Изобретение относится к области аналитической химии, а именно к идентификации и количественному определению 10-хлор-9,10-дигидрофенарсазина.
К настоящему времени известен ограниченный ряд методов, позволяющих достоверно осуществлять количественный анализ 10-хлор-9,10-дигидрофенарсазина (адамсита, I). Проведение такого вида анализа осуществляется в рамках экологического мониторинга за содержанием (I) в объектах окружающей среды в районе полигона его хранения, расположенного на территории Саратовской области.
Наиболее чувствительными являются методы, основанные на реакциях с колориметрическим окончанием. Один из таких методов основан на образовании окрашенного в интенсивный красный цвет соединения, при действии концентрированной серной кислоты на кристалл испытуемого вещества (или остаток после выпаривания экстракта) (Франке З., Франц П., Варнке В., Химия отравляющих веществ. Т.2., Пер. с нем. М.: Химия, 1973. 406 С.). Данный метод используется для целей индикации (I) и не применим для его количественного определения. Это связано с неоднозначным ходом химической реакции, что вносит большой процент ошибки в процесс анализа. Одновременно некоторые соединения, например бромбензилцианид, в условиях реакции могут также образовывать окрашенные вещества. В литературе описан метод количественного определения (I), основанный на его способности поглощать излучение в УФ-области спектра (Mohler H. et al.//Helv. Chem. Acta. 1940. V. 100. P. 104). Для этих целей использовалась полоса с характерным максимумом поглощения в районе 198 нм и высоким коэффициентом молярного светопоглощения. Чувствительность метода составляет 5•10-4 мг/мл.
К недостаткам данного метода следует отнести то, что, во-первых, используемая полоса проявляется при применении в качестве растворителей соединений, не поглощающих на этой длине волны (предельные углеводороды, например, гексан, гептан и др.). Во-вторых, метод не применим при анализе экстрактов проб, в составе которых могут находиться примеси, имеющие в своей структуре хромофорные группировки, поглощающие в данной УФ-области спектра. В-третьих, метод является не специфичным при анализе проб с одновременным присутствием (I) и 10,10'-окси-бис-9,10-дигидрофенарсазина в связи с их схожими проявлениями в УФ-области спектра.
Метод, описанный в литературе (Bruckner G., Parkany M., Magyar//Kem. Fol. 1962. V.69 Р. 164), основан на реакции получения интенсивно окрашенной в красный цвет натриевой соли продукта нитрования (I). Чувствительность данного метода составляет 0,5-1,0 мкг/мл. Количественное определение (I) по методике, основанной на реакции нитрования затруднено в сложных многокомпонентных смесях. Это обусловлено рядом причин, одной из которых является увеличение оптической плотности пробы в результате побочных, в ходе реакции нитрования, неконтролируемых процессов. Однако данный способ по своему техническому решению наиболее близок заявляемому объекту и поэтому выбран в качестве прототипа.
В настоящее время широкое распространение, для анализа многокомпонентных сложных смесей, получили инструментальные газохроматографические (ГХ) методы анализа. Основным их достоинством является возможность хроматографического разделения компонентов анализируемой смеси с последующей их детекцией. Однако, как показывает практика, анализ (I) ГХ-методами является проблематичным ввиду его низкой летучести. Для проведения ГХ-анализа соединений с низкой летучестью (органические кислоты, амины, амиды) используются различные методы предварительной химической модификации (дериватизации) анализируемых соединений с целью получения хроматографируемых летучих производных.
Задачей настоящего изобретения являлась разработка способа дериватизации (I) и последующего его количественного определения методом газовой хроматографией с атомно-эмиссионным детектированием.
В качестве реакции дериватизации (I) была выбрана реакция получения 9-ацетил-10-хлор-9,10-дигидрофенарсазина (II) (Соборовский Л.Э., Эпштейн Г.Ю., Химия и технология боевых химических веществ. М.: НКОП СССР, 1938. 586 С.).
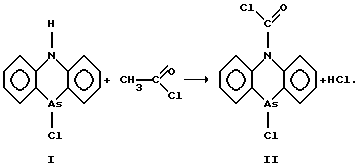
Для решения поставленной задачи исследовали возможность проведения реакции ацилирования (I) в бензоле. Приготовление реакционной смеси осуществлялось следующим способом.
Навеску адамсита растворяли в предварительно приготовленной смеси с концентрацией хлористого ацетила в бензоле 5 моль•л-1.
Реакционную смесь выдерживали в течение 3-х часов при (60±5)oС. Затем раствор охлаждали до (20±5)oС. Анализ содержания ацетильной производной адамсита, N, N'-дифениламина (III) и ацетамид-N,N'-дифенила (IV) осуществлялся методом хромато-масс-спектрометрии. Результаты анализа представлены в табл. 1.
Анализ реакционной смеси показал образование в пробе целевого продукта, массовое содержание которого составило 96-98%, что является приемлемым для использования данной реакции в аналитических целях (Березкин В.Г. Химические методы в газовой хроматографии. М.: Химия, 1980. 256 С.).
Масс-спектр целевого продукта представлен на фиг.1. Наличие пика молекулярного иона с m/z 319 а.е.м., имеющего нечетную массу, свидетельствует о наличии в структуре соединения одного атома азота. Образование ион-фрагмента с m/z 277 а.е.м. свидетельствует об отрыве ацетильного радикала с брутто-формулой [С2Н3О] .. Дальнейшая фрагментация анализируемого соединения протекает аналогично фрагментации (I). Таким образом, полученный масс-спектр подтверждает структуру (II).
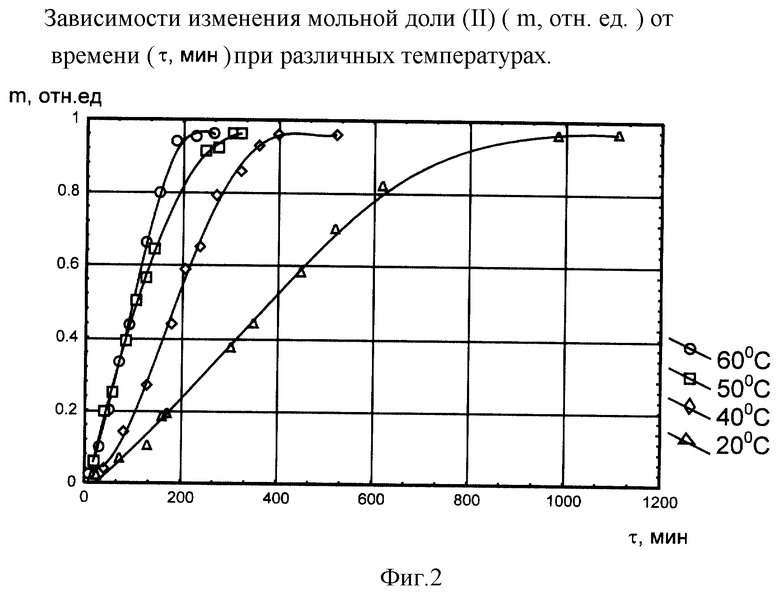
Для оптимизации условий проведения реакции ацилирования (I) проводились исследования времени протекания реакции в интервале температур 20-60oС. Использование температуры проведения реакции выше 60oС не представляется целесообразным. Это связано с испарением реагентов, используемых при приготовлении реакционной смеси (tкип хлористого ацетила составляет 60oС, tкип бензола - 80oС). На фиг. 2 представлены зависимости изменения мольной доли (II) (m, отн.ед) от времени (t, мин) при различных температурах.
Исследование реакции ацилирования (I) проводилось для пяти параллельных измерений при каждой температуре. Как видно из данных, представленных на фиг. 2, нагревание увеличивает скорость реакции ацилирования. По нашему мнению, оптимальное значение температуры реакционной смеси для осуществления реакции ацилирования (I) составляет 60±5oС, при данной температуре время протекания реакции составляет 180±30 мин.
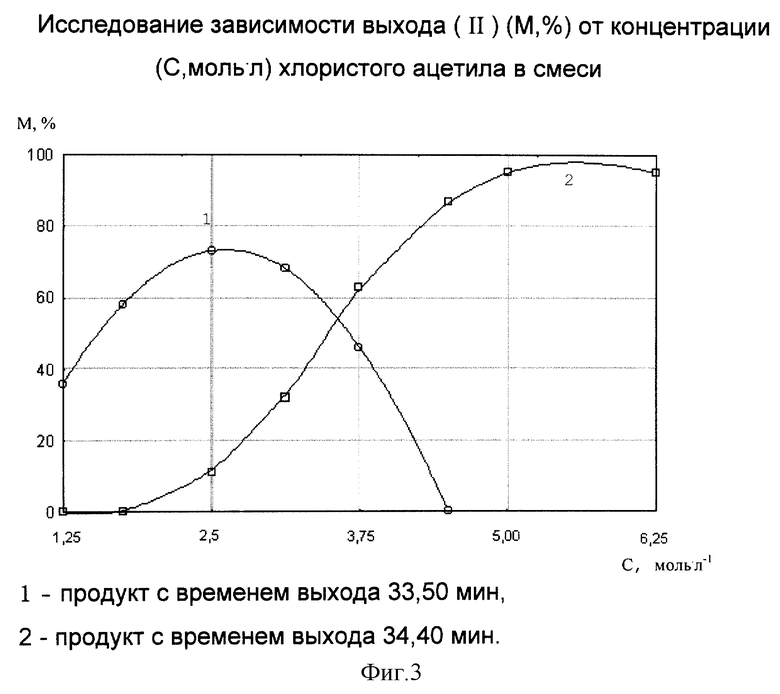
Для оптимизации состава реакционной смеси исследовали выход целевого продукта от концентрации хлористого ацетила. В результате хромато-масс-спектрального анализа проб было зафиксировано образование двух продуктов со схожими масс-спектрами, аналогичными масс-спектру целевого продукта, но различными временем выхода (33,50 и 34,40 мин соответственно). Причем обнаружена зависимость образования этих продуктов от количества хлористого ацетила в реакционной смеси. Результаты исследований представлены на фиг.3. Для рассматриваемой системы, как видно из анализа данных, представленных на фиг.3, получение целевым образом одного из изомеров с выходом продукта реакции 96-98% возможно при использовании реакционной смеси, с концентрацией хлористого ацетила в бензоле не менее 5 моль•л-1.
Использование для приготовления реакционной смеси больших количеств хлористого ацетила является нецелесообразным, так как, во-первых, выход целевого продукта в результате реакции не увеличивается, во-вторых, при увеличении содержания хлористого ацетила в анализируемой смеси наблюдается снижение чувствительности детектирования, которое обусловлено активным воздействием ацилирующего агента на неподвижную жидкую привитую фазу колонки.
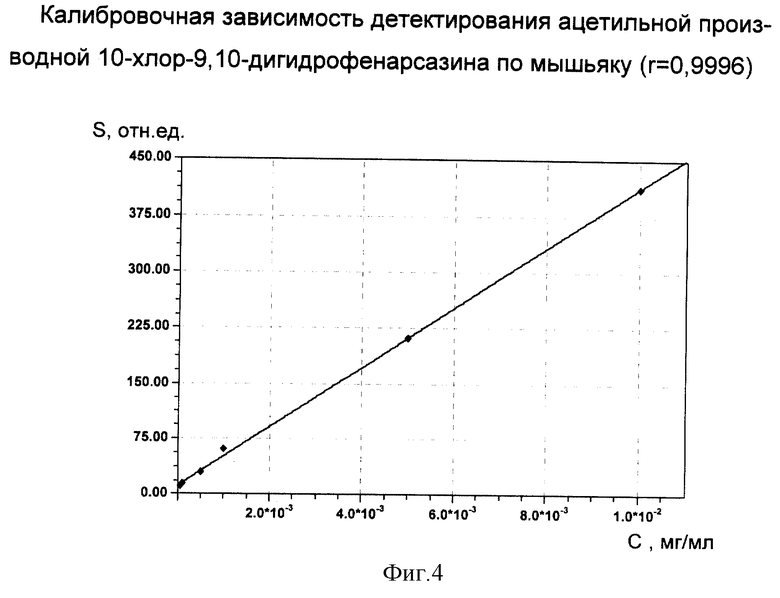
Установление калибровочной зависимости стандартных растворов (I) в реакционной смеси по его ацетильной производной с концентрациями 5•10-5, 1•10-5, 5•10-4, 1•10-3, 5•10-3, 1•10-2 мг/мл проводилось на газовом хроматографе HP 5890 Series II с атомно-эмиссионным детектором HP 5921А фирмы "Hewlett-Packard". Основные параметры хроматографического разделения:
кварцевая капиллярная колонка: типа НР-5 (длина - 25 м, диаметр - 0,32 мм, толщина НПЖФ - 0,17 мкм);
температура испарителя 280oС;
температура детектора 280oС;
режим прогрева хроматографической колонки - программируемый;
начальная температура колонки 60oС;
время выдержки начальной температуры 2 мин;
скорость подъема температуры прогрева колонки 20oС/мин;
конечная температура колонки 250oС;
время выдержки конечной температуры 5 мин;
калибровочная зависимость площади хроматографического пика детектирования ацетильной производной (I) по мышьяку (S, отн.ед.) от концентрации (С, мг/мл) вещества в растворе представлена на фиг.4.
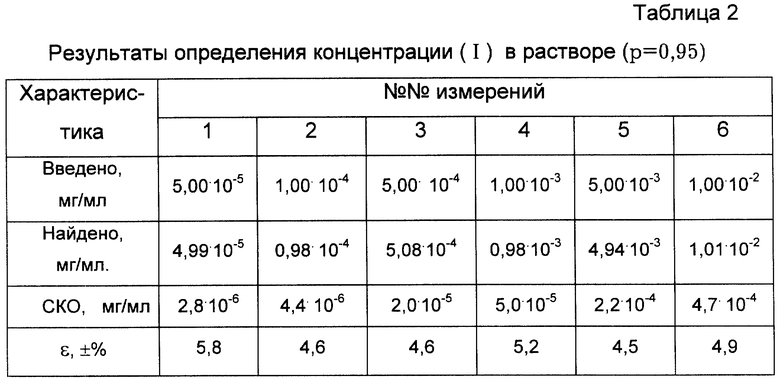
Проверку правильности определения концентрации данным методом проводили методом введено-найдено по 6 измерениям каждой концентрации. В табл.2 представлены результаты экспериментального определения (I) в реакционной смеси.
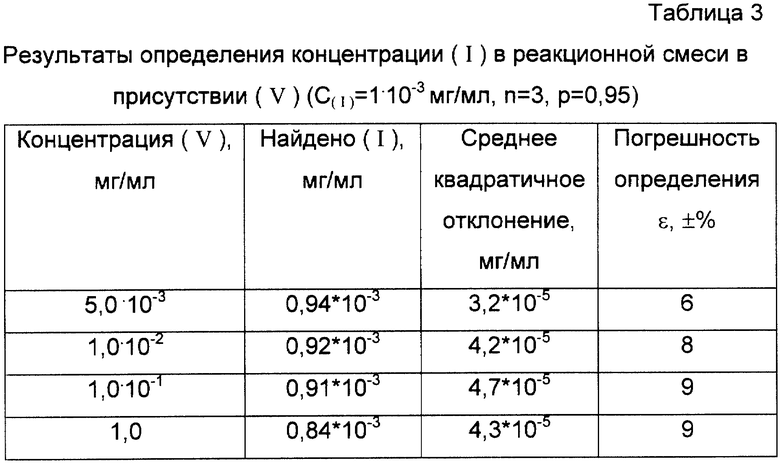
Аналогичные результаты были получены при количественном определении (I) в различных сложных смесях с одновременным присутствием в пробе таких соединений, как (III), 10,10'-оксибис-9,10-дигидрофенарсазин (V).
Одной из наиболее вероятных примесей в техническом адамсите является (V). Близкое строение этих веществ обусловливает определенное сходство их спектральных и химических свойств. Это, в свою очередь, делает невозможным селективное определение адамсита в присутствии (V), например, фотометрическими методами по собственному поглощению или по реакции нитрования. Применение реакционной газовой хроматографии оказывается эффективным для разрешения подобных затруднений. Это связано во-первых, с разным отношением адамсита и (V) к хлористому ацетилу в условиях методики и во-вторых, с возможностью разделения веществ на газохроматографической колонке и последующего детектирования индивидуальных веществ. Для проверки возможности определения адамсита в присутствии (V) с помощью разработанной методики в растворы с известным содержанием (I) добавляли различные количества (V). Результаты определения адамсита в реакционной смеси представлены в табл.3.
Анализ данных, представленных в табл. 3, показал, что в присутствии 1000-кратного избытка (V) (по весу) относительная погрешность определения (I) вблизи нижнего предела определения не превысила 10%.
Таким образом, разработанный способ позволяет вести количественное определение концентрации (I) по его ацетильной производной в присутствии 1000 кратного избытка (V) с относительной погрешностью не более 10%.

Использование: для количественного определения 10-хлор-9,10-дигидрофенарсазина при проведении экологического мониторинга. Технический результат изобретения заключается в разработке высокочувствительного способа количественного определения 10-хлор-9,10-дигидрофенарсазина в растворе бензола. Сущность: используют химическую модификацию анализируемого соединения путем обработки хлористым ацетилом в бензоле при концентрации 5 моль/л при (60±5)oС за интервал времени (2,5±0,5) ч с образованием хроматографируемого производного, регистрируемого с чувствительностью 5•10-5 мг/мл методом газовой хроматографии с атомно-эмиссионным детектированием. 4 ил., 3 табл.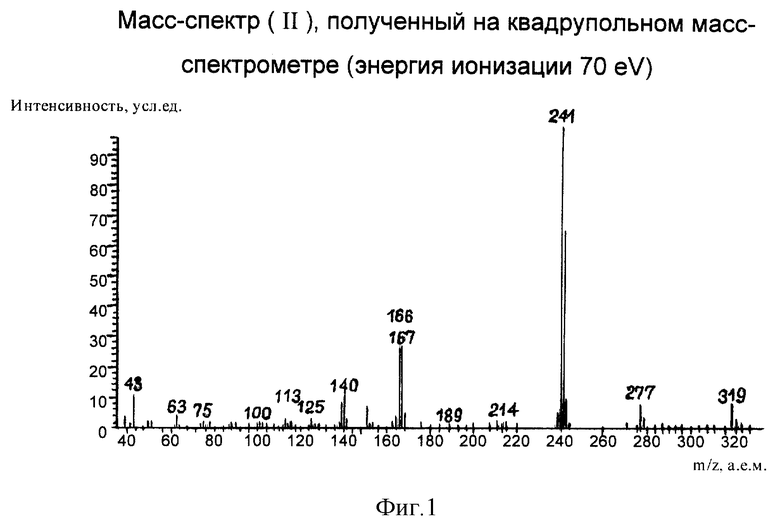
Способ количественного определения 10-хлор-9,10-дигидрофенарсазина в растворе по его производной, отличающийся тем, что используют химическую модификацию анализируемого соединения путем обработки хлористым ацетилом в бензоле при концентрации 5 моль/л при (60±5)oС за интервал времени (2,5±0,5) ч с образованием хроматографируемого производного, регистрируемого с чувствительностью 5•10-5 мг/мл методом газовой хроматографии с атомно-эмиссионным детектированием.
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ДИФЕНИЛАМИНОХЛОРАРСИНА | 1993 |
|
RU2084870C1 |
| RU 95106355 A1, 20.01.1997 | |||
| Франке З., Франц П., Варнке В | |||
| Химия отравляющих веществ | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| - М.: Химия, 1973, 406 с. | |||
