидролиз этих уретановых производных, например, минеральной кислотой, такой как соляная кислота, или путем обработки в подходящих случаях цинком, приводит к соединениям формулы (III).
Соли в соответствии с настоящим изобретением могут быть получены путем обработки соединения формулы (I) подходящим основанием, например гидроксидом щелочного металла, щелочноземельного металла или аммония, или подходящей органической или неорганической кислотой, такой как соляная, фумаровая или малеиновая кислота.
Настоящее изобретение также включает все новые промежуточные соединения, описанные выше, и, в частности соединения формулы (II). Примерами промежуточных соединений в соответствии с настоящим изобретением являются:
α-(4-фторфенил)-α-(4-хлор-2-тиенил)-1-метил-4-пиперидинметанол;
α-(4-фторфенил)-α-(4-метил-2-тиенил)-1-метил-4-пиперидинметанол;
α-(4-фторфенил)-α-(4,5-дихлор-2-тиенил)-1-метил-4-пипери-инметанол;
α-(4-фторфенил)-α-(5-этил-2-тиенил)-1-метил-4-пиперидинметанол;
α-(4-фторфенил)-α-(4-этил-2-тиенил)-1-метил-4-пиперидинметанол;
α-(4-фторфенил)-α-(2-бензотиенил)-1-метил-4-пиперидинметанол;
α-(4-фторфенил)-α-(5-фтор-2-бензотиенил)-1-метил-4-пиперидинметанол;
α-(4-фторфенил)-α-(6-метокси-2-бензотиенил)-1-метил-4-пиперидинметанол;
α-(4-фторфенил)-α-(2-бензофуранил)-1-метил-4-пиперидинметанол;
α-(4-фторфенил)-α-(4, 5-диметил-2-фуранил)-1-метил-4-пиперидинметанол;
α-(4-фторфенил)-α-(4-метил-2-тиенил)-1-трифенилметил-4-пиперидинметанол.
Следующие примеры предназначены только для иллюстрации изобретения и ни коим образом не ограничивают его изобретения.
Пример 1
1. Получение 2-хлор-4-(4-фторбензоил)-тиофена
Измельченный хлорид алюминия (24,3 г) добавляют при перемешивании к раствору 2-(4-фторбензоил)тиофена (15,0 г) в сухом дихлорметане (150 мл) при приблизительно 5oС и через 1 час по каплям добавляют раствор хлора (5,16 г в четыреххлористом углероде (26,3 мл)), поддерживая температуру приблизительно 5oС. По окончании добавления температуре реакционной смеси дают подняться приблизительно до 12oС в течение 1,5 часа и перемешивают при этой температуре в течение 1 часа. Раствор снова охлаждают приблизительно до 5oС и добавляют дополнительное количество раствора хлора (1,3 г) в четыреххлористом углероде (6,8 мл). Полученный раствор перемешивают при этой температуре в течение еще 1 часа, а затем выдерживают в течение ночи приблизительно при 20oС. Раствор снова охлаждают до 5oС, добавляют дополнительное количество хлора (2,6 г) в четыреххлористом углероде (13 мл), температуру повышают приблизительно до 20oС и смесь перемешивают в течение 2 часов. Смесь охлаждают на ледяной бане, добавляют воду (200 мл), а затем эфир (400 мл) и слои разделяют. Эфирный слой промывают до нейтральной реакции водой, сушат Na2SO4 и упаривают, получают коричневую смолу (18,9 г), которую кристаллизуют из смеси эфир/гексан. Получают 2-хлор-4-(4-фторбензоил)тиофен (10,3 г), выход 77,6% (ГЖХ).
2. Получение 2,3-дихлор-4-(4-фторбензоил)-тиофен
Маточную жидкость из предыдущего примера хроматографируют на двуокиси кремния. При элюировании смесью толуол/гексан (4:1) получают фракцию, которую упаривают, остаток кристаллизуют из смеси эфир/гексан и получают названное соединение (1,9 г), т.пл. 104-105oС.
Пример 2
А. 1-Метил-4-[(4-хлор-2-тиенил)-(4-фторфенил)]метиленпиперидин(фумарат, соль)
4-Хлор-1-метилпиперидин НС1 (18,7 г) подщелачивают водным гидроксидом аммония и раствор экстрагируют эфиром, экстракт промывают рассолом, сушат (Na2SO4) и упаривают, получают 4-хлор-1-метилпиперидин (14,7 г) в виде почти бесцветного масла.
К суспензии стружек магния (2,32 г) в перегнанном тетрагидрофуране (ТГФ) (10,0 г) при перемешивании добавляют кристалл йода, затем добавляют этилбромид (0,16 мл) и после исчезновения йодного окрашивания (приблизительно 5 мин) по каплям в течение приблизительно 30 минут добавляют 4-хлор-1-метилпиперидин (14,4 г) в ТГФ (96 мл). Реакционную смесь немного нагревают так, чтобы она слегка кипела. По окончании добавления смесь кипятят с обратным холодильником в течение еще 1,5 часа, затем охлаждают приблизительно до 0oС. К раствору по каплям в течение 40 минут добавляют раствор 2-хлор-4-(4-фторбензоил)тиофена (10,3 г, пример 1) в ТГФ (25 мл) и смесь кипятят с обратным холодильником в течение 2,5 часов.
К охлажденной смеси добавляют насыщенный раствор хлорида аммония (100 мл), затем эфир (500 мл). Нерастворимые вещества отфильтровывают через дикалит, слои разделяют. Эфирный слой промывают водой, сушат (Na2SO4) и упаривают. Получают коричневую смолу (18,2 г).
Смесь соляной кислоты (5 н., 60 мл) и соляной кислоты
(2 н., 60 мл) добавляют к коричневой смоле (10,8 г) и при перемешивании смесь кипятят с обратным холодильником в течение 0,75 часа. После полного растворения реакционную смесь охлаждают приблизительно до 5oС и подщелачивают с помощью водного раствора гидроксида аммония. Продукт экстрагируют дихлорметаном, экстракт промывают водой, сушат (Na2SO4) и упаривают. Получают коричневую смолу (8,2 г), которую хроматографируют на двуокиси кремния. При элюировании смесью дихлорметан/метанол (элюирование с градиентом 1-10% метанола) выделяют фракцию, которую упаривают, и получают α-(4-фторфенил)-α-(4-хлор-2-тиенил)-1-метил-4-пиперидинметанол (1,07 г), содержащий некоторое количество примеси перехлорированного продукта.
К суспензии сырого продукта (0,78 г) в растворе гидроксида натрия (4 н., 7,8 мл) добавляют порошок дуста (1,56 г) и смесь кипятят с обратным холодильником в течение 2,5 часов. Смесь охлаждают, добавляют воду (15 мл) и продукт экстрагируют дихлорметаном (30 мл). Экстракт промывают водой (3•30 мл), сушат (Na2SO4) и упаривают, получают коричневую смолу (0,75 г). Этот сырой материал растворяют в метаноле, к метанольному раствору добавляют раствор фумаровой кислоты и полученный раствор упаривают для уменьшения объема. Добавляют эфир и полученные кристаллы собирают, получают чистое названное соединение в виде бледного твердого вещества (0,71 г), т.пл. 209-210oС.
Б. Сукцинат 1-метил-4-[(4-хлор-2-тиенил)-(4-фторфенил)] метиленпиперидина получают аналогично; т.пл. 161-169oС.
В. Аналогичным образом, но без необходимости удаления перехлорированного материала, 2,3-дихлор-4-(4-фторбензоил)-тиофен превращают в фумарат 1-метил-4-[(4,5-дихлор-2-тиенил)-(4-фторфенил)]-метиленпиперидина: т.пл 220oС.
Г. Метил-4-[(4-хлор-2-тиенил)-(4-фторфенил)]метиленпиперидин (альтернативный способ получения)
I. 2-Бром-3-хлортиофен
Раствор N-бромсукцинимида (221,7 г) в диметилформамиде (550 мл) при перемешивании в течение 75 минут по каплям добавляют к раствору 3-хлортиофена (143,4 г) и перхлорной кислоты (70%, 5,8 мл), охлажденному на водно-ледяной бане до 15oС. Температуру реакционной смеси постепенно в течение 30 минут повышают до 40oС и затем охлаждают до 11oС. Охлаждение убирают и реакционную смесь перемешивают в течение еще 2 часов. Реакционную смесь выливают в воду и экстрагируют метил-трет-бутиловым эфиром. Органический экстракт последовательно промывают водой, водным раствором гидросульфата натрия и водой, сушат (Na2S04) и упаривают досуха. Полученное масло (246 г) перегоняют под вакуумом на масляной бане с температурой 80-90oС. Получают 2-бром-3-хлортиофен в виде масла (202 г, выход 97%); т. кип. 42oС (1 мм. рт. ст.).
II. 2-Бром-3-хлор-5-(4-фторбензоил)тиофен
К раствору 2-бром-3-хлортиофена (148,5 г) и 4-фторбензоилхлорида (169,7 г) в дихлорметане (2230 мл), охлажденному до 18oС на водяной бане со льдом, добавляют при перемешивании хлорид железа (301,2 г). Температуру реакционной смеси повышают до 22oС и затем охлаждают до 12oС. Охлаждение убирают и реакционную смесь перемешивают в течение еще 50 минут, в течение которых температура реакционной смеси повышается до 16oС. Реакционную смесь снова охлаждают до 10oС, осторожно добавляют воду (700 мл), поддерживая температуру смеси ниже 15oС, и полученную смесь перемешивают в течение 1 часа. Дихлорметановый слой отделяют, водный слой промывают дихлорметаном. Объединенный дихлорметановый слой промывают водой и насыщенным раствором бикарбоната натрия. Дихлорметан отделяют, водный слой фильтруют через дикалит и экстрагируют дихлорметаном. Экстракт промывают водой, сушат (Na2S04) и упаривают досуха. Получают 2-бром-3-хлор-5-(4-фторбензоил)-тиофен в виде твердого вещества (233,3 г).
III. 4-Хлор-2-(4-фторбензоил)тиофен
К раствору описанного выше 2-бром-3-хлор-5-(4-фторбензоил)-тиофена (299,4 г) в этаноле (3400 мл), содержащему ацетат натрия (77,0 г), в атмосфере азота добавляют суспензию 5% палладия на угле (20 г) в этаноле (100 мл) и полученную смесь гидрируют в течение 3 часов при комнатной температуре и комнатном давлении (суммарное поглощение водорода составляет 22,994 мл). Реакционную смесь фильтруют через слой дикалита, и этот слой промывают этанолом (2•200 мл). Фильтрат упаривают для уменьшения объема (приблизительно до 2 л) и добавляют воду (17 л). Осадок отфильтровывают и влажным растворяют в дихлорметане (1 л), затем объем раствора уменьшают приблизительно до 500 мл. Добавляют метанол (500 мл) и остаток дихлорметана отгоняют. Полученные кристаллы отфильтровывают и сушат, получают 4-хлор-2-(4-фторбензоил)тиофен в виде белого твердого вещества (169 г). Выделяют вторую порцию (19,2 г) продукта и перекристаллизовывают из смеси дихлорметан/метанол, получая более чистый продукт (12,9 г). Выделенные порции используют на следующей стадии.
IV. α-(4-Фторфенил)-α-(4-хлор-2-тиенил)-1-метил-4-пиперидинметанол
Суспензию магниевой стружки (36,7 г) в тетрагидрофуране (470 мл) нагревают в атмосфере азота до 55oС и добавляют аликвоту (20,0 мл) раствора N-метил-4-хлорпиперидина (221,6 г) в тетрагидрофуране (1400 мл), а затем кристалл йода и этилбромид (14,0 мл). Почти сразу же наблюдается повышение температуры до 64oС, которое сопровождается исчезновением окраски, обусловленной йодом, и началом кипения. Затем в течение 75 минут добавляют остаток раствора N-метил-4-хлорпиперидина, поддерживая мягкое кипение. По окончании добавления смесь перемешивают при 64oС в течение еще 90 минут, после чего в реакционной смеси остается только несколько крупинок магния. Раствор охлаждают на водяной бане со льдом до 20oС и затем в течение 60 минут подают насосом при повышенном давлении азота в раствор 4-хлор-2-(4-фторбензоил)тиофена (140,0 г) в тетрагидрофуране (1,4 л), который предварительно охлажден до 0oС на бане со льдом и солью; температуру смеси при этом поддерживают ниже 8oС. Раствор в течение 40 минут добавляют к холодному (5oС) насыщенному раствору хлорида аммония (4,6 л), поддерживая температуру ниже 15oС. Смесь экстрагируют этилацетатом, экстракт промывают водой, сушат (Na2SO4) и упаривают досуха. Получают коричневую смолу (202 г). Смолу кристаллизуют из смеси дихлорметан/метил-трет-бутиловый эфир, получают твердый продукт, который фильтруют и промывают холодным метил-трет-бутиловым эфиром. Твердый продукт фильтруют и сушат. Полученный продукт (79,2 г) перекристаллизовывают из смеси дихлорметан/метил-трет-бутиловый эфир. Получают α-(4-фторфенил)-α-(4-хлор-2-тиенил)-1-метил-4-пиперидинметанол(61,6 г).
V. 1-Метил-4-[(4-хлор-2-тиенил)-(4-фторфенил)]метиленпиперидин
Описанный выше спирт (73,8 г) в течение 50 минут кипятят с обратным холодильником в смеси 2 н. соляной кислоты (440 мл) и 5 н. соляной кислоты (440 мл). Реакционную смесь охлаждают на водяной бане со льдом, полученный белый осадок отфильтровывают и промывают водой, получают белые блестящие кристаллы. Влажные кристаллы суспендируют в смеси этилацетата (300 мл) и воды (300 мл) и при перемешивании подщелачивают с помощью 4 н. раствора гидроксида натрия (200 мл). Этилацетатный слой отделяют, и водный слой экстрагируют этилацетатом. Объединенные экстракты промывают водой, сушат (Na2SO4) и упаривают. Получают 1-метил-4-[(4-хлор-2-тиенил)-(4-фторфенил)]-метиленпиперидин в виде коричневого масла (66,8 г), 98% (ГЖХ).
VI. Сукцинат 1-метил-4-[(4-хлор-2-тиенил)-(4-фторфенил)]метиленпиперидина
Описанный выше амин (64,6 г) растворяют в метаноле (640 мл), добавляют янтарную кислоту (24,88 г), раствор упаривают досуха и полученный твердый остаток (89,4 г) растворяют в горячем этаноле (1,1 л). Объем раствора уменьшают (200 мл), охлаждают на водяной бане со льдом, образующийся твердый продукт отфильтровывают и промывают этанолом (200 мл). Твердый продукт сушат в вакууме при 50oС, получают сукцинат 1-метил-4-[(4-хлор-2-тиенил)-(4-фторфенил)]-метиленпиперидина (1:1, соль) (78,2 г) в виде почти белого твердого вещества; т. пл. 171oС.
Пример 3
А. Получение гидрохлорида 1-метил-4-[(4-метил-2-тиенил)-(4-фторфенил)] метиленпиперидина
К раствору н-бутиллития в гексане (1,6 н., 20 мл) добавляют раствор 3-метилтиофена (3,5 г) в сухом ТГФ (40 мл). Полученный раствор кипятят с обратным холодильником в течение 2 часов. Раствор охлаждают до 0oС и в течение 10 минут добавляют раствор 1-метил-4-(4-фторбензоил)пиперидина (2,1 г) в сухом ТГФ (40 мл), поддерживая температуру приблизительно 0oС. Раствору дают нагреться до комнатной температуры, осторожно добавляют воду (20 мл) и смесь экстрагируют этилацетатом. Экстракт снова экстрагируют соляной кислотой (1 н. ), водный экстракт подщелачивают водным гидроксидом аммония и продукт экстрагируют этилацетатом. Этот экстракт промывают водой, сушат (Na2SO4), упаривают, получают масло (2,6 г). Раствор этого масла (1,7 г) в пиридине (17 мл), содержащий оксихлорид фосфора (0,1 мл), нагревают при 115-120oС в течение 8,5 часов. Раствор охлаждают, добавляют воду, смесь подщелачивают водным гидроксидом аммония и экстрагируют этилацетатом. Экстракт промывают водой, сушат (Na2SO4) и упаривают, получают сырой продукт в виде свободного основания, которое очищают путем фильтрования на двуокиси кремния. Обработка раствора свободного основания эфирным раствором хлористого водорода дает твердый продукт, который кристаллизуют из смеси метанол/эфир. Получают 1-метил-4-[(4-метил-2-тиенил-(4-фторфенил)] метиленпиперидин НС1 (1,45 г); т. пл. 235oС.
Б. Следующие соединения получают по аналогичной методике:
- из 2-этилтиофена получают малеат 1-метил-4-[(5-этил-2-тиенил)-(4-фторфенил)]метиленпиперидина; т. пл. 175-176oС.
- из 3-этилтиофена получают малеат 1-метил-4-[(4-этил-2-тиенил)-(4-фторфенил)]метиленпиперидина; т. пл. 76,8oС.
из бензотиофена получают малеат 1-метил-4-(2-бензотиенил)-(4-фторфенил)] метиленпиперидина: т. пл. 197-214oС.
- из 5-фторбензотиофена получают малеат 1-метил-4-[(5-фтор-2-бензотиенил)-(4-фторфенил)]метиленпиперидина; т. пл. 152-155oС.
- из 6-метоксибензотиофена получают малеат 1-метил-4-[(6-метокси-2-бензотиенил)-(4-фторфенил)]метиленпиперидина; т. пл. 150-153oС.
- из бензофурана получают малеат 1-метил-4-[(2-бензофуранил)-(4-фторфенил)]метиленпиперидина: т. пл. 196,2oС.
- из 2,3-диметилфурана получают малеат 1-метил-4-[(4,5-диметил-2-фуранил)-(4-фторфенил)]метиленпиперидина; т. пл. 197-200oС.
Пример 4
Гидрохлорид 4-[(4-Фторфенил)-2-(4-метилтиенил)метиленпиперидина
К раствору 4-(1-ацетилпиперидинил)хлорида (50 г) в дихлорметане (690 мл) в атмосфере азота при температуре -25oС последовательно при перемешивании добавляют порошкообразный хлорид алюминия (71 г), затем в течение 17 минут раствор 2-бром-3-метилтиофена (50 г) в дихлорметане (300 мл). Через 30 минут к реакционной смеси по каплям добавляют воду (240 мл), позволяя температуре реакционной смеси повыситься приблизительно до +20oС. После перемешивания в течение еще 30 минут неорганические компоненты отфильтровывают через слой дикалита. Слои разделяют, органический слой дважды промывают водой, сушат (Na2SO4) и упаривают при пониженном давлении. Сырой продукт (73 г) очищают хроматографией, получают 2-(5-бром-4-метилтиенил)-4-(1-ацетилпиперидин)метанон (62,2 г); т. пл. 105-108,5oС (разл.).
Суспензию порошка цинка (22 г), йодида натрия (11 г), трифенилфосфина (16,5 г) и гексагидрата хлорида никеля (2,56 г) в очищенном от кислорода метаноле (340 мл) (получают кипячением метанола в потоке азота в течение 2 часов) перемешивают в атмосфере азота при 60oС в течение 15 минут. К этой смеси добавляют раствор полученного выше бромсодержащего соединения (62,2 г) в очищенном от кислорода метаноле (150 мл) и реакционную смесь кипятят с обратным холодильником в атмосфере азота в течение 22 часов. Реакционную смесь охлаждают, и неорганические компоненты отфильтровывают через слой дикалита. Фильтрат упаривают, и остаток растворяют в дихлорметане. Раствор промывают разбавленной минеральной кислотой и затем водой до нейтральной реакции, сушат (Na2SO4) и упаривают досуха при пониженном давлении. Сырой продукт (61,3 г) очищают быстрой хроматографией, кристаллизуют из смеси дихлорметан/эфир и получают в две порции 2-(4-метилтиенил)-4-(1-ацетилпиперидин)метанон (41,2 г); т. пл. 120-125oС. Раствор полученного метанона (41,2 г) в 5 н. водном растворе соляной кислоты (140 мл) кипятят с обратным холодильником в течение 16 часов и затем упаривают при пониженном давлении, оставшуюся воду удаляют азеотропной отгонкой с толуолом. После растирания остатка с диэтиловым эфиром получают сырой продукт (38,8 г), который выделяют фильтрованием. После перекристаллизации из смеси метанол/диэтиловый эфир получают гидрохлорид 2-(4-метилтиенил)-4-пиперидинметанона в две порции (29,5 г); т. пл. 217,5-218,5oС (изменения в кристаллической форме начинаются от температуры выше 200oС).
Раствор полученного выше гидрохлорида (28 г) в воде подщелачивают и раствор основания (24,1 г) в дихлорметане (240 мл) и триэтиламине (48 мл) перемешивают при 0oС в атмосфере азота. По порциям добавляют трифенилметилхлорид (33,7 г) с такой скоростью, чтобы поддерживать температуру реакции 0±2oС. Через 30 минут смесь осторожно добавляют водой (240 мл) и экстрагируют дихлорметаном. Экстракт промывают, сушат (Na2SO4) и упаривают при пониженном давлении, частично заменяя дихлорметан гептаном, и оставляют кристаллизоваться. Кристаллы отфильтровывают и промывают смесью гептана и дихлорметана, 4:1, получают 2-(4-метилтиенил)-4-(1-трифенилметилпиперидин)-метанон (46,9 г): т. пл. 219-221oС (разл.).
К суспензии магниевой стружки (6,4 г) в сухом диэтиловом эфире (100 мл), содержащей кристаллик йода, при перемешивании добавляют бромэтан (1,5 мл). Температуру экзотермичной реакции поддерживают в интервале 32-36oС на всем протяжении реакции, пока осторожно добавляют раствор 4-бромфторбензола (29 мл) в сухом диэтиловом эфире (170 мл). Полученную смесь мягко кипятят с обратным холодильником в течение 30 минут и затем охлаждают до 0oС.
К этой смеси в течение 15 минут по каплям добавляют раствор полученного выше метанона (23,5 г) в сухом диэтиловом эфире (280 мл), поддерживая температуру в интервале 0-5oС. Затем реакционной смеси дают нагреться до комнатной температуры в течение 30 минут и продукт экстрагируют этилацетатом. Экстракты промывают водой, сушат (Na2SO4) и упаривают при пониженном давлении, получают смолу (32,4 г), которую растворяют в смеси уксусной кислоты (261 мл) и воды (130 мл). Полученный раствор кипятят с обратным холодильником в течение 18 часов. Добавляют воду (130 мл) и реакционную смесь охлаждают до температуры <5oС. Твердое вещество (трифенилметиловый спирт) отфильтровывают, и фильтрат упаривают при пониженном давлении до небольшого объема. Остаток подщелачивают концентрированным раствором гидроксида аммония, и продукт экстрагируют этилацетатом. Экстракты промывают водным раствором хлорида натрия, сушат (Na2SO4) и упаривают досуха при пониженном давлении, получают смолистый остаток (15,0 г). К раствору этого продукта в диэтиловом эфире добавляют раствор соляной кислоты в метаноле, и раствор оставляют для кристаллизации, получают гидрохлорид 4-[(4-фторфенил)-2-(4-метилтиенил)-метиленпиперидина (9,0 г); т. пл. 191-206oС (разл.).
Пример 5
Фумарат 1-бутил-4-[(5-хлор-2-тиенил)-(4-фторфенил)]-метиленпиперидина
Раствор 4-[(5-хлор-2-тиенил)-(4-фторфенил)] метиленпиперидина (0,69 г), 1-бромбутана (0,48 мл) и триэтиламина (1,56 мл) в толуоле (20 мл) при перемешивании кипятят с обратным холодильником в течение 24 часов. Реакционной смеси дают охладиться, переносят в делительную воронку и промывают толуолом (30 мл) и водой (30 мл). Смесь встряхивают, слой толуола отделяют, промывают водой, сушат (Na2SO4) и упаривают, получают вязкое масло (0,71 г), которое хроматографируют на двуокиси кремния. После элюирования смесью дихлорметан/метанол, 24:1, получают фракцию, которую упаривают, получают вязкое масло (0,53 г). Полученное масло (0,48 г) растворяют в метаноле (5 мл) и к раствору добавляют раствор фумаровой кислоты (0,16 г) в метаноле (5 мл). Раствор упаривают до небольшого объема. Добавляют эфир и полученные кристаллы отфильтровывают, получают названное соединение (0,51 г); т. пл. 167-172oС.
Пример 6
По методике, описанной в примере 5, но с использованием (бромметил)циклопропана в качестве алкилирующего агента, получают фумарат 1-циклопропилметил -4-[(5-хлор-2-тиенил)-(4-фторфенил)]метиленпиперидина: т. пл. 171-172oС.
Пример 7
Фумарат 4-(4-фторфенил-2-нафртил)-1-метилпиперидина
В атмосфере азота к суспензии магниевой стружки (0,37 г) в сухом перегнанном тетрагидрофуране (10 мл), содержащем кристаллик йода, при перемешивании добавляют раствор 2-бромнафталина (3,13 г) в сухом тетрагидрофуране (10 мл). Реакционную смесь нагревают до 55oС на водяной бане, причем нагревание прекращают через 3 минуты после начала реакции. При добавлении последних порций температуру реакционной смеси поддерживают в интервале 50-55oС с помощью водяной бани. Через 15 минут реакционную смесь охлаждают до 5oС и затем добавляют раствор 4-(4-фторбензоил)-N-метилпиперидина (0,81 г) в сухом тетрагидрофуране (4 мл), поддерживая температуру в интервале 5-10oС. Смесь нагревают при 50oС в течение 1 часа и затем охлаждают до 5oС, добавляют водный раствор хлорида аммония (15 мл). Продукт экстрагируют диэтиловым эфиром, экстракты промывают водой, сушат (Na2SO4) и упаривают, получают белое твердое вещество (2,41 г), которое хроматографируют на силикагеле. После элюирования смесью дихлорметан/метанол (95:5, об./об.) получают α- (4-фторфенил) -α- (2-нафтил)-1-(метил)-4-пиперидинметанол, который кристаллизуется из дихлорметана при добавлении эфира (0,27 г); т. пл. 164-169oС.
Суспензию полученного выше спирта (0,81 г) в 3,5 н. соляной кислоте (10 мл) кипятят с обратным холодильником в течение 2,5 часов, охлаждают до 20oС и затем подщелачивают водным раствором аммиака (5 мл). Продукт экстрагируют дихлорметаном (50 мл), экстракт промывают водой, сушат (Na2SO4) и упаривают досуха, получают 4-(4-фторфенил-2-нафтил)-1-метилпиперидин в виде бледно-желтой смолы (0,81 г). Это соединение переводят в фумарат по описанной выше методике и кристаллизуют из метанола при добавлении эфира, получают названное соединение (0,89 г); т. пл. 214-218oС.
Пример 8
Тест на индуцированный апоморфином рефлекс лазания у мышей
Способность антагонистов рецепторов допамина ингибировать поведенческие рефлексы у грызунов, вызванные агонистами допамина, такими как апоморфин, представляет собой хорошо изученный критерий для определения антипсихотической эффективности таких лекарственных средств для человека (см., например, W. C. Bowman, M.J. Rand, Textbook of Pharmacology, 2-nd ed., 1980, 15, 6). Особенно подходящим в данном случае испытанием является тест на индуцированный апоморфином рефлекс лазания у мышей (АТС-apomorphine cliving test), в котором оценивается способность антагонистов ингибировать рефлекс лазания у мышей, вызванный путем подкожного или перорального введения апоморфина. Активность, выявленная в этом тесте и проявляющаяся после системного или перорального введения, широко используется в качестве показателя антипсихотической активности, то есть, антишизофренической активности (см., например, J. T. Strupczewski et al., J.Med.Chem., 1995, 38, 1119). Мыши, обработанные гидрохлоридом апоморфина, имеют тенденцию принимать вертикальное положение вдоль стенки цилиндра из проволоки с отверстиями, оставаясь при этом неподвижными или залезая на стенку. Такое лазание, как считают, является результатом опосредуемой апоморфином стимуляции допаминовых рецепторов. Большое число лекарственных средств вызывает поведение, проявляющееся в лазании, однако антагонисты допамина в дозах, не влияющих на самопроизвольную моторную активность и/или моторную координацию у мышей, обычно ингибируют такое поведение. Испытываемые соединения, которые модулируют указанный рефлекс лазания, могут иметь антипсихотическую активность.
Мышей случайным образом подвергают различным вариантам обработок. Каждый опыт включает 1+n испытуемых групп: 1 группа представляет собой контрольную группу из 12 мышей, которым вводят апоморфин и растворитель подкожно; или представляет собой контрольную группу из 12 мышей, которым вводят подкожно апоморфин и перорально растворитель; n-группа (обычно 4) представляет собой группу для испытываемых соединений, состоящую из 12 мышей, которым вводят апоморфин и испытываемое соединение подкожно, или группу для испытываемых соединений из 12 мышей, которым вводят подкожно апоморфин и перорально испытываемое соединение.
Опыты проводят в 3 сериях по 20 мышей каждая. Мышей помечают и взвешивают. Испытываемые соединения и растворитель вводят подкожно, и мышей помещают в небольшие клетки Макролона (17х11х13 см), по 5 мышей в клетку. Или испытываемое соединение и растворитель вводят перорально, и мышей помещают в клетки Макролона (29х11х13 см), по 5 мышей в клетку. Через 30 минут подкожно вводят 0,75 мг/кг гидрохлорида апоморфина мышам, обработанным подкожно растворителем или испытываемым соединением, или 0,75 мг/кг гидрохлорида апоморфина вводят подкожно мышам, обработанным перорально растворителем или испытываемым соединением, а мышей помещают по отдельности в проволочный цилиндр с отверстиями (диаметр 12 см, высота 14 см).
Через 10 минут после обработки апоморфином оценивают проявление рефлекса лазания для каждой мыши и выражают его в баллах в соответствии со следующей шкалой;
4 лапы на полу - 0 баллов
1 или 2 лапы находятся на стенке - 1 балл
3 или 4 лапы на стенке - 2 балла
Через 20 минут после обработки апоморфином снова оценивают рефлекс лазания в баллах. Для каждой из обработанных групп определяют среднее значение. Балл для контрольной группы должен быть равен, по меньшей мере, 1,0; если нет, то испытание забраковывают. Конечные результаты для группы выражают в процентах относительно контрольной группы.
Полученные в этом опыте результаты для испытанных соединений представлены в Таблице I (при подкожном и пероральном введении испытываемых соединений).
Соединения Примера 2,1-метил-4-[(4-хлор-2-тиенил)-(4-фторфенил)] метиленпиперидин, и Примера 3, 1-метил-4-[(4-метил-2-тиенил)-(4-фторфенил)]метиленпиперидин, сравнивают с 1-метил-4-[(2-тиенил)-(4-фторфенил)]метиленпиперидином (Соединение А), которое подпадает под объем притязаний патента США 2739968, 1-метил-4-(2-тиенил)фенилметиленпиперидином (Соединение Б) в соответствии с патентом США 2739968 и с 1-метил-4-[(5-хлор-2-тиенил)-фенил]метиленпиперидином (Соединение С). Соединения в соответствии с настоящим изобретением обладают высокой активностью, в особенности высокой пероральной активностью по сравнению с Соединением А.
Пример 9
Каталепсия у крыс: В опытах по оценке каталепсии используют самцов крыс Wistar (100-125 г, Olac UK). Каталепсию оценивают в соответствии с описанной ранее методикой (Broekkamp et al., Naunyn-Schmiedeberg's Arch. Pharmacol. 338, 191, 1988). Крыс испытывают в 6 различных опытах, при которых животных ставят в аномальные позы и оценивают достоверно с одной точки сохранение навязанного положения в течение 10 сек. Навязанными положениями были: вертикальное цепляние за решетку, вертикальная стойка с высокой подпоркой для передних лап, растяжение задних лап, положение на спине, со шпателем во рту и вращение в проволочном цилиндре.
Теоретически может быть получен максимальный балл 6. Каталепсию определяют через 60 и 120 минут после введения лекарства. Результаты оценивают с помощью 2-х ходового анализа вариантов ANOVAR, за которым следует Newman Kools post hoc тест, и рассчитывают ЕД50 (Таблица II).


Изобретение относится к новым производным пиперидина формулы I, где R1 означает бензотиенил, бензофуранил, нафтил, которые могут быть замещены галогеном, С1-С6-алкилом, С1-С6-алкоксигруппой, замещенный тиенил или замещенный фуранил, которые замещены галогеном, С1-С6-алкилом, С3-С6-циклоалкилом или С1-С6-алкенилом, R2 означает галоген и R3 означает С1-С6-алкил или С3-С6-циклоалкилметил, или их фармацевтически приемлемым солям, или сольватам. Соединения формулы I обладают антипсихотической активностью и могут найти применение в медицине. 2 с. и 6 з.п.ф-лы, 2 табл.
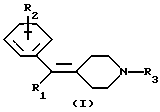

где заместитель R1 представляет собой бензотиенил, бензофуранил, нафтил (где бензотиенильный, бензофуранильный или нафтильный фрагмент может быть необязательно замещен одним или несколькими заместителями, выбираемыми из атома галогена, C1-6-алкоксигруппы, C1-6-алкила и C1-6-алкенила), замещенный тиенил или замещенный фуранил (где тиенильный или фуранильный фрагмент замещены одним или несколькими заместителями, выбираемыми из атома галогена, C1-6-алкила, С3-6-циклоалкила и C1-6-алкенила);
заместитель R2 представляет собой атом галогена;
заместитель R3 представляет собой С1-6-алкил или С3-6-циклоалкилметил,
или их фармацевтически приемлемые соли, или сольваты.
| ОПТОВОЛОКОННЫЙ ФЛУОРИМЕТР С ПОГРУЖАЕМОЙ ТЕРМОКАМЕРОЙ | 2019 |
|
RU2739968C1 |
| Устройство для подачи ленточного материала в штамп | 1973 |
|
SU465254A1 |
| Хлоргидрат 2-/2-/2- @ 4-(дифенилметилен)-1-пиперидинил @ -этокси/-этокси/-уксусной кислоты,обладающий антигистаминной и бронхолитической активностью | 1982 |
|
SU1108090A1 |

