Область техники
Настоящее изобретение относится к гидроксикарбонильным-галогеналкильным производным, разработанным для значительного повышения пероральной активности соединений, обладающих низкой активностью при пероральном введении, соединений, обладающих противоопухолевой активностью, соединений, обладающих эстрогенной активностью или соединений, обладающих антиэстрогенной активностью.
Предпосылки создания изобретения
При лечении заболеваний, вызванных аномальным ростом ткани, который зависит от некоторых половых стероидных гормонов, таких как эстроген, очень важно ингибировать в значительной степени, более предпочтительно исключить полностью, индуцируемое гормоном действие. С этой целью желательно понизить уровень гормона, способного воздействовать на рецепторный сайт стероидного гормона. Например, антиэстрогенные агенты обычно вводят при альтернативной или комбинационной терапии для ограничения продуцирования эстрогена до количества, меньше того, которое требуется для активации сайта рецептора. Однако такие обычные способы блокирования продуцирования эстрогена не могут в достаточной степени ингибировать действие, индуцируемое посредством эстрогенового рецептора. Практически, даже когда эстроген отсутствует полностью, некоторые из рецепторов могут быть активированными. Вследствие этого полагают, что антагонисты эстрогена могут обеспечить лучшее терапевтическое действие в сравнении со способами только блокирования продуцирования полового стероидного гормона. Таким образом, были разработаны многочисленные антагонисты эстрогена. Например, во многих патентных публикациях, включая патенты США №№4760061, 4732912, 4904661, 5395842 и WO 96/22092, описаны различные антиэстрогенные соединения. Однако иногда антагонисты предшествующего уровня техники сами могут действовать в качестве агонистов и, следовательно, скорее активировать рецептор, чем его блокировать. Например, в качестве антиэстрогенного агента наиболее широко используется Тамоксифен. Однако данный агент обладает тем недостатком, что он проявляет эстрогенную активность в некоторых органах (см. М.Harper и A.Walpole, J.Reprod. Fertile., 1967, 13, 101).
В качестве другого нестероидного антиэстрогенного соединения в WO 93/10741 описано производное бензопирана, имеющее аминоэтоксифенильный заместитель(и) (Эндоречерче, Endorecherche), типичным соединением в ряду которых является ЕМ-343, имеющий следующую структуру:

Указанное соединение также обладает агонистическим действием. Следовательно, необходимо разработать антиэстрогенное соединение, которое в достаточной степени или полностью не обладает агонистическим действием и которое может эффективно блокировать эстрогеновый рецептор.
Кроме того, известно, что 7α-замещенные производные эстрадиола, например 7α-(CH2)10CONMeBu производные, являются стероидными антиэстрогенными агентами без агонистического действия (см. ЕР-А 0138504, патент США 4659516). Кроме того, производное эстрадиола, имеющее 7α-(CH2)9SOC5H6F5 заместитель, также описано в качестве 7α-замещенного производного эстрадиола (см. Wakeling et al. Cancer.Res., 1991, 51, 3867).
Нестероидные антиэстрогенные агенты, не обладающие агонистическим действием, впервые были описаны Wakeling et al. в 1987 г. (см. A. Wakeling and Bowler, J. Endocrinol., 1987, 112, R7). Между тем, в патенте США 4904661 описаны производные фенола, обладающие антиэстрогенной активностью. Такие производные фенола обычно имеют нафталиновый каркас и включают, обычно, следующие соединения:
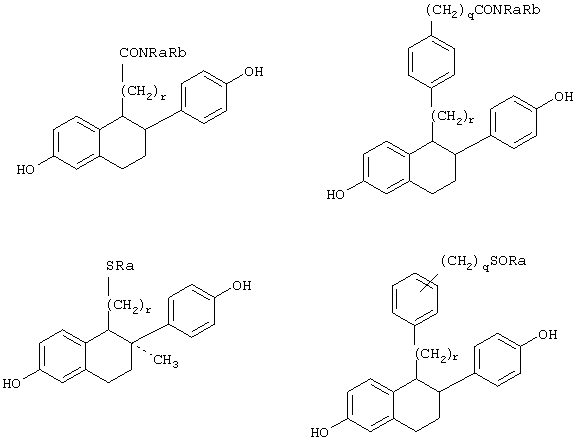
Сообщалось о некоторых производных хромана и тиохромана в качестве антиэстрогенных соединений, не обладающих агонистическим действием (WO 98/25916). Хотя существующие антиэстрогенные соединения, не обладающие агонистическим действием, показывают значительный терапевтический эффект при введении путем внутривенной или подкожной инъекции, они проявляют значительно сниженное терапевтическое действие при пероральном введении вследствие их низкой биодоступности при пероральном пути введения. Следовательно, для удобства при введении желательно разработать антиэстрогенные соединения, которые демонстрируют достаточное действие при пероральном введении и в то же время не обладают агонистическим действием. Также обычно требуется разработать агенты, которые демонстрируют достаточное действие при пероральном введении.
Описание изобретения
Задача настоящего изобретения заключается в разработке гидроксикарбонил-галогеналкильных производных, созданных для значительного повышения пероральной активности соединений, обладающих низкой активностью при пероральном введении, соединений, обладающих противоопухолевой активностью, соединений, обладающих эстрогенной активностью или соединений, обладающих антиэстрогенной активностью, путем усиления их абсорбции из кишечного тракта и/или улучшения их стабильности в отношении метаболизма.
Исследовательская работа авторов данного изобретения была направлена на достижение вышеуказанной задачи, и ими установлено, что боковая цепь общей формулы (1), если она присоединена к исходному каркасу соединений, дает возможность эстрогенным соединениям проявить существенно повышенную активность при пероральном пути введения. Настоящее изобретение было осуществлено на основании этих сведений.
А именно, настоящее изобретение относится к соединению, состоящему из фрагмента и группы, химически связанной с указанным фрагментом, где указанный фрагмент содержит соединение, обладающее низкой активностью при пероральном введении, или его исходный каркас, и указанная группа имеет следующую общую формулу (1):

в которой R1 представляет атом водорода или солеобразующий металл,
R2 представляет прямую или разветвленную C1-C7 галогеналкильную группу,
m представляет целое чисто от 2 до 14 и
n представляет целое число от 2 до 7,
или энантиомерам первоначально указанного соединения, или гидратам, или фармацевтически приемлемым солям соединения или его энантиомеров.
Настоящее изобретение также относится к соединению, состоящему из фрагмента и группы, химически связанной с указанным фрагментом, где указанный фрагмент содержит соединение, обладающее противоопухолевой активностью, или его исходный каркас, и указанная группа имеет следующую общую формулу (1):

в которой R1 представляет атом водорода или солеобразующий металл,
R2 представляет прямую или разветвленную C1-C7 галогеналкильную группу,
m представляет целое чисто от 2 до 14 и
n представляет целое число от 2 до 7,
или энантиомерам первоначально указанного соединения, или гидратам, или фармацевтически приемлемым солям соединения или его энантиомеров.
Кроме того, настоящее изобретение относится к соединению, состоящему из фрагмента и группы, химически связанной с фрагментом, где указанный фрагмент содержит соединение, обладающее эстрогенной активностью, или его исходный каркас, или соединение, обладающее антиэстрогенной активностью, или его исходный каркас, и указанная группа имеет следующую общую формулу (1):

в которой R1 представляет атом водорода или солеобразующий металл,
R2 представляет прямую или разветвленную C1-C7 галогеналкильную группу,
m представляет целое чисто от 2 до 14 и
n представляет целое число от 2 до 7,
или энантиомерам первоначально указанного соединения, или гидратам, или фармацевтически приемлемым солям соединения или его энантиомеров.
Еще, помимо этого, настоящее изобретение относится к соединению, имеющему следующую общую формулу (2):

в которой R1 представляет атом водорода или солеобразующий металл,
R2 представляет прямую или разветвленную C1-C7 галогеналкильную группу,
m представляет целое чисто от 2 до 14,
n представляет целое число от 2 до 1 и
А представляет группу, выбранную из следующих формул (3)-(8) и (10)-(26):


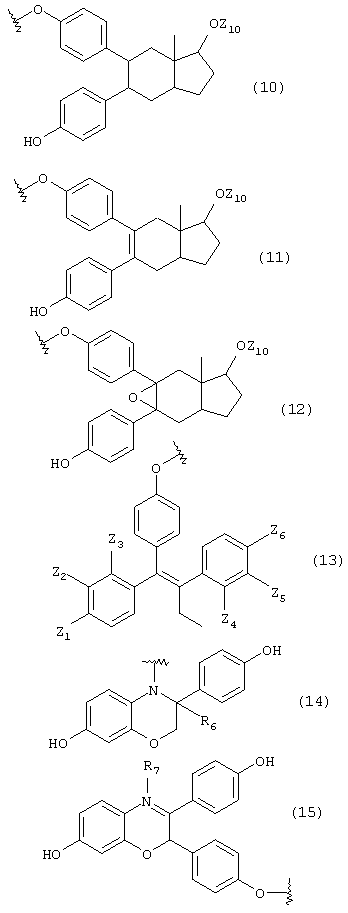


где в формулах (6), (7), (14) и (24) каждый из R3 и R6 представляет прямую или разветвленную C1-C5 алкильную группу, в формулах (10), (11) и (12) Z10 представляет атом водорода или ацильную группу, в формулах (13), (21) и (22) каждый из Z1, Z2, Z3, Z4, Z5 и Z6 независимо представляет атом водорода, гидроксильную группу или прямую или разветвленную C1-C5 алкильную группу, в формуле (15) R7 представляет атом водорода или прямую или разветвленную C1-C5 алкильную группу, в формуле (16) каждый из Z7, Z8 и Z9 независимо представляет атом водорода или гидроксильную группу, в формулах (18) и (20) R8 представляет прямую или разветвленную C1-C5 алкильную группу, прямую или разветвленную C2-C5 алкенильную группу или прямую или разветвленную C2-C5 алкинильную группу, в формуле (23) каждый из R21, R22, R23 и R24 независимо представляет атом водорода, прямую или разветвленную C1-C5 алкильную группу, прямую или разветвленную C1-C7 галогеналкильную группу, атом галогена или ацильную группу и в формулах (25) и (26) Х представляет атом галогена, или энантиомерам соединения, или гидратам, или фармацевтически приемлемым солям соединения или его энантиомеров.
Кроме того, настоящее изобретение относится к фармацевтической композиции, включающей соединение общей формулы (2) в качестве активного ингредиента. Настоящее изобретение также относится к антиэстрогенной фармацевтической композиции, включающей указанное выше соединение в качестве активного ингредиента. Далее настоящее изобретение относится к терапевтическому агенту против рака молочной железы, включающему соединение общей формулы (2) в качестве активного ингредиента.
Как использовано в данном описании, термин “исходный каркас(ы)” относится к частичной структуре, являющейся общей для класса соединений, обладающих одинаковым или подобным фармакологическим действием или физико-химическими свойствами. Исходные каркасы включают, но не ограничиваются ими, следующие структуры: стероид, индол, нафталин, бензофуран, бензотиофен, бензопиран, бензоксазин, 3,4-дифенил[4.3.0]нонан, 4-(1,2-дифенил-1-бутенил)фенол, флавон, эритромицин, алкалоид, цефалоспорин, β-лактам и их производные.
Соединения, обладающие низкой активностью при пероральном введении, относятся к таким соединениям, которые не способны проявлять адекватную активность для желаемого фармакологического действия при пероральном введении, поскольку они слабо абсорбируются из кишечного тракта или быстро метаболизируют в теле. Примеры включают некоторые типы противоопухолевых соединений, некоторые типы эстрогенных соединений (например, эстрадиол) и антиэстрогенные соединения.
Соединения, обладающие противоопухолевой активностью включают все типы соединений, способных ингибировать рост опухоли. Настоящее изобретение имеет особые преимущества для соединений, проявляющих низкую активность при пероральном пути введения.
Соединения, обладающие эстрогенной активностью, относятся к тем соединениям, которые обладают аффинностью к эстрогеновому рецептору и усиливают сигнал, опосредованный эстрогеновым рецептором. Примеры включают эстрадиол.
Соединения, обладающие антиэстрогенной активностью, относятся к тем соединениям, которые обладают антагонистической активностью против фармакологических действий эстрогена. Примеры включают соединения, описанные в упомянутом выше предшествующем уровне развития данной области.
Настоящее изобретение относится к соединениям, где фрагмент химически связан с группой, при этом указанный фрагмент содержит соединение, обладающее низкой активностью при пероральном введении, соединение, обладающее противоопухолевой активностью, соединение, обладающее эстрогенной активностью или соединение, обладающее антиэстрогенной активностью, или исходные каркасы данных соединений, и указанная группа имеет общую формулу (1). Как использовано в данном описании, термин “химически связанный” означает, что группа связана посредством ковалентной связи и тому подобное, включая С-С связь, С-O связь, C-N связь и т.д. Фрагмент, содержащий указанные выше соединения или их исходные каркасы может иметь любую структуру пока возможны данные связи. Для улучшения стабильности против метаболизма и, следовательно, активности при пероральном пути введения предпочтительно используют С-С связь.
Солеобразующие металлы в качестве R1 включают, но не ограничиваются ими, щелочные металлы, такие как натрий и калий, щелочноземельные металлы, такие как магний и кальций, редкоземельные металлы, такие как церий и самарий, а также цинк и олово. Среди них щелочные металлы и щелочноземельные металлы являются предпочтительными.
R1 предпочтительно может представлять атом водорода, щелочной металл и щелочноземельный металл.
Галогены в прямых или разветвленных C1-C7 галогеналкильных группах в качестве R2 включают фтор, хлор, бром и иод, где фтор является предпочтительным. R2 может содержать один или несколько атомов галогена. Когда R2 содержит два или более атомов галогена, они могут быть одинаковыми или различными, предпочтительно одинаковыми атомами галогена. В частности, R2 предпочтительно представляет пергалогеналкильную группу. Алкилы в рассматриваемых прямых или разветвленных C1-C7 галогеналкильных группах включают, но не ограничиваются ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, 1-метилбутил, 2-метилбутил, 3-метилбутил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, н-гексил и н-гептил. Предпочтительными являются прямые или разветвленные C1-C4 алкилы, т.е. метил, этил, н-пропил, изопропил и н-бутил.
Примеры прямой или разветвленной C1-C7 пергалогеналкильной группы в качестве R2 включают перечисленные выше прямые или разветвленные C1-C7 алкильные группы при условии, что они являются пергалогенированными, предпочтительно перфторированными. Также предпочтительными являются пергалогенированные прямые или разветвленные C1-C5 алкильные группы, и группа следующей общей формулы (9):
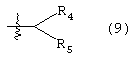
в которой каждый из R4 и R5, которые могут быть одинаковыми или различными, представляют прямую или разветвленную C1-C3 пергалогеналкильную группу. Из них предпочтительными являются перфторированные группы. Более конкретно, особенно предпочтительными являются перфторметильная группа, перфторэтильная группа, перфтор-н-пропильная группа и перфтор-н-бутильная группа.
В случае, когда R2 в общей формуле (2) представляет группу общей формулы (9), примеры прямой или разветвленной C1-C3 пергалогеналкильной группы в качестве R4 и R5 включают перечисленные выше прямые или разветвленные C1-C3 алкильные группы, при условии, что они являются пергалогенированными, предпочтительно перфторированными. Кроме того, пергалогенированные C1 алкильные группы являются предпочтительными и перфторированная группа является особенно предпочтительной. Более конкретно, перфторметильная группа является предпочтительной.
В случае, когда R2 в общей формуле (2) представляет группу общей формулы (9), R2 предпочтительно представляет 1,1,1,3,3,3-гексафторизопропильную группу.
С учетом данного выше определения, R2 предпочтительно представляет перфторэтильную группу, пертфор-н-пропильную группу, перфтор-н-бутильную группу и 1,1,1,3,3,3-гексафторизопропильную группу.
Примеры прямой или разветвленной C1-C5 алкильной группы, как использовано в данном описании, включают, но не ограничиваются ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, 1-метилбутил, 2-метилбутил, 3-метилбутил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил и 1-этилпропил.
Примеры прямой или разветвленной C2-C5 алкенильной группы, как использовано в данном описании, включают, но не ограничиваются ими, винил, аллил, 1-бутенил, 2-бутенил и 3-бутенил.
Примеры прямой или разветвленной C2-C5 алкинильной группы, как использовано в данном описании, включают, но не ограничиваются ими, этинил, пропинил, 1-бутинил, 2-бутинил и 3-бутинил.
Примеры ацильной группы, как использовано в данном описании, включают, но не ограничиваются ими, алкилкарбонильные группы, такие как формил, ацетил, пропионил, бутирил, изобутирил, валерил, изовалерил, пивалоил, капроил и фенилацетил; алкенилкарбонильные группы, такие как акрилоил, пропиолоил, метакрилоил, кротоноил и изокротоноил, и арилкарбонильные гурппы, такие как бензоил.
Примерами прямой или разветвленной C1-C7 галогеналкильной группы в качестве R21, R22, R23 и R24 могут быть те же группы, как перечисленные выше для R2.
Группа А предпочтительно может представлять любую из групп, имеющих формулы (3)-(8) и (10)-(23), особенно группы, имеющие формулы (3)-(6), (17)-(20) и (23), и более конкретные группы имеют формулы (3), (4) и (17)-(20).
m предпочтительно может представлять целое число от 4 до 10.
n предпочтительно может представлять целое число от 2 до 7.
Группа общей формулы (1), которая представляет собой один из компонентов соединения согласно настоящему изобретению, имеет асимметрический центр, в то время как другой компонент может иметь асимметрический центр. Кроме того, соединение общей формулы (2) согласно настоящему изобретению может иметь асимметрический центр в группе А в дополнение к асимметрическому центру в группе общей формулы (1). По этой причине соединения настоящего изобретения имеют энантиомеры. Предполагается, что все индивидуальные энантиомеры и их смеси включены в объем настоящего изобретения. Когда группа А, имеющая асимметрический центр в стероидном каркасе, представлена одной из формул (3), (4) и (17)-(20), группа общей формулы (1) предпочтительно присоединена к стероидному исходному каркасу в 7α- или 11β-положении.
Также в общих формулах (1) и (2) предпочтительными являются оба соединения с R- и S-конфигурацией асимметрического углерода, к которому присоединена карбоновая кислота или ее металлическая соль.
Среди соединений общей формулы (2) предпочтительными являются те соединения, в которых R1 представляет атом водорода, щелочной металл или щелочноземельный металл; R2 представляет перфторэтильную группу, пертфор-н-пропильную группу, перфтор-н-бутильную группу или 1,1,1,3,3,3-гексафторизопропильную группу, m представляет целое число от 4 до 10 и n представляет целое число от 2 до 6.
Соединения настоящего изобретения могут быть получены в виде гидратов.
Фармацевтически приемлемые соли включают, но не ограничены ими, указанные выше соли металлов, например соли натрия, калия и кальция.
Соединения согласно настоящему изобретению можно вводить в виде фармацевтической композиции в любой дозированной форме, подходящей для предназначенного пути введения, в сочетании с одним или несколькими фармацевтически приемлемыми разбавители, смачивающими агентами, эмульгаторами, диспергирующими агентами, вспомогательными агентами, консервантами, буферами, связующими веществами, стабилизаторами и тому подобное. Соединения и композиции можно вводить парентерально или перорально.
Доза соединения может быть определена подходящим образом в соответствии с физическими данными, возрастом и физическим состоянием пациента, серьезностью подвергаемого лечению заболевания, временем, прошедшим после начала заболевания и т.д. Поскольку предполагается, что соединение настоящего изобретения будет обладать существенно более высокой активностью при пероральном пути введения, его обычно используют в количестве от 0,1 до 500 мг/день при пероральном введении и в количестве от 0,1-1000 мг/день до 0,1-1000 мг/месяц при парентеральном введении (внутривенном, внутримышечном или подкожном путях введения) для взрослого пациента.
Лучший способ осуществления изобретения
Соединение общей формулы (1), в частности соединение общей формулы (2), может быть получено в соответствии с любой из следующих реакционных схем А-К и 1-19. В данных схемах реакций А-К и 1-19 (то есть способы А-К и с 1 по 19) R2, R3, R6, R7, Z1, Z2, Z3, Z4, Z5, Z6, Z7, Z8, Z9, Z10, m и n являются такими, как определено выше для общих формул (1) и (2); каждый из R11, R12, R13 и R16 представляет защитную группу; R33 представляет прямую или разветвленную алкильную группу; каждый из Y1, Y2, Y3, Y4, Y5 и Y6 независимо представляет атом водорода, алкильную группу (например, прямую или разветвленную C1-C5 алкильную группу) или ОR11; каждый из L1 и L2 представляет уходящую группу; Х представляет атом галогена; m1 равно m-2; R8 представляет прямую или разветвленную C1-C5 алкильную группу, прямую или разветвленную C2-C5 алкенильную группу или прямую или разветвленную C2-C5 алкинильную группу.
Соединение по настоящему изобретению может включать различные стереоизомеры, поскольку оно содержит один или несколько асимметрических атомов углерода. Для получения отдельного стереоизомера имеется два способа, в одном из которых используется хиральная колонка для разделения смеси стереоизомеров, а другой включает асимметрический синтез. Способ с применением хиральной колонки может быть осуществлен с использованием колонки, коммерчески доступной от DAICEL, например, под торговой маркой CHIRALPAK-OT(+), ОР(+) или AD, или CHIRALCEL-OA, OB, OJ, ОК, ОС, OD, OF или OG. Что касается асимметрического синтеза, способы 14-16 иллюстрируют асимметрический синтез соединения изобретения, касающийся асимметрического атома углерода, к которому присоединена боковая цепь карбоксильной группы.
Реакционная схема А (Способ А)

Примечание. Соединение (I) может быть синтезировано способом, описанным в J. Org. Chem., 60 (1995) 5316-5318.
Реакционная схема В (Способ В)



Примечание. Соединение (XXI) может быть синтезировано способом, описанным в патенте Германии DE4218743A1.
Реакционная схема F (Способ F)













в которой R8 представляет прямую или разветвленную C1-C5 алкильную группу, прямую или разветвленную C2-C5 алкенильную группу или прямую или разветвленную C2-C5 алкинильную группу и
М представляет металл.

в которой R8 представляет прямую или разветвленную C1-C5 алкильную группу, прямую или разветвленную C2-C5 алкенильную группу или прямую или разветвленную C2-C5 алкинильную группу и
М представляет металл.

в которой каждый из Y1, Y2 и Y3 независимо представляет атом водорода, алкильную группу (например, прямую или разветвленную C1-C5 алкильную группу) или ОR11 и каждый из Z1, Z2 и Z3 независимо представляет атом водорода, гидроксильную группу или прямую или разветвленную C1-C5 алкильную группу.
Реакционная схема 13(Способ 13)
в которой каждый из Y1, Y2, Y3, Y4, Y5 и Y6 независимо представляет атом водорода, алкильную группу (например, прямую или разветвленную C1-C5 алкильную группу) или ОR11 и каждый из Z1, Z2, Z3, Z4, Z5 и Z6 независимо представляет атом водорода, гидроксильную группу или прямую или разветвленную C1-C5 алкильную группу.
 Примеры R* включают
Примеры R* включают


Примеры R* включают

В указанных выше реакционных схемах 14 и 15 (способы 14 и 15) R2, R11, R12, X, m, n, X, L1 и L2 являются такими, как определено выше, R* представляет хиральный вспомогательный радикал и m и m3 представляют целые числа, которые удовлетворяют отношению m=m3+3.
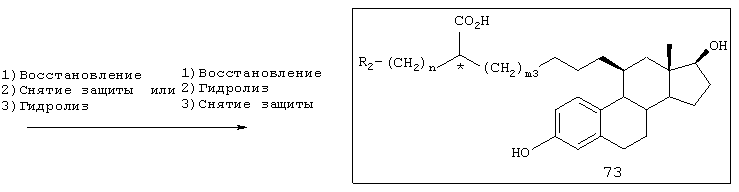 Примеры R* включают
Примеры R* включают

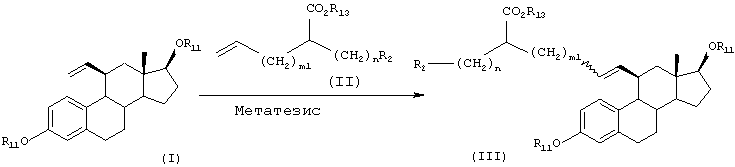
[Способ А]
Способ А иллюстрирует синтез соединения (VI) исходя из соединения (I). Соединение (I) может быть синтезировано способом, описанным в J. ORg. Chem., 60 (1995), 5316-5318.
Стадия 1: Получение соединения (III)
В присутствии катализатора, такого как бензилиденбис(трициклогексилфосфин)дихлоррутений, соединение (I) подвергают взаимодействию с соединением (II) в растворителе (например, хлористом метилене, хлороформе, бензоле, толуоле, ксилоле, диоксане, тетрагидрофуране, диметилсульфоксиде или диметилформамиде) при температуре в диапазоне от -78°С до температуры кипения реакционной смеси, предпочтительно при температуре кипения реакционной смеси, давая соединение (III).
Стадия 2: Получение соединения (IV)
С использованием катализатора (например, палладия на активированном углероде, гидроксида палладия, оксида платины или катализатора Вилкинсона (Wilkinson)) соединение (III) гидрируют в инертном растворителе (например, метаноле, этаноле, этилацетате, тетрагидрофуране, диоксане, дихлорметане, дихлорэтане, хлороформе или бензоле) при температуре в диапазоне от комнатной температуры до температуры кипения реакционной смеси, предпочтительно при температуре кипения реакционной смеси, получая соединение (IV).
Стадия 3: Получение соединения (V)
Когда R11 представляет, например, метильную группу, соединение (IV) обрабатывают кислотой (например, хлористым водородом, серной кислотой, бромистым водородом, гидрохлоридом пиридина или трехбромистым бором) при температуре в диапазоне от -78°С до температуры кипения реакционной смеси, получая соединение (V).
Стадия 4: Получение соединения (VI)
Соединение (V) обрабатывают гидроксидом натрия или гидроксидом калия в растворителе (например, воде, этаноле, метаноле или смеси вода/этанол, или смеси вода/метанол) при температуре в диапазоне от комнатной температуры до температуры кипения реакционной смеси, предпочтительно при температуре кипения реакционной смеси, получая соединение (VI).
[Способ В]
Как показано ниже, соединение (VI), полученное способом А, также может быть получено исходя из соединения (I) следующим образом.
Стадия 1: Получение соединения (VIII)
В присутствии катализатора, такого как бензилиденбис(трициклогексилфосфин)дихлоррутений, соединение (I) подвергают взаимодействию с соединением (VII) в растворителе (например, хлористом метилене, хлороформе, бензоле, толуоле, ксилоле, диоксане, тетрагидрофуране, диметилсульфоксиде или диметилформамиде) при температуре в диапазоне от -78°С до температуры кипения реакционной смеси, предпочтительно при температуре кипения реакционной смеси, получая соединение (VIII).
Стадия 2: Получение соединения (IX)
С использованием катализатора (например, палладия на активированном углероде, гидроксида палладия, оксида платины или катализатора Вилкинсона (Wilkinson)) соединение (VIII) гидрируют в инертном растворителе (например, метаноле, этаноле, этилацетате, тетрагидрофуране, диоксане, дихлорметане, дихлорэтане, хлороформе или бензоле) при температуре в диапазоне от комнатной температуры до температуры кипения реакционной смеси, предпочтительно при температуре кипения реакционной смеси, получая соединение (IX).
Стадия 3: Получение соединения (X)
Соединение (IX) обрабатывают гидроксидом натрия или гидроксидом калия в растворителе (например, воде, этаноле, метаноле или смеси вода/этанол, или смеси вода/метанол) при температуре в диапазоне от комнатной температуры до температуры кипения реакционной смеси, предпочтительно при температуре кипения реакционной смеси, получая соединение (X).
Стадия 4: Получение соединения (XI)
В растворителе (например, диметилсульфоксиде, диметилформамиде, бензоле, толуоле, ксилоле, диоксане или тетрагидрофуране) и, при необходимости, в присутствии кислоты (например, хлористого водорода, серной кислоты или п-толуолсульфоновой кислоты) соединение (X) нагревают до температуры в диапазоне от 50°С до температуры кипения реакционной смеси, получая соединение (XI).
Стадия 5: Получение соединения (VI)
Когда R11 представляет, например, метильную группу, соединение (XI) обрабатывают кислотой (например, хлористым водородом, серной кислотой, бромистым водородом, гидрохлоридом пиридина или трехбромистым бором) при температуре в диапазоне от -78°С до температуры кипения реакционной смеси, получая соединение (VI).
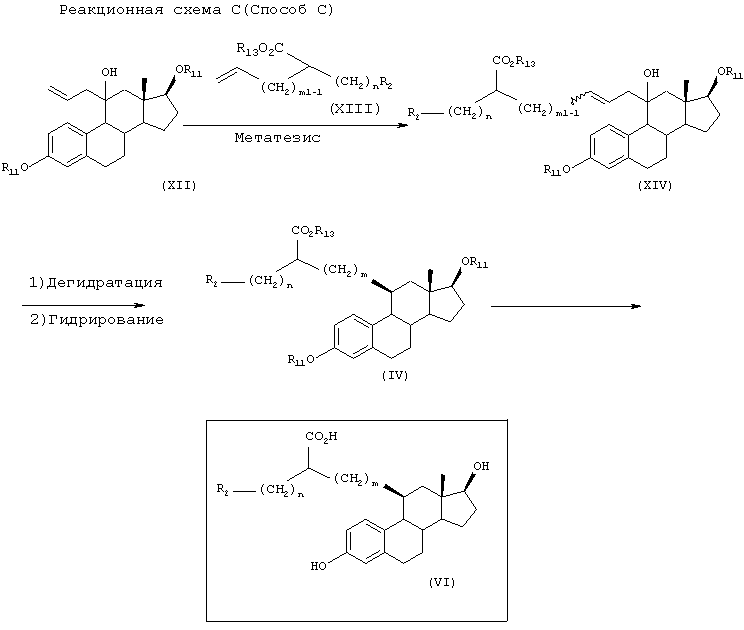
[Способ С]
Как показано ниже, соединение (VI), полученное способами А и В, также может быть получено, исходя из соединения (XII) следующим образом.
Стадия 1: Получение соединения (XIV)
В присутствии катализатора, такого как бензилиденбис(трициклогексилфосфин)дихлоррутений, соединение (XII) подвергают взаимодействию с соединением (XIII) в растворителе (например, хлористом метилене, хлороформе, бензоле, толуоле, ксилоле, диоксане, тетрагидрофуране, диметилсульфоксиде или диметилформамиде) при температуре в диапазоне от -78°С до температуры кипения реакционной смеси, предпочтительно при температуре кипения реакционной смеси, получая соединение (XIV).
Стадия 2: Получение соединения (IV)
Соединение (XIV) дегидратируют с использованием кислоты (например, хлористоводородной кислоты, бромистоводородной кислоты, смеси бромистоводородная кислота/уксусная кислота) в инертном растворителе (например, метаноле, этаноле) при температуре в диапазоне от комнатной температуры до температуры кипения реакционной смеси, предпочтительно при 50°С, и дополнительно подвергают гидрированию аналогично способу А, получая соединение (IV).
Стадия 3: Получение соединения (VI)
Соединение (IV) подвергают гидролизу и снятию защитных групп аналогично способам А или В, получая соединение (VI).
[Способ D]
Как показано ниже, соединение (VI), полученное способами А, В и С, также может быть получено исходя из соединения (XII) следующим образом.
Стадия 1: Получение соединения (XVI)
В присутствии катализатора, такого как бензилиденбис(трициклогексилфосфин)дихлоррутений, соединение (XII) подвергают взаимодействию с соединением (XV) в растворителе (например, хлористом метилене, хлороформе, бензоле, толуоле, ксилоле, диоксане, тетрагидрофуране, диметилсульфоксиде или диметилформамиде) при температуре в диапазоне от -78°С до температуры кипения реакционной смеси, предпочтительно при температуре кипения реакционной смеси, получая соединение (XVI).
Стадия 2: Получение соединения (XVII)
Соединение (XVI) дегидратируют с использованием кислоты (например, хлористоводородной кислоты, бромистоводородной кислоты, смеси бромистоводородная кислота/уксусная кислота) в инертном растворителе (например, метаноле, этаноле) при температуре в диапазоне от комнатной температуры до температуры кипения реакционной смеси, предпочтительно при 50°С, и дополнительно подвергают гидрированию аналогично способу А, получая соединение (XVII).
Стадия 3: Получение соединения (VI)
Соединение (XVII) подвергают гидролизу, декарбоксилированию и снятию защитной группы аналогично способам А или В, получая соединение (VI).
Соединение (XII), используемое в качестве исходного вещества в способах С и D, может быть получено в соответствии со способом, описанным в Tetrahedron., 30 (1977), pp.609-616.
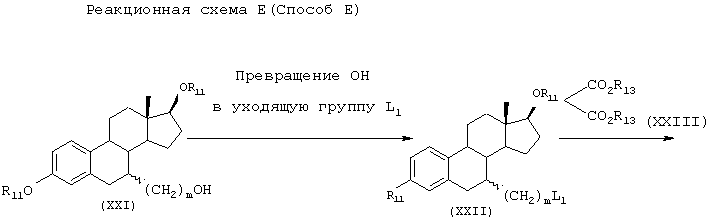
[Способ Е]
Способ Е иллюстрирует синтез соединения (XXIX), исходя из соединения (XXI).
Стадия 1: Получение соединения (XXII)
В присутствии органического основания (например, триэтиламина или пиридина) соединение (XXI) обрабатывают хлорангидридом кислоты (например, метансульфонилхлоридом или п-толуолсульфонилхлоридом) в инертном растворителе (например, тетрагидрофуране, диоксане, дихлорметане, дихлорэтане или хлороформе, предпочтительно в дихлорметане) при температуре в диапазоне от комнатной температуры до температуры кипения реакционной смеси, предпочтительно при комнатной температуре, превращая (CH2)mOH в соединении (XXI), например (CH2)m-L1, где L1 представляет -О-SО2СН3 или -O-SO2-C6H4-п-CH3. Полученное таким образом соединение затем обрабатывают галогенидом металла (например, иодидом натрия или иодидом калия) в инертном растворителе (например, ацетоне, тетрагидрофуране, диоксане, дихлорметане, дихлорэтане или хлороформе, предпочтительно ацетоне) при температуре в диапазоне от комнатной температуры до температуры кипения реакционной смеси, предпочтительно при температуре кипения реакционной смеси, получая соединение (XXII).
Стадия 2: Получение соединения (XXIV)
В присутствии основания (например, гидрида натрия, гидроксида натрия или трет-бутоксида калия) соединение (XXII) подвергают взаимодействию с малоновым эфиром (XXIII) (например, диэтилмалонатом или диметилмалонатом) в инертном растворителе (например, тетрагидрофуране, диоксане, дихлорметане, дихлорэтане или хлороформе, предпочтительно тетрагидрофуране) при температуре в диапазоне от комнатной температуры до температуры кипения реакционной смеси, получая соединение (XXIV).
Стадия 3: Получение соединения (XXVI)
В присутствии основания (например, гидрида натрия, гидроксида натрия или трет-бутоксида калия) соединение (XXIV) подвергают взаимодействию с галоидным алкилом (XXV), в котором L2 представляет атом галогена, в инертном растворителе (например, тетрагидрофуране, диоксане, дихлорметане, дихлорэтане или хлороформе, предпочтительно тетрагидрофуране) при температуре в диапазоне от комнатной температуры до температуры кипения реакционной смеси, получая соединение (XXVI).
Стадия 4: Получение соединения (XXVII)
Соединение (XXVI) обрабатывают гидроксидом натрия или гидроксидом калия в растворителе (например, воде, этаноле, метаноле, смеси вода/этанол или смеси вода/метанол) при температуре в диапазоне от комнатной температуры до температуры кипения реакционной смеси, предпочтительно при температуре кипения реакционной смеси, получая соединение (XXVII).
Стадия 5: Получение соединения (XXVIII)
В растворителе (например, диметилсульфоксиде, диметилформамиде, бензоле, толуоле, ксилоле, диоксане или тетрагидрофуране) и, при необходимости, в присутствии кислоты (например, хлористого водорода, серной кислоты или п-толуолсульфоновой кислоты) соединение (XXVII) нагревают до температуры в диапазоне от 50°С до температуры кипения реакционной смеси, получая соединение (XXVIII).
Стадия 6: Получение соединения (XXIX)
Когда R11 представляет, например, метальную группу, соединение (XXVIII) обрабатывают кислотой (например, хлористым водородом, серной кислотой, бромистым водородом, гидрохлоридом пиридина или трехбромистым бором) при температуре в диапазоне от -78°С до температуры кипения реакционной смеси, получая соединение (XXIX).
Соединение (XXI), использованное в качестве исходного вещества в способе Е, может быть получено в соответствии со способом, описанным в DE421874A1.
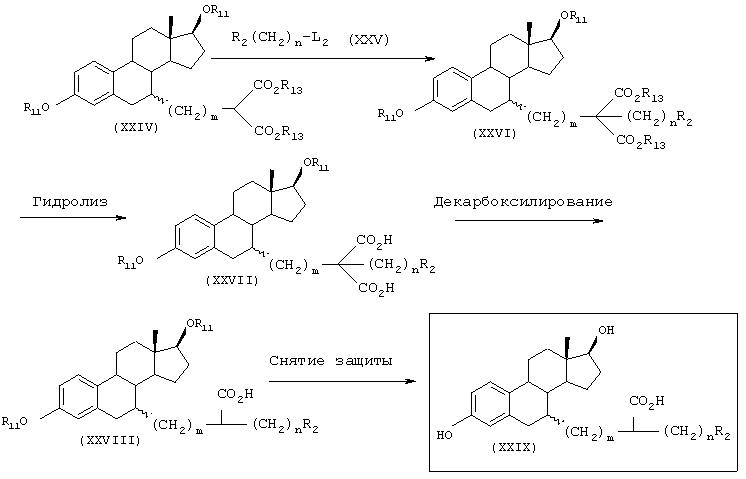
[Способ F]
Соединение (XXIX), полученное способом Е, также может быть получено, исходя из соединения (XXII), в соответствии со следующими стадиями.
Стадия 1: Получение соединения (XXXI)
В присутствии основания (например, гидрида натрия, гидроксида натрия или трет-бутоксида калия) соединение (XXII) подвергают взаимодействию с соединением (XXX) в инертном растворителе (например, тетрагидрофуране, диоксане, дихлорметане, дихлорэтане или хлороформе, предпочтительно, тетрагидрофуране) при температуре в диапазоне от -78°С до температуры кипения реакционной смеси, получая соединение (XXXI).
Стадия 2: Получение соединения (XXIX)
Соединение (XXXI) превращают в соединение (XXIX) способом, аналогичным способу Е.
[Способ G]
Способ G иллюстрирует синтез соединения (ILVIII) исходя из соединения (ILI).
Стадия 1: Получение соединения (ILIII)
В присутствии основания (например, карбоната натрия, карбоната калия, гидроксида натрия, гидроксида калия, гидроксида бария, гидроксида лития, гидрида натрия, предпочтительно карбоната калия) соединение (ILI) подвергают взаимодействию с соединением (ILII) в инертном растворителе (например, ацетоне, метилэтилкетоне, тетрагидрофуране, предпочтительно в ацетоне) при температуре в диапазоне от -78°С до температуры кипения реакционной смеси, предпочтительно при комнатной температуре, получая соединение (ILIII).
Стадия 2: Получение соединения (ILIV) перегруппировкой Кляйзена
Соединение (ILIII) растворяют в инертном растворителе (например, N,N-диметиланилине, N,N-диэтиланилине, нитробензоле, дихлорбензоле, дибромбензоле, предпочтительно N,N-диметиланилине) и затем нагревают до температуры в диапазоне от 180°С до температуры кипения реакционной смеси, предпочтительно от 180 до 200°С, получая соединение (ILIV).
Стадия 3: Получение соединения (ILV)
В присутствии основания (например, триэтиламина, диэтилизопропиламина, пиридина, карбоната натрия, карбоната калия, гидроксида натрия, гидроксида калия, гидрида натрия, предпочтительно пиридина) соединение (ILIV) подвергают взаимодействию с Tf2O (ангидридом трифторметансульфоновой кислоты) в инертном растворителе (например, дихлорметане, хлороформе, бензоле, толуоле, предпочтительно дихлорметане) при температуре в диапазоне от 0°С до комнатной температуры, получая соединение (ILV).
Стадия 4: Получение соединения (ILVII)
В присутствии палладиевого или никелевого катализатора соединение (ILV) подвергают взаимодействию с соединением (ILVI) в инертном растворителе (например, простом эфире, тетрагидрофуране, диоксане, диметилформамиде, воде, предпочтительно диоксане) при температуре в диапазоне от комнатной температуры до температуры кипения реакционной смеси, предпочтительно при температуре кипения реакционной смеси, получая соединение (ILVII).
Стадия 5: Получение соединения (ILVIII)
Соединение (ILVII) превращают в соединение (ILVIII) аналогично способам А, В, С или D.
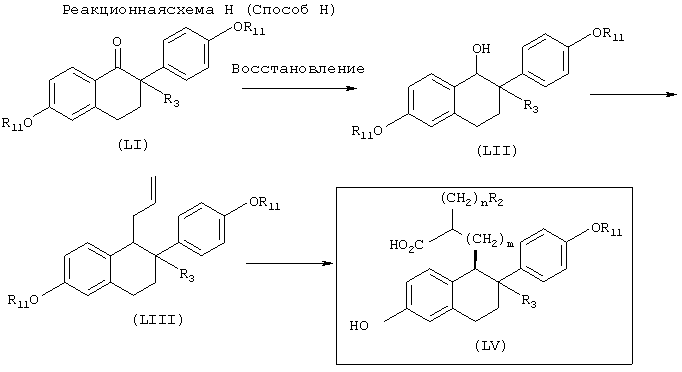
[Способ Н]
Способ Н иллюстрирует синтез соединения (LIV), исходя из соединения (LI), синтезированного способом, описанным в патенте США 4904661.
Стадия 1: Получение соединения (LII)
Соединение (LI) подвергают взаимодействию с восстанавливающим агентом (например, литийалюминийгидридом, диизобутилалюминийгидридом, боргидридом натрия) в инертном растворителе (например, тетрагидрофуране, диоксане, диэтиловом эфире) при температуре в диапазоне от 0°С до температуры кипения реакционной смеси, предпочтительно от 0 до 50°С, получая соединение (LII).
Стадия 2: Получение соединения (LIII)
В присутствии подходящей кислоты (например, иодистого цинка, трехфтористого бора) соединение (LII) подвергают взаимодействию с аллилтриметилсиланом в инертном растворителе (например, дихлорэтане, дихлорметане, хлороформе) при температуре в диапазоне от 0°С до температуры кипения реакционной смеси, предпочтительно от 0 до 50°С, получая соединение (LIII).
Стадия 3: Получение соединения (LIV)
Соединение (LIII) подвергают аналогичным процедурам, способов А, В, С или D, то есть метатезису, восстановлению, гидролизу, декарбоксилированию, снятию защитных групп и т.д., получая соединение (LIV).
[Способ I]
Соединение (LIV) также может быть синтезировано следующим образом, исходя из соединения (LI).
Стадия 1: Получение соединения (LVI)
В присутствии основания (например, гидрида натрия, н-бутиллития, трет-бутиллития, диизопропиламида лития,трет-бутоксида калия) соединение (LI) подвергают взаимодействию с соединением (LV) в инертном растворителе (например, тетрагидрофуране, диоксане, диэтиловом эфире) при температуре в диапазоне от -78°С до температуры кипения реакционной смеси, предпочтительно от -78 до 0°С, получая соединение (LVI).
Стадия 2: Получение соединения (LVII)
В присутствии подходящей кислоты (например, иодистого цинка, трехфтористого бора) соединение (LVI) подвергают взаимодействию с цианоборгидридом натрия в инертном растворителе (например, дихлорэтане, дихлорметане, хлороформе) при температуре в диапазоне от 0°С до температуры кипения реакционной смеси, предпочтительно от 0 до 50°С, получая соединение (LVII).
Стадия 3: Получение соединения (LVIII)
В присутствии катализатора (например, палладия на активированном углероде, гидроксида палладия, оксида платины) соединение (LVII) гидрируют в инертном растворителе (например, метаноле, этаноле, этилацетате, тетрагидрофуране, диоксане, предпочтительно тетрагидрофуране, этилацетате) при температуре в диапазоне от комнатной температуры до температуры кипения реакционной смеси, предпочтительно при комнатной температуре, получая соединение (LVIII). Соединение (LVIII) может быть непосредственно получено из соединения (LVI) гидрированием с использованием катализатора (например, палладия на активированном углероде, гидроксида палладия, оксида платины) в инертном растворителе (например, метаноле, этаноле, этилацетате, тетрагидрофуране, диоксане, предпочтительно тетрагидрофуране, этилацетате) при температуре в диапазоне от комнатной температуры до температуры кипения реакционной смеси, предпочтительно при комнатной температуре.
Стадия 4: получение соединения (LIV)
Соединение (LVIII) подвергают взаимодействию в соответствии с методикой способов Е или F, получая соединение (LIV).
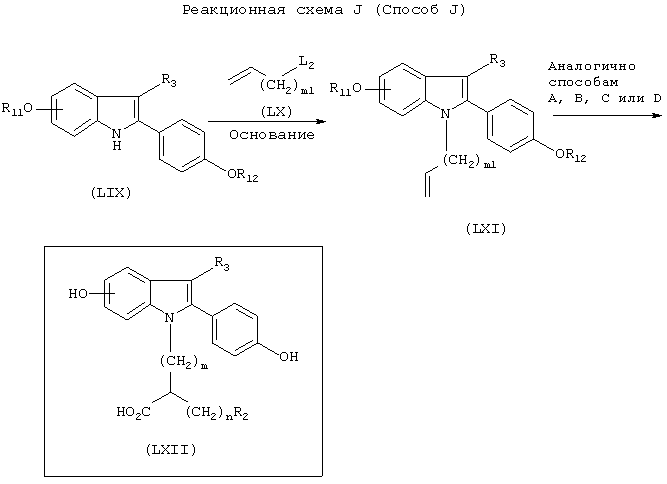
[Способ J]
Способ J иллюстрирует синтез соединения (LXII), исходя из соединения (LIX).
Стадия 1: Получение соединения (LXI)
В присутствии основания (например, гидрида натрия, н-бутиллития, трет-бутоксида калия) соединение (LIX) подвергают взаимодействию с соединением (LX) в инертном растворителе (например, диметилформамиде, тетрагидрофуране, диоксане, диэтиловом эфире, диметилсульфоксиде) при температуре в диапазоне от 0°С до температуры кипения реакционной смеси, предпочтительно от 0 до 50°С, получая соединение (LXI).
Стадия 2: Получение соединения (LXII)
Соединение (LXIV) подвергают метатезису, восстановлению, гидролизу и снятию защитных групп аналогично методике способов А, В, С или D, получая соединение (LXII).
[Способ К]
Соединение (LXII) также может быть получено, исходя из соединения (LIX), следующим образом.
В присутствии основания (например, гидрида натрия, н-бутиллития, трет-бутоксида калия) соединение (LIX) подвергают взаимодействию с соединением (LXIII) в инертном растворителе (например, диметилформамиде, тетрагидрофуране, диоксане, диэтиловом эфире, диметилсульфоксиде) при температуре в диапазоне от 0°С до температуры кипения реакционной смеси, предпочтительно от 0 до 50°С, получая соединение (LXIV).
Стадия 2: Получение соединения (LXII)
Соединение (LXIV) подвергают взаимодействию аналогично методике способов Е или F, получая соединение (LXII).
Соединение общей формулы (2), в котором группа А представлена формулой (8), может быть получено, например, как показано в примерах 6-10, таким же или эквивалентным способом.
[Способ 1]
Соединение 2 получают, исходя из соединения 1, аналогично методике способа Е. Соединение 1, используемое в качестве исходного вещества, может быть получено в соответствии со способом, описанным в WO 99/64393.
[Способ 2]
Соединение 4 получают, исходя из соединения 3, аналогично методике способа Е. Соединение 5 может быть получено окислением соединения 4 в соответствии со способом, описанным в WO 99/64393. Соединение 3, используемое в качестве исходного вещества, может быть получено в соответствии со способом, описанным в WO 99/64393.
[Способ 3]
Способ 3 иллюстрирует синтез соединения 9, исходя из соединения 6. Соединение 6, используемое в качестве исходного вещества, может быть синтезировано, например, способами, описанными в J. Org. Chem., 50(1985), 2121-2123, и J. Org. Chem., 61 (1996), 3890-3893.
Стадия 1: Получение соединения 8
В присутствии основания (например, гидрида натрия, н-бутиллития, трет-бутоксида калия) соединение 6 подвергают взаимодействию с соединением 7 в инертном растворителе (например, диметилформамиде, тетрагидрофуране, диоксане, диэтиловом эфире, диметилсульфоксиде) при температуре в диапазоне от 0°С до температуры кипения реакционной смеси, предпочтительно от 0 до 50°С, получая соединение 8.
Стадия 2: Получение соединения 9
Соединение 8 подвергают взаимодействию аналогично методике способа Е, получая соединение 9.
[Способ 4]
Способ 4 иллюстрирует синтез соединения 20, исходя из соединения 10.
Стадия 1: Получение соединения 11
В присутствии кислотного катализатора, такого как серная кислота, соединение 10 нагревают в спирте (например, метаноле, этаноле) при температуре в диапазоне от -78°С до температуры кипения реакционной смеси, предпочтительно при температуре кипения реакционной смеси, получая соединение 11.
Стадия 2: Получение соединения 12
Амино и гидроксильную группы соединения 11, полученного на стадии 1, защищают, получая соединение 12.
Стадия 3: Получение соединения 13
Соединение 12 обрабатывают восстанавливающим агентом (например, боргидридом лития и т.д.) в растворителе (например, метаноле, этаноле или смеси этанол/ тетрагидрофуран) при температуре в диапазоне от -78°С до температуры кипения реакционной смеси, предпочтительно при комнатной температуре, получая соединение 13.
Стадия 4: Получение соединения 15
Соединение 13 подвергают реакции Мицунобу с соединением 14, получая соединение 15.
Стадия 5: Получение соединения 16
Соединение 15 подвергают снятию защиты аминогруппы, получая соединение 16.
Стадия 6: Получение соединения 17
В присутствии основания (например, карбоната калия, трет-бутоксида калия, трет-бутоксида натрия) соединение 16 подвергают взаимодействию путем добавления металлического катализатора, такого как палладий, вместе с лигандом, таким как дифенилфосфиноферроцен или 2,2-бис(дифенилфосфино)-1,1'-бинафтил, предпочтительно, добавляя катализатор трис(дибензилиденацетон)дипалладий вместе с 2,2-бис(дифенилфосфино)-1,1'-бинафтилом, в инертном растворителе (например, бензоле, толуоле, ксилоле, диоксане или тетрагидрофуране) при температуре в диапазоне от -78°С до температуры кипения реакционной смеси, предпочтительно при 100°С, получая соединение 17.
Стадия 7: Получение соединения 19
В присутствии основания (например, гидрида натрия, н-бутиллития, трет-бутоксида калия, карбоната калия) и, при необходимости, с добавлением реагента, такого как иодид натрия, соединение 17 подвергают взаимодействию с соединением 18 в инертном растворителе (например, диметилформамиде, тетрагидрофуране, диоксане, диэтиловом эфире, диметилсульфоксиде, ацетоне) при температуре в диапазоне от 0°С до температуры кипения реакционной смеси, предпочтительно при температуре кипения реакционной смеси, получая соединение 19.
Стадия 8: Получение соединения 20
Соединение 19 подвергают взаимодействию аналогично способам А или В, получая соединение 20.
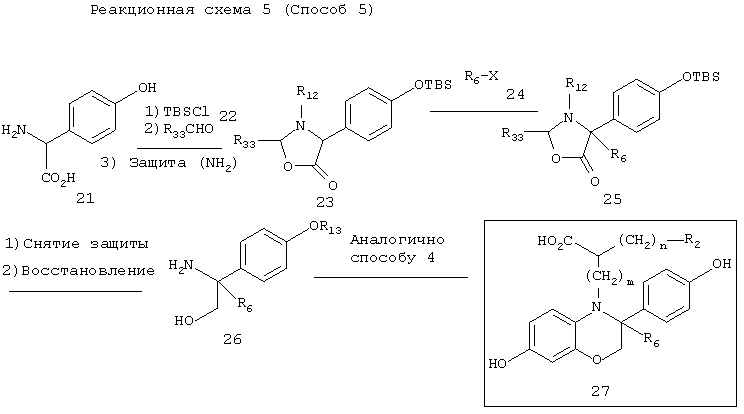
[Способ 5]
Способ 5 иллюстрирует синтез соединения 27, исходя из соединения 21.
Стадия 1: Получение соединения 23
Соединение 21 подвергают защите с использованием TBS,затем подвергают взаимодействию с альдегидом 22 и затем защищают его аминогруппу, получая соединение 23.
Стадия 2: Получение соединения 25
Соединение 23, полученное на стадии 1, алкилируют соединением 24, получая соединение 25.
Стадия 3: Получение соединения 26
Соединение 25 подвергают снятию защиты аминогруппы и затем обрабатывают восстанавливающим агентом (например, литийалюминийгидридом и т.д.) в растворителе (например, тетрагидрофуране, простом эфире) при температуре в диапазоне от -78°С до температуры кипения реакционной смеси, предпочтительно при комнатной температуре, получая соединение 26.
Стадия 4: Получение соединения 27
Соединение 26 подвергают взаимодействию аналогично способу 4, получая соединение 27.
[Способ 6]
Способ 6 иллюстрирует синтез соединения 35, исходя из соединения 28.
Соединение 35 можно синтезировать, исходя из соединения 28, следующим образом.
Стадия 1: Получение соединения 29
Соединение 29 получают из соединения 28 способом, описанным в Synthesis, 12 (1995), 1493-1495, или эквивалентным способом.
Стадия 2: Получение соединения 32
В присутствии основания (например, гексаметилдисилазида лития, гексаметилдисилазида натрия, гексаметилдисилазида калия, гидрида натрия, н-бутиллития, трет-бутиллития, диизопропиламида лития, трет-бутоксида калия, водного гидроксида калия, водного гидроксида натрия) соединение 29 подвергают взаимодействию с соединением 30 или 31 в инертном растворителе (например, 1,2-диметоксиэтане, тетрагидрофуране, диоксане, трет-бутилметиловом эфире, диэтиловом эфире, диметилсульфоксиде, N,N-диметилформамиде, N,N-диметилацетамиде, толуоле) при температуре в диапазоне от -78°С до температуры кипения реакционной смеси, предпочтительно от -78 до 0°С, получая соединение 32.
Стадия 3: Получение соединения 33
Соединение 32 изомеризуют в основных условиях (например, в смеси фторид тетрабутиламмония/тетрагидрофуран, метоксид натрия/метанол, этоксид натрия/этанол, метоксид калия/метанол, метоксид натрия/пропанол, водный гидроксид калия, водный гидроксид натрия) с последующим снятием защиты группы R12 и очисткой перекристаллизацией, получая отдельный изомер формулы 33. В случае, когда R12 представляет трет-бутилдиметилсилильную группу, соединение 32 изомеризуют одновременно с удалением TBS при обработке фторидом тетрабутиламмония и далее очищают перекристаллизацией, получая отдельный изомер формулы 33.
Стадия 4: Получение соединения 34
В присутствии подходящей кислоты (например, трифторуксусной кислоты, эфирата трехфтористого бора, тетрахлорида титана, хлорида алюминия, трифторметансульфоновой кислоты, хлористоводородной кислоты, серной кислоты), соединение 33 подвергают взаимодействию с триэтилсиланом в инертном растворителе (например, дихлорэтане, дихлорметане, хлороформе, трет-бутилметиловом эфире, толуоле) при температуре в диапазоне от 0°С до температуры кипения реакционной смеси, предпочтительно от 0°С до комнатной температуры, получая соединение 34.
Стадия 5: Получение соединения 35
Соединение 34 подвергают взаимодействию аналогично способам А или В, получая соединение 35.
[Способ 7]
Способ 7 иллюстрирует синтез соединения 38, исходя из соединения 36.
Соединение 38 может быть синтезировано, исходя из соединения 37, аналогично методике способа 3. Соединения 36 и 37, используемые в качестве исходных веществ, могут быть синтезированы способами, описанными в J. Med. Chem., 40 (1997), 2117-2122, и J. Med. Chem., 33 (1990), 3222-3229, или эквивалентными способами.
[Способ 8]
Способ 8 иллюстрирует синтез соединения 40, исходя из соединения 39.
Соединение 40 может быть синтезировано, исходя из соединения 39, аналогично методике способа 3. Соединение 39, используемое в качестве исходного вещества, может быть синтезировано, например, способами, описанными в ЕР0826670А1 и J. Org. Chem., 60 (1995), 739-741.
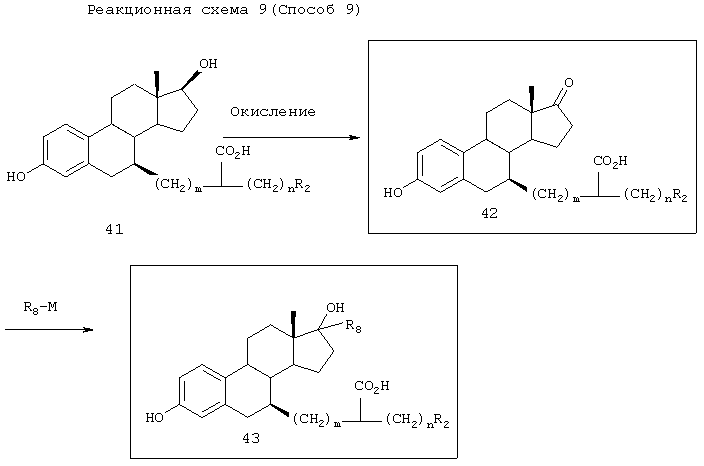
[Способ 9]
Соединения 42 или 43 могут быть синтезированы следующим образом.
Соединение 42 может быть синтезировано из соединения 41 окислением по Джонсу (Jones), PCC окислением, окислением по Сверну (Swern) или рутениевым окислением (например, ТРАР) 17-гидроксильной группы. Соединение 42 далее подвергают взаимодействию с R8-M, где R8 представляет низшую алкильную группу или низшую алкенильную группу, или низшую алкинильную группу и М представляет металл, такой как литий, натрий, калий, магний, кальций или алюминий, в инертном растворителе (например, диметилсульфоксиде, тетрагидрофуране, простом эфире, диметилформамиде) при температуре в диапазоне от -78°С до температуры кипения реакционной смеси, предпочтительно от 0°С до комнатной температуры, получая соединение 43.
Соединение 41, используемое в качестве исходного вещества, может быть синтезировано способами Е, F или 6.
[Способ 10]
Соединения 45 или 46 могут быть синтезированы следующим образом.
Соединение 45 может быть синтезировано из соединения 44 окислением по Джонсу (Jones), PCC окислением, окислением по Сверну (Swern) или рутениевым окислением (например, ТРАР) 17-гидроксильной группы. Соединение 45 далее подвергают взаимодействию с R8-M, где R8 представляет низшую алкильную группу или низшую алкенильную группу, или низшую алкинильную группу и М представляет металл, такой как литий, натрий, калий, магний, кальций или алюминий, в инертном растворителе (например, диметилсульфоксиде, тетрагидрофуране, простом эфире, диметилформамиде) при температуре в диапазоне от -78°С до температуры кипения реакционной смеси, предпочтительно от 0°С до комнатной температуры, получая соединение 46.
Соединение 44, используемое в качестве исходного вещества, может быть синтезировано способами А, В, С или D.
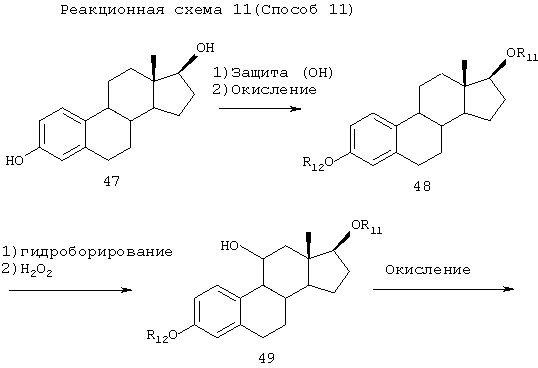
[Способ 11]
Соединения 53 может быть синтезировано следующим образом.
Соединение 47 подвергают защите его гидроксильных групп, а затем окисляют между 9 и 11-положениями с использованием DDQ (2,3-дихлор-5,6-дицианобензохинона) и тому подобное, получая соединение 48.
Соединение 48 преобразовывают в соединение 49 способом, описанным в J. Org. Chem., 1995, 60, 5316-5318.
Соединение 49 подвергают окислению по Сверну, окислению по Джонсу, РСС окислению или рутениевому окислению (например, ТРАР), получая соединение 50.
Соединение 50 подвергают взаимодействию с металлоорганическим реагентом (например, аллилмагнийгалогенидом) в инертном растворителе (например, тетрагидрофуране, простом эфире) при температуре в диапазоне от -78°С до температуры кипения реакционной смеси, предпочтительно от -48°С до комнатной температуры, получая соединение 51.
Соединение 51 дегидратируют для удаления его гидроксильной группы с использованием смеси хлористый тионил/пиридин и тому подобное, получая соединение 52.
Соединение 52 может быть преобразовано в соединение 53 способами А, В, С или D.
[Способ 12]
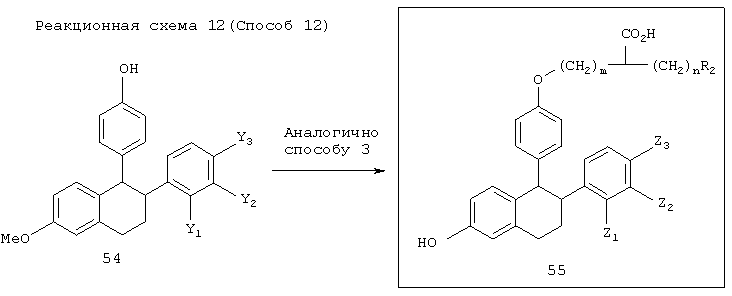
Соединение 55 может быть синтезировано, подвергая соединения 54 реакциям, аналогично методике способа 3. Соединение 54 может быть синтезировано в соответствии с описанными способами (Drugs Future, 1978, 3, 211-215; J. Med. Chem., 1967, 10,78-84; J. Med. Chem.,1998, 41, 2928-2931).
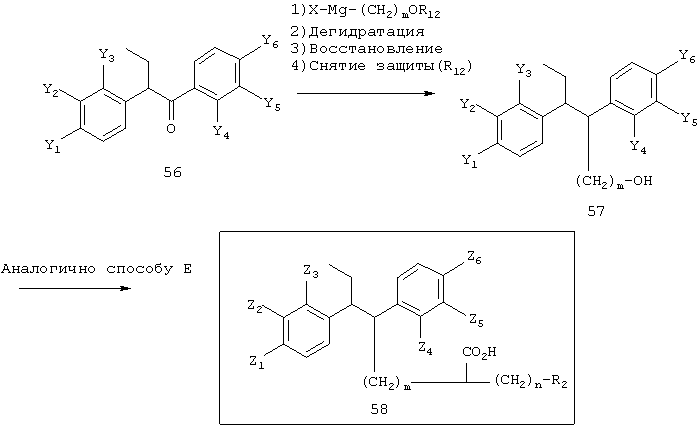
[Способ 13]
Соединение 56 может быть преобразовано в соединение 57 с использованием следующих стадий: 1) 1,2-присоединения X-Mg-(CH2)mOR12, 2) дегидратации, 3) восстановления и 4) снятия защиты (R12), и затем введения в реакцию, аналогично схеме Е, давая соединение 58.
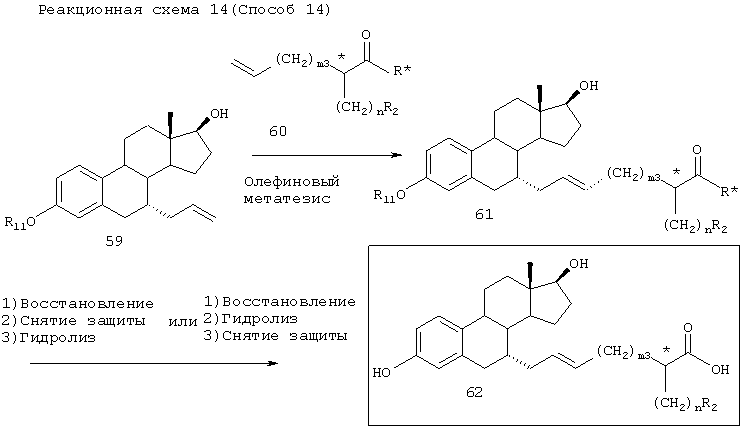
[Способ 14]
В присутствии катализатора, такого как бензилиденбис(трициклогексилфосфин)дихлоррутений, соединение 59 подвергают взаимодействию с хиральным олефином 60 в растворителе (например, хлористом метилене, хлороформе, бензоле, толуоле, ксилоле, диоксане, тетрагидрофуране, диметилсульфоксиде или диметилформамиде) при температуре в диапазоне от -78°С до температуры кипения реакционной смеси, предпочтительно при температуре кипения реакционной смеси, давая соединение 61. Соединение 61 затем подвергают следующим реакциям в указанном порядке: (а) восстановления, снятия защиты и гидролиза или (b) восстановления, гидролиза и снятия защиты, получая соединение 62.
(а) Восстановление, снятие защиты и гидролиз
1) Восстановление
В присутствии катализатора (например, палладия на активированном углероде, гидроксида палладия, оксида платины или катализатора Вилкинсона) соединение 61 гидрируют в инертном растворителе (например, метаноле, этаноле, этилацетате, тетрагидрофуране, диоксане или бензоле) при температуре в диапазоне от 0°С до температуры кипения реакционной смеси, предпочтительно при комнатной температуре, с получением продукта восстановления
2) Снятие защиты
Затем осуществляют снятие защиты фенольной гидроксильной группы с получением незащищенного продукта.
3) Гидролиз
В качестве примера, если R* представляет группу формулы 63, продукт после снятия защиты дополнительно обрабатывают гидроксидом лития, гидроксидом натрия, гидроксидом лития плюс пероксид водорода, гидроксидом натрия плюс пероксид водорода или гидроксидом тетрабутиламмония плюс пероксид водорода в растворителе (например, смеси тетрагидрофуран/вода, смеси диэтиловый эфир/вода, смеси диоксан/вода, смеси диметоксиэтан/вода, смеси метанол/вода, смеси этанол/вода) при температуре в диапазоне от комнатной температуры до температуры кипения реакционной смеси, предпочтительно при комнатной температуре, получая соединение 62.
(b) Восстановление, гидролиз и снятие защиты
1) Восстановление
В присутствии катализатора (например, палладия на активированном углероде, гидроксида палладия, оксида платины или катализатора Вилкинсона) соединение 61 гидрируют в инертном растворителе (например, метаноле, этаноле, этилацетате, тетрагидрофуране, диоксане или бензоле) при температуре в диапазоне от 0°С до температуры кипения реакционной смеси, предпочтительно при комнатной температуре, с получением продукта восстановления.
2) Гидролиз
В качестве примера, если R* представляет группу формулы 63, продукт дополнительно обрабатывают гидроксидом лития, гидроксидом натрия, гидроксидом лития плюс пероксид водорода, гидроксидом натрия плюс пероксид водорода или гидроксидом тетрабутиламмония плюс пероксид водорода в растворителе (например, смеси тетрагидрофуран/вода, смеси диэтиловый эфир/вода, смеси диоксан/вода, смеси диметоксиэтан/вода, смеси метанол/вода, смеси этанол/вода) при температуре в диапазоне от комнатной температуры до температуры кипения реакционной смеси, предпочтительно при комнатной температуре, получая карбоновую кислоту.
3) Снятие защиты
Затем осуществляют снятие защиты фенольной гидроксильной группы, получая соединение 62.
Хиральный олефин формулы 60, используемый в способе выше, может быть получен, как показано на реакционной схеме 15.
[Способ 15]
[Синтез хирального олефина]
В присутствии основания (например, диизопропиламида лития, гексаметилдисилазида лития, гексаметилдисилазида натрия, бутиллития) и НМРА соединение 67 подвергают взаимодействию с R2(CH2)n-L1 в инертном растворителе (например, тетрагидрофуране, толуоле, диэтиловом эфире, гексане, предпочтительно в тетрагидрофуране) при температуре в диапазоне от -78°С до температуры кипения реакционной смеси, предпочтительно от -30°С до комнатной температуры, получая соединение 60.
Хиральный олефин 60 также можно синтезировать следующим образом.
В присутствии основания (например, диизопропиламида лития, гексаметилдисилазида лития, гексаметилдисилазида натрия, бутиллития) и НМРА соединение 67 подвергают взаимодействию с соединением 69 в инертном растворителе (например, тетрагидрофуране, толуоле, диэтиловом эфире, гексане, предпочтительно в тетрагидрофуране) при температуре в диапазоне от -78°С до температуры кипения реакционной смеси, предпочтительно от -30°С до комнатной температуры, получая соединение 60.
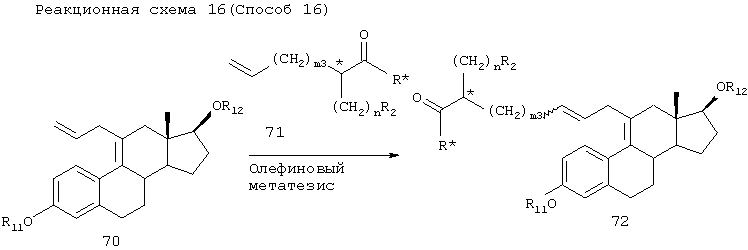
[Способ 16]
Соединение 70 может быть преобразовано в соединение 73 аналогично методике способа 14.
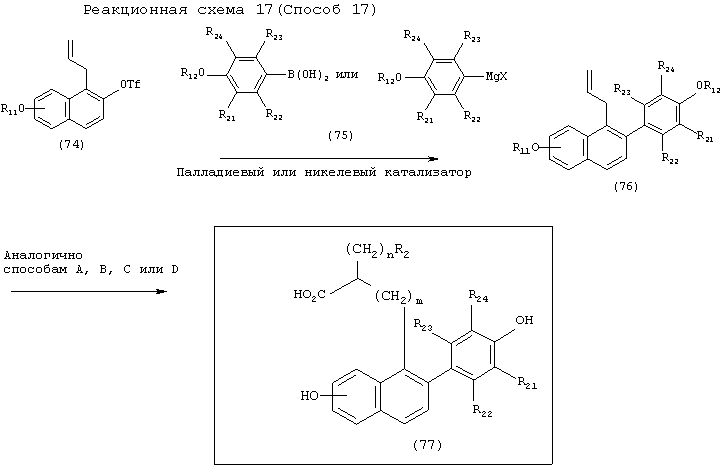
[Способ 17]
Соединение 75, имеющее заместители R21, R22, R23 и R24 в своем бензольном кольце, может быть преобразовано в соединение 77 с использованием методики, аналогичной способу G. Каждый из R21, R22, R23 и R14 независимо представляет атом водорода, прямую или разветвленную C1-C5 алкильную группу, прямую или разветвленную C1-C7 галогеналкильную группу, атом галогена или ацильную группу.
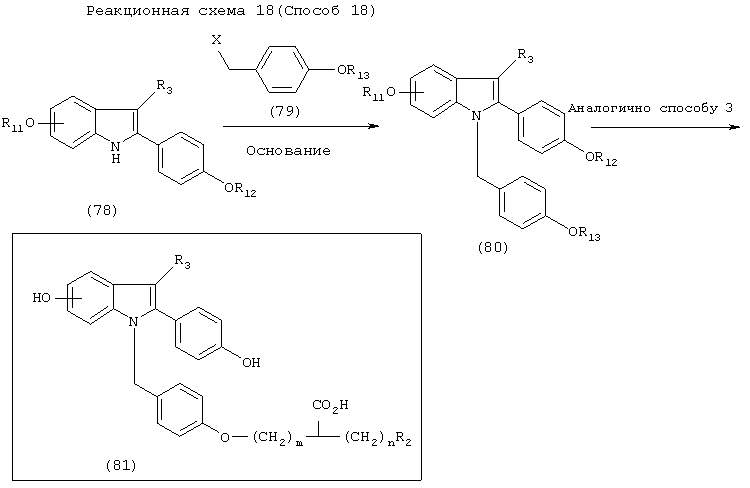
[Способ 18]
Соединение 78 подвергают взаимодействию с соединением 79 в присутствии основания, получая соединение 80. Соединение 81 может быть синтезировано из соединения 80 в соответствии со способами 3 и К.
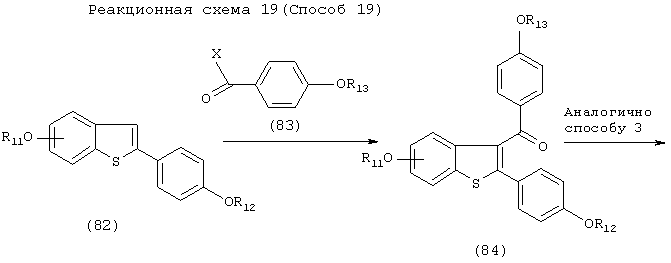
[Способ 19]
Соединение 82, синтезированное согласно способу, описанному в J. Med. Chem., 1057 (1984), подвергают реакции Фриделя-Крафтса с соединением 83, а затем обрабатывают в соответствии со способом 3.
Примеры
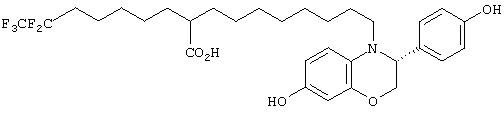
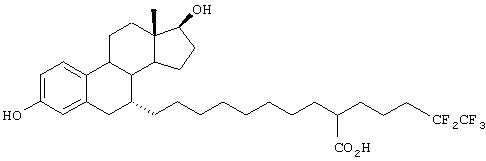
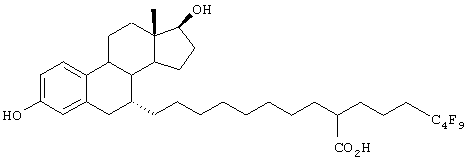
Настоящее изобретение более конкретно объяснено с помощью следующих примеров. Однако следует понимать, что настоящее изобретение не ограничено каким-либо образом данными примерами. Для того чтобы объяснить эффективность соединений согласно настоящему изобретению, иллюстративные соединения были испытаны на их антиэстрогенную активность в приведенном ниже примере испытаний. В таблице 1 показаны химические структуры полученных в примерах соединений.






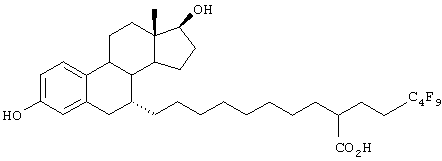
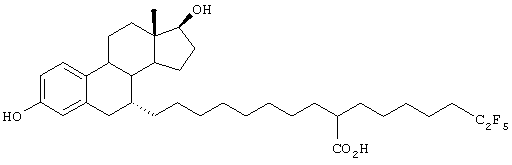






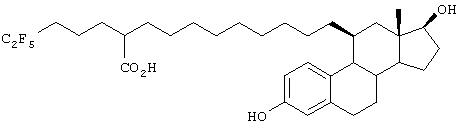

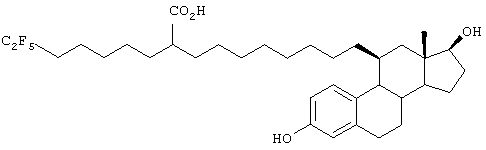







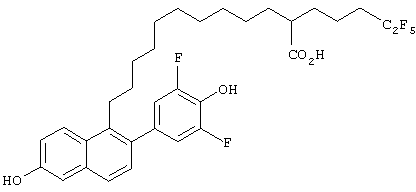
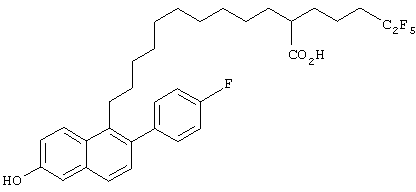
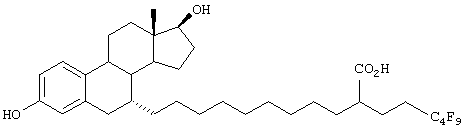


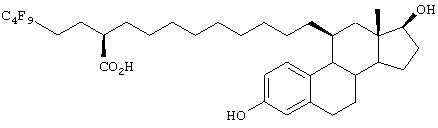
Пример 1
Синтез 6-метокси-2-(4-метоксифенил)-1-(2-пропенил)нафталина
(Стадия 1)
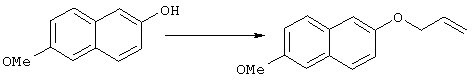
6-Метокси-2-нафтол (22,1 г, 127,0 ммоль) растворяли в ацетоне (200 мл). К полученному раствору добавляли карбонат калия (70,2 г, 508,0 ммоль) и бромистый аллил (16,5 мл, 191,0 ммоль) с последующим перемешиванием в течение 2 дней при комнатной температуре. После фильтрования реакционной смеси органический растворитель отгоняли при пониженном давлении. К остатку добавляли воду, затем экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом натрия. Органический растворитель опять отгоняли, получая 6-метокси-2-(2-пропенилокси)нафталин (24,9 г, выход 91%) в виде сырого продукта.
1Н-ЯМР (270 МГц, СDСl3): δ 7,66-7,60 (м, 2Н, Аr-Н), 7,18-7,10 (м, 4Н, Аr-Н), 6,20-6,05 (м, 1Н, CH2=CHCH2-), 5,45 (дд, J=18,8, 1,3 Гц, 1Н, СН2=СНСН2-), 5,31 (дд, J=10,5, 1,3 Гц, 1Н, СН2=СНСН2-), 4,62 (д, J=5,3 Гц, 2H, СН2=СНСН2-), 3,90 (с, 3Н, -ОСН3).
(Стадия 2)

6-Метокси-2-(2-пропенилокси)нафталин (24,9 г, 116,2 ммоль) растворяли в N,N-диметиланилине (100 мл) с последующим нагреванием при кипении с обратным холодильником в течение 15 часов. После отгонки органического растворителя при пониженном давлении к остатку добавляли 2 н. водную хлористоводородную кислоту, затем экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом натрия. После отгонки растворителя остаток очищали колоночной хроматографией на силикагеле (элюент: этилацетат/гексан=1/4) с последующей перекристаллизацией из смеси этилацетат/ гексан, получая 6-метокси-1-(2-пропенил)-2-нафтол (20,3 г, выход 82%).
1Н-ЯМР (270 МГц, СDСl3): δ 7,80 (д, J=9,3 Гц, 1H, Ar-H), 7,56 (д, J=8,9 Гц, 1H, Ar-H), 7,16 (дд, J=9,3, 2,7 Гц, 1Н, Ar-H), 7,11 (д, J=2,7 Гц, 1H, Ar-H), 7,07 (д, J=8,9 Гц, 1H, Ar-H), 6,13-5,98 (м, 1H, CH2=CHCH2-), 5,10 (дд, J=10,0, 1,3 Гц, 1H, CH2=CHCH2-), 5,04 (дд, J=17,5, 1,3 Гц, 1H, CH2=CHCH2-), 4,93 (с, 1H, -ОН), 3,90 (с, 3Н, -ОСН3). 3,79 (д, J=5,9 Гц, 2H, CH2=CHCH2-).
(Стадия 3)

6-Метокси-1-(2-пропенил)-2-нафтол (18,77 г, 87,6 ммоль) растворяли в дихлорметане (300 мл). К данному раствору добавляли по каплям при 0°С пиридин (21,3 мл, 262,8 ммоль) и ангидрид трифторметансульфоновой кислоты (22,1 мл, 131,4 ммоль) и полученную смесь перемешивали в течение 30 минут. После завершения реакции к реакционной смеси добавляли воду при 0°С, затем экстрагировали этилацетатом. Органический слой промывали разбавленной соляной кислотой и насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия. После отгонки растворителя,остаток очищали колоночной хроматографией на силикагеле (элюент: этилацетат: гексан=1/9), получая 6-метокси-1-(2-пропенил)-2-нафтилтрифторметансульфонат (30,8 г, выход 100%).
1Н-ЯМР (270 МГц, СDСl3): δ 7,95 (д, J=9,3 Гц, 1H, Ar-H), 7,69 (д, J=8,9 Гц, 1H, Ar-H), 7,35 (д, J=8,9 Гц, 1H, Ar-H), 7,25 (дд, J=9,3, 2,7 Гц, 1H, Ar-H), 7,17 (д, J=2,7 Гц, 1H, Ar-H), 6,07-5,04 (м, 1H, СН2=СНСН2-), 5,10 (дд, J=10,0, 1,3 Гц, 1H, СН2=СНСН2-), 5,02 (дд, J=17,2, 1,3 Гц, 1H, CH2=CHCH2-), 3,93 (с, 3Н, -ОСН3), 3,89 (д, J=5,6 Гц, 2H, CH2=CHCH2-).
(Стадия 4)
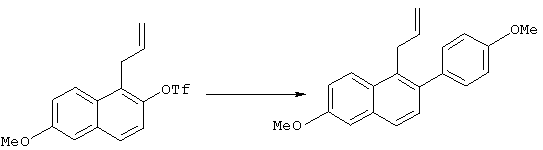
4-Метоксифенилбороновую кислоту (10,15 г, 66,8 ммоль), гидрат трикалийфосфата (77,6 г, 278,3 ммоль) и тетракис(трифенилфосфин)палладий(0) (1,93 г, 1,67 ммоль, 3 мол.%) добавляли к раствору 6-метокси-1-(2-пропенил)-2-нафтилтрифторметансульфоната (19,3 г, 55,66 ммоль) в диоксане (300 мл) с последующим нагреванием при кипении с обратным холодильником в течение 8 часов в атмосфере аргона. К реакционной смеси добавляли воду, затем экстрагировали этилацетатом, промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом натрия. После отгонки растворителя остаток очищали колоночной хроматографией на силикагеле (элюент: этилацетат/гексан=1/9) с последующей перекристаллизацией из гексана, получая 6-метокси-2-(4-метоксифенил)-1-(2-пропенил)нафталин (12,65 г, выход 75%).
1Н-ЯМР (270 МГц, СDСl3): δ 7,95 (д, J=9,8 Гц, 1H, Ar-H), 7,65 (д, J=8,5 Гц, 1H, Ar-H), 7,35 (д, J=8,9 Гц, 1H, Ar-H), 7,34-7,16 (м, 4Н, Ar-H), 6,96 (д, J=8,6 Гц, 2H, Ar-H), 6,16-6,02 (м, 1H, СН2=СНСН2-), 5,06 (дд, J=10,2, 1,6 Гц, 1H, СН2=СНСН2), 4,83 (дд, J=17,2, 1,6 Гц, 1H, СН2=СНСН2-), 3,94 (с, 3Н, -ОСН3), 3,87 (с, 3Н, -ОСН3), 3,73 (д, J=5,3 Гц, 2H, СН2=СНСН2-).
Пример 2
Синтез диэтил-2-(5-гексенил)-2-(4,4,5,5,5-пентафторпентил)малоната
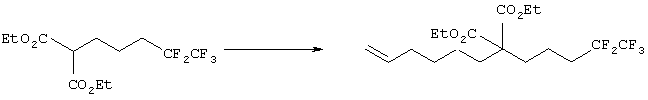
Раствор диэтил-2-(4,4,5,5,5-пентафторпентил)малоната (4,0 г, 12,5 ммоль) в диметилсульфоксиде (30 мл) охлаждали до 10°С. К данному раствору добавляли 60% гидрид натрия (600 мг, 15 ммоль) и полученную смесь перемешивали в течение 1 часа при комнатной температуре. К реакционной смеси медленно добавляли по каплям 6-бром-1-гексен (2,5 мл, 18,75 ммоль) с последующим перемешиванием в течение 3 часов при комнатной температуре. К реакционной смеси добавляли воду, затем экстрагировали этилацетатом. Органический слой промывали водой и насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия. После отгонки растворителя остаток очищали колоночной хроматографией на силикагеле (элюент: этилацетат/гексан=1/9), получая диэтил-2-(5-гексенил)-2-(4,4,5,5,5-пентафторпентил)малонат (3,86 г, выход 77%).
1Н-ЯМР (270 МГц, СDСl3): δ 5,82-5,72 (м, 1Н, -CH=CH2), 5,02-4,92 (м, 2Н, -CH=CH2), 4,19 (кв, J=7,3 Гц, 4H, -CO2CH2CH2), 2,10-1,86 (м, 8Н), 1,53-1,34 (м, 6Н), 1,26 (т, J=7,3 Гц, 6H, -CO2CH2CH3).
Пример 3
Синтез 9-[6-гидрокси-2-(4-гидроксифенил)нафт-1-ил]-2-(4,4,5,5,5-пентафторпентил)нонановой кислоты
(Стадия 1)

Диэтил-2-(5-гексенил)-2-(4,4,5,5,5-пентафторпентил)малонат, полученный в примере 2, (1,83 г, 4,55 ммоль) и бензилиденбис(трициклогексилфосфин)дихлоррутений (94 мг, 0,11 ммоль) добавляли к раствору 6-метокси-2-(4-метоксифенил)-1-(2-пропенил)нафталина (692 мг, 2,28 ммоль) в дихлорметане (10 мл), с последующим нагреванием при кипении с обратным холодильником в течение 20 часов в атмосфере аргона. После отгонки растворителя остаток очищали колоночной флеш-хроматографией на силикагеле (элюент: гексан/этилацетат=10/1), получая целевой олефин (1,8 г) в виде смеси цис- и транс-форм и димера боковой цепи. Данную смесь растворяли в этилацетате (20 мл) и к полученному раствору добавляли 10% палладия-на-углероде (236 мг) с последующим перемешиванием в течение 2 часов при комнатной температуре в атмосфере водорода. Катализатор удаляли фильтрованием и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флеш-хроматографией на силикагеле (элюент: гексан/этилацетат=4/1), получая диэтил-2-[7-[6-метокси-2-(4-метоксифенил)нафт-1-ил]гептил]-2-(4,4,5,5,5-пентафторпентил)малонат (1,05 г, выход 68%).
1Н-ЯМР (270 МГц, СDСl3): δ 7,96 (д, J=9,3 Гц, 1Н, Ar-H), 7,59 (д, J=8,2 Гц, 1Н, Ar-H), 7,30-7,21 (м, 4Н, Ar-H), 7,18-7,15 (м, 1Н, Ar-H), 6,97 (д, J=8,6 Гц, 2Н, Ar-H), 4,18 (кв, J=7,0 Гц, 4Н, -СO2СН2СН2), 3,94 (с, 3Н, -ОСН3), 3,88 (с, 3Н, -ОСН3), 2,97-2,91 (м, 2Н, нафтил-CH2-), 2,09-2,03 (м, 2Н, -CH2CF3), 1,99-1,82 (м, 4Н, алкил-Н), 1,55-1,45 (м, 6Н, алкил-Н), 1,23 (т, J=7,0 Гц, 6Н, -CO2CH2CH3), 1,10-1,04 (м, 6Н, алкил-Н).
(Стадия 2)
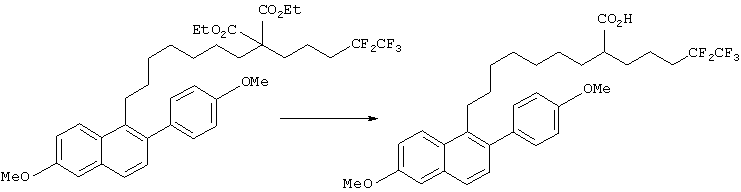
Диэтил-2-[7-[6-метокси-2-(4-метоксифенил)нафт-1-ил]гептил]-2-(4,4,5,5,5-пентафторпентил)малонат (1,02 г, 1,5 ммоль) растворяли в этаноле (10 мл). К данному раствору добавляли гидроксид натрия (1,2 г, 30 ммоль) и воду (1 мл) и полученную смесь нагревали при кипении с обратным холодильником в течение 3 часов. Разбавленную соляную кислоту добавляли к реакционной смеси, которую затем экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом натрия. Растворитель отгоняли, получая 2-[7-[6-метокси-2-(4-метоксифенил)нафт-1-ил]гептил]-2-(4,4,5,5,5-пентафторпентил)малоновую кислоту (1,0 г).
Далее, полученную 2-[7-[6-метокси-2-(4-метоксифенил)нафт-1-ил]гептил]-2-(4,4,5,5,5-пентафторпентил)малоновую кислоту (1,0 г) растворяли в диметилсульфоксиде (10 мл) и смесь нагревали в течение 4 часов при 120°С. К реакционной смеси добавляли воду, затем экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом натрия. После отгонки растворителя остаток очищали колоночной хроматографией на силикагеле (элюент: этилацетат/гексан=1/1), получая 9-[6-метокси-2-(4-метоксифенил)нафт-1-ил]-2-(4,4,5,5,5-пентафторпентил)нонановую кислоту (820 мг, выход 94%).
1Н-ЯМР (270 МГц, СDСl3): δ 7,97 (д, J=8,9 Гц, 1H, Ar-H), 7,59 (д, J=8,2 Гц, 1H, Ar-H), 7,30-7,21 (м, 4Н, Ar-H), 7,18-7,15 (м, 1H, Ar-H), 6,97 (д, J=8,6 Гц, 2Н, Ar-H), 3,94 (с, 3Н, ОСН3), 3,88 (с, 3Н, -ОСН3), 2,97-2,91 (м, 2Н, нафтил-CH2-), 2,38-2,35 (м, 1H, -CHCO2), 2,09-1,94 (м, 2Н, -CH2CF2), 1,73-1,41 (м, 8Н, алкил-Н), 1,29-1,18 (м, 8Н, алкил-Н).
(Стадия 3)

Раствор трехбромистого бора в дихлорметане (1,0 М, 8,5 мл, 8,47 ммоль) добавляли по каплям к раствору 9-[6-метокси-2-(4-метоксифенил)нафт-1-ил]-2-(4,4,5,5,5-пентафторпентил) нонановой кислоты (820 мг, 1,41 ммоль) в дихлорметане (20 мл) при -78°С в атмосфере аргона. Реакционную смесь нагревали при перемешивании до 0°С в течение 5 часов. К реакционной смеси добавляли воду, затем экстрагировали этилацетатом. Органический слой промывали водой и насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния. После отгонки растворителя остаток очищали колоночной хроматографией на силикагеле (элюент: этилацетат/гексан=3/2) с последующей колоночной хроматографией на силикагеле с обращенной фазой RP-18 (элюент: ацетонитрил, содержащий 0,1% трифторуксусную кислоту/вода=3/2), получая 9-[6-гидрокси-2-(4-гидроксифенил)нафт-1-ил]-2-(4,4,5,5,5-пентафторпентил)нонановую кислоту (633 мг, выход 81%).
1Н-ЯМР (270 МГц, СDСl3): δ 7,93 (д, J=9,9 Гц, 1Н, Аr-Н), 7,47 (д, J=8,2 Гц, 1Н, Аr-Н), 7,18-7,11 (м, 5Н, Аг-Н), 6,85 (д, J=9,3 Гц, 2Н, Аr-Н), 2,97-2,91 (м, 2Н, нафтил-CH2-), 2,34-2,29 (м, 1Н, -CHCO2), 2,20-1,99 (м, 2Н, -CH2CF2), 1,62-1,41 (м, 6Н, алкил-Н), 1,27-1,18 (м, 10Н, алкил-Н).
Пример 4
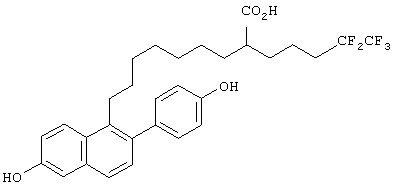
Синтез 11-[6-гидрокси-2-(4-гидроксифенил)нафт-1-ил]-2-(4,4,5,5,5-пентафторпентил)ундекановой кислоты

Повторяли те же методики, как описано в примерах 1, 2 и 3, получая 11-[6-гидрокси-2-(4-гидроксифенил)нафт-1-ил]-2-(4,4,5,5,5-пентафторпентил)ундекановую кислоту.
1Н-ЯМР (270 МГц, СDСl3): δ 7,98 (д, 1Н), 7,54 (д, 1Н), 7,33-7,03 (м, 5Н), 6,88 (д, 2Н), 2,93 (т, 2Н), 2,5 (м, 1Н), 2,2-1,0 (м, 22Н).
Пример 5
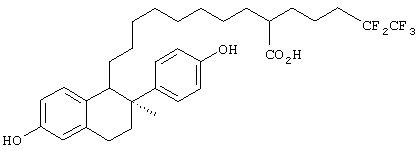
Синтез 10-[(1RS, 2RS)-6-гидрокси-2-(4-гидроксифенил)-2 метил-1,2,3,4-тетрагидро-1-нафтил]-2-(4,4,5,5,5-пентафторпентил)декановой кислоты
(Стадия 1)
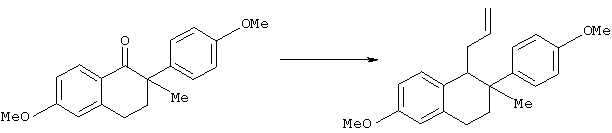
6-Метокси-2-(4-метоксифенил)-2-метил-1,2,3,4-тетрагидронафталин-1-он синтезировали способом, описанным в патенте США No. 4904661. Раствор данного соединения (1,5 г, 5,1 ммоль), растворенного в безводном тетрагидрофуране (25 мл), добавляли по каплям к раствору литийалюминийгидрида в безводном тетрагидрофуране (1М в ТГФ, 2,6 мл, 2,6 ммоль) при -78°С в атмосфере аргона. Реакцию проводили в течение 1,5 часов. Затем реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение 8 часов. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония, затем экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором бикарбоната натрия, водой и насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем упаривали, удаляя растворитель. Полученный остаток растворяли в 1,2-дихлорэтане (35 мл). К данному раствору добавляли при 0°С в атмосфере аргона иодистый цинк (2,02 г, 6,31 ммоль) и аллилтриметилсилан (1,67 мл, 10,52 ммоль) и полученную смесь перемешивали в течение 12 часов при комнатной температуре. К реакционной смеси добавляли воду, затем экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором бикарбоната натрия, водой и насыщенным водным раствором хлорида натрия, а затем сушили над безводным сульфатом магния. После отгонки растворителя остаток очищали колоночной флеш-хроматографией на силикагеле (элюент: гексан/этилацетат=60/1), получая (1RS,2RS)-6-метокси-2-(4-метоксифенил)-2-метил-1-(2-пропенил)-1,2,3,4-тетрагидронафталин (1,27 г, выход 78%).
1Н-ЯМР (270 МГц, СDСl3): δ 7,31 (д, 2Н, J=7,5 Гц), 6,97 (д, 1Н, J=7,9 Гц), 6,88 (д, 2Н, J=8,7 Гц), 6,68-6,65 (м, 2Н), 5,53 (м, 1Н), 4,76-4,57 (м, 2Н), 3,81 (с, 3Н), 3,78 (с, 3Н), 2,99-2,78 (м, 2Н), 2,81 (м, 1Н), 2,28 (м, 1Н), 1,98-1,92 (м, 2Н), 1,71 (м, 1Н), 1,17 (с, 3Н).
(Стадия 2)
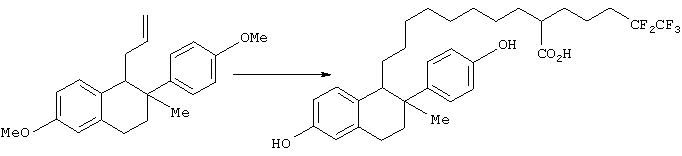
Полученный таким образом (1RS, 2RS)-6-метокси-2-(4-метоксифенил)-2-метил-1-(2-пропенил)-1,2,3,4-тетрагидронафталин превращали в 10-[(1RS, 2RS)-6-гидрокси-2-(4-гидроксифенил)-2-метил-1,2,3,4-тетрагидро-1-нафтил]-2-(4,4,5,5,5-пентафторпентил)декановую кислоту способом, аналогичным примеру 3.
1Н-ЯМР (300 МГц, СDСl3): δ 7,23 (д, 2Н, J=7,5 Гц), 6,90 (д, 1Н, J=7,9 Гц), 6,80 (д, 2Н, J=8,7 Гц), 6,58 (м, 2Н), 2,90 (м, 2Н), 2,60 (д, 1Н, J=8,7 Гц), 2,37 (м, 1Н), 2,22 (м, 1Н), 2,02 (м, 2Н), 1,87 (м, 1Н), 1,37-1,75 (м, 6Н), 0,86-1,26 (м, 17Н).
Масс-спектр (ESI): 585 (М+1).
Пример 6
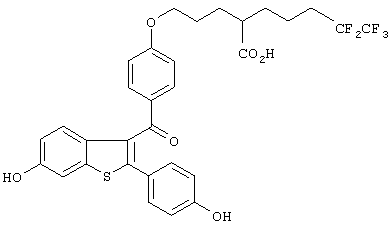
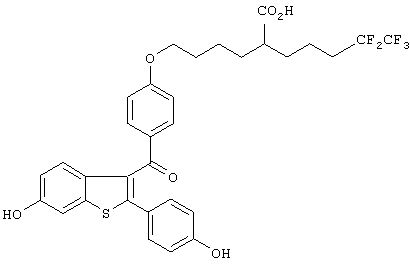
Синтез 2-[5-[4-[(6-гидрокси-2-(4-гидроксифенил)бензо[b] тиофен-3-ил)кар0онил]фенокси]пентил]-6,6,7,7,7-пентафторгептановой кислоты
(Стадия 1)

4-Метоксибензойную кислоту (450 мг, 2,96 ммоль), хлористый тионил (3 мл, 44,4 ммоль) и безводный диметилформамид (1 капля) добавляли к безводному хлороформу (10 мл), полученную смесь нагревали при кипении с обратным холодильником в течение 3 часов в атмосфере аргона и затем охлаждали до комнатной температуры. Затем реакционную смесь концентрировали при пониженном давлении, остаток растворяли в безводном дихлорметане и добавляли к полученному раствору 6-метокси-2-(4-метоксифенил)бензо[b]тиофен (760 мг, 2,8 ммоль), синтезированный способом, описанным в J. Med. Chem. 1057 (1984), и хлорид алюминия (2,37 г, 17,76 ммоль) с последующим перемешиванием в течение 4 часов при комнатной температуре. Для того чтобы остановить реакцию, добавляли тетрагидрофуран и лед и реакционную смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния. После отгонки растворителя фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле (элюент: дихлорметан/гексан=1/1), получая [6-метокси-2-(4-метоксифенил)бензо[b]тиофен-3-ил](4-метоксифенил)метанон (405 мг, выход 39%) в виде желтого масла.
1Н-ЯМР (270 МГц, СDСl3): δ 7,80-7,75 (м, 2Н), 7,52 (д, 1Н, J=8,6 Гц), 7,37-7,28 (м, 3Н), 6,95 (дд, 1Н, J1=8,9 Гц, J2=2,2 Гц), 6,78-6,74 (м, 4Н), 3,91 (с, 3Н), 3,85 (с, 3Н), 3,77 (с, 3Н).
(Стадия 2)
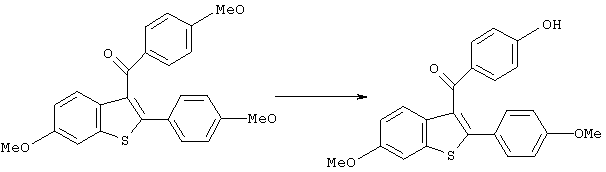
[6-Метокси-2-(4-метоксифенил)бензо[b]тиофен-3-ил]-(4-метоксифенил)метанон (410 мг, 1,02 ммоль) растворяли в безводном диметилформамиде (15 мл) и добавляли к полученному раствору этантиолят натрия (170 мг, 2,04 ммоль) с последующим перемешиванием в течение 1,5 часов при температуре от 90 до 100°С в атмосфере аргона. Реакционную смесь охлаждали до комнатной температуры и, после добавления воды, экстрагировали этилацетатом. Органический слой промывали водой и насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния. После отгонки растворителя остаток очищали колоночной хроматографией на силикагеле (элюент: этилацетат/гексан=1/1), получая (4-гидроксифенил)[6-метокси-2-(4-метоксифенил)бензо[b]тиофен-3-ил]метанон (323 мг, выход 81,6%) в виде желтого масла.
1Н-ЯМР (300 МГц, СDСl3): δ 7,70 (д, 2Н, J=9,0), 7,51 (д, 1Н, J=8,7), 7,33-7,26 (м, 3Н), 6,95 (дд, 1Н, J1=8,9 Гц, J2=2,6 Гц), 6,75-6,65 (м, 4Н), 3,87 (с, 3Н), 3,72 (с, 3Н).
(Стадия 3)

Диэтил-2-(4,4,5,5,5-пентафторпентил)малонат (3 г, 9,37 ммоль) растворяли в диметилсульфоксиде (20 мл) и к полученному раствору добавляли гидрид натрия (60%, 525 мг, 13,11 ммоль) с последующим перемешиванием в течение 1 часа при комнатной температуре. К реакционной смеси добавляли 5-бром-1-хлорпентан (7,4 мл, 56,2 ммоль) с последующим перемешиванием в течение 1,5 часов при комнатной температуре. Реакционную смесь разбавляли водой, экстрагировали этилацетатом, промывали водой и насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле (элюент: дихлорметан/гексан=1/4), получая диэтил-2-(5-хлорпентил)-2-(4,4,5,5,5-пентафторпентил)малонат (3,3 г, выход 83%) в виде бесцветного масла.
1Н-ЯМР (300 МГц, СDСl3): δ 4,14 (кв, 4Н, J=7,1 Гц), 3,46 (т, 2Н, J=6,7 Гц), 2,06-1,64 (м, 8Н), 1,50-1,36 (м, 4Н), 1,24-1,10 (м, 2Н), 1,21 (т, 6Н, J=7,1 Гц).
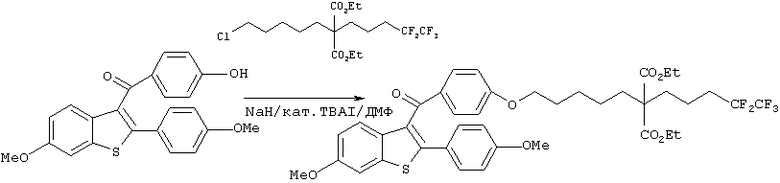
(4-Гидроксифенил)[6-метокси-2-(4-метоксифенил)бензо[b]тиофен-3-ил]метанон (1 г, 2,56 ммоль) растворяли в диметилформамиде (15 мл) и добавляли к полученному раствору гидрид натрия (60%, 143 мг, 3,59 ммоль) с последующим перемешиванием в течение 1 часа при комнатной температуре.
К реакционной смеси добавляли диэтил-2-(5-хлорпентил)-2-(4,4,5,5,5-пентафторпентил)малонат (1,85 г, 4,35 ммоль), иодид натрия (769 мг, 5,13 ммоль) и иодид тетрабутиламмония (189 мг, 0,51 ммоль) с последующим перемешиванием в течение 24 часов при 60°С. Затем реакционную смесь охлаждали до комнатной температуры, к реакционной смеси добавляли насыщенный водный раствор хлорида аммония, затем экстрагировали этилацетатом, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле (элюент: этилацетат/гексан = 1/9), получая диэтил-2-[5-[4-[(6-метокси-2-(4-метоксифенил) бензо[b]тиофен-3-ил)карбонил]фенокси]пентил]-2-(4,4,5,5,5-пентафторпентил)малонат (1,4 г, выход 70%) в виде желтого масла.
1H-ЯМР (300 МГц, CDCl3): δ 7,73 (д, 2Н, J=9,1 Гц), 7,48 (д, 1Н, J=8,5 Гц), 7,32 (д, 2Н, J=8,6 Гц), 7,27 (д, 1Н, J=2,3 Гц), 6,91 (дд, 1Н, J1=8,8 Гц, J2=2,3 Гц), 6,74-6,67 (м, 4Н), 4,15 (кв, 4H, J=7,1 Гц), 3,88 (т, 2Н, J=6,3 Гц), 3,83 (с, 3Н), 3,70 (с, 3Н), 2,08-1,82 (м, 6Н), 1,75-1,71 (м, 2Н), 1,53-1,37 (м, 6Н), 1,21 (т, 6Н, J=7,1 Гц).
(Стадия 5)
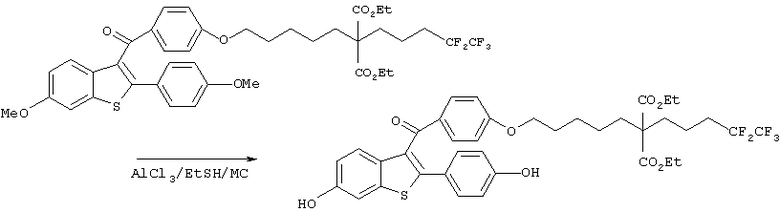
Диэтил-2-[5-[4-[(6-метокси-2-(4-метоксифенил)бензо[b] тиофен-3-ил)карбонил]фенокси]пентил]-2-(4,4,5,5,5-пентафторпентил)малонат (1,58 г, 2,03 ммоль) растворяли в дихлорметане (40 мл) и добавляли к полученному раствору хлорид алюминия (1,62 г, 12,2 ммоль) и этантиол (0,25 мл, 10,15 ммоль) с последующим перемешиванием в течение 1,5 часов при комнатной температуре. Затем реакционную смесь охлаждали до 0°С, медленно добавляли тетрагидрофуран (30 мл), затем разбавляли водой и экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле (элюент: этилацетат/гексан=1/2), получая диэтил-2-[5-[4-[(6-гидрокси-2-(4-гидроксифенил)бензо[b]тиофен-3-ил)карбонил]фенокси]пентил]-2-(4,4,5,5,5-пентафторпентил)малонат (1,2 г, выход 79%) в виде коричневой пены.
1H-ЯМР (300 МГц, СDСl3): δ 7,73 (д, 2Н, J=9,1 Гц), 7,38 (д, 1Н, J=8,6 Гц), 7,18 (д, 1Н, J=2,3 Гц), 7,14 (д, 2Н, J=8,6 Гц), 6,79 (дд, 1Н, J1=8,6 Гц, J2=2,3 Гц), 6,72 (д, 2Н, J=9,1 Гц), 6,57 (д, 2Н, J=8,6 Гц), 4,17 (кв, 4Н, J=7,2 Гц), 3,91 (т, 2Н, J=6,1 Гц), 2,10-1,78 (м, 6Н), 1,74-1,68 (м, 2Н), 1,54-1,36 (м, 4Н), 1,26-1,13 (м, 2Н), 1,21 (т, 6Н, J=7,2 Гц).
(Стадия 6)
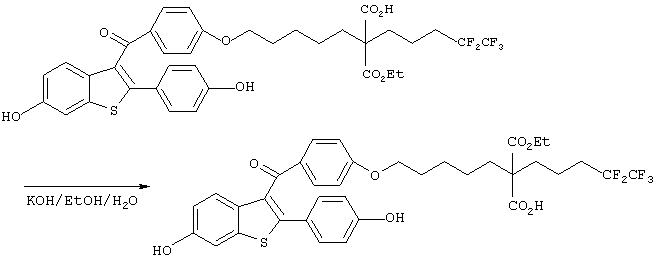
Диэтил-2-[5-[4-[(6-гидрокси-2-(4-гидроксифенил)бензо[b] тиофен-3-ил)карбонил]фенокси]пентил]-2-(4,4,5,5,5-пентафторпентил)малонат (1,197 г, 1,59 ммоль) растворяли в этаноле (20 мл) и затем добавляли гидроксид калия (3,58 г, 63,8 ммоль), растворенный в воде (10 мл). После перемешивания в течение 24 часов при 80°С реакционную смесь охлаждали до комнатной температуры, концентрировали при пониженном давлении для удаления этанола, доводили до рН 3 с помощью 3 н. водной хлористоводородной кислоты и затем экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, получая 2-[5-[4-[(6-гидрокси-2-(4-гидроксифенил)бензо[b]тиофен-3-ил)карбонил]фенокси]пентил]-2-(4,4,5,5,5-пентафторпентил)малоновую кислоту (1,1 г) в виде коричневого продукта, который затем использовали в последующей реакции без дополнительной очистки.
1H-ЯМР (300 МГц, СDСl3): δ 7,68 (д, 2Н, J=9,0 Гц), 7,38 (д, 1Н, J=9,0 Гц), 7,24 (д, 1Н, J=2,0 Гц), 7,18 (д, 2Н, J=8,6 Гц), 6,85 (дд, 1H, J1=9,0 Гц, J2=2,0 Гц), 6,80 (д, 2H, J=8,6 Гц), 6,63 (д, 2Н, J=8,6 Гц), 3,95 (т, 2H, J=6,0 Гц), 2,21-1,80 (м, 6Н), 1,74-1,70 (м, 2Н), 1,58-1,21 (м, 6Н).
(Стадия 7)
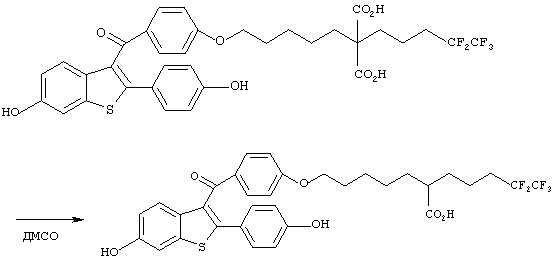
2-[5-[4-[(6-Гидрокси-2-(4-гидроксифенил)бензо[b]тиофен-3-ил)карбонил]фенокси]пентил]-2-(4,4,5,5,5-пентафторпентил)малоновую кислоту (1,1 г, 1,58 ммоль) растворяли в диметилсульфоксиде (10 мл) и перемешивали в течение 3 часов при 120°С. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой и затем экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле (элюент: этилацетат/гексан=1/1), получая 2-[5-[4-[(6-гидрокси-2-(4-гидроксифенил)бензо [b]тиофен-3-ил)карбонил]фенокси]пентил]-6,6,7,7,7-пентафторгептановую кислоту (732 мг, выход 71%) в виде желтого твердого вещества.
1H-ЯМР (300 МГц, СDOD): δ 7,68 (д, 2Н, J=8,8 Гц), 7,38 (д, 1Н, J=8,8 Гц), 7,25 (д, 1Н, J=2,3 Гц), 7,18 (д, 2Н, J=8,6 Гц), 6,85 (дд, 1Н, J1=8,8 Гц, J2=2,3 Гц), 6,79 (д, 2Н, J=8,9 Гц), 6,63 (д, 2Н, J=8,6 Гц), 3,95 (т, 2Н, J=6,5 Гц), 2,85 (м, 1Н), 2,15-1,94 (м, 2Н), 1,78-1,29 (м, 12Н).
Масс-спектр (ESI): 651 (М+1).
Пример 7
Синтез 8-[4-[(6-гидрокси-2-(4-гидроксифенил)бензо[b] тиофен-3-ил)карбонил]фенокси]-2-(4,4,5,5,5-пентафторпентил) октановой кислоты

Повторяли ту же методику, как описано в примере 1, получая 8-[4-[(6-гидрокси-2-(4-гидроксифенил)бензо[b]тиофен-3-ил)карбонил]фенокси]-2-(4,4,5,5,5-пентафторпентил)октановую кислоту.
1H-ЯМР (300 МГц, СDСl3): δ 7,68 (д, 2Н, J=8,6 Гц), 7,38 (д, 1Н, J=8,7 Гц), 7,24-7,16 (м, 3Н), 6,86-6,78 (м, 3Н), 6,62 (д, 2Н, J=8,3 Гц), 3,95 (т, 2Н, J=6,4 Гц), 2,35 (м, 1Н), 2,15-1,95 (м, 2Н), 1,77-1,25 (1м, 4Н).
Масс-спектр (ESI): 665 (М+1).
Пример 8
Синтез 2-[2-[4-[(6-гидрокси-2-(4-гидроксифенил)бензо[b] тиофен-3-ил)карбонил]фенокси]этил]-6,6,7,7,7-пентафторгептановой кислоты
Повторяли ту же методику, как описано в примере 1, получая 2-[2-[4-[(6-гидрокси-2-(4-гидроксифенил)бензо[b]тиофен-3-ил)карбонил]фенокси]этил]-6,6,7,7,7-пентафторгептановую кислоту.
1H-ЯМР (300 МГц, СDСl3+CD3OD): δ 7,70 (д, 2Н, J=8,9 Гц), 7,50 (д, 1Н, J=8,8 Гц), 7,26 (д, 1Н, J=2,2 Гц), 7,20 (д, 2Н, J=8,6 Гц), 6,90 (дд, 1Н, J1=8,8 Гц, J2=2,2 Гц), 6,70 (д, 2Н, J=8,9 Гц), 6,65 (д, 2Н, J=8,6 Гц), 4,10-3,90 (м, 2Н), 2,58 (м, 1Н), 2,18-1,84 (м, 4Н), 1,82-1,52 (м, 4Н).
Пример 9
Синтез 2-[3-[4-[(6-гидрокси-2-(4-гидроксифенил)бензо[b] тиофен-3-ил)карбонил]фенокси]пропил]-6,6,7,7,7-пентафторгептановой кислоты

Повторяли ту же методику, как описано в примере 1, получая 2-[3-[4-[(6-гидрокси-2-(4-гидроксифенил)бензо[b]тиофен-3-ил)карбонил]фенокси]пропил]-6,6,7,7,7-пентафторгептановую кислоту.
1H-ЯМР (300 МГц, СDСl3): δ 7,73 (д, 2Н, J=8,8 Гц), 7,47 (д, 1Н, J=8,8 Гц), 7,27 (д, 1Н, J=2,2 Гц), 7,21 (д, 2Н, J=8,6 Гц), 6,86 (дд, 1Н, J1=8,7 Гц, J2=2,3 Гц), 6,73 (д, 2Н, J=8,9 Гц), 6,67 (д, 2Н, J=8,6 Гц), 3,94 (т, 2Н, J=6,1 Гц), 2,39 (м, 1Н), 2,10-1,97 (м, 2Н), 1,82-1,55 (м, 8Н).
Масс-спектр (ESI): 623 (М+1).
Пример 10
Синтез 2-[4-[4-[(6-гидрокси-2-(4-гидроксифенил)бензо[b] тиофен-3-ил)карбонил]фенокси]бутил]-6,6,7,7,7-пентафторгептановой кислоты
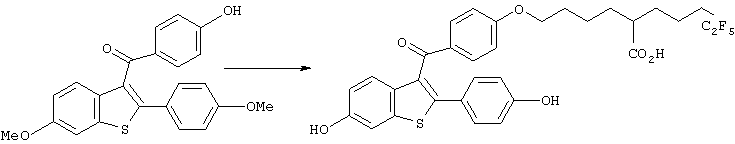
Повторяли ту же методику, как описано в примере 1, получая 2-[4-[4-[(6-гидрокси-2-(4-гидроксифенил)бензо[b]тиофен-3-ил)карбонил]фенокси]бутил]-6,6,7,7,7-пентафторгептановую кислоту.
1H-ЯМР (300 МГц, СDСl3): δ 7,71 (д, 2Н, J=8,8 Гц), 7,54 (д, 1Н, J=8,7 Гц), 7,26 (д, 1Н, J=2,3 Гц), 7,19 (д, 2Н, J=8,6 Гц), 6,88 (дд, 1Н, J1=8,8 Гц, J2=2,3 Гц), 6,69 (д, 2Н, J=8,8 Гц), 6,64 (д, 2Н, J=8,6), 3,93 (т, 2Н, J=6,1 Гц), 2,38 (м, 1Н), 2,15-1,47 (м, 12Н).
Масс-спектр (ESI): 637 (М+1).
Пример 11 Синтез 10-[(R)-7-гидрокси-3-(4-гидроксифенил)-3,4-дигидро-2Н-бензо[1,4]оксазин-4-ил]-2-(6,6,7,7,7-пентафторгептил)декановой кислоты
(Стадия 1)

Хлористый тионил (0,65 мл) добавляли к (R)-4-гидроксифенилглицину (1,00 г, 5,98 ммоль) в метаноле (10 мл) с последующим перемешиванием в течение ночи при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении и остаток растворяли в этилацетате (20 мл). К полученному раствору добавляли насыщенный водный раствор бикарбоната натрия (20 мл) и ди-трет-бутилдикарбоната (1,57 г, 7,19 ммоль) с последующим перемешиванием в течение 4 часов при комнатной температуре. Затем реакционную смесь разделяли на органический и водный слои, органический слой промывали последовательно водой и насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия. Затем органический слой концентрировали при пониженном давлении, полученные твердые вещества промывали смесью этилацетат/гексан, получая метиловый эфир (R)-N-(трет-бутоксикарбонил)-4-гидроксифенилглицина (1,47 г, выход 87%).
1H-ЯМР (300 МГц, СDСl3): δ 7,18 (2Н, д, J=8,6 Гц), 6,74 (2Н, д, J=8,6 Гц), 5,71 (1Н, ушир. с), 5,50-5,60 (1H, м), 5,18-5,26 (1H, м), 3,71 (3Н, с), 1,43 (9Н, с).
(Стадия 2)

Бромистый бензил (0,66 мл, 5,55 ммоль) добавляли к метиловому эфиру (R)-(N-трет-бутоксикарбонил)-4-гидроксифенилглицина (1,42 г, 5,05 ммоль) и карбонату калия (768 мг, 5,56 ммоль) в ацетоне (5 мл). Полученную смесь перемешивали в течение ночи при комнатной температуре и затем нагревали при кипении с обратным холодильником в течение 1 часа. После охлаждения реакционную смесь концентрировали при пониженном давлении. К полученному остатку добавляли этилацетат, затем промывали последовательно водой и насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом натрия. Затем органический слой концентрировали при пониженном давлении, полученные твердые вещества промывали смесью этилацетат/гексан, получая метиловый эфир (R)-N-(трет-бутоксикарбонил)-4-бензилоксифенилглицина (1,67 г, выход 89%).
1H-ЯМР (270 МГц, СDСl3): δ 7,30-7,45 (5Н, м), 7,27 (2Н, д, J=8,6 Гц), 6,95 (2Н, д, J=8,6 Гц), 6,40-6,55 (1Н, м), 6,20-6,30 (1H, м), 5,05 (2Н, с), 3,71 (3Н, с), 1,43 (9Н, с).
(Стадия 3)
К метиловому эфиру (R)-N-(трет-бутоксикарбонил)-4-бензилоксифенилглицина (200 мг, 0,538 ммоль) и тетрагидроборату лития (23 мг, 1,06 ммоль) в тетрагидрофуране (2 мл) добавляли этанол (4 мл) с последующим перемешиванием в течение ночи при комнатной температуре. Реакционную смесь подкисляли (рН 4) 10%-ной лимонной кислотой и концентрировали при пониженном давлении. К полученному остатку добавляли этилацетат, затем промывали последовательно водой и насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом натрия. Органический слой концентрировали при пониженном давлении, получая (R)-2-(трет-бутоксикарбонил)амино-2-(4-бензилоксифенил)этанол (187 мг, выход 100%).
1H-ЯМР (270 МГц, СDСl3): δ 7,27-7,45 (5Н, м), 7,21 (2Н, д, J=8,6 Гц), 6,96 (2Н, д, J=8,6 Гц), 5,05-5,20 (1Н, м), 5,05 (2Н, с), 5,65-5,80 (1Н, м), 3,75-3,85 (2Н, м), 2,35 (1Н, ушир. с), 1,43 (9Н, с).
(Стадия 4)

Диизопропилазодикарбоксилат (0,10 мл, 0,603 ммоль) добавляли к (R)-2-(трет-бутоксикарбонил)амино-2-(4-бензилоксифенил)этанолу (161 мг, 0,469 ммоль), 5-бензилокси-2-бромфенолу (131 мг, 0,469 ммоль) и трифенилфосфину (160 мг, 0,610 ммоль) в тетрагидрофуране (3 мл) в токе азота с последующим перемешиванием в течение 5 часов при комнатной температуре, к реакционной смеси добавляли диизопропилазодикарбоксилат (0,05 мл, 0,302 ммоль) с последующим перемешиванием в течение 3 часов при комнатной температуре. К реакционной смеси добавляли воду, затем экстрагировали этилацетатом. Органический слой промывали последовательно водой и насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (элюент: этилацетат/гексан=1/4, об/об), получая (R)-N-(трет-бутоксикарбонил)-2-(5-бензилокси-2-бромфенокси)-1-(4-бензилоксифенил)этиламин (214 мг, выход 75%).
1H-ЯМР (270 МГц, СDСl3): δ 7,25-7,45 (13Н, м), 6,95 (2Н, д, J=8,6 Гц), 6,40-6,55 (2Н, м), 5,35-5,50 (1Н, м), 5,05 (2Н, с), 5,00 (2Н, с), 4,95-5,05 (1Н, м), 4,05-4,25 (2Н, м), 1,42 (9Н, м).
(Стадия 5)
Трифторуксусную кислоту (1 мл) добавляли к (R)-N-(трет-бутоксикарбонил)-2-(5-бензилокси-2-бромфенокси)-1-(4-бензилоксифенил)этиламину (200 мг, 0,331 ммоль) в хлористом метилене (1 мл) с последующим перемешиванием в течение 1 часа при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении, подщелачивали насыщенным водным раствором бикарбоната натрия и затем экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (элюент: этилацетат/гексан=2/1 об/об), получая (R)-2-(5-бензилокси-2-бромфенокси)-1-(4-бензилоксифенил)этиламин (127 мг, выход 76%).
1H-ЯМР (270 МГц, СDСl3): δ 7,25-7,45 (13Н, м), 6,97 (2Н, д, J=8,6 Гц), 6,42-6,53 (2Н, м), 5,07 (2Н, с), 5,00 (2Н, с), 4,42 (1Н, дд, J=8,9, 3,6 Гц), 3,87 (1Н, дд, J=8,9, 8,9 Гц), 1,77 (2Н, ушир.с).
(Стадия 6)

Раствор (R)-2-(5-бензилокси-2-бромфенокси)-1-(4-бензилоксифенил)этиламина (120 мг, 0,238 ммоль), трис(дибензилиденацетон)дипалладия (11 мг, 0,012 ммоль), 2,2'-бис(дифенилфосфино)-1,1'-бинафтила (15 мг, 0,024 ммоль) и трет-бутоксида калия 937 мг, 0,330 ммоль) в толуоле (2,5 мл) перемешивали в течение 3 часов при 100°С в токе азота. После охлаждения к реакционной смеси добавляли воду, затем экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (элюент: этилацетат/гексан=1/5 об/об), получая (R)-7-бензилокси-3-(4-бензилоксифенил)-3,4-дигидро-2Н-бензо[1,4]оксазин (55,4 мг, выход 55%).
1H-ЯМР (270 МГц, СDСl3): δ 7,25-7,50 (12Н, м), 6,98 (2Н, д, J=8,6 Гц), 6,58 (1Н, д, J=8,6 Гц), 6,55 (1Н, д, J=2,6 Гц), 6,48 (1Н, дд, J=8,6, 2,6 Гц), 5,07 (2Н, с), 4,99 (2Н, с), 4,39 (1Н, дд, J=8,9, 2,6 Гц), 4,22 (1Н, дд, J=10,6, 2,6 Гц), 3,96 (1Н, дд, 10,6, 8,9 Гц), 3,73 (1Н, ушир.с).
(Стадия 7).

Раствор (R)-7-бензилокси-3-(4-бензилоксифенил)-3,4-дигидро-2Н-бензо[1,4]оксазина (47,6 мг, 0,112 ммоль), иодида натрия (67 мг, 0,450 ммоль), карбоната калия (31 мг, 0,224 ммоль) и бромистого аллила (0,04 мл, 0,473 ммоль) в ацетоне (1 мл) перемешивали в течение 3 часов при 50°С в токе азота и затем нагревали при кипении с обратным холодильником в течение 7 часов. После охлаждения к реакционной смеси добавляли воду, затем экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (элюент: этилацетат/гексан=1/5, об/об), получая (R)-4-аллил-7-бензилокси-3-(4-бензилоксифенил)-3,4-дигидро-2Н-бензо[1,4]оксазин (44 мг, выход 85%).
1H-ЯМР (270 МГц, СDСl3): δ 7,25-7,45 (10Н, м), 7,20 (2Н, д, J=8,6 Гц), 6,95 (2Н, д, J=8,6 Гц), 6,74 (1Н, д, J=9,6 Гц), 6,50-6,56 (2Н, м), 5,68-5,85 (1Н, м), 5,06-5,17 (2Н, м), 5,05 (2Н, с), 4,98 (2Н, с), 4,33 (1Н, д, J=6,9, 3,0 Гц), 4,19 (1Н, дд, J=10,9, 3,0 Гц), 4,11 (1Н, дд, J=10,9, 6,9 Гц), 4,85-4,98 (1Н, м), 3,47 (1Н, дд, J=16,8, 6,3 Гц).
(Стадия 8)

1)Раствор (R)-4-аллил-7-бензилокси-3-(4-бензилоксифенил)-3,4-дигидро-2Н-бензо[1,4]оксазина (177 мг, 0,382 ммоль), этилового эфира 2-(6,6,7,7,7-пентафторгептил)нон-8-еновой кислоты (285 мг, 0,765 ммоль) и бензилиденбис(трициклогексилфосфин)дихлоррутения (16 мг, 0,019 ммоль) в дихлорметане (2 мл) нагревали при кипении с обратным холодильником в течение 5 часов в токе азота. Реакционную смесь дополнительно смешивали с этиловым эфиром 2-(6,6,7,7,7-пентафторгептил)нон-8-еновой кислоты (71 мг, 0,190 ммоль) и бензилиденбис(трициклогексилфосфин)дихлоррутением (16 мг, 0,019 ммоль) и затем нагревали при кипении с обратным холодильником в течение 2 часов. После охлаждения реакционную смесь концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (элюент: хлороформ/гексан=3/1, об/об), получая масло (197 мг).
2) Смесь масла, полученного выше в 1), и 10% Pd-C (13 мг, 0,012 ммоль) в смеси этанол/метанол (1:1, 3 мл) перемешивали в течение 13 часов при комнатной температуре в токе водорода. После фильтрования реакционной смеси через целит маточный раствор концентрировали при пониженном давлении. Полученный остаток подвергали двум дополнительным реакциям восстановления, как указано выше. Полученный остаток дополнительно очищали колоночной хроматографией на силикагеле (элюент: этилацетат/гексан -1/2 1/1, об/об), получая этил-10-[(R)-7-гидрокси-3-(4-гидроксифенил)-3,4-дигидро-2Н-бензо [1,4]оксазин-4-ил]-2-(6,6,7,7,7-пентафторгептил)деканоат (60,2 мг, выход 26%).
1H-ЯМР (270 МГц, СD3OD): δ 7,13 (2Н, д, J=8,6 Гц), 6,78 (2Н, д, J=8,6 Гц), 6,65 (1Н, д, J=8,6 Гц), 6,36 (1Н, дд, J=8,6, 2,6 Гц), 6,29 (1H, д, J=2,6 Гц), 4,27 (1H, дд, J=6,3, 3,0 Гц), 4,00-4,20 (4Н, м), 3,20-3,35 (1H, м), 2,80-2,95 (1H, м), 2,30-2,45 (1H, м), 2,00-2,23 (2Н, м), 1,15-1,70 (25Н, м).
(Стадия 9)

Водный раствор гидроксида натрия (1 н., 1 мл) добавляли к этил-10-[(R)-7-гидрокси-3-(4-гидроксифенил)-3,4-дигидро-2Н-бензо[1,4]оксазин-4-ил]-2-(6,6,7,7,7-пентафторгептил)деканоату (56,8 мг, 0,0902 ммоль) в этаноле (1 мл) в токе азота с последующим перемешиванием в течение 7 часов при 50°С. После охлаждения реакционную смесь подкисляли 1 н. водной хлористоводородной кислотой и экстрагировали этилацетатом. Органический слой промывали последовательно водой и насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (элюент: этилацетат/гексан=1/1, об/об), получая 10-[(R)-7-гидрокси-3-(4-гидроксифенил)-3,4-дигидро-2Н-бензо[1,4]оксазин-4-ил]-2-(6,6,7,7,7-пентафторгептил)декановую кислоту (41,4 мг, выход 76%).
1H-ЯМР (270 МГц, СD3OD): δ 7,13 (2Н, д, J=8,6 Гц), 6,78 (2Н, д, J=8,6 Гц), 6,64 (1H, д, J=8,6 Гц), 6,37 (1H, дд, J=8,6, 2,6 Гц), 6,29 (1H, д, J=2,6 Гц), 4,21-4,35 (1H, м), 4,12 (1H, дд, J=10,6, 3,0 Гц), 4,05 (1H, дд, J=10,6, 6,6 Гц), 3,20-3,35 (1H, м), 2,82-2,95 (1H, м), 2,25-2,38 (1H, м), 2,00-2,21 (2Н, м), 1,10-1,70 (22Н, м).
Пример 12
Синтез 10-[3,17β-дигидроксиэстра-1,3,5(10)-триен-11β-ил]2-(6,6,7,7,7-пентафторгептил)декановой кислоты
(Стадия 1)
Раствор 3,17β-бис(бензилокси)эстра-1,3,5(10)-триен-11-она (148,8 мг, 0,318 ммоль) в безводном тетрагидрофуране (2,5 мл) охлаждали до -10°С в атмосфере аргона. К данному раствору добавляли по каплям 1,0 М раствор аллилмагнийбромида в безводном простом эфире (1,5 мл, 1,5 ммоль) и полученную смесь перемешивали в течение 15 часов при комнатной температуре. Реакционную смесь охлаждали до 0°С, добавляли затем воду и насыщенный водный раствор хлорида аммония. Затем реакционную смесь экстрагировали этилацетатом, органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем фильтровали. После отгонки растворителя остаток очищали флеш-хроматографией на силикагеле (элюент: гексан/этилацетат=6/1), получая 3,17β-бис(бензилокси)-11α-(2-пропенил)эстра-1,3,5(10)-триен-11β-ол (150,4 мг, выход 93%).
1H-ЯМР (270 МГц, СDСl3): δ 7,79 (д, J=10 Гц, 1Н, Cl-H), 7,44-7,29 (м, 10Н), 6,82-6,76 (м, 2Н, С2 и С4-Н), 6,00-5,85 (м, 1Н, олефиновый Н), 5,20-5,12 (м, 2Н, олефиновый Н), 5,04 (с, 2Н, Ph-CH2), 4,55 (с, 2Н, Ph-CH2), 3,44 (т, J=8 Гц, 1Н, С17-Н), 2,88 (дд, J=14,8 Гц, 1Н, аллильный СН2), 2,78-2,58 (м, 2Н), 2,50 (дд, J=14,7 Гц, 1Н, аллильный СН2), 2,23 (д, J=11 Гц, 1Н), 2,11 (д, J=14 Гц, 1Н), 2,08-1,95 (м, 1Н), 1,90-1,15 (м, 9Н), 1,07 (с, 3Н, С18-Н).
(Стадия 2)

Бензилиденбис(трициклогексилфосфин)дихлоррутений (5,9 мг, 0,00717 ммоль) добавляли к раствору 3,17β-бис(бензилокси)-11α-(2-пропенил)эстра-1,3,5(10)-триен-11β-ола (65,5 мг, 0,129 ммоль) и этил-2-(6,6,7,7,7-пентафторгептил)-8-ноненоата (101,3 мг, 0,272 ммоль) в дихлорметане (0,5 мл) с последующим нагреванием при кипении с обратным холодильником в течение 2,5 часов в атмосфере аргона. После охлаждения к реакционной смеси добавляли этил-2-(6,6,7,7,7-пентафторгептил)-8-ноненоат (100 мг, 0,269 ммоль) и бензилиденбис(трициклогексилфосфин)дихлоррутений (6 мг, 0,00729 ммоль), затем нагревали при кипении с обратным холодильником в течение 3 часов в атмосфере аргона. После охлаждения к реакционной смеси дополнительно добавляли этил-2-(6,6,7,7,7-пентафторгептил)-8-ноненоат (100 мг, 0,269 ммоль) и бензилиденбис (трициклогексилфосфин)дихлоррутений (6 мг, 0,00729 ммоль), затем нагревали при кипении с обратным холодильником в течение 6,5 часов в атмосфере аргона и оставляли охлаждаться. Помимо этого бензилиденбис(трициклогексилфосфин)дихлоррутений (6,8 мг, 0,00826 ммоль) добавляли к раствору 3,17β-бис(бензилокси)-11α-(2-пропенил)эстра-1,3,5(10)-триен-11β-ола (84,5 мг, 0,166 ммоль) и этил-2-(6,6,7,7,7-пентафторгептил)-8-ноненоата (124 мг, 0,333 ммоль) в дихлорметане (0,5 мл) с последующим нагреванием при кипении с обратным холодильником в течение 2,5 часов в атмосфере аргона. После охлаждения к реакционной смеси добавляли этил-2-(6,6,7,7,7-пентафторгептил)-8-ноненоат (124 мг, 0,333 ммоль) и бензилиденбис(трициклогексилфосфин)дихлоррутений (6,8 мг, 0,00826 ммоль), затем нагревали при кипении с обратным холодильником в течение 3 часов в атмосфере аргона. После охлаждения к реакционной смеси дополнительно добавляли этил-2-(6,6,7,7,7-пентафторгептил)-8-ноненоат (124 мг, 0,333 ммоль) и бензилиденбис(трициклогексилфосфин)дихлоррутений (6,8 мг, 0,00826 ммоль), затем нагревали при кипении с обратным холодильником в течение 6,5 часов в атмосфере аргона и оставляли охлаждаться. Полученные таким образом две реакционные смеси объединяли и концентрировали при пониженном давлении. Остаток очищали флеш-хроматографией на силикагеле (элюент; гексан/этилацетат=5/1), получая этил-10-[3,17β-бис(бензилокси)-11β-гидроксиэстра-1,3,5(10)-триен-11α-ил]-2-(6,6,7,7,7-пентафторгептил)-8-деценоат (186,4 мг, выход 74%).
1H-ЯМР (270 МГц, СDСl3): δ 7,79 (д, J=10 Гц, 1H), 7,45-7,25 (м, 10Н), 6,82-6,72 (м, 2Н), 5,62-5,40 (м, 2Н, олефиновый Н), 5,04 (с, 2Н, Ph-CH2), 4,55 (с, 2Н, Ph-CH2), 4,13 (кв, J=7 Гц, 2Н, СОО-СН2), 3,42 (т, J=8 Гц, 1H), 2,95-1,14 (м, 40Н), 1,07 (с, 3Н).
(Стадия 3)

30%-ный раствор НВr в уксусной кислоте (2 мл) добавляли к раствору этил-10-[3,17β-бис(бензилокси)-11β-гидроксиэстра-1,3,5(10)-триен-11α-ил]-2-(6,6,7,7,7-пентафторгептил)-8-деценоата (155,4 мг, 0,182 ммоль) в этаноле (8 мл) с последующим перемешиванием в течение 24 часов при 50°С. После охлаждения реакционную смесь выливали в насыщенный водный раствор бикарбоната натрия и экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем фильтровали. После концентрирования при пониженном давлении полученный остаток очищали флеш-хроматографией на силикагеле (элюент: гексан/этилацетат=20/1), получая масло. Масло растворяли в смешанном растворителе, состоящем из этанола (5 мл) и метанола (5 мл), 10%-ный палладий-на-углероде (78,8 мг) добавляли к полученному раствору с последующим перемешиванием в течение 14 часов при комнатной температуре в атмосфере водорода. После продувания азотом к реакционной смеси добавляли 10%-ный палладий-на-углероде (74,0 мг) с последующим перемешиванием в течение 15 часов при комнатной температуре в атмосфере водорода. Реакционную смесь фильтровали и концентрировали, остаток растворяли в метаноле (10 мл). Опять к реакционной смеси добавляли 10%-ный палладий-на-углероде (80 мг) с последующим перемешиванием в течение 2 дней при комнатной температуре в атмосфере водорода. После фильтрования реакционной смеси и концентрирования остаток очищали флеш-хроматографией на силикагеле (элюент: гексан/этилацетат=10/1), получая масло. Масло дополнительно очищали с использованием пластины с силикагелем (проявляющий растворитель: гексан/этилацетат=2/1), получая этил-10-[3,17β-дигидроксиэстра-1,3,5(10)-триен-11β-ил]-2-(6,6,7,7,7-пентафторгептил)деканоат (14,6 мг, выход 12%).
1H-ЯМР (270 МГц, СDСl3): δ 7,00 (д, J=8,6 Гц, 1Н), 6,62 (дд, J=8,6, 2,6 Гц, 1Н), 6,55 (д, J=2,6 Гц, 1Н), 5,28 (ушир.с, 1Н, Ar-OH), 4,15 (кв, J=7,3 Гц, 2Н, СОО-СН2), 3,70 (т, J=8,6 Гц, 1Н), 2,90-1,10 (м, 45Н), 0,91(с, 3Н).
Значение Rf: 0,45 (пластина с силикагелем, проявляющий растворитель: гексан/этилацетат=2/1).
(Стадия 4)

Этил-10-[3,17β-дигидроксиэстра-1,3,5(10)-триен-11β-ил]-2-(6,6,7,7,7-пентафторгептил)деканоат (11,1 мг, 0,0168 ммоль) растворяли в смешанном растворителе, состоящем из этанола (0,5 мл) и тетрагидрофурана (0,5 мл). К данному раствору добавляли 1 н. водный раствор NaOH (0,5 мл) и полученную смесь нагревали при кипении с обратным холодильником в течение 5 часов. После охлаждения к реакционной смеси добавляли насыщенный водный раствор хлорида аммония, затем экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем фильтровали. После концентрирования при пониженном давлении полученный остаток очищали с использованием пластины с силикагелем (проявляющий растворитель: гексан/этилацетат=1/1), получая 10-[3,17β-дигидроксиэстра-1,3,5(10)-триен-11β-ил]-2-(6,6,7,7,7-пентафторгептил)декановую кислоту (5,3 мг, выход 50%).
1H-ЯМР (270 МГц, СDСl3): δ 7,00 (д, J=8,3 Гц, 1Н), 6,62 (д, J=8,3 Гц, 1Н), 6,54 (с, 1Н), 3,73 (т, J=8,1 Гц, 1Н) 2,90-1,07 (м, 42Н), 0,92 (с, 3Н).
Масс-спектр (ESI): 653 (M+Na).
Значение Rf: 0,22 (силикагелевая пластина, проявляющий растворитель: гексан/этилацетат=1/1).
Пример 13
Синтез 11-(3,17β-дигидроксиэстра-1,3,5(10)-триен-7α-ил)-2-(4,4,5,5,5-пентафторпентил)ундекановой кислоты
(Стадия 1)
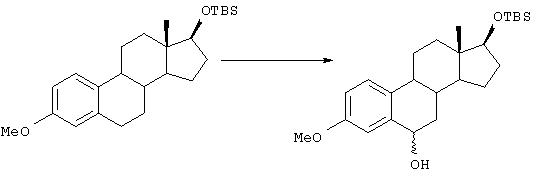
н-Бутиллитий (2,5 М в гексане, 40 мл, 100 ммоль) добавляли к трет-бутоксиду калия (1 М в тетрагидрофуране, 100 мл, 100 ммоль) при -70°С в атмосфере азота с последующим добавлением диизопропиламина (19,1 г, 100 ммоль) при той же температуре. После перемешивания смеси в течение 10 минут добавляли по каплям при -70°С в течение 10-15 минут 17β-(трет-бутилдиметилсилокси)-3-метоксиэстра-1,3,5(10)-триен (10 г, 25 ммоль), синтезированный способом, описанным в Tetrahedron Lett., 3223 (1988), и растворенный в тетрагидрофуране (40 мл), и полученную смесь перемешивали в течение 4 часов. К реакционной смеси добавляли триметилборат (15,6 г, 150 ммоль), затем нагревали до температуры льда, перемешивали в течение 1 часа и дополнительно перемешивали с 30% пероксидом водорода (35 мл) в течение 1 часа при комнатной температуре. Реакционную смесь опять охлаждали на льду и для того, чтобы остановить реакцию, добавляли 10%-ный водный раствор тиосульфата натрия (150 мл). Затем реакционную смесь экстрагировали простым эфиром, органический слой промывали водой и насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем фильтровали. После отгонки растворителя остаток очищали флеш-хроматографией на силикагеле (элюент: гексан/этилацетат=5/1 3/1), получая 17β-(трет-бутилдиметилсилокси)-3-метоксиэстра-1,3,5(10)-триен-6-ол (8,38 г, выход 82%).
1H-ЯМР (300 МГц, СDСl3): δ 7,18 (д, J=8,5 Гц, 1H, Cl-CH), 7,11 (д, J=2,7 Гц, 1H, С4-СН), 6,77 (дд, J=8,5, 2,7 Гц, 1H, С2-СН), 4,8 (м, 1H), 3,78 (с, 3Н, С3-ОСН3), 3,62 (м, 1H), 2,3-2,2 (м, 3Н), 2,0-1,8 (м, 2Н), 1,7-1,1 (м, 8Н), 0,87 (с, 9Н), 0,71 (с, 3Н, С18-СН3), 0,04 (с, 3Н), 0,03 (с, 3Н).
(Стадия 2)

17β-(трет-Бутилдиметилсилокси)-3-метоксиэстра-1,3,5(10)-триен-6-ол (14,4 г, 34,5 ммоль) растворяли в дихлорметане (250 мл). К полученному раствору добавляли диоксид марганца (29 г) и порошкообразные молекулярные сита 4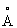 (7,2 г) с последующим перемешиванием в течение 1 часа при комнатной температуре. Реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Остаток перекристаллизовывали из гексана, получая 17β-(трет-бутилдиметилсилокси)-3-метоксиэстра-1,3,5(10)-триен-6-он (11,36 г, выход 79%).
(7,2 г) с последующим перемешиванием в течение 1 часа при комнатной температуре. Реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Остаток перекристаллизовывали из гексана, получая 17β-(трет-бутилдиметилсилокси)-3-метоксиэстра-1,3,5(10)-триен-6-он (11,36 г, выход 79%).
1H-ЯМР (300 МГц, СDСl3): δ 7,55 (д, J=3,0 Гц, 1H, С4-СН), 7,34 (д, J=8,5 Гц, 1H, Cl-CH), 7,10 (дд, J=8,5, 3,0 Гц, 1H, С2-СН), 3,84 (с, 3Н, С3-ОСН3), 3,66 (м, 1Н), 2,74 (дд, J=16,8, 3,3 Гц, 1Н), 2,5-2,3 (м, 2Н), 2,19 (дд, J=16,8, 13,2 Гц, 1Н), 2,0-1,8 (м, 3Н), 1,7-1,2 (м, 5Н), 0,89 (с, 9Н), 0,75 (с, 3Н, С18-СН3), 0,04 (с, 3Н), 0,03 (с, 3Н).
Т.пл. 159-160°С.
(Стадия 3)

Раствор 17β-(трет-бутилдиметилсилокси)-3-метоксиэстра-1,3,5(10)-триен-6-она (2,12 г, 5 ммоль) в безводном 1,2-диметоксиэтане (30 мл) охлаждали до -70°С в атмосфере азота и к полученному раствору добавляли гексаметилдисилазид калия в твердом виде (1,1 г, 5,5 ммоль) с последующим перемешиванием в течение 1 часа, поддерживая температуру при -70°С. Затем добавляли перегнанный иодистый аллил (1,68 г, 10 ммоль) при -70°С с использованием шприца и реакционную смесь нагревали до 0°С в течение 1 часа. Часом позже к реакционной смеси добавляли воду при 0°С, затем экстрагировали простым эфиром. Органический слой промывали водой и насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем фильтровали. После отгонки растворителя остаток очищали флеш-хроматографией на силикагеле (элюент: гексан/этилацетат=20/1 15/1), получая 17β-(трет-бутилдиметилсилокси)-3-метокси-7αβ-(2-пропенил) эстра-1,3,5(10)-триен-6-он (2,0 г, выход 88%) (соотношение 7α/7β примерно 1/6).
Указанную выше смесь растворяли в растворе метоксида натрия (0,1 М) и нагревали при кипении с обратным холодильником в течение 2 часов, получая смесь, имеющую соотношение 7α/7β, равное примерно 7/1.
1H-ЯМР (300 МГц, СDСl3, спектр 7α-замещенного соединения): δ 7,53 (д, 1Н, J=2,8 Гц, C4-CH), 7,32 (д, 1Н, J=8,5 Гц, Cl-CH), 7,09 (дд, J=8,5, 2,8Гц, 1Н, С2-СН), 5,87-5,72 (м, 1Н), 5,02-4,9 (м, 2Н), 3,84 (с, 3Н, С3-ОСН3), 3,68 (м, 1Н), 2,77-2,64 (м, 1Н), 2,6-2,54 (м, 1Н), 2,4-2,3 (м, 2Н), 2,22-2,06 (м, 2Н), 2,0-1,85 (м, 2Н), 1,65-1,2 (м, 6Н), 0,9 (с, 9Н), 0,75 (с, 3Н, С18-СН3), 0,05 (с, 3Н), 0,03 (с, 3Н).
(Стадия 4)

17β-(трет-Бутилдиметилсилокси)-3-метокси-7αβ-(2-пропенил)эстра-1,3,5(10)-триен-6-он (5,78 г, 12,7 ммоль) растворяли в тетрагидрофуране (30 мл). К данному раствору добавляли фторид тетра-н-бутиламмония (1 М в тетрагидрофуране, 60 мл) и полученную смесь нагревали при кипении с обратным холодильником в течение 4 часов в атмосфере азота. После охлаждения к реакционной смеси добавляли воду, затем экстрагировали простым эфиром. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем фильтровали. Фильтрат концентрировали при пониженном давлении, получая 17β-гидрокси-3-метокси-7αβ-(2-пропенил)эстра-1,3,5(10)-триен-6-он в виде смеси диастереомеров, которую затем перекристаллизовывали из диизопропилового эфира (70 мл), получая 17β-гидрокси-3-метокси-7α-(2-пропенил)эстра-1,3,5(10)-триен-6-он (2,61 г, выход 60%) в виде отдельного изомера.
1H-ЯМР (270 МГц, СDСl3,): δ 7,53 (д, J=3,0 Гц, 1H, C4-CH), 7,32 (д, J=8,5 Гц, 1H, Cl-CH), 7,09 (дд, J=8,5, 3,0 Гц, 1H, С2-СН), 5,84-5,74 (м, 1H), 5,01-4,92 (м, 2Н, олефиновый Н), 3,84 (с, 3Н, С3-ОСН3), 3,84-3,75 (м, 1H, С17-СН), 2,80-2,68 (м, 1H, С9-СН), 2,63-2,52 (м, 1H, С7-СН), 2,51-2,38 (м, 2Н, аллил-СН2 и C11-CH2), 2,24-2,05 (м, 3Н, аллил-СН2 и С8-СН и С16-СН2), 2,02-1,91 (м, 1H, C11-CH2), 1,71-1,33 (м, 7Н), 0,79 (с, 3Н, С18-СН3).
Т.пл. 122-123°С.
(Стадия 5)
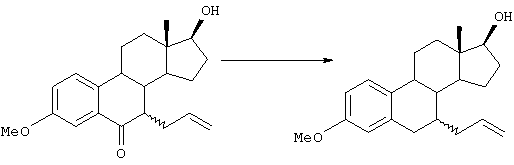
Триэтилсилан (5 мл) и диэтилэфират трехфтористого бора (5 мл) добавляли к раствору 17β-гидрокси-3-метокси-7α-(2-пропенил)эстра-1,3,5(10)-триен-6-она (1,0 г, 2,9 ммоль) в дихлорметане (20 мл) при 0°С. Полученную смесь нагревали до комнатной температуры и перемешивали в течение 18 часов. После завершения реакции к реакционной смеси добавляли 10%-ный водный карбонат калия и затем экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем фильтровали. После концентрирования при пониженном давлении полученный остаток очищали колоночной хроматографией на силикагеле (элюент: гексан/этилацетат=2/1), получая 3-метокси-7α-(2-пропенил)эстра-1,3,5(10)-триен-17β-ол (935 мг, выход 98%).
1H-ЯМР (300 МГц, СDСl3,): δ 7,20 (д, J=8,6 Гц, Cl-1H), 6,71 (дд, J=8,6, 2,4 Гц, C2-1H), 6,60 (д, J=2,4 Гц, C4-1H), 5,86-5,72 (м, 1H), 5,00-4,90 (м, 2Н, олефин Н), 3,77 (с, 3Н, С3-ОСН3), 3,77-3,71 (м, 1H, С17-СН), 2,80-2,68 (м, 1H, С9-СН), 2,63-2,52 (м, 1H, С7-СН), 2,51-2,38 (м, 2Н, аллил-CH2 и C11-CH2), 2,24-2,05 (м, 3Н, аллил-СН2 и С8-СН и С16-СН2), 2,02-1,91 (м, 1H, C11-CH2), 1,71-1,33 (м, 7Н), 0,79 (с, 3Н, С18-СН3).
(Стадия 6)

Бензилиденбис(трициклогексилфосфин)дихлоррутений (98 мг, 0,11 ммоль) добавляли к раствору 3-метокси-7α-(2-пропенил)эстра-1,3,5(10)-триен-17β-ола (723 мг, 2,21 ммоль) и этил-2-(4,4,5,5,5-пентафторпентил)-9-деценоата (1,59 г, 4,43 ммоль) в дихлорметане (20 мл) с последующим нагреванием при кипении с обратным холодильником в течение 20 часов в атмосфере аргона. После охлаждения реакционную смесь концентрировали при пониженном давлении и полученный остаток очищали флеш-хроматографией на силикагеле (элюент: гексан/этилацетат=4/1), получая этил-11-[17β-гидрокси-3-метоксиэстра-1,3,5(10)-триен-7α-ил]-2-(4,4,5,5,5-пентафторпентил)-9-ундеценоат (973 мг, выход 67%).
1H-ЯМР (270 МГц, СDСl3,): δ 7,20 (д, J=8,6 Гц, 1H, Cl-CH), 6,76-6,69 (м, 1H, C2-CH), 6,63-6,58 (м, 1H, С4-СН), 5,42-5,27 (м, 2Н, олефин-Н), 4,15 (кв, J=7,1 Гц, 2H, СОО-СН2), 3,78-3,70 (м, 4Н, С17-СН и С3-ОСН3), 2,90-2,63 (м, 2H), 2,41-2,22 (м, 3Н), 2,20-1,16 (м, 34Н), 0,78 (с, 3Н, С18-СН3).
(Стадия 7)

Этил-11-[17β-гидрокси-3-метоксиэстра-1,3,5(10)-триен-7α-ил]-2-(4,4,5,5,5-пентафторпентил)-9-ундеценоат (970 мг, 1,48 ммоль) растворяли в этилацетате (30 мл) и к полученному раствору добавляли 10%-ный палладий-на-углероде (300 мг) с последующим перемешиванием в течение 4 часов при комнатной температуре в атмосфере водорода. Реакционную смесь фильтровали и концентрировали, получая этил 11-[17β-гидрокси-3-метоксиэстра-1,3,5(10)-триен-7α-ил]-2-(4,4,5,5,5-пентафторпентил)ундеканоат (946 мг, выход 97%) в виде масла.
1H-ЯМР (270 МГц, СDСl3,): δ 7,20 (д, J=8,6 Гц, 1H, Cl-CH), 6,76-6,69 (м, 1H, С2-СН), 6,63-6,58 (м, 1H, С4-СН), 4,14 (кв, J=7,1 Гц, 2H, COO-CH2), 3,77 (с, 3Н, С3-ОСН3), 3,74 (т, J=8,4 Гц, 1H, С17-СН), 2,94-2,70 (м, 2H), 2,40-2,24 (м, 3Н), 2,20-1,84 (м, 4Н), 1,80-0,96 (м, 34Н), 0,78 (с, 3Н, С18-СН3).
(Стадия 8)
Этил-11-[17β-гидрокси-3-метоксиэстра-1,3,5(10)-триен-7α-ил]-2-(4,4,5,5,5-пентафторпентил)ундеканоат (946 мг, 1,43 ммоль) растворяли в дихлорметане (18 мл). К данному раствору добавляли по каплям при -78°С 1 М раствор трехбромистого бора в дихлорметане (4,3 мл, 4,3 ммоль). Реакционную смесь медленно нагревали и перемешивали в течение 3 часов при 0°С. Для того чтобы остановить реакцию, добавляли воду и реакционную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем фильтровали. После концентрирования при пониженном давлении полученный остаток очищали колоночной хроматографией на силикагеле (элюент: гексан/этилацетат=2/1, получая этил-11-[3,17β-дигидроксиэстра-1,3,5(10)-триен-7α-ил]-2-(4,4,5,5,5-пентафторпентил)ундеканоат (663 мг, выход 72%).
1H-ЯМР (270 МГц, СDСl3,): δ 7,14 (д, J=8,4 Гц, 1H, Cl-CH), 6,66-6,59 (м, 1H, C2-CH), 6,57-6,53 (м, 1Н, С4-СН), 5,10 (ушир.с, 1H, С3-ОН), 4,15 (кв, J=7,1 Гц, 2H, СОО-СН3), 3,75 (т, J=8,4 Гц, 1H, С17-СН), 2,94-2,68 (м, 2H), 2,40-2,22 (м, 3Н), 2,20-1,84 (м, 4Н), 1,80-0,96 (м, 34Н), 0,78 (с, 3Н, С18-СН3).
(Стадия 9)
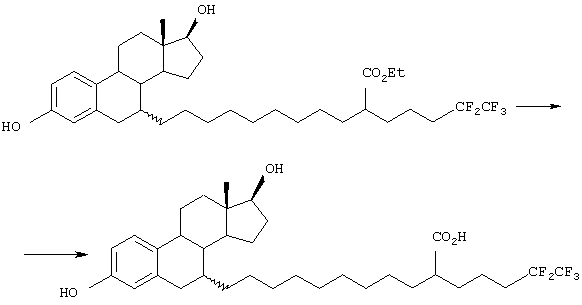
Этил-11-[3,17β-дигидроксиэстра-1,3,5(10)-триен-7α-ил]-2-(4,4,5,5,5-пентафторпентил)ундеканоат (660 мг, 1,03 ммоль) растворяли в смешанном растворителе, состоящем из этанола (10 мл) и воды (2,0 мл). К данному раствору добавляли NaOH (820 мг, 20,5 ммоль) и полученную смесь нагревали в течение 4 часов при 60°С. После охлаждения к реакционной смеси добавляли 2 н. водную хлористоводородную кислоту, затем экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем фильтровали. После концентрирования при пониженном давлении полученный остаток очищали колоночной хроматографией на силикагеле (элюент: гексан/этилацетат=1/1), получая 11-[3,17β-дигидроксиэстра-1,3,5(10)-триен-7α-ил]-2-(4,4,5,5,5-пентафторпентил)ундекановую кислоту (613 мг, выход 97%).
1H-ЯМР (270 МГц, СD3OD): δ 7,07 (д, J=8,4 Гц, 1H, Cl-CH), 6,54 (дд, J=8,4, 2,3 Гц, 1H, C2-CH), 6,45 (д, J=2,3 Гц, 1H, С4-СН), 3,67 (т, J=8,3 Гц, 1H, C17-CH), 2,85-2,62 (м, 2Н), 2,39-1,84 (м, 7Н), 1,80-0,96 (м, 33Н), 0,78 (с, 3Н, С18-СН3).
Пример 14
Синтез 10-[3,17β-дигидроксиэстра-1,3,5(10)-триен-7α-ил]-2-(4,4,5,5,5-пентафторпентил)декановой кислоты
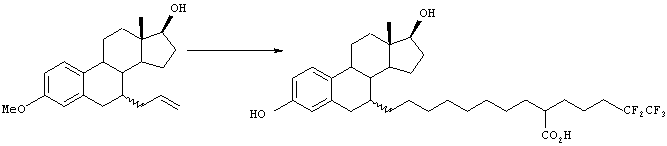
Исходя из 3-метокси-7α-(2-пропенил)эстра-1,3,5(10)-триен-17β-ола, полученного в примере 13, и этил-2-(4,4,5,5,5-пентафторпентил)-8-ноненоата, полученного отдельно, повторяли ту же методику, как описано в примере 13, получая 10-[3,17β-дигидроксиэстра-1,3,5(10)-триен-7α-ил]-2-(4,4,5,5,5-пентафторпентил)декановую кислоту.
1H-ЯМР (300 МГц, СDСl3,): δ 7,16 (д, J=8,4 Гц, 1H, Cl-CH), 6,63 (дд, J=8,4 Гц, J=2,7 Гц, 1H, C2-CH), 6,55 (с, 1H, С4-СН), 3,75 (т, J=8,5 Гц, 1H, C17-CH), 3,60-3,35 (ушир.с, 1H, С3-ОН), 2,82-2,73 (м, 2Н), 2,45-2,23 (м, 4Н), 2,20-0,96 (м, 31Н), 0,78 (с, 3Н, С18-СН3).
Пример 15
Синтез 10-[3,17β-дигидроксиэстра-1,3,5(10)-триен-7α-ил]-2-(4,4,5,5,6,6,7,7,7-нонафторгептил)декановой кислоты

Исходя из 3-метокси-7α-(2-пропенил)эстра-1,3,5(10)-триен-17β-ола, полученного в примере 13, и этил-2-(4,4,5,5,6,6,7,7,7-нонафторгептил)-8-ноненоата, полученного отдельно, повторяли ту же методику, как описано в примере 13, получая 10-[3,17β-дигидроксиэстра-1,3,5(10)-триен-7α-ил]-2-(4,4,5,5,6,6,7,7,7-нонафторгептил)декановую кислоту.
1H-ЯМР (300 МГц, СDСl3,): δ 7,12 (д, J=8,4 Гц, 1H), 6,68 (дд, J=8,5, 2,4 Гц, 1H), 6,53 (д, J=2,6 Гц, 1H), 3,68 (т, J=8,5 Гц, 1H), 2,95-2,60 (м, 2Н), 2,47-2,19 (м, 4Н), 2,18-1,03 (м, 31Н), 0,69 (с, 3Н).
Пример 16
Синтез 10-[3,17β-дигидроксиэстра-1,3,5(10)-триен-7α-ил]-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)декановой кислоты

Исходя из 3-метокси-7α-(2-пропенил)эстра-1,3,5(10)-триен-17β-ола, полученного в примере 13, и этил-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)-8-ноненоата, полученного отдельно, повторяли ту же методику, как описано в примере 13, получая 10-[3,17β-дигидроксиэстра-1,3,5(10)-триен-7α-ил]-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)декановую кислоту.
1H-ЯМР (300 МГц, СDСl3): δ 7,14 (д, J=8,5 Гц, 1H), 6,62 (дд, J=8,2, 2,1 Гц, 1H), 6,54 (д, J=2,6 Гц, 1H), 3,77 (т, J=8,1 Гц, 1H), 2,84-2,56 (м, 2Н), 2,44 (м, 1H), 2,31 (м, 2Н), 2,18-1,02 (м, 30Н), 0,74 (с, 3Н).
Пример 17
Синтез 10-[3,17β-дигидроксиэстра-1,3,5(10)-триен-7α-ил]-2-(6,6,7,7,7-пентафторгептил)декановой кислоты
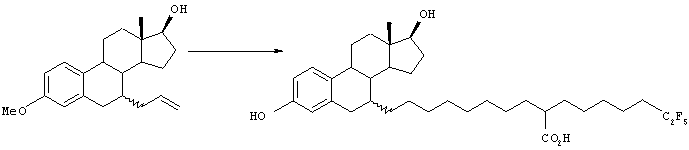
Исходя из 3-метокси-7α-(2-пропенил)эстра-1,3,5(10)-триен-17β-ола, полученного в примере 13, и этил-2-(6,6,7,7,7-пентафторгептил)-8-ноненоата, полученного отдельно, повторяли ту же методику, как описано в примере 13, получая 10-[3,17β-дигидроксиэстра-1,3,5(10)-триен-7α-ил]-2-(6,6,7,7,7-пентафторгептил)декановую кислоту.
1H-ЯМР (300 МГц, СDСl3): δ 7,05 (д, J=8,4 Гц, 1H), 6,65 (дд, J=8,6, 2,6Гц, 1H), 6,52 (д, J=2,5 Гц, 1H), 3,75 (т, J=8,4 Гц, 1H), 2,92-2,62 (м, 2Н), 2,33-2,12 (м, 4Н), 2,09-1,03 (м, 35Н), 0,75 (с, 3Н).
Пример 18
Синтез 11-[3,17β-дигидроксиэстра-1,3,5(10)-триен-7α-ил]-2-(4,4,5,5,6,6,7,7,7-нонафторгептил)ундекановой кислоты

Исходя из 3-метокси-7-(2-пропенил)эстра-1,3,5(10)-триен-17β-ола, полученного в примере 13, и этил-2-(4,4,5,5,6,6,7,7,7-нонафторгептил)-9-деценоата, полученного отдельно, повторяли ту же методику, как показано в примере 13, получая 11-[3,17β-дигидроксиэстра-1,3,5(10)-триен-7α-ил]-2-(4,4,5,5,6,6,7,7,7-нонафторгептил)ундекановую кислоту.
1H-ЯМР (300 МГц, СDСl3,): δ 7,13 (д, J=8,4 Гц, 1H, Cl-CH), 6,62 (дд, J=8,4, 2,3 Гц, 1H, C2-CH), 6,53 (д, J=2,3 Гц, 1H, C4-CH), 3,75 (т, J=8,1 и 8,4 Гц, 1H, С17-СН), 2,85 (дд, J=4,8 и 16,7Гц, 1H), 2,70(м, 1H), 2,39-1,88 (м, 7Н), 1,80-0,96 (м, 33Н), 0,78 (с,3Н, С18-СН3).
Пример 19
Синтез 11-[3,17β-дигидроксиэстра-1,3,5(10)-триен-7α-ил]-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)ундекановой кислоты

Исходя из 3-метокси-7α-(2-пропенил)эстра-1,3,5(10)-триен-17β-ола, полученного в примере 13, и этил-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)-9-деценоата, полученного отдельно, повторяли ту же методику, как показано в примере 13, получая 11-[3,17β-дигидроксиэстра-1,3,5(10)-триен-7α-ил]-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)ундекановую кислоту.
1H-ЯМР (270 МГц, СD3OD): δ 7,07 (д, J=8,4 Гц, 1H, Cl-CH), 6,53 (дд, J=8,4, 2,2 Гц, 1H, C2-CH), 6,45 (д, J=2,2 Гц, 1H, C4-CH), 3,67 (т, J=8,1 Гц, 1H, C17-CH), 2,87-2,62 (м, 2Н), 2,43-0,95 (м, 35Н), 0,78 (с,3Н, С18-СН3).
Пример 20
Синтез 11-(3,17β-дигидроксиэстра-1,3,5(10)-триен-11β-ил)-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)ундекановой кислоты
(Стадия 1)

Диметилформамид (11 мл) добавляли к β-эстрадиолу (1,08 г, 4,0 ммоль) в атмосфере азота с последующим охлаждением на льду. К реакционной смеси добавляли гидрид натрия (480 мг 60%-ной суспензии) с последующим перемешиванием в течение 10 минут на льду и затем перемешиванием в течение 1 часа при комнатной температуре. После повторного охлаждения на льду к реакционной смеси добавляли бромистый бензил (2,05 г, 12 ммоль) с последующим перемешиванием в течение 10 минут на льду и затем перемешиванием в течение 20 часов при комнатной температуре. Реакционную смесь гасили охлажденной льдом водой и экстрагировали этилацетатом. Органический слой промывали водой и насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем фильтровали. Фильтрат концентрировали при пониженном давлении с использованием роторного испарителя и полученный остаток растирали с метанолом для осаждения твердых веществ. Твердые вещества собирали фильтрованием и сушили в вакууме, получая 3,17β-бис(бензилокси)эстра-1,3,5(10)-триен (1,72 г, выход 95%).
1H-ЯМР (300 МГц, СDСl3,): δ 7,45-7,20 (м, 11Н), 6,79 (дд, J=9,0 и 3,0 Гц, 1Н, С3-СН), 6,72 (д, J=2,7 Гц, 1H, С4-СН), 5,04 (с, 2Н), 4,56 (С, 2Н), 3,51 (т, J=8,1, 1H), 2,88-2,83 (м, 2Н), 2,36-1,15 (м, 13Н), 0,88 (с, 3Н, С18-СН3).
(Стадия 2)
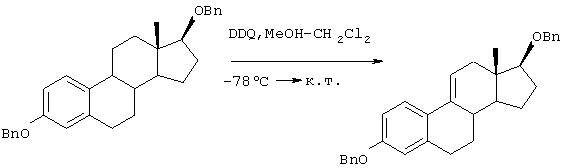
К 3,17β-бис(бензилокси)эстра-1,3,5(10)-триену (45,2 мг, 0,1 ммоль) в атмосфере азота добавляли дихлорметан (0,2 мл) и метанол (0,1 мл) с последующим охлаждением до -78°С. К реакционной смеси добавляли 2,3-дихлор-5,6-дицианобензохинон (22,7 мг, 0,1 ммоль) с последующим перемешиванием в течение 10 минут при -78°С и затем перемешиванием в течение 3 часов при комнатной температуре. Реакционную смесь гасили насыщенным водным раствором бикарбоната натрия и экстрагировали этилацетатом. Органический слой промывали водой и насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем фильтровали. Фильтрат концентрировали при пониженном давлении с использованием роторного испарителя и полученный остаток очищали колоночной хроматографией на силикагеле (Merck Kieselgel 60, элюент: гексан/этилацетат=10/1 → 8/1), получая целевое соединение - 3,17β-бис(бензилокси)эстра-1,3,5(10),9(11)-тетраен (35,7 мг, выход 80%).
1H-ЯМР (300 МГц, СDСl3,): δ 7,54 (д, J=9,0 Гц, 1Н, Cl-CH), 7,27-7,45 (м, 10Н), 6,80 (дд, J=8,7 и 2,7 Гц, 1Н, С3-СН), 6,69 (д, J=2,4 Гц, 1Н, С4-СН), 6,12 (м, 1Н), 5,05 (с, 2Н), 4,56 (с, 2Н), 4,22 (м, 1Н), 3,60 (т, J=8,7 Гц, 1Н), 2,97-2,70 (м, 2Н), 2,43-1,09 (м, 9Н), 0,87 (с, 3Н, С18-СН3).
(Стадия 3)

Катехолборан (1,0 М в тетрагидрофуране, 66,33 мл) добавляли к 3,17β-бис(бензилокси)эстра-1,3,5(10),9(11)-тетраену (27,17 г, 60,30 ммоль) при комнатной температуре в атмосфере азота. Далее к реакционной смеси при комнатной температуре добавляли боргидрид лития (1,31 г, 60,30 ммоль) с последующим перемешиванием в течение 10 часов. При охлаждении льдом данную смесь прибавляли к смеси гидроксида натрия (24,12 г, 603,0 ммоль), воды (70 мл), этанола (150 мл) и 30% пероксида водорода (150 мл) с последующим перемешиванием в течение 1 часа и 45 минут на льду, а затем перемешивали в течение 3 часов при комнатной температуре. К реакционной смеси добавляли простой эфир и воду, затем экстрагировали простым эфиром. Органический слой промывали последовательно 10%-ным водным раствором гидроксида натрия, водой и насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем фильтровали. Фильтрат концентрировали при пониженном давлении с использованием роторного испарителя. Остаток очищали колоночной хроматографией на силикагеле (Merck Kieselgel 60, элюент: гексан/этилацетат=6/1 5,5/1), получая целевое соединение -3,17β-бис(бензилокси)эстра-1,3,5(10)-триен-11α-ол (21,56 г, выход 76%).
1H-ЯМР (300 МГц, СDСl3,): δ 7,87 (д, J=8,4 Гц, 1H, Cl-СН), 7,27-7,45 (м, 10Н), 6,81 (дд, J=8,7 и 2,7 Гц, 1H, С3-СН), 6,74 (д, J=2,7Гц, 1H, С4-СН), 5,05 (с, 2Н), 4,58 (с, 2Н), 4,22 (м, 1H), 3,53 (т, J=8,1, 1H), 2,82 (м, 2Н), 2,43 (дд, J=12,2 и 5,4 Гц, 1H), 2,18-2,02 (м, 2Н), 1,91-1,84 (м, 1H), 1,74-1,59 (м, 2Н), 1,49-1,27 (м, 5Н), 0,86 (с, 3Н, С18-СН3).
(Стадия 4)

Дихлорметан (92,25 мл) добавляли к оксалилдихлориду (3,54 мл, 40,59 ммоль) в атмосфере азота и полученную смесь охлаждали до -78°С. К смеси добавляли по каплям раствор диметилсульфоксида (5,76 мл, 81,18 ммоль), разбавленного дихлорметаном (18,45 мл). Затем смесь перемешивали в течение 2 минут при -78°С, добавляли по каплям раствор 3,17β-бис(бензилокси)эстра-1,3,5(10)-триен-11α-ола (17,29 г, 36,90 ммоль) в дихлорметане (36,90 мл) с последующим перемешиванием в течение 15 минут при -78°С. К реакционной смеси по каплям добавляли триэтиламин (25,7 мл, 184,5 ммоль), затем перемешивали в течение 5 минут при -78°С и нагревали до комнатной температуры. Реакционную смесь опять охлаждали на льду, гасили добавлением льда и воды и затем экстрагировали дихлорметаном. Органический слой промывали водой и насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем фильтровали. Фильтрат концентрировали при пониженном давлении с использованием роторного испарителя, получая неочищенный 3,17β-бис(бензилокси)эстра-1,3,5(10)-триен-11-он (неочищенного 18,76 г, количественный для сырого продукта 1H-ЯMP].
1H-ЯМР (300 МГц, СDСl3,): δ 7,41-7,20 (м, 11Н), 6,82 (дд, J=8,4 и 2,7 Гц, 1Н, С3-СН), 6,70 (д, J=2,7 Гц, 1Н, С4-СН), 5,03 (с, 2Н), 4,54 (с, 2Н), 3,72 (т, J=8,1 Гц, 1Н), 3,46 (д, J=10,2 Гц, 1Н), 2,84-2,66 (м, 3Н), 2,47 (д, J=11,4 Гц, 1H), 2,23-2,13 (м, 1H), 1,96-1,48 (м, 8Н), 0,86 (с, 3Н, С18-СН3).
(Стадия 5)

Тетрагидрофуран (280 мл) добавляли к сырому 3,17β-бис(бензилокси)эстра-1,3,5(10)-триен-11-ону (35,7 г, 76,5 ммоль) в атмосфере азота и полученную смесь охлаждали до -40°С. К смеси по каплям добавляли аллилмагнийхлорид (2,0 М в тетрагидрофуране, 50,0 мл, 100 ммоль), затем перемешивали в течение 10 минут при -40°С и в течение 1 часа при комнатной температуре. Реакционную смесь опять охлаждали на льду и гасили добавлением льда, воды и насыщенного водного раствора хлорида аммония. Затем реакционную смесь экстрагировали этилацетатом, органический слой промывали водой и насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем фильтровали. Фильтрат концентрировали при пониженном давлении с использованием роторного испарителя и к полученному остатку добавляли гексан для осаждения твердых веществ. Твердые вещества собирали фильтрованием и сушили в вакууме, получая 3,17β-бис(бензилокси)-11α-(2-пропенил)эстра-1,3,5(10)-триен-11β-ол (35,81 г, выход 90% по 3,17β-бис(бензилокси)эстра-1,3,5(10)-триен-11α-олу).
1H-ЯМР (300 МГц, СDСl3,): δ 7,82 (д, J=9,6 Гц, 1Н, Сl-Н), 7,44-7,33 (м, 10Н), 6,83-6,79 (м, 2Н, С2 и С4-Н), 6,01-5,87 (м, 1Н, олефиновый Н), 5,21-5,13 (м, 2Н, олефиновый Н), 5,06 (с, 2Н), 4,57 (с, 2Н), 3,46 (т, J=8,7 Гц, 1Н, С17-Н), 2,90 (дд, J=14,0 и 8,4 Гц, 1Н, аллильный СН2), 2,74-2,63 (м, 2Н), 2,52 (дд, J=14,1 и 7,0 Гц, 1Н, аллильный СН2), 2,25 (д, J=10,8 Гц, 1Н), 2,13 (д, J=14,1 Гц, 1Н), 2,06-1,98 (м, 1Н), 1,88-1,14 (м, 9Н), 1,09 (с, 3Н, С18-Н).
(Стадия 6)
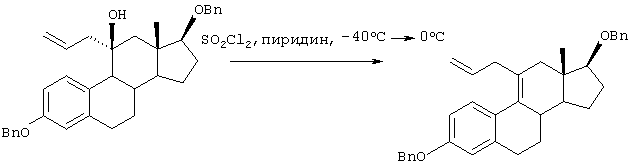
После охлаждения раствора 3,17β-бис(бензилокси)-11α-(2-пропенил)эстра-1,3,5(10)-триен-11β-ола (17,20 г, 33,80 ммоль) в пиридине (135 мл) до -40°С в атмосфере азота добавляли по каплям хлористый тионил (3,7 мл, 50,7 ммоль), затем перемешивали в течение 10 минут при -40°С и в течение 1 часа на льду. Реакционную смесь гасили добавлением льда и воды и затем экстрагировали смешанным растворителем, состоящим из гексана и трет-бутилметилового простого эфира (гексан/трет-бутилметиловый простой эфир=4/1). Органический слой промывали водой и насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем фильтровали. Фильтрат концентрировали при пониженном давлении с использованием роторного испарителя и полученный остаток растирали с метанолом для осаждения твердых веществ. Твердые вещества собирали фильтрованием и сушили в вакууме, получая 3,17β-бис(бензилокси)-11-(2-пропенил)эстра-1,3,5(10),9(11)-тетраен (14,92 г, выход 90%).
1H-ЯМР (300 МГц, СDСl3,): δ 7,46-7,25 (м, 11Н), 6,78-6,75 (м, 2Н, С2 и С4-Н), 6,01-5,89 (м, 1Н, олефиновый Н), 5,18-5,09 (м, 2Н, олефиновый Н), 5,06 (с, 2Н), 4,58 (с, 2Н), 3,58 (т, J=8,4 Гц, 1Н, С17-Н), 3,30 (дд, J=15,8 и 5,1 Гц, 1Н, аллильный СН2), 2,81-2,72 (м, 3Н), 2,48 (д, J=17,4 Гц, 1Н, аллильный СН2), 2,16-1,36 (м, 9Н), 0,90 (с, 3Н, С18-Н).
(Стадия 7)

Бензилиденбис(трициклогексилфосфин)дихлоррутений (90 мг, 0,1 ммоль) добавляли к раствору 3,17β-бис(бензилокси)-11-(2-пропенил)эстра-1,3,5(10),9(11)-тетраена (1,0 г, 2,0 ммоль) и этил-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)-9-деценоата (1,81 г, 4,1 ммоль) в дихлорметане (20 мл) с последующим нагреванием при кипении с обратным холодильником в течение 5 часов в атмосфере азота. После охлаждения реакционную смесь концентрировали при пониженном давлении и полученный остаток очищали флеш-хроматографией на силикагеле (элюент: гексан/этилацетат=4/1), получая этил-11-[3,17β-бис(бензилокси)эстра-1,3,5(10),9(11)-тетраен-11-ил]-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)-9-ундеценоат (863 мг, выход 48%).
1H-ЯМР (270 МГц, СDСl3,): δ 7,46-7,21 (м, 11Н), 6,78-6,73 (м, 2Н), 5,50-5,42 (м, 2Н), 5,05 (с, 2Н), 4,57 (д, J=2,5 Гц, 2Н), 4,15 (кв, J=7,1 Гц, 2Н, СОО-СН2), 3,60-3,52 (м, 1Н, С17-СН), 3,20-3,11 (м, 1Н), 2,82-2,68 (м, 3Н), 2,51-2,22 (м, 2Н), 2,20-1,16 (м, 28Н), 0,87 (с, 3Н, С18-СН3).
(Стадия 8)
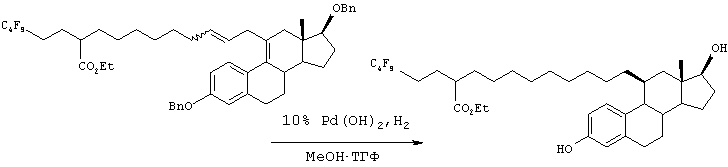
Этил-11-[3,17β-бис(бензилокси)эстра-1,3,5(10),9(11)-тетраен-11-ил]-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)-9-ундеценоат (500 мг, 0,55 ммоль) растворяли в смешанном растворителе, состоящем из метанола (10 мл) и тетрагидрофурана (1 мл) с последующим добавлением гидроксида палладия-на-углероде (150 мг) при комнатной температуре. После продувания водородом реакционную смесь перемешивали в течение 23 часов при комнатной температуре и затем фильтровали. Растворитель концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат=4/1 3/1 2/1), получая этил-11-(3,17β-дигидроксиэстра-1,3,5(10)-триен-11β-ил)-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)ундеканоат (287 мг, выход 71%).
1H-ЯМР (270 МГц, СDСl3,): δ 7,00 (д, J=8,6 Гц, 1H, Cl-CH), 6,62 (д, J=8,5 Гц, 1H, C2-CH), 6,54 (д, J=2,3 Гц, 1H, C4-CH), 5,03 (ушир.с, 1H, С3-ОН), 4,17 (кв, J=7,1 Гц, 2H, СОО-СН2), 3,70 (т, J=7,9 Гц, 1H, С17-СН), 2,83-2,60 (м, 2H), 2,58-2,30 (м, 3Н), 2,24-1,74 (м, 7Н), 1,74-1,11 (м, 29Н), 0,92 (с, 3Н, С18-СН3).
(Стадия 9)
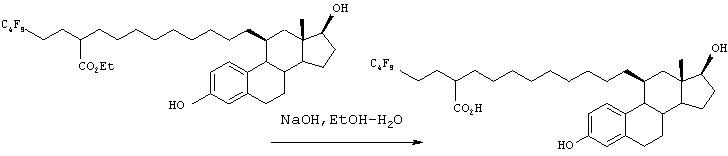
Этил-11-(3,17β-дигидроксиэстра-1,3,5(10)-триен-11β-ил)-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)ундеканоат (747 мг, 1,02 ммоль) растворяли в смешанном растворителе, состоящем из этанола (5 мл) и воды (5 мл). К данному раствору добавляли NaOH (82 мг, 2,04 ммоль) и полученную смесь нагревали в течение 15 часов при 60°С. После охлаждения к реакционной смеси добавляли 2 н. водную хлористоводородную кислоту, затем экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем фильтровали. После концентрирования при пониженном давлении полученный остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат=4/1 3/1 2/1), получая 11-(3,17β-дигидроксиэстра-1,3,5(10)-триен-11β-ил)-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)ундекановую кислоту (600 мг, выход 84%).
1H-ЯMP (270 МГц, CD3OD): δ 6,95 (д, J=8,3 Гц, 1H, Cl-CH), 6,55 (дд, J=8,3, 2,3Гц, 1H, C2-CH), 6,47 (д, J=2,3 Гц, 1H, C4-CH), 3,63 (т, J=8,6 Гц, 1H, С17-СН), 2,85-2,58 (м, 2Н), 2,55-2,34 (м, 3Н), 2,30-1,93 (м, 2Н), 1,91-1,75 (м, 3Н), 1,75-1,10 (м, 26Н), 0,92 (с, 3Н, С18-СН3).
Пример 21
Синтез 10-(3,17β-дигидроксиэстра-1,3,5(10)-триен-11β-ил)-2-(4,4,5,5,5-пентафторпентил)декановой кислоты

Исходя из 3,17β-бис(бензилокси)-11-(2-пропенил)эстра-1,3,5(10),9(11)-тетраена, полученного в примере 20, и этил-2-(4,4,5,5,5-пентафторпентил)-8-ноненоата, полученного отдельно, повторяли ту же методику, как описано в примере 20, получая 10-(3,17β-дигидроксиэстра-1,3,5(10)-триен-11β-ил)-2-(4,4,5,5,5-пентафторпентил)декановую кислоту.
1H-ЯМР (300 МГц, СDСl3,): δ 6,99 (д, J=8,7 Гц, 1H, Cl-CH), 6,62 (д, J=8,2 Гц, 1H, С2-СН), 6,55 (д, J=2,3 Гц, 1H, C4-CH), 4,87-3,82 (ушир.с, 1H, С3-ОН), 3,73 (т, J=7,4 Гц, 1H, С17-СН), 2,84-2,65 (м, 2Н), 2,51 (м, 1H), 2,39 (м, 2Н), 2,24-1,12 (м, 32Н), 0,91 (с, 3Н, С18-СН3).
Пример 22
Синтез 10-(3,17β-дигидроксиэстра-1,3,5(10)-триен-11β-ил)-2-(4,4,5,5,6,6,7,7,7-нонафторгептил)декановой кислоты

Исходя из 3,17β-бис(бензилокси)-11-(2-пропенил)эстра-1,3,5(10),9(11)-тетраена, полученного в примере 20, и этил-2-(4,4,5,5,6,6,7,7,7-нонафторгептил)-8-ноненоата, полученного отдельно, повторяли ту же методику, как описано в примере 20, получая 10-(3,17β-дигидроксиэстра-1,3,5(10)-триен-11β-ил)-2-(4,4,5,5,6,6,7,7,7-нонафторгептил)декановую кислоту.
1H-ЯМР (300 МГц, СDСl3,): δ 7,11 (д, J=8,3 Гц, 1H, Cl-CH), 6,62 (д, J=8,6 Гц, 1H, С2-СН), 6,54 (д, J=2,7 Гц, 1H, C4-CH), 3,94-3,07 (ушир.с, 1H, С3-ОН), 3,72 (т, J=7,4 Гц, 1H, С17-СН), 2,82-2,62 (м, 2Н), 2,52 (м, 1H), 2,40 (м, 2Н), 2,23-1,11 (м, 32Н), 0,91 (с, 3Н, С18-СН3).
Пример 23
Синтез 10-(3,17β-дигидроксиэстра-1,3,5(10)-триен-11β-ил)-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)декановой кислоты

Исходя из 3,17β-бис(бензилокси)-11-(2-пропенил)эстра-1,3,5(10),9(11)-тетраена, полученного в примере 20, и этил-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)-8-ноненоата, полученного отдельно, повторяли ту же методику, как описано в примере 20, получая 10-(3,17β-дигидроксиэстра-1,3,5(10)-триен-11β-ил)-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)декановую кислоту.
1H-ЯМР (300 МГц, СDСl3,): δ 7,07 (д, J=8,5 Гц, 1H), 6,65 (дд, J=8,2, 2,1 Гц, 1H), 6,47 (д, J=2,6 Гц, 1H), 3,82 (т, J=8,2 Гц, 1H), 2,91-2,58 (м, 2Н), 2,53-2,23 (м, 3Н), 2,19-1,89 (м, 4Н), 1,85-1,02 (м, 26Н), 0,92 (с, 3Н).
Пример 24
Синтез 11-(3,17β-дигидроксиэстра-1,3,5(10)-триен-11β-ил)-2-(4,4,5,5,5-пентафторпентил)ундекановой кислоты
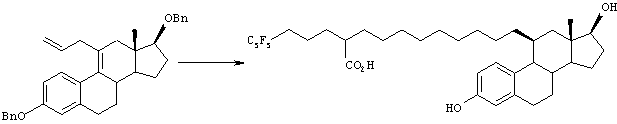
Исходя из 3,17β-бис(бензилокси)-11-(2-пропенил)эстра-1,3,5(10),9(11)-тетраена, полученного в примере 20, и этил-2-(4,4,5,5,5-пентафторпентил)-9-деценоата, полученного отдельно, повторяли ту же методику, как описано в примере 20, получая 11-(3,17β-дигидроксиэстра-1,3,5(10)-триен-11β-ил)-2-(4,4,5,5,5-пентафторпентил)ундекановую кислоту.
1H-ЯМР (270 МГц, СDСl3,): δ 7,00 (д, J=8,4 Гц, 1H), 6,59 (дд, J=8,7, 2,7Гц, 1H), 6,54 (д, J=2,7 Гц, 1H), 3,73-3,71 (м, 1H), 2,88-2,82 (м, 2Н), 2,58-2,33 (м, 3Н), 2,24-1,18 (м, 34Н), 0,92 (с, 3Н).
Пример 25
Синтез 11-(3,17β-дигидроксиэстра-1,3,5(10)-триен-11β-ил)-2-(4,4,5,5,6,6,7,7,7-нонафторгептил)ундекановой кислоты
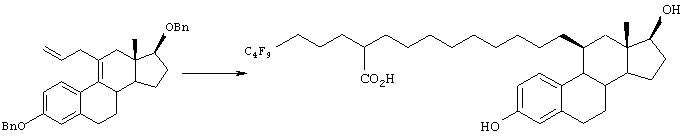
Исходя из 3,17β-бис(бензилокси)-11-(2-пропенил)эстра-1,3,5(10),9(11)-тетраена, полученного в примере 20, и этил-2-(4,4,5,5,6,6,7,7,7-нонафторгептил)-9-деценоата, полученного отдельно, повторяли ту же методику, как описано в примере 20, получая 11-(3,17β-дигидроксиэстра-1,3,5(10)-триен-11β-ил)-2-(4,4,5,5,6,6,7,7,7-нонафторгептил)ундекановую кислоту.
1H-ЯМР (270 МГц, СD3OD): δ 7,07 (д, J=8,4 Гц, 1H, Cl-CH), 6,54 (дд, J=8,4, 2,3 Гц, 1H, C2-CH), 6,45 (д, J=2,3 Гц, 1H, C4-CH), 3,67 (т, J=8,3 Гц, 1H, С17-СН), 2,85-2,62 (м, 2Н), 2,05-1,80 (м, 7Н), 1,80-0,96 (м, 32Н), 0,91(с, 3Н, С18-СН3).
Пример 26
Синтез 10-(3,17β-дигидроксиэстра-1,3,5(10)-триен-11β-ил)-2-(6,6,7,7,7-пентафторгептил)декановой кислоты
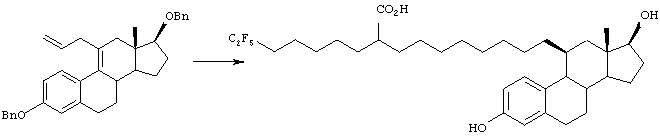
10-(3,17β-Дигидроксиэстра-1,3,5(10)-триен-11β-ил)-2-(6,6,7,7,7-пентафторгептил)декановая кислота, полученная в примере 12, также может быть синтезирована способом, аналогично описанному в примере 20, исходя из 3,17β-бис(бензилокси)-11-(2-пропенил)эстра-1,3,5(10),9(11)-тетраена, и отдельно полученного этил-2-(6,6,7,7,7-пентафторгептил)-8-ноненоата.
Пример 27
Синтез 12-[6-гидрокси-2-(4-гидроксифенил)нафт-1-ил]-2-(4,4,5,5,5-пентафторпентил)додекановой кислоты
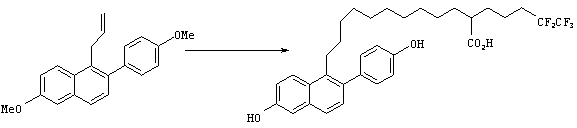
Исходя из 6-метокси-2-(4-метоксифенил)-1-(2-пропенил)нафталина и полученного отдельно диэтил-2-(8-ноненил)-2-(4,4,5,5,5-пентафторпентил)малоната, повторяли те же методики, как описано в примерах 1, 2 и 3, получая 12-[6-гидрокси-2-(4-гидроксифенил)нафт-1-ил]-2-(4,4,5,5,5-пентафторпентил)додекановую кислоту.
1H-ЯМР (270 МГц, СDСl3,): δ 7,98 (д, J=9 Гц, 1Н, Ar-H), 7,53 (д, J=8 Гц, 1Н, Ar-H), 7,28-7,12 (м, 5Н, Ar-H), 6,89 (д, J=9 Гц, 2Н, Ar-H), 2,96-2,90 (м, 2Н, нафтил-СН2-), 2,44-2,42 (м, 1Н, -CHCO2), 2,18-1,18 (м, 24Н, алкил-Н).
Пример 28
Синтез 11-[6-гидрокси-2-(4-гидроксифенил)нафт-1-ил]-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)ундекановой кислоты

Исходя из 6-метокси-2-(4-метоксифенил)-1-(2-пропенил)нафталина и отдельно полученного диэтил-2-(7-октенил)-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)малоната, повторяли те же методики, как описано в примерах 1, 2 и 3, получая 11-[6-гидрокси-2-(4-гидроксифенил)нафт-1-ил]-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)ундекановую кислоту.
1H-ЯМР (300 МГц, СDСl3,): δ 7,98 (д, J=8,9 Гц, 1H, Ar-H), 7,53 (д, J=8,4 Гц, 1Н, Ar-H), 7,13-7,28 (м, 5Н, Ar-H), 6,89 (д, J=8,5 Гц, 2H, Ar-H), 2,92 (т, J=8,1 Гц, 2Н, нафтил-СН2), 2,46-2,48 (м, 1Н, СНСO2Н), 1,95-2,20 (м, 2H, CH2CF2), 1,18-2,11 (м, 18Н, алкил-Н).
Масс-спектр (ESI): 667 (М+1).
Пример 29
Синтез 12-[6-гидрокси-2-(4-гидрокси-2-метилфенил)нафт-1-ил]-2-(4,4,5,5,5-пентафторпентил)додекановой кислоты
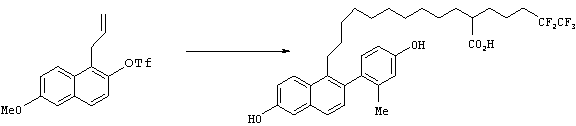
Исходя из 6-метокси-1-(2-пропенил)-2-нафтилтрифторметансульфоната, полученного в примере 1, и 4-метокси-2-метилфенилбороновой кислоты, полученной отдельно, повторяли те же методики, как описано в примерах 1, 2 и 3, получая 12-[6-гидрокси-2-(4-гидрокси-2-метилфенил)нафт-1-ил]-2-(4,4,5,5,5-пентафторпентил)додекановую кислоту.
1H-ЯМР (300 МГц, СDСl3,): δ 7,97 (д, J=8,9 Гц, 1H), 7,53 (д, J=8,4 Гц, 1H), 7,19-7,10 (м, 3Н), 7,02 (д, J=8,3 Гц, 1H), 6,77 (д, J=2,1 Гц, 1H), 6,70 (дд, J=8,3, 2,1 Гц, 1H), 4,8 (ушир., 3Н), 3,0-2,8 (м, 1H), 2,7-2,5 (м, 1H), 2,5-2,3 (м, 1H), 2,1-1,9 (м, 2H), 1,99 (с, 3Н, СН3), 1,8-1,0 (м, 22Н).
Пример 30
Синтез 12-[2-(2-этил-4-гидроксифенил)-6-гидроксинафт-1-ил]-2-(4,4,5,5,5-пентафторпентил)додекановой кислоты

Исходя из 6-метокси-1-(2-пропенил)-2-нафтилтрифторметансульфоната, полученного в примере 1, и 2-этил-4-метоксифенилбороновой кислоты, полученной отдельно, повторяли те же методики, как описано в примерах 1, 2 и 3, получая 12-[2-(2-этил-4-гидроксифенил)-6-гидроксинафт-1-ил]-2-(4,4,5,5,5-пентафторпентил)додекановую кислоту.
1H-ЯМР (300 МГц, СDСl3): δ 7,97 (д, J=9,1 Гц, 1Н, Ar-H), 7,52 (д, J=8,4 Гц, 1Н, Ar-H), 7,06-7,23 (м, 3Н, Ar-H), 6,85 (д, J=8,2 Гц, 1Н, Ar-H), 6,79 (д, J=2,6 Гц, 1Н, Ar-H), 6,61 (дд, J=8,2, 2,6 Гц, 1Н, Ar-H), 2,81-2,93 (м, 1Н, нафтил-СН2), 2,49-2,75 (м, 1Н, нафтил-СН2), 2,39-2,50 (м, 1Н, CHCO2H), 2,18-2,29 (м, 2Н, АrСН2СН3), 1,91-2,12 (м, 2Н, CH2CF2), 0,95-1,72 (м, 25Н, алкил-Н).
Масс-спектр (ESI): 623 (М+1).
Пример 31
Синтез 12-[2-(2-фтор-4-гидроксифенил)-6-гидроксинафт-1-ил]-2-(4,4,5,5,5-пентафторпентил)додекановой кислоты

Исходя из 6-метокси-1-(2-пропенил)-2-нафтилтрифторметансульфоната, полученного в примере 1, и 2-фтор-4-метоксифенилбороновой кислоты, полученной отдельно, повторяли те же методики, как описано в примерах 1, 2 и 3, получая 12-[2-(2-фтор-4-гидроксифенил)-6-гидроксинафт-1-ил]-2-(4,4,5,5,5-пентафторпентил)додекановую кислоту.
1H-ЯМР (300 МГц, СDСl3,): δ 7,97 (д, J=9,1 Гц, 1H, Ar-H), 7,52 (д, J=8,5 Гц, 1H, Ar-H), 7,06-7,25 (м, 4Н, Ar-H), 6,64-6,69 (м, 2Н, Ar-H), 2,81-2,90 (м, 2Н, нафтил-CH2), 2,40-2,50 (м, 1H, CHCO2H), 1,90-2,10 (м, 2Н, CH2CF2), 1,05-1,75 (м, 22Н, алкил-Н).
Масс-спектр (ESI): 613 (М+1).
Пример 32
Синтез 12-[6-гидрокси-2-(4-гидрокси-2-трифторметилфенил)нафт-1-ил]-2-(4,4,5,5,5-пентафторпентил)додекановой кислоты
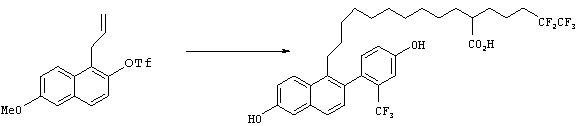
Исходя из 6-метокси-1-(2-пропенил)-2-нафтилтрифторметансульфоната, полученного в примере 1, и 4-метокси-2-трифторметилфенилбороновой кислоты, полученной отдельно, повторяли те же методики, как описано в примерах 1, 2 и 3, получая 12-[6-гидрокси-2-(4-гидрокси-2-трифторметилфенил)нафт-1-ил]-2-(4,4,5,5,5-пентафторпентил) додекановую кислоту.
1H-ЯМР (270 МГц, СDСl3,): δ 7,96 (д, J=9 Гц, 1H, Ar-H), 7,50 (д, J=8 Гц, 1H, Ar-H), 7,24-7,01 (м, 6Н, Ar-H), 2,89-2,84 (м, 1H), 2,51-2,42 (м, 2Н), 2,11-1,15 (м, 24Н, алкил-Н).
Пример 33
Синтез 12-[2-(3-фтор-4-гидроксифенил)-6-гидроксинафт-1-ил]-2-(4,4,5,5,5-пентафторпентил)додекановой кислоты

Исходя из 6-метокси-1-(2-пропенил)-2-нафтилтрифторметансульфоната, полученного в примере 1, и 3-фтор-4-метоксифенилбороновой кислоты, полученной отдельно, повторяли те же методики, как описано в примерах 1, 2 и 3, получая 12-[2-(3-фтор-4-гидроксифенил)-6-гидроксинафт-1-ил]-2-(4,4,5,5,5-пентафторпентил)додекановую кислоту.
1H-ЯМР (270 МГц, СDСl3,): δ 7,96 (д, J=9 Гц, 1H, Ar-H), 7,51 (д, J=8 Гц, 1Н, Ar-H), 7,24-6,95 (м, 6Н, Ar-H), 2,94-2,88 (м, 2Н, нафтил-СН2-), 2,39 (м, 1Н, -СНСО2), 2,16-1,18 (м, 24Н, алкил-Н).
Пример 34
Синтез 12-[2-(3,5-дифтор-4-гидроксифенил)-6-гидроксинафт-1-ил]-2-(4,4,5,5,5-пентафторпентил)додекановой кислоты

Исходя из 6-метокси-1-(2-пропенил)-2-нафтилтрифторметансульфоната, полученного в примере 1, и 3,5-дифтор-4-метоксифенилбороновой кислоты, полученной отдельно, повторяли те же методики, как описано в примерах 1, 2 и 3, получая 12-[2-(3,5-дифтор-4-гидроксифенил)-6-гидроксинафт-1-ил]-2-(4,4,5,5,5-пентафторпентил)додекановую кислоту.
1H-ЯМР (300 МГц, СDСl3,): δ 7,98 (д, J=9,9 Гц, 1H, Ar-H), 7,53 (д, J=8,5 Гц, 1H, Ar-H), 7,14-7,26 (м, 3Н, Ar-H), 6,85-6,90 (м, 2Н, Ar-H), 2,88-2,94 (м, 2Н, нафтил-СН2), 2,39-2,48 (м, 1H, СНСО2Н), 2,01-2,12 (м, 2Н, CH2CF2), 1,24-1,88 (м, 21Н, алкил-Н).
Масс-спектр (ESI): 631(М+1).
Пример 35
Синтез 12-[2-(4-фторфенил)-6-гидроксинафт-1-ил]-2-(4,4,5,5,5-пентафторпентил)додекановой кислоты
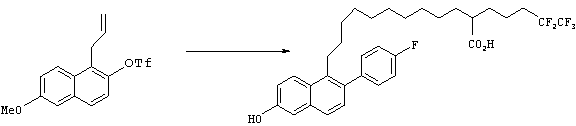
Исходя из 6-метокси-1-(2-пропенил)-2-нафтилтрифторметансульфоната, полученного в примере 1, и 4-фторфенилбороновой кислоты, полученной отдельно, повторяли те же методики, как описано в примерах 1, 2 и 3, получая 12-[2-(4-фторфенил)-6-гидроксинафт-1-ил]-2-(4,4,5,5,5-пентафторпентил)додекановую кислоту.
1H-ЯМР (270 МГц, СDСl3,): δ 7,98 (д, J=9 Гц, 1H, Ar-H), 7,54 (д, J=8 Гц, 1Н, Ar-H), 7,31-7,07 (м, 7Н, Ar-H), 2,93-2,87 (м, 2Н, нафтил-СН2-), 2,46-2,35 (м, 1Н, -СНСO2), 2,15-1,14 (м, 24Н, алкил-Н).
Пример 36
Синтез (2R)-11-(3,17β-дигидроксиэстра-1,3,5(10)-триен-7α-ил)-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)ундекановой кислоты
(Стадия 1)
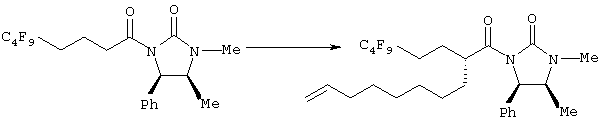
Безводный тетрагидрофуран (10 мл) добавляли к (4S, 5R)-3,4-диметил-1-(5,5,6,6,7,7,8,8,8-нонафтороктаноил)-5-фенилимидазолидин-2-ону (1,20 г, 2,5 ммоль) в атмосфере азота и полученную смесь охлаждали до -78°С. К смеси добавляли бис(триметилсилил)амид лития (2,75 мл, 1,0 М в тетрагидрофуране, 2,75 ммоль), затем перемешивали в течение 1 часа. После добавления 8-бром-1-октена (714 мг, 3,0 ммоль) и НМРА (1,25 мл) при -78°С реакционную смесь нагревали при перемешивании до -50°С в течение 2 часов и до 0°С в течение 30 минут и затем перемешивали в течение 12 часов при 0°С. Реакционную смесь гасили насыщенным водным раствором хлорида аммония при 0°С и затем экстрагировали смешанным растворителем, состоящим из этилацетата и н-гексана (3:7). Органический слой промывали последовательно насыщенным водным раствором бисульфата калия, насыщенным водным раствором хлорида натрия, насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем фильтровали. Фильтрат концентрировали при пониженном давлении с использованием роторного испарителя и полученный остаток очищали колоночной хроматографией на силикагеле (Kanto Kagaku, силикагель 60 (сферический, нейтральный), 40-100 мкм, элюент: этилацетат/н-гексан=1/5 3/7), получая (4S,5R)-3,4-диметил-1-[(2R)-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)-9-деценоил]-5-фенилимидазолидин-2-он (1,37 г, выход 93%).
Оптическая чистота: 95,5% de (диастереомерный избыток), как измерено с помощью ВЭЖХ (колонка: Daicel Chiralpack AD, φ 0,46×25 см, растворитель: н-гексан/изопропанол=97/3, скорость потока: 0,5 мл/мин, детектируемая длина волны: 206 нм).
1H-ЯМР (270 МГц, СDСl3,): δ 7,32-7,13 (м, 5Н), 5,87-5,72 (м, 1Н), 5,34 (д, J=8,9 Гц, 1Н), 5,02-4,91 (м, 2Н), 4,10-3,86 (м, 2Н), 2,84 (с, 3Н), 2,19-1,08 (м, 16Н), 0,82 (д, J=6,5 Гц, 3Н).
(Стадия 2)

3-Метокси-7α-(2-пропенил)эстра-1,3,5(10)-триен-17β-ол (377 мг, 1,16 ммоль) и (4S, 5R)-3,4-диметил-1-[(2R)-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)-9-деценоил]-5-фенилимидазолидин-2-он (1,36 г, 2,31 ммоль) растворяли в безводном дихлорметане (12 мл) при комнатной температуре в атмосфере азота. К данному раствору добавляли бензилиденбис(трициклогексилфосфин)дихлоррутений (47,5 мг, 5,78×10-2 ммоль) и полученную смесь нагревали при кипении с обратным холодильником в течение 5 часов в атмосфере азота. После охлаждения реакционную смесь фильтровали через слой оксида алюминия. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали флеш-хроматографией на силикагеле (Kanto Kagaku, силикагель 60 (сферический нейтральный), 40-100 мкм, элюент: этилацетат/н-гексан=1/1), получая смесь (4S, 5R)-3,4-диметил-1-[(2R, 9Е)-11-(17β-гидрокси-3-метоксиэстра-1,3,5(10)-триен-7α-ил)-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)-9-ундеценоил]-5-фенилимидазолидин-2-он и (4S, 5R)-3,4-диметил-1-[(2R,9Z)-11-(17β-гидрокси-3-метоксиэстра-1,3,5(10)-триен-7α-ил)-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)-9-ундеценоил]-5-фенилимидазолидин-2-он (579 мг, выход 57%).
1H-ЯМР (300 МГц, СDСl3,): δ 7,33-7,12 (м, 6Н), 6,73-6,69 (м, 1H), 6,61-6,56 (м, 1Н), 5,42-5,25 (м, 3Н), 4,05-3,87 (м, 2Н), 3,77-3,70 (м, 4Н), 2,90-2,71 (м, 5Н), 2,38-1,06 (м, 31Н), 0,83 -0,78 (м, 6Н).
(Стадия 3)
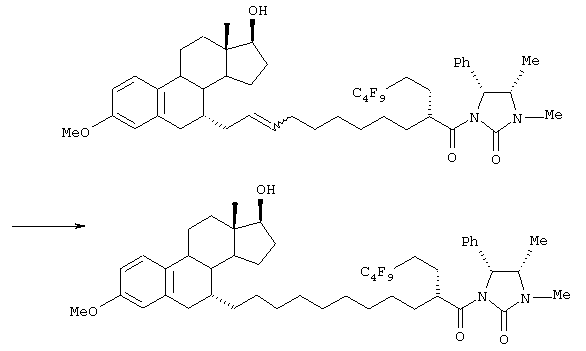
Смесь (4S, 5R)-3,4-диметил-1-[(2R,9Е)-11-(17β-гидрокси-3-метоксиэстра-1,3,5(10)-триен-7α-ил)-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)-9-ундеценоил]-5-фенилимидазолидин-2-он и (4S, 5R)-3,4-диметил-1-[(2R,9Z)-11-(17β-гидрокси-3-метоксиэстра-1,3,5(10)-триен-7α-ил)-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)-9-ундеценоил]-5-фенилимидазолидин-2-он (579 мг, 653 мкмоль) растворяли в этилацетате (14 мл) с последующим добавлением 10%-ного палладия-на-углероде (58 мг) при комнатной температуре. После продувания водородом реакционную смесь перемешивали в течение 19 часов при комнатной температуре. После фильтрования реакционной смеси через целит растворитель концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (Kanto Kagaku, силикагель 60 (сферический, нейтральный), 40-100 мкм, элюент: этилацетат/н-гексан=1/1), получая (4S,5R)-3,4-диметил-1-[(2R)-11-(17β-гидрокси-3-метоксиэстра-1,3,5(10)-триен-7α-ил)-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)ундеканоил]-5-фенилимидазолидин-2-он (579 мг, выход 100%).
1H-ЯМР (300 МГц, СDСl3,): δ 7,33-7,11 (м, 6Н), 6,73-6,70 (м, 1Н), 6,69-6,62 (м, 1Н), 5,33 (д, J=7,8 Гц, 1Н), 4,05-3,86 (м, 2Н), 3,79-3,71 (м, 1Н), 3,77 (с, 3Н), 2,94-2,72 (м, 2Н), 2,84 (с, 3Н), 2,38-0,99 (м, 35Н), 0,82 (д, J=5,9 Гц, 3Н), 0,78 (с, 3Н).
(Стадия 4)

(4S, 5R)-3,4-Диметил-1-[(2R)-11-(17β-гидрокси-3-метоксиэстра-1,3,5(10)-триен-7α-ил)-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)ундеканоил]-5-фенилимидазолидин-2-он (445 мг, 0,50 ммоль) растворяли в безводном диметиловом эфире этиленгликоля (5 мл) в атмосфере азота и затем охлаждали до 0°С. К данному раствору добавляли раствор гидроксида тетра-н-бутиламмония (40% вес/вес, 649 мг, 1,0 ммоль) и водный пероксид водорода (30% вес/вес, 113 мг, 1,0 ммоль) и полученную смесь перемешивали в течение 1,5 часов при комнатной температуре. Реакционную смесь гасили насыщенным водным раствором тиосульфата натрия, подкисляли насыщенным водным раствором бисульфата калия и затем экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем фильтровали. Фильтрат концентрировали при пониженном давлении с использованием роторного испарителя и полученный остаток очищали колоночной хроматографией на силикагеле (Wako gel C-200, элюент: этилацетат/н-гексан=4/6 8/2), получая (2R)-11-(17β-гидрокси-3-метоксиэстра-1,3,5(10)-триен-7α-ил)-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)ундекановую кислоту (360 мг, выход 100%) и (4R, 5S)-1,5-диметил-4-фенилимидазолидин-2-он (93 мг, выход 98%).
1H-ЯМР (300 МГц, СDСl3,): δ 7,21-7,18 (м, 1Н), 6,73-6,69 (м, 1Н), 6,62-6,61 (м, 1H), 6,30 (ушир.с, 1Н), 3,79-3,71 (м, 1H), 3,77 (с, 3Н), 2,93-2,72 (м, 2Н), 2,47-1,01 (м, 36Н), 0,78 (с, 3Н).
(Стадия 5)
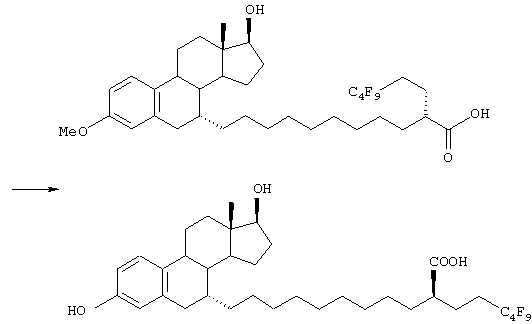
Безводный дихлорметан (10 мл) добавляли к (2R)-11-(17β-гидрокси-3-метоксиэстра-1,3,5(10)-триен-7α-ил)-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)ундекановой кислоте (358 мг, 0,50 ммоль) в атмосфере азота и полученную смесь охлаждали до -78°С. К смеси по каплям добавляли трибромид бора (1,0 М в тетрагидрофуране, 3 мл, 3,0 ммоль), затем перемешивали на льду в течение 2,5 часов. Реакционную смесь опять охлаждали до -78°С и гасили насыщенным водным раствором бикарбоната натрия в течение 1 часа. После экстракции реакционной смеси этилацетатом органический слой промывали последовательно насыщенным водным раствором бисульфата калия, насыщенным водным раствором хлорида натрия, насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем фильтровали. Фильтрат концентрировали при пониженном давлении с использованием роторного испарителя и полученный остаток очищали колоночной хроматографией на силикагеле (Wako gel С-200, элюент: этилацетат/н-гексан=4/6), получая (2R)-11-(3,17β-дигидроксиэстра-1,3,5(10)-триен-7α-ил)-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)ундекановую кислоту (231 мг, выход 60%) в виде желтой аморфной массы.
Химическая чистота: 99,05% и 98,63% при обнаружении при длине волны 280 и 219 нм соответственно, как измерено с помощью ВЭЖХ (колонка: YMC-Pack ODS-A, А-312 ⊘0,6×15 см, растворитель: H2O/MeCN/ТФУК=30/70/0,1, скорость потока: 1,0 мл/мин).
Оптическая чистота: 96,3%de (диастереомерный избыток), как измерено с помощью ВЭЖХ (колонка: Daicel Chiralpack AD, ⊘0,46×25 см, растворитель: н-гексан/изопропанол/ТФУК=90/10/0,1, скорость потока: 0,5 мл/мин, длина волны обнаружения: 280 нм).
1H-ЯМР (270 МГц, СDСl3,): δ 7,16-7,13 (м, 1Н), 6,64-6,60 (м, 1Н), 6,55-6,54 (м, 1Н), 3,74 (т, J=8,6 Гц, 1Н), 2,91-2,67 (м, 2Н), 2,50-1,01 (м, 36Н), 0,78 (с, 3Н).
Пример 37
Синтез (2R)-10-(3,17β-дигидроксиэстра-1,3,5(10)-триен-7α-ил)-2-(4,4,5,5,6,6,7,7,7-нонафторгептил)декановой кислоты
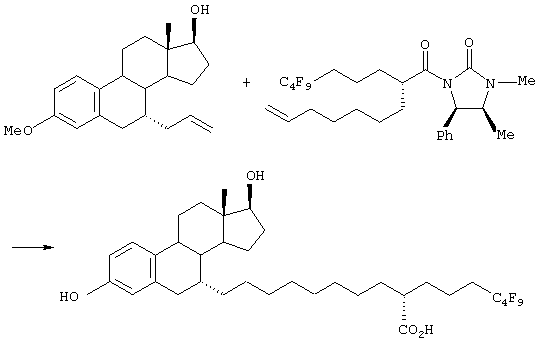
Исходя из 3-метокси-7α-(2-пропенил)эстра-1,3,5(10)-триен-17β-ола и (4S,5R)-3,4-диметил-1-[(2R)-2-(4,4,5,5,6,6,7,7,7-нонафторгептил)-8-ноненоил]-5-фенилимидазолидин-2-она, полученного отдельно по методике примера 36, повторяли методику, аналогично примеру 36, получая (2R)-10-(3,17β-дигидроксиэстра-1,3,5(10)-триен-7α-ил)-2-(4,4,5,5,6,6,7,7,7-нонафторгептил)декановую кислоту.
1H-ЯМР (300 МГц, СDСl3,): δ 7,12 (д, J=8,4 Гц, 1H), 6,68 (дд, J=8,5, 2,4 Гц, 1H), 6,53 (д, J=2,6 Гц, 1H), 3,68 (т, J=8,5 Гц, 1H), 2,95-2,60 (м, 2Н), 2,47-2,19 (м, 4Н), 2,18-1,03 (м, 31Н), 0,69 (с, 3Н).
Пример 38
Синтез (2S)-10-(3,17β-дигидроксиэстра-1,3,5(10)-триен-7α-ил)-2-(4,4,5,5,6,6,7,7,7-нонафторгептил)декановой кислоты

Исходя из 3-метокси-7α-(2-пропенил)эстра-1,3,5(10)-триен-17β-ола и (4R,5S)-3,4-диметил-1-[(2S)-2-(4,4,5,5,6,6,7,7,7-нонафторгептил)-8-ноненоил]-5-фенилимидазолидин-2-она, полученного отдельно способом примера 36, повторяли методику, аналогично примеру 36, получая (2S)-10-(3,17β-дигидроксиэстра-1,3,5(10)-триен-7α-ил)-2-(4,4,5,5,6,6,7,7,7-нонафторгептил)декановую кислоту.
1H-ЯМР (300 МГц, СDСl3,): δ 7,12 (д, J=8,4 Гц, 1Н), 6,68 (дд, J=8,5, 2,4 Гц, 1H), 6,53 (д, J=2,6 Гц, 1H), 3,68 (т, J=8,5 Гц, 1H), 2,95-2,60 (м, 2Н), 2,47-2,19 (м, 4Н), 2,18-1,03 (м, 31Н), 0,69(с, 3Н).
Пример 39
Синтез (2R)-11-[3,17β-дигидроксиэстра-1,3,5(10)-триен-11β-ил]-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)ундекановой кислоты
(Стадия 1)

3,17β-Бис(бензилокси)-11-(2-пропенил)эстра-1,3,5(10),9(11)-тетраен (491 мг, 1,00 ммоль) и (4S,5R)-3,4-диметил-1-[(2R)-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)-9-деценоил]-5-фенилимидазолидин-2-он (1,18 г, 2,00 ммоль), полученный в примере 36, растворяли в безводном дихлорметане (10 мл) при комнатной температуре в атмосфере азота, смешивали с бензилиденбис(трициклогексилфосфин)дихлоррутением (41 мг, 0,05 ммоль) и затем нагревали при кипении с обратным холодильником с последующим перемешиванием в течение 6 часов в атмосфере азота. После охлаждения реакционную смесь концентрировали при пониженном давлении и полученный остаток очищали флэш-хроматографией на силикагеле (элюент: этилацетат/н-гексан = 3/7), получая смесь (4S, 5R)-1-{(2R, 9Е)-11-[3,17β-бис(бензилокси)эстра-1,3,5(10),9(11)-тетраен-11-ил]-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)-9-ундеценоил}-3,4-диметил-5-фенилимидазолидин-2-она и (4S,5R)-1-{(2R,9Z)-11-[3,17β-бис(бензилокси)эстра-1,3,5(10), 9(11)-тетраен-11-ил]-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)-9-ундеценоил)-3,4-диметил-5-фенилимидазолидин-2-она (664 мг, выход 63%).
1H-ЯМР (270 МГц, СDСl3,): δ 7,45-7,10 (м, 16Н), 6,78-6,70 (м, 2Н), 5,58-5,40 (м, 2Н), 5,32 (д, J=8,7 Гц, 1H), 5,04 (с, 2Н), 4,65-4,50 (м, 2Н), 4,10-3,80 (м, 2Н), 3,62-3,50 (м, 1H), 3,22-3,05 (м, 1H), 2,83 (с, 3Н), 2,80-2,60 (м, 3Н), 2,52-2,30 (м, 1H), 2,20-1,0 (м, 25Н), 0,87 (с, 3Н, С18-СН3), 0,81 (д, J=6,6 Гц, 3Н).
(Стадия 2)

Смесь (4S,5R)-1-{(2R,9Е)-11-[3,17β-бис(бензилокси)эстра-1,3,5(10),9(11)-тетраен-11-ил]-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)-9-ундеценоил}-3,4-диметил-5-фенилимидазолидин-2-она и (4S,5R)-1-{(2R,9Z)-11-[3,17β-бис(бензилокси)эстра-1,3,5(10),9(11)-тетраен-11-ил]-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)-9-ундеценоил}-3,4-диметил-5-фенилимидазолидин-2-она (283 мг, 0,27 ммоль) растворяли в смешанном растворителе, состоящем из метанола (12 мл) и тетрагидрофурана (1,2 мл), с последующим добавлением гидроксида палладия на углероде (85 мг) при комнатной температуре. После продувания водородом реакционную смесь перемешивали в течение 1 дня при комнатной температуре. После фильтрования реакционной смеси растворитель концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан = 3/7), получая (4S,5R)-1-{(2R)-11-[3,17β-дигидроксиэстра-1,3,5(10)-триен-11β-ил]-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)ундеканоил}-3,4-диметил-5-фенилимидазолидин-2-он (164 мг, выход 70%).
1H-ЯМР (300 МГц, СDСl3,): δ 7,36-7,12 (м, 5Н), 6,94-6,88 (м, 1Н), 6,56-6,52 (м, 1Н), 6,44-6,36 (м, 1Н), 5,64 (с, 1Н), 5,35 (д, J=8,7 Гц, 1H), 5,70-5,15 (м, 3Н), 4,38 (с, 3Н), 4,40-2,60 (м, 36Н), 2,43 (с, 3Н), 2,34 (д, J=6,6 Гц, 3Н).
(Стадия 3)
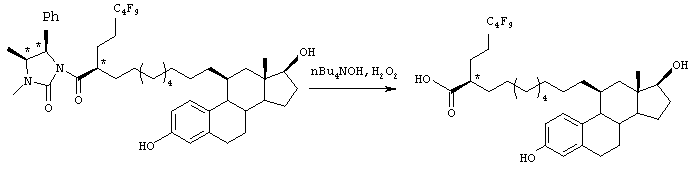
(4S,5R)-1-{(2R)-11-[3,17β-Дигидроксиэстра-1,3,5(10)-триен-11β-ил]-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)ундеканоил}-3,4-диметил-5-фенилимидазолидин-2-он (140 мг, 0,16 ммоль) растворяли в ДМЭ (3 мл). К данному раствору добавляли раствор гидроксида тетра-н-бутиламмония (40% вес/вес, 312 мг, 0,48 ммоль) и водный пероксид водорода (30% вес/вес, 56 мг, 0,48 ммоль) и полученную смесь перемешивали в течение 2 часов при комнатной температуре. Реакционную смесь гасили 10%-ным водным раствором сульфита натрия, подкисляли 2 н. соляной кислотой и затем экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле (Wako gel C-200, элюент: этилацетат/н-гексан = 3/7), затем препаративной ВЭЖХ (YMC-ODS-5-B (3×25 см), элюент: ацетонитрил/вода/трифторуксусная кислота = 90/10/0,1, скорость потока: 18 мл/мин), получая целевое соединение (2R)-11-[3,17β-дигидроксиэстра-1,3,5(10)-триен-11β-ил]-2-(3,3,4,4,5,5,6,6,6-нонафторгексил)ундекановую кислоту (58 мг, выход 52%).
1H-ЯМР (300 МГц, СDСl3,): δ 7,00 (д, J=8,4 Гц, 1H, Cl-CH), 6,55 (дд, J=8,4, 2,4 Гц, 1H, C2-CH), 6,54 (д, J=2,4 Гц, 1H, C4-CH), 3,75 (т, J=7,5 Гц, 1H), 2,85-1,10 (м, 38Н), 0,92 (с, 3Н, С18-СН3).
Химическая чистота: 98,4%, как измерено с помощью ВЭЖХ (колонка: YMC-Pack ODS-A, А-312 ⊘0,6×15 см, растворитель: H2O/MeCN/ТФУК=30/70/0,1, скорость потока: 1,0 мл/мин, длина волны обнаружения: 220 нм).
Оптическая чистота: 95,7%de (диастреомерный избыток), как измерено с помощью ВЭЖХ (колонка: Daicel Chiralpack AD, ⊘0,46×25 см, растворитель: н-гексан/изопропанол/ТФУК = 92/8/0,08, скорость потока: 0,5 мл/мин, длина волны обнаружения: 280 нм).
Пример испытаний 1: Антиэстрогенная активность (пероральное введение)
Тестируемые соединения исследовали на их пероральную антиэстрогенную активность следующим образом. В данном эксперименте в качестве тестируемых соединений использовали соединения, полученные в примерах 5, 12, 14-33, 37 и 38. В качестве контрольных соединений использовали те, которые имеют такую же структуру исходного каркаса, что и тестируемые соединения, то есть ZM189154 для примеров 5 и 27-33 и ICI182780 для примеров 12, 14-26, 37 и 38.
Для определения антиэстрогенной активности мышам (ICR, вес 30±2 г), которых овариэктомировали за 2 недели до этого, вводили подкожно 17β-эстрадиолбензоат (Sigma) в количестве 0,1 мкг/мышь в течение 3 дней и измеряли степень, в которой тестируемое соединение ингибировало увеличение веса матки. В данном эксперименте каждое из тестируемых и контрольных соединений суспендировали в 5%-ном растворе аравийской камеди и вводили перорально в течение 3 дней из расчета один раз в день. Через 24 часа после последнего введения тестируемых животных умерщвляли, матку удаляли и взвешивали. Полученные результаты представлены в таблице 2.
Антиэстрогенная активность для овариэктомированных мышей при введении 17β-эстрадиола (пероральное введение, 3 дня)
Результаты, приведенные в таблице 2, показывают, что соединения, имеющие боковую цепь общей формулы (1) согласно настоящему изобретению, демонстрируют превосходную ингибирующую активность по отношению к индуцированному эстрадиолом увеличению веса матки в сравнении с антиэстрогенными контрольными соединениями ZM189154 и ICI182780, которые имели такой же исходный каркас, но не имели подобной боковой цепи.
Промышленная применимость
Соединения по настоящему изобретению имеют боковую цепь общей формулы (1). Такая боковая цепь дает возможность соединениям по изобретению демонстрировать улучшенную биодоступность и значительно высокую активность при пероральном введении в сравнении с обычными соединениями, не имеющими такой боковой цепи, такими как соединения, обладающие низкой активностью при пероральном введении, соединения, обладающие противоопухолевой активностью, соединения, обладающие эстрогенной активностью или соединения, обладающие антиэстрогенной активностью. Вследствие этого соединения по настоящему изобретению являются преимущественными при фармацевтическом применении.
| название | год | авторы | номер документа |
|---|---|---|---|
| 17-ДЕЗОКСИ-1,3,5(10)-ЭСТРАТРИЕН, СПОСОБЫ ЛЕЧЕНИЯ, СПОСОБ СИНТЕЗА 7АЛЬФА-МЕТИЛЭСТРОНА, СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2233288C2 |
| ПРОИЗВОДНЫЕ 19-НОРСТЕРОИДОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1992 |
|
RU2111213C1 |
| СОЕДИНЕНИЯ, НАБОР, АНДРОГЕННАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2242479C2 |
| ПРОИЗВОДНЫЕ ЭСТРА-1,3,5(10)-ТРИЕНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2139885C1 |
| ПРОИЗВОДНЫЕ БЕНЗОПИПЕРИДИНА, БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1992 |
|
RU2142945C1 |
| РОДСТВЕННОЕ ВИТАМИНУ A СОЕДИНЕНИЕ И СПОСОБЫ ЕГО ПОЛУЧЕНИЯ | 1998 |
|
RU2188193C2 |
| ДИАЦЕТАТ РАЦЕМИЧЕСКОГО 3,17β-ДИГИДРОКСИ-7α,18-ДИМЕТИЛ-6-ОКСАЭСТРА-1,3,5( 10 ),8(9)-ТЕТРАЕНА, ОБЛАДАЮЩИЙ ОСТЕОПРОТЕКТОРНЫМ, ГИПОХОЛЕСТЕРИНЕМИЧЕСКИМ И АНТИОКСИДАНТНЫМ ДЕЙСТВИЕМ | 2009 |
|
RU2422455C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛИДИН-2,4-ДИОНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННОГО СРЕДСТВА ПРОТИВ РАКА | 2010 |
|
RU2497812C2 |
| 15,15-ДИАЛКИЛСТЕРОИДНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СОЕДИНЕНИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2147306C1 |
| 17 БЕТА-ОКСИЭСТРАТРИЕНЫ | 2002 |
|
RU2339643C2 |
Изобретение относится к новому соединению, имеющему следующую общую формулу (2), и способу его получения:
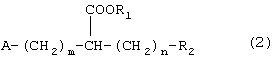
в которой R1 представляет атом водорода или солеобразующий металл, R2 представляет прямую или разветвленную C1-C7 галогеналкильную группу, m представляет целое число от 2 до 14, n представляет целое число от 2 до 7 и А представляет группу, выбранную из следующих формул (3)-(6), (17)-(20), (23), (25) и (26):
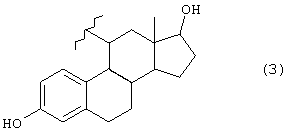

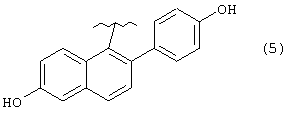
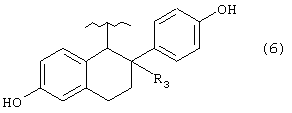


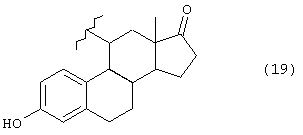
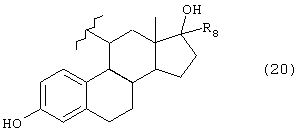

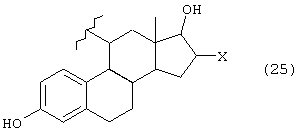
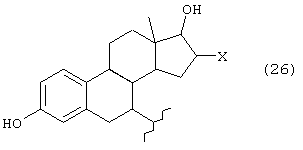
где в формуле (6) R3 представляет прямую или разветвленную C1-С5 алкильную группу,
в формулах (18) и (20) R8 представляет прямую или разветвленную C1-C5 алкильную группу, прямую или разветвленную C2-C5 алкенильную группу или прямую или разветвленную С2-С5 алкинильную группу, в формуле (23) каждый из R21, R22, R23 и R24 независимо представляет атом водорода, прямую или разветвленную C1-C5 алкильную группу, прямую или разветвленную C1-C7 галогеналкильную группу, атом галогена или ацильную группу, и в формулах (25) и (26) Х представляет атом галогена, или энантиомеры соединения, или гидраты, или фармацевтически приемлемые соли соединения или его энантиомеров. Изобретение также относится к фармацевтической композиции, обладающей антиэстрогенным действием, содержащей указанное соединение в качестве активного ингредиента, и терапевтическому агенту против рака молочной железы на его основе. 5 н. и 5 з.п. ф-лы, 2 табл.

в которой
R1 представляет атом водорода или солеобразующий металл,
R2 представляет прямую или разветвленную C1-C7 галогеналкильную группу,
m представляет целое число от 2 до 14, и
n представляет целое число от 2 до 7, и
А представляет группу, выбранную из следующих формул (3)-(6), (17)-(20), (23), (25) и (26):


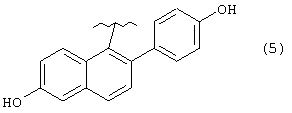
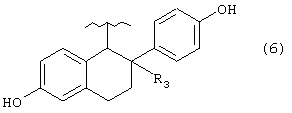
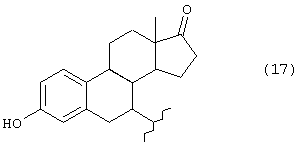



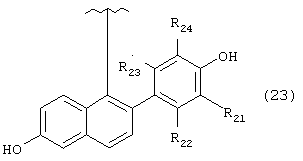


где
в формуле (6) R3 представляет прямую или разветвленную C1-С5 алкильную группу,
в формулах (18) и (20) R8 представляет прямую или разветвленную C1-C5 алкильную группу, прямую или разветвленную C2-C5 алкенильную группу или прямую или разветвленную С2-С5 алкинильную группу,
в формуле (23) каждый из R21, R22, R23 и R24 независимо представляет атом водорода, прямую или разветвленную C1-C5 алкильную группу, прямую или разветвленную C1-C7 галогеналкильную группу, атом галогена или ацильную группу, и
в формулах (25) и (26) Х представляет атом галогена, или энантиомеры соединения, или гидраты, или фармацевтически приемлемые соли соединения или его энантиомеров.
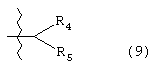
в которой каждый из R4 и R5, которые могут быть одинаковыми или различными, представляет прямую или разветвленную C1-С3 пергалогеналкильную группу.
i) олефинового метатезиса соединения формулы

где R11 и R12 представляют защитную группу, с соединением формулы
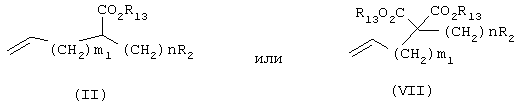
где R13 представляет защитную группу, m1 представляет целое число m-2, и R2, m и n имеют значения, указанные в п.1;
ii) гидрирования двойной связи;
iii) удаления защитной группы -OR11;
iv) гидролиза группы -CO2R13; и
v) декарбоксилирования одной карбоксильной группы, при условии, если на стадии i) используется соединение формулы (II), то стадия v) из способа исключается.
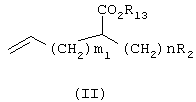
где R13 представляет карбоксизащитную группу, m1 представляет целое число m-2, и R2 представляет фторалкильную группу, m и n имеют значения, указанные в п.1.
Приоритет по пунктам и признакам:
| ЕР 0670162 А1, 06.09.1995, M.J.K | |||
| HARPER et al | |||
| “A NEW DERIVATIVE OF TRIPHENYLETHYLENE: EFFECT ON IMPLANTATION AND MODE OF ACTION IN RATS”, JOURNAL OF REPRODUCTION AND FERTILITY, 13,1967, p | |||
| Приспособление для записи звуковых явлений на светочувствительной поверхности | 1919 |
|
SU101A1 |
| Wakeling et al | |||
| “STEROIDAL RURE ANTIESTROGENS”, Journal of Endocrinology, vol | |||
| Прялка для изготовления крученой нити | 1920 |
|
SU112A1 |
| Wakeling et al | |||
| “A Potent Specific Pure | |||

