Изобретение относится к углеводным производным, обладающим антитромботической активностью, к фармацевтическим композициям, содержащим их, а также к использованию указанных углеводных производных для получения лекарственных средств.
В настоящее время известен ряд углеводных производных, обладающих антитромботической активностью, в частности сульфатированное гликозамингликановое производное, раскрытое в патенте ЕР 84999. Другие углеводные производные, относящиеся к сульфатированным гликозамингликанам, описаны в ЕР 529715 и характеризуются улучшенными фармакологическими свойствами. У этих углеводных производных отсутствуют характерные для гликозамингликанов функциональные группы, такие как гидроксильные группы, N-сульфатные и N-ацетильные группы.
Найдено, что углеводные производные настоящего изобретения, имеющие формулу I,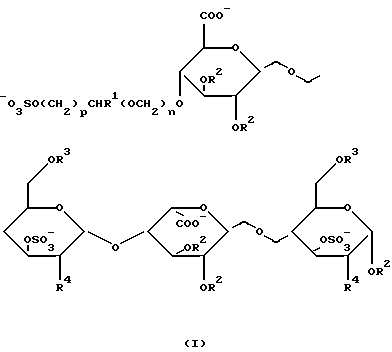
где R1 обозначает Н или CH2OSO3 -;
R2 и R3 представляют собой независимо друг от друга Н, (1-6С)алкил или SO3 -;
R4 обозначает OSO3 - или NНSО3 -;
n принимает значение 0 или 1;
р принимает значение 1 или 2;
или фармацевтически приемлемая соль его обладает анти-Ха активностью, которая существенно выше такой активности сахаридов, не имеющих на невосстановленнном конце 4-м положении глицериноподобной или гликолеподобной группы.
Фактор Ха играет важную роль в цепи реакций свертывания крови. Он является катализатором образования тромбина, который регулирует последнюю стадию в цепи процесса коагуляции. Первой функцией тромбина является расщепление фибриногена с образованием фибриновых мономеров, которые за счет поперечных связей образуют нерастворимый гель, фибриновый сгусток.
Соединения настоящего изобретения пригодны для лечения и профилактики заболеваний, опосредованных тромбином и связанных с ним. Такие заболевания включают множество тромботических и протромботических состояний, при которых происходит активация процесса коагуляции и которые включают, не ограничиваясь ими, тромбоз глубоких вен, тромбоэмболию легочной артерии, тромбофлебит, окклюзию артерий вследствие тромбоза или эмболии, артериальную реокклюзию, происходящую во время или после проведения пластики сосудов или тромболизиса, рестеноз, наступающий после повреждения артерий или инвазивных кардиологических процедур, послеоперационные тромбоз или эмболия вен, острый или хронический атеросклероз, кровоизлияние, инфаркт миокарда, рак и метастазы, а также нейродегенеративные заболевания. Углеводные производные настоящего изобретения могут также применяться в качестве ингибиторов пролиферации гладкомышечных клеток и для лечения ангиогенеза, рака и ретровирусных инфекций, таких как ВИЧ-инфекция.
Кроме того, соединения настоящего изобретения могут использоваться в качестве антикоагулянтов и антикоагулирующих покрытий в аппарате экстракорпорального кровообращения, применяемого в процессе диализа и оперативного вмешательства.
Соединения настоящего изобретения могут использоваться также in vitro или ex vivo как антикоагулянты.
Предпочтительными производными углеводов, имеющих формулу I, в соответствии с настоящим изобретением являются такие производные, в которых R2 представляет собой (1-6С)алкил, R3 обозначает SО3 -, R4 представляет собой OSO3 -, а R1, n и р соответствуют данным ранее определениям; или их фармацевтически приемлемая соль.
Более предпочтительны такие углеводные производные формулы I, в которых R1 является метилом. Особенно предпочтительны углеводные производные формулы I, в которых n равно 1 и р равно 1. И наиболее предпочтительны такие углеводные производные формулы I, в которых R1 представляет собой СН2ОSО3 -.
Термин (1-6С)алкил относится к разветвленной или неразветвленной алкильной группе, включающей от 1 до 6 атомов углерода, такой, как метил, этил, изопропил, т-бутил, изопентил, гексил и др. Предпочтительными алкильными группами являются (1-4С)алкильные группы, такие, как метил, этил, (изо)пропил, н-бутил и т-бутил. Наиболее предпочтительной алкильной группой является метил.
Противоионы, компенсирующие заряженные части молекулы, относятся к фармацевтически приемлемым противоионам, таким, как водород, или, что более предпочтительно, ионы щелочных или щелочноземельных металлов, таким, как натрий, кальций или магний.
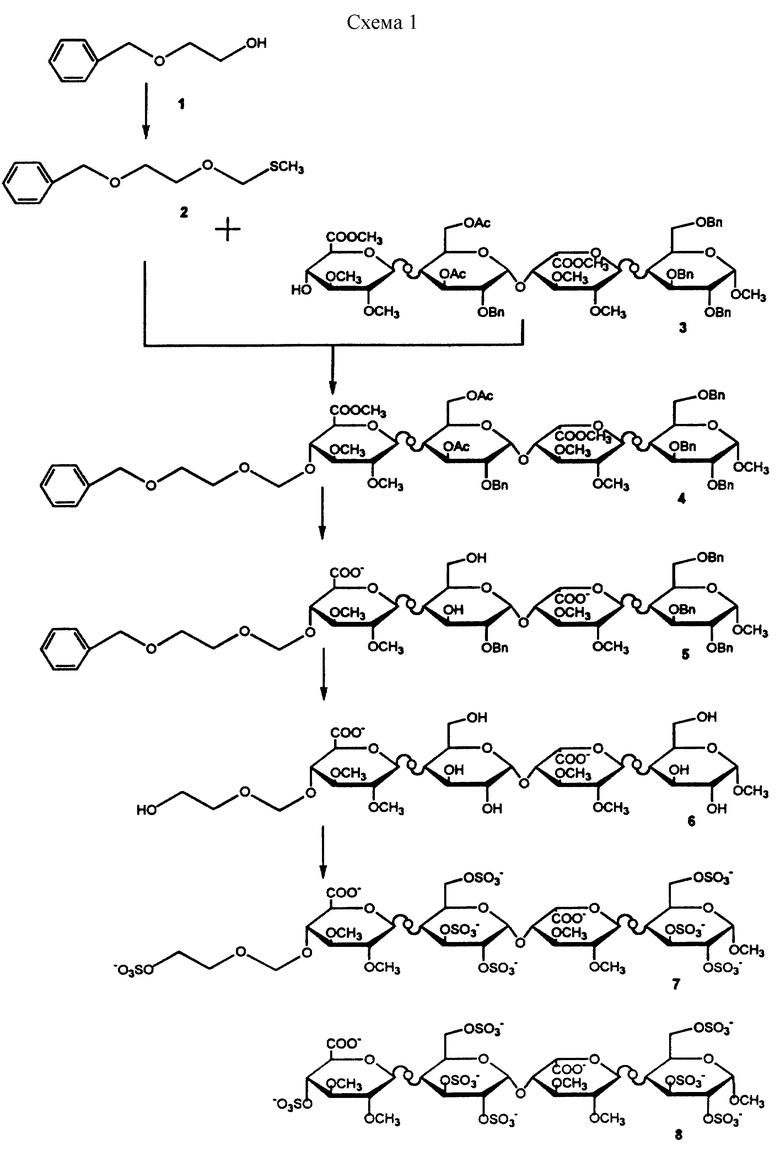
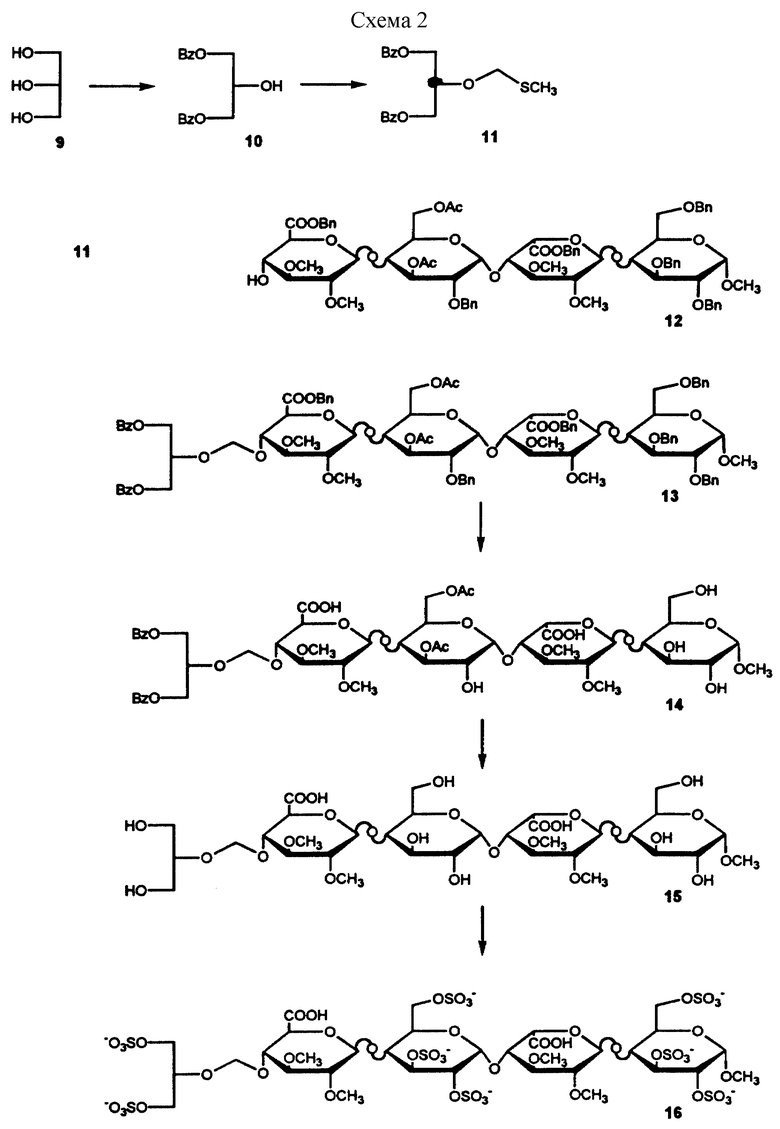
Углеводные производные настоящего изобретения могут быть получены при взаимодействии защищенной глицериноподобной или гликолеподобной части молекулы с 4-гидроксигруппой невосстановленного конца также защищенного тетрасахарида, который может быть получен методом, описанным в литературе (Westerduin P. , Bioorg. and Med.Chem., 1994, 2, 1267-1280). В дальнейшем защищающие группы удаляют с последующим сульфированием соединения, приводящим к образованию углеводного производного формулы I.
Для лечения тромбоза вен или при ингибировании пролиферации гладкомышечных клеток соединения настоящего изобретения могут вводиться энтерально или парентерально, при этом в случае людей предпочтительно использовать дневную дозу в 0,001-10 мг на 1 кг веса тела. Смешанные с фармацевтически приемлемыми добавками (приведенными в основном руководстве Gennaro et al., Remington's Pharmaceutical Sciences (18th ed. , Mack Publishing Company, 1990, см. в особенности часть 8: Pnarmaceutical Preparations and their Manufacture) эти соединения в случае активности их при пероральном, трансбуккальном или сублингвальном использовании могут быть спрессованы с получением твердых дозированных форм, таких, как пилюли, таблетки, или же могут быть подвергнуты иной обработке для изготовления капсул или суппозиториев. С помощью фармацевтически приемлемых жидкостей соединения можно применять также в виде препарата для инъекции, представляющего собой раствор, суспензию, эмульсию или в виде спрэя, в частности назального спрэя.
Для изготовления единичных дозированных форм, например таблеток, показано применение традиционных добавок, таких, как наполнители, красители, полимерные связующие вещества и др. В целом может использоваться любая фармацевтически приемлемая добавка, которая не мешает действию активного соединения.
Подходящие носители, вместе с которыми могут вводиться рассматриваемые композиции, включают лактозу, крахмал, производные целлюлозы и др., а также их смеси, используемые в соответствующих количествах.
Ниже приведены примеры, иллюстрирующие настоящее изобретение.
(В примерах даны ссылки на схемы 1 и 2. Промежуточные и конечные продукты имеют обозначения, отсылающие к соответствующему номеру на схемах).
Пример 1
Получение соединений 7 и 8
Получение 2
К охлажденному раствору (0oС) 2-бензилоксиэтанола 1 (2,84 мл) и хлорметилметилсульфида (1,59 мл) в диметиловом эфире этиленгликоля (25 мл) в атмосфере азота добавляют гидрид натрия в виде 60% дисперсии в минеральном масле (1,2 г) и перемешивают полученную смесь в течение 20 ч при комнатной температуре. К реакционной смеси добавляют метанол и продолжают перемешивание еще в течение 15 мин. Далее смесь разбавляют этилацетатом (100 мл), последовательно промывают водным раствором кислого карбоната натрия и водой, после чего высушивают органический слой над сульфатом магния и концентрируют в вакууме. Сырой продукт чистят затем хроматографированием на колонке с силикагелем с получением в результате 2 в количесте 2,5 г.
Получение 3
Синтез 3 описан в литературе (Westerduin P. et а1., Bioorganic and Medicinal Chemistry, vol. no. 11, pp. 1267-1280, 1994).
Получение 4
Смесь 3 (125 мг), 2 (64 мг) и порошка молекулярных сит (4 ) в дихлорметане (1,5 мл) перемешивают в течение 15 мин в атмосфере азота. Раствор охлаждают (5oС) и вносят в него свежеприготовленный раствор, содержащий N-йодсукцинимид (68 мг) и трифторметансульфоновую кислоту (2,7 мкл) в 1,5 мл 1,2-дихлорэтана-диоксана (1/1, объем/объем). Через 10 мин фильтруют красную реакционную смесь, разбавляют дихлорметаном, промывают последовательно водным раствором тиосульфата натрия и водным раствором кислого карбоната натрия. Органический слой высушивают над сульфатом магния и концентрируют в вакууме. Полученный остаток чистят вытеснительной хроматографией на Сефадексе LH-20, суспендированным в смеси дихлорметана-метанола (2/1, объем/объем) до получения в результате 4, 108 мг.
) в дихлорметане (1,5 мл) перемешивают в течение 15 мин в атмосфере азота. Раствор охлаждают (5oС) и вносят в него свежеприготовленный раствор, содержащий N-йодсукцинимид (68 мг) и трифторметансульфоновую кислоту (2,7 мкл) в 1,5 мл 1,2-дихлорэтана-диоксана (1/1, объем/объем). Через 10 мин фильтруют красную реакционную смесь, разбавляют дихлорметаном, промывают последовательно водным раствором тиосульфата натрия и водным раствором кислого карбоната натрия. Органический слой высушивают над сульфатом магния и концентрируют в вакууме. Полученный остаток чистят вытеснительной хроматографией на Сефадексе LH-20, суспендированным в смеси дихлорметана-метанола (2/1, объем/объем) до получения в результате 4, 108 мг.
Получение 5
К охлажденному (-5oС) раствору 4 (105 мг) в тетрагидрофуране (7,3 мл) добавляют 30%-ный водный раствор перекиси водорода (3,8 мл), перемешивают в течение 10 мин, после чего добавляют раствор гидроксида лития (1,25 М, 1,7 мл). Смесь перемешивают в течение 2 ч при температуре -5oС, после чего поднимают температуру до 0oС. После перемешивания в течение 20 ч повышают температуру до 20oС и продолжают перемешивание еще в течение 24 ч. Затем реакционную смесь охлаждают (0oС) и добавляют последовательно метанол (7,0 мл) и водный раствор гидроксида натрия (4 М, 2,0 мл). Перемешивают смесь в течение 1 ч, после чего снова повышают температуру до 20oС и продолжают перемешивание еще в течение 20 ч. После этого смесь охлаждают (0oС), подкисляют до рН 3 соляной кислотой (2 н.) и проводят экстракцию дихлорметаном. Органический слой промывают водным раствором сульфита натрия (5%), высушивают над сульфатом магния и концентрируют в вакууме. Сырой продукт чистят колоночной хроматографией на силикагеле с получением 5, 80 мг.
Получение 6
К раствору 6 (80 мг) в смеси воды (7 мл) и 2-метил-2-пропанола (7 мл) добавляют 80 мг палладия на угле (10%). Реакционную смесь помещают на 16 ч в атмосферу водорода. Катализатор удаляют фильтрованием и промывают смесью 2-метил-2-пропанола/воды. Фильтрат и промывные жидкости концентрируют в вакууме с последующей лиофилизацией, которая дает 6 в количестве 38 мг.
Получение 7 и 8
Раствор 6 (38 мг) в воде (0,8 мл) элюируют водой через колонку Дауэкс 50WХ8Н+ и выпаривают досуха объединенные фракции. После выпаривания вместе с N, N-диметилформамидом остаток растворяют в N,N-диметилформамиде (2,5 мл), помещают в атмосферу азота и добавляют комплекс триэтиламина и триоксида серы (SO3) (287 мг). Смесь перемешивают в течение ночи при температуре 50oС, охлаждают до 0oС и добавляют водный раствор кислого карбоната натрия (533 мг). Смесь перемешивают при температуре 20o в течение 1 ч, концентрируют до малого объема и обессоливают на колонке с Сефадексом G-25, суспендированным в смеси вода : ацетонитрил, 9 : 1 (объем/объем). Сырой продукт элюируют на колонке Дауэкс 50WX8Na+ и чистят анионообменной хроматографией на колонке (ВЭЖХ, Моно-Q 5/5, градиент хлорида натрия) с получением 12 мг соединения 7 {[α]
Пример 2
Получение соединения 16
Получение 10
К охлажденной (-20oС) смеси глицерина (10 г) и пиридина (109 мл) добавляют по каплям в течение 1 ч раствор бензоилхлорида (26,3 мл) в сухом диоксане (26 мл). Полученную смесь перемешивают в течение 16 ч при температуре 0oС, после чего добавляют воду. Перемешивают смесь в течение 15 мин и затем концентрируют ее до 1/5 объема, разбавляют дихлорметаном и промывают водой, водным раствором кислого карбоната натрия и водой. Органический слой высушивают над сульфатом магния и концентрируют в вакууме. Полученное масло чистят хроматографией на колонке с силикагелем с получением 10 в количестве 21 г.
Получение 11
К раствору 10 (600 мг) в смеси ацетонитрила : дихлорметана (1:1, объем/объем) добавляют метилсульфид (1,45 мл). Реакционную смесь охлаждают до 0oС и добавляют к ней по каплям смесь сухого бензоилпероксида (3,63 г) в смеси ацетонитрила: дихлорметана (1:1, объем/объем; 10 мл). После перемешивания в течение 16 ч при температуре 20oС смесь разбавляют дихлорметаном и промывают водным раствором кислого карбоната натрия и водой. Органический слой высушивают над сульфатом магния и концентрируют в вакууме. После хроматографирования на колонке с силикагелем получают 400 мг соединения 11.
Получение 12
Соединение 12 получают с помощью процедуры, аналогичной той, что была описана в работе Вестердуина с соавт. (Westerduin P. et al., Bioorganic and Medicinal Cnemistry, 1994, vol. 2, pp.1267-1280). На этапах получения 12 уроновые кислоты защищают бензильными группами, а не метильными группами.
Получение 13
Соединение 13 получают по методу, сходному с тем, что применялся для получения 4 посредством объединения 11 и 12.
Получение 14
К раствору 1,3 (480 мг) в 2-метил-2-пропаноле (60 мл) добавляют суспензию кислого карбоната натрия (142 мг) в воде (2 мл) и Pd/C (400 мг). Смесь помещают в атмосферу водорода на 16 ч. Затем катализатор удаляют фильтрованием и промывают его смесью 2-метил-2-пропанол/вода. Объединенные фильтрат и промывные жидкости концентрируют под вакуумом с получением 315 мг 14, который затем используют без дальнейшей очистки.
Получение 15
Соединение 14 (315 мг) растворяют в 0,35 н. водном растворе гидроксида натрия (10 мл). В течение ночи перемешивают реакционную смесь, после чего ее рН доводят до значения 8,5 с помощью 1 н. соляной кислоты. Далее смесь обессоливают на колонке с Сефадексом G-25, суспендированным в смеси вода : ацетонитрил : триэтиламин (90 : 10 : 0,1; объем/объем/объем). Объединяют соответствующие фракции и концентрируют их в вакууме. Затем продукт снова подвергают восстановлению методом, аналогичным приведенному для соединения 14. После выполнения процедуры и концентрирования фильтрата и промывных жидкостей смесь обессоливают на колонке с Сефадексом G-25, суспендированном в смеси вода:ацетонитрил : триэтиламин (90 : 10 : 0,1; объем/объем/объем). Объединяют соответствующие фракции, концентрируют их в вакууме, наносят на колонку Дауэкс 50WХ8Nа+, элюируют водой и в конце проводят лиофилизацию с получением 165 мг соединения 15.
Получение 16
Раствор 15 (165 мг) выпаривают вместе с N,N-диметилформамидом и растворяют в N,N-диметилформамиде (11,0 мл). Далее к реакционной смеси добавляют комплекс триэтиламина и триоксида серы (1,31 г). Смесь перемешивают при температуре 50oС в течение 16 ч, затем охлаждают ее (0oС), добавляют водный раствор кислого карбоната натрия (2,43 г) и продолжают перемешивание еще в течение 1 ч при температуре 20oС, после чего раствор концентрируют в вакууме. Полученный остаток обессоливают на колонке с Сефадексом G-25, суспендированным в смеси вода : ацетонитрил (9 : 1, объем/объем), объединяют соответствующие фракции и концентрируют их в вакууме. Затем продукт наносят на колонку Дауэкс 50WX8Na+, элюируя с нее, и чистят продукт на колонке с сефазорой (Q-Sepharose High Load) с применением градиента хлорида натрия, что дает в результате 160 мг соединения 16:
{[α]
Активированный фактор Х (Ха) представляет собой коагуляционный каскад; его активность слегка ингибируется антитромбином 111 (AT-111). Антикоагулянты могут ингибировать Ха непосредственно или аналогично гепарину посредством увеличения ингибиторной активности AT-111. Анти-Ха активность может быть оценена посредством определения степени гидролиза хромогенного субстрата S-2222 в присутствии АТ-111.
Настоящее исследование используется для определения анти-Ха активности образца человеческой плазмы, содержащей, по крайней мере, 0,8 АТ-111 Ед/мг (клинический образец).
Среда исследования: человеческая плазма, разведенная 3 объемами буфера Каби.
Соединение сравнения: межд. стандартный гепарин. Orgaran: партия АК, содержащая 11,4 анти-Ха Ед/мл
Реагенты: 1. Буфер Каби
состав: NaCl 10,17 г (174 ммоль)
эдетат динатрий дигидрат 3,26 г (9,6 ммоль)
трометамин (tris) 6,11 г (50,4 ммоль)
доведен до объема 1 л дистиллированной водой.
рН раствора устанавливают до 8,4 с помощью НСI (0,10 моль/л).
2. Ха-раствор
Бычий Ха-фактор (Каби Диагностика, Стокгольм, Швеция) растворяют в буфере с получением раствора, содержащего 1 Ед/мл (0,5 нКат/мл).
3. S-2222 раствор
S-2222 (Каби Диагностика) растворяют в дистиллированной воде с получением раствора, содержащего 0,374 мг/мл.
4. Разбавленные объединенные образцы плазмы. Плазма бралась от здоровых добровольцев, которые не принимали никаких лекарств в течение 10 дней (Bloedbank Nijmegen, The Netherlands). Ее объединяли и хранили при -20oС. Перед использованием эту плазму разбавляли 3 объемами буфера Каби.
5. Стандартный раствор калибровочного образца.
Стандартный гепарин или Orgaran партии АК растворяют в разбавленной объединенной плазме с получением стандартного раствора, содержащего приблизительно 0,1 анти-Ха Ед/мл.
6. Контрольные образцы
Требуемые количества стандартного гепарина или Orgaran партии АК растворяют в объединенной плазме, взятой от здоровых добровольцев, с получением раствора, содержащего:
а) стандартный гепарин: приблизительно 0,125 (низкая доза) и 0,50 (высокая доза) анти-Ха Ед/мл.
б) Orgaran: партия АК: приблизительно 0,15 (низкая доза) и 0,60 (высокая доза) анти-Ха Ед/мл.
Приготовление опытных образцов
Если требуется, человеческий AT-111 (Каби Диагностика) добавляют к клиническим образцам для получения образцов, содержащих, по крайней мере, 0,8 АТ-111 Ед/мл. Получают 4 следующих образца:
Образец А: 1 объем клинического образца +3 объема Каби
буфера (фактор разбавления 1/4)
Образец В: 1 объем образца А + 0,5 объема разведенной объединенной плазмы (фактор разбавления 1/6)
Образец С: 1 объем образца В + 0,5 объема разведенной
объединенной плазмы (фактор разбавления 1/9)
Образец Д: 1 объем образца С + 0,5 объема разведенной объединенной плазмы (фактор разбавления 2/9)
Разведенные образцы В, С и Д получают для того, чтобы ограничить требуемое количество клинического образца.
Если требуется, все 4 опытных образца разбавляют в том же соотношении разведенной объединенной плазмой с получением образцов, содержащих 0,02-0,08 АТ-Ха Ед/мл.
Пиковые контрольные образцы получают, используя ту же процедуру, как и для клинических образцов (разведенные пиковые контрольные образцы).
Определение Ха-активности
Каждый опытный образец/разведенный пиковый контрольный образец (0,05 мл) пепитируют в лунку титрационной микропланшеты при комнатной температуре. В каждую лунку добавляют Ха-раствор (0,05), используя шейкер Вари.
Точно через 2 мин в каждую лунку пепитируют Ха-раствор, 0,1 мл S-2222 раствора и планшета вновь встряхивается. Оставшееся количество Ха катализирует гидролиз S-2222, степень которого измеряется фотометрически.
Последующие периоды инкубации 2 и 22 мин соответственно при комнатной температуре, абсорбция каждого образца измеряется при 405 нм, используя Reader Microelisa model 310 (Organon Teknika, Oss, The Netherlands) и рассчитывается увеличение абсорбции (Δ ОД).
Каждый опытный образец/разведенный пиковый контрольный образец определяли дважды. Контрольная проба (0,05 мл разведенной обобщенной плазмы) была включена вместе со всеми 10 образцами.
Калибровочная кривая
Из аликвоты стандартного раствора калибровочного образца осуществили ряд разбавлений (фактор разбавления 1,3 для образцов гепарина или 1,4 для Orgaran образцов). Полученные стандартные образцы (приблизительно 10 образцов) должны содержать 0,01-0,10 анти-Ха Ед/мл. В каждой серии опытов тестировались 0,05 мл каждого стандартного образца, по крайней мере, 3 раза, как указано при определении Ха активности. Калибровочную кривую получают путем подбора прямой линии к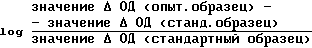
против значений log анти-Ха Ед/мл, используя площадь наименьших квадратов.
Для каждого клинического образца и контрольного пикового образца значение анти-Ха активности в Ед/мл определяли, используя калибровочную кривую. Значение найденное для клинического образца должно быть помножено на поправочный коэффициент. Поправка для повседневного изменения теста проводят с помощью контрольных пиковых образцов.
Поправочный коэффициент для:
а) низкой дозы стандартного гепаринового образца или Orgaran контрольного образца является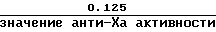
или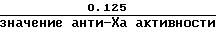
в) высокой дозы стандартного гепаринового образца или Orgaran контрольного образца является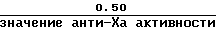
или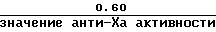
Поправочный коэффициент для клинического образца представляет собой среднее значение релевантных а) и в).
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПЕНТАСАХАРИДЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1999 |
|
RU2193040C2 |
| ПРОИЗВОДНЫЕ 3-ДЕЗОКСИОЛИГОСАХАРИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2133752C1 |
| ПЕНТАСАХАРИДНЫЙ КОНЪЮГАТ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2000 |
|
RU2266913C2 |
| ИНГИБИТОРЫ СЕРИНПРОТЕАЗЫ | 1997 |
|
RU2191193C2 |
| ИНГИБИТОРЫ ТРОМБИНА | 1996 |
|
RU2178796C2 |
| ИНГИБИТОРЫ ПРОТЕАЗЫ СЕРИНА | 1997 |
|
RU2178419C2 |
| СИНТЕТИЧЕСКИЕ ПОЛИСАХАРИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2167163C2 |
| ИНГИБИТОРЫ СЕРИНОВЫХ ПРОТЕАЗ | 1998 |
|
RU2183642C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗО[1,5-А]ПИРИДИНА КАК ИНГИБИТОРЫ СЕРИНПРОТЕАЗ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2175327C2 |
| ПРОИЗВОДНОЕ 11-(ФЕНИЛЗАМЕЩЕННЫЙ)ЭСТРА-4,9-ДИЕНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2135514C1 |
Изобретение относится к углеводному производному общей формулы I, где R1= H или СН2ОSO3 -; R2 и R3 независимо равны Н, (1-6С) алкил или SO3 -; R4= OSO3 -; n= 0 или 1; р=1 или 2; или его фармацевтически приемлемой соли. Соединения по изобретению обладают антитромботической активностью. Также предложена фармацевтическая композиция для лечения и профилактики заболеваний, опосредованных или связанных с тромбином, включающая указанное углеводное производное. 2 с. и 6 з.п.ф-лы. 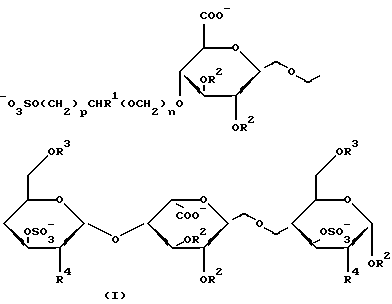
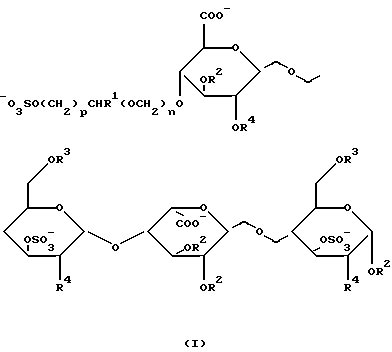
где R1 представляет собой Н или СН2OSO3 -;
R2 и R3 представляют собой независимо друг от друга Н, (1-6С) алкил или SO3 -;
R4 представляет собой OSO3 - или NHSO3 -;
n принимает значение 0 или 1;
р принимает значение 1 или 2;
или его фармацевтически приемлемая соль.
| УСТРОЙСТВО для ТРАНСПОРТИРОВАНИЯ и ПОДКЛЮЧЕНИЯ | 0 |
|
SU347964A1 |
| Машина для разрезания ленточных материалов | 1932 |
|
SU30099A1 |
| ПРОИЗВОДНЫЕ 3-ДЕЗОКСИОЛИГОСАХАРИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2133752C1 |

