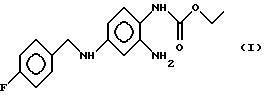
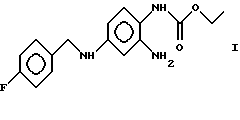
Изобретение относится к новым модификациям 2-амино-4-(4-фторбензиламино)-1-этоксикарбонил-аминобензола формулы I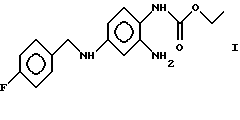
способам их получения и их применению в фармацевтических композициях.
Уровень техники
Вещества формулы I и способы их получения описаны в патенте DE 4200259. Эти вещества обладают противосудорожным, жаропонижающим и анальгетическим действием и вследствие этого могут применяться в фармацевтических композициях.
Однако при кристаллизации вещества формулы I образуют кристаллы различной формы и размеров и зачастую содержат различные побочные примеси. Смесь различных кристаллических модификаций представляет большую проблему при приготовлении фармацевтических композиций. В случае лекарственных форм с высоким содержанием действующего вещества негомогенность субстанции особенно отрицательно сказывается на соблюдении постоянных условий получения галеновых препаратов.
Кроме того могут наблюдаться существенные колебания в стабильности, чистоте и стандартности готовых продуктов, в результате чего могут в недостаточной степени быть выполнены требования, предъявляемые к фармацевтическому качеству действующих веществ.
Вследствие этого представляет интерес получение веществ формулы I в однородной кристаллической форме.
Сущность изобретения
Целью изобретения является получение вещества формулы I в однородной кристаллической форме, соответствующей фармацевтическим требованиям.
Неожиданно было найдено, что вещество формулы I может быть получено в трех различных достаточно чистых кристаллических модификациях. При этом физически гомогенные вещества формулы I могут быть получены для приготовления фармацевтических готовых продуктов противосудорожного, жаропонижающего и анальгетического действия.
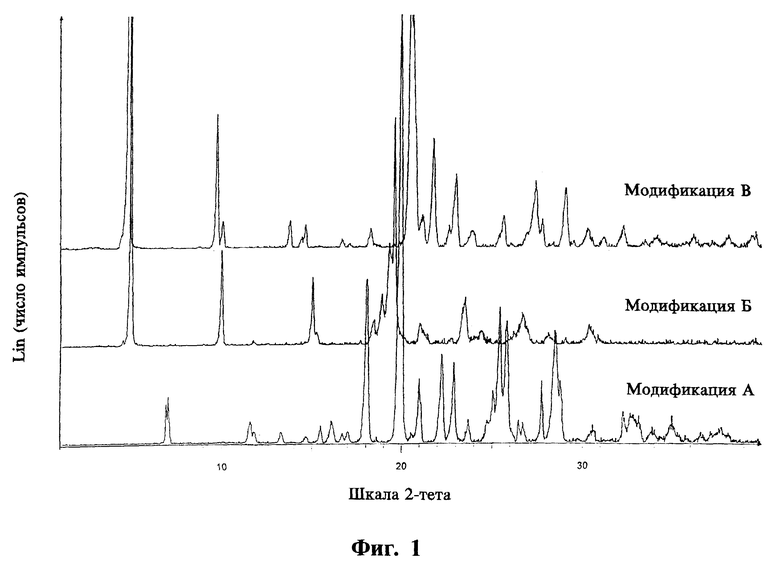
Модификации, названные А, Б и В, обладают различными физико-химическими свойствами. Для идентификации этих трех модификаций соединения формулы I были использованы дифракционные картины (рентгенограммы). Модификации различаются также графиками дифференциальной сканирующей колориметрии (ДСК) и отчасти ИК-спектрами, а также формой кристаллов.
В изобретении предложен способ получения чистой кристаллической формы модификации А соединения формулы I путем кристаллизации из пересыщенного раствора соединения формулы I в протонном, биполярном апротонном или неполярном растворителе при температуре от -20oС до 110oС, предпочтительно при температуре в диапазоне от 20oС до 50oС. При проведении кристаллизации при пониженной, предпочтительно при комнатной, температуре в качестве исходного вещества используют соединение формулы I в виде модификаций Б и В.
Другим объектом изобретения является способ получения чистой кристаллической формы модификации Б соединения формулы I, который проводят путем кристаллизации из насыщенного раствора соединения формулы I в неполярном растворителе при температуре от 50oС до 110oС.
Согласно еще одному варианту способа чистую кристаллическую форму модификации Б соединения формулы I получают посредством термического фазового перехода при температуре выше 80oС с использованием в качестве исходного вещества соединения формулы I в виде модификации А.
Еще одним объектом изобретения является способ получения чистой кристаллической формы модификации В соединения формулы I путем кристаллизации из насыщенного раствора соединения I в протонном растворителе при температуре от 50oС до 70oС, предпочтительно в диапазоне от 60oС до 70oС.
Перечень фигур чертежей и иных материалов
На фиг.1 представлены дифракционные рентгенограммы, полученные на дифрактометре с рентгеновской порошковой камерой и CuK-излучением.
На фиг. 2 показаны графики ДСК, полученные при скорости нагревания 10 К/мин. При различных температурах наблюдаются максимумы интенсивности.
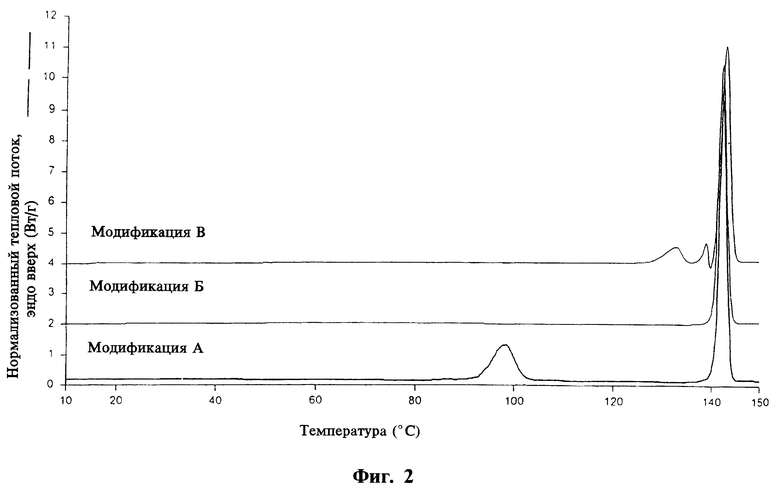
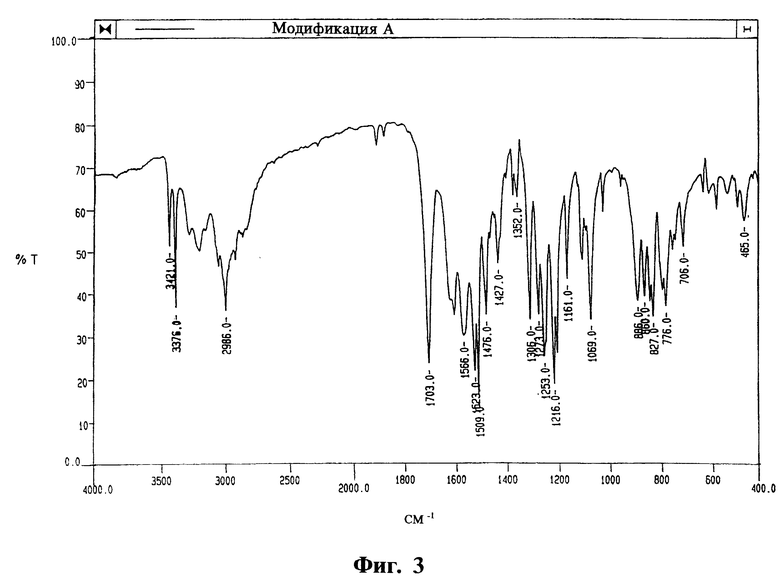
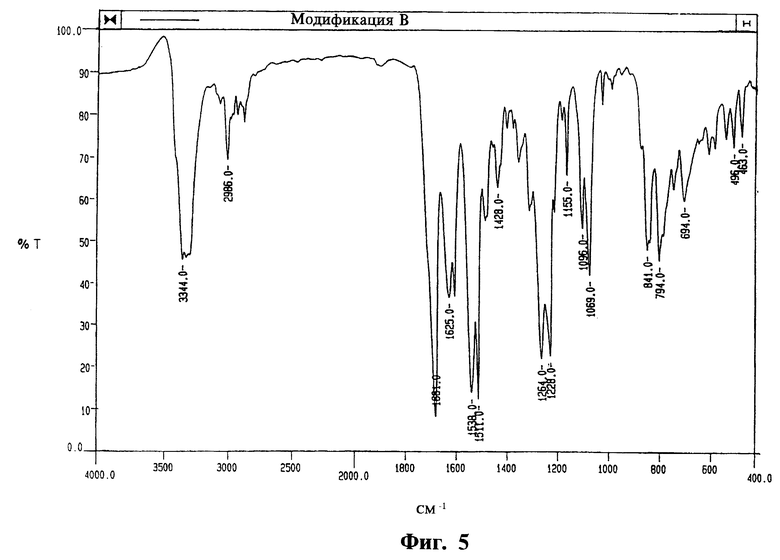
На фиг. 3, 4 и 5 приведены ИК-спектры, соответственно, модификаций А, Б и В, полученные на спрессованных таблетках КВг.
Сведения, подтверждающие возможность осуществления изобретения
Модификация А характеризуется следующими параметрами:
- дифракционной рентгенограммой, на которой наблюдаются рефлексы, не совпадающие с рефлексами двух других модификаций, в том числе при 6.97o2θ (12.67 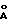 ), 18.02o2θ (4.92
), 18.02o2θ (4.92 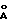 ) и 19.94o2θ (4.45
) и 19.94o2θ (4.45 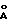 ),
),
- эндотермическим А-В-переходным эффектом на графике ДСК при 97oС (максимум) ниже эффекта плавления модификации Б при 142oС,
- а также ИК-спектром, отличающимся от двух других модификаций интенсивными частотами при 3421 см-1 (v N-H), 3376 см-1 (v N-H), 1703 см-1 (v C=O) и 886 см-1 (у С-Н),
- образованием преимущественно от почти изометрических до короткостолбчатых кристаллов.
Модификация Б характеризуется следующими параметрами:
- дифракционной рентгенограммой, на которой наблюдаются не совпадающие с другими двумя модификациями рефлексы, в том числе при 15.00o2θ (5.90 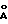 ), 19.29o2θ (4.60
), 19.29o2θ (4.60 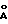 ) и 19.58o2θ (4.53
) и 19.58o2θ (4.53 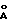 ),
),
- отсутствием термического эффекта на графике ДСК ниже 142oС, где наблюдается эффект плавления,
- образованием преимущественно от продолговатых таблитчатых до стеблевидных кристаллов.
Модификация В характеризуется следующими параметрами:
- дифракционной рентгенограммой, на которой наблюдаются не совпадающие с другими двумя модификациями рефлексы, в том числе при 9.70o2θ (9.11 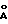 ) и 21.74oθ (4.09
) и 21.74oθ (4.09 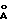 ),
),
- двумя эндотермическими эффектами на графике ДСК, связанными с фазовым переходом в модификацию Б, в области между примерно 130oС и температурой плавления модификации Б около 142oС,
- преимущественно таблитчатыми кристаллами.
Получение трех модификаций вещества согласно формуле I можно осуществлять по следующим способам, причем существенно важным фактором является строгое соблюдение условий. Модификации могут быть получены из неочищенного продукта, содержащего соединение формулы I, или путем взаимопревращения этих модификаций.
Получение модификации А:
Модификация А может быть получена из модификаций Б и В путем перемешивания в растворителе.
Кристаллизацию модификации А проводят предпочтительно путем перемешивания пересыщенного раствора вещества I в протонном, биполярном апротонном или неполярном растворителе.
В качестве протонных растворителей можно использовать низшие спирты, такие как этанол, 2-пропанол, н-бутанол, в качестве биполярных апротонных растворителей - ацетонитрил или ацетон, а в качестве неполярного растворителя - толуол.
Кристаллизацию предпочтительно проводят в присутствии низших спиртов. Кристаллизацию проводят из раствора при температуре от -20oС до 110oС. Предпочтительно кристаллизацию чистой модификации А осуществляют из раствора в определенном растворителе, например, в н-бутаноле при температуре вплоть до 110oС. Наиболее предпочтительно чистую модификацию А получают путем кристаллизации из раствора при температуре от 20oС до 50oС.
Получение модификации Б:
Кристаллизацию модификации Б осуществляют путем медленного охлаждения насыщенного раствора вещества I. В качестве растворителя используют протонный (вода) или апротонный (толуол) растворитель.
Предпочтительно кристаллизацию проводят в присутствии толуола.
Кристаллизация проходит из раствора при температуре от 50oС до 110oС, предпочтительно от 80oС до 100oС.
Модификация Б может также быть получена предпочтительно путем термического фазового перехода предпочтительно модификации А при температуре выше 80oС.
Получение модификации В:
Модификацию В кристаллизуют при медленном охлаждении насыщенного раствора вещества I в протонном растворителе, например, этаноле или 2-пропаноле, или апротонном растворителе, например, толуоле, нагретого до 30-80oС.
Предпочтительно кристаллизацию осуществляют из раствора, нагретого при температуре 50-70oС.
Каждая из модификаций вещества I может выпускаться в виде галеновых препаратов, удовлетворяющих фармацевтическим требованиям.
Настоящее изобретение относится также к применению модификаций А, Б и В вещества I для получения фармацевтических готовых форм, являющихся высокоэффективными антиэпилептиками и нейропротекторами.
Фармацевтические готовые формы могут содержать в одной дозе от 10 до 200 мг по крайней мере одной из модификаций вещества I. Предпочтительной готовой формой являются таблетки.
Для изготовления готовых форм модификации вещества формулы I можно смешивать с подходящими носителями или вспомогательными веществами.
Для производства галеновых препаратов наиболее подходящими свойствами обладает модификация А вещества I.
Кристаллическая структура устойчива до температуры около 80oС. Кроме того, при длительном хранении модификации А при температуре до 60oС и относительной влажности воздуха до 70% не наблюдается изменений кристаллической решетки.
Кристаллическая решетка модификации А не претерпевает никаких изменений при контакте с растворителями, например, такими как вода, этанол, ацетон или толуол.
Благодаря почти изометрической или короткостолбчатой форме кристаллов модификация А представляет собой гранулярную форму, подходящую для приготовления галеновых препаратов.
Модификации Б и В можно использовать для получения специальных лекарственных форм, таких как капсулы или препараты, высушенные в ампулах. Так, например, модификация В образуется преимущественно в виде тонкодисперсного и вследствие этого легко растворимого порошка, наиболее пригодного для получения сухих ампульных форм.
Более детально способы получения отдельных модификаций описаны в приведенных примерах.
Пример 1
Модификация А
2,34 кг соединения I растворяют в смеси 0,16 кг активированного угля в 7,0 л этанола при перемешивании и при нагревании в сосуде объемом 16 л. Горячий раствор фильтруют под давлением в охлажденный кристаллизатор объемом 32 л, содержащий 0,5 л этанола, причем температуру внутри кристаллизатора поддерживают при < 45oС. Затем сосуд, в котором растворяли соединение I, промывают 0,75 л горячего этанола и полученный раствор фильтруют под давлением в кристаллизатор, суспензию в котором постоянно охлаждают. Полученную суспензию перемешивают еще в течение 0,5 часов при 5-12oС и фильтруют в вакууме в инертных условиях. Продукт промывают три раза по 1,2 л охлажденного этанола. Кристаллический продукт высушивают в вакуум-сушильном шкафу при 50-55oС до постоянного веса. При этом получают 2,04 кг (87% от теории) очищенного соединения модификации А.
Пример 2
Модификация А
2 г соединения модификации В перемешивают с 6 мл этанола при комнатной температуре в течение 2 дней. Получают соединение модификации А с количественным выходом.
Пример 3
Модификация А
5 г соединения модификации Б или В перемешивают с 50 мл толуола при комнатной температуре в течение 2 дней. Получают соединение модификации А с количественным выходом.
Пример 4
Модификация А
3 г соединения модификации Б перемешивают с 1,5 мл ацетона при комнатной температуре в течение 2 дней. Получают соединение модификации А с количественным выходом.
Пример 5
Модификация А
10 г соединения I растворяют при нагревании в 5 мл н-бутанола. Продукт кристаллизуют из раствора при 105-110oС, охлаждают до 20oС и кристаллы промывают н-бутанолом с последующим фильтрованием в вакууме. Получают соединение модификации А с количественным выходом.
Пример 6
Модификация Б
10 г соединения I в 20 мл толуола быстро нагревают с обратным холодильником и растворяют. Продукт кристаллизуют из раствора при 90-100oС, отфильтровывают в вакууме и промывают 5 мл толуола. После высушивания получают 9,8 г (98% от теории) продукта в виде иглообразных кристаллов.
Пример 7
Модификация Б
10 г соединения модификации А выдерживают в сушильном шкафу при 100oС в течение 8 ч. Получают соединение модификации Б с количественным выходом.
Пример 8
Модификация В
3,0 кг соединения I растворяют в смеси 0,2 кг активированного угля в 19,6 л изопропанола при перемешивании и при нагревании в сосуде объемом 32 л. Горячий раствор фильтруют под давлением в охлажденный кристаллизатор объемом 32 л, причем температуру внутри кристаллизатора поддерживают при 60-65oС. Затем сосуд, в котором растворяли соединение I, промывают 2,5 л горячего изопропанола (приблизительно при 70oС) и полученный раствор фильтруют под давлением в кристаллизатор. Полученную суспензию после начала кристаллизации перемешивают при 60-65oС. Затем суспензию постоянно охлаждают, перемешивают при 5-12oС и фильтруют в вакууме в инертных условиях. Продукт промывают три раза по 2,5 л охлажденного изопропанола. Кристаллический продукт высушивают в вакуум-сушильном шкафу при 50-55oС до постоянного веса. При этом получают 2,64 кг (88% от теории) соединения модификации В.
Описывается 2-амино-4-(4-фторбензиламино)-1 -этоксикарбониламинобензол формулы I в виде кристаллических модификаций А, Б и В, каждая из которых характеризуется дифракционной рентгенограммой, на которой наблюдаются рефлексы, не совпадающие с рефлексами двух других модификаций. Кристаллические модификации А, Б, В предназначены для приготовления фармацевтических композиций противосудорожного, жаропонижающего и анальгетического действия. Описываются также способы получения указанных модификаций. 5 с. и 8 з.п. ф-лы, 5 ил.
в виде кристаллических модификаций А, Б и В, каждая из которых характеризуется дифракционной рентгенограммой, на которой наблюдаются рефлексы, не совпадающие с рефлексами двух других модификаций, в том числе при 6,97o2θ (12,67 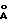 ), 18,02o2θ (4,92
), 18,02o2θ (4,92 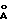 ) и 19,94o2θ (4,45
) и 19,94o2θ (4,45 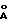 ) для модификации А, при 15,00o2θ (5,90
) для модификации А, при 15,00o2θ (5,90 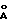 ), 19,29o2θ (4,60
), 19,29o2θ (4,60 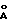 ) и 19,58o2θ (4,53
) и 19,58o2θ (4,53 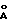 ) для модификации Б и при 9.70o2θ (9,11
) для модификации Б и при 9.70o2θ (9,11 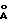 ) и 21,74oθ (4,09
) и 21,74oθ (4,09 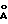 ) для модификации В.
) для модификации В.
| Гербицидное средство | 1978 |
|
SU735151A3 |
| DE 4200259 A1, 15.07.1993 | |||
| DE 3917113 A1, 29.11.1990 | |||
| УСТРОЙСТВО ДЛЯ ИНДИКАЦИИ | 1972 |
|
SU424194A1 |

