Изобретение относится к ортозамещенным фенильным соединениям, пригодным для использования в качестве ингибиторов простагландинсинтазы, лекарственным препаратам, содержащим эти соединения, и методам применения указанных соединений в качестве противовоспалительных и жаропонижающих средств.
Нестероидные противовоспалительные средства (НПВС) на протяжении 2 столетий использовали в качестве основных противоревматических и противовоспалительных препаратов (Weissman, G. , Scientific American, стр. 84-90, 1991). Механизм действия НПВС связан с ингибированием биосинтеза простагландина (Vane, J.R., Nature-New Biology вып. 231, стр. 232-235, 1971). В частности указанные средства действуют в качестве ингибиторов циклооксигеназы (простагландин-G/Н-синтазы). Циклооксигеназа является первым ферментом в каскаде биотрансформаций арахидоновой кислоты, приводящих к образованию простагландинов подтипа D2, E2, и F2a. Кроме того, простациклин (ПГI2) и тромбоксаны A2 и В2 образуются из промежуточных продуктов биосинтеза ПГНS2 из циклооксигеназы. (Prostaglandins and Related Substances - A Practical Approach (1987). Benedetto, С., McDonald-Gibson, R.G., и Nigam, S., и Slater, T.F., из-во. ZRL Press, Washington, D.C). Указанные метаболиты арахидоновой кислоты участвуют в реализации процессов, выражаемых болевым синдромом, лихорадочным состоянием, агрегацией тромбоцитов и воспалением. Кроме того, простагландины ответственны за целосность слизистой оболочки желудочно-кишечного тракта (Cryer, В., и Feldman, M., Arch Intern. Med., вып. 152, 1145-1155, 1992) и функционирование почек, особенно в состояниях стресса (Whelton, А. , и Hamilton. C.W., J. Clin. Pharmacol. вып. 31, стр. 588-598, 994). Таким образом, средства, подавляющие циклооксигеназную активность, обладают противовоспалительным и анальгезирующим эффектами вследствие блокады синтеза медиатора боли и воспаления, однако из-за механизма своего действия эти же самые агенты могут оказывать побочные эффекты на желудочно-кишечный тракт и почки. Минимизация или устранение таких побочных эффектов в новом методе лечения позволяет осуществлять рациональный поиск "безопасных" НПВС с улучшенными профилем их распределения в желудочно-кишечном тракте и почках (Vane, J.R., Nature, вып. 367, стр. 215-216, 1994).
Вплоть до настоящего времени предполагали, что только лишь один изофермент циклооксигеназа ответственнен за простагландин-G/Н2-синтазную активность. Однако недавно появились сообщения о другой идентифицированной митоген-индуцируемой форме этого фермента, обозначенной как циклооксигеназа 2 (Сох 2) (Xie, W., Chipman, J.G., Robertson, D.L., Erickson, R.L., и Simmons, D. L. , Proc. Natl. Acad. Sci., вып. 88, стр. 2692-2696, 1991; Kujubu, D.A., Fletcher, B. S., Varnum, B.C., Lim, R.W., и Herschman, H.R., J. Biol Chem., вып. 266(20) стр. 12866-12872, 1991; Н1а, Т., и Neilson, К., Рrос. Natl. Acad. Sci. 89, стр. 7384-7388, 1991; Xie, W., Robertson, D.L., и Simmons, D.L. , Drug Development Research, вып. 25, стр. 249-265, 1992). Сох 2 демонстрирует физико-биологические свойства, отличные от классического типа циклооксигеназы Сох 1. Полагают, что механизм распределения продукции Сох 2 между тканями и клетками организма наряду с ее регулируемой генной экспрессией связан с ее медиаторной функцией в воспалительных реакциях и патологических состояниях, например ревматоидный артрит, в то время как генная экспрессия Сох 1 связана с конституитивными функциями. В соответствии с этим различием между Сох 1 и Сох 2 высказываемые ранее предположения о механизме действия НПВС, которые базируются на эффекте только лишь одного изофермента, вызывают сомнение. В частности нельзя принять точку зрения о том, что противовоспалительный и анальгетический эффекты НПВС связаны исключительно с ингибированием активности изофермента Сох 1. В самом деле, возможно, более вероятной покажется гипотеза о том, что механизм противовоспалительного и анальгетического действия большинства НПВС в ответ на хронический раздражитель может быть связан с ингибированием митоген-индуцируемой формы Сох 2, в то время как механизм воздействия имеющихся в настоящее время НПВС на желудочно-кишечный тракт и почки связан с ингибированием активности конституитивно экспрессируемого фермента Сох 1 (Vane, J.R., Nature, вып. 367, стр. 215-216, 1994). Таким образом, полагают, что лекарственные вещества, обладающие избирательным или специфическим ингибирующим действием относительно Сох 2, позволят снизить ее вредное воздействие на желудочно-кишечный тракт и почки, но одновременно сохранить высокий уровень их противовоспалительной, жаропонижающей и анальгезирующей активности.
Возможность использования более безопасных НПВС через механизм избирательного ингибирования ускорило проведение исследований по оценке влияния соединений на препараты из очищенного фермента. Избирательное ингибирование как изофермента, так и фактора с аналогичной ингибиторной активностью оценивали с использованием набора терапевтически пригодных НПВС (DeWitt, D.L., Meade, Е. А. , и Smith, W.L., Amer. J. Меd., вып. 95 (Suppl.2A), 40S-44S, 1993). Однако только лишь одно соединение в этом наборе продемонстрировало избирательность к Сох 2, то есть 6-метоксинафтилуксусная кислота (6MNA) - активный метаболит небуметона. Известен ряд других веществ, обладающих такой же селективностью к Сох-2, в том числе фактор BF389 (Mitchell, J.A., Akarasereenot, P., Thiemermann, С., Flower, R.J., и Vane, J.R., Proc. Natl. Acad. Sci., вып. 90, стр. 11693-11697, 1994) и фактор NS-398 (Futaki, N.,) Takahashi, S. , Yokayama, M, Arai, I., Higuchi, S., и Otomo, S., Prostaglandins, вып. 47, стр. 55-59, 1994; Masferrer, J.L., Zuieifel, B.S., Maiming, P.T., Hauser, S.D., Leaky, K.M., Smith, W.G., Isakson, P.C., и Seibert, K., Proc. Natl. Acad. Sci., вып. 91, стр. 3228-3232, 1994). В случае последнего соединения избирательное ингибирование Сох-2 приводило к блокаде синтеза in vivo предшественника простагландина, вызывающего аллергическую реакцию в ответ на каррагинан, однако не отмечено ни подавления синтеза простагландина, способствующего выделению желудочного сока, ни патологических изменений слизистой желудка (Masferrer и другие, см. выше).
Эти данные подтверждают предположение о том, что избирательные ингибиторы синтеза Сох 2 обладают мощной противовоспалительной активностью и имеют улучшенные показатели их безопасности для больного. Детальные механистические исследования показали, что фактор NS-398, а также второй избирательно действующий ингибитор Сох-2, то есть фактор DuP 697, обеспечивают свою селективность через уникальный биопроцесс (отчет, представленный Copeland R.A., Williams, J.M., Giannaras, J., Nurnberg, S., Covington, M., Pinto, D., Pick, S. , и Trzaskos, J. M. Mechanism of Selective Inhibition of the Inducible Isoform of Prostaglandin G/H Synthase (Механизм избирательного ингибирования индуцируемой изоформы простагландин-G/H-синтазы). Указанное ингибирование является конкурентным относительно обоих изоферментов, однако демонстрирует избирательный времязависимый эффект относительно Сох-2, приводящий к более мощному подавлению ее активности в течение более продолжительного периода. Времязависимое воздействие обеспечивает ингибирование реакции специфического связывания, которая может быть обратимой только после денатурации фермента и экстрагирования его из органической фазы.
Newkome G. R. и другие (J. Org. Chem. 1980, вып. 45, стр. 4380) сообщают о получении бис (5-карбокси-2-пиридил)бензолов, но не приводят никакой информации относительно назначения этих соединений.
Bushby и другие. (J. Chem. Soc. Perkin Trans., вып. I, стр. 721, 1986) раскрывают синтез замещенных терфенилов, включая примеры, приведенные далее в описании.
Hori M. и другие (Chem. Pharm. Bull., вып. 22(9), стр. 2020, 1974) сообщают о синтезе терфенилов, в том числе 2-фенил-2'-метилтио-1-дифенила.

Кеmр и другие (J. Org. Chem. , вып. 46, стр. 5441, 1981) сообщают о синтезе 4-метоксифенил-(4'-алкилфенил)бензолов.
Floyd и другие раскрывают в патенте США 4613611 α-гидрокси-β-оксо-[1,1': 2', 1''-терфенил] -4-этансульфоновую кислоту в форме мононатриевой соли для лечения сахарного диабета.
Имеется сообщение о том, что орто-бис(диметоксифенил)-бензолкарбоксамиды можно использовать в качестве антагонистов фактора активации тромбоцитов (Tilley, и другие. J. Med. Chem. вып. 32, стр. 1814, 1989).
В заявке на Европатент 130045 А1, опубликованной 01.02.85 раскрываются замещенные бис-(метоксифенил)бензолы, пригодные для использования в качестве анальгетиков и противовоспалительных средств.
В патенте США 3624142 раскрываются 4-метилсульфонилдифенилуксусные кислоты, пригодные в качестве противовоспалительных средств.
Ни в одной из вышеуказанных ссылок не раскрываются или предлагаются метилсульфонилы настоящего изобретения. Таким образом, цель настоящего изобретения направлена на создание соединений, относящихся к ингибиторам синтеза простагландинсинтазы, в том числе соединений, являющихся селективными ингибиторами Сох 2, которые можно использовать в качестве новых противовоспалительных средств с улучшенным терапевтическим профилем для лечения ревматических и воспалительных заболеваний, а также в пирогенной терапии.
Настоящее изобретение относится к ортозамещенным фенильным соединениям формулы I, раскрываемым далее в описании, в качестве ингибиторов простагландинсинтазы, к лекарственным препаратам на их основе и способам использования указанных соединений в медицинской практике в качестве противовоспалительных и жаропонижающих средств.
Настоящее изобретение предлагает любое соединение формулы I: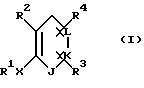
или их фармацевтическую соль или пролекарственную форму,
где J, К, и L каждый независимо означает CR3, CR4 или N;
Х означает простую связь (то есть радикал Х отсутствует), -(CHR5)2-, -CH=CR5-, -CR5=CH-, 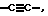 -(CHR5)pZ-, -Z(CHR5)p-, -С(=O)СН2 или -СН2С(=O)-;
-(CHR5)pZ-, -Z(CHR5)p-, -С(=O)СН2 или -СН2С(=O)-;
Z означает О или S;
R1 означает: фенил, замещенный 0-2 заместителями, выбранными из радикала R7, 2-нафтил, замещенный 0-2 заместителями, выбранными из радикала R7, С5-С7 циклоалкил, замещенный 0-1 заместителями, выбранными из радикала R9, С5-С7 циклоалкенил при условии, что, когда R1 связан непосредственно с гетероатомом, то указанный гетероатом не связан с атомом углерода, содержащим двойную связь в циклоалкеновом кольце, 5-10-членную гетероциклическую ароматическую систему, выбранную из группы фурила, тиенила, пирролила, тиазолила, оксазолила, N-метилпирролила, изоксазолила, изотиазолила, пиразолила, 3-пиридинила, пиразинила, бензофуранила, бензотиенила, бензотиазолила, бензоксазолила, бензотриазолила, бензоизотиазолила, бензизоксазолила, хинолинила, изохинолинила или липеридинила, причем указанная гетероциклическая ароматическая система замещена 0-2 заместителями, выбранными из радикала R7;
R2 означает: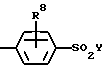
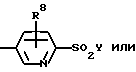
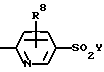
Y означает -СН3 или NH2.
R3 означает Н, F, Br, Cl, I, CN, C1-C4 алкил, замещенный 0-1 заместителями, выбранными из радикала R12, C1-C4 галогеналкил, С1-С4 алкенил, замещенный 0-1 заместителями, выбранными из радикала R13, NО2, NR15R16, SOmR11, SO2NR15aR16, -C(=O)R6, -COOR17, -C(=O)NR15aR16, или OR18;
R4 означает H, F, Br, Cl, I, C1-C2 алкил, C1-C2 алкокси, C1-С2 галогеналкил, -СF3, -SR10a;
В альтернативном варианте, когда R3 и R4 означают заместители у соседних атомов углерода, то R3 и R4, взятые вместе атомами углерода, с которыми они непосредственно связаны, образуют 5-7-членную карбоциклическую или гетероциклическую ароматическую систему, причем указанная гетероциклическая ароматическая система содержит от 1 до 3 гетеротомов, выбранных из N, О или S;
R5 означает C1-C2 алкил, C1-C2 алкокси или C1-С2 галогеналкил;
R6 означает водород, C1-C6 алкил, замещенный 0-1 заместителями, выбранными из радикала R14, фенил, замещенный 0-2 заместителями, выбранными из радикала R9, С5-С7 циклоалкил, замещенный 0-1 заместителями, выбранными из радикала R9, 5-10-членную гетероциклическую ароматическую систему, выбранную из группы фурила, тиенила, тиазолила, оксазолила, N-метилпирролила, изоксазолила, изотиазолила, пиразолила, пиридинила, пиридазинила, пиразинила или пиримидинила, причем указанная гетероциклическая ароматическая система замещена 0-2 заместителями, выбранными из радикала R7;
R7 означает заместитель у атома углерода, выбранный из группы, состоящей из Н, F, Br, Cl, I, C1-C4 алкила, фенила, СН2ОН, СН2ОСН3, C1-C4 алкокси, C1-C4 галогеналкила, -SR10, NR15R16, -C(=0)Rl0a, CH2COOR17 или OR19; при условии, что когда Х означает простую связь, R7 не может быть в ортоположении относительно X.
R8 означает Н, F, Br, C1, I, C1-C4 алкил, C1-C4 алкокси;
R9 означает Н, F, Br, C1, I, гидрокси, C1-C4 алкил или C1-C4 алкокси;
R10 означает Н или C1-C4 алкил;
R10a означает C1-C4;
R11 означает C1-C4 алкил, C1-C2 фторалкил, фенил или бензил;
R12 означает F, OR18; NR15R16, фенил, замещенный 0-2 заместителями, выбранными из радикала R9, -CN, -C(=O)R6, -COOR17, -C(=O)NR15aR16, или гетероциклическую ароматическую систему, выбранную из группы морфолинила, пиперидинила, пирролидинила, фурила, тиенила, пиридинила, пиперидазинила, пиримидинила, пиразинила, или тетрагидропиридинила, причем указанная гетероциклическая ароматическая система замещена 0-2 заместителями, выбранными из радикала R9;
R13 означает -CN, -C(=O)R6, -COOR17, -NO2, или NR15R16;
R14 означает F, ОН, C1-C4 алкокси, -NH2, фенил, замещенный 0-2 заместителями, выбранными из радикала R9, алкилкарбонил, арилкарбонил, -COOR17 или -C(=O)NH2;
R15 означает Н, C1-C4 алкил, замещенный 0-1 заместителями, выбранными из радикала R23, C6-С10 арил, С3-С7 циклоалкил, С4-С11 циклоалкилалкил, С2-С4 алкенил, C1-C4 алкокси, C1-C6 алкилкарбонил, C1-C6 алкоксикарбонил, C7-C14 арилалкоксикарбонил, С6-С10 арилоксикарбонил, C1-C6 алкиламинокарбонил, C6-С10 арилкарбонил, C1-C6 алкилсульфонил, C6-C10 арилсульфонил, C7-C14 алкиларилсульфонил, С7-C14 арилалкилсульфонил;
R15a означает Н, C1-C4 алкил, замещенный 0-1 заместителями, выбранными из радикала R23, С6-С10 арил, С3-С7 циклоалкил, С4-С11 циклоалкилалкил, С2-С4 алкенил или C1-C4 алкоксигруппу;
R16 означает Н или C1-C4 алкил; или
в альтернативном варианте, R15 и R16, взятые вместе, означают группу -(СН2)4-, -(СН2)5-, -(СН2)2O(СН2)2-, или -(CH2)2NR21(CH2)2-,
R17 означает C1-C4 алкил или арилалкил;
R18 означает C1-C4 алкил, замещенный 0-2 заместителями, выбранными из радикала R24, C6-C10 арил, С3-С7 циклоалкил, C1-C6 алкилкарбонил, C1-C6 алкиламинокарбонил, C7-C14 арилалкилкарбонил или C6-C10 арилкарбонил, замещенный 0-2 заместителями, выбранными из радикала R9;
R19 означает C1-C4 алкил, C1-C4 галогеналкил, C1-C4 алкоксиалкил, C1-C6 алкилкарбонил, C1-C6 алкиламинокарбонил, C7-C14 арилалкилкарбонил или С6-С10 арилкарбонил, замещенный 0-2 заместителями, выбранными из радикала R9;
R20 означает C1-C4 алкил, C1-C4 галогеналкил, C1-C4 алкоксиалкил, C6-С10 арил, С3-С7 циклоалкил, C1-C6 алкилкарбонил, C1-C6 алкиламинокарбонил, C7-C14 арилалкилкарбонил или C6-C10 арилкарбонил, замещенный 0-2 заместителями, выбранными из радикала R9;
R21 означает C1-С4 алкил или бензил;
R22 означает Н, R2, R1, C1-C4 алкил, С4-С10 циклоалкилалкил, C7-C14 арилалкил, или C6-С10 гетероарилалкил;
R23 означает Н, F, фенил, замещенный 0-2 заместителями, выбранными из радикала R9, -C(= O)R6, -COOR17, -C(=O)NHR16, или гетероциклическую ароматическую систему, выбранную из группы морфолинила, пиперидинила, пирролидинила, фурила, тиенила или тетрагидропиридинила, причем указанная гетероциклическая ароматическая система замещена 0-2 заместителями, выбранными из радикала R9;
R24 означает Н, F, NR15R16, фенил, замещенный 0-2 заместителями, выбранными из радикала R9, C1-C4 алкокси, C1-C4 алкилкарбонилокси, -C(=O)R6, -COOR17, -C(=O)NHR15R16 или гетероциклическую ароматическую систему, выбранную из группы морфолинила, пиперидинила, пирролидинила, фурила, тиенила или тетрагидропиридинила, причем указанная гетероциклическая ароматическая система замещена 0-2 заместителями, выбранными из радикала R9;
m означает 0-2; а
р означает 0-1,
при условии, что когда J и L оба означают атом азота, а К означает CR4, то R4 не может принимать значение SR10.
К предпочтительным соединениям формулы I или их фармацевтически приемлемым солям или пролекарственным формам относятся соединения, где:
J означает СН или N;
К и L каждый независимо означает CR3 или CR4;
Х означает простую связь, (то есть радикал Х отсутствует), 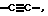 или -(CHR5)pZ-;
или -(CHR5)pZ-;
R3 означает группу, состоящую из Н, F, Br, CN, C1-C4 алкила, замещенного 0-1 заместителями, выбранными из радикала R12, C1-C4 галогеналкил, NO2, SOmR11, -C(O=)R6 или OR18;
R4 означает Н, F, СН3 или в альтернативном варианте, когда R3 и R4 имеют заместители при соседних атомах углерода, R3 и R4, взятые вместе с атомами углерода, с которыми они непосредственно связаны, образуют 5-7-членную карбоциклическую ароматическую систему;
R6 означает водород, C1-C6 алкил, замещенный 0-1 заместителями, выбранными из радикала R14, или фенил, замещенный 0-2 заместителями, выбранными из радикала R9, a
R7 означает заместитель при атоме углерода, выбранный из группы, состоящей из Н, F, Вr, С1-С4 алкила, СН2ОН, СН2ОСН3, C1-C4 алкокси, C1-C4 галогеналкила, NR15R16, -C(=O)R10;
в котором все другие заместители в структуре соединения формулы I имеют вышеуказанные значения.
К особенно предпочтительным соединениям формулы I относятся соединения или их фармацевтически приемлемые соли или пролекарства, в которых:
R8 означает Н;
R9 означает Н;
R12 означает F, OR18, -CN, -COOR17;
R13 означает -CN, -C(=O)R6, -COOR17, -NO2, или NR15R16;
R14 означает Н;
R15 означает Н или C1-C4 алкил;
R16 означает Н или C1-C4 алкил;
R18 означает Н или C1-C4 алкил;
R19 означает C1-C4 алкил.
К самым предпочтительным соединениям или их фармацевтически приемлемым солям или пролекарствам относятся соединения, выбранные из группы, состоящей из:
(а) соединений формулы Iа: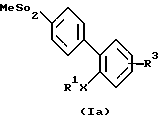
где: R1X означает фенил, a R3 означает водород,
R1X означает фенил, а R3 означает 4-ОН,
R1X означает фенил, а R3 означает 4-NO2
R1X означает фенил, а R3 означает 5-NO2,
R1X означает фенил, а R3 означает 4-СН3С(=O),
R1X означает 4-фторфенил, а R3 означает Н,
R1X означает 4-метоксифенил, а R3 означает Н,
R1X означает 4-метилфенил, а R3 означает Н,
R1X означает 3-метоксифенил, а R3 означает Н,
R1X означает 3,4-диметоксифенил, а R3 означает Н,
R1X означает 4-гидроксиметилфенил, а R3 означает Н,
R1X означает 4-метоксиметилфенил, а R3 означает Н,
R1X означает 4-диметиламинофенил, а R3 означает Н,
R1X означает 4-формилфенил, а R3 означает Н,
R1X означает 2-нафтил, a R3 означает Н,
R1X означает 5-метокси-2-нафтил, а R3 означает Н,
R1X означает 3-хинолинил, а R3 означает Н,
R1X означает 2-хинолинил, а R3 означает Н,
R1X означает 5-бензотиенил, а R3 означает Н,
R1X означает 2-бензотиенил, а R3 означает Н,
R1X означает 3-пиридил, а R3 означает Н,
R1X означает  а R3 означает Н,
а R3 означает Н,
R1X означает фенокси, а R3 означает Н,
R1X означает 1-циклогексенил, а R3 означает Н,
R1X означает циклогексил, а R3 означает Н,
R1X означает 4-фторфенокси, а R3 означает Н,
R1X означает циклогексилокси, а R3 означает Н,
R1X означает бензилокси, а R3 означает Н,
R1X означает 1-пиперидинил, а R3 означает Н,
R1X означает 1-пирролил, а R3 означает Н,
(b) соединение формулы I, которое представляет собой
2-(4-метилсульфонилфенил)-3-фенилнафталин,
(c) соединение формулы I, которое представляет собой
3-(4-метилсульфонилфенил)-2-фенилпиридин и
(d) соединение формулы I, которое представляет собой
2-(4-аминосульфонилфенил)-1-дифенил.
Настоящее изобретение также предлагает лекарственные препараты, содержащие соединение формулы I и фармацевтически приемлемый носитель.
Соединения, раскрываемые выше, можно использовать в качестве противовоспалительных и жаропонижающих средств при введении их в форме лекарственных препаратов в организм млекопитающего в случае необходимости его лечения с использованием таких противовоспалительных и жаропонижающих средств. В объем настоящего изобретения входят лекарственные препараты, содержащие эффективное количество вышеуказанных соединений формулы I, обладающих эффектом ингибитора активности ПГНS2, противовоспалительного или жаропонижающего средства. В объем настоящего изобретения входят также способы лечения артрита и других воспалительных болезней у млекопитающего, заключающийся в том, что в организм млекопитающего вводят терапевтически эффективное количество соединения формулы I, раскрываемого выше.
Соединения предлагаемого изобретения можно также вводить в комбинации с одним или несколькими дополнительными лекарственными средствами. Назначение соединения формулы I предлагаемого изобретения в комбинации с таким дополнительным лекарственным средством имеет преимущество относительно раздельного их приема, поскольку позволяет повысить терапевтическую эффективность препарата и при этом использовать каждый из них в меньших дозах. Использование препарата в меньшей дозировке позволяет свести к минимуму возможность возникновения побочных эффектов, увеличивая тем самым порог его переносимости.
Под термином "терапевтически эффективное количество" следует понимать количество соединения формулы I, вводимого в чистом виде или в сочетании с дополнительным лекарственным средством в клетку или организм млекопитающего, которое является эффективным для ингибирования активности ПГНS2, в такой степени, чтобы предупредить или облегчить состояния больного, обусловленное воспалительным процессом, или блокировать развитие болезни.
Под используемом в данном описании определении "принимаемый в сочетании" или "комбинированное лечение" следует понимать соединение формулы I и один или несколько дополнительных лекарственных средств, которые вводят параллельно в организм млекопитающего в ходе его лечения. При комбинированной терапии каждый компонент можно принимать одновременно или последовательно в любом порядке в разные интервалы времени. Таким образом, каждый компонент можно принимать по отдельности, но через достаточно близкий промежуток времени, чтобы получить желаемый лечебный эффект.
Предлагаемые соединения могут иметь асимметричные центры. Если не указано иначе, все хиральные, диастереизомерные и рацемические формы предлагаемых соединений включены в объем настоящего изобретения. Многие геометрические изомеры олефинов, сопряженные связи C=N и тому подобное могут также присутствовать в предлагаемых соединениях, и каждая из таких устойчивых изомерных форм рассматривается в объеме настоящего изобретения. Ясно, что соединения предлагаемого изобретения могут содержать ассиметрически замещенные атомы углерода, и их можно выделить в форме оптически активных или рацемических соединений. Специалисты в данной области техники хорошо знакомы с методами получения оптически активных соединений, например, путем разделения рацемических смесей или синтезом их из исходных оптически активных соединений. Если не оговорено особо, конкретная стереохимия или изомерная форма, все хиральные, диастереоизомерные, рацемические формы и все геометрические изомеры могут быть представлены в структуре предлагаемых соединений.
Если любая переменная появляется более 1 раза в любой составляющей или в любой формуле, определение ее при каждом появлении не зависит от ее определения при каждом другом появлении. Так, например, если указано, что группа замещена 0-3 заместителями, выбранными из R6, то эта группа может необязательно иметь до трех заместителей, выбранных из R6, при этом значение R6 при каждом появлении переменной выбирают независимо от установленного перечня возможных значений R6. Можно привести также в качестве примера группу -N(R5a)2, каждый из двух заместителей радикала R5a которой при атоме N выбирают самостоятельно из установленного перечня возможных значений радикала R5a. Аналогичным образом, например в случае группы -C(R7)2-, каждый из двух заместителей из R7 при атоме С выбирают самостоятельно из установленного перечня возможных значений радикала R7.
В случае когда указано, что связь с заместителем проходит через связь, соединяющую два атома в кольце, такой заместитель может быть связан с любым атомом в кольце.
Если заместитель приводится без указания атома, через который указанный заместитель связан с остальными группами углеродной цепи соединения формулы I, то указанный заместитель может быть связан через любой свой атом. Например, если заместитель означает пиперазинил, пиперидинил или тетразолил, то любая указанная пиперазинильная, пиперидинильная или тетразолильная группа, если не оговорено особо, может быть связана с остальными группами углеродной цепи соединения формулы I через любой свой атом.
Допускаются комбинации заместителей и/или переменных только при условии, что такие комбинации приведут с получению стабильных соединений. Под стабильным соединением или стабильной структурой следует понимать в данном описании соединение, которое обладает достаточной устойчивостью, чтобы выдержать операцию выделения его из реакционной смеси с требуемой степенью чистоты и последующую технологию приготовления из него эффективного лекарственного средства.
Под используемом в настоящей заявке термином "замещенный" следует понимать любой один или несколько атомов водорода при обозначенном атоме, замещенный группой, выбранной из установленного перечня значений при условии, что главная валентность обозначенного атома не является избыточной и что в результате такого замещения получают стабильное соединение. В случае когда заместитель означает кето-группировку (то есть =O), 2 атома водорода могут быть замещены при обозначенном атоме.
Используемый в данном описании термин "алкил" означает радикалы ряда насыщенных алифатических углеводородов как с разветвленной, так и нормальной цепью, содержащие определенное число атомов углерода (например, "C1-С10" означает алкил с 1-10 атомами углерода); термин "галогеналкил" означает радикалы ряда насыщенных алифатических углеводородов как с разветвленной, так и нормальной цепью, содержащие определенное число атомов углерода, замещенные 1 или несколькими атомами галогена (Например, CvFw, где v=1-3, a w=1-(2v+l)); "алкокси" означает алкильную группу с указанным количеством атомов углерода в цепи, связанных через кислородный мостик; "алкилтио" означает алкильную группу с указанным количеством атомов углерода в цепи, связанных через серный мостик; "диалкиламино" означает атом N, замещенный 2 алкильными группами с указанным количеством атомов углерода; "циклоалкил" означает радикалы ряда непредельных циклических углеводородов, в том числе моно-, би- или полициклические ароматические системы, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил и адамантил; и "бициклоалкил" означает радикалы ряда предельных бициклических углеводородов, например [3.3.0]бициклооктан, [4.3.0]бициклононан, [4.4.0]бициклодекан (декалин), [2.2.2] бициклооктан и так далее. Под термином "алкенил" следует понимать цепи из атомов углерода как неразветвленной, так и разветвленной конфигурации с одной или несколькими ненасыщенными углерод-углеродными связями, которые могут присутствовать в цепи в любой устойчивой ее точке, например этенил, пропенил и тому подобные; и "алкинил" означает цепи из атомов углерода как неразветвленной, так и разветвленной конфигурации с одной или несколькими тройными углерод-углеродными связями, которые могут присутствовать в цепи в любой устойчивой ее точке, например этинил, пропинил и тому подобные.
Термины "алкилен", "алкенилен", "фенилен" и тому подобные означают алкильные, алкенильные и фенильные группы соответственно, которые связаны через двойные связи с остальным группам циклической структуры соединения формулы I. Так например, "алкилен", "алкенилен", "фенилен" и тому подобные в данном описании могут иметь как альтернативное, так и эквивалентное обозначения "-(алкил)-", "-(алкенил)-", "-(фенил)-" и тому подобное.
Используемые в данном описании термины "гало" или "галоген" означают фторо, хлоро, бромо и йодо; а "противоион" означает небольшие отрицательно заряженные молекулярные частицы, например, хлорид, бромид, гидроксид, ацетат, сульфат и тому подобные.
Используемый в данном описании термин "арил" или "остаток углеводорода ароматического ряда" означает фенил или нафтил; термин "арилалкил" означает арильную группу, связанную через алкильный мостик.
Используемый в данном описании термин "карбоцикл" или "карбоциклический радикал" означает любой устойчивый 3-7-членный моноциклический или бициклический карбоцикл, или 7-14-членный бициклический или трициклический карбоцикл, или полициклический карбоцикл, содержащий до 26 атомов углерода, каждый из которых может быть насыщенным, частично ненасыщенным или ароматическим. В качестве примера таких карбоциклов могут служить, но не ограничивая их объема, циклопропил, циклопентил, циклогексил, фенил, дифенил, нафтил, инданил, адамантил или тетрагидронафтил(тетралин).
Под используемыми в данном описании терминами "гетероцикл" или "гетероарил" или "гетероциклический" следует понимать любой устойчивый 5-7-членный моноциклический или бициклический гетероцикл или 7-10-членный бициклический гетероцикл, каждый из которых может быть насыщенным, частично ненасыщенным или ароматическим, состоящий из атомов углерода и 1 до 4 гетеротомов, независимо выбранных из группы N, О и S, при этом гетероатомы N и S могут быть при желании окислены, а атомы N кватернизованы, включая любую бициклическую группировку, в которой любой из вышеуказанных гетероциклов может быть конденсирован с бензольным кольцом. Гетероцикл может быть связан с боковой группой при любом гетероатоме или атоме углерода, обеспечивая тем самым получение стабильной структуры. Гетероциклы, раскрываемые в настоящем описании, могут быть замещены у атома углерода или азота при условии, что полученное соединение стабильно. В качестве примера таких гетероциклов могут служить, но не ограничивая их объема, пиридил (пиридинил), пиримидинил, фуранил (фурил), тиазолил, тиенил, пирролил, пиразолил, имидазолил, тетразолил, бензофуранил, бензотиофенил, индолил, хинолинил, изохинолинил, бензимидазолил, пиперидинил, пирролидинил, 2-пирролинил, тетрагидрофуранил, тетрагидрохинолинил, тетрагидроизохинолинил, декагидрохинолинил, октагидроизохинолинил, пиранил, изобензофуранил, 2Н-пирролил, изотиазолил, изоксазолил, оксазолил, пиразинил, пиридазинил, индолизинил, изоиндолил, 3Н-индолил, 12-индазолил, пирролидинил, пирролинил, имидазолидинил, имидазолинил, пиразолидинил. пиразолинил, пиперидинил, пиперазинил, индолинил, изоиндолинил, морфолинил или оксазолидинил. В объем настоящего изобретения входят также соединения с конденсированным кольцом и спиро-соединения, содержащие, например, вышеуказанные гетероциклы.
Под используемом в данном описании термином "фармацевтически приемлемые соли" следует понимать производные раскрываемых соединений, в которых любое полученное соединение формулы I модифицируют путем получения его в форме соли присоединения кислоты или основания. В качестве примера фармацевтически пригодных солей можно привести, но не ограничивая их объема, соли минеральных и органических кислот с остатками оснований, например аминов; щелочные или органические соли с остатками кислот, например карбоновых кислот; и тому подобное.
Под используемом в данном описании изобретения термином "Пролекарства" следует понимать любые ковалентно связанные носители, которые высвобождают in vivo активное соединение формулы I из исходного лекарственного средства при введении указанной пролекарственной формы в организм млекопитающего. Пролекарственную форму соединений формулы I получают путем модификации содержащихся в них функциональных групп, при которой происходит трансформация их при расщеплении как вследствие рутинных манипуляций, так и in vivo, в исходные активные соединения. В объем пролекарственных форм входят соединения формулы I, гидроксильная, амино-, сульфгидрильная или карбоксильная группы которых связаны с любой группой, которая при введении в организм млекопитающего отщепляется с образованием свободной гидроксильной, амино-, сульфгидрильной или карбоксильной группы соответственно. В качестве примеров таких пролекарственных форм можно привести, но без ограничения их объема ацетатные, формиатные, бензоатные производные спирта и функциональные аминогруппы в соединениях формулы I и тому подобные.
В объем фармацевтически приемлемых солей соединений формулы I входят традиционные нетоксичные соли или четвертичные соли аммония соединений формулы I, образованные, например, из нетоксичных неорганических или органических кислот. В качестве таких традиционных нетоксичных солей, могут служить, например, соли, образованные из неорганических кислот, например соляной, бромистоводородной, серной, сульфаминовой, фосфорной, азотной и тому подобных; и соли, полученные из органических кислот, например уксусной, пропионовой, янтарной, гликолевой, стеариновой, молочной, яблочной, винной, лимонной, аскорбиновой, памовой, малеиновой, гидроксималеиновой, фенилуксусной, глутаминовой, бензойной, салициловой, сульфаниловой, 2-ацетоксибензойной, фумаровой, толуолсульфокислоты, метансульфокислоты, этандисульфокислоты, щавелевой, изэтионовой и тому подобных.
Фармацевтически приемлемые соли настоящего изобретения можно получить из соединений формулы I, содержащих остаток основания или кислоты стандартными методами органического синтеза. Такие соли обычно получают путем взаимодействия свободного основания или кислоты со стехиометрическими количествами или избытком соответствующей солеобразующей неорганической или органической кислоты или основания в подходящем растворителе или различных смесях растворителей.
Фармацевтически приемлемые соли присоединения кислот настоящего изобретения можно получить путем взаимодействия соединений формулы I с соответствующим количеством неорганического основания, например гидроксидами щелочных и щелочноземельных металлов, например гидроксидами натрия, калия, лития, кальция или магния, или органического основания, например аминовым основанием, например дибензилэтилендиамином, триметиламином, пиперидином, пирролидином, бензиламином и тому подобных, или гидроксидом четвертичного аммония, например гидроксидом тетраметиламмония и тому подобное.
Как указывалось ранее, фармацевтически приемлемые соли соединений формулы I настоящего изобретения можно получить путем взаимодействия указанных соединений в форме свободной кислоты или основания со стехиометрическим количеством подходящего основания или кислоты соответственно в воде или органическом растворителе или смеси из двух растворителей; обычно предпочтительно проведение реакции в безводной среде, например, такой как простой диэтиловый эфир, этилацетат, этанол, изопропанол или ацетонитрил. Перечень пригодных для использования солей можно найти в справочнике Remington Pharmaceutical Sciences, 17th изд., Mack Publishing Company, Easton, PA, 1985, стр. 1418, который указан в данном описании в качестве ссылочного материала.
Синтез
Соединения предлагаемого изобретения можно получить различными способами, которые общеизвестны специалистам в области органического синтеза. Соединения настоящего изобретения можно получить в соответствии со способами, описываемыми ниже, в сочетании с методами, общеизвестными в области органического синтеза, или их модификациями, которые очевидны специалистам. К предпочтительным способам синтеза предлагаемых соединений относятся способы, раскрываемые ниже, которые, однако, не должны служить ограничением объема. Все противопоставленные источники литературы включены в данное описание в качестве ссылочного материала, и их следует рассматривать как единое целое.
Новые соединения формулы I можно получить путем проведения различных реакций и используя методы, описываемые в данном разделе. Указанные реакции проводят в растворителях, которые совместимы с используемыми реагентами и исходными материалами и пригодны для осуществляемых превращений. Кроме того, при осуществлении способов синтеза, раскрываемых ниже, необходимо иметь в виду, что все предлагаемые реакционные условия, включая растворитель, реакционную среду, температуру проведения реакции, продолжительность проведения эксперимента, должны подбираться в соответствии с условиями, типичными для данного типа реакции, которая должна быть очевидна любому специалисту в данной области техники. Специалист в области органического синтеза должен понимать, что функциональные группы, присутствующие в различных участках высвобождаемой молекулы, должны быть совместимы с предлагаемыми реагентами и реакциями. Не все соединения формулы I, подпадающие под данный класс, могут быть совместимы с некоторыми реакционными условиями, которые необходимы в некоторых из раскрываемых способов. Такие ограничения, касающиеся заместителей, которые должны быть совместимы с реакционными условиями, станут очевидны специалисту в данной области, и тогда необходимо использовать альтернативные методы.
Соединения формулы I, где R1 означает замещенный арил, Х означает простую связь (то есть радикал Х отсутствует), R2 означает 4-метилсульфонилфенил, a R3, R4, R7 и R8 имеют вышеуказанные значения, можно получить в соответствии с общей методикой синтеза, проиллюстрированной в Схеме 1.
Взаимодействие замещенной надлежащим образом фенилбороновой кислоты с орто-дибромбензолом в соответствии с методологией, предложенной Suzuki (A. Suzuki и другие, J. Am. Chem. Soc., 1989, вып. 11, стр. 513 и V.N.Kalinin, Russ. Chem. Rev. , 1991, вып. 60, стр. 173) дает смесь, состоящую из 2-бромдифенила А и 1,2-диарилбензола. В качестве примера растворителей, пригодных для использования в этой реакции сочетания, могут служить, но не ограничивая их объема, толуол, диметилформамид, диоксан и этанол. Реакцию проводят в присутствии палладиевого катализатора, например тетракис(трифенилфосфин)палладий или бис(трифенилфосфин)палладий дихлорид. Продукт реакции сочетания можно выделить с использованием стандартных методов хроматографии, известных специалистам в области органического синтеза, с получением требуемого полупродукта синтеза дифенилов. Взаимодействие методом Suzuki полученного 2-бромдифенила с 4-фенилбороновой кислотой при использовании реакционных условий, описанных выше, дает 2-(4'-метилтио)фенил-1-дифенил. В результате последующего окисления метилтиогруппы до соответствующей метилсульфонильной группы получают соединение формулы I. Указанную реакцию окисления можно проводить с использованием любого из реагентов, известных специалистам в данной области для восстановления меркаптанов до сульфонов. В качестве примера таких реагентов могут служить, но не ограничивая их объема, оксон в смеси метанола с водой (Trost и другие. Теt. Lett. вып. 22 (14), стр. 1287, 1981), перекись водорода, м-хлорнадбензойная кислота или монопероксифталевая кислота, соль магния.
В альтернативном варианте соединения формулы I, где R1 означает замещенный арил, Х означает простую связь, a R2 означает 4-метилсульфонилфенил, можно также получить из коммерчески доступных 2-бромфенолов, как показано в схеме 2. Реакцию сочетания по Suzuki 2-бромфенола с фенилбороновой кислотой можно проводить при вышеописанных условиях с использованием как свободной, так и защищенной соответствующим образом группой фенола или соответствующего трифлата. Вторая реакция сочетания по Suzuki между промежуточным трифлатом и 4-метилтиофенилбороновой кислотой с последующим окислением по ранее описанной методике дает соединение формулы I.
Соединения формулы I, где R2 означает 4-метилсульфонилфенил, Х означает простую связь, а R1 означает циклоалкенильный или циклоалкильный остаток, получают из 2-бром-(4'-метилтио)дифенилов серией реакционных стадий, показанных в Схеме 3. Требуемые дифенильные соединения, используемые в качестве исходных материалов, получают реакцией сочетания по Suzuki 1,2-дибромбензола с 4-метилтиофенилбороновой кислотой при использовании вышеописанных условий.
Обработка 2-бром-(4'-метилтио)дифенила сильным основанием при низкой температуре с последующим прибавлением соответствующего циклоалканона дает промежуточное соединение (1-гидроксициклоалкил)-дифенил. В качестве примера подходящих сильных оснований для использования в этой реакции могут служить н-бутиллитий, трет-бутиллитий или метиллитий. Реакцию проводят в апротонном растворителе, например тетрагидрофуране, простом эфире, гексане или 1,4-диоксане. Дегидратацию полученного третичного спирта можно легко обеспечить путем его обработки каталитическим количеством сильной кислоты, например п-толуолсульфокислоты, в подходящем растворителе, например толуоле. При последующем окислении метилтиогруппы до метилсульфонильной группы по методике, описанной выше, образуется соединения формулы I, где R1 означает циклоалкенил. Каталитическая гидрогенизация указанных циклоалкенильных производных через походящий катализатор, например оксид платины, в соответствующем полярном растворителе, например метаноле, приводит к получению соединения формулы I, где R1 означает циклоалкил. В альтернативном варианте циклоалкильные соединения можно получить из промежуточного спирта вначале окислением метилтиогруппы до метилсульфона с последующей непосредственной гидрогенизацией третичных спиртов при
использовании аналогичных условий дегидрогенизации, которые описаны выше для восстановления олефина.
Соединения формулы I, где Х означает кислород, R1 означает замещенный или незамещенный фенил, а R2 означает 4-метилсульфонилфенил, можно получить из 2-гидрокси-(4'-метилтио)дифенила, как проиллюстрировано в Схеме 4.
Обработка 2-гидрокси-1-(4'-метилсульфонил)-дифенила (полученного через реакцию синтеза, проиллюстрированную в Схеме 3) подходящим основанием, например гидридом натрия, с последующим добавлением 4-фтор-1-нитробензола приводит к образованию промежуточного соединения синтеза 2-(4-нитрофенокси)-дифенила. При восстановлении нитрогруппы (см. "Compendium of Organic Synthetic Methods" том. 1, стр. 266, 1971) образуется соединение формулы I, где R7= NH2. Удаление аминогруппы можно осуществлять по методу Cadogan, J.I.G. и другие. (J. Chem. Soc. Perkin. Trans. вып. I, cтp. 541, 1973). В альтернативном варианте аминогруппу можно превратить в другие функциональные группы через промежуточную соль диазония при использовании методов, которые известны специалистам в области органического синтеза. При использовании этой методологии синтеза можно легко получить другие замещенные надлежащим образом простые арильные эфиры формулы I.
Соединения формулы 2, где R2 означает 4-метилсульфонилгетероарил, можно получить катализированной палладием реакцией сочетания по Suzuki 2-дифенилбороновой кислоты с замещенным надлежащим образом 4-метилтиогетероарилбромидом или трифлатом (см. Схему 5). Избирательное окисление оксона обеспечивает выход требуемого метилсульфонильного соединения.
2-Метилтио-5-бром-пиридиновые реагенты, используемые в Схеме 5, можно получить за одну стадию из коммерчески доступных 2,5-дибромпиридинов, как проиллюстрировано в Схеме 6, путем обработки солью щелочного металла метилмеркаптана, например метилтиолата натрия в любом полярном апротонном растворителе, например безводном диметилформамиде.
Другие бром- или гидроксизамещенные метилтиогетероарилы, которые можно использовать в качестве исходных соединений в реакции сочетания с 2-дифенилбороновой кислотой по методике Suzuki, можно легко получить аналогичным образом из коммерчески доступных реактивов.
Например, 2-бром-5-метилтиопиридин можно получить путем обработки 2-метокси-5-бромпиридина (Shiao. M. J. и другие. Syn. Comm. вып. 20 (19), стр. 2971, 1990) н-бутиллитием в безводном тетрагидрофуране при температуре -78oС и последующим гашением реакции диметилдисульфидом с выходом 2-метокси-5-метилтиопиридина. В результате реакции деметилирования образуется 2-гидрокси-5-метилтиопиридин, который при взаимодействии с форсфороксибромидом дает требуемое исходное соединение 2-бром-5-метилтиопиридин (Схема 7).
Соединения формулы I, где X означает простую связь, a R1 означает ароматический гетероцикл, можно получить заменой на бромгетероарил бромбензола, используемого в реакциях сочетания по Suzuki, описанных в вышеприведенных Схемах. В качестве примера пригодных для использования бромгетероарилов могут служить, но не ограничивая объема, 2- или 3-бромфуран, 2- или 3-бромтиофен, 3-бромпиридин, 2-бромбензофуран (Baciocchi, E. и другие. J. Perk. Trans. , вып. II, 1976, стр. 266) и 5-бромбензотиофен (Worden и другие. J. Het. Chem. вып. 25, стр. 1271, 1988).
Соединения формулы I, где R8 имеет значения кроме Н можно получить при использовании в качестве исходного материала замещенные надлежащим образом 4-метилтиофенолы. Такие фенолы можно получить из коммерчески доступных реактивов методами, известными в области органического синтеза. Один такой способ получения проиллюстрирован в Схеме 8, где при избирательном деметилировании 3-метил-4-метилтиоанизола получают соответствующий фенол, который после обработки трифлиновым ангидридом в присутствии 2,6-лутидина в метиленхлориде (Gerlach. U. и другие. Tet. Lett. 33(38), 5499, 1992), дает трифлат, пригодный для использования в вышеописанных палладий-катализированных реакциях сочетания. Полученное метилтиозамещенное промежуточное соединение можно превратить в соединение формулы I путем окисления соответствующего сульфона по описанной выше методике.
Соединения формулы I, где R3 имеет значения кроме водорода, можно получить при использовании замещенных надлежащим образом коммерчески доступных бромбензолов в качестве исходных материалов для реакций сочетания по Suzuki, описанных выше. При проведении стандартных манипуляций с функциональными группами полученного соединения с использованием методов, которые хорошо знает любой специалист в области органического синтеза, можно получить дополнительные заместители радикала R3, для которых не подходят имеющиеся в продаже исходные материалы. Следующие Схемы служат в качестве иллюстрации способов получения соединения соединения формулы I с широким разнообразием заместителей для радикала R3.
Катализированная палладием реакция сочетания по Suzuki 3-нитро-4-бромацетофенона с фенилбороновой кислотой обеспечивает выход 3-нитро-1-ацетдифенила. При восстановлении нитрогруппы раствором хлорида олова в соляной кислоте образуется амин, который можно превратить в фторборат диазония путем обработки раствором изо-амилнитрита и трифторборат эфират в метиленхлориде (Doyle, M.P. et. al., J. Org. Chem. вып. 44, стр. 1572, 1979). Соль диазония можно затем превратить непосредственно в трифлат путем обработки трифторуксусной кислоты (Yoneda, N. и другие. Chem. Lett. 1991, стр. 459). При взаимодействии полученного трифлата с 4-метилтиозамещенной фенилбороновой кислотой по методике, описанной выше, с последующим окислением избытком МХНБК (м-хлорнадбензойная кислота) получают соединение формулы I, где R3 означает ОН (Схема 9).
Это соединение может служить в качестве исходного материала для получения соединений формулы I, проиллюстрированных в Схеме 10. Превращение гидроксильной группы в простую эфирную связь можно обеспечить реакцией алкилирования с использованием раствора гидрида натрия и соответствующего галоидного алкила в безводном тетрагидрофуране. Гидроксильную группу можно также превратить в трифлат путем обработки трифлинового ангидрида в присутствии 2,6-лутидина при использовании метиленхлорида в качестве растворителя. Полученный трифлат можно подвергнуть катализированной палладием реакции сочетания по Suzuki (Cacchi и другие. Теt Lett. вып. 27(33), стр. 3931, 1986; Kalinin, V. Synthesis стр. 413, 1992) или сочетания по Stille (Stille, J.К.J. Am. Chem. Soc., 1988, 110, 1557) с выходом замещенных производных алкенилов, кетонов и карбоновых кислот.
Кроме превращений, показанных в Схеме 10, при использовании методов, известных в области органического синтеза, сложные эфиры могут быть омылены до карбоновых кислот, которые в свою очередь можно превратить в замещенные амиды, кетоны или гидроксаматы. Сложные алкеновые эфиры можно также восстановить каталитической гидрогенизацией с выходом насыщенных сложных эфиров при использовании палладиевой черни в качестве катализатора.
Соединения формулы I, где R3 означает функциональную аминогруппу, можно получить из промежуточного соединения 2-[4-метилтиофенил]-4-ацето-1-дифенила, полученного в Схеме 9, по методике, проиллюстрированной в Схеме 11. Бекмановская перегруппировка (Donaruma, L. G. и другие, Organic Reactions, том 11, стр. 1-156, 1960) кетона с последующим гидролизом полученного амида приводит к образованию амина, который затем может быть превращен в амиды, дизамещенные амины или замещенные амиды общеизвестными методами в области органического синтеза. При окислении метилтиогруппы по методике, описанной ранее, получают соединения формулы I. В альтернативном варианте соединения, где R3 означает функциональную аминогруппу, можно получить из карбоновых кислот через перегруппировку Курциуса (Banthorpe, D.V. "The Chemistry of the Azido Group, " Palai, S. Ed., Interscience, New York, 1971. стр. 397-405), как проиллюстрировано в Схеме 12.
Соединения формулы I, где R3 и R4 оба имеют значения кроме водорода, можно получить через множество различных реакций методами, общеизвестными в данной области. Один из таких реакционных путей проиллюстрирован в Схеме 13.
Превращение 3-(4'-метилтио)фенил-1-гидрокси-4-дифенила в N,N-диметилкарбамат можно осуществить путем взаимодействия гидрида натрия с N,N-диметилкарбамоилхлоридом в безводном тетрагидрофуране. Направленное ортометаллирование (Snieckus, V. Chemical Reviews, 1990, 879) при использовании втор-бутиллития в безводном тетрагидрофуране с последующим гашением полученного аниона подходящим электрофильным реагентом (например, йодистым метилом) дает промежуточное соединение, которое можно превратить в различные соединения формулы I при использовании методов, описанных выше, или общеизвестными методами в области органического синтеза.
Соединения формулы I, в которых одна или несколько групп из радикалов J, К или L означает атом водорода, можно получить путем замещения гетероцикла с соответствующей функциональной группой на монобромзамещенные или дибромзамещенные бензолы, полученные в вышеуказанных Схемах. Например, в случае когда J означает атом азота, соединение формулы I получают по методике, проиллюстрированной в Схеме 14.
Катализируемая палладием реакция сочетания по Suzuki 2-бром-3-гидроксипиридина с замещенной надлежащим образом фенилбороновой кислотой дает 2-фенил-3-гидроксипиридин. В результате превращения гидроксильной группы в трифлат при использовании вышеописанных реакционных условий с последующим проведением реакции сочетания по Suzuki с 4-метилтиофенилбороновой кислотой в безводной среде в присутствии палладиевого катализатора получают 2,3-диарилпиридин. В качестве растворителя для этой реакции сочетания можно использовать безводный 1,4-диоксан. Избирательное окисление метилтиогруппы можно обеспечить путем обработки оксоном с получением соединения формулы I, где J означает N.
Соединения формулы I, где Z означает простую связь, a R1 означает 1-пиперидинил или 1-пирролил можно получить из 2-броманилина, как показано в Схеме 15. Взаимодействие по Suzuki 2-броманилина с 4-тиометилфенилбороновой кислотой по методике, описанной выше, с последующей конденсацией 2-(4-метилтиофенил)анилина с дибромпентаном в присутствии аминового основания, например триэтиламина, дает соответствующий 1-[2-(4-метилтиофенил)фенил]пиперидин. Реакция окисления, если требуется, метилтиогруппы до метилсульфонила при использовании методов, описанных выше, дает соединение формулы I, где R1 означает 1-пиперидинил. В альтернативном варианте исходный 2-броманилин можно превратить в 1-[(2-бромфенил)фенил] пиррол путем обработки раствором 2,5-диметокситетрагидрофурана в ледяной уксусной кислоте. Взаимодействие по Suzuki полученного промежуточного соединения с 4-метилтиофенилбороновой кислотой с последующей реакцией окисления по методике. описанной выше, приводит к образованию 1-[2-(4-метилсульфонилфенил)фенил]пиррола.
Для лучшего понимания соединений предлагаемого изобретения и способов их получения ниже приводятся конкретные примеры, которые служат только в качестве иллюстрации и ни в коей мере ограничением объема изобретения.
Примеры
Все температуры плавления приведены без корректировки. За исключением особо оговоренных случаев все реакции проведены в атмосфере азота. Все коммерчески доступные химические реагенты использовали сразу же после их получения. Хроматографию осуществляли на силикагеле 60 фирмы Merck (230-400 меш). Элюенты для хроматографии даются в виде соотношения (по объему). За исключением случаев, оговоренных особо, органические фазы из сольвент-сольвентных экстрактов сушат обычно сульфатом магния. За исключением случаев, оговоренных особо, растворители обычно удаляют упариванием при пониженном давлении на роторном выпаривателе. Пиковые положения спектров 1Н ЯМР даны в частях на млн (δ) в направлении поля из тетраметилсилана в качестве внутреннего стандарта. Приняты следующие аббревиатуры для спектров 1Н ЯМР: с= синглет, д=дублет, м=мултиплет, дд=двойной дублет. Масс-спектры получены при использовании химической ионизации аммиаком в качестве газа для реагентов. Микроанализ осуществляли с использованием аппаратуры фирмы Quantitative Technologies Inc., Bound Brook, N.J.
Пример 1
2-[(4-метилтио)фенил]-1-дифенил (Способ 1)
А. Получение 4-метилтиофенилбороновой кислоты.
К суспензии магниевых опилок (4,3 г, 180 ммоль), охлажденной до 0oС, медленно прибавляют 1М раствор, содержащий боран-тетрагидрофурановый комплекс (600 мл, 600 ммоль). К полученной смеси прибавляют по каплям суспензию 4-бромтиоанизола (30 г, 148 ммоль) в тетрагидрофуране (75 мл). Затем добавляют несколько йодных кристалликов и реакционной смеси дают нагреться до комнатной температуры, а затем перемешивают в течение 72 часов. Реакционную смесь осторожно сливают в 500 г измельченного льда. Раствор подкисляют (рН 3) 1 н. раствором соляной кислоты и оставляют на ночь. Кислый раствор экстрагируют диэтиловым эфиром. Диэтиловый эфир экстрагируют 1 н. раствором гидроксида натрия. Щелочной слой подкисляют и затем экстрагируют диэтиловым эфиром. После упаривания растворителя получают бесцветные кристаллы, которые после перекристаллизации из этилацетата с небольшим количеством воды дают 12,5 г 4-метилтиофенилбороновой кислоты; 1Н ЯМР (DMSO) δ: 7,73 (д, J=8,42 Гц, 2Н), 7,21 (д, J=8,42 Гц, 2Н), 2,47 (с, 3Н). Масс-спектр (Cl, CH4), m/z 195 (М+Н+) сложный этиленгликолевый эфир).
В. Получение 2-бром-1-(4'-метилтиофенил) бензола.
Смесь, состоящую из 4-метилтиофенилбороновой кислоты (31,1 г, 185 ммоль), 1,2-дибромбензола (35 г, 148 ммоль), и тетрабутиаммония бромида (1 г, 3,10 ммоль) в смеси этанола (125 мл) и толуола (250 мл), дегазируют путем барботирования азота через смесь в течение 15 минут. 2 М раствор карбоната натрия (148 мл, 296 ммоль) дегазируют и прибавляют к реакционной смеси. К реакционной смеси прибавляют тетракис(трифенилфосфин)палладий (0,35 г, 0,303 ммоль) и смесь кипятят с обратным холодильником в течение 24 часов. Реакционную смесь охлаждают до комнатной температуры и фильтруют для удаления твердого осадка. Фильтрат упаривают и затем разбавляют водой и этилацетатом. Водный слой экстрагируют этилацетатом. Органические слои объединяют, промывают солевым раствором и сушат на сульфате натрия. Этилацетат упаривают и остаток осаждают. При добавлении диэтилового эфира (200 мл) образуется дополнительное количество твердого осадка. Выпавший осадок отфильтровывают и фильтрат упаривают с получением неочищенного масла. После очистки методом колоночной хроматографии на силикагеле с использованием гексана в качестве элюента получают требуемый продукт (25,75 г, 62%), который выдерживают для солюбилизации, т. пл. 33-35oC; 1Н ЯМР (СDС13) δ: 7,66 (д, J=8,05 Гц, 1Н), 7,36-7,28 (м, 6Н), 7,21 (м, 1Н), 2,52 (с, 3Н). Масс-спектр, m/z 279,1, 281,1 (М+Н).
Элементный анализ для C13H11BrS.
Вычислено: С 55,92%, Н 3,97%, Вr 28,62%.
Найдено: С 56,24%, Н 4,04%, Вr 28,96%.
С. Получение 2-Бром-1-(4'-метилсульфонилфенил)бензола.
Соединение, полученное в Примере 1, в разделе В (5,2 г, 18,7 ммоль), растворяют в дихлорметане (100 мл) и охлаждают до температуры 0oС. 3-Хлорнадбензойную кислоту (8,9 г, 41,2 ммоль) прибавляют к реакционной смеси и смесь перемешивают при комнатной температуре в течение 18 часов. Реакционную смесь разбавляют дихлорметаном и промывают последовательно бикарбонатом натрия, разбавляют бисульфитом натрия, сушат на сульфате натрия, фильтруют и упаривают. После очистки остатка методом хроматографии на силикагеле с использованием смеси гексан/этилацетат 7: 1 в качестве элюента получают бесцветные кристаллы, которые после перекристаллизации (дихлор/метан) дают чистый продукт (4,02 г, 69%), т. пл. 155-157oС; 1Н ЯМР (CDC13) δ: 8,02 (д, J= 8,42 Гц, 2Н), 7,71 (д, J=6,96 Гц, 1Н), 7,63 (д, J=8,42 Гц, 2Н), 7,43 (м, 1Н), 7,32 (м, 2Н), 3,13 (с, 3Н); ИК (КВr): 1306, 1142 см-1.
Элементный анализ для C13H11BrO2S.
Вычислено: С 50,17%, Н 3,56%, S 10,30%.
Найдено: С 50,09%, Н 3,41%, S 10,52%.
D. Получение 2-[(4-метилсульфонил)фенил]-1-дифенила.
2-Бром-1-(4'-метилсульфонилфенил)бензол (4 г, 12,8 ммоль), фенилбороновую кислоту (1,72 г, 14 ммоль), и тетрабутиламмония бромид (0,21 г, 0,65 ммоль) растворяют в смеси толуола (70 мл) и этанола (35 мл) и полученную смесь барботируют азотом в течение 15 минут. Затем прибавляют дегазированный 2М раствор, содержащий бикарбонат натрия (14 мл, 28 ммоль) и тетракис(трифенилфосфин)палладий (0,074 г, 0,064 ммоль) и полученную смесь кипятят с обратным холодильником в течение 4 часов. Реакционную смесь упаривают и разбавляют водой и этилацетатом. Образовавшиеся слои разделяют и водный слой экстрагируют этилацетатом. Объединенные органические слои промывают солевым раствором, сушат, фильтруют и упаривают. После очистки методом колоночной хроматографией на силикагеле с использованием смеси гексан/этилацетат 3:1 в качестве элюента и перекристаллизации в смеси дихлор/метан получают 2,55 г (65%) названного соединения в виде бесцветных кристаллов, т. пл. 136-138oС; 1H ЯМР (СDС13) δ: 7,79 (д, J=8,42 Гц, 2Н), 7,47 (м, 3Н), 7,42 (м, 1Н), 7,34 (д, J=8,7 Гц, 2Н), 7,23 (м, 3Н), 7,11 (м, 2Н) 3,04 (с, 3Н). Масс-спектр (С1, CH4), m/z 309 (М+Н), 337 (М+С2Н5).
Элементный анализ для C19H16O2S.
Вычислено: С 74,00%, Н 5,23%, S 10,40%.
Найдено: С 74,01%, Н 5,13%, S 10,63%.
Пример la
2-[(4'-метилтио)фенил]дифенил (Способ 2)
А. Получение 2-Фенил-1-фенокситрифторметансульфоната.
Смесь, состоящую из 2-фенилфенола (5 г, 29,4 ммоль), N,N-диметиламинопиридина (0,61 г, 4,99 ммоль) и 2,6-лутидина (4,1 мл, 35,0 ммоль) в дихлорметане (180 мл), охлаждают до температуры -30oС. Ангидрид трифторметансульфокислоты (5,90 мл, 35,0 ммоль) прибавляют к реакционной смеси и удаляют охлаждающую ванну. После выдерживания в течение 1 часа при комнатной температуре смесь затем промывают последовательно 0,5 н раствором НС1, водой, насыщенным бикарбонатом натрия и солевым раствором. Смесь сушат, фильтруют и упаривают с получением требуемого трифлата (8,80 г, 99%) в виде желтого масла; 1H ЯМР (CDC13) δ: 7,35-7,50 (м, 9Н). Масс-спектр: (Cl, CH4), m/z 303 (М+Н), 331 (М+С2Н4).
В. Получение 2-[(4'-метилтио)фенил]-1-дифенила.
2-Фенил-1-фенокситрифторметансульфонат (13,75 г, 45,5 ммоль), 4-метилтиобензолбороновой кислоты (8,4 г, 50,0 ммоль), и трикалия фосфат (12,6 г, 59,0 ммоль) суспендируют в 1,4-диоксане и дегазируют путем пропускания азота в течение 30 минут. Тетракис(трифенилфосфин)палладий (1,30 г, 1,14 ммоль) прибавляют к реакционной смеси и смесь кипятят с обратным холодильником в течение 24 часов. Смесь охлаждают, фильтруют и упаривают. Остаток растворяют в этилацетате, промывают затем водой и солевым раствором и сушат. После очистки смеси методом хроматографии на силикагеле с использованием гексана в качестве элюента и перекристаллизации (EtOH) получают требуемый продукт (4,27 г) в виде белых кристаллов, т. пл. 42-44oС. После упаривания маточного раствора получают дополнительно 4,98 г названного соединения; 1H ЯМР (CDC13) δ: 7,41 (с, 4Н), 7,23 (м, 3Н), 7,16 (м, 2Н), 7,13-7,04 (м, 4Н), 2,45 (с, 3Н). Масс-спектр, m/z 277,1 (М+Н), 294,1 (M+NH4).
Элементный анализ для C19H16S.
Вычислено: С 82,56%, Н 5,84%, S 11,60%.
Найдено: С 82,39%, Н 5,77%, S 11,60%.
С. Получение 2-[(4-метилсульфонил)фенил]-1-дифенила.
4'-Метилтиофенил-2-фенилбензол (2,0 г, 7,30 ммоль) растворяют в дихлорметане (60 мл) и охлаждают до температуры 0oС. 3-Хлорнадбензойную кислоту (3,40 г, 15,9 ммоль) прибавляют к реакционной смеси и смесь перемешивают в течение 3 часов. Смесь промывают бикарбонатом натрия, бисульфитом натрия, солевым раствором и сушат. После очистки остатка методом хроматографии на силикагеле с использованием смеси гексан/этилацетат 4:1 в качестве элюента и перекристаллизации (дихлор/метан) получают названное соединение (0,64 г, 28,6%) в виде кристаллического продукта, т. пл. 135-137oС; 1Н ЯМР (CDC13) δ: 7,79 (д, J=8,42 Гц, 2Н), 7,47 (м, 3Н), 7,42 (м, 1Н), 7,34 (д, J=8,7 Гц, 2Н), 7,23 (м, 3Н), 7,11 (м, 2Н), 3,04 (с, 3Н). Масс-спектр, m/z 309 (М+Н), 326 (M+NH4); ИК (КВr): 1312, 1154, 760 см-1.
Элементный анализ для C19H16O2S.
Вычислено: С 74,00%, Н 5,23%, S 10,40%.
Найдено: С 74,07%, Н 5,17%, S 10,37%.
Пример 109
1-циклогексен-2-(4'-метилсульфонилфенил) бензол
А. Получение 2-(4'-метилтиофенил)-1-(1-гидрокси-1-циклогексил)бензола.
2-Бром-(4'-метилтиофенил) бензол (3,02 г, 10,8 ммоль) растворяют в тетрагидрофуране (35 мл), охлаждают до температуры -78oС и затем медленно прибавляют н-бутиллитий (4,5 мл, 11,3 ммоль). Полученную бледно-желтую смесь перемешивают в течение 2 часов при температуре -78oС, а затем добавляют циклогексанон (1,3 мл, 12,9 ммоль). Реакционную смесь перемешивают в течение 18 часов и затем доводят до комнатной температуры. Реакционную смесь разбавляют водой и этилацетатом. Водный слой экстрагируют этилацетатом и объединенные органические слои сушат, фильтруют и упаривают. После очистки остатка методом хроматографии на силикагеле с использованием смеси гексан/этилацетат 6: 1 в качестве элюента получают требуемый продукт (2,51 г, 77%) в виде прозрачного масла; 1H ЯМР (CDC13) δ: 7,58 (д, 1Н), 7,36 (м, 2Н), 7,27 (м, 4Н), 7,04 (дд, 1Н), 2,53 (с, 3Н), 2,34 (т, 1Н), 1,83-1,10 (м, 10Н). Масс-спектр (высокое разрешение, EI/DEP): вычислено М+298,139137; найдено М+298,138665.
В. Получение 1-циклогексен-2-(4'-метилтиофенил)бензола.
Соединение, полученное в Примере 109, часть А (2,17 г, 7,27 ммоль), растворяют в толуоле (30 мл) и прибавляют каталитическое количество п-толуолсульфокислоты (0,05 г). Смесь кипятят с обратным холодильником. Через 4 часа смесь охлаждают и промывают бикарбонатом натрия, сушат, фильтруют и упаривают. После очистки остатка методом колоночной хроматографией на силикагеле с использованием смеси гексан/этилацетат, 4:1, в качестве элюента и перекристаллизации (метанол) получают циклоалкен (1,29 г, 65%) в виде белых кристаллов, т. пл. 71-73oС. После упаривания маточного раствора получают еще 0,15 г требуемого продукта; 1H ЯМР (CDC13) δ: 7,37 (д, J=8,42 Гц, 2Н), 7,28 (м, 6Н), 5,67 (м, 1Н), 2,52 (с, 3Н), 2,09 (м, 2Н), 1,83 (м, 2Н), 1,53 (м, 4Н).
Элементный анализ для C19H2ОS.
Вычислено: С 81,38%, Н 7,19%, S 11,43%.
Найдено: С 81,17%, Н 7,16%, S 11,53%.
С. Получение 1-циклогексен-2-(4'-метилсульфонилфенил)бензола.
Соединение, полученное в Примере 109, часть В (1,35 г, 4,80 ммоль), суспендируют в метаноле (125 мл), охлаждают до температуры 0oС, и прибавляют оксонTM (8,30 г, 13,0 ммоль) в воде (50 мл). Образовавшуюся густую суспензию нагревают до комнатной температуры и затем перемешивают в течение 18 часов. Реакционную смесь разводят водой (200 мл) и выпавшие белый кристаллический продукт собирают. Полученный продукт промывают водой, разбавляют бисульфитом натрия и водой и сушат в вакууме. После очистки остатка методом хроматографии на силикагеле с использованием смеси гексан/этилацетат 4:1 в качестве элюента и перекристаллизации (в метаноле) получают названное соединение (0,524 г, 35%) в виде бесцветных кристаллов, т. пл. 126-128oС. После упаривания маточного раствора получают дополнительно 0,278 г требуемого продукта; 1H ЯМР (CDC13) δ: 7,95 (д, J=8,42 Гц, 2Н), 7,63 (д, J=8,42 Гц, 2Н), 7,36-7,25 (м, 4Н), 5,63 (м, 1Н), 3,10 (с, 3Н), 2,06 (м, 2Н), 1,84 (м, 2Н), 1,51-1,45 (м, 4Н).
Элементный анализ для С19Н20О2S.
Вычислено: С 73,04%, Н 6,45%, S 10,26%.
Найдено С 73,22%, Н 6,47%, S 10,46%.
Пример 130
3-(4'-метилсульфонилфенил)-4-фенилфенол
А. Получение 3-нитро-4-фенилацетофенона.
Смесь, состоящую из 4-бром-3-нитроацетофенона (2,0 г, 8,19 ммоль), фенилбороновой кислоты (1,2 г, 9,83 ммоль), и тетрабутиаммония бромида (0,13 г, 0,41 ммоль) в 2М растворе карбоната натрия (35 мл) в смеси этанола (20 мл) и толуола (65 мл), дегазируют путем барботирования азотом в течение 30 минут. Реакционную смесь кипятят с обратным холодильником в течение 4 часов. Затем смесь охлаждают и образовавшиеся слои разделяют. Водный слой экстрагируют этилацетатом и объединенные органические слои сушат, фильтруют и упаривают. После очистки остатка методом хроматографии на силикагеле при использовании смеси гексан/этилацетат 4:1 в качестве элюента получают требуемый продукт (1,98 г, 89%) в виде желтого порошка; 1H ЯМР (CDC13) δ: 8,39 (д, 1H), 8,16 (дд, 1H), 7,57 (д, 1H), 7,43 (м, 3Н), 7,32 (дд, 2Н), 2,69 (с, 3Н). Масс-спектр 242,1 (М+Н).
В. Получение 3-амино-4-фенилацетофенона.
Смесь, состоящую из продукта, полученного в Примере 130, часть А (2,0 г, 8,29 ммоль), хлорида олова (8,23 г, 36,48 ммоль), этанола (30 мл) и концентрированной соляной кислоты (7 мл), кипятят с обратным холодильником в течение 2,5 часов. Реакционную смесь охлаждают до 0oС, затем подщелачивают (рН 10) 6М раствором NaOH и экстрагируют этилацетатом. Экстракты сушат и фильтруют через силикагель. Фильтрат упаривают и вновь фильтруют через силикагель при использовании хлороформа в качестве элюента. После упаривания растворителя получают амин (1,20 г, 69%) в виде желтого порошка; 1Н ЯМР (CDC13) δ: 7,47 (д, 1Н), 7,46 (с, 3Н), 7,38 (дд, 2Н), 7,36 (д, 1Н), 7,20 (д, 1H), 3,90 (с, 2Н), 2,60 (с, 3Н). Масс-спектр, m/z 212,1 (М+Н).
С. Получение 5-Ацето-2-фенилбензолдиазония тетрафторбората.
Соединение, полученное в Примере 130, часть В (0,50 г, 2,36 ммоль), растворяют в дихлорметане (3 мл) и медленно прибавляют к раствору трифторборат эфират в дихлорметане (10 мл) при -15oС. Затем прибавляют раствор, содержащий изоамилнитрит (0,35г, 2,60 ммоль) в дихлорметане (3 мл), ванну со льдом удаляют с выпадением коричневого осадка. К реакционному раствору прибавляют пентан (20 мл) и полученную смесь опять охлаждают до температуры -15oС в течение 20 минут. После фильтрования получают соль диазония (0,76 г) в виде светло-коричневого порошка; 1H ЯМР (СDС13) δ: 9,55 (д, 1Н), 8,71 (дд, 1H), 7,90 (д, 1H), 7,69 (с, 5Н), 2,79 (с, 3Н).
D. Получение 5-Ацето-2-фенилбензола трифторметансульфоната.
5-Ацето-2-фенилбензолдиазония тетрафторборат (1,46 г, 4,79 ммоль) медленно прибавляют к трифторметансульфокислоте (10 мл) при температуре -15oС. Смесь нагревают до температуры 50oС в течение 20 минут и затем выливают в ванну со льдом (25 г). Водный слой экстрагируют этилацетатом, сушат, фильтруют и упаривают. После очистки остатка методом хроматографии на силикагеле с использованием смеси гексан/этилацетат 4:1 в качестве элюента получают требуемый трифлат (0,428 мг, 77%) в виде коричневого сиропа; lH ЯМР (CDC13) δ: 8,04 (дд, 1H), 7,96 (д, 1Н), 7,62 (д, 1Н), 7,48 (с, 5Н), 2,67 (с, 3Н). Масс-спектр, m/z 345 (М+Н).
Е. Получение 3-(4'-Метилтиофенил)-4-фенилацетофенона.
Смесь, состоящую из соединения, полученного в Примере 130, часть D (1,22 г, 3,54 ммоль), 4-метилтиофенилбороновой кислоты (0,71 г, 4,25 ммоль), и трехосновной калия фосфат (1,13 г, 5,32 ммоль) в 1,4-диоксане дегазируют путем барботирования азотом в течение 15 минут.
К реакционному раствору прибавляют тетракис(трифенилфосфин)-палладий (0,10 г, 0,089 ммоль) и полученную смесь кипятят с обратным холодильником в течение 18 часов. Смесь охлаждают, фильтруют и упаривают. После очистки остатка методом хроматографии на силикагеле при использовании смеси гексан/этилацетат 4: 1 в качестве элюента получают требуемый продукт (1,02 г, 90%) в виде коричневого сиропа; 1H ЯМР (CDC13) δ: 7,99 (д, 2Н), 7,53 (д, 1H), 7,48 (с, 2Н), 7,27 (д, с, 2Н), 7,17 (дд, 2Н), 7,14 (кв, 3Н). Масс-спектр, m/z 319 (М+Н).
F. Получение 3-(4'-Метилсульфонилфенил)-4-фенилфенола.
К соединению, полученному в Примере 130, часть Е (0,30 г, 0,942 ммоль), прибавляют надбензойную кислоту (10 мл) и затем концентрированную серную кислоту (0,25 мл). Реакционную смесь перемешивают при комнатной температуры в течение 48 часов. Затем выливают ее в смесь из льда и 20% бисульфита натрия (10 мл). Водную фазу экстрагируют этилацетатом и органические слои сушат, фильтруют и упаривают. После очистки остатка методом хроматографии на силикагеле при использовании смеси гексан/этилацетат 2:1 в качестве элюента получают названное соединение (0,064 г, 21%) в виде белого порошка; 1H ЯМР (CDC13) δ: 7,79 (д, 2Н), 7,35 (д, 1Н), 7,34 (д, 2Н), 7,21 (д, 1Н), 7,19 (д, 2Н), 7,06 (м, 2Н), 6,97 (дд, 1Н), 6,90 (д, 1Н), 4,96 (с, 1Н), 3,05 (с, 3Н). Масс-спектр высокого разрешения, m/z вычислено: 342,1, найдено: 342,116391 (M+NH4).
Пример 151
1-[2-(4-метилсульфонилфенил)фенил]пиперидин
А. Получение 2[(4-метилтио)фенил)анилина.
Смесь, состоящую из 2-броманилина (2,0 г, 11,62 ммоль), 4-метилтиофенилбороновой кислоты (2,3 г, 13,69 ммоль), тетрабутиламмония бромида (0,19 г, 0,58 ммоль) и 2М раствора карбоната натрия (12 мл) в 85 мл смеси толуол/этанол 2:1, дегазируют путем барботирования через нее азота в течение 10 минут. К реакционной смеси прибавляют тетракис(трифенилфосфин) палладий (54 мг, 0,047 ммоль) и полученную смесь кипятят с обратным холодильником в течение 5 часов. Реакционную смесь охлаждают, упаривают и разбавляют этилацетатом и водой. Водный слой экстрагируют этилацетатом и объединенные органические слои сушат (MgSО4), фильтруют и упаривают. После очистки полученного нечищенного продукта методом хроматографии (смесь гексан/этилацетат) получают твердое вещество (1,4 г, 56%). Т. пл. 70-72oС: ЯМР (CDC13) δ: 7,41-7,32 (м, 4Н), 7,18-7,09 (м, 2Н), 6,85-6,75 (м, 2Н), 3,75 (шир. м, 2Н), 2,53 (с, 3Н) ч/млн. Масс-спектр (NH3-С1), m/z 215,9 (М+Н+, 100%).
В. Получение 1-[2-(4-метилтиофенил)фенил]пиперидина.
К смеси, состоящей из продукта, полученного выше, часть А (0,3 г, 1,39 ммоль), этанола (10 мл) и триэтиламина (0,39 мл, 2,77 ммоль), прибавляют 1,5-дибромпентан (0,29 мл, 2,08 ммоль). Реакционную смесь кипятят с обратным холодильником в течение 48 часов, затем упаривают и хроматографируют (смесь гексанов) с получением розового масла (0,147 г, 37%). ЯМР (CDC13) δ: 7,73 (д, 2Н), 7,39 (д, 2Н) 7,36-7,30 (м, 2Н), 7,15-7,10 (м, 2Н), 2,87-2,85 (м, 4Н), 2,62 (с, 3Н), 1,55 (с, 6Н). Масс-спектр (NH3-Cl), m/z 284,2 (M+H+, 100%).
С. Получение 1-[2-(4-метилсульфонилфенил)фенил]пиперидина.
К раствору, содержащему соединение, полученное в Примере 195, часть С (0,145 г, 0,512 ммоль), в метаноле (15 мл), охлажденному до температуры 0oС, прибавляют оксонTM (0,79 г, 1,28 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Затем реакционную смесь разбавляют метиленхлоридом и экстрагируют. Объединенные органические слои промывают бикарбонатом натрия, бисульфитом натрия, солевым раствором и сушат (MgSО4). Неочищенный продукт хроматографируют (смесь гексаны/этилацетат) и после перекристаллизации (метиленхлорид/гексаны) получают твердое вещество (50 мг, 31%), т. пл. 140-140,5oС. 1H ЯМР (CDC13) δ: 7,97-7,85 (дд, 4Н), 7,36 (т, 1Н), 7,23-7,20 (дд, 1Н), 7,10-7,05 (м, 2Н), 3,10 (с, 3Н), 2,75 (м, 4Н), 1,43 (м, 6Н). Масс-спектр высокого разрешения: вычислено для C18H21NSО2: 316,137126; найдено: 316,136504.
Пример 153
1-[2-(4'-метилсульфонилфенил)фенил]пиррол
А. Получение 1-(2-бромфенил)пиррола.
Смесь, состоящую из 2-броманилина (1,72 г, 10 ммоль), 2,5-диметокситетрагидрофурана (1,32 г, 10 ммоль) и ледяной уксусной кислоты (4,5 мл), кипятят с обратным холодильником при перемешивании в течение 2 часов в атмосфере азота. Смесь охлаждают до комнатной температуры. Растворитель отгоняют при пониженном давлении и остаток очищают флэш-хроматорафией (смесь гексаны/этилацетат 9:1) с выходом требуемого пиррола (1,85 г, 8,33 ммоль, 83.3%) в виде прозрачной жидкости. 1H ЯМР (ССD13) δ: 7,70-6,35 (м, 8Н); ИК (КВr) 3102, 1588 см-1; Масс-спектр, m/z 221,9 (М+Н)+.
В. Получение 1-(2-(4-Метилтиофенил)фенил)пиррола.
Смесь, состоящую из 1-(2-бромфенил)пиррола (0,666 г, 3,0 ммоль), 4-метилтиофенилбороновой кислоты (0,554 г, 1,1 экв.), 2М водного раствора карбоната натрия (6 мл) и толуола (30 мл), перемешивают при комнатной температуре в атмосфере азота. Азот барботируют через реакционный раствор в течение 20 мин. К полученной смеси прибавляют тетракис(трифенилфосфин)палладий (100 мг, каталитическое количество) и смесь кипятят при перемешивании с обратным холодильником в течение 4 часов. Полученную смесь охлаждают до комнатной температуры и затем выливают в 100 мл воды. Смесь экстрагируют тремя 100 мл порциями этилацетата. Объединенные органические слои сушат на безводном сульфате магния, фильтруют и растворитель отгоняют при пониженном давлении. Остаток очищают флэш-хроматографией (29:1, смесь гексанов/этилацетат). Получают продукт реакции в виде масла (0,74 г, 2,79 ммоль, 92,9%). 1Н ЯМР (CDC13) δ: 7,44-6,16 (м, 12Н) 2,46 (с, 3Н); ИК (чистый): 2918, 1596 см-1. Масс-спектр, m/z 266,0 (М+Н)+.
С. Получение 1-[2-(4-метилсульфонилфенил)фенил]пиррола.
Смесь, состоящую из 1-(2-(4-метилтиофенил)фенил)пиррола (0,74 г, 2,788 ммоль), и метиленхлорида (35 мл), перемешивают и охлаждают на водяной бане со льдом и солью в атмосфере азота. Затем к реакционной смеси прибавляют в одной порции 3-хлорнадбензойную кислоту (50-60% раствор, 1,924 г, >2 экв.). Полученный реакционный раствор доводят до комнатной температуры и перемешивают в течение ночи. Смесь выливают в насыщенный раствор бисульфита натрия и экстрагируют тремя порциями по 50 мл каждая хлористого метилена. Объединенные органические слои промывают насыщенным раствором бикарбоната натрия, сушат на безводном сульфате магния, фильтруют и растворитель упаривают при пониженном давлении. После очистки остатка методом флэш-хроматографии (2:1, смесь гексанов/этилацетат) получают названное соединение в виде грязновато-белого порошка (16 г, 0,538 ммоль, 19,2%). 1H ЯМР (CDC13) δ: 7,88-6,15 (м, 12Н) 3,06 (с, 3Н); ИК (KBr): 2922, 1602 см-1. Масс-спектр, m/z 298,0 (М+Н)+.
Пример 201
1-фенокси-2-(4'-метилсульфонилфенил)бензол
А. Получение 2-(4'-метилтиофенил)фенола.
Смесь, состоящую из 2-бромфенола (3,0 г, 17,0 ммоль), 4-метилтиобензолбороновой кислоты (3,5 г, 20,8 ммоль), и тетрабутиламмония бромида (0,28 г, 0,867 ммоль) в толуоле (100 мл), этанола (25 мл), и 2М раствор бикарбоната натрия (50 мл), дегазируют барботированием азотом в течение 30 минут. Тетракис(трифенилфосфин)палладий (0,06 г, 0,052 ммоль) прибавляют к реакционной смеси и смесь кипятят с обратным холодильником в течение 2,5 часов. Реакционную смесь охлаждают до комнатной температуры и слои разделяют. Водный слой экстрагируют этилацетатом и объединенные органические слои сушат, фильтруют и упаривают. После очистки остатка методом хроматографии на силикагеле с использованием смеси 4: 1 гексан/этилацетат в качестве элюента получают продукт реакции (3,03 г, 81%) в виде желтого порошка; 1H ЯМР (СDС13) δ: 7,42 (м, 4Н), 7,25 (м, 2Н), 7,01 (т, 4Н), 5,13 (с, 1Н), 2,57 (с, 3Н). Масс-спектр m/z 217 (М+Н).
В. Получение 2-(4''-нитрофенокси)-1-(4'-метилтиофенил)бензола.
2-(4'-Метилтиофенил)фенол (0,4 г, 1,9 ммоль) и 1-фтор-4-нитробензол (0,27 г, 1,94 ммоль) растворяют в диметилформамиде (2 мл) и охлаждают до 0oС. Гидрид натрия (80% дисперсия в масле, 0,063 г, 2,1 ммоль) прибавляют к реакционной смеси и смесь доводят до комнатной температуры, а затем перемешивают в течение 18 часов. Реакционную смесь разбавляют этилацетатом и водой. Водный слой экстрагируют этилацетатом. Объединенные органические слои промывают солевым раствором, сушат, фильтруют и упаривают. После очистки остатка методом хроматографии на силикагеле с использованием смеси гексан/этилацетат 6:1 в качестве элюента и перекристаллизации (дихлор/метан) получают названный продукт (0,59, г, 96%) в виде желтых кристаллов, т. пл. 70-72oС; 1Н ЯМР (CDC13) δ: 8,11 (д, J=9,15 Гц, 2Н), 7,51 (дд, 1Н), 7,41-7,36 (м, 4Н), 7,20 (д, J=8,42 Гц, 2Н), 7,14 (дд, 1Н), 6,88 (д, J=9,15 Гц, 2Н), 2,46 (с, 3Н); ИК (КВr): 1514, 1342 см-1.
Элементный анализ для C19H15NО3S.
Вычислено: С 67,64%, Н 4,48%, N 4,15%.
Найдено: С 7,60%, Н 4,39%, N 4,09%.
С. Получение 2-фенокси-1-(4'-метилтиофенил)бензола.
Смесь, состоящую из соединения Примера 201, часть В (0,18 г, 0,53 ммоль), железной пудры (0,1 г, 1,8 ммоль), ледяной уксусной кислоты (0,3 мл, 5 ммоль) и этанола (10 мл), кипятят с обратным холодильником в течение 4 часов. Реакционную смесь охлаждают, фильтруют и упаривают в вакууме. К полученному неочищенному амину прибавляют тетрагидрофуран (11 мл) и полученную смесь нагревают. Изоамилнитрит (0,143 мл, 1,06 ммоль) прибавляют к смеси и реакционную смесь кипятят с обратным холодильником в течение 4 часов. После упаривания реакционной смеси и хроматографии на силикагеле с использованием смеси гексан/дихлорметан в качестве элюента получают требуемый продукт (0,096 г, 61%) в виде желтого масла; 1H ЯМР (СDС13) δ: 7,49 (м, J=8,42 Гц, 2Н), 7,45 (дд, 1Н), 7,30-7,19 (м, 6Н), 7,05 (м, 2Н) 6,94 (д, J=8,42 Гц, 2Н), 2,48 (с, 3Н). Масс-спектр m/z 293 (М+Н).
D. Получение 1-фенокси-2-(4'-метилсульфонил)фенилбензола.
Продукт, полученный в Примере 201, часть С (0,096 г, 0,35 ммоль), растворяют в дихлорметане (5 мл) и охлаждают до температуры 0oС. К полученному раствору прибавляют 3-хлорнадбензойную кислоту (0,15 г, 0,73 ммоль) и полученную смесь перемешивают при комнатной температуре в течение 18 часов. Реакционную смесь разбавляют дихлорметаном, промывают последовательно бикарбонатом натрия, бисульфитом натрия, солевым раствором и затем сушат, отфильтровывают и упаривают. Полученный остаток очищают хроматографией на силикагеле с использованием смеси гексан/этилацетат 4:1 в качестве элюента и после перекристаллизации (дихлорметан/гексан) получают названное соединение (0,063 г, 56%), т. пл. 130-131oС. После упаривания маточного раствора получают дополнительно 0,02 г требуемого продукта; 1H ЯМР (CDC13) δ: 7,94 (д, J= 8,79 Гц, 2Н), 7,77 (д, J=8,79 Гц, 2Н), 7,46 (дд, 1Н), 7,37 (м, 4Н), 7,09 (м, 2Н), 6,94 (дд, 2Н), 3,06 (с, 3Н). Масс-спектр, m/z 325 (М+Н), 342 (M+NH4).
Элементный анализ для C19H16O3S.
Вычислено: С 70,35%, Н 4,97%, S 9,88%.
Найдено: С 70,28%, Н 4,89%, S 9,99%.
При использовании вышеописанных методов или их модификаций, которые очевидны специалистам в области химического синтеза, можно также получить соединения, приведенные ниже в Таблицах 1-3.
Промышленная применимость
Соединения формулы I являются ингибиторами простагландинсинтазы, и следовательно, могут найти применение при лечении воспалительных заболеваний, а также в качестве жаропонижающих средств. Ингибиторная активность соединений предлагаемого изобретения относительно простагландин-G/Н-синтазы продемонстрирована в тестах, описываемых ниже, проведенных для определения активности ингибиторов простагландин-G/Н-синтазы. Предпочтительные соединения настоящего изобретения обеспечивают избирательное ингибирование активности ПГНS2 и продукции ПГЕ2 в человеческих моноцитах, как демонстрируют результаты клеточного теста, описанного ниже.
Соединения формулы I обладают способностью подавлять пирексию in vivo, например, как продемонстрировано на модели экспериментального животного, описанной ниже. Соединения настоящего изобретения обладают in vivo противовоспалительной активностью, как продемонстрировано при использовании стандартных моделей животных с индуцированным острым и хроническим воспалением, описанных ниже. Соединения настоящего изобретения также обладают способностью подавлять или ингибировать синдром боли in vivo, как демонстрируют результаты, полученные на модели аналгезии у животного, по методике, описанной ниже.
Используемые в данном описании термины "мкг" обозначает микрограмм, "мг" - миллиграмм, "г" - грамм, "мкл" обозначает микролитр, "мл" обозначает, миллилитр, "л" обозначает литр, "нм" обозначает наномоль, "мкмоль" обозначает микромоль, "ммоль" обозначает миллимоль, "м" обозначает моль, и "нм" обозначает нанометр. "Sigma" означает Sigma-Aldrich Corp. of St. Louis, МО.
Любое соединение считается активным в тесте на ингибирование простагландин-G/Н-синтазы, описанном ниже, если оно подавляет активность простагландин-G/H-синтазы при ИК50<300 мкмоль. Избирательные ингибиторы ПГНS2 демонстрируют отношение ИК50: ПГНS-1/ИК50:ПГНS-2, которое составляет >1.
Тест на Ингибирование Простагландин-G/Н-Синтазы
Активность простагландин-G/Н-синтазы (циклооксигеназы, ПГНS, Сох) определяли методом спектрофотометрии по методике, описанной Kulmacz и другими (используемого в данном описании в качестве ссылочного материала). В этом тесте используют TMPD (4,4,4',4'-тетраметилфенилдиамин) в качестве восстанавливающего агента, который при окислении дает интенсивные синие полосы, наблюдаемые при 610 нм. Пробу калибровали по формату 96-луночного титрационного микропланшета по методике? описанной ниже. Испытуемые соединения инкубировали с ферментным источником, ПГНS-1 или ПГНS-2, в 125 мкл буфера (40 мкмоль Трис-малеата, 0,8% Твин-20, 1,2 мкМ гема, 0,4 мг/мл желатина, рН 6,5) в течение двух минут при комнатной температуре с инициацией в течение этого промежутка времени ферментативной реакции путем добавления 125 мкл арахидоновой кислоты в буфере (0,1 М Трис/HCl, 0,2% Твин-20, рН 8,5) до получения конечной концентрации арахидоната 100 мкМ. Агглютационный планшет сразу же помещали в микротитрационнй самописец и данные считывали при 610 нм в течение 1,5 минуты через каждые 3 секунды. Значения скорости реакции рассчитывали по кривизне кривой линейного участка полосы поглощения в зависимости от времени. Скорости реакции контрольных образцов, в которые не добавляли ингибиторов ферментов использовали для расчета % ингибирования ферментативной активности каждым из испытуемых соединений. Результаты представлены как показатель ИК50, которая является концентрацией введенного соединения, которое приводит к 50% ингибированию скорости реакции Контроля.
Сравнительный анализ способности к избирательному ингибированию ПГНS-2 относительно ПГНS-1, осуществляли путем сравнения значений ИК50, полученных по двум изоформам фермента. Отношение ИК50 ПГНS-1/ИК50ПГНS-2 выражается как степень сродства к специфическому ферменту. Соединения с более высоким показателем специфического сродства характеризуют соединения с более сильным эффектом ингибирования изоформы ПГНS-2 фермента.
В Таблице А, приведенной ниже, представлены данные по активности испытуемых соединений настоящего изобретения, полученные в вышеописанном тесте по ингибированию простагландин-G/Н-синтазы. В указанной Таблице показатели ИК50 обозначены как +++=ИК50<10 мкМ, ++=ИК50 10-50 мкМ, и +=ИK50 50-300 мкМ (мкМ=микромолярная концентрация).
Клеточный Тест
Моноциты периферической крови человека получали из образцов крови, взятой у здоровых доноров методом лейкоцитофореза и выделения путем отмучивания. Моноциты суспендировали в среде RPMI при концентрации 2•106 кл/мл и полученную клеточную суспензию вносили в 96-луночный титрационный микропланшет при концентрации 200 мкл/лунка. К клеточной культуре добавляли растворы, содержащие испытуемые соединений в диметилсульфоксиде DMSO при соответствующей концентрации, так чтобы получить конечную концентрацию их в культуральной среде 0,5%. Клеточные суспензии с испытуемым соединением или только с DMSO инкубировали в течение 1 часа при температуре 37oС и при стимуляции клеток путем добавления 1 мкг/мл LPS-среды (липосахариды, Salmonella typehrium, 5 мг/мл в 0,1% водном растворе тетраэтиламмония (TEA)) для индуцирования активности фермента ПГНS-2 и синтеза простагландина. Клетки инкубировали в течение 17,5 часов при температуре 37oС в атмосфере с 95% содержанием O2 и 5% СО2. Культуральный надосадок отделяли и использовали для определения уровня продукции простагландина Е2 (ПГЕ2) иммуноферментным методом анализа (фирмы PerSeptive Diagnostics). Способность испытуемых соединений, приводящая к 50% ингибированию синтеза ПГЕ2, сопоставимую с культурами, обработанными только DMSO, выражается как ИК50, и представляет собой критерий оценки мощности эффекта относительно изофермента ПГНS-2.
В Таблице В, приведенной ниже, представлены данные по активности испытуемых соединений настоящего изобретения, полученные в вышеописанном клеточном тесте. В указанной Таблице показатели ИК50 обозначены как +++=ИК50<10 нмоль, ++= ИК50 10-50 нмоль и +=ИK50 51-100 нмольМ (нмоль=наномолярная концентрация).
Антипирогенная проба у крыс
Жаропонижающую активность испытуемых соединений оценивали по методу Smith и Hambourger (J. Pharmacol. Exp. Ther. вып. 54, стр. 346-351, (1935)). В первый день эксперимента крыс-самцов уравновешивают в испытательном отсеке в течение 7 часов, во время которых у крыс убирают еду и вводят им (подкожно) 20% раствор пивных дрожжей по Schiff (в физиологическом растворе), вызывающий лихорадочное состояние. Контрольную группу, получающую только солевой раствор, содержали также в аналогичных условиях. На 2 день через 19 часов после провокации у крыс измеряют температуру, а затем вводят перорально, подкожно, внутрибрюшинно или внутривенно соответствующие дозы испытуемых соединений или носитель. После введения температуру снимают в течение шести часов через каждый час. Степень пирексии определяют по изменению средних значений ректальной температуры у контрольной группы и животных с дрожжевой провокацией. Антипирогенная активность выражается как степень снижения средней ректальной температуры под действием испытуемых соединений у тех животных, которым вводили соединение относительно животных, получающих только носитель. ЕD50 рассчитывают как дозу соединения, необходимую для ослабления пирексии на 50%.
Соединения настоящего изобретения были проверены в вышеупомянутой антипирогенной пробе на крысах и имели ЕD50≤30 мг/кг.
Каррагенановая проба, индуцирующая отек лапки у крысы
Противовоспалительную активность испытуемых соединений определяли методом Winter, С.A., Risley, Е.А., и Nuss, G.N. (Proc. Soc. Exp. Biol. Med., 111, 544547 (1962)), который вкратце можно представить следующим образом. Крысам-самцам Lewis инъецируют 0,1 мл 1% раствора каррагенана (в физиологическом растворе) в подошвенную поверхность задней лапки. Крысам контрольной группы вводят только физиологический раствор. Через три часа по степени опухания лапки оценивают воспалительную реакцию. За час до инъекции провокационной пробы в подушечку лапки животным вводят перорально, подкожно, внутрибрюшинно или внутривенно испытуемые соединения или носитель. Уменьшение в задней лапке отека под воздействием испытуемых соединений относительно контрольной группы с введенным носителем является критерием оценки противовоспалительной активности. ЕD30 рассчитывают как дозу активного соединения, необходимую для уменьшения отека лапки, на 30%.
Адъювантная проба, индуцирующая артрит у крысы
Противовоспалительную активность испытуемых соединений определяли методом Pearson, C.M. (Proc. Soc. Exp. Biol. Med., вып. 91, стр. 95-101 (1956)), который вкратце можно представить следующим образом. Крысам-самцам Lewis вводили в подушечку задней лапки инъекцию полного адъюванта Фрейнда (0,1 мл 5 мг/мл в светлом минеральном масле) или только одно минеральное масло (0,1 мл). На 18 день после инъекции по степени воспалительной суставной припухлости, определенной при сравнении с контрольной группой животных, которым вводили только инъекцию минерального масла, оценивают воспалительную реакцию. Со дня 0 по день 18 животным вводят перорально, подкожно, внутрибрюшинно или внутривенно испытуемые соединения или носитель. Уменьшение суставной припухлости под воздействием испытуемых соединений относительно контрольной группы с введенным носителем является критерием оценки противовоспалительной активности. ED50 рассчитывают как дозу соединения, необходимую для уменьшения степени суставной припухлости на 50%.
Провокационная проба у крысы по Randall Selitto
Анальгетическую активность испытуемых соединений оценивали в тесте с дрожжевой провокацией в крысиную лапку, модифицированном в соответствии с методом Randall, L.O. И Selitto, J.J. (Arch. Int. Pharmacodyn. Ther. вып. 3, стр. 409-419 (1957)) при использование анальгезиометра Ugo Basile (Stoelting). Зафиксированных крыс-самцов на обе задние лапки отбирали по их ответной реакции на индуцированную дрожжами боль (писк или признаки сопротивления), которые проползли менее 15 см по поверхности анальгезиометра. В правой задней лапке затем индуцировали воспалительную реакцию путем инъекции в подошвенную поверхность (0,1 мл) 20% водной суспензии активных сухих дрожжей Fleischmann. Испытуемые соединения вводят животным перорально, подкожно, внутрибрюшинно или внутривенно через 2 часа после инъекции дрожжевой суспензии. Пороги болевой чувствительности определяют через 0,5, 1, 2 и 4 часа. ЕD30 рассчитывают как дозу соединения, необходимую для повышения болевого порога на 30% по сравнению с Контролем.
Состав и лекарственные формы
Соединения настоящего изобретения можно принимать перорально при использовании любой фармацевтически приемлемой формы выпуска, известной в данной области техники. Активный ингредиент может быть введен в организм в твердой лекарственной форме, например в виде порошков, гранул, таблеток или капсул, или в жидкой форме, например в виде сиропов или водных суспензий. Активный ингредиент можно принимать в чистом виде, но обычно его вводят в смеси с фармацевтически приемлемым носителем. Ценную информацию относительно форм выпуска лекарственных препаратов можно найти в справочнике Remington, Pharmaceutical Sciences, Mack Publishing.
Соединения настоящего изобретения можно принимать в лекарственных формах для внутреннего использования, например таблетки, капсулы (каждая из которых содержит состав с замедленным или быстрым высвобождением активного ингредиента), пилюли, порошки, гранулы, эликсиры, настойки, суспензии, сиропы, и эмульсии. Аналогичным образом предлагаемые соединения можно принимать в лекарственной форме для внутривенного (в виде болюса или быстрого вливания), внутрибрюшинного, подкожного или внутримышечного введения, причем каждая из указанных лекарственных форм общеизвестна в области фармации. Эффективное, но нетоксичное количество соединения можно использовать в качестве противовоспалительного средства или жаропонижающего.
Соединения предлагаемого изобретения можно вводить в организм млекопитающего любым способом, который обеспечивает взаимодействие активного вещества с сайтом его воздействия, то есть ПГНS-2. Предлагаемые соединения можно вводить в организм любыми традиционным биодоступными способами в комбинации с фармацевтическими составами как в виде индивидуальных лекарственных веществ, так и в комбинации с другими лекарственными средствами. Их можно принимать в чистом виде, но, как правило, в смеси с фармацевтически приемлемым носителем, подбираемым на основе выбранной схемы приема и стандартной фармацевтической технологии.
Схема приема соединений настоящего изобретения, разумеется, меняется в зависимости от известных факторов, например фармакодинамические характеристики конкретного лекарственного вещества, а также метода и пути введения; типа, возраста, пола, состояния и тяжести заболевания, а также веса больного; характера и симптоматики заболевания; типа лечения сопутствующего заболевания; частоты повторных курсов лечения; схемы приема, функции почек и печени у больного и желаемого эффекта. Обычно дипломированный врач или ветеринар может легко определить и назначить эффективную дозу препарата, необходимую для профилактики предупреждения, нормализации или блокирования развития болезни.
В качестве общего руководства суточная доза для приема внутрь каждого активного вещества, если оно используется для достижения вышеуказанных эффектов, варьируется примерно от 0,001 до 1000 мг/кг массы тела, предпочтительно примерно от 0,01 до 100 мг/кг массы тела в сутки, и наиболее предпочтительно от приблизительно 1,0 до 20 мг/кг/сут. Для нормального взрослого мужчины с массой тела примерно 70 кг это соответствует дозе от 70 до 1400 мг/сут. При внутривенном введении наиболее предпочтительная доза составляют примерно от 1 до приблизительно 10 мг/кг/мин при постоянной скорости вливания.
В предпочтительном варианте соединения настоящего изобретения можно принимать в однократной суточной дозе или общую суточную дозу можно поделить на два, три, или четыре приема в день.
Соединения предлагаемого изобретения можно принимать в интраназальной лекарственной форме при использовании подходящих интраназальных носителей для наружного применения, или их можно вводить трансдермально, например при нанесении их на кожу в форме пластыря, которые общеизвестны специалистам в этой области. При трансдермальной системе доставки поступление лекарственного вещества, разумеется, происходит, по схеме непрерывного, а не дробного введения.
В способах предлагаемого изобретения соединения, раскрытые подробно в данном описании, могут образовывать активное начало, и обычно их вводят в смеси с фармацевтически приемлемыми разбавителями, наполнителями, или носителями (обозначенные в данном описании общим термином вещества-носители), выбранные соответствующим образом в зависимости от лекарственной формы, предназначенной для приема, то есть таблеток, капсул, эликсиров, сиропов и тому подобные для перорального введения и совместимые с традиционными фармацевтическими действиями.
Например, для перорального введения в форме таблетки или капсулы активный ингредиент лекарственного средства можно использовать в смеси с нетоксичным фармацевтически приемлемым инертным носителем, например, таким как лактоза, крахмал, сахароза, глюкоза, метилцеллюлоза, магния стеарат, дикальция фосфат, кальция сульфат, маннит, сорбит и тому подобные; для перорального введения в жидкой форме ингредиенты лекарственного препарата для внутреннего приема могут быть смешаны с любым нетоксичным фармацевтически приемлемым инертным носителем, например этанолом, глицерином, водой и тому подобные. Кроме того, при желании или в случае необходимости подходящие связующие, замасливатели, дезинтеграторы и красители можно также вводить в смесь. В качестве связующих можно использовать, например, крахмал, желатин, натуральные сахара, например глюкозы или бета-лактозы, сахаристые вещества из кукурузы, естественные и синтетические смолы, например акациевая камедь, трагакантовая камедь или натрия алгинат, карбоксиметилцеллюлоза, полиэтиленгликоль, воск и тому подобные. В качестве замасливателей, которые пригодны для использования в указанных лекарственных формах, могут служить, например, натрия олеат, натрия стеарат, магния стеарат, натрия бензоат, натрия ацетат, натрия хлорид и тому подобные. В качестве дезинтеграторов можно использовать, например, но не ограничивая их объема, крахмал, метилцеллюлозу, агар, бентонит, ксантамовую смолу и тому подобные.
Соединения настоящего изобретения можно также вводить в виде систем доставки с использованием синтетических липосом, например небольших липосомных, больших однослойных липосомных везикул и многослойных липосомных везикул. Липосомы можно получить из широкого разнообразия фосфолипидов, например холестерина, стеариламина или фосфатидилхолинов.
Соединения настоящего изобретения можно также использовать в форме конъюгата с растворимыми полимерами в качестве носителей лекарственного вещества к объекту-мишени. В качестве таких полимеров можно использовать, например, поливинилпирролидон, сополимер на основе пирана, полигидроксипропилметакриламидофенол, полигидроксиэтиласпартамидодифенол или полиэтиленоксидполилизин, замещенный пальмитоильными группами. Кроме того, соединения настоящего изобретения можно использовать в связанной с биорасщепляемыми полимерами форме для обеспечения контролируемого высвобождения лекарственного вещества, например с такими как полиуксусная кислота, полигликолевая кислота, сополимеры полимолочной и полигликолевой кислоты, полиэпсилонкапролактон, полигидроксимасляная кислота, сложные полиортоэфиры, полиацетали, полидигидропираны, полицианоацилаты, и сшитые или амфотерные блок-сополимеры гидрогелей.
Лекарственные формы (фармацевтические составы), пригодные для введения, могут содержать примерно от 1 до 100 мг активного ингредиента на стандартную единицу. В таких лекарственных составах содержание активного ингредиента обычно составляет примерно 0,5-95 мас.% от всей фармацевтической композиции.
Активный ингредиент можно вводить перорально как в твердых лекарственных формах, например капсулах, таблетках и порошках, так и жидких лекарственных формах, например эликсирах, сиропах, и суспензиях. Активный ингредиент можно вводить парентерально в форме стерильных растворов. Желатиновые капсулы могут содержать активный ингредиент и порошкообразные носители, например лактозу, крахмал, производные целлюлозы, магния стеарат, стеариновую кислоту и тому подобные. Аналогичные наполнители можно использовать для получения прессованных таблеток. Как таблетки, так и капсулы можно получать в виде продукта с длительным высвобождением активного вещества для обеспечения непрерывного лечебного эффекта препарата в течение продолжительного периода времени. Прессованные таблетки можно изготавливать в форме с сахаристым покрытием, или они могут содержать пленочное покрытие для маскировки любого неприятного вкуса и защиты таблетки от воздействия окружающей среды или содержать энтерсолюбильное покрытие для избирательной распадаемости препарата в желудочно-кишечном тракте. Жидкие лекарственные формы для перорального введения могут содержать красящие вещества и ароматизирующие добавки для повышения восприятия больного к препарату. В качестве носителей для приготовления растворов для парентерального введения обычно можно использовать воду, соответствующее масло, солевой раствор, водный раствор декстрозы (глюкозы) и растворы родственных соединений класса сахаров, а также гликолей, например пропиленгликоля или полиэтиленгликоля.
Парентеральные растворы предпочтительно содержат активное вещество в виде водорастворимой соли, подходящие стабилизаторы и при необходимости буферные вещества. Антиокислители, например, такие как натрия бисульфит, натрия сульфит или аскорбиновая кислота, подходят для использования в качестве стабилизаторов, как по отдельности, так и в комбинации друг с другом. Возможно также использование лимонной кислоты и натриевой соли ЭДТА. Кроме того, растворы для парентерального введения могут содержать консерванты, например бензалкония хлорид, метил- или пропил-парабен, и хлорбутанол. Информацию по фармацевтически пригодным носителям можно найти в Remington Pharmaceutical Sciences, фирмы Mack Publishing Company, стандартном библиографическом справочнике в этой области.
Пригодные для использования лекарственные формы для введения соединений настоящего изобретения можно проиллюстрировать следующим примерами
Капсулы
Капсулы получают традиционными методами так, что стандартная таблетка содержит 500 мг активного ингредиента, 100 мг целлюлозы и 10 мг стеарата магния.
Большое количество унифицированных капсул можно также получить путем заполнения разделенных на две части капсул из твердого желатина со стандартной дозой, каждая часть которых содержит 100 мг порошкообразного активного ингредиента, 150 мг лактозы, 50 мг целлюлозы и 6 мг стеарата магния.
Сироп
Компоненты - Содержание, мас.%:
Активный ингредиент - 10
Сахарный сироп - 50
Сорбит - 20
Глицерин - 5
Ароматизатор, краситель и консервант - При необходимости
Вода - При необходимости
Конечный объем доводят до 100% путем прибавления дистиллированной воды.
Водная суспензия
Компоненты - Содержание, мас.%:
Активный ингредиент - 10
Сахарин натрия - 0,01
Келтрол® (пищевая ксантановая камедь) - 0,2
Сахарный сироп - 5
Ароматизатор, краситель и консервант - При необходимости
Вода - При необходимости
Ксантановую камедь медленно прибавляют к дистиллированной воде перед введением активного ингредиента и остальных ингредиентов состава. Полученную суспензию пропускают через гомогенизатор для придания эстетичности конечной продукции.
Ресуспендированный порошок
Компоненты - Содержание, мас.%:
Активный ингредиент - 50,0
Лактоза - 35,0
Сахар - 10,0
Акация - 4,7
Натриевая соль карбоксиметилцеллюлозы - 0,3
Каждый ингредиент тонко измельчают, а затем смешивают до получения гомогенной смеси. В альтернативном варианте порошок можно получить в виде суспензии, которую затем сушат методом распыления.
Полутвердый гель
Компоненты - Содержание, мас.%:
Активный ингредиент - 10
Сахарин натрия - 0,02
Желатин - 2
Ароматизатор, краситель и консервант - При необходимости
Вода - При необходимости
Желатин разводят в горячей воде. Тонко измельченный активный ингредиент суспендируют в желатиновом растворе, а затем добавляют остальные ингредиенты состава. Полученной суспензией заполняют подходящий упаковочный контейнер с получением геля после остывания.
Полутвердая паста
Компоненты - Содержание, мас. %:
Активный ингредиент - 10
Гелкарин® (каррагениновая камедь) - 1
Сахарин натрия - 0,01
Желатин - 2
Ароматизатор, краситель и консервант - При необходимости
Вода - При необходимости
Гелкарин® разводят в горячей воде (температура около 80oС) и затем тонко измельченный активный ингредиент суспендируют в полученном растворе. К суспензии добавляют сахарин натрия и остальные ингредиенты состава, пока она еще не остыла. Суспензию гомогенизируют и полученным гомогенатом заполняют подходящие контейнеры.
Эмульгируемая паста
Компоненты - Содержание, мас.%:
Активный ингредиент - 30
Твин 80 и Спэн 80 - 6
Келтрол® (пищевая ксантановая камедь) - 0,5
Минеральное масло - 63,5
Все ингредиенты тщательно перемешивают до получения гомогенной пасту.
Мягкие желатиновые капсулы
Активный ингредиент смешивают с растительным маслом, например соевым, хлопковым или оливковым, и закачивают объемным насосом в желатин с получением мягких желатиновых капсул, содержащих 100 мг активного ингредиента. Капсулы промывают и сушат.
Таблетки
Таблетки можно получить традиционными методами так, что стандартная таблетка содержит 500 мг активного ингредиента, 150 мг лактозы, 50 мг целлюлозы и 10 мг стеарата магния.
Большое количество таблеток можно также получить в соответствии с традиционными методами так, что унифицированная таблетка содержит 100 мг активного ингредиента, 0,2 мг коллоидной окиси кремния, 5 мг стеарата магния, 275 мг микрокристаллической целлюлозы, 11 мг крахмала и 98,8 мг лактозы. Для улучшения биодоступности и замедления всасываемости можно использовать соответствующие покрытия.
Состав для парентерального введения
Парентеральный состав, пригодный для его введения в виде инъекции, получают путем перемешивания 1,5 мас.% активного ингредиента в 10 об.% пропиленглиголя с водой. При добавлении натрия хлорида получают изотонический раствор, который затем стерилизуют.
Суспензия
Водную суспензию для приема внутрь получают в стандартной дозе 5 мл, которая содержит 100 мг тонкоизмельченного активного ингредиента, 200 мг натриевой соли карбоксиметилцеллюлозы, 5 мг натрия бензоата, 1,0 г раствора сорбита (по фармакопее США) U.S.Р. и 0,025 мл ванилина.
Соединения настоящего изобретения можно принимать в комбинации со вторым лекарственным средством. Соединение формулы I и дополнительное терапевтическое средство можно принимать как раздельно, так и в смеси, содержащей однократную дозу, в любой лекарственной форме и различными способами введения, как описано выше.
Соединение формулы I можно составлять в рецептуре вместе со вторым лекарственным средством в стандартной отднократной дозе (то есть в виде смеси в одной капсуле, таблетке, порошке, или жидком носителе и тому подобное). Если соединение формулы I и второе лекарственное средство используют по отдельности, то соединение формулы I и указанное второе средство можно принимать одновременно или параллельно в любой последовательности; например, можно принимать вначале соединение формулы I, а затем второе лекарственное средство. При неодновременном приеме предпочтительно, чтобы промежуток между приемом соединения формулы I и второго лекарственного средства составлял менее примерно 1 часа, более предпочтительно менее примерно от 5 до 30 минут.
Предпочтительно, чтобы соединение формулы I вводилось перорально. Хотя и предпочтительно введение соединения формулы I и второго лекарственного средства одним и тем же способом (т.е., например, перорально), при желании каждый из них можно принимать различными способами и в различных лекарственных формах (то есть один компонент можно принимать перорально, а другой компонент вводить внутривенно). Доза соединения формулы I как при раздельном приеме, так и в комбинации со вторым лекарственным средством может меняться в зависимости от различных факторов, например, таких как фармакодинамические характеристики конкретного лекарственного вещества, а также метода и схемы приема, возраста, состояния и веса больного, характера и симптоматики заболевания, вида лечения сопутствующего заболевания; частоты повторных курсов лечения и желаемого эффекта, как указывалось ранее. В частности при введении в одной лекарственной форме существует вероятность химического взаимодействия между используемыми в комбинации действующими началами. По этой причине, при одновременном введении соединения формулы I и второго лекарственного средства, они формулируются так, что несмотря на то что эти действующие начала скомбинированы для приема в унифицированной дозе, физический контакт между активными веществами должен быть минимальным. Например, одно активное вещество может содержать энтеросолюбильное покрытие.
Использование одного из активных ингредиентов с энтеросолюбильным покрытием позволяет не только свести к минимуму контакт между используемыми в комбинации активными веществами, но и также обеспечивает регулируемое высвобождение одного из таких компонентов в желудочно-кишечном тракте, при котором происходит высвобождение одного активного вещества в желудке, а другого в тонкий кишечник. Один из активных компонентов может быть также покрыт материалом, который обеспечивает замедленное высвобождение действующего начала, приводя тем самым к равномерному распределению по всему желудочно-кишечному тракту, а также свести к минимуму физический контакт между используемой комбинацией активных веществ. Кроме того, компонент с замедленным высвобождением может дополнительно содержать энтеросолюбильное покрытие, что обеспечивает его высвобождение только в тонкую кишку. В еще одном варианте предлагается состав комбинированного препарата, в котором один его компонент покрыт полимерным материалом, обеспечивающим пролонгированное его высвобождение и/или высвобождение в тонкую кишку, и другой компонент может также содержать полимерное покрытие, например, из маловязкой гидроксипропилметицеллюлозы (НРМС) или других подходящих материалов, общеизвестных в данной области техники, для разделения активных ингредиентов. Полимерное покрытие позволяет обеспечить дополнительный барьер для взаимодействия с другим компонентом.
Вышеуказанные, а также другие способы обеспечения минимального контакта между компонентами комбинированного препарата настоящего изобретения независимо от того, вводятся ли они в одной лекарственной форме или формах, вводимых раздельно, однако параллельно одним и тем же способом, станут очевидны специалистам в данной области техники при знакомстве с настоящим описанием сущности изобретения.
В объем предлагаемого изобретения также входят лекарственные комплекты, например, используемые в терапии или профилактике воспалительных заболеваний, которые включают один или несколько контейнеров с лекарственным составом, содержащим терапевтически эффективное количество соединения формулы I. В такой комплект может также входить при желании один или несколько компонентов различных традиционных фармацевтических наборов, например контейнер с одним или несколькими фармацевтически приемлемыми носителями, дополнительные контейнеры и тому подобное, которые легко понятны для специалистов в данной области техники. В комплект могут также входить инструкции, такие как вкладыши, или как маркировочные ярлыки, где даны количественные характеристики вводимых компонентов, а также указания по схеме введения и комбинации компонентов.
Необходимо понимать, что в настоящем описании раскрываемые материалы и условия играют важное значение в практической реализации изобретения, однако материалы и условия, которые не охарактеризованы, не должны исключаться из объема до тех пор, пока они не препятствуют получению положительного эффекта при осуществлении настоящего изобретения.
Термин "состоящий из", когда его используют в настоящем описании, имеет свое традиционное значение; то есть все указанные материалы и условия очень важны в практической реализации предлагаемого изобретения, однако материалы и условия, которые не охарактеризованы, не должны исключаться из объема до тех пор пока они не препятствуют получению положительного эффекта при осуществлении настоящего изобретения.
В предлагаемом описании сущности изобретения содержится вся необходимая информация, чтобы специалист в данной области техники смог реализовать на практике заявляемое изобретение. Поскольку противопоставленные материалы могут дать дополнительно полезную информацию, то следует понимать, что эти материалы включены в описание в качестве ссылки.
Хотя настоящее изобретение описано выше на конкретных примерах его осуществления, необходимо понимать, что изобретение не ограничивается изложенными вариантами его осуществления или отдельными их деталями и что возможны различные его модификации, не выходящие за рамки существа и объема предлагаемого изобретения.
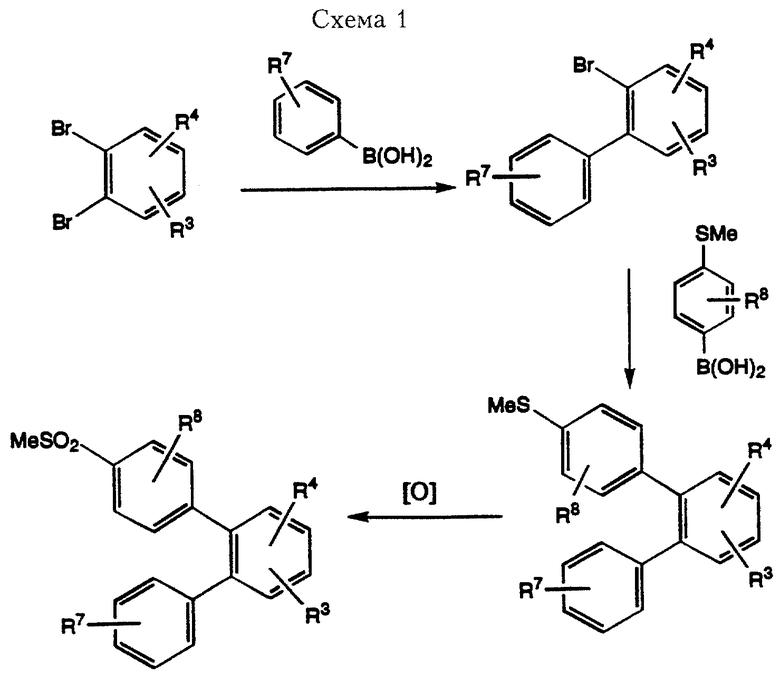
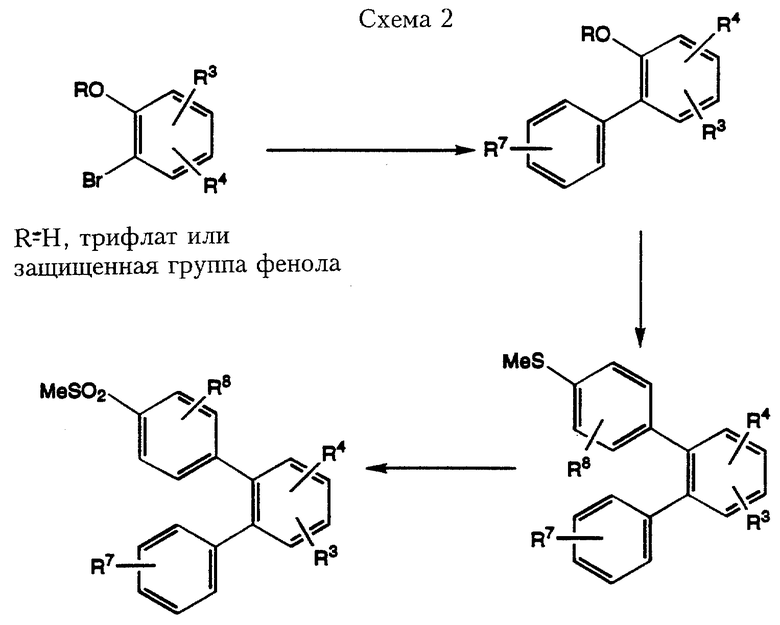
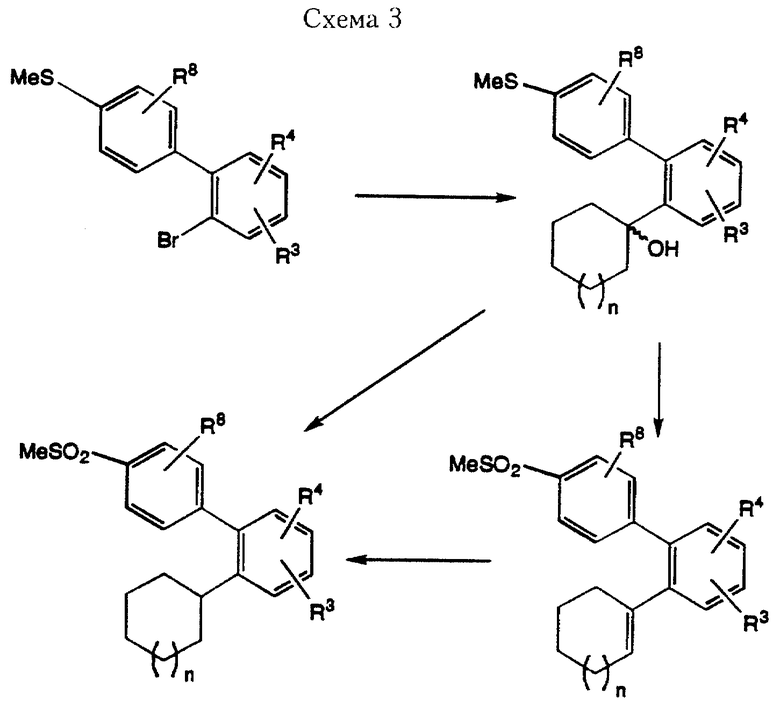
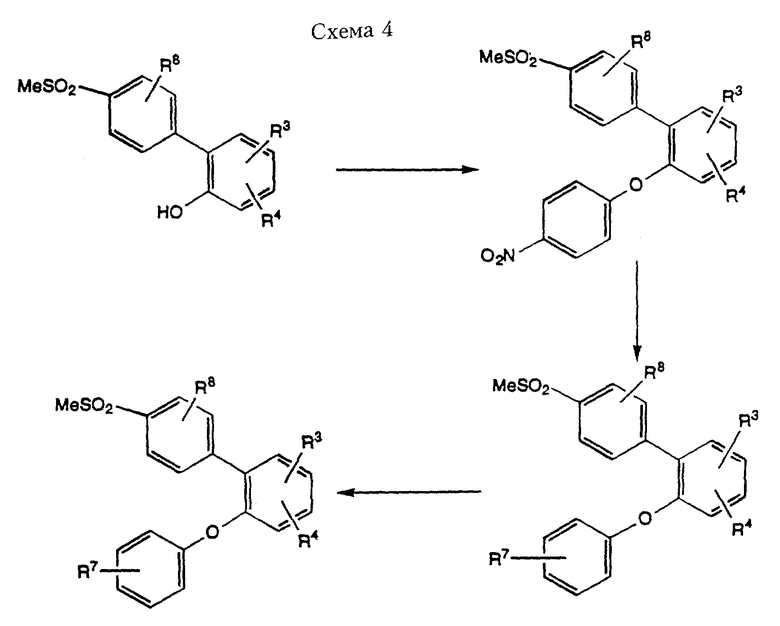

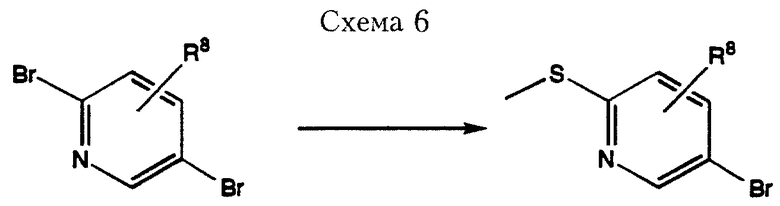
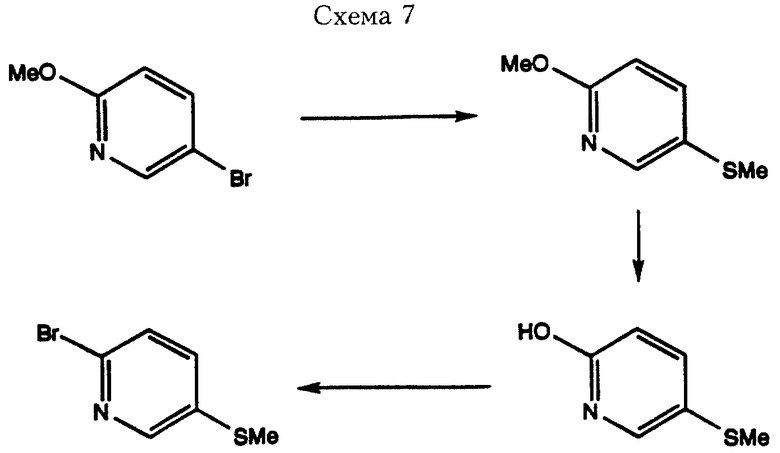
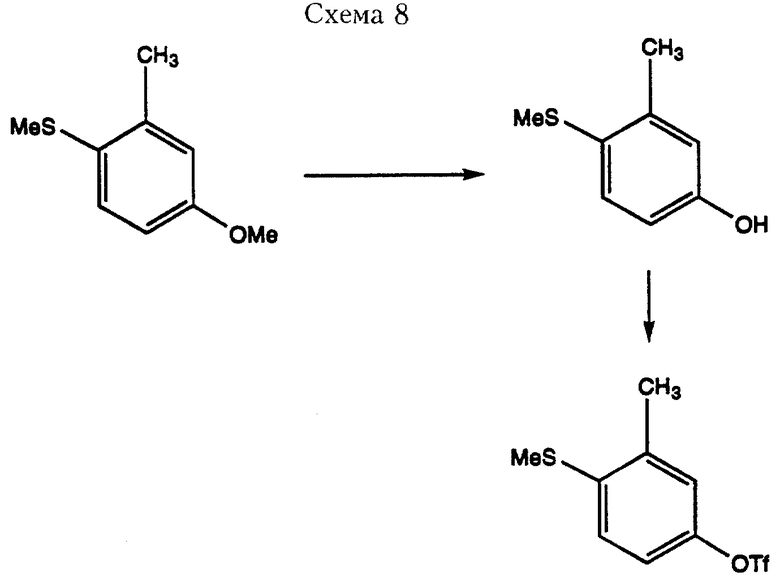
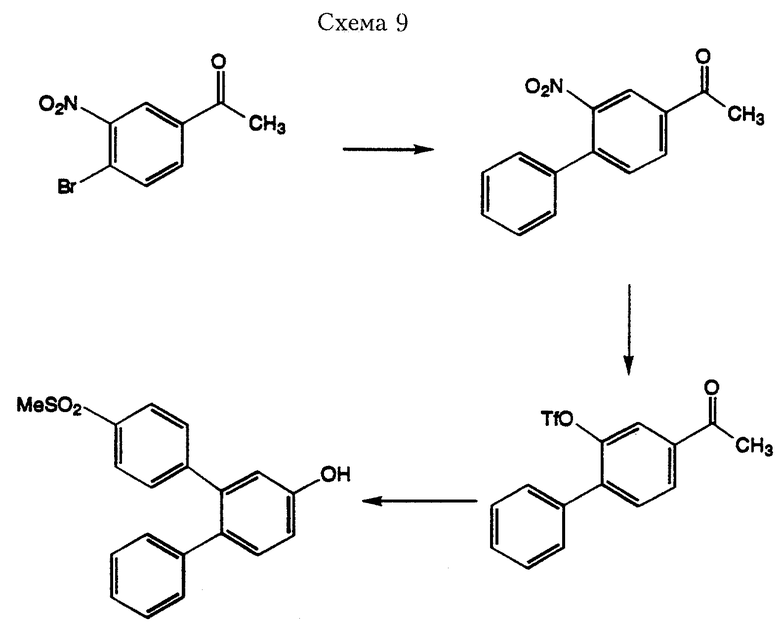
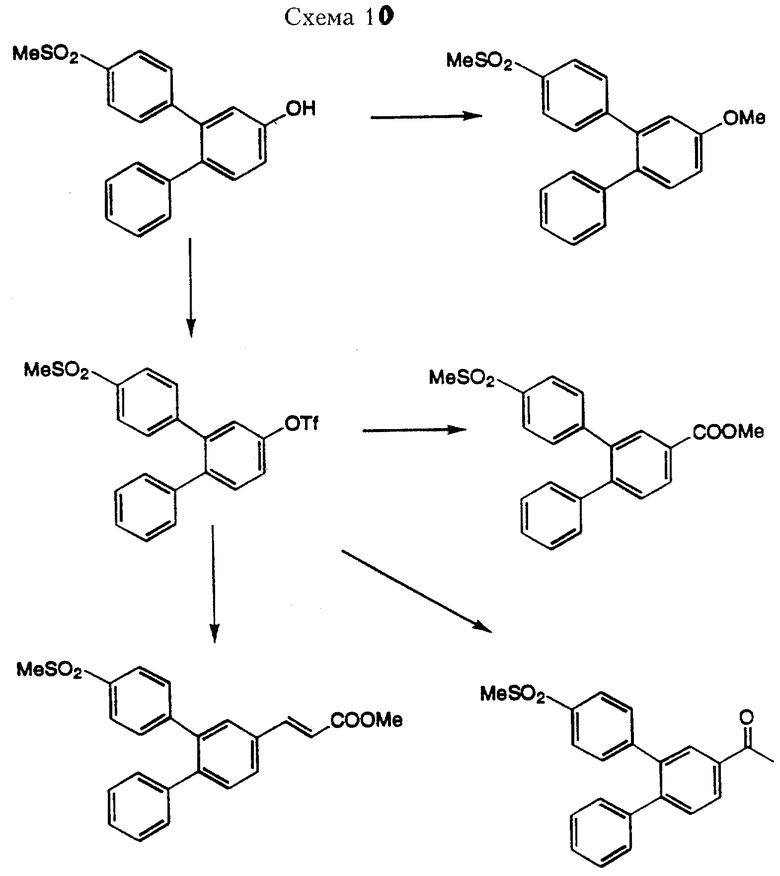
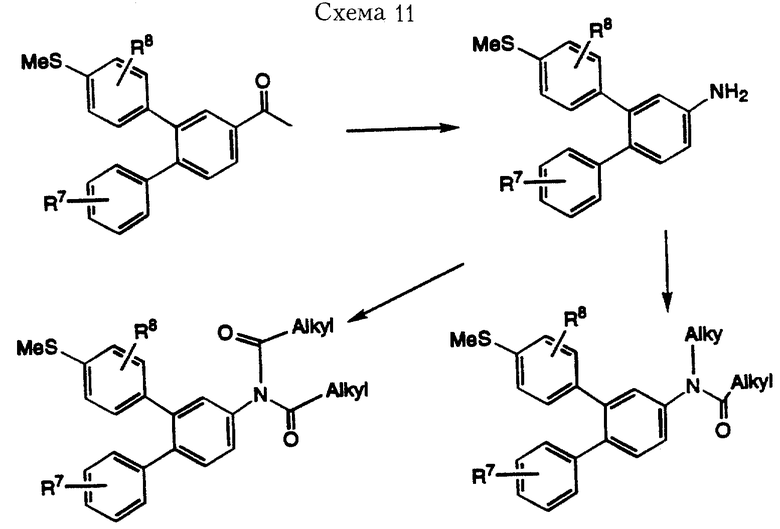
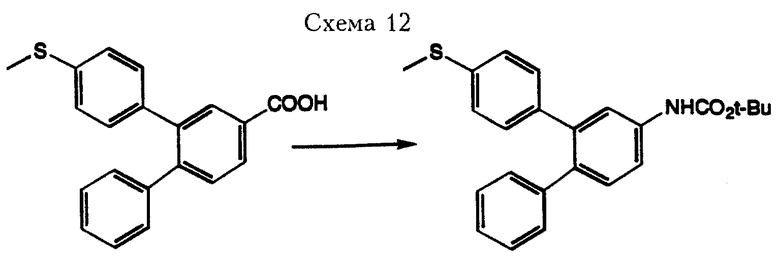
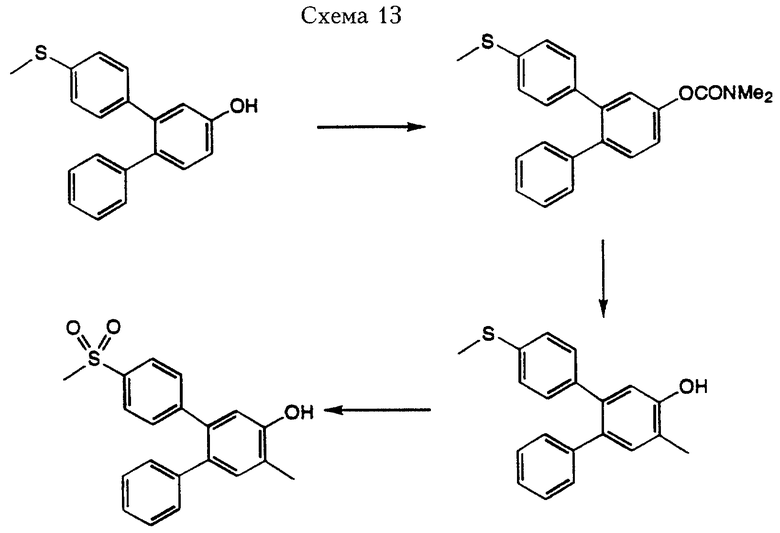
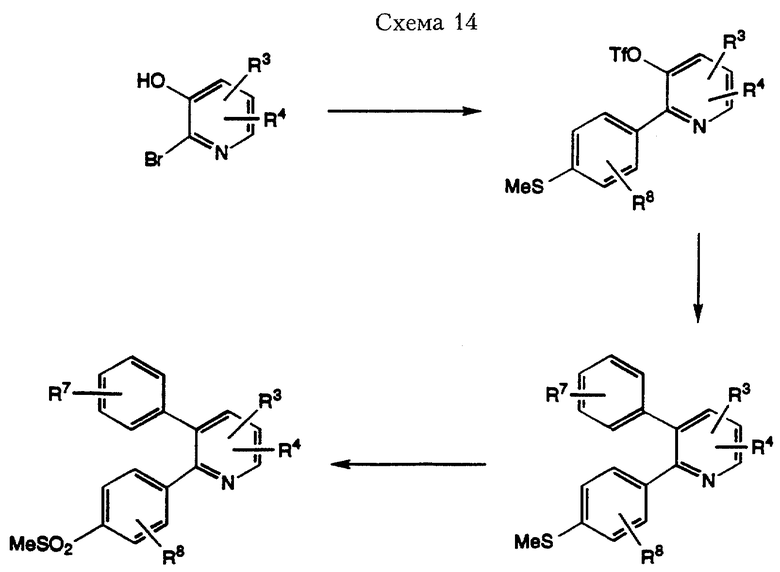
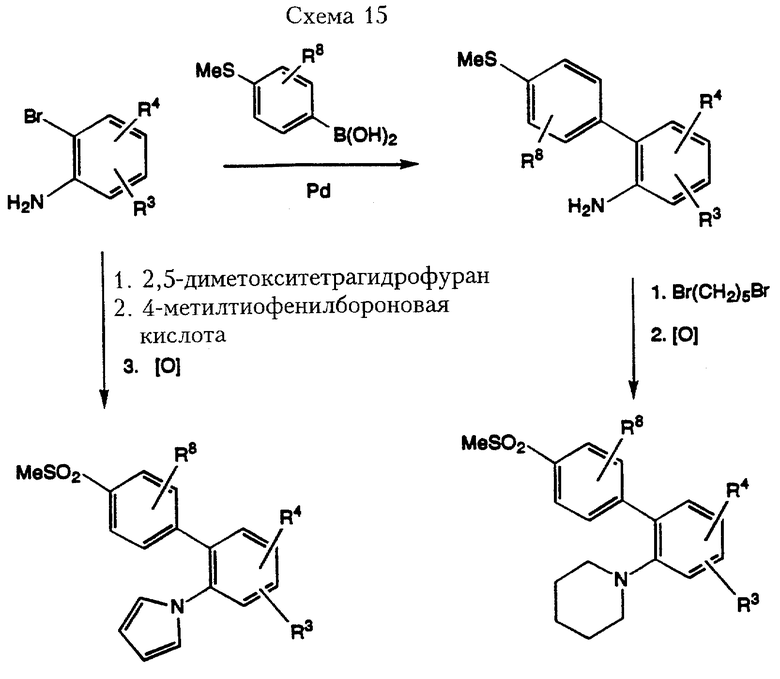
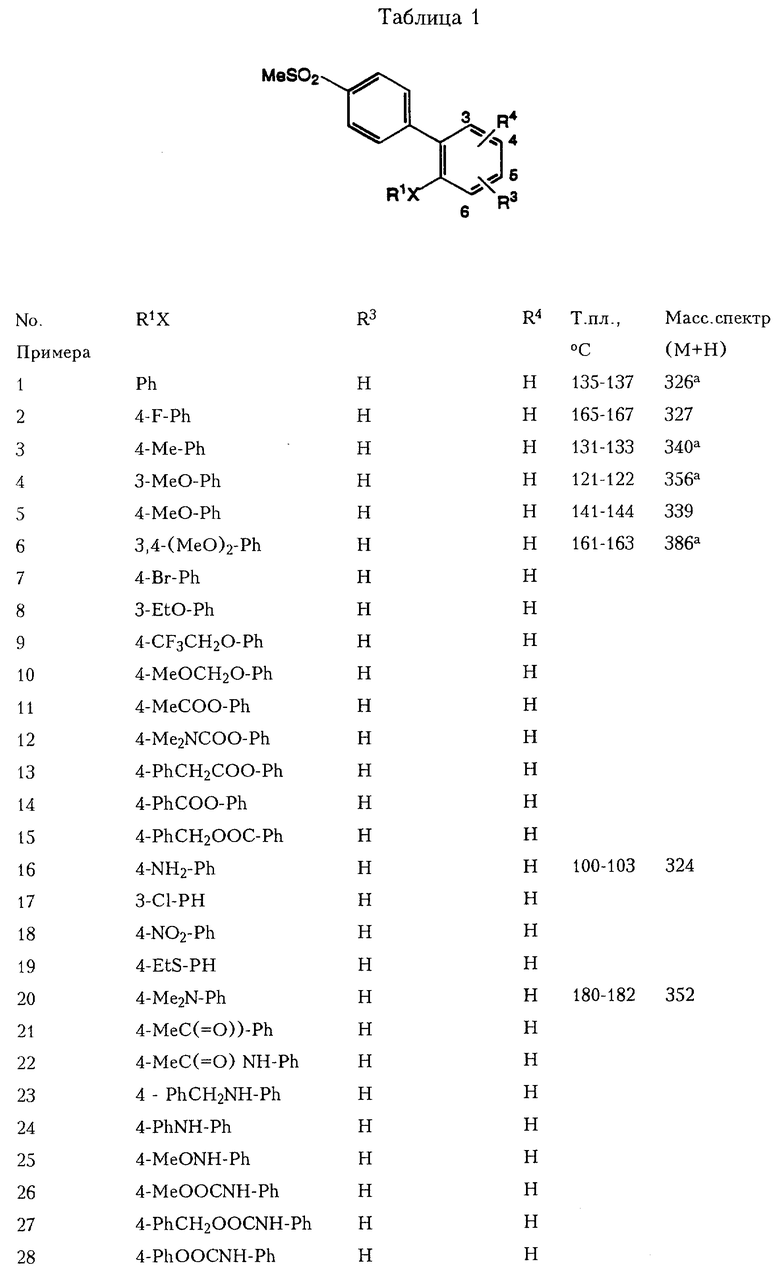

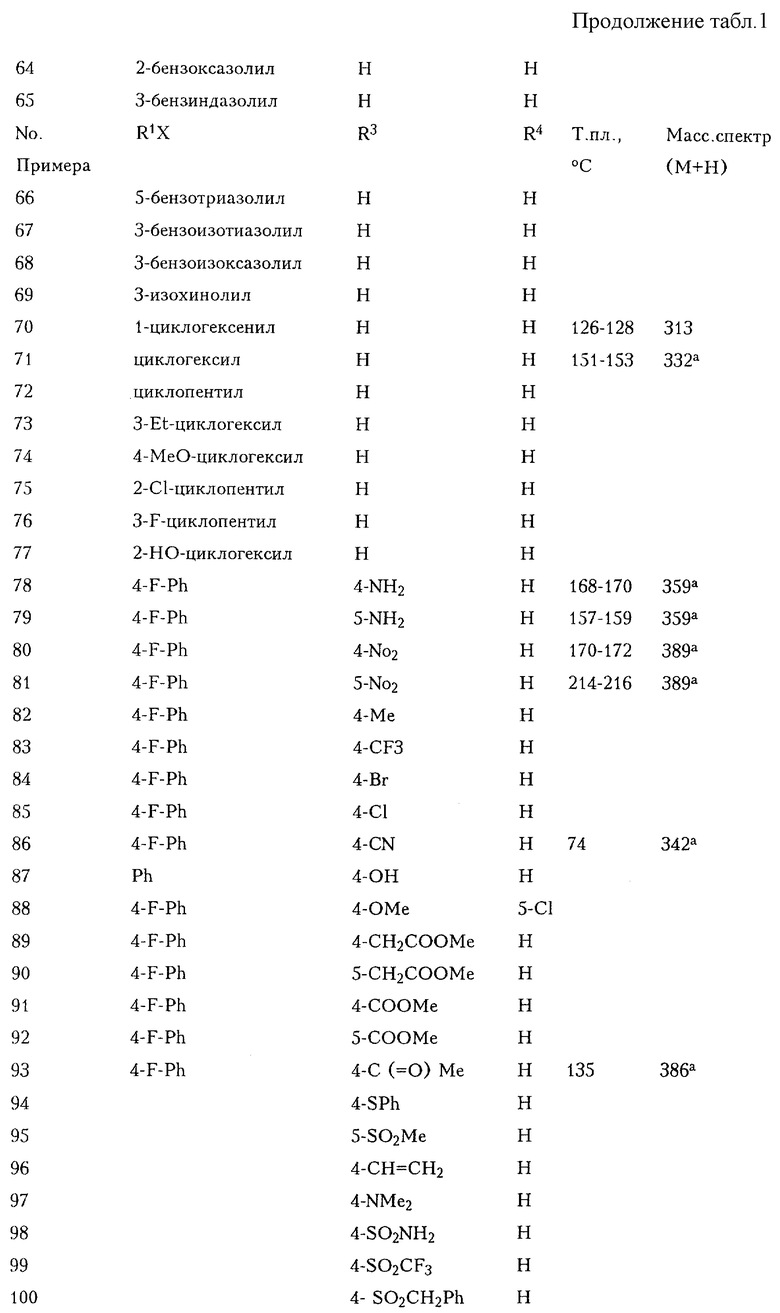


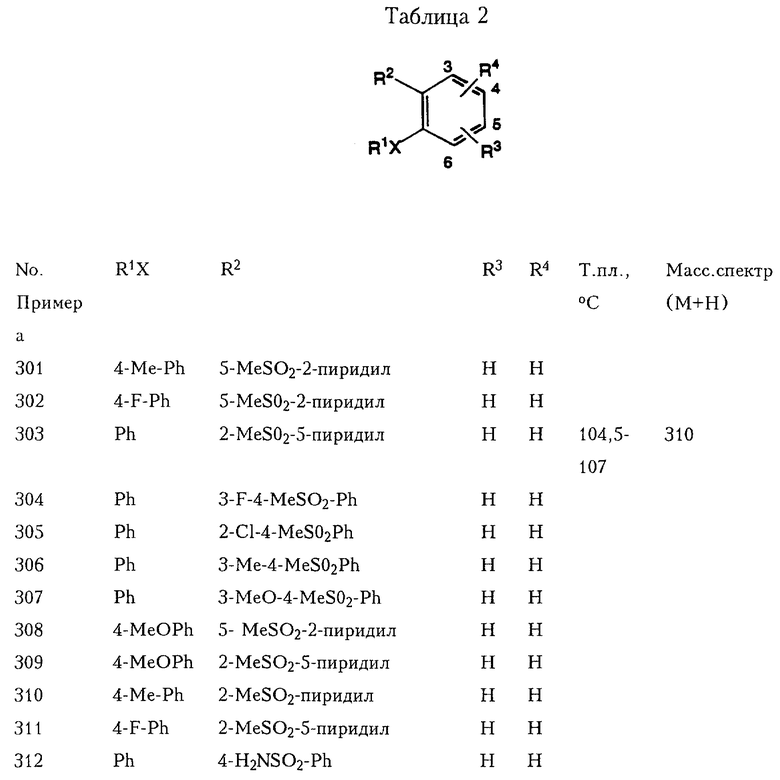




Изобретение относится к ортозамещенным соединениям формулы I или их фармацевтически приемлемым солям, которые являются ингибиторами простагландин Н синтазы. В соединениях формулы I J представляет СН или N, K и L представляют СН; Х представляет простую связь,  -Z(СНR5)p; Z представляет О; R1 представляет фенил или 2-нафтил, замещенный 0-2 заместителями R7, С5-С7 циклоалкил или С5-С7 циклоалкенил, при условии, что R1 связан непосредственно с гетероатомом, указанный гетероатом не связан с атомом углерода, содержащим двойную связь в циклоалкеновом кольце, или 5-10-членную гетероциклическую систему, выбранную из пирролила, бензотиенила, 3-пиридила, необязательно замещенного С1-С4 алкилом, хинолинила или пиперидинила; R2 представляет ниже приведенные группы 1) или 2), Y представляет метил; R3 представляет Н, F, Br, Cl, OH, NO2, NR15R16, C(=O)R6; R4 представляет водород или альтернативно, когда R3 и R4 являются заместителями у соседних атомов углерода, R3 и R4, взятые вместе с атомами углерода, к которым они присоединены, образуют 5-7-членную карбоциклическую систему; R5 представляет водород; R6 представляет С1-С6 алкил; R7 представляет заместитель у атома углерода, выбранный из группы, состоящей из фтора, брома, хлора, йода, С1-С4 алкила, СН2ОН, СН2OCH3, С1-С4 алкокси, СНО, NR15R16; R8 представляет водород; R15 представляет водород или С1-С4 алкил; R16 представляет водород или С1-С4 алкил; p = 0 или 1. Предложены также фармацевтические композиции для ингибирования простагландин Н синтазы и способ ингибирования простагландин Н синтазы. 6 с. и 3 з.п. ф-лы, 5 табл.
-Z(СНR5)p; Z представляет О; R1 представляет фенил или 2-нафтил, замещенный 0-2 заместителями R7, С5-С7 циклоалкил или С5-С7 циклоалкенил, при условии, что R1 связан непосредственно с гетероатомом, указанный гетероатом не связан с атомом углерода, содержащим двойную связь в циклоалкеновом кольце, или 5-10-членную гетероциклическую систему, выбранную из пирролила, бензотиенила, 3-пиридила, необязательно замещенного С1-С4 алкилом, хинолинила или пиперидинила; R2 представляет ниже приведенные группы 1) или 2), Y представляет метил; R3 представляет Н, F, Br, Cl, OH, NO2, NR15R16, C(=O)R6; R4 представляет водород или альтернативно, когда R3 и R4 являются заместителями у соседних атомов углерода, R3 и R4, взятые вместе с атомами углерода, к которым они присоединены, образуют 5-7-членную карбоциклическую систему; R5 представляет водород; R6 представляет С1-С6 алкил; R7 представляет заместитель у атома углерода, выбранный из группы, состоящей из фтора, брома, хлора, йода, С1-С4 алкила, СН2ОН, СН2OCH3, С1-С4 алкокси, СНО, NR15R16; R8 представляет водород; R15 представляет водород или С1-С4 алкил; R16 представляет водород или С1-С4 алкил; p = 0 или 1. Предложены также фармацевтические композиции для ингибирования простагландин Н синтазы и способ ингибирования простагландин Н синтазы. 6 с. и 3 з.п. ф-лы, 5 табл.
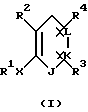
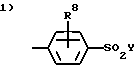
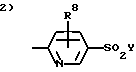
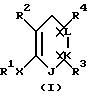
или их фармацевтически приемлемые соли,
где J представляет СН или N;
К и L представляют СН;
Х представляет прямую связь, 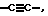 -Z(CHR5)p;
-Z(CHR5)p;
Z представляет О;
R1 представляет фенил или 2-нафтил, замещенный 0-2 заместителями R7, С5-С7 циклоалкил, или С5-С7 циклоалкенил, при условии, что, когда R1 связан непосредственно с гетероатомом, указанный гетероатом не связан с атомом углерода, содержащим двойную связь в циклоалкеновом кольце, или 5-10 членную гетероциклическую систему, выбранную из пирролила, бензотиенила, 3-пиридила, необязательно замещенного C1-C4 алкилом, хинолинила или пиперидинила;
R2 представляет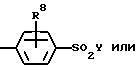
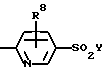
Y представляет метил;
R3 представляет Н, F, Br, Сl, ОН, NО2, NR15R16, C(= O)R6;
R4 представляет водород, или альтернативно, когда R3 и R4 являются заместителями у соседних атомов углерода, R3 и R4, взятые вместе с атомами углерода, к которым они присоединены, образуют 5-7 членную карбоциклическую систему;
R5 представляет водород;
R6 представляет C1-C6 алкил;
R7 представляет заместитель у атома углерода, выбранный из группы, состоящей из фтора, брома, хлора, иода, C1-C4 алкила, СН2ОН, СН2ОСН3, C1-C4 алкокси, СНО, NR15R16;
R8 представляет водород;
R15 представляет водород или C1-C4 алкил;
R16 представляет водород или C1-C4 алкил;
р = 0 или 1.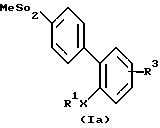
в которой R1X представляет фенил, 4-фторфенил, 3-метоксифенил, 4-метоксифенил, 3,4-диметоксифенил, 4-гидроксиметилфенил, 4-метоксиметилфенил, 4-диметиламинофенил, 4-формилфенил, 2-нафтил, 5-метокси-2-нафтил, 2-хинолинил, 3-хинолинил, 2-бензотиенил, 5-бензотиенил, 3-пиридил, необязательно замещенный C1-C4 алкилом, фенилацетиленил, фенокси, циклогексенил, циклогексил, 4-фторфенокси, циклогексилокси, бензилокси, 1-пирролил или 1-пиперидинил;
R3 представляет Н, 5-хлор, 4-гидрокси-, 4-нитро-, 5-нитро- или 4-ацето-;
или их фармацевтически приемлемые соли.
| C.F.H | |||
| ALLEN ET | |||
| AL | |||
| The chemistry o-terphenyl.III Sulfonic acids, J | |||
| Org | |||
| Chem., vol | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Экономайзер | 0 |
|
SU94A1 |
| Арил-( -алкил-п-метоксибензил)сульфиды, проявляющие противовоспалительную активность | 1976 |
|
SU556599A1 |
