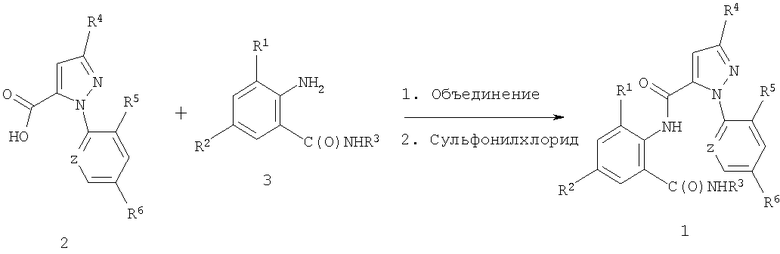
Данное изобретение относится к способу получения N-фенилпиразол-1-карбоксамидов связыванием карбоновых кислот с антраниламидами и к соединениям антраниламидов, пригодным для способа.
Патентная публикация PCT WO 03/015518 раскрывает использование в качестве антроподицитов N-ацил производных антраниловой (о-аминобензойной) кислоты формулы i

где A и B независимо представляют O или S; R1 представляет H; R2 представляет H, C1-C6 алкил, C2-C6 алкоксикарбонил или C2-C6 алкилкарбонил; R3 представляет, наряду с прочим, H или C1-C6 алкил; R4 представляет, наряду с прочим, H или C1-C6 алкил; R5 представляет Н, C1-C6 алкил или галоген; R6 представляет Н, C1-C6 алкил, C1-C6 галогеналкил, галоген, CN, C1-C4 алкокси или C1-C4 галогеналкокси; R7 представляет, наряду с прочим, кольцо фенила, кольцо бензила, 5- или 6-членное гетероароматическое кольцо, кольцевую систему нафтила, каждое кольцо или кольцевая система необязательно замещены 1-3 заместителями; и R8 представляет, наряду с прочим, H. Этот документ раскрывает различные способы получения соединений формулы i. Однако остается потребность в новых способах, менее дорогих, более эффективных, более гибких или более удобных для работы.
Сущность изобретения
Данное изобретение относится к способу получения соединения формулы 1

где R1 означает CH3 или Cl;
R2 означает Br, Cl, I или CN;
R3 означает H или C1-C4 алкил;
R4 означает Cl, Br, CF3, OCF2H или OCH2CF3;
R5 означает F, Cl или Br;
R6 означает Н, F или Cl;
Z означает CR7 или N и
R7 означает Н, F, Cl или Br.
Способ включает объединение (1) соединения карбоновой кислоты формулы 2

(2) соединения анилина формулы 3

и (3) сульфонилхлорида с получением соединения формулы 1.
Данное изобретение также относится к соединению анилина формулы 3, где
R1 представляет CH3 или Cl;
R2 представляет Br, Cl, I или CN и
R3 представляет H или C1-C4 алкил,
при условии, что
(a) когда R1 и R2 представляют Cl, тогда R3 не является H, CH2CH3 или CH(CH3)CH2CH3;
(b) когда R1 представляет CH3 и R2 представляет Cl, Br или CN, тогда R3 не является CH3 или CH(CH3)2;
(c) когда R1 представляет Cl и R2 представляет Cl или Br, тогда R3 не является CH3 или CH(CH3)2, и
(d) когда R1 представляет CH3 и R2 представляет CN, тогда R3 не является H.
Подробное описание изобретения
Используемые в описании термины "содержит", "содержащий", "включает", "включающий", "имеет", "имеющий" или любые другие их вариации предназначаются для охвата неэксклюзивного добавления. Например, состав, процесс, способ, изделие или аппарат, который содержит перечень элементов, не должен ограничиваться только этими элементами, но может включать другие элементы, четко не перечисленные или свойственные такому составу, процессу, способу, изделию или аппарату. Далее, если четко не указано противоположного, "или" относится к инклюзивному или, а не к эксклюзивному или. Например, условие А или В удовлетворяет одному из следующего: А является истинным (или присутствует) и В является ложным (или отсутствует), А является ложным (или отсутствует) и В является истинным (или присутствует), и оба А и В являются истинными (или присутствуют).
Объединение реагентов относится к взаимодействию их друг с другом.
К тому же неопределенные артикли "а" и "an" перед элементом или компонентом изобретения предполагают неограничительное рассмотрение числа отдельных примеров (т.е. случаев) элемента или компонента. Поэтому "a" или "an" следует понимать как включающие один или по меньшей мере один, и единственное число слова, обозначающего элемент или компонент, также включающие множественное число, если число явно не должно быть единственным.
Радикал на основе углерода относится к одновалентному молекулярному компоненту, содержащему атом углерода, который соединяет радикал с остальной химической структурой через одинарную связь. Радикалы на основе углерода необязательно могут содержать насыщенные, ненасыщенные и ароматические группы, цепи, кольца и кольцевые системы и гетероатомы. Хотя радикалы на основе углерода не подвергаются какому-либо конкретному ограничению по размеру, в контексте данного изобретения они обычно содержат от 1 до 16 атомов углерода и от 0 до 3 гетероатомов. Предпочтительными являются радикалы на основе углерода, выбранные из C1-C6 алкила, C1-C4 галогеналкила и фенила, необязательно замещенных 1-3 заместителями, выбранными из C1-C3 алкила, галогена и нитро.
В перечислениях в описании сокращение "Ph" означает фенил. Алкил может быть с прямой или разветвленной цепью. Термин "галоген", или один, или в сложных словах, таких как "галогеналкил", включает фтор, хлор, бром или йод. К тому же при использовании в сложных словах, таких как "галогеналкил", указанный алкил может быть частично или полностью замещенным атомами галогена, которые могут быть одинаковыми или разными. Примеры "галогеналкила" включают F3C, ClCH2, CF3CH2 и CF3CCl2.
Варианты воплощения данного изобретения включают:
Вариант воплощения M1. Способ, где молярное отношение соединения формулы 2 к соединению формулы 3 от около 1,2:1 до около 1:1,2.
Вариант воплощения M2. Способ по варианту воплощения M1, где молярное отношение соединения формулы 2 к соединению формулы 3 от около 1:1 до около 1:1,2.
Вариант воплощения M3. Способ по варианту воплощения M2, где молярное отношение соединения формулы 2 к соединению формулы 3 около 1:1,1.
Вариант воплощения M4. Способ, где молярное отношение сульфонилхлорида к соединению формулы 2 по меньшей мере около 1:1.
Вариант воплощения M5. Способ по варианту воплощения M4, где молярное отношение сульфонилхлорида к соединению формулы 2 от около 1:1 до около 2,5:1.
Вариант воплощения M6. Способ по варианту воплощения M5, где молярное отношение сульфонилхлорида к соединению формулы 2 от около 1,1:1 до около 1,4:1.
Вариант воплощения M7. Способ по варианту воплощения M6, где когда R2 представляет Br, Cl или I, тогда молярное отношение сульфонилхлорида к соединению формулы 2 около 1,2:1.
Вариант воплощения M8. Способ по варианту воплощения М6, где когда R2 представляет CN, тогда молярное отношение сульфонилхлорида к соединению формулы 2 около 1,4:1.
Вариант воплощения M9. Способ, где сульфонилхлорид представлен формулой 4
R8S(O)2Cl,
где R8 представляет углеродсодержащий радикал.
Вариант воплощения M10. Способ по варианту воплощения M9, где R8 представляет C1-C4 алкил, C1-C2 галогеналкил или фенил, необязательно замещенные 1-3 заместителями, выбранными из группы, состоящей из галогена, C1-C3 алкила и нитро.
Вариант воплощения M11. Способ по варианту воплощения M10, где R8 представляет C1-C2 алкил, CF3, фенил или 4-метилфенил.
Вариант воплощения M12. Способ по варианту воплощения M11, где R8 представляет C1-C2 алкил, фенил или 4-метилфенил.
Вариант воплощения M13. Способ по варианту воплощения M12, где R8 представляет CH3.
Вариант воплощения M14. Способ, где карбоновую кислоту формулы 2, анилин формулы 3 и сульфонилхлорид объединяют при температуре между около -70 и 100°С.
Вариант воплощения M15. Способ по варианту воплощения M14, где температура между около -20 и 40°С.
Вариант воплощения M16. Способ по варианту воплощения MI5, где температура между около -10 и 20°С.
Вариант воплощения M17. Способ, где карбоновую кислоту формулы 2 объединяют с анилином формулы 3, чтобы получить смесь, и затем смесь объединяют с сульфонилхлоридом.
Вариант воплощения M18. Способ по варианту воплощения M17, где основание объединяют со смесью или до, или после объединения с сульфонилхлоридом.
Вариант воплощения M19. Способ по варианту воплощения M17, где основание объединяют с соединениями формул 2 и 3, чтобы получить смесь, до объединения с сульфонилхлоридом.
Вариант воплощения M20. Способ, где основание объединяют с соединениями формул 2 и 3 и сульфонилхлоридом.
Вариант воплощения M21. Способ по любому из вариантов воплощения M18-M20, где количество основания равно по меньшей мере приблизительно 2 эквивалентам по отношению к сульфонилхлориду.
Вариант воплощения M22. Способ по варианту воплощения M21, где количество равно по меньшей мере приблизительно 2,1 эквивалента по отношению к сульфонилхлориду.
Вариант воплощения M23. Способ по варианту воплощения M22, где количество основания равно от около 2,1 до 2,2 эквивалента по отношению к сульфонилхлориду.
Вариант воплощения M24. Способ по любому из вариантов воплощения M18-M20, где основание выбрано из третичных аминов (включая необязательно замещенные пиридины).
Вариант воплощения M25. Способ по варианту воплощения M24, где основание выбрано из необязательно замещенных пиридинов и их смесей.
Вариант воплощения M26. Способ по варианту воплощения M25, где основание выбрано из 2-пиколина, 3-пиколина, 2,6-лутидина, пиридина и их смесей.
Вариант воплощения M27. Способ по варианту воплощения M26, где основанием является 3-пиколин.
Вариант воплощения M28. Способ, где растворитель объединяют с соединениями формул 2 и 3 и сульфонилхлоридом.
Вариант воплощения M29. Способ по варианту воплощения M17, где растворитель объединяют с соединениями формул 2 и 3 с получением смеси перед объединением с сульфонилхлоридом.
Вариант воплощения M30. Способ по варианту воплощения M28 или M29, где растворитель выбран из нитрилов (например, ацетонитрил, пропионитрил), сложных эфиров (например, метилацетат, этилацетат, бутилацетат), кетонов (например, ацетон, метилэтилкетон, метилбутилкетон), галогеналканов (например, дихлорметан, трихлорметан), простых эфиров (например, этиловый простой эфир, метил-трет-бутиловый простой эфир, тетрагидрофуран, п-диоксан), ароматических углеводородов (например, бензол, толуол, хлорбензол, дихлорбензол), третичных аминов (например, триалкиламины, диалкиланилины, необязательно замещенные пиридины) и их смесей.
Вариант воплощения M31. Способ по варианту воплощения M30, где растворитель выбран из третичных аминов (например, триалкиламины, диалкиланилины, необязательно замещенные пиридины) и их смесей.
Вариант воплощения M32. Способ по варианту воплощения M30, где растворитель выбран из нитрилов (например, ацетонитрил, пропионитрил), сложных эфиров (например, метилацетат, этилацетат, бутилацетат), кетонов (например, ацетон, метилэтилкетон, метилбутилкетон), галогеналканов (например, дихлорметан, трихлорметан), простых эфиров (например, этиловый простой эфир, метил-трет-бутиловый простой эфир, тетрагидрофуран, п-диоксан), ароматических углеводородов (например, бензол, толуол, хлорбензол, дихлорбензол) и их смесей.
Вариант воплощения M33. Способ по варианту воплощения M32, где растворителем является ацетонитрил.
Вариант воплощения C1. Соединение формулы 3, где R1 представляет CH3.
Вариант воплощения C2. Соединение формулы 3, где R2 представляет Br или Cl.
Вариант воплощения C3. Соединение формулы 3, где R2 представляет I.
Вариант воплощения C4. Соединение формулы 3, где R2 представляет CN.
Вариант воплощения C5. Соединение формулы 3, где R3 представляет H или CH3.
Вариант воплощения С6. Соединение формулы 3, где R3 представляет CH3.
Достойны внимания соединения формулы 3, где R1 представляет CH3, R2 представляет Cl и R3 представляет H, CH2CH3, CH2CH2CH3, CH2CH2CH2CH3, CH2CH(CH3)2, CH(CH3)CH2CH3 или C(CH3)3. Следует отметить также соединения формулы 3, где R1 представляет CH3, R2 представляет Br и R3 представляет Н, CH2CH3, CH2CH2CH3, CH2CH2CH2CH3, CH2CH(CH3)2, CH(CH3)CH2CH3 или C(CH3)3. Также значительными являются соединения формулы 3, где R1 представляет CH3, R2 означает I и R3 представляет Н, CH3, CH2CH3, CH2CH2CH3, CH(CH3)2, CH2CH2CH2CH3, CH2CH(CH3)2, CH(CH3)CH2CH3 или C(CH3)3. Также значительными являются соединения формулы 3, где R1 представляет CH3, R2 представляет CN и R3 представляет CH2CH3, CH2CH2CH3, CH2CH2CH2CH3, CH2CH(CH3)2, CH(CH3)CH2CH3 или C(CH3)3. Также значительными являются соединения формулы 3, где R1 представляет Cl, R2 представляет Cl и R3 представляет CH2CH2CH3, CH2CH2CH2CH3, CH2CH(CH3)2 или C(CH3)3. Также значительными являются соединения формулы 3, где R1 представляет Cl, R2 представляет Br и R3 представляет Н, CH2CH3, CH2CH2CH3, CH2CH2CH2CH3, CH2CH(CH3)2, CH(CH3)CH2CH3 или C(CH3)3. Также значительными являются соединения формулы 3, где R1 представляет Cl, R2 представляет I и R3 представляет Н, CH3, CH2CH3, CH2CH2CH3, CH(CH3)2, CH2CH2CH2CH3, CH2CH(CH3)2, CH(CH3)CH2CH3 или C(CH3)3. Также значительными являются соединения формулы 3, где R1 представляет Cl, R2 представляет CN и R3 представляет Н, CH3, CH2CH3, CH2CH2CH3, CH(CH3)2, CH2CH2CH2CH3, CH2CH(CH3)2, CH(CH3)CH2CH3 или C(CH3)3.
На следующих схемах значения R1, R2, R3, R4, R5 и R6 в соединениях нижеследующих формул 1-34 являются такими же, как указано выше в разделе "Сущность изобретения" и описании вариантов воплощения, если не указано иначе.
Как показано на схеме 1, данное изобретение относится к способу получения соединений формулы 1 путем взаимодействия карбоновых кислот формулы 2 с антраниламидами формулы 3 с использованием сульфонилхлорида, обычно в присутствии основания и растворителя.
Схема 1

Таким образом, в данном способе пиразолкарбоновую кислоту формулы 2, анилин формулы 3 и сульфонилхлорид объединяют (т.e. взаимодействуют) с получением соответствующего N-фенил-5 пиразол-1-карбоксамида формулы 1.
Возможен широкий диапазон отношений реагентов, однако обычно используют молярное соотношение соединения формулы 3 к соединению формулы 2, составляющее от около 0,9 до 1,1, и предпочтительно около 1,0, так что оба соединения могут быть полностью израсходованы. Данный способ может быть осуществлен в широком диапазоне температур, но обычно его проводят при температурах в пределах от -70°С до +100°С. Предпочтительными являются температуры от -20°С до +40°С. Наиболее пригодны для удобства работы, благоприятной скорости и селективности реакции и высокого выхода процесса температуры от -10°С до +20°С.
Соединение сульфонилхлорида используют в качестве реагента для облегчения связывания карбоновой кислоты с антраниламидом, чтобы получить N-фенилпиразол-1-карбоксамид. Обычно молярное соотношение сульфонилхлорида к соединению формулы 2 составляет от около 1,0 до 2,5 и предпочтительно от около 1,1 до 1,4, при этом побочная реакция циклизации, описанная ниже, происходит в незначительной степени (т.e. 0-10%). Сульфонилхлориды представлены, в основном, формулой R8S(O)2Cl (формула 4), где R8 представляет радикал на основе углерода. Обычно для данного способа R8 представляет C1-C4 алкил, C1-C2 галогеналкил или фенил, необязательно замещенные 1-3 заместителями, выбранными из группы, состоящей из галогена, C1-C3 алкила и нитро. Соединения сульфонилхлорида, предпочтительные для данного способа, вследствие их коммерческой доступности, включают метансульфонилхлорид (R8 представляет CH3), пропансульфонилхлорид (R8 представляет (CH2)2CH3), бензолсульфонилхлорид (R8 представляет Ph) и п-толуолсульфонилхлорид (R8 представляет 4-CH3-Ph). Метансульфонилхлорид является наиболее предпочтительным вследствие низкой стоимости, легкости присоединения и/или снижения отходов.
В данном способе сульфонилхлорид объединяют с пиразолкарбоновой кислотой формулы 2 и анилином формулы 3. Реагенты могут быть объединены в разных последовательностях, таких как объединение сульфонилхлорида с карбоновой кислотой формулы 2 для получения смеси и последующее объединение смеси с анилином формулы 3. Однако для получения конкретных N-фенилпиразол-1-карбоксамидов формулы 1 наиболее предпочтительным является объединение карбоновой кислоты формулы 2 с анилином формулы 3 с получением смеси и последующее объединение сульфонилхлорида со смесью (например, добавление сульфонилхлорида к смеси соединений формул 2 и 3), потому что такой порядок добавления позволяет удобно контролировать процесс связывания. Скорость реакции легко регулируют простым регулированием скорости добавления соединения сульфонилхлорида. Поэтому предпочтительный вариант воплощения данного способа включает последовательные стадии (1) объединения карбоновой кислоты формулы 2 и анилина формулы 3 с получением смеси и (2) последующее объединение смеси с сульфонилхлоридом. Хотя при добавлении сульфонилхлорида к смеси, содержащей анилин формулы 2, потенциально могут присходить нежелательные побочные реакции, было обнаружено, что конкретные стереоэлектронные профили соединений формул 2 и 3 позволяют достичь удивительно высоких выходов соединений формулы 1 при использовании данного способа.
Соединение формулы 1 получают, когда исходные соединения формул 2 и 3 и сульфонилхлорид взаимодействуют друг с другом в объединенной жидкой фазе, в которой каждый из них по меньшей мере частично растворим. В частности, так как исходные реагенты формул 2 и 3 обычно являются твердыми веществами при обычных температурах окружающей среды, способ наиболее успешно проводят, используя растворитель, в котором исходные соединения хорошо растворяются. Так, обычно способ осуществляют в жидкой фазе, содержащей растворитель. В некоторых случаях карбоновая кислота формулы 2 может иметь только слабую растворимость, но ее соль с присоединенным основанием может иметь более хорошую растворимость в растворителе. Подходящие для этого способа растворители включают нитрилы, такие как ацетонитрил и пропионитрил; сложные эфиры, такие как метилацетат, этилацетат и бутилацетат; кетоны, такие как ацетон, метилэтилкетон (MEK) и метилбутилкетон; галогеналканы, такие как дихлорметан и трихлорметан; простые эфиры, такие как этиловый простой эфир, метил-трет-бутиловый простой эфир, тетрагидрофуран (THF) и п-диоксан; ароматические углеводороды, такие как бензол, толуол, хлорбензол и дихлорбензол; третичные амины, такие как триалкиламины, диалкиланилины и необязательно замещенные пиридины; и их смеси. Представляющие интерес растворители включают ацетонитрил, пропионитрил, этилацетат, ацетон, MEK, дихлорметан, метил-трет-бутиловый простой эфир, THF, п-диоксан, толуол и хлорбензол. Особо предпочтительным в качестве растворителя является ацетонитрил, так как он часто обеспечивает продукты с превосходным выходом и/или чистотой.
Так как в результате реакции в способе по данному изобретению образуется в качестве побочного продукта хлорид водорода, который иначе мог бы связываться с имеющими основный характер центрами на соединениях формул 1, 2 и 3, способ наиболее удовлетворительно осуществляют в присутствии по меньшей мере одного добавленного основания. Основание может также облегчать конструктивное взаимодействие карбоновой кислоты с соединением сульфонилхлорида и антраниламидом. В результате взаимодействия добавленного основания с карбоновой кислотой формулы 2 образуется соль, которая может иметь более хорошую растворимость, чем карбоновая кислота, в реакционной среде. Хотя основание и может быть добавлено одновременно, с чередованием или даже после добавления сульфонилхлорида, обычно основание добавляют перед добавлением сульфонилхлорида. Некоторые растворители, такие как третичные амины, также служат в качестве оснований и, когда их используют в качестве растворителей, они должны быть в большом стехиометрическом избытке в качестве оснований. Когда основание не используют в качестве растворителя, номинальное молярное соотношение загруженного основания к загруженному сульфонилхлориду обычно составляет от около 2,0 до 2,2 и предпочтительно от около 2,1 до 2,2. Предпочтительными основаниями являются третичные амины, включая замещенные пиридины. Более предпочтительные основания включают 2-пиколин, 3-пиколин, 2,6-лутидин и пиридин. Особо предпочтительным основанием является 3-пиколин, так как его соли с карбоновыми кислотами формулы 2 часто являются высокорастворимыми в растворителях, таких как ацетонитрил.
Признаки данного способа обеспечивают эффективное получение N-фенилпиразол-1-карбоксамида формулы 1, в то же время ограничивая количества карбоновой кислоты, сульфонилхлорида и антраниламида, которые расходуются во время образования N-фенилпиразол-1-карбоксамида, и уменьшение отходов. Данный способ позволяет удобно регулировать процесс соединения и обеспечивает способ с вовлечением меньшего числа и более простых операций по сравнению с ранее известными процессами получения N-фенилпиразол-1-карбоксамидов, таких как представленные формулой 1.
Предпочтительный вариант воплощения данного способа объединяет пиразолкарбоновую кислоту формулы 2, антраниловую кислоту формулы 3 и подходящее основание в подходящем растворителе с последующим добавлением соединения сульфонилхлорида (или одного, или в смеси с подходящим растворителем).
Полученные N-фенилпиразол-1-карбоксамиды формулы 1 могут быть выделены из реакционных смесй способами, известными специалистам в этой области, включая кристаллизацию, фильтрование и экстракцию. Как показано на схеме 2, в некоторых случаях в условиях реакции взаимодействия происходит частичная циклизация амидов 1 до иминобензоксазинов формулы цикло-1.
Схема 2

В таких случаях часто предпочтительно превращать соединение формулы цикло-1 обратно в амид формулы 1 перед выделением продукта реакции. Такое превращение может быть осуществлено обработкой реакционной смеси водной кислотой. В качестве варианта, смесь иминобензоксазина формулы цикло-1 и амида формулы 1 может быть выделена и эта смесь затем может быть превращена в амид формулы 1, например, обработкой разбавленной водной кислотой необязательно в присутствии подходящего органического растворителя.
В предпочтительных условиях этого процесса побочная реакция циклизации, превращающая желательный продукт формулы 1 в соединение формулы цикло-1, обычно происходит только до незначительной степени, если вообще происходит, в таких случаях предпочтительные соотношения сульфонилхлорида и основания являются достаточными для завершения реакции соединения. Однако для некоторых пиразолкарбоновых кислот формулы 2, антраниловых кислот формулы 3 (таких, где R2 представляет CN) и условий реакции (например, с использованием стерически затрудненных замещенных пиридинов, таких как 2,6-лутидин, в качестве оснований) превращение желательного продукта формулы 1 в соединение формулы цикло-1 может происходить до более значительной степени или может быть доминирующей реакцией. В таких случаях применение более высоких соотношений сульфонилхлорида и основания может облегчить завершение реакции взаимодействия. Побочная реакция циклизации стехиометрически потребляет эквивалент сульфонилхлорида дополнительно к эквиваленту сульфонилхлорида, израсходованному на реакцию взаимодействия. Поэтому если бы происходила 100% циклизация, стехиометрически было бы необходимо молярное соотношение 2:1 сульфонилхлорида к соединению формулы 2 для достижения полного расходования исходных реагентов, и обычно было бы использовано молярное соотношение вплоть до около 2,5:1 сульфонилхлорида к соединению формулы 2 в сравнении с молярным соотношением около 1,4:1 сульфонилхлорида к соединению формулы 2, когда циклизация происходит только на 5-10% (как это происходит обычно с большинством оснований, когда R2 представляет CN) и молярным соотношением около 1,2:1 сульфонилхлорида к соединению формулы 2, когда побочная реакция циклизации незначительная (как это происходит обычно с большинством оснований, когда R2 представляет Br, Cl или I). Дополнительные количества сульфонилхлорида и основания могут быть добавлены в течение реакции, если наблюдается реакция циклизации.
Вышесказанное показывает ценный признак этого процесса, который заключается в том, что дополнительные количества любого из компонентов процесса могут быть добавлены в любое время, как требуется для завершения превращения. Другое пояснение ценности этого признака касается ситуации, когда или компонент формулы 2, или компонент формулы 3 непредумышленно недогружен в реакционную смесь. Эта недогрузка может быть определена анализом реакционной смеси с использованием любого из разнообразных методов, которые широко известны и доступны, включая ЖХВР и ЯМР. При обнаружении недогрузка может быть скорректирована дополнительным добавлением соответствующего компонента к реакционной смеси. Это может быть особенно ценно для производства в больших масштабах, так как позволяет исправить ошибку при загрузке и предотвратить отходы дорогостоящего промежуточного соединения, которые иначе могли бы образовываться.
Пиразолкарбоновые кислоты формулы 2 могут быть получены с использованием методов гетероциклического синтеза, известных в литературе, включая ссылки, находящиеся в следующем списке: Rodd's Chemistry of Chemistry of Carbon Compounds, Vol. IVa to IVl, S. Coffey editor, Elsevier Scientific Publishing, New York, 1973; Comprehensive Heterocyclic Chemistry, Vol.1-7, A.R. Katritzky и C.W. Rees editors, Pergamon Press, New York, 1984; Comprehensive Heterocyclic Chemistry II, Vol.1-9, A.R.Katritzky, C.W. Rees, и E.F. Scriven editors, Pergamon Press, New York, 1996; и серии The Chemistry of Heterocyclic Compounds, E.C. Taylor, editor, Wiley, New York. Разнообразные гетероциклические кислоты (включая пиразолкарбоновые кислоты) и общие способы их синтеза находятся в патентной публикации PCT WO 98/57397.
Один особенно используемый способ получения пиразолкарбоновых кислот формулы 2a показан на схеме 3
Схема 3

Взаимодействие пиразола формулы 6 с 2-галогенпиридином формулы 7 позволяет получить высокие выходы 1-пиридинилпиразола формулы 8 с хорошей специфичностью в отношении желательной региохимии. Металлирование соединения формулы 8 диизопропиламидом лития (LDA) с последующим гашением соли лития диоксидом углерода дает 1-(2-пиридинил)пиразолкарбоновую кислоту формулы 2a. Приводимая ссылка на этот способ: см. патентную публикацию PCT WO 03/015519.
Как показано на схеме 4, пиразолкарбоновые кислоты формулы 2b могут быть получены посредством 3+2 циклоприсоединения соответствующим образом замещенного иминогалогенида формулы 9, или с замещенными пропиолатами формулы 10, или с акрилатами формулы 11.
Схема 4
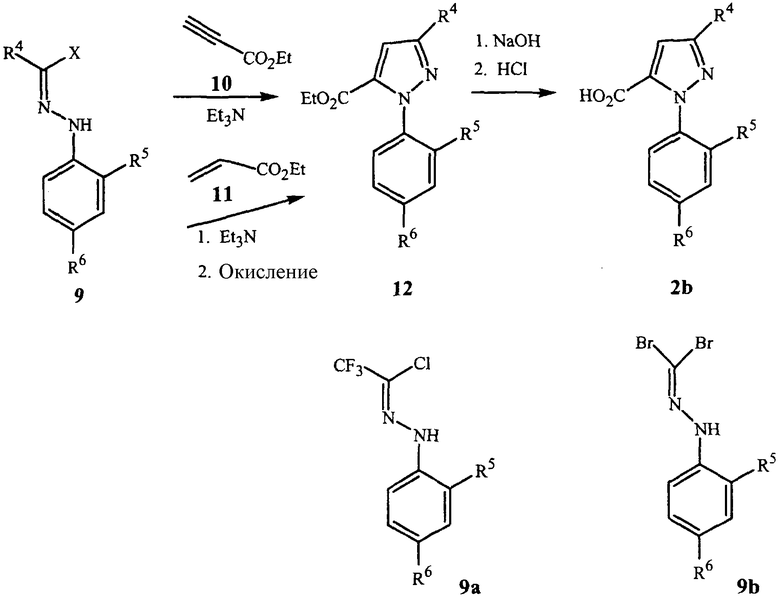
Циклоприсоединение с акрилатами требует дополнительного окисления промежуточного пиразолина до пиразола. Гидролиз сложного эфира формулы 12 дает пиразолкарбоновые кислоты формулы 2b. Предпочтительные иминогалогениды для этой реакции включают трифторметилиминохлорид формулы 9a и иминодибромид формулы 9b. Соединения, такие как представленные формулой 9a, известны (J. Heterocycl. Chem. 1985, 22(2), 565-8). Другие соединения формулы 9, такие как соединения формулы 9b, доступны известными способами (Tetrahedron Letters 1999, 40, 2605).
Другой способ получения пиразолкарбоновых кислот формулы 2b показан на схеме 5.
Схема 5

Пиразолы формулы 13 могут быть конденсированы с арилиодидами с использованием способов, таких как те, о которых сообщалось A. Klapars, J.C. Antilla, X. Huang и S.L. Buchwald, J. Am. Chem. Soc. 2001, 123, 7727-7729, или с арилбороновыми кислотами с использованием способов, таких как те, о которых сообщалось P.Y.S. Lam, C.G. Clark, S. Saubern, J. Adams, M.P. Winters, D.M.T. Chan и A. Combs, Tetrahedron Lett. 1998, 39, 2941-2944. Полученные аддукты формулы 15 могут быть окислены окислителями, такими как перманганат калия, до образования пиразолкарбоновых кислот формулы 2b.
Исходные пиразолы формул 6 и 13 являются известными соединениями или могут быть получены согласно известным способам. Например, пиразол формулы 6a (соединение формулы 6, где R4 представляет CF3) может быть получен путем описанных в литературе процедур (J. Fluorine Chem. 1991, 55(1), 61-70). Пиразолы формулы 6b (соединения формулы 6, где R4 представляет Cl или Br) могут быть получены путем процедуры, описанной в Chem. Ber. 1966, 99(10), 3350-7.
Применимый альтернативный способ получения соединения формулы 6b изображен на схеме 6.
Схема 6

Металлирование сульфамоилпиразола формулы 16 н-бутиллитием с последующим непосредственным галогенированием аниона или гексахлорэтаном (для R4, обозначающего Cl), или 1,2-дибромтетрахлорэтаном (для R4, обозначающего Br) дает галогенированные производные формулы 17a. Удаление сульфамоилгруппы трифторуксусной кислотой (TFA) при комнатной температуре происходит четко и с хорошим выходом с образованием пиразолов формулы 6c. Специалисту в этой области будет понятно, что формула 6c является таутомером формулы 6b.
Пиразолкарбоновые кислоты 2 могут быть также получены окислением пиразолина формулы 18 до получения пиразола формулы 19 с последующим гидролизом до карбоновой кислоты, как показано на схеме 7.
Схема 7
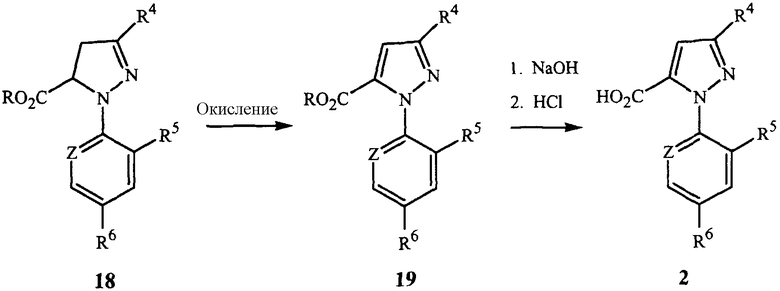
Окислителем может быть пероксид водорода, органические пероксиды, персульфат калия, персульфат натрия, персульфат аммония, моноперсульфат калия (например, Oxone®) или перманганат калия. Такое окисление может быть проведено в присутствии растворителя, предпочтительно простого эфира, такого как тетрагидрофуран, п-диоксан и тому подобное, органического сложного эфира, такого как этилацетат, диметилкарбонат и тому подобное, или полярного апротонного органического соединения, такого как N,N-диметилформамид, ацетонитрил и тому подобное.
Галогенпиразолины 18, где R4 представляет Cl или Br, могут быть получены из пиразолонов формулы 20 обработкой соответствующим галогенирующим агентом, как показано на схеме 8.
Схема 8

Галогенирующие реагенты, которые могут быть использованы, включают оксигалогениды фосфора, тригалогениды фосфора, пентагалогениды фосфора, тионилхлорид, дигалогентриалкилфосфораны, дигалогентрифенилфосфораны, оксалилхлорид и фосген. Предпочтительны оксигалогениды фосфора и пентагалогениды фосфора. Типичные растворители для этого галогенирования включают галогенированные алканы, такие как дихлорметан, хлороформ, хлорбутан и тому подобное, ароматические растворители, такие как бензол, ксилол, хлорбензол и тому подобное, простые эфиры, такие как тетрагидрофуран, п-диоксан, диэтиловый простой эфир и тому подобное, и полярные апротонные растворители, такие как ацетонитрил, N,N-диметилформамид и тому подобное. Необязательно может быть добавлено органическое основание, такое как триэтиламин, пиридин, N,N-диметиланилин или тому подобное. Возможно также добавление катализатора, такого как N,N-диметилформамид.
В альтернативном случае соединения формулы 18, где R4 представляет галоген, могут быть получены обработкой соответствующих соединений формулы 18, где R4 представляет другой галоген (например, Cl для получения соединения формулы 18, где R4 представляет Br) или сульфогруппу, такую как метансульфонат, бензолсульфонат или п-толуолсульфонат, бромистым водородом или хлористым водородом соответственно. Этим способом галоген или сульфозаместитель R4 исходного соединения формулы 18 замещают на Br или Cl от бромистого водорода или хлористого водорода соответственно. Исходные соединения формулы 18, где R4 представляет Cl или Br, могут быть получены из соответствующих соединений формулы 20, как уже описано. Исходные соединения формулы 18, где R4 представляет сульфогруппу, могут быть получены подобным образом из соответствующих соединений формулы 20 стандартными способами, такими как обработка сульфонилхлоридом (например, метансульфонилхлоридом, бензолсульфонилхлоридом или п-толуолсульфонилхлоридом) и основанием, таким как третичный амин (например, триэтиламин), в подходящем растворителе, таком как дихлорметан.
Пиразолкарбоновые кислоты формулы 2c, где R4 представляет OCHF2 или OCH2CF3, могут быть получены способом, охарактеризованным в общих чертах на схеме 9.
Схема 9

В этом способе вместо галогенирования, как показано на схеме 8, соединение формулы 20 окисляют до соединения формулы 21. Условия реакции для этого окисления являются такими, как уже описано для превращения соединения формулы 18 в соединение формулы 19 на схеме 7. Соединение формулы 21 может быть затем алкилировано, чтобы получить соединение формулы 22 путем контактирования с дифторкарбеном, полученным in situ из CHClF2 в присутствии основания. Соединение формулы 21 также может быть алкилировано, чтобы получить соединение формулы 24, путем контактирования с алкилирующим агентом CF3CH2Lg в присутствии основания. Реакцию алкилирования обычно проводят в растворителе, который может содержать простые эфиры, такие как тетрагидрофуран или диоксан, и полярные апротонные растворители, такие как ацетонитрил, N,N-диметилформамид, и тому подобное. Основание может быть выбрано из неорганических оснований, таких как карбонат калия, гидроксид натрия или гидрид натрия. Предпочтительно реакцию проводят, используя карбонат калия с N,N-диметилформамидом или ацетонитрилом в качестве растворителя. В алкилирующем агенте CF3CH2Lg Lg представляет нуклеофуг (т.e. уходящую группу), такой как галоген (например, Br, I), OS(O)2CH3 (метансульфонат), OS(O)2CF3, OS(О)2Ph-п-CH3 (п-толуолсульфонат) и тому подобное. Продукт формулы 22 может быть выделен обычными техническими приемами, такими как экстракция. Сложные эфиры могут быть затем превращены в карбоновые кислоты формулы 2c способами, уже описанными для превращения формулы 12 в формулу 2b на схеме 4.
Соединения формулы 20 могут быть получены из соединений формулы 25, как показано в общих чертах на схеме 10.
Схема 10
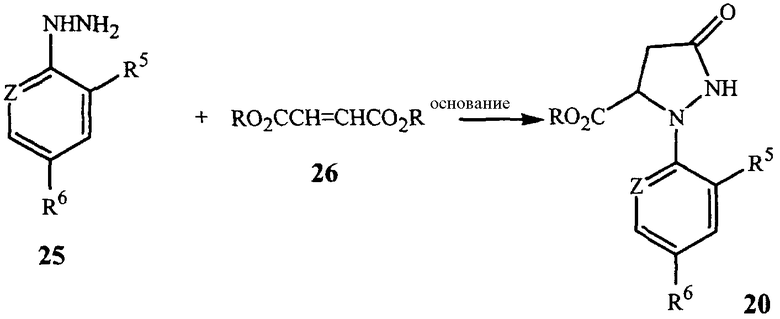
В этом способе соединение гидразина формулы 25 приводят в контакт с соединением формулы 26 (может быть использован сложный эфир фумаровой кислоты (фумарат), или сложный эфир малеиновой кислоты (малеат), или их смесь) в присутствии основания и растворителя. Основанием обычно является соль алкоксид металла, такая как метоксид натрия, метоксид калия, этоксид натрия, этоксид калия, трет-бутоксид калия, трет-бутоксид лития и тому подобное. Могут быть использованы полярные протонные и полярные апротонные органические растворители, такие как спирты, ацетонитрил, тетрагидрофуран, N,N-диметилформамид, диметилсульфоксид и тому подобное. Предпочтительными растворителями являются спирты, такие как метанол и этанол. Особенно предпочтительно, чтобы спирт соответствовал (т.e. был таким же, как и составляющие) сложному эфиру фумарату или малеату и алкоксидному основанию. В зависимости от условий реакции и способов выделения функциональная группа -CO2R соединения формулы 20 может быть гидролизована до -CO2H; например, присутствие воды в реакционной смеси может промотировать такой гидролиз. Если образуется карбоновая кислота (-CO2H), она может быть превращена обратно в -CO2R, где R представляет C1-C4 алкил, с использованием методов этерификации, хорошо известных в технике. Желательный продукт, а именно соединение формулы 20, может быть выделен способами, известными специалистам, такими как кристаллизация, экстракция или дистилляция.
Другой аспект данного изобретения относится к антраниламидам формулы 3, которые являются важными промежуточными соединениями в способе по данному изобретению. Образцы антраниламидов формулы 3 применимы также в качестве аналитических стандартов для определения присутствия антраниламидов.
Антраниламиды формулы 3 могут быть получены при взаимодействии ангидридов N-карбоксиантраниловой кислоты формулы 27 с аммиаком или алкиламинами, как показано на схеме 11, с использованием процедур, таких как те, которые описаны L.H. Sternbach et al., J. Org. Chem. 1971,36, 777-781.
Схема 11

Ангидриды N-карбоксиантраниловой кислоты формулы 27 могут быть получены различными известными способами, которые документально подтверждены в химической литературе. Например, ангидриды N-карбоксиантраниловой кислоты доступны из соответствующих антраниловых кислот посредством циклизации при реакции антраниловой кислоты с фосгеном или эквивалентом фосгена. Для приведения ссылок на способы см. Coppola, Synthesis 1980, 505 и Fabis et al., Tetrahedron, 1998, 10789.
Синтез ангидридов N-карбоксиантраниловой кислоты формулы 27 может быть также выполнен из изатинов формулы 30, как показано в общих чертах на схеме 12.
Схема 12
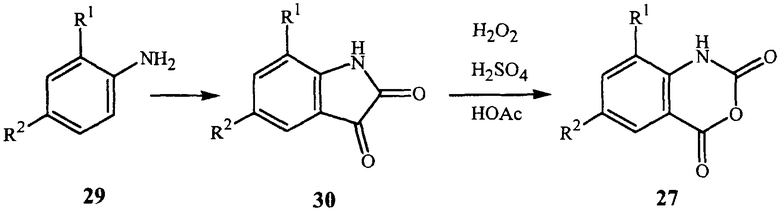
Изатины формулы 30 могут быть получены из производных анилина формулы 29, следуя описанным в литературе процедурам, например, F.D. Popp, Adv. Heterocycl. Chem. 1975, 18, 1-58 и J.F.M. Da Silva et al., Journal of the Brazilian Chemical Society 2001, 12(3), 273-324. Окисление изатина 30 пероксидом водорода обычно дает хорошие выходы соответствующего ангидрида N-карбоксиантраниловой кислоты формулы 28 (G. Reissenweber and D. Mangold, Angew. Chem. Int. Ed. Engl. 1980, 19, 222-223).
Как показано на схеме 13, изатины формулы 30, где R2 представляет Cl, Br или I, также могут быть получены из 5-незамещенных изатинов формулы 31 путем галогенирования. Замещение цианидом может затем обеспечить изатины формулы 30a (формулы 30, где R2 представляет CN).
Схема 13

Реакция галогенирования может быть проведена с использованием многих реагентов и процедур, известных в литературе. Подходящие реагенты включают элементарные галогены (хлор, бром или йод), "позитивно галогеновые" реагенты, такие как трихлоризоциануровая кислота, N-хлорсукцинимид (NCS), N-бромсукцинимид (NBS) или N-йодсукцинимид (NIS), и галогенирующие реагенты, такие как смеси, содержащие пероксид водорода и галогенид водорода. Галоген при положении 5 изатинов формулы 30, где R2 представляет Cl, Br или I, может быть замещен цианидом при использовании способов, известных в литературе. Такие способы включают применение соли цианида, обычно с использованием соединения металла, и часто в присутствии лиганда, такого как замещенный фосфин или замещенный бисфосфиноалкан. Подходящие способы включают способы с использованием соединений палладия, такие как способы, которые описаны P.E. Maligres et al., Tetrahedron Letters 1999, 40, 8193-8195 и M. Beller et al., Chem. Eur. J. 2003, 9(8), 1828-1836; способы с использованием соединений меди, такие как способы, описанные S.L. Buchwald в J. Am. Chem. Soc. 2003, 125, 2890-2891; и способы с использованием соединений никеля, такие как способы, описанные в европейском патенте 384392 и K. Sasaki в Bull. Chem. Soc. Japan 2004, 77, 1013-1019 и R.K. Arvela and N.E. Leadbeater в J. Org. Chem. 2003, 68, 9122-9125. Специалист будет учитывать, что когда R1 представляет Cl, R2 формулы 27 представляет предпочтительно Br или I, чтобы достичь селективности в процессе цианирования (т.e. замещения галогена цианидом).
Как показано на схеме 14, антраниламиды формулы 3 обычно можно получить из соответствующих 2-нитробензойных кислот (или сложных эфиров) формулы 32 посредством каталитического гидрирования нитрогруппы с последующей реакцией антранилового сложного эфира формулы 33 с аммиаком или алкиламином.
Схема 14

Типичные процедуры восстановления предусматривают восстановление водородом в присутствии металлического катализатора, такого как палладий-на-углероде или оксид платины, в гидроксильных растворителях, таких как этанол и изопропанол. Восстановление может быть также проведено в присутствии цинка в уксусной кислоте. Такие методы восстановления нитрогрупп хорошо документированы в химической литературе. Многие способы взаимного превращения карбоновых кислот, сложных эфиров и амидов также документально подтверждены в химической литературе.
Как показано на схеме 15, антраниламиды формулы 3 также могут быть получены из 5-незамещенных антраниламидов формулы 34 галогенированием до образования антраниламидов формулы 3, где R2 представляет Br, Cl или I, необязательно с последующим замещением цианидом, чтобы обеспечить антраниламиды формулы 3a (формулы 3, где R2 представляет CN).
Схема 15

Подходящие способы и процедуры известны в литературе и подобны тем, которые описаны для процедур галогенирования и замещения цианидом, показанных на схеме 13. Специалисту будет понятно, что галогенирование и цианирование могут быть также проведены на других стадиях получения антраниламидов формулы 3.
Признано, что некоторые реагенты и условия реакции, описанные выше для получения соединений формул 2 и 3, могут быть несовместимыми с конкретными функциональными группами, присутствующими в промежуточных соединениях. В таких случаях введение в синтез последовательностей защиты/устранения защиты или взаимных превращений функциональных групп будет способствовать достижению желательных продуктов. Применение и выбор защитных групп будут очевидны для специалиста в области химического синтеза (см., например, Greene, T.W.; Wuts, P.G.M. Protective Groups in Organic Synthesis, 2nd ed.; Wiley: New York, 1991). Специалист в этой области будет осознавать, что в некоторых случаях после введения заданного реагента, как это обозначено на каждой отдельной схеме, может быть необходимо осуществить дополнительные рутинные стадии синтеза, не описанные подробно, чтобы завершить синтез соединений формул 2 и 3. Специалисту будет также понятно, что может быть необходимо осуществить комбинацию стадий, поясняемых на указанных схемах, в ином порядке, чем предполагаемый конкретной представленной последовательностью, чтобы получить соединения формул 2 и 3. Специалисту будет также понятно, что соединения формул 2 и 3 и промежуточные соединения, раскрытые в описании, могут быть подвергнуты различным электрофильным, нуклеофильным, радикальным, металлоорганическим, окислительным и восстановительным реакциям для добавления заместителей или модификации существующих заместителей.
Предполагается, что без дополнительной разработки специалист в этой области сможет использовать данное изобретение во всей его полноте. Поэтому следующие примеры следует толковать только как пояснительные и не ограничивающие раскрытие каким-либо образом. Стадии в следующих примерах иллюстрируют процедуру для каждой стадии в общем синтетическом преобразовании, и исходный материал для каждой стадии может быть необязательно получен конкретным способом получения, процедура которого описана в других примерах или стадиях. Процентные доли даны по массе за исключением смесей хроматографических растворителей или где это указано иначе. Части и процентные доли смесей хроматографических растворителей даны по объему, если не указано иначе. Спектры 1H ЯМР представлены в м.д. нижней области от тетраметилсилана; s - синглет, d - дублет, t - триплет, q - квартет, m - мультиплет, dd: дублет дублетов, dt - дублет триплетов, br s - широкий синглет. Количественную ЖХВР продукта осуществляли с использованием хроматографической колонки Ace Cl8 или C4 Ultra Inert® (колонка с обращенной фазой, производимая MacMod Analytical Inc., Chadds Ford, PA 19317) (размер частиц 3 мкм, 4,6 мм×15 см, элюент 5-80% ацетонитрил/pH 3 фосфатный буфер).
ПРИМЕР 1
Получение 2-амино-5-хлор-N,3-диметилбензамида
К суспензии 6-хлор-8-метил-2Н-3,1-бензоксазин-2,4(1Н)-диона (211,6 г, 1000 ммоль) в ацетонитриле (700 мл) добавляют уксусную кислоту (7,3 г, 122 ммоль). Затем добавляют по каплям в течение 30 мин при 25-30°С 40% водный метиламин (104 мл). Перемешивание продолжают в течение 2 час и затем медленно добавляют воду (700 мл). Полученную суспензию охлаждают до 5°С и перемешивают в течение 30 мин при этой температуре. Суспензию затем фильтруют и твердые вещества промывают водой (3×200 мл) и сушат в атмосфере азота, получая указанное в заголовке соединение в виде не совсем белых игольчатых кристаллов, 172,8 г (выход 87,0%), т.пл. 141-143°С.
1H ЯМР (ДМСО-d6) δ 2,08 (с, 3H), 2,72 (д, J=4,5 Гц, 3H), 6,34 (ушир.с, 2H), 7,12 (д, J=2,4 Гц, 1H), 7,39 (д, J=2,4 Гц, 1H), 8,31 (ушир.д, 1H).
ПРИМЕР 2
Получение метил 2-амино-5-хлор-3-метилбензоата
Стадия A: Получение 2-амино-5-хлор-3-метилбензойной кислоты
К раствору 2-амино-3-метилбензойной кислоты (Aldrich, 15,0 г, 99,2 ммоль) в N,N-диметилформамиде (50 мл) добавляют N-хлорсукцинимид (13,3 г, 99,2 ммоль) и реакционную смесь нагревают до 100°С в течение 30 мин. Нагревание прекращают и реакционную смесь охлаждают до комнатной температуры и дают возможность отстояться в течение ночи. Реакционную смесь затем медленно выливают на воду со льдом (250 мл), чтобы осадить белое твердое вещество. Твердое вещество отфильтровывают и промывают четыре раза водой и затем переносят в этилацетат (900 мл). Этилацетатный раствор сушат (MgSO4) и выпаривают при пониженном давлении и остаточное твердое вещество промывают простым эфиром, получая желательное промежуточное соединение в виде белого твердого вещества, 13,9 г (выход 75,4%).
1H ЯМР (ДМСО-d6) δ 2,11 (с, 3H), 7,22 (с, 1H), 7,55 (с, 1H).
Стадия В: Получение метил 2-амино-5-хлор-3-метилбензоата
К суспензии 2-амино-5-хлор-3-метилбензойной кислоты (т.e. продукта стадии A) (92,8 г, 500 ммоль) в ацетонитриле (500 мл) при 0-5°С добавляют 1,4-диазабицикло[5,4,0]ундец-7-ен (DBU, 90 мл, 92 г, 600 ммоль) и затем диметилсульфат (57 мл, 76 г, 600 ммоль) добавляют по каплям при 0-5°С. После перемешивания в течение 3 час при этой температуре дополнительно добавляют DBU (15 мл) и диметилсульфат (10 мл). После перемешивания еще в течение 3 час при этой температуре дополнительно еще добавляют DBU (15 мл) и диметилсульфат (10 мл). После перемешивания в течение следующих 2 час при этой температуре добавляют по каплям при 0-10°С концентрированную соляную кислоту (60 мл, 720 ммоль). Полученную суспензию перемешивают в течение 30 мин при 0-5°С, затем фильтруют и твердые вещества промывают охлажденной льдом смесью 2:1 вода-ацетонитрил (3×100 мл) и сушат в атмосфере азота. Неочищенный продукт суспендируют в метаноле (250 мл), добавляют воду (1000 мл) и смесь перемешивают при комнатной температуре в течение 1 час. Затем твердые вещества отфильтровывают, промывают смесью 4:1 вода-метанол (100 мл), затем водой (3×100 мл) и сушат в атмосфере азота, получая указанное в заголовке соединение в виде низкоплавкого белого твердого вещества, 87,6 г (выход 87,8%). ЖХВР твердого продукта показала 99,7% по площади указанного в заголовке сложного эфира.
1H ЯМР (CDC13) δ 2,15 (с, 3H), 3,87 (с, 3H), 5,82 (ушир.с, 2H), 7,15 (д, J=2,7 Гц, 1H), 7,74 (д, J=2,7 Гц, 1H).
ПРИМЕР 3
Получение 2-амино-5-хлор-N,3-диметилбензамида
К суспензии метил 2-амино-5-хлор-3-метилбензоата (т.e. продукта из примера 2) (4,03 г, 20,2 ммоль) в ацетонитриле (12,4 г) добавляют раствор метиламина (3,1 г, 0,10 моль) в этиленгликоле (12,4 г). Смесь нагревают при 60°С в течение 23 час и затем охлаждают до комнатной температуры. Воду (25 мл) добавляют по каплям и полученную суспензию охлаждают до 5°С и перемешивают в течение 10 мин при этой температуре. Смесь фильтруют и твердые вещества промывают водой (3×10 мл) и сушат в атмосфере азота, получая указанное в заголовке соединение в виде белых игольчатых кристаллов, 3,43 г (выход 85,5%).
ПРИМЕР 4
Получение 2-амино-5-хлор-N,3-диметилбензамида
Стадия A: Получение метил 2-амино-3-метилбензоата
Метил 3-метил-2-нитробензоат (98,5 г, 505 ммоль), 5% Pd/C (Degussa CE 105 XRC/W, 1,0 г) и ацетонитрил (300 мл) объединяют в 600-мл автоклаве. Смесь нагревают до 70°С и гидрируют при давлении 65 фунт на кв. дюйм (450 кПа) в течение 8 час. Добавляют еще 5% Pd/C (1,0 г) и гидрирование продолжают при 100 фунт на кв. дюйм (690 кПа) в течение 8,5 час. Затем реакционную смесь охлаждают, продувают азотом и фильтруют через диатомовый фильтр Celite®, промывая ацетонитрилом (3×25 мл). Объединенные фильтраты частично выпаривают до массы ~160 г и затем разбавляют ацетонитрилом до общей массы 200 г. Количественная ЖХВР этого раствора показала 40,3% масс. указанного в заголовке соединения (80,6 г, выход 97,5%).
Стадия B: Получение метил 2-амино-5-хлор-3-метилбензоата
Раствор, полученный на стадии A (195 г, 475 ммоль), разбавляют ацетонитрилом (50 мл) и нагревают до 50°С. Затем в течение 3,25 час при 50-55°С добавляют раствор сульфурилхлорида (70,6 г, 523 ммоль) в ацетонитриле (100 мл). Сразу после завершения добавления смесь охлаждают до 5°С, добавляют воду (150 г) и pH раствора доводят до 6,0 медленным добавлением 50% водного гидроксида натрия (103 г). После перемешивания в течение 10 мин при этой температуре органический слой отделяют и водный слой экстрагируют ацетонитрилом (50 мл). Органические слои объединяют, сушат (MgSO4) и частично выпаривают до массы 193,7 г. Количественная ЖХВР этого раствора показала 41,5% масс. указанного в заголовке соединения (80,4 г, выход 84,8%).
Стадия C: Получение 2-амино-5-хлор-N,3-диметилбензамида
Раствор, полученный на стадии B (96,2 г, 200 ммоль), разбавляют ацетонитрилом (60,0 г) и этиленгликолем (180 г) и сушат азеотропной перегонкой при атмосферном давлении с дистилляционной насадкой Кляйзена, чтобы убрать ~72 мл летучих веществ. Затем дистилляционную насадку заменяют охлаждаемым сухим льдом холодильником, остающийся раствор охлаждают до 0-5°С и газообразный метиламин (31,1 г, 1000 ммоль) добавляют ниже поверхности реакционной смеси. Смесь нагревают при 70°С в течение 17,5 час и затем медленно добавляют воду (400 мл), чтобы осадить продукт. Смесь охлаждают медленно до 5°С, перемешивают в течение 15 мин при этой температуре, фильтруют и твердые вещества промывают водой и сушат в атмосфере азота, получая указанное соединение (36,36 г, выход 91,5%). ЖХВР показала чистоту 99,3% по площади.
ПРИМЕР 5
Получение 2-амино-5-хлор-N,3-диметилбензамида
Стадия A: Получение 2-амино-N,3-диметилбензамида
Смесь 8-метил-2Н-3,1-бензоксазин-2,4(1H)-диона (патентная публикация PCT WO 00/27831) (18 г, 0,1 моль) и уксусной кислоты (1,2 г, 0,02 моль) в этилацетате (200 мл) нагревают до 35°С и водный метиламин (40%, 9,0 г, 0,12 моль) добавляют по каплям в течение 50 мин при 35-37°С. Затем еще добавляют водный метиламин (40%, 0,9 г, 12 ммоль) и смесь перемешивают дополнительные 2,5 час при 36°С. Затем добавляют воду (20 мл), слои разделяют и органический слой промывают водой, сушат (MgSO4) и выпаривают, получая указанное в заголовке соединение, 15,45 г (92%).
1H ЯМР (CDCl3) δ 2,14 (с, 3Н), 2,94 (д, 3H, J=5 Гц), 5,37 (ушир.с, 2H), 6,21 (ушир.с, 1H), 6,56 (т, J=7,5 Гц, 1H), 7,10 (дд, J=7,5 Гц, 7,5 Гц, 1H), 7,18 (дд, J=7,5 Гц, 7,5 Гц, 1H).
Стадия B: Получение 2-амино-5-хлор-N,3-диметилбензамида
Смесь 2-амино-N,3-диметилбензамида (т.e. продукта стадии A) (16,6 г, 100 ммоль) и N,N-диметилформамида (15,0 г) охлаждают до 10°С и медленно добавляют концентрированную соляную кислоту (70 г, 700 ммоль). Затем смесь нагревают до 30°С и 30% и добавляют по каплям в течение 15 мин при 30-35°С водный пероксид водорода (18,5 г, 160 ммоль). После перемешивания при около 35°С в течение 3 час смесь охлаждают до около 10°С и затем добавляют воду (200 мл). Добавляют сульфит натрия (7,56 г, 60 ммоль) и затем pH доводят до 2,2 медленным добавлением 50% водного гидроксида натрия (38,1 г). После перемешивания при 10°С в течение 15 мин смесь фильтруют и твердые вещества промывают водой (2×50 мл) и сушат в вакуумной печи, получая указанное в заголовке соединение в виде розового твердого вещества, 14,61 г (выход 72,7%). Количественная ЖХВР твердого продукта показала 99,1% масс. указанного в заголовке соединения.
ПРИМЕР 6
Получение 2-амино-5-циано-N,3-диметилбензамида
Стадия A: Получение 2-амино-5-бром-N,3-диметилбензамида
Смесь 2-амино-N,3-диметилбензамида (т.e. продукта стадии A примера 5) (14 г, 85 ммоль), уксусной кислоты (50 мл) и воды (50 мл) охлаждают до 12°С и концентрированную бромистоводородную кислоту (28,5 г, 0,34 моль) добавляют в течение 10 мин при этой температуре. Затем 30% водный пероксид водорода (9 г, 0,08 моль) добавляют в течение 5 мин при 10-11°С и смеси позволяют медленно нагреваться до комнатной температуры при перемешивании в течение 2,5 час. Затем еще добавляют концентрированную бромистоводородную кислоту (2,9 г) и смесь перемешивают в течение ночи при комнатной температуре. К смеси затем добавляют воду (50 мл) и бисульфит натрия (1,5 г) и затем pH доводят до 5-6 добавлением 50% водного гидроксида натрия (~15 мл). Смесь фильтруют и твердые вещества промывают водой и сушат в вакууме, получая указанное в заголовке соединение, 19,5 г (94%).
1H ЯМР (CDCl3) δ 2,14 (с, 3H), 2,95 (д, J=5 Гц, 3H), 5,55 (ушир.с, 2H), 6,01 (ушир.с, 1H), 7,21 (м, 1H), 7,30 (д, J=2 Гц, 1H).
Стадия B: Получение 2-амино-5-циано-N,3-диметилбензамида
Колбу продувают сухим азотом и загружают в нее ацетат палладия (II) (370 мг, 1,64 ммоль), 1,4-бис(дифенилфосфино)бутан (850 мг, 2 ммоль), активированный цинковый порошок (500 мг, 7,64 ммоль), цианид цинка (II) (51 г, 434 ммоль) и 2-амино-5-бром-N,3-диметилбензамид (т.e. продукт стадии A) (200 г, 820 ммоль). Затем добавляют свежедегазированный N,N-диметилформамид (500 мл) и смесь нагревают при 130°С в течение 25,5 час. Затем температуру понижают до 95°С и добавляют уксусную кислоту (200 мл). Смесь разбрызгивают азотом, чтобы удалить цианид водорода, через газопромыватели, загруженные водным гидроксидом натрия и раствором гипохлорита натрия, при охлаждении до комнатной температуры. Затем добавляют воду (1500 мл) в течение 1,5 час и разбрызгивание азотом продолжают в течение ночи. Затем смесь фильтруют и твердые вещества промывают водой и сушат в вакуумной печи, получая указанное в заголовке соединение в виде хлопьевидного светло-желтого твердого вещества, 141,5 г (выход 90,9%).
1H ЯМР (CDCl3) δ 2,16 (c, 3H), 2,98 (д, J=4,8 Гц, 3H), 6,17 (ушир.с, 3H), 7,34 (д, J=1,8 Гц, 1H), 7,56 (д, J=1,8 Гц, 1H).
ПРИМЕР 7
Получение 3-бром-N-[4-хлор-2-метил-6-[(метиламино)карбонил]фенил]-1-(3-хлор-2-пиридинил)-1H-пиразол-5-карбоксамида
К смеси 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты (получение: см. патентную публикацию PCT WO 03/015519) (чистота 93,6%, 16,16 г, 50,0 ммоль) и 2-амино-5-хлор-N,3-диметилбензамида (т.e. продукта из примеров 1, 3, 4 и 5) (10,43 г, 52,5 ммоль) в ацетонитриле (35 мл) добавляют 3-пиколин (12,65 мл, 12,11 г, 130 ммоль). Смесь охлаждают до -5°С и затем добавляют по каплям при температуре от -5 до 0°С раствор метансульфонилхлорида (4,64 мл, 6,89 г, 60 ммоль) в ацетонитриле (10 мл). Смесь перемешивают в течение 15 мин при этой температуре и затем в течение 3 час при комнатной температуре. Затем воду (15 мл) добавляют по каплям и смесь охлаждают до 0°С в течение 1 час. Смесь фильтруют и твердые вещества промывают смесью 3:1 ацетонитрил-вода (2×10 мл) и затем ацетонитрилом (2×10 мл) и сушат в атмосфере азота, получая указанное в заголовке соединение в виде слегка коричневатого порошка, 23,98 г (выход без учета поправки 92,9%), т.пл. 239-240°С.
1H ЯМР (CDCl3) δ 2,18 (с, 3Н), 2,95 (с, 3Н), 6,21 (м, 1H), 7,10 (с, 1H), 7,24 (м, 2H), 7,39 (м, 1H), 7,80 (д, 1H), 8,45 (д, 1H).
ПРИМЕР 8
Получение 3-бром-N-[4-хлор-2-метил-6-[(метиламино)карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида с использованием пиридина в качестве основания
К смеси 3-бром-1-(3-хлор-2-пиридинил)-1H-пиразол-5-карбоновой кислоты (получение: см. патентную публикацию PCT WO 03/015519) (6,05 г, 20,0 ммоль) и 2-амино-5-хлор-N,3-диметилбензамида (т.e. продукта из примеров 1, 3, 4 и 5) (4,17 г, 21,0 ммоль) в ацетонитриле (18 мл) добавляют пиридин (4,20 мл, 4,11 г, 52 ммоль). Смесь охлаждают до -5°С и затем метансульфонилхлорид (1,86 мл, 2,75 г, 24 ммоль) добавляют по каплям при температуре от -5 до 0°С. Смесь перемешивают в течение 1 час при этой температуре и затем в течение 3 час при комнатной температуре. Затем добавляют по каплям воду (6 мл) и смесь перемешивают при комнатной температуре в течение 1 час. Смесь фильтруют и твердые вещества промывают смесью 3:1 ацетонитрил-вода (2×4 мл) и затем ацетонитрилом (2×4 мл) и сушат в атмосфере азота, получая указанное в заголовке соединение в виде не совсем белого порошка, 9,35 г (выход без учета поправки 96,8%).
ПРИМЕР 9
Получение 3-бром-N-[4-хлор-2-метил-6-[(метиламино)карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида с использованием "смешанных пиколинов" в качестве основания
К смеси 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты (получение: см. патентную публикацию PCT WO 03/015519) (6,05 г, 20,0 ммоль) и 2-амино-5-хлор-N,3-диметилбензамида (т.e. продукта из примеров 1, 3, 4 и 5) (4,17 г, 21,0 ммоль) в ацетонитриле (18 мл) добавляют 3-пиколин (2,53 мл, 2,42 г, 26 ммоль) с последующим добавлением 4-пиколина (2,53 мл, 2,42 г, 26 ммоль). Смесь становится значительно гуще после добавления 4-пиколина. Смесь охлаждают до -5°С и затем добавляют по каплям при температуре от -5 до 0°С метансульфонилхлорид (1,86 мл, 2,75 г, 24 ммоль). Смесь перемешивают в течение 2 час при температуре от 0 до 5°С. Затем добавляют по каплям воду (6 мл) и смесь перемешивают при 0°С в течение 1 час. Смесь фильтруют и твердые вещества промывают смесью 3:1 ацетонитрил-вода (2×4 мл) и затем ацетонитрилом (2×4 мл) и сушат в атмосфере азота, получая указанное в заголовке соединение в виде желтого порошка, 9,15 г (94,7% выход без учета поправки).
ПРИМЕР 10
Получение 3-бром-N-[4-хлор-2-метил-6-[(метиламино)карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида в ацетоне
К смеси 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты (получение: см. патентную публикацию PCT WO 03/015519) (6,05 г, 20,0 ммоль) и 2-амино-5-хлор-N,3-диметилбензамида (т.e. продукта из примеров 1, 3, 4 и 5) (4,17 г, 21,0 ммоль) в ацетоне (18 мл) добавляют 3-пиколин (5,06 мл, 4,84 г, 52 ммоль). Смесь охлаждают до -5°С и затем добавляют по каплям при температуре от -5 до 0°С метансульфонилхлорид (1,86 мл, 2,75 г, 24 ммоль). Смесь перемешивают в течение 3 час при 0-5°С. Затем добавляют по каплям воду (9 мл) и смесь перемешивают при 0°С в течение 1 час. Смесь фильтруют и твердые вещества промывают охлажденной льдом смесью 2:1 ацетон-вода (2×4 мл) и сушат в атмосфере азота, получая указанное в заголовке соединение в виде почти белого порошка, 9,32 г (выход без учета поправки 96,4%).
ПРИМЕР 11
Получение 3-бром-N-[4-хлор-2-метил-6-[(метиламино)карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида в тетрагидрофуране
К смеси 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты (получение: см. патентную публикацию PCT WO 03/015519) (6,05 г, 20,0 ммоль) и 2-амино-5-хлор-N,3-диметилбензамида (продукт из примеров 1, 3, 4 и 5) (4,17 г, 21,0 ммоль) в тетрагидрофуране (THF, 18 мл) добавляют 3-пиколин (5,06 мл, 4,84 г, 52 ммоль). Смесь охлаждают до -5°С и затем добавляют по каплям при температуре от -5 до 0°С метансульфонилхлорид (1,86 мл, 2,75 г, 24 ммоль). Смесь перемешивают в течение 3 час при температуре от 0 до 5°С. Затем добавляют по каплям воду (9 мл) и смесь перемешивают при 0°С в течение 1 час. Смесь фильтруют и твердые вещества промывают охлажденной льдом смесью 2:1 THF-вода (2×4 мл) и сушат в атмосфере азота, получая указанное в заголовке соединение в виде почти белого порошка, 6,93 г (выход без учета поправки 71,7%).
ПРИМЕР 12
Получение 3-бром-N-[4-хлор-2-метил-6-[(метиламино)карбонил]фенил]-1-(3-хлор-2-пиридинил)-1H-пиразол-5-карбоксамида в дихлорметане
К смеси 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты (получение: см. патентную публикацию PCT WO 03/015519) (6,05 г, 20,0 ммоль) и 2-амино-5-хлор-N,3-диметилбензамида (т.e. продукта из примеров 1, 3, 4 и 5) (4,17 г, 21,0 ммоль) в дихлорметане (18 мл) добавляют 3-пиколин (5,06 мл, 4,84 г, 52 ммоль). Смесь охлаждают до -5°С и затем добавляют по каплям при температуре от -5 до 0°С метансульфонилхлорид (1,86 мл, 2,75 г, 24 ммоль). Смесь перемешивают в течение 3 час при температуре от 0 до 5°С. Затем добавляют по каплям воду (9 мл). Еще добавляют дихлорметан (18 мл) для перемешивания густой суспензии и смесь перемешивают при 0°С в течение 1 час. Смесь фильтруют и твердые вещества промывают охлажденной льдом смесью 2:1 дихлорметан-вода (2×4,5 мл) и сушат в атмосфере азота, получая указанное в заголовке соединение в виде почти белого порошка, 8,86 г (выход без учета поправки 91,7%).
ПРИМЕР 13
Получение 3-бром-N-[4-хлор-2-метил-6-[(метиламино)карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида в пропионитриле
К смеси 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты (получение: см. патентную публикацию PCT WO 03/015519) (6,05 г, 20,0 ммоль) и 2-амино-5-хлор-N,3-диметилбензамида (т.e. продукта из примеров 1, 3, 4 и 5) (4,17 г, 21,0 ммоль) в пропионитриле (18 мл) добавляют 3-пиколин (5,06 мл, 4,84 г, 52 ммоль). Смесь охлаждают до -5°С и затем добавляют по каплям при температуре от -5 до 0°С метансульфонилхлорид (1,86 мл, 2,75 г, 24 ммоль). Смесь перемешивают в течение 1 час при температуре от 0 до 5°С и затем в течение 3 час при комнатной температуре. Затем добавляют по каплям воду (9 мл) и смесь перемешивают при комнатной температуре в течение 1 час. Смесь фильтруют и твердые вещества промывают смесью 3:1 пропионитрил-вода (2×4 мл), затем пропионитрилом (2×4 мл) и сушат в атмосфере азота, получая указанное в заголовке соединение в виде почти белого порошка, 9,37 г (выход без учета поправки 97,0%).
ПРИМЕР 14
Получение 3-бром-N-[4-хлор-2-метил-6-[(метиламино)карбонил]фенил]-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоксамида в метилэтилкетоне
К смеси 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты (получение: см. патентную публикацию PCT WO 03/015519) (6,05 г, 20,0 ммоль) и 2-амино-5-хлор-N,3-диметилбензамида (продукта из примеров 1, 3, 4 и 5) (4,17 г, 21,0 ммоль) в метилэтилкетоне (MEK, 18 мл) добавляют 3-пиколин (5,06 мл, 4,84 г, 52 ммоль). Смесь охлаждают до -5°С и затем добавляют по каплям при температуре от -5 до 0°С метансульфонилхлорид (1,86 мл, 2,75 г, 24 ммоль). Смесь перемешивают в течение 3 час при температуре от 0 до 5°C. Затем добавляют по каплям воду (9 мл) и смесь перемешивают при комнатной температуре в течение 1 час. Смесь фильтруют и твердые вещества промывают смесью 3:1 MEK-вода (2×4 мл), затем MEK (2×4 мл) и сушат в атмосфере азота, получая указанное в заголовке соединение в виде почти белого порошка, 9,27 г (выход без учета поправки 95,9%).
ПРИМЕР 15
Получение 3-бром-1-(3-хлор-2-пиридинил)-N-[4-циано-2-метил-6-[(метиламино)карбонил]фенил]-1Н-пиразол-5-карбоксамида
К смеси 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты (получение: см. патентную публикацию PCT WO 03/015519) (чистота 95,4%, 15,85 г, 50,0 ммоль) и 2-амино-5-циано-N,3-диметилбензамида (т.e. продукта из примера 6) (9,93 г, 52,5 ммоль) в ацетонитриле (120 мл) добавляют 3-пиколин (17,5 мл, 16,7 г, 180 ммоль). Смесь охлаждают до -10°С и затем добавляют по каплям при температуре от -10 до -5°С раствор метансульфонилхлорида (5,4 мл, 8,0 г, 70 ммоль). Смесь перемешивают в течение 5 мин при этой температуре и затем в течение 3 час при температуре от 0 до 5°С. Затем добавляют по каплям воду (55 мл). Смесь перемешивают в течение 15 мин, затем добавляют по каплям концентрированную хлористоводородную кислоту (5,0 мл, 60 ммоль) и смесь перемешивают при температуре от 0 до 5°С в течение 1 час. Затем смесь фильтруют и твердые вещества промывают смесью 2:1 ацетонитрил-вода (2×10 мл) и затем ацетонитрилом (2×10 мл) и сушат в атмосфере азота, получая указанное в заголовке соединение в виде не совсем белого порошка, 24,70 г (выход без учета поправки 99,5%), т.пл. 177-181°С (разложение).
Кристаллизация неочищенного продукта (5,00 г) из 1-пропанола (50 мл) дает указанное в заголовке соединение в виде белых кристаллов, 4,44 г (извлечение 88,8%), т.пл. 217-219°С.
1H ЯМР (ДМСО-d6) δ 2,21 (с, 3H), 2,67 (д, J=4,8 Гц, 3H), 7,41 (с, 1H), 7,60 (м, 1H), 7,76 (д, J=1,8 Гц, 1H), 7,87 (д, J=1,8 Гц, 1H), 8,16 (дд, 1H), 8,36 (м, 1H), 8,49 (дд, 1H).
ПРИМЕР 16
Получение 3-бром-1-(3-хлор-2-пиридинил)-N-[4-циано-2-метил-6-[(метиламино)карбонил]фенил]-1Н-пиразол-5-карбоксамида
с использованием пиридина в качестве основания
К смеси 3-бром-1-(3-хлор-2-пиридинил)-1H-пиразол-5-карбоновой кислоты (получение: см. патентную публикацию PCT WO 03/015519) (чистота 95,4%, 15,85 г, 50,0 ммоль) и 2-амино-5-циано-N,3-диметилбензамида (продукта из примера 6) (9,93 г, 52,5 ммоль) в ацетонитриле (120 мл) добавляют пиридин (14,6 мл, 14,3 г, 180 ммоль). Смесь охлаждают до -10°С и затем добавляют по каплям при температуре от -10 до -5°С раствор метансульфонилхлорида (5,4 мл, 8,0 г, 70 ммоль). Смесь перемешивают в течение 5 мин при этой температуре и затем в течение 3 час при температуре от 0 до 5°С. Затем смесь нагревают до комнатной температуры и добавляют по каплям воду (85 мл). Смесь перемешивают в течение 15 мин, затем добавляют по каплям концентрированную хлористоводородную кислоту (5,0 мл, 60 ммоль) и смесь перемешивают в течение 1 час. Затем смесь фильтруют и твердые вещества промывают смесью 4:3 ацетонитрил-вода (2×10 мл) и затем ацетонитрилом (2×10 мл) и сушат в атмосфере азота, получая указанное в заголовке соединение в виде не совсем белого порошка, 24,29 г (выход без учета поправки 97,9%).
ПРИМЕР 17
Получение 3-бром-1-(3-хлор-2-пиридинил)-N-[4-циано-2-метил-6-[(метиламино)карбонил]фенил]-1Н-пиразол-5-карбоксамида
с использованием 2-пиколина в качестве основания
К смеси 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты (получение: см. патентную публикацию PCT WO 03/015519) (чистота 96,7%, 15,64 г, 50,0 ммоль) и 2-амино-5-циано-N,3-диметилбензамида (т.e. продукта из примера 6) (9,93 г, 52,5 ммоль) в ацетонитриле (120 мл) добавляют 2-пиколин (17,8 мл, 16,8 г, 180 ммоль). Смесь охлаждают до -10°С и затем добавляют по каплям при температуре от -10 до -5°С раствор метансульфонилхлорида (5,4 мл, 8,0 г, 70 ммоль). Смесь перемешивают в течение 5 мин при этой температуре, затем в течение 3 час при температуре от 0 до 5°С и затем в течение 18 час при комнатной температуре. Затем добавляют по каплям воду (25 мл). Смесь перемешивают в течение 15 мин, затем добавляют по каплям концентрированную хлористоводородную кислоту (5,0 мл, 60 ммоль) и смесь перемешивают в течение 1 час. Затем смесь фильтруют и твердые вещества промывают смесью 4:1 ацетонитрил-вода (2×10 мл) и затем ацетонитрилом (2×10 мл) и сушат в атмосфере азота, получая указанное в заголовке соединение в виде не совсем белого порошка, 22,52 г (выход без учета поправки 92,0%).
ПРИМЕР 18
Получение 3-бром-1-(3-хлор-2-пиридинил)-N-[4-циано-2-метил-6-[(метиламино)карбонил]фенил]-1Н-пиразол-5-карбоксамида
с использованием 2,6-лутидина в качестве основания
К смеси 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты (получение: см. патентную публикацию PCT WO 03/015519) (чистота 97,6%, 15,50 г, 50,0 ммоль) и 2-амино-5-циано-N,3-диметилбензамида (т.e. продукта из примера 6) (9,93 г, 52,5 ммоль) в ацетонитриле (120 мл) добавляют 2,6-лутидин (21,0 мл, 19,3 г, 180 ммоль). Смесь охлаждают до -10°С и затем добавляют по каплям при температуре от -10 до -5°С раствор метансульфонилхлорида (5,4 мл, 8,0 г, 70 ммоль). Смесь перемешивают в течение 5 мин при этой температуре, затем в течение 1 час при температуре от 0 до 5°С и затем в течение 1 час при комнатной температуре. Анализ ЯМР реакционной массы показывает, что указанного в заголовке соединения мало, но образуется 10,3% циклизованного производного. Добавляют дополнительный 2,6-лутидин (11,7 мл, 10,8 г, 100 ммоль) и метансульфонилхлорид (3,9 мл, 5,8 г, 50 ммоль) и смесь перемешивают в течение 22 час при комнатной температуре. Анализ ЯМР реакционной массы показывает, что образуется 9,6% указанного в заголовке соединения и 89,8% циклизованного производного. Добавляют по каплям воду (55 мл). Смесь перемешивают в течение 15 мин, затем добавляют по каплям концентрированную хлористоводородную кислоту (5,0 мл, 60 ммоль) и смесь перемешивают в течение 1 час. Затем смесь фильтруют и твердые вещества промывают смесью 2:1 ацетонитрил-вода (2×10 мл) и затем ацетонитрилом (2×10 мл) и сушат в атмосфере азота, получая светло-желтый порошок, 21,92 г. Это твердое вещество суспендируют в ацетонитриле (60 мл) и воде (10 мл) и добавляют хлористоводородную кислоту (1 н., 10 мл) и смесь перемешивают при комнатной температуре в течение 30 мин. Затем смесь фильтруют и твердые вещества промывают смесью 3:1 ацетонитрил-вода (2×10 мл) и затем ацетонитрилом (2×10 мл) и сушат в вакуумной печи, получая указанное в заголовке соединение в виде не совсем белого порошка, 20,72 г (выход без учета поправки 85,4%).
ПРИМЕР 19
Получение 3-бром-1-(3-хлор-2-пиридинил)-N-[4-циано-2-метил-6-[(метиламино)карбонил]фенил]-1Н-пиразол-5-карбоксамида в ацетоне
К смеси 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты (получение: см. патентную публикацию PCT WO 03/015519) (чистота 97,6%, 15,50 г, 50,0 ммоль) и 2-амино-5-циано-N,3-диметилбензамида (т.e. продукта из примера 6) (9,93 г, 52,5 ммоль) в ацетоне (120 мл) добавляют 3-пиколин (17,5 мл, 16,7 г, 180 ммоль). Смесь охлаждают до -10°С и затем добавляют по каплям при температуре от -10 до -5°С раствор метансульфонилхлорида (5,4 мл, 8,0 г, 70 ммоль). Смесь перемешивают в течение 5 мин при этой температуре, затем в течение 3 час при температуре от 0 до 5°С. Затем добавляют по каплям воду (55 мл). Смесь перемешивают в течение 15 мин, затем добавляют по каплям концентрированную хлористоводородную кислоту (5,0 мл, 60 ммоль) и смесь перемешивают при температуре от 0 до 5°С в течение 1 час. Затем смесь фильтруют и твердые вещества промывают смесью 2:1 ацетон-вода (3×10 мл) и сушат в атмосфере азота, получая указанное в заголовке соединение в виде не совсем белого порошка, 24,07 г (выход без учета поправки 99,2%). Титрование по Karl Fisher (KFT) этого твердого вещества показывает, что оно содержит 5,5% масс. воды. Часть твердого вещества (23,35 г) сушат в вакуумной печи, получая указанное в заголовке соединение в виде не совсем белого порошка, 22,16 г, теперь содержащего 0,76% масс. воды по KFT.
ПРИМЕР 20
Получение 3-бром-1-(3-хлор-2-пиридинил)-N-[4-циано-2-метил-6-[(метиламино)карбонил]фенил]-1H-пиразол-5-карбоксамида
в пропионитриле
К смеси 3-бром-1-(3-хлор-2-пиридинил)-1Н-пиразол-5-карбоновой кислоты (получение: см. патентную публикацию PCT WO 03/015519) (чистота 97,6%, 15,50 г, 50,0 ммоль) и 2-амино-5-циано-N,3-диметилбензамида (продукта из примера 6) (9,93 г, 52,5 ммоль) в пропионитриле (120 мл) добавляют 3-пиколин (17,5 мл, 16,7 г, 180 ммоль). Смесь охлаждают до -10°С и затем добавляют по каплям при температуре от -10 до -5°С раствор метансульфонилхлорида (5,4 мл, 8,0 г, 70 ммоль). Смесь перемешивают в течение 5 мин при этой температуре, затем в течение 4 час при температуре от 0 до 5°С. Затем добавляют по каплям воду (55 мл). Смесь перемешивают в течение 15 мин, затем добавляют по каплям концентрированную хлористоводородную кислоту (5,0 мл, 60 ммоль) и смесь перемешивают при температуре от 0 до 5°С в течение 1 час. Затем смесь фильтруют и твердые вещества промывают смесью 2:1 пропионитрил-вода (2×10 мл), затем пропионитрилом (2×10 мл) и сушат в атмосфере азота, получая указанное в заголовке соединение в виде не совсем белого порошка, 21,85 г (выход без учета поправки 90,1%). Титрование по Karl Fisher (KFT) этого твердого вещества показывает, что оно содержит 5,4% масс. воды. Часть твердого вещества (21,03 г) сушат в вакуммной печи, получая указанное в заголовке соединение в виде не совсем белого порошка, 20,07 г, содержащего теперь 0,9% масс. воды по KFT.
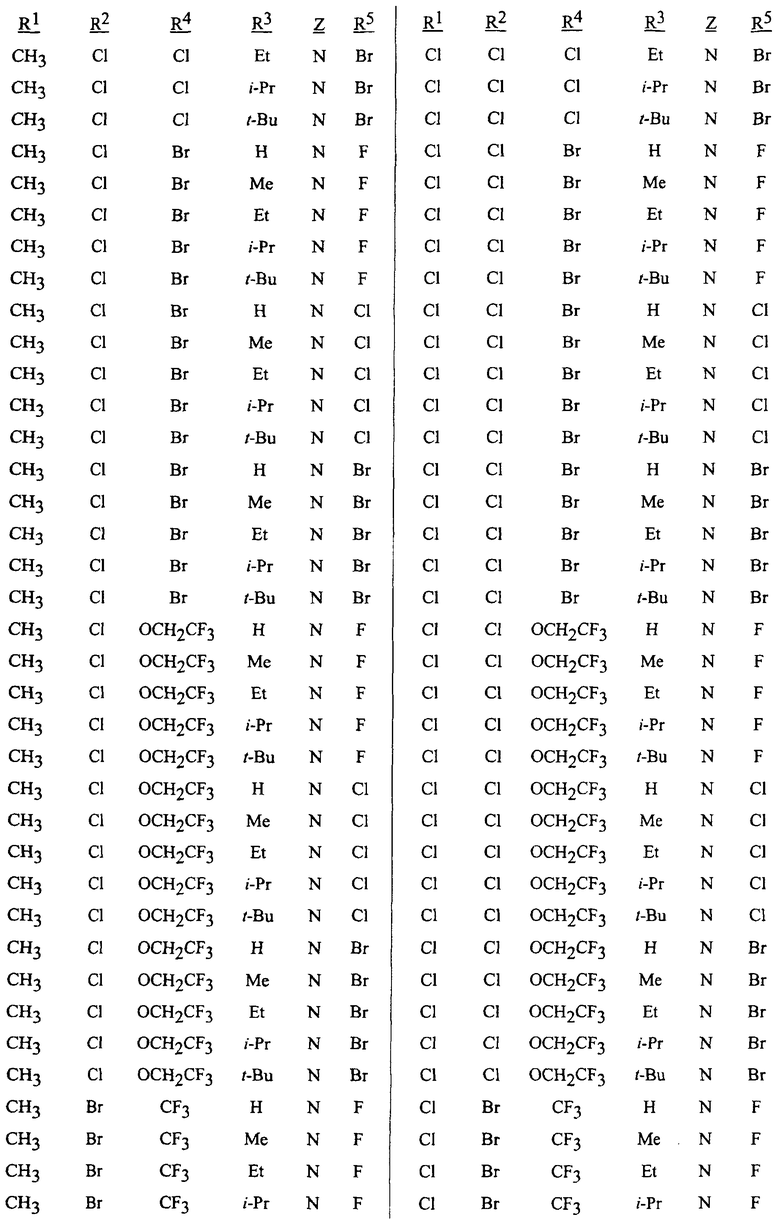
Следующие аббревиатуры использованы в таблицах: t означает третичный, s означает вторичный, n означает нормальный, i означает изо. Me означает метил, Et означает этил, Pr означает пропил, -i-Pr означает изопропил и Bu означает бутил. Путем описанных в описании процедур вместе с известными в технике способами могут быть получены следующие соединения таблицы 1 и использованы в способе по данному изобретению.

Таблица 2 поясняет конкретные преобразования для получения соединений формулы 1 согласно способу данного изобретения.













Изобретение относится к способу получения соединений формулы (I), который включает взаимодействие соединений формул (II) и (III) и сульфонилхлорида в подходящем органическом растворителе в присутствии основания с образованием соединения формулы (I), причем растворитель или/и основание объединяется с соединениями формул (II) и (III). Описаны также соединения формулы (III), которые применимы в качестве исходных соединений для этого способа. Радикалы R1-R6 имеют значения, указанные в формуле изобретения. Технический результат - разработка менее дорогого и более эффективного, гибкого и удобного в работе способа получения соединений формулы (I). 2 н. и 11 з.п. ф-лы, 2 табл.

1. Способ получения соединения формулы 1
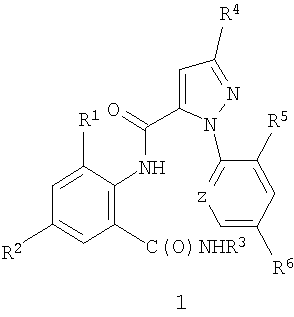
где R1 представляет СН3 или Cl;
R2 представляет Br, Cl, I или CN;
R3 представляет Н или С1-С4алкил;
R4 представляет Cl, Br, CF3, OCF2H или OCH2CF3;
R5 представляет F, Cl или Br;
R6 представляет Н, F или Cl;
Z представляет CR7 или N и
R7 представляет Н, F, Cl или Br, включающий взаимодействие (1) соединения карбоновой кислоты формулы 2
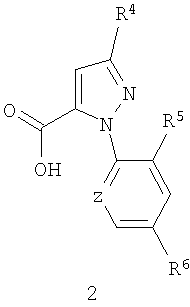
(2) соединения анилина формулы 3
и (3) сульфонилхлорида в подходящем органическом растворителе в присутствии основания с образованием соединения формулы 1, причем растворитель или/и основание объединяется соединениями формулы 2 и 3.
2. Способ по п.1, где сульфонилхлорид представлен формулой 4

где R8 представляет С1-С4алкил, С1-С2галогеналкил или фенил, необязательно замещенные 1-3 заместителями, выбранными из группы, состоящей из галогена, С1-С3алкила и нитро.
3. Способ по п.2, где сульфонилхлоридом является метансульфонилхлорид.
4. Способ по п.1, где карбоновую кислоту формулы 2 объединяют с анилином формулы 3 для получения смеси, и затем смесь объединяют с сульфонилхлоридом.
5. Способ по п.4, где основание объединяют с соединениями формул 2 и 3, чтобы получить смесь перед объединением с сульфонилхлоридом.
6. Способ по п.5, где основание выбрано из третичных аминов.
7. Способ по п.6, где основание выбрано из, необязательно, замещенных пиридинов.
8. Способ по п.7, где основание выбрано из 2-пиколина, 3-пиколина, 2,6-лутидина и пиридина.
9. Способ по п.8, где сульфонилхлорид представлен формулой 4
где R8 представляет С1-С4алкил, C1-С2галогеналкил или фенил, необязательно замещенные 1-3 заместителями, выбранными из группы, состоящей из галогена, С1-С3алкила и нитро.
10. Способ по п.1 или 4, где растворитель объединяют с соединениями формул 2 и 3 и сульфонилхлоридом.
11. Способ по п.10, где растворителем является ацетонитрил.
12. Соединение формулы 3
где R1 представляет СН3 или Cl;
R2 представляет Br, Cl, I или CN и R3 представляет Н или С1-С4алкил;
при условии, что
(a) когда R1 и R2 представляют Cl, тогда R3 не является Н, СН2СН3 или СН(СН3)СН2СН3;
(b) когда R1 представляет СН3 и R2 представляет Cl, Br или CN, тогда R3 не является СН3 или СН(СН3)2;
(c) когда R1 представляет Cl и R2 представляет Cl или Br, тогда R3 не является СН3 или СН(СН3)2; и
(d) когда R1 представляет СН3 и R2 представляет CN, тогда R3 не является Н.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Устройство В.Г.Вохмянина для защиты трехфазного потребителя от обрыва фазы | 1986 |
|
SU1410178A1 |
| RU 2002118334 A, 20.12.2003. | |||

