Данное изобретение касается способа и устройства для проведения манипуляций на последовательностях нуклеиновых кислот с применением системы твердого носителя, а также носителей для применения в способе и устройстве.
Известны различные способы манипулирования последовательностями нуклеиновых кислот. Например, ЕР-А-0200362 /Cetus/ описывает жидкофазный способ амплификации, обнаружения и/или/ клонирования последовательностей нуклеиновых кислот. Способ предусматривает следующие стадии:
/i/ обработку отдельных комплементарных цепей последовательности нуклеиновой кислоты двумя олигонуклеотидными праймерами, каждый из которых гибридизуется с одной из этих цепей;
/ii/ удлинение праймеров с образованием двухцепочечных последовательностей нуклеиновой кислоты;
/iii/ денатурацию продукта на /ii/ с образованием одиночных цепей нуклеиновой кислоты;
/iv/ применение одиночных цепей из /iii/ для повторения стадий /i/-/iii/, причем весь процесс повторяют столько раз, сколько это необходимо.
Особенностью этого предшествующего способа является то, что обе комплементарные цепи применяют в качестве матриц для второй и последующих стадий амплификации. Кроме того, в способе ЕР-А-0200362 смесь реагентов содержит негибридизованную нуклеиновую кислоту-мишень /целевую нуклеиновую кислоту/, негибридизованную копию мишени и негибридизованный праймер, что делает эту систему очень неэффективной. В дополнение к этому любая ошибка при копировании на любой стадии приводит к ошибке, переносимой в "полимеразную цепную реакцию".
Способы амплификации с применением систем твердофазных носителей также известны, например, с применением магнитных гранул DYNAL /Trade Mark/, описанных в ЕР-А-0091453 и ЕР-А-0106873. При применении магнитных гранул ДНК синтезируется на них и затем отщепляется от гранул гидроконцом аммония. Затем гранулы можно отделить от синтезированной ДНК при помощи магнита. Альтернативно, к одному концу синтезированной или природной ДНК может быть добавлен биотин и эту последовательность можно извлечь при помощи магнитной гранулы, предварительно компьюгированной со стрептавидином /аттрагируя биотики для извлечения ДНК/. Проблема, возникающая с этим типом твердого носителя, заключается в том, что связь биотина со стрептавидином является биодеградируемой.
Другими известными системами твердых носителей являются пористые кремнеземы. Наличие пор может создавать проблемы, т.к. рост нуклеотидной цепи происходит внутри пор, что приводит к неэффективному вымыванию и оставлению остатков внутри пор, что опять-таки снижает выход и приводит к относительно неэффективному связыванию.
ЕР-А-0184056 описывает способ широкомасштабного получения зондов ДНК при помощи твердого субстрата. Способ этой предшествующей спецификации предусматривает ковалентное связывание с твердым субстратом полинуклеотида, комплементарного получаемому зонду, и последующую гибридизацию этого полинуклеотида с олигонуклеотидом, действующим в качестве праймера. Затем олигонуклеотид удлиняется в направлении от субстрата /с применением полинуклеотида в качестве матрицы/ с образованием удлиненной последовательности, комплементарной связанному с субстратом полинуклеотиду. После этого гибридизованные полинуклеотид и удлиненный олигонуклеотид денатурируют для высвобождения удлиненного олигонуклеотида из твердого субстрата для коллекции. Удлиненный олигонуклеотид можно использовать в качестве аналитического зонда. Например, полинуклеотидом, исходно связанным с носителем, может быть ген, а удлиненный олигонуклеотид можно применять в качестве зонда для обнаружения этого гена в биологической пробе.
Однако имеется ряд неблагоприятных моментов, связанных со способом, описанным в ЕР-А-0184056. В частности, если полинуклеотид, который следует связать с носителем, не является "чистым" и содержит другие полинуклеотиды, то они будут также связываться с носителем. В результате олигонуклеотид может также гибридизоваться с этими другими полинуклеотидами, так что полученный в конечном счете удлиненный олигонуклеотид может фактически быть смесью продуктов. Поэтому этот способ не очень хорош для получения "чистых" проб нуклеоиновых кислот.
Поэтому объектом данного изобретения является устранение или ослабление вышеупомянутых неблагоприятных моментов.
Согласно первому аспекту данного изобретения обеспечен способ осуществления манипуляции последовательностью нуклеиновой кислоты, который предусматривает
/а/ обеспечение системы твердого носителя, с которым связан одноцепочечный олигонуклеотид, комплементарный специфической последовательности на нуклеиновой кислоте-мишени, которая имеет большую длину, чем олигонуклеотид,
/b/ добавление источника одноцепочечной нуклеиновой кислоты-мишени к системе твердого носителя,
/с/ гибридизацию нуклеиновой кислоты-мишени с олигонуклеотидом и
/d/ осуществление манипуляции на гибридизованной нуклеиновой кислоте-мишени.
Данный способ приводит к тому, что целевая нуклеиновая кислота изобретательно гибридизуется с олигонуклеотидом на носителе. Любые примеси, присутствующие в системе /например, введенные в пробе, содержащей целевую нуклеиновую кислоту/, можно удалить промыванием, что оставляет "чистую" пробу целевой нуклеиновой кислоты, на которой можно выполнять манипуляцию. Промывание проводят при температуре, при которой не происходит плавления целевой нуклеиновой кислоты с отделением ее от олигонуклеотида.
Нуклеиновая кислота-мишень может, например, быть очищенной или неочищенной, нативной или синтезированной нуклеиновой кислотой. Мишенью может быть любая ДНК или РНК из вирусного, бактериального, животного или растительного источника.
Олигонуклеотид, связанный с носителем, должен содержать, как правило, не менее 8 нуклеотидов. Обычно полинуклеотид, который должен гибридизоваться с олигонуклеотидом, содержит 1000-2000 оснований и значительно длиннее, чем олигонуклеотид.
Олигонуклеотид может быть связан с носителем в результате реакции между пригодными для этого реактивными группами, обеспеченными на носителе, и олигонуклеотидом. Альтернативно олигонуклеотид может быть синтезирован in situ на носителе.
Олигонуклеотид может быть связан с твердым носителем в ориентации 31-51 или 51-31.
Любые защитные группы на олигонуклеотиде удаляют перед стадией гибридизации /с/.
В способе данного изобретения одноцепочечная нуклеиновая кислота-мишень гибридизуется с олигонуклеотидом, связанным с носителем. Последовательность олигонуклеотида должна быть очень специфичной для гибридизуемой мишени. В результате гибридизацию можно проводить при температуре, которая очень специфичная для гибридизации между этим олигонуклеотидом и мишенью.
Гибридизацию нуклеиновой кислоты-мишени идеально проводить при температуре едва ниже /например, 1-3oС/ температуры /Тп/, при которой нуклеиновая кислота-мишень отсоединяется путем плавления от олигонуклеотида. В виде общего правила Тп можно рассчитать по формуле, известной в данной области исследований
Тп =2 (А+Т) +3 (Г+Ц)
где А, Т, Г, Ц обозначают соответственно число остатков аденина, тимина, гуанина и цитозина, присутствующих в олигонуклеотиде, связанном с носителем. Формула обычно применима для последовательностей длиной до 30 нуклеотидов. После стадии гибридизации носитель можно промыть при температуре, слегка более низкой, чем Тп, для удаления негибридизовавшейся мишени и примесей /например, белков при нежелательных последовательностей нуклеиновых кислот/. Поэтому можно добавить к системе твердого носителя смесь полинуклеотидов /которые могут присутствовать в биологической пробе/, но только специфический целевой полинуклеотид /т.е. мишень/ удержится на носителе. Таким образом, после промывания манипуляции полинуклеотидом можно проводить на его "чистой" пробе.
Последовательные манипуляции можно проводить на гибридизовавшейся целевой нуклеиновой кислоте. Альтернативно исходное манипулирование можно выполнять для получения копии мишени, связанной с носителем. Эту копию можно затем применять для последующих операций манипулирования.
Между каждыми двумя манипуляциями можно промывать, если необходимо, для удаления примесей, после чего собирают требуемый продукт. После дальнейшего промывания носителя /если необходимо/ манипуляцию можно повторять.
Манипуляция может представлять собой, например, копирование, амплификацию последовательности нуклеиновой кислоты-мишени, транскрипцию in vitro или очистку ДНК-связывающих белков. Манипуляцией может быть также обнаружение присутствия мишени тестированием на гибридизацию меченого праймера с мишенью. Манипуляции на полинуклеотидной последовательности можно выполнять при помощи ранее известных способов. Например, в способах манипулирования можно использовать ДНК- полимеразу 1, ее фрагменты Кленова, термостабильную полимеразу и/или/ обратную транскриптазу вместе с дезоксинуклеотидтрифосфатами. Нуклеотиды могут, если нужно, быть мечеными, например, 33s, 33р, хромофорами, биотином, пероксидазой, биофатазой или любым другими биологическим маркером по выбору.
Способ изобретения, в частности, применим для анализа проб /например, медицинских проб/ для обнаружения присутствия или отсутствия определенной нуклеиновой кислоты. Если, например, пробу тестируют на состояние болезни, при котором наблюдается относительно высокое количество определенной нуклеиновой кислоты /мишени/, то обнаружение этой нуклеиновой кислоты можно выполнять следующим образом. Если того как мишень /если она присутствует/ гибридизовалась с носителем и носитель промыли для оставления на нем "чистой" пробы мишени, меченый олигонуклеотидный праймер, комплементарный последовательности на мишени, добавляют к носителю при условиях, в которых праймер будет гибридизоваться с мишенью. Метка может быть, например, радиоактивной меткой, хромофором или реагентом, соединенным с ферментом. Затем носитель промывают для удаления негибридизовавшегося праймера, причем промывание проводят при температуре, более низкой, чем та, при которой праймер, гибридизовавшийся с целевой нуклеиновой кислотой, отсоединяется плавлением от мишени. Затем температуру носителя можно повысить для отсоединения гибридизовавшегося праймера /либо с целевой ДНК, либо без нее/. Для обнаружения присутствия праймера можно использовать детектор. Если праймер детектируется, то это является подтверждением того, что нуклеиновая кислота-мишень присутствует в пробе.
Однако, если проба содержит лишь относительно низкое количество целевой нуклеиновой кислоты, то можно провести повторяющиеся реакции амплификации /см. ниже, каждая из которых продуцирует меченые копии амплифицированного продукта, если исходно предполагаемая мишень присутствует в пробе. Затем можно провести операцию детектирования для определения, присутствует ли меченый продукт амплификации. Если это так, то это является подтверждением того, что исходно предполагаемая мишень присутствует в пробе. Конечно, желательно, чтобы можно было бы провести любое количество амплификаций, так чтобы тест был очень чувствительным и давал возможность определения присутствия очень низких количеств мишени в исходной пробе.
Важной особенностью данного изобретения является то, что обнаружение присутствия мишени в исходной пробе может достигаться без необходимости разделения продуктов на гелях.
Особенно предпочтительно в соответствии с данным изобретением обеспечение носителя в проточном сосуде /например, в колонке/, что облегчает промывание носителя, а также последующую манипуляцию, что будет видно из дальнейшего описания.
Применение проточного сосуда является важной особенностью и поэтому согласно второму аспекту данного изобретения обеспечен проточный сосуд, имеющий входное и выходное отверстия и содержащий систему твердого носителя со связанным с ней одноцепочечным олигонуклеотидом, комплементарным специфической последовательности на нуклеиновой кислоте-мишени.
Сосуд может быть размером, например, на 150-250 мкл.
Манипулирование можно выполнять при помощи устройства /в котором помещен проточный сосуд/ для подачи растворов реагентов к сосуду и для сбора и обнаружения продукта манипулирования.
Согласно третьему аспекту данного изобретения обеспечено устройство для выполнения манипулирования на последовательности нуклеиновой кислоты, содержащее
проточный сосуд,
средства для хранения растворов, удаляемых из сосуда во время промывания или других процедур, перед тем как эти растворы возвращаются в колонку,
детекторные средства для детектирования продуктов манипулирования нуклеиновыми кислотами и
контрольные средства для отведения растворов в сосуд и из сосуда, в зоны отходов или хранения и к детекторным средствам.
В предпочтительной форме устройства выходное отверстие сосуда может избирательно сообщаться с /i/ участком хранения реагентов, /ii/ участком детектирования продукта манипулирования и /iii/ выпускным отверстием для отходов. Участок сбора реагентов позволяет раствору реагентов, исходно подаваемому к сосуду, через его входное отверстие, проходить к участку сбора реагентов для последующего возврата к сосуду, где должны выполняться повторные манипуляции на той же самой нуклеиновой кислоте-мишени /или на ее копии/. Продукт из каждой операции манипулирования может затем проходить к детекторному участку для детектирования. Если нужно, устройство может быть таким, что продукт из последовательных манипуляций собирают перед его прохождением к детектору.
Устройство содержит, конечно, клапанное приспособление /например, управляемые соленоидом клапаны/, для того чтобы растворы, продукт и т.д. могли перемещаться/ предпочтительно под давлением газа/ через устройство.
Твердый носитель может содержать непористые частицы, имеющие размер 100-200 микронов. Применение непористого материала имеет особые преимущества, т.к. оно позволяет избежать некоторых проблем, связанных с пористыми носителями, применяемыми ранее, а именно: рост нуклеотидной цепи внутри пор приводит к неэффективному промыванию и оставлению остатков внутри пор, что снижает выход и ведет к относительно неэффективному связыванию. Носитель может представлять собой кальцинированные сферические частицы с диаметром 100-200 микронов. Носитель может быть, например, непористым силикагелем.
Носитель может иметь реактивные группы /например, эпоксигруппы/ для применения их в иммобилизации олигонуклеотидной последовательности на носителях.
Предпочтительно, чтобы носитель имел сшитый поперечными связями силоксановый матрикс, имеющий реактивные группы, которые могут быть использованы для обеспечения иммобилизованного олигонуклеотида на носителе. Такой носитель является также важной особенностью и поэтому согласно четвертому аспекту данного изобретения обеспечена система твердого носителя для иммобилизации последовательности нуклеиновой кислоты, имеющего сшитый поперечными связями силоксановый матрикс с реактивными группами, которые могут быть использованы для обеспечения иммобилизованной нуклеиновой кислоты на этом носителе.
Согласно пятому аспекту данного изобретения обеспечен способ получения носителя для иммобилизации последовательности нуклеиновой кислоты, предусматривающий реакцию твердого носителя, имеющего свободные гидроксигруппы, с предшественником силоксанового матрикса, который реагирует со свободными гидроксигруппами и имеет группу, которая может быть использована для иммобилизации последовательности нуклеиновой кислоты на носителе, и нагревание продукта с образованием силоксанового матрикса.
Применение силоксанового матрикса особенно предпочтительно благодаря устойчивости к кислотам и основаниям. Это является преимуществом при сравнении этого матрикса с прежними носителями, содержащими связь биотин-стрептавидин, которые являются биодеградируемыми. Силоксановый матрикс делает возможным проведение широкого диапазона манипуляций без удаления олигонуклеотида из носителя. Например, аммиак можно применять в качестве реагента для депротектирования олигонуклеотида, который остается на носителе. Нормальные связи с янтарной кислотой, применяемые для иммобилизации олигонуклеотида, лабильны в присутствии щелочей, что вызывает отделение олигонуклеотида от носителя.
Предпочтительной реактивной группой, которая может применяться для иммобилизации последовательности нуклеиновой кислоты, является эпоксигруппа.
Если требуется, свободные гидроксигруппы, присутствующие на носителе, после реакции с предшественником силоксанового матрикса, но перед образованием сшитого поперечными связями силоксанового матрикса могут быть блокированы, например, при помощи хлорсилана.
Предпочтительно предшественник силоксанового матрикса представляет собой глицидоксисоединение формулы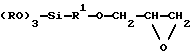
где R представляет собой С1-С4-алкил, a R1 представляет собой алкилен. Наиболее предпочтительно R обозначает метил, a R1 обозначает -(СН2)3-.
Синтетический олигонуклеотид может быть ковалентно связан с носителем через эпоксигруппу. Альтернативно, натриевая соль нуклеотида может быть ковалентно связана с носителем и может быть проведен синтез олигонуклеотида /с применением β-цианоэтилфосфоамидита/.
Далее изобретение будет описано при помощи примера, только со ссылкой на соответствующие чертежи, в которых
фиг. 1 представляет в виде схемы принцип изобретения;
фиг.2 изображает продольный разрез проточной колонки, содержащей систему олиго-твердое вещество;
фиг. 3 иллюстрирует путь реакции для получения твердого носителя для иммобилизации олигонуклеотида;
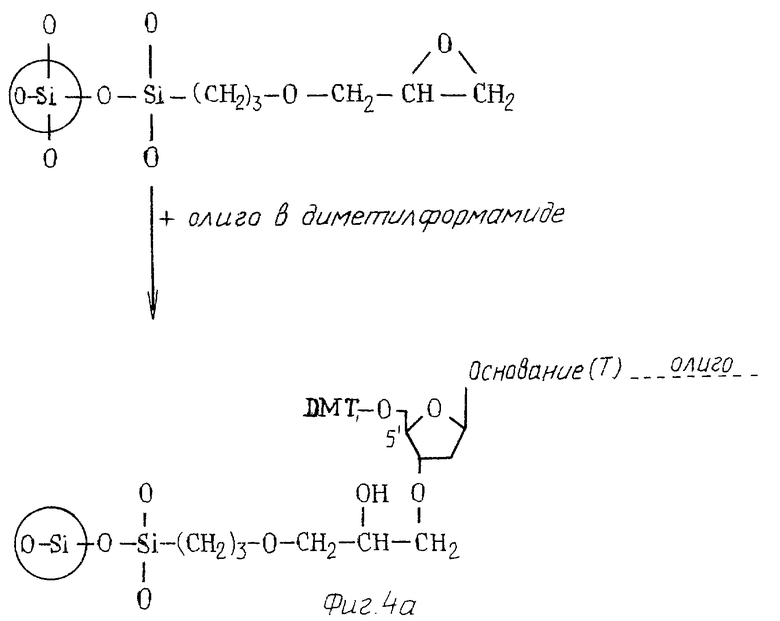
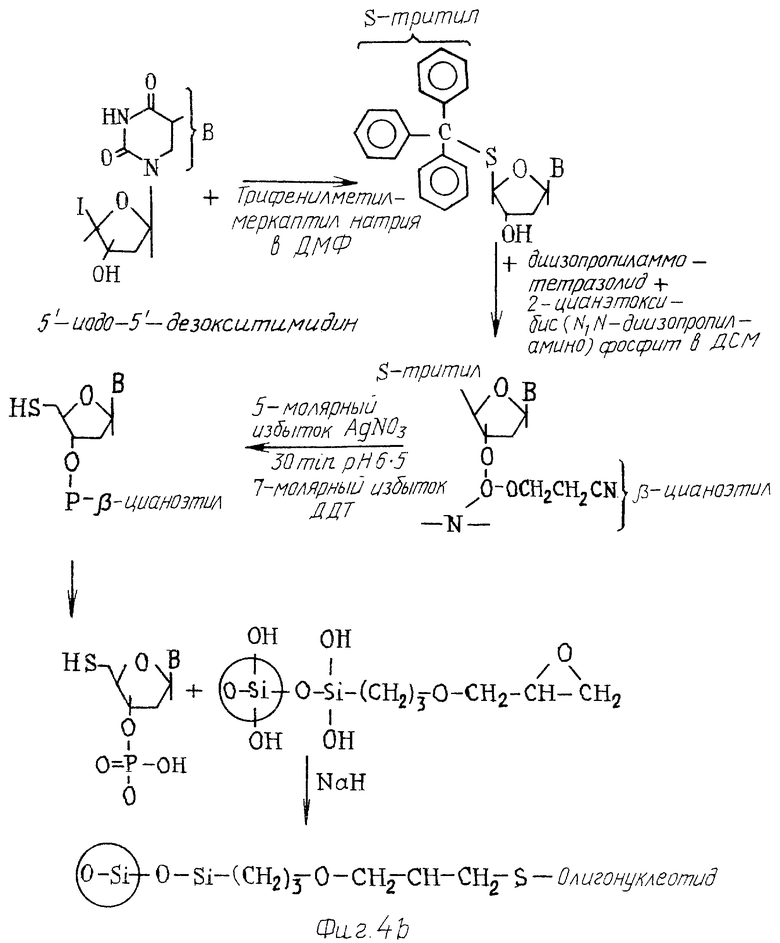
фиг. 4а дают схематическое изображение системы твердого и 4в носителя и реакций, участвующих в связывании с ней олигонуклеотида;
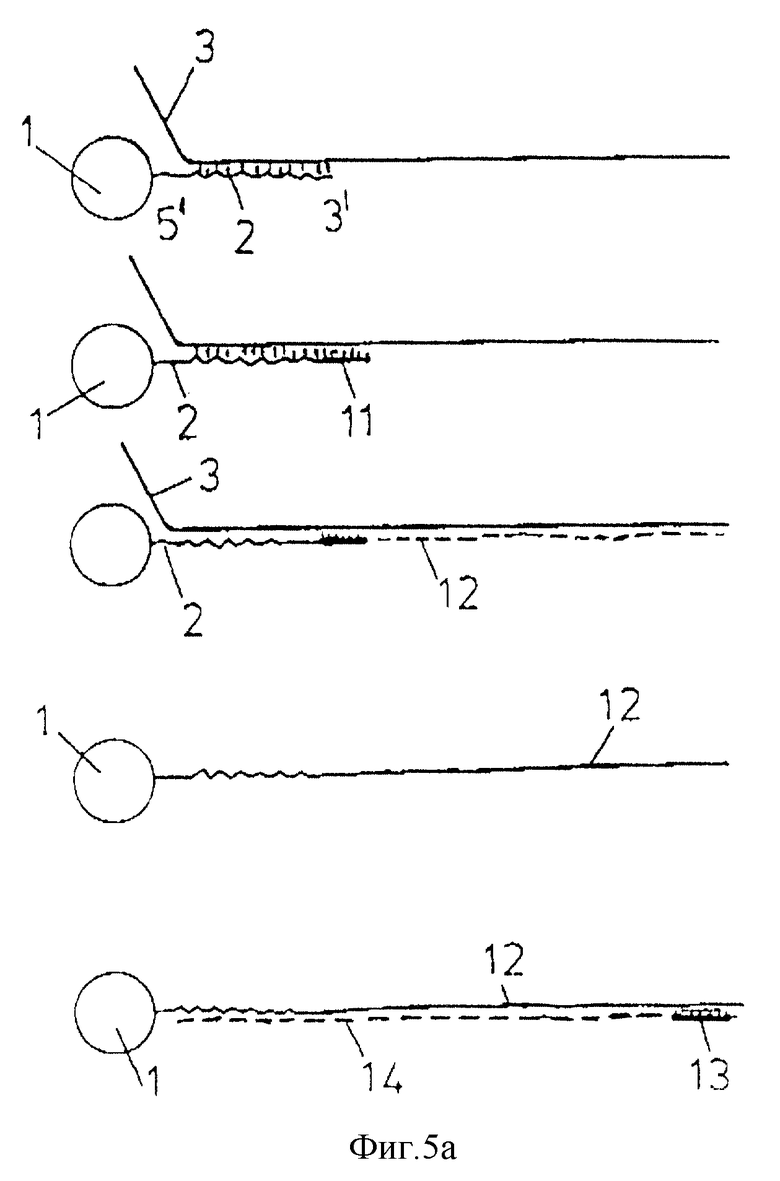
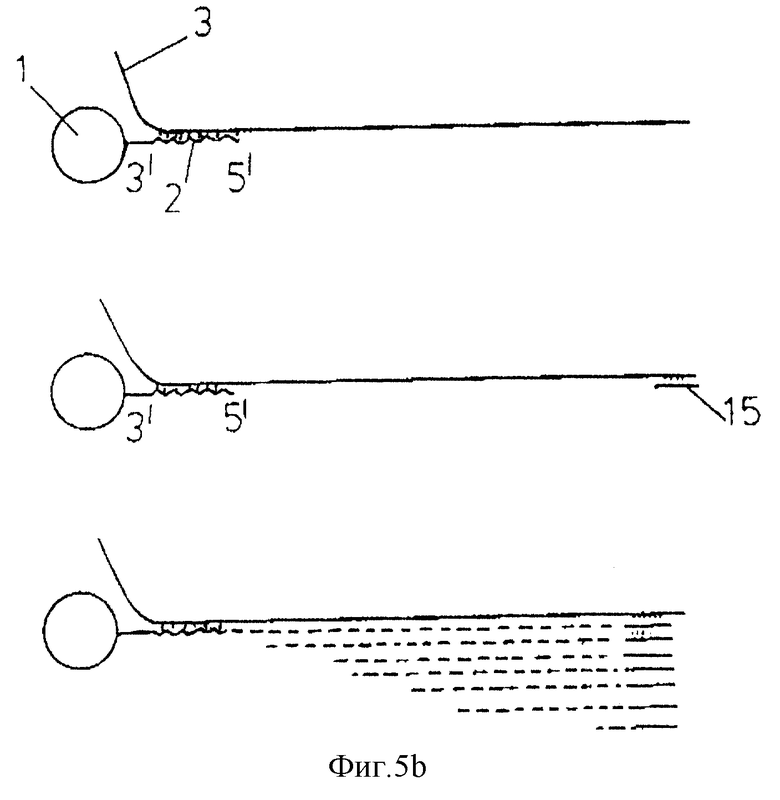
фиг. 5а-5d дают схематические изображения стадий, участвующих в амплификации нуклеиновой кислоты-мишени;
фиг. 6 иллюстрирует устройство для проведения манипулирования в соответствии с изобретением, и
фиг.7 - радиоавтограф, показывающий результаты примера 3.
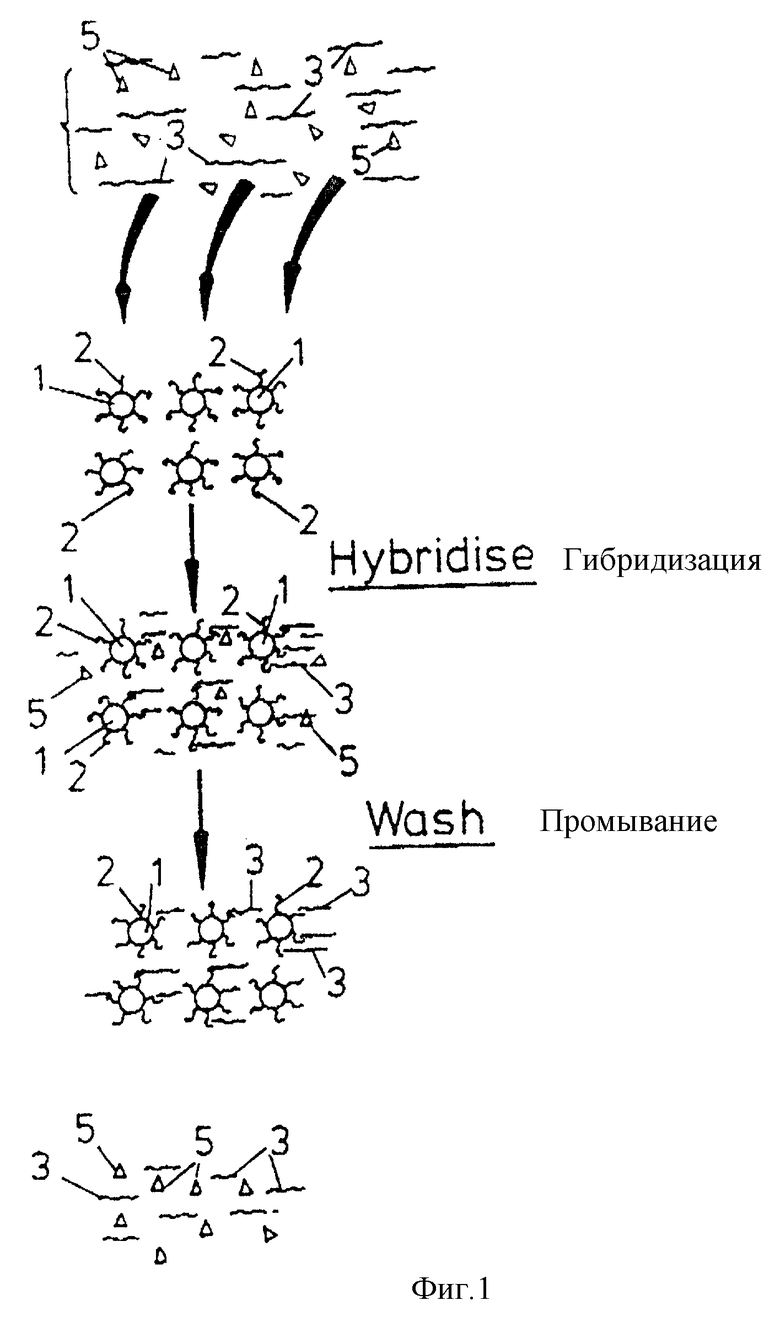
На фиг.1 иллюстрировано /в сильно увеличенном масштабе/ множество частиц 1 материала твердого носителя, каждая из которых несет иммобилизованные на ней идентичные олигонуклеотидные последовательности 2, которые специфичны относительно одноцепочечной последовательности нуклеиновой кислоты-мишени 3, содержащейся внутри пробы 4, которая содержит также нежелательные примеси /например, другие последовательности нуклеиновых кислот, белки и т.д./, представленные треугольными 5.
Пробу 4 добавляют к носителю 1 при гибридизационных условиях, так что часть последовательностей нуклеиновой кислоты 3 связывается с олигонуклеотидом, а часть этих последовательностей остается внегибридизованном виде вместе с примесями 5 в жидкой фазе. Во время последующего промывания негибридизованная нуклеиновая кислота 3 вместе с примесями удаляется из носителя 1, как показано, оставляя чистую пробу целевой нуклеиновой кислоты 3 на носителе в связанном виде.
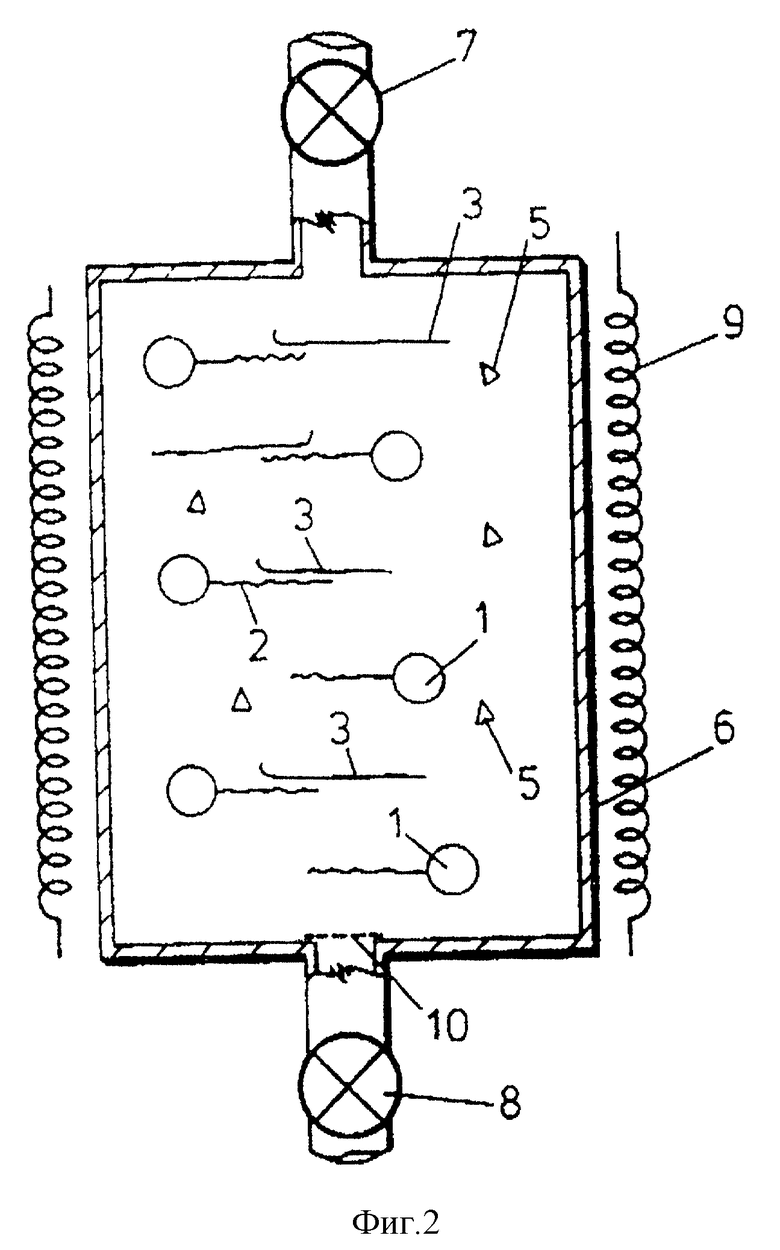
Особенно предпочтительно, чтобы частицы 1 носителя находились внутри проточной колонки 6 /см. фиг.2/, имеющей входной и выходной клапаны 7 и 8, как показано на чертеже, а также нагреватель 9. Частицы могут удерживаться внутри колонки пористыми элементами 10. Для удобства частицы 1, изображенные в фиг.2, представлены как имеющие только одну иммобилизованную на них олигонуклеотидную последовательность.
Нуклеиновую кислоту-мишень добавляют в проточную колонку через входной клапан 7 в отношении, например, 1000:1 /олигонуклеотид:мишень/, в подходящем буфере, например, в высокосоленом буфере и гибридизацию проводят при условиях, специфических для связанного с носителем олигонуклеотида и нуклеиновой кислоты-мишени. Если источником нуклеиновой кислоты-мишени является двухцепочечная ДНК, то ее сначала разделяют на одиночные цепи обычными способами либо перед нанесением на колонку, либо когда она уже находится на колонке.
Проба 4 удерживается в колонке благодаря тому, что выпускной клапан 8 закрыт. После стадии гибридизации нежелательные примеси 5 /которые не иммобилизуются на носителе/ можно удалить промыванием /с открытым клапаном 8/, тогда как чистая проба нуклеиновой кислоты-мишени остается на носителе.
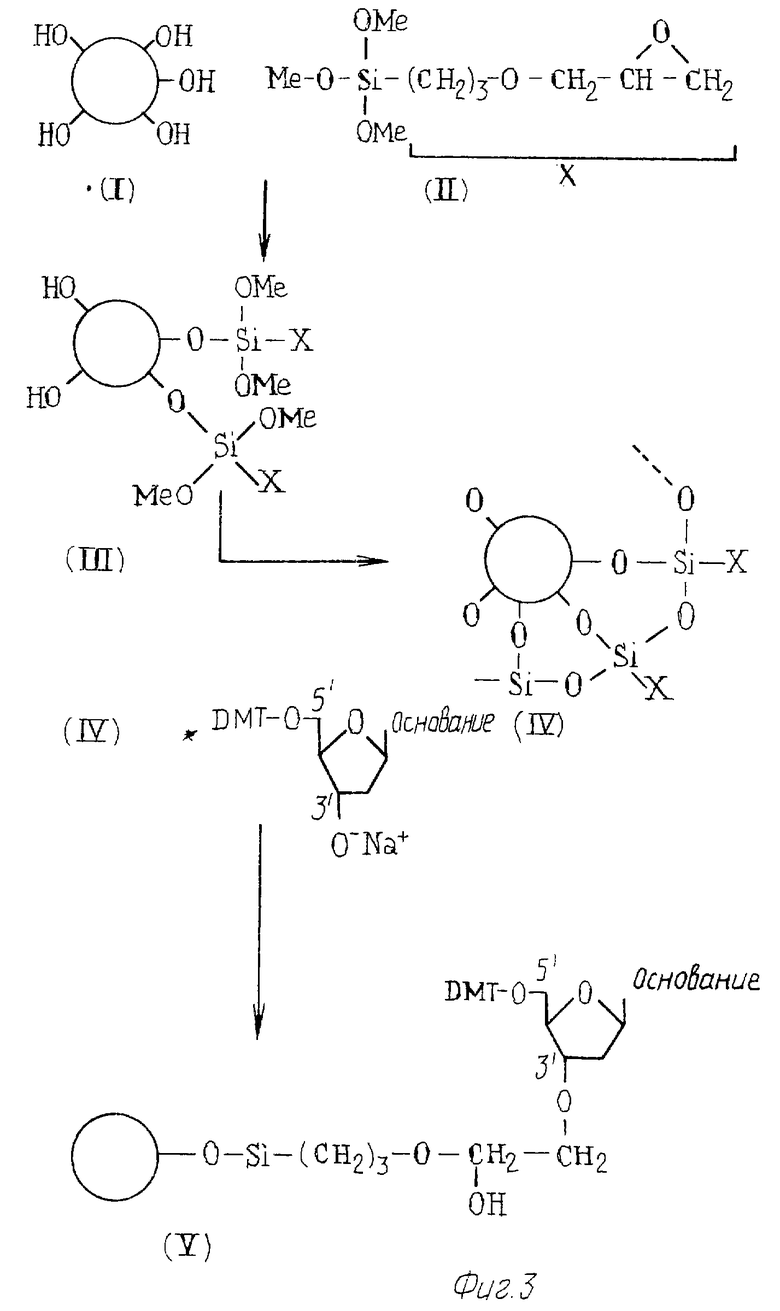
На фиг. 3-5 изображен способ, при помощи которого готовят твердый носитель /с иммобилизованным олигонуклеотидом/.
Сначала сделана ссылка на фиг.3, который иллюстрирует один вариант пути реакции для получения носителя с реакционными группами, при помощи которых может быть иммобилизован олигонуклеотид.
Процедура начинается с частиц 1 силикагеля, имеющих поверхностные гидроксигруппы, как показано на чертеже. В первой стадии реакции частицы обрабатывают 3-глицидоксипропилтриметоксисиланом /II/, например, при температуре не выше 95oС в течение 2 часов в атмосфере азота. В результате происходит реакция конденсации, вследствие которой остатки /II/ становятся связанными с частицами кремнезема /I/ с образованием продукта, изображенного как /III/. Хотя чертеж показывает только два остатка, связанных с частицей диоксида кремния /кремнезема/, это сделано лишь для ясности. На практике значительно большее количество остатков соединяется с частицей диоксида кремния.
Обычно уровень связывания составляет 40-45 микромолей /I/ на грамм диоксида кремния.
Затем продукт /III/ промывают последовательно сухим толуолом, сухим метанолом и сухим эфиром.
Следующая стадия предусматривает нагревание продукта для проведения сшивания остатков поперечными связями. Как правило, эту реакцию сшивания проводят при температуре 110oС в течение по меньшей мере 2 часов. Образующийся продукт представлен в /IV/, где видно, что атомы кремния соединены вместе через атомы кислорода.
Свободные гидроксигруппы, оставшиеся на поверхности частиц диоксида кремния, могут, если нужно, быть блокированы хлорсиланом. Блокирующим агентом может быть, например, триметилхлорсилан и реакцию проводят в пиридине в течение двух часов при комнатной температуре. После блокирования носитель может быть подвергнут операции промывания.
Полученный таким образом носитель имеет свободные эпоксигруппы, которые могут быть применены для иммобилизации олигонуклеотида.
Можно, например, при помощи известных процедур синтезировать in situ на носителе олигонуклеотид, комплементарный специфической последовательности на целевой нуклеиновой кислоте. Так, натриевая соль защищенного диметокситритилом /DMT/ нуклеотида может реагировать с носителем посредством того, что - 0 часть в 31- положении нуклеотида реагирует со свободной эпоксигруппой с образованием продукта /Y/. Натриевую соль можно приготовить из DMT-нуклеотида /который был высушен над F2O5/ путем растворения в сухом DMF, выдерживания этого раствора в безводной атмосфере и добавления гидрида натрия. Затем гидрид натрия отфильтровывают, получая натриевую соль.
После удаления ДМТ-защитной группы целевой олигонуклеотид можно синтезировать из отдельных нуклеотидов при помощи известных процедур синтеза олигонуклеотидов.
Альтернативно можно связать с носителем заранее синтезированный олигонуклеотид /комплементарный специфической последовательности на целевой нуклеиновой кислоте и предпочтительно содержащий также сайт расщепления рестриктазы/.
Имеется в распоряжении ряд синтетических олигонуклеотидов, комплементарных ряду последовательностей нуклеиновых кислот-мишеней и применимых в качестве зондов для тестирования на присутствие определенных бактерий, вирусов, паразитов и т.п., см. приложение А. Изобретение можно применять, например, для детектирования следующих организмов, для которых на носителе должны быть иммобилизованы представленные ниже олигонуклеотидные последовательности.
Вирус Эпштейна-Барра ГАН ААЦ ТЦГ ГЦЦ ГТЦ АТГ ГА
Тохор lasma gongii ЦГА АЦТ ГЦА ТЦЦ ГТТ ПАТ ГАГ
N rypa nosom a Brucei Brucei ЦГА АТГ ААТ ААА ЦАА ТГЦ
ГЦА
Олигонуклеотид может быть связан на носителе либо в ориентации 31-51, либо в ориентации 31-31. Связанные в ориентации 31-51 может быть достигнуто реакцией эпоксигруппы на носителе с первичной гидроксигруппой натриевой соли нуклеотида (например, тимидина (диметокситритил-дезоксирибонуклеозида) (дТ) см. фиг.4а)) с образованием связи, которая устойчива как к кислотам, так и к щелочам в отличие от применяемых в настоящее время соединительных групп.
Связанные в ориентации 51-31 может быть достигнуто следующим образом /см. также фиг. 4/. 51-иод-51-дезокситимидин реагирует с трифенилметилмеркаптилом натрия в ДМФ с образованием соединения S-тритила. Далее это соединение реагирует с диизопропиламмо-тетразолидом и 2-цианэтокси-бас N,N-диизопропиламино)-фосфоамидитом в DCM с образованием β-цианоэтила. S-тритил удаляют восстановительными реакциями известными способами /Connoly, Nucleic Acid Research.
13,12,1985; Chu, Nac L eic Acid Research, 16, 9, 1988;
Guar, Nucleic Acid Research, 17, 11, 1989/
перед соединением с эпокси-замещенным носителем при помощи гидрида натрия. Известны и другие способы приготовления олигонуклеотидов для соединения в 51-31 ориентации /Anisorge, Nucleic Acid Research, 15, 11, 1987; Sproat, Nucleic Acid Research.
15, 12, 1987; Zuckevman, Nucleic Acid Reseach.
13, 5305, 1987; Verheeden, JOREGA
CHEM. 35, 2319, 1970/, хотя этот перечень не является исчерпывающим и любой другой способ, известный исследователям в этой области, может быть применен для получения олигонуклеотидов в ориентации либо 31-51, либо 51-31 для связи с системой твердого носителя данного изобретения.
Если олигонуклеотид имеет защитные группы в результате реакции синтеза, например, бутил/изопропил группы на основаниях /Г, А, Ц/ 31-51-олигонуклеотидов и β-цианоэтил в 51-31-oлигoнyклeoтидax, они должны быть удалены после синтеза. Носитель с олигонуклеотидом /олиготвердый носитель/ можно нагреть до 55oС в течение 2 часов в 38% NH4CH для удаления защитных групп и образования незащищенного олигонуклеотида.
Примерам манипуляции, пригодной для этой системы, является амплификация целевой последовательности нуклеиновой кислотой.
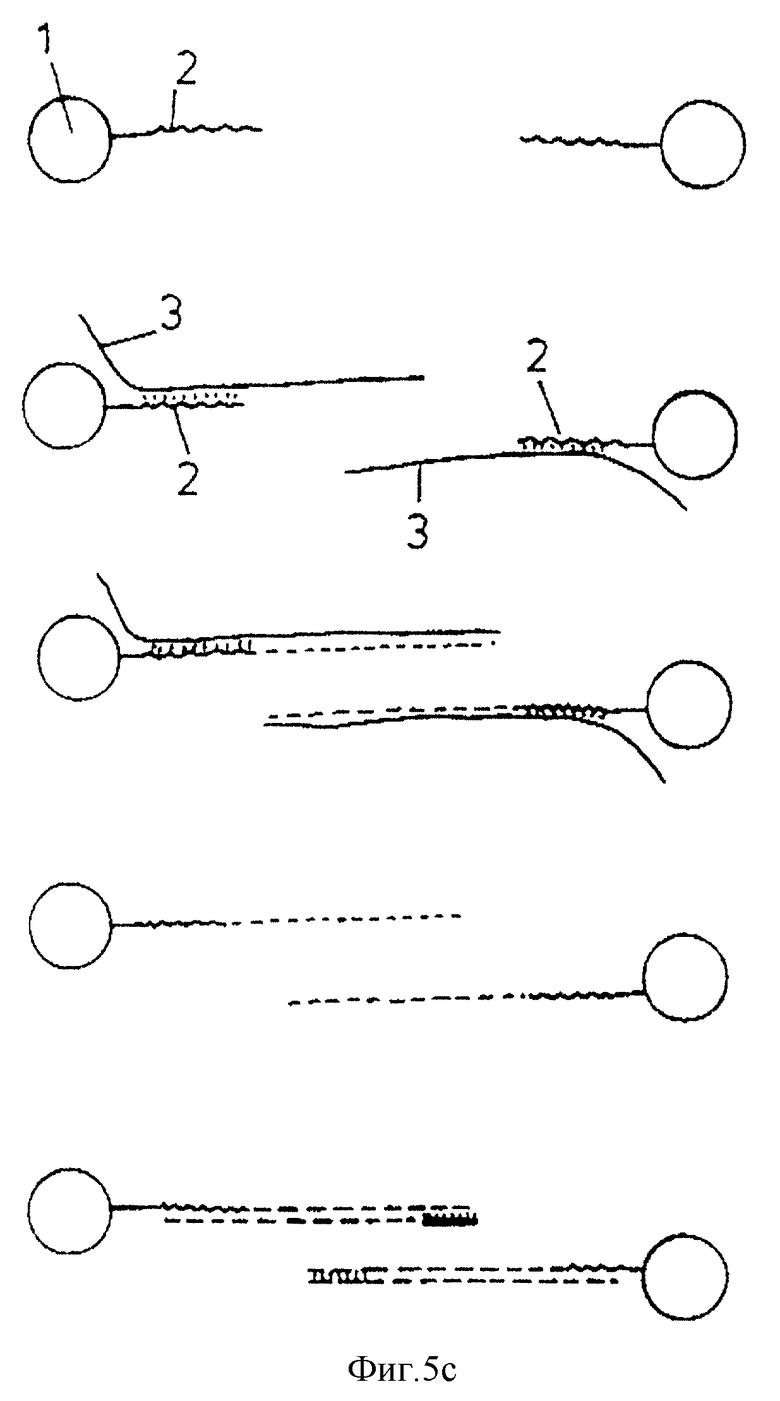
Фиг. 5а-5с схематически изображают стадии, участвующие в амплификации, в зависимости от места праймера /затравки/, ориентации олигонуклеотида /т.е. 31-51 или 51-31/ и, следовательно целевой последовательности нуклеиновой кислоты и присутствии одной или двух олигонуклеотидных последовательностей /т. е. олигонуклеотидов с обратной ориентацией/. Стадии амплификации известны, т. е. добавление праймера, удлинение праймера, добавление второго праймера для обратного копирования копии мишени и т.д.
В варианте, представленном в фиг.5а, 51 - конец олигонуклеотида может быть связан с носителем и манипуляцией может быть амплификация нуклеиновой кислоты-мишени, которая гибридизуется с олигонуклеотидом. В этом случае олигонуклеотид может служить в качестве праймера или отдельный праймер 11 может быть легирован с олигонуклеотидом перед стадией гибридизации. В любом случае синтезируется продукт удлинения праймера 12 /с использованием нуклеиновой кислоты-мишени в качестве матрицы/, начиная с праймера и до конца целевой нуклеиновой кислоты. Таким образом получена последовательность нуклеиновой кислоты, связанная с носителем и комплементарная нуклеиновой кислоте-мишени. Для удобства эту комплементарную последовательность называют здесь "копией мишени".
Затем носитель можно промыть, а гибридизованные цепи нуклеиновой кислоты /т. е. мишень и копию мишени/ можно впоследствии денатурировать при помощи известных способов /например, нагреванием при определенной температуре/, чтобы на твердом носителе осталась в связанном через олигонуклеотид виде только копия мишени. Для удобства твердый носитель со связанным олигонуклеотидом будет называться в дальнейшем "олиготвердый носитель".
Систему носителя можно промыть, а копию мишени, которая остается на нем, можно затем применить для синтеза новых количеств исходной нуклеиновой кислоты -мишени. Такой синтез предусматривает гибридизацию праймера 13 с копией мишени, промывание носителя для удаления негибридизовавшегося праймера, проведение удлинения праймера /с использованием копии мишени в качестве матрицы/ и денатурацию и сбор синтезированной последовательности нуклеиновой кислоты 14. Этот процесс можно повторять столько раз, сколько требуется, до желаемой степени амплификации исходной нуклеиновой кислоты-мишени. Предпочтительно, чтобы в каждой стадии амплификации применяли один и тот же набор молекул копии мишени.
Процедура, изображенная в фиг.5а, пригодна, в частности, для обнаружения присутствия низких количеств нуклеиновой кислоты-мишени в медицинской пробе. Праймер 13 может быть помещен известными способами и операцию детектирования проводят для обнаружения соответственно меченой последовательности нуклеиновой кислоты 14. Детектирование метки является подтверждением того, что нуклеиновая кислота-мишень 3 присутствует в исходной пробе.
В модификации, описанной в фиг.5а, процедура двухцепочечная нуклеиновая кислота может содержать сайт рестрикции /например, обеспеченный праймером, лигированным с олигонуклеотидом/ и для отделения двухцепочечной молекулы от носителя применяют в этом случае подходящую рестриктазу.
В альтернативном варианте изобретения /см. фиг.5/, в котором с носителем связан 31- конец олигонуклеотида, манипуляцией может быть секвенирование гибридизованной нуклеиновой кислоты-мишени при помощи стандартной методологии /например, Sanger DNA sequencing technique/. Так, например 15 может быть гибридизован с нуклеиновой кислотой-мишенью/, которая гибридизована с олигонуклеотидом на носителе/. В этом случае пригоден праймер, который будет гибридизоваться с мишенью при температуре, при которой мишень не отсоединяется плавлением от олигонуклеотида. Реакцию синтеза цепи проводят затем со смесью четырех дезоксинуклеотидов /дАТФ, тТТФ, дТГФ, дЦТФ/ вместе с одним дидезоксинуклеотидом. Синтез идет в направлении 51-31 с праймера в направлении олигонуклеотида.
Реакцию проводят 4 раза с использованием разных дидезоксинуклеотидов каждый раз. Как известно, присутствие дидезоксинуклеотида позволяет получать последовательности нуклеиновых кислот разной длины, как это обозначено прерывистыми линиями. Продукты четырех реакций разделяют параллельно на подходящем геле и последовательности определяют по положению полос на четырех треках. Этот вариант данного изобретения пригоден, в частности, для определения точковых мутаций в нуклеиновой кислоте /например, в случае медицинской пробы/ путем сравнения полученной последовательности с последовательностью контрольной пробы, с которой известно, что она является "нормальной" последовательностью.
Вариант, в котором с носителем связан 31 - конец олигонуклеотида, может быть применен также для амплификации последовательности нуклеиновой кислоты-мишени. В этом случае пригоден праймер, который гибридизуется с кислотой при температуре более низкой, чем температура, при которой мишень отсоединяется плавлением от олигонуклеотида. Удлинение праймера проводят затем при известных условиях в направлении олигонуклеотида. Полученный продукт - копия может быть отсоединен от нуклеиновой кислоты-мишени плавления при условии, в которых нуклеиновая кислота-мишень остается гибридизованной с носителем и может применяться для следующих реакций амплификации.
Во всех описанных выше реакциях амплификации одна и та же матрица остается связанной с олиго-твердым носителем и применяется для последующих реакций амплификации. Это является существенным отличием данного изобретения от способа, описанного в ЕР-А-0200362, т.к. в нем в качестве матриц использовали обе цепи нуклеиновой кислоты-мишени и после первого копирования в качестве матриц для второй и последующих стадий амплификации использовали как цепи мишени, так и цепи-копии. Поэтому, если на какой-либо из стадий происходила ошибка в копировании, эта ошибка копировалась в "цепной реакции".
Данное изобретение избегает этой проблемы либо благодаря тому, что проводят одну стадию копирования и сохраняют полученную копию во всех последующих амплификациях, либо сохраняют исходную последовательность мишени для всех амплификаций. Данное изобретение также более эффективно, чем описанный в ЕР-А-0200362 способ, т.к. в данном изобретении негибридизовавшиеся праймеры и последовательности мишени для копий удаляют из системы промыванием, а в ЕР-А-0200362 негибридизовавшиеся мишень, праймер и продукт - копия присутствуют при проведении всего способа, что делает системы неэффективной.
Еще в одном варианте /см. фиг.5с/ могут быть добавлены два праймера, которые комплементарны различным частям мишени или копии мишени, в ориентации либо 51-31, либо 31-51, и использованы для проверки последовательности на точковые мутации согласно известным способам (ДНК-лигазная реакция отжига (гибридизации)). В таком анализе праймеры обычно являются мечеными и тест применим для быстрой диагностики, например, мутации Р53 опухолевого супрессорного гена, поскольку 75% всех больных с раком толстой кишки имеют точковую делецию в этом гене.
Из описанного выше должно быть понятно, что данный способ отличается от известных способов тем, что твердофазная система и устройство данного изобретения делают возможными большую эффективность и больший выход амплифицированной нуклеиновой кислоты. Это связано с возможностью проведения способа в виде отдельных стадий. Колонку можно промывать после каждой гибридизации для удаления негибридизовавшейся ДНК и р. и "очищать" систему, что значительно повышает эффективность. Компоненты реакционного раствора на колонке, т. е. вновь амплифицированная последовательность-мишень, могут быть отведены в детектор, такой как оптическая ячейка, соединенный с колонкой, и присутствие или отсутствие последовательности - мишени быстро подтверждается. Это, в частности, применимо для диагностики проб, взятых у больных, для определения присутствия любого вызывающего заболевание организма в пробе при помощи быстрого и надежного способа, не доступного ранее.
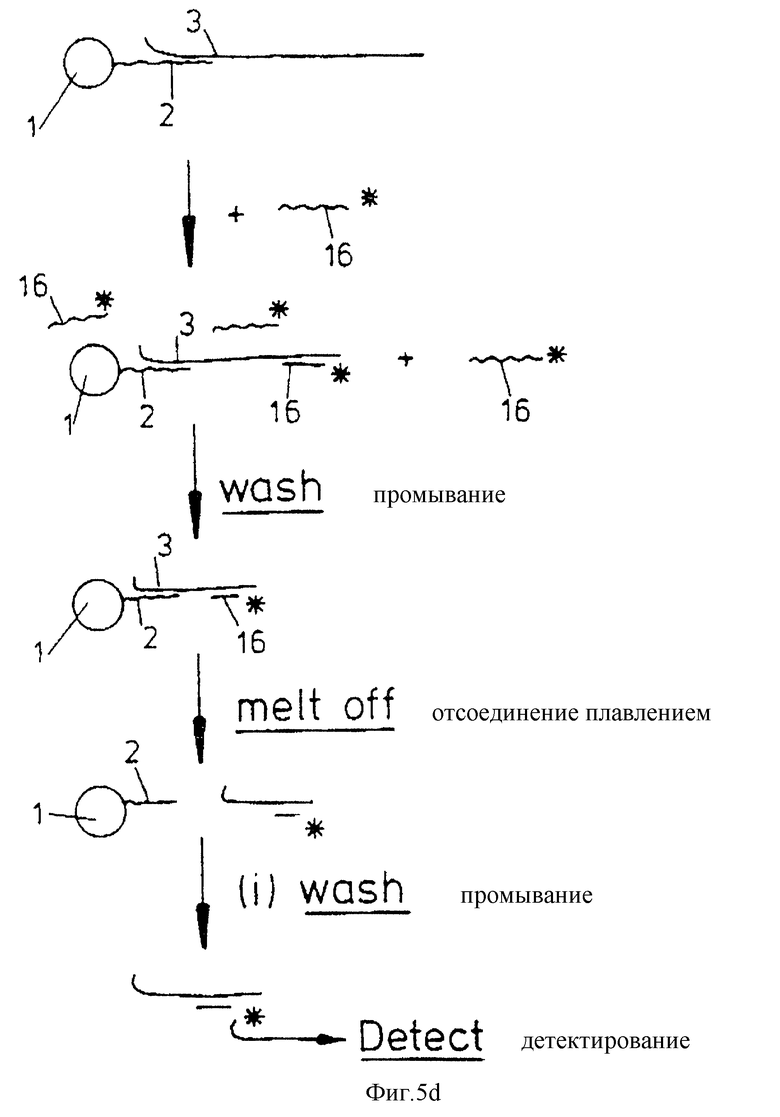
Следующий вариант изобретения /который не касается амплификации/ представлен на фиг.5d. Вариант применим для тестирования проб на присутствие или отсутствие относительно большого количества нуклеиновой кислоты-мишени. В варианте фиг. 5d и системе носителя добавляют меченый праймер, который гибридизуется с нуклеиновой кислотой-мишенью при температуре, более низкой, чем температура, при которой мишень отсоединяется от олигонуклеотида плавлением. Если нуклеиновая кислота-мишень 3 присутствует, то праймер гибридизуется с ней. Затем носитель может быть промыт при температуре, более низкой, чем температура, при которой праймер 6 отсоединяется плавлением от нуклеиновой кислоты-мишени, для удаления негибридизовавшегося праймера 16. В следующей стадии твердый носитель нагревают до температуры, при которой праймер 16 отсоединяется /либо с нуклеиновой кислотой-мишенью, либо без нее/, носитель промывают и элюированный продукт направляют к детектору.
Если метка детектируется, это свидетельствует о присутствии нуклеиновой кислоты-мишени в исходной пробе.
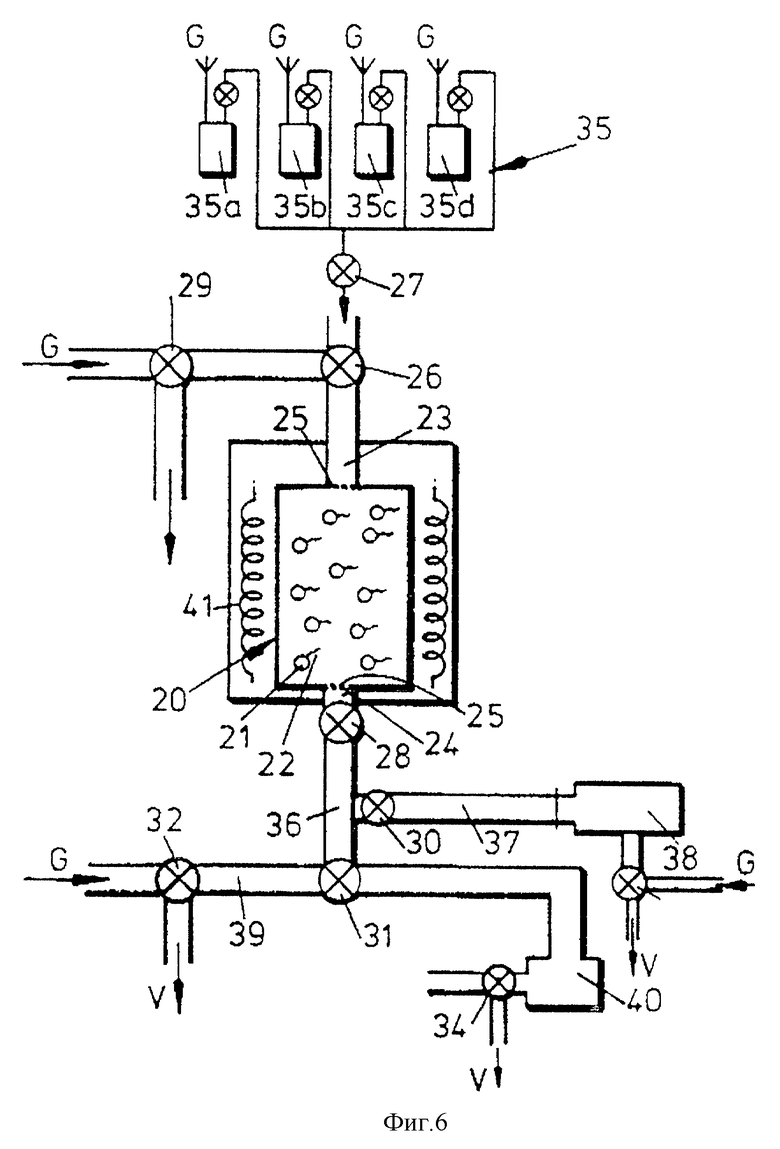
На фиг. 6 изображено устройство, в котором можно проводить гибридизацию нуклеиновой кислоты-мишени с олигонуклеотидом, связаны с носителем, а также реакции амплификации.
Устройство, изображенное на фиг.6, содержит колонку 20 /эквивалентную колонке 6 - фиг.2/, предварительно загруженную частицами носителя 21 со связанным с ними олигонуклеотидом. Обычно колонка 20 имеет объем приблизительно 200 мкл и имеет входное 23 и выходное 24 отверстия, снабженные пористыми удерживающими элементами 25 для удерживания частиц 21 внутри колонки. Устройство имеет клапанное приспособление 26-34, как показано на чертеже, и также обеспечено источником сжатого газа, который может подаваться в соответствии со стрелками G. Газ применяют для перемещения реагентов и продуктов внутри устройства. Некоторые клапаны приспособлены для обеспечения выброса газа, как указано стрелками V.
Клапан 26 делает возможным селективное сообщение внутреннего пространства колонки 20 с узлом подачи реагентов 35, имеющим отдельные сосуды 35а-35, содержащие растворы /например, пробы, праймеры, буферы и т.д./, которые подаются к колонке 20 через открытые клапаны 26 и 27 при помощи давления газа. Клапан 29 можно применять во время этой процедуры для снятия избытка давления газа.
На выходной стороне клапана 28 имеется передаточная зона 36, которая может селективно сообщаться /через клапан 30/ с зоной сбора продукта 37, соединенной с детектором 38, который может быть например, оптическим детектором или детектором для радиоактивной метки. Можно применять также и другие типы детекторов.
Передаточная зона 36 может также селективно сообщаться /через клапан 31/ с удерживающей раствор зоной 39 или приемником отходов 40. Зона сбора раствора 39 обеспечена одной из трубчатых линий устройства и имеет объем, по меньшей мере равный объему колонки 20.
Вокруг колонки обеспечен нагреватель 41, как описано на чертеже.
В устройстве могут применяться, например, тефлоновые клапаны /zero dead Teflon seated valves из Generak Valve, U.S.А/, а трубки, применяемые в устройстве, могут быть тефлоновыми трубками диаметром 1,5 мм.
При использовании устройства пробу вместе с подходящими буферами подают к колонке 20 под давлением газа, как указано ранее. Гибридизацию нуклеиновой кислоты-мишени проводят при подходящей температуре с последующим промыванием колонки раствором, подаваемым из узла 35. Промывные растворы могут проходить к приемнику отходов 40 через клапаны 28 и 31. Клапан 34 может быть открыт во время этой процедуры для сбора избыточного давления.
После этого проводят манипуляцию на нуклеиновой кислоте-мишени, находящейся на носителе. Для этого соответствующие растворы реагентов подают из узла 36 через ряд клапанов /как для подачи жидкости, так и для сброса давления/.
После проведения манипуляции раствор реагентов может проходить из колонки 20 к зоне удерживания раствора 39.
Далее продукт манипуляции отсоединяют плавлением от носителя 21 и направляют его к зоне сбора продукта 37 либо для непосредственного детектирования, либо для хранения.
Если на нуклеиновой кислоте-мишени /или ее копии/, находящейся на носителе, должна проводиться следующая манипуляция, то раствор, удерживаемый в зоне 39, может быть возвращен к колонке 20 под давлением газа через клапан 32. Описанный выше процесс может быть после этого повторен.
Продукт манипуляции может быть собран так часто, как это необходимо, в зоне 37. Во время сбора продукта клапан 33 должен быть поставлен на выброс. Для предотвращения немедленного прохождения продукта к детектору клапан 33 может быть закрыт после короткого периода времени, так что собранный продукт не может проходить достаточно далеко вдоль зоны 37, чтобы достичь детектора 38. Подача газа к детектору 38 позволяет очистить его в случае необходимости.
Описанное выше устройство может быть автоматизировано путем обеспечения электронного контроля для клапанов.
Устройство может иметь одну колонку, как это показано на фиг.6, или может содержать несколько колонок для тестирования нескольких олигонуклеотидных зондов на одной пробе или для тестирования одного олигонуклеотидного конца /например, ВИЧ-1/ на нескольких пробах.
Кроме того, колонка 20 не должна быть фиксированной частью устройства. Можно, например, обеспечить съемные колонки 20, предварительно загруженные твердым носителем с присоединенным к нему олигонуклеотидом, и затем помещать колонку в устройство. Это позволяет избежать необходимости удаления твердого носителя из колонки, которая возникает для фиксированной колонки.
Изобретение будет описано далее не ограничивающими его примерами. Олигонуклеотиды, применяемые в этих примерах /как связанные с колонкой, так и отщепленные от нее пробы праймеров/, были синтезированы по стандартной программе связывания на синтезаторе ДНК Biosearch Model 8500. Для синтеза использовали β-цианоэтил-замещенные фосфоамидиты. Отщепленные от колонки пробы праймеров были полностью деблокированы /все группы ДМГр удалены/ и отщеплялись автоматически при помощи NH4OH от GPG-колонок.
Связанные с колонкой олигонуклеотиды не отщеплялись NH4ОН и конечная группа ДМТр не удалялась, если это не указано.
После синтеза как связанные с колонкой, так и отщепленные олигонуклеотиды переносили в пробирки с завинчивающимися крышками с 38% NH4OH /1 мл/. Пробирки помещали в фиксаторах на 1 час в печь /56oС/ для удаления защитных rpegg/. Затем пробирки и фиксаторы переносили в морозильник /-20oС/ на 10 мин. Супернатант с NH4OH удаляли из связанных с колонкой олигонуклеотидов и выбрасывали. Пробы сушили замораживанием в центрифужном сушителе Sav ant и хранили в эксикаторе. Каждый отщепленный от колонки олигонуклеотид делили на три пробирки и очищали присутствующий неочищенный олигонуклеотид.
Пример 1
Приготовление носителя из силикагеля
1 г кальцинированного носителя Spherisorb GC сушили над P2O5 в колбе с круглым дном с 40 мл безводного толуола. Содержимое колбы перемешивали при помощи магнитной мешалки и образующуюся суспензию нагревали до 90-95oС в масляной бане. Безводный 3-глицидосипропил-триметоксисилан /1,5 мл/ добавляли к суспензии и продолжали реакцию при перемешивании в течение 3 часов при 90-95oС. При этом следили, чтобы температура не превышала 95oС. После трех часов реакционную смесь охлаждали до комнатной температуры, фильтровали и промывали безводным толуолом /40 мл/, метанолом /40 мл/ и эфиром /20 мл/. Содержащий функциональные группы силикагель сушили над Р2O5 и диоксидом кремния в течение ночи.
Пример 2
Синтез замещенного нуклеотидом цитозином носителя
/а/ Получение натриевой соли производного цитозина: 1. диметокситритил-дезоксирибонуклеизида цитозина /ДМТр- дЦ/ сушили над P2O5 и диоксидом кремния в течение нескольких дней. Высушенный ДМТр- дЦ растворяли в безводном N, N-диметилформамиде /ДМФ/ /20 мл/ в атмосфере азота. После завершения растворения добавляли 0,6 г гидрида натрия и проводили реакцию с ДМТр- дЦ в безводном АМФ в течение 20 мин при перемешивании. NaH затем отфильтровывали от реакционной среды.
/а/ Реакция приготовленного силикагеля и производного нуклеозида: высушенный носитель из силикагеля добавляли непосредственно к отфильтрованной натриевой соли нуклеозида и смесь перемешивали с обратным холодильником в течение двух часов. Смесь связанного нуклеозида и носителя охлаждали до комнатной температуры в течение ночи. Замещенный носитель из диоксида кремния фильтровали через фильтр из силицированного матированного стекла и промывали 20 мл каждого из растворителей: безводного ДМФ, безводного толуола, метанола и смеси метанола с эфиром. Затем твердый носитель сушили и хранили в эксикаторе.
51 - кислород сахарной части производного нуклеозида защищали группой ДМТр, которая лабильна в кислоте, и может быть удалена безводными кислотами, такими как дихлоруксусная кислота, с образованием ярко-оранжевого катиона ДМТр. Часть высушенного замещенного носителя обрабатывали дихлоруксусной кислотой /1 мл 2% дихлоруксусной кислоты в дихлорметане/. Ярко-оранжевая окраска свидетельствовала о том, что связывание носителя и производного цитозина было успешным.
Пример 3
Была разработана процедура выделения клона из смеси бактериальной и вирусной ДНК, применимого в качестве тест-системы. Целевая последовательность /клонированный ген/ был помещен внутрь бактериофага М13mр и содержал две следующие последовательности
ГПГ ГГТ ЦПЦ АААА ГГГ ТЦА ГТГ ЦТГ /1 /
МГТ ГТГ ТЦЦ ТТТ ГТЦ ГАТ АЦТГ /2/
М13 mp является стандартным вирусом, применяемым в молекулярной биологии в качестве рутинного секвенирующего вектора.
На носителе, приготовленном в примере 2, был синтезирован олигонуклеотид следующей последовательности, которая гомологична последовательности /1/
ЦГЦ ЦЦА ГГГ ТТТ ФГТ ЦАЦ ГАЦ /3/
В носитель, полученный в примере 2, был включен остаток цитозина на левом конце этой последовательности.
15 мг твердого носителя со связанным с ним олигонуклеотидом помещали в проточную колонку, после чего на колонку наносили смесь E. coli продукт из 10 клеток /и 1 мкг М13 ДНК после денатурации нагреванием. Кислоту выдерживали при температуре (Тп-2)oС, где Тп - температура плавления последовательности /3/, рассчитанная по формуле
Тп=2 (А+Т) +3 (Г+Ц)
где А, Т, Г и Ц обозначают соответственно количество остатков аденина, тимина, гуанина и цитозина в последовательности /3/.
Поскольку колонку поддерживают при температуре слегка ниже температуры плавления /Тп/, вирусная ДНК способна гибридизоваться со связанным олигонуклеотидом.
Колонку поддерживали при (Тп-2)oС и промывали 2,5 мл гибридизационного буфера /10 мМ MgCl2 и 10 мМ Трис-HCl, pH 7,5/. Промывные растворы из колонки собирали в пробирки по фракциям 200 мкл /всего 11 фракций/.
Каждую из собранных фракций подвергали затем стандартной реакции секвенирования /способ Sanger Coulson/ путем добавления 1 кг последовательности /3/ и каждой из пробирок вместе с ДНК-полимеразой /фрагментом Кленова/ и дезоаксирибонуклеотидами и дидезоксирибонуклеотидами в стандартной реакционной смеси.
Реакцию секвенирования проводили также на материале, иммобилизованном на колонке. Связанный с колонкой материал отсоединяли плавлением и собирали. Секвенирование проводили с применением той же самой реакционной смеси, которую применяли в секвенировании фазы раствора.
Продукты реакций секвенирования как связанной с носителем фазы, так и фазы раствора разделяли и проявляли на стандартном 8%-ном полиакриламидном геле с использованием стандартных процедур. Результаты представлены на радиоавтографе фиг. 7, где
Дорожка /трек/ - Объяснение
1 - Последовательность, полученная на связанном с твердым носителем материале после промывания 2,5 мл гибридизационной смеси
2 - Последовательность, полученная для первых 200 мкл
3 - Последовательность, полученная для следующих 200 мкл
4 - И т.д.
5 - "
6 - "
7 - "
8 - "
9 - "
10 - "
11 - "
12 - "
Дорожка 1 показывает, что нуклеиновая кислота-мишень /вирусная ДНК/ действительно была связана с носителем /что подтверждено тем фактом, что праймерная последовательность /3/ была способна гибридизоваться с нуклеиновой кислотой-мишенью и делала возможной осуществление реакции секвенирования/. Дорожки 1-10 также демонстрируют, что не было перекрестной реакции или размытости от ДНК E. coli. Следовательно, вирусная ДНК избирательно удерживалась на носителе. Кроме того, связанная с носителем ДНК - мишень стабильно удерживалась на носителе. Не было стерического препятствия для фрагмента Кленова у самого носителя, т.к. реакции секвенирования проводили на связанной с носителем ДНК.
Дорожки 2-12 представляют прогрессивно уменьшающиеся количества ДНК-мишени /т. е. вирусной ДНК/ в промывных растворах из колонки. Следовательно, к колонке был добавлен избыток вирусной ДНК, который не гибридизовался с олигонуклеотидной последовательностью /3/. Поэтому связывающую способность /емкость/ колонки можно "настрить" на концентрацию нуклеиновой кислоты-мишени.



Способ применим, в частности для анализа медицинских проб. Способ проведения манипуляции последовательностью нуклеиновой кислоты предусматривает обеспечение системы твердого носителя, с которым связан одноцепочечный олигонуклеотид, комплементарный специфической последовательности на нуклеиновой кислоте-мишени, более длинной, чем данный олигонуклеотид, добавление источника одноцепочечной нуклеиновой кислоты-мишени к системе твердого носителя, гибридизацию нуклеиновой кислоты-мишени с олигонуклеотидом и проведение манипуляции на гибридизованной нуклеиновой кислоте-мишени. Манипуляция может представлять собой, например, копирование или амплификацию последовательности нуклеиновой кислоты-мишени. В предпочтительном варианте обеспечен носитель в проточном сосуде, который облегчает промывание носителя для удаления примесей и оставления "чистой" пробы нуклеиновой кислоты-мишени, на которой может быть проведена манипуляция. Предпочтительно носитель имеет силоксановый матрикс, с которым связан олигонуклеотид. Это обеспечивает стабильную связь между олигонуклеотидом и носителем. 6 с. и 41 з.п. ф-лы, 11 ил.
(I) денатурацию нуклеиновой кислоты-мишени от указанной иммобилизованной копии нуклеиновой кислоты-мишени, ковалентно связанной с носителем,
(II) выполнение проточной промывки системы твердого носителя для удаления из нее указанной нуклеиновой кислоты-мишени,
(III) гибридизацию праймера с указанной иммобилизованной копией нуклеиновой кислоты-мишени, ковалентно связанной с носителем, и
(IV) удлинение праймера в 5'-3' направлении назад к указанному твердому носителю при использовании указанной копии нуклеиновой кислоты-мишени в качестве матрицы, посредством чего получают удлиненный продукт праймера.
Приоритет по пунктам:
24.12.1991 по пп.1-8, 11-28, 31-37, 41-46;
08.12.1992 по пп. 9,29,38,40,47;
23.12.1992 по пп.10.30.39.
| Мотальный механизм для крестовой намотки | 1972 |
|
SU455905A1 |
| Пюпитр для работы на пишущих машинах | 1922 |
|
SU86A1 |
| US 4483964, 20.11.1984 | |||
| US 4517338, 14.05.1985 | |||
| US 4598049, 01.07.1986. | |||

