Изобретение относится к технологии получения галоидпроизводных алифатических и алициклических углеводородов, а именно к способу получения хлорциклогексана, используемого в тонком органическом синтезе в качестве растворителя и исходного вещества для получения различных органических соединений, например, циклогексена, дициклогексилдисульфида, капролактама, а также для получения средств защиты растений и фармацевтических препаратов.
Известен способ получения хлорциклогексана с выходом более 95% путем взаимодействия циклогексена с концентрированной соляной кислотой без катализатора и без растворителя при температурах ниже температуры кипения циклогексена, в частности, при 40oС (Заявка ФРГ 19604566, МПК6 С 07 С 23/00, 1997). Недостатком этого способа является использование в качестве исходного субстрата - относительно труднодоступного чистого циклогексена.
Известен способ получения хлорциклогексана с количественным выходом взаимодействием циклогексанола с жидким хлористым водородом при низких температурах (193-253 К) и давлении до 14 атм (Журнал общей химии, 1998, т.68, вып. 12, с.2004). Недостатками этого способа являются использование в качестве реагента жидкого хлористого водорода и связанные с этим технические сложности (поддержание низкой температуры и относительно высокого давления, использование специальной аппаратуры), а также использование большого избытка хлористого водорода.
Наиболее близким по технической сущности является способ получения хлорциклогексана взаимодействием циклогексанола с концентрированной соляной кислотой при 70-80oС в присутствии безводного хлорида кальция, взятого в количестве 50% от массы циклогексанола (Журнал общей химии, 1961, т.31, вып.7, с. 2156). Образующийся органический слой отделяют, промывают холодной серной кислотой, затем несколько раз промывают водой, сушат над безводным хлоридом кальция и перегоняют. Выход хлорциклогексана составляет 90% от теории.
Недостатками этого способа являются относительно невысокий выход целевого продукта, использование значительного количества хлорида кальция и образование большого количества неутилизируемого отработанного солянокислого раствора хлорида кальция.
Задачей предлагаемого изобретения является увеличение выхода целевого продукта, снижение количества используемого хлорида кальция и утилизация отработанного солянокислого раствора хлорида кальция.
Это достигается тем, что взаимодействие циклогексанола с соляной кислотой осуществляют при 40-77oС и атмосферном или избыточном давлении до 0,7 ати в присутствии хлорида кальция, взятого в количестве 10-30% от массы циклогексанола и в присутствии поверхностно-активного вещества ионогенного или неионогенного типа, взятого в количестве 0,04-0,4% от массы исходного циклогексанола и/или в присутствии газообразного хлористого водорода, взятого в количестве 80-140% от количества, теоретически необходимого для полной конверсии циклогексанола, и при использовании соляной кислоты с массовой долей хлористого водорода в пределах 25-36%. Взаимодействие обычно осуществляют при объемном соотношении циклогексанол : соляная кислота в пределах 1:1,5-1: 4. Для снижения продолжительности процесса и достижения высокой конверсии циклогексанола используют соляную кислоту с массовой долей хлористого водорода не менее 33,0%. Образующуюся после разделения фаз водную фазу, представляющую собой раствор хлорида кальция в соляной кислоте с массовой долей хлористого водорода в пределах 25-36%, повторно используют в процессе в качестве исходной соляной кислоты.
В качестве ПAB ионогенного типа используют четвертичную аммониевую соль (ЧАС), выбранную из группы: триметилбензиламмоний хлорид, триэтилбензиламмоний хлорид, трибутилбензиламмоний хлорид, тетрабутиламмоний бромид, триоктилбензиламмоний хлорид, или их смеси. В качестве ПАВ неионогенного типа используют соединение, выбранное из группы: полиэтиленгликоль, поливиниловый спирт, оксиэтилированные алкил- или диалкилфенолы, например, вспомогательные вещества ОП-7 или ОП-10 (смеси полиоксиэтиленалкил- и диалкилфениловых эфиров с числом оксиэтильных групп 7-9 и 10-12 соответственно).
Оптимальными концентрациями хлористого водорода (НСl) в соляной кислоте являются концентрации не менее 33,0 мас.%, что обеспечивает высокую конверсию (более 98%) исходного циклогексанола и сокращает продолжительность взаимодействия. Оптимальными концентрациями НС1 в соляной кислоте при проведении синтеза в присутствии газообразного хлористого водорода являются концентрации в пределах 25-36%, что обеспечивает стабильно высокую конверсию циклогексанола и почти полную утилизацию образующегося после деления фаз раствора хлорида кальция в соляной кислоте. Этот же эффект достигается и путем использования газообразного хлористого водорода в количестве 80-140% от теоретически необходимого для полной конверсии циклогексанола. Использование больших количеств хлористого водорода приводит к увеличению его проскока через реакционную смесь и вызывает необходимость утилизации абгазов. Объемные соотношения циклогексанола и соляной кислоты в пределах 1:1,5-1:4 являются оптимальными, поскольку при иных соотношениях увеличивается продолжительность синтеза и снижается конверсия циклогексанола, или снижается производительность технологического оборудования (при увеличении количества соляной кислоты).
Использование ПАВ в количествах меньших, чем 0,04% от массы циклогексанола, не приводит к ожидаемому эффекту, а использование количеств ПАВ больших, чем 0,4%, технически нецелесообразно.
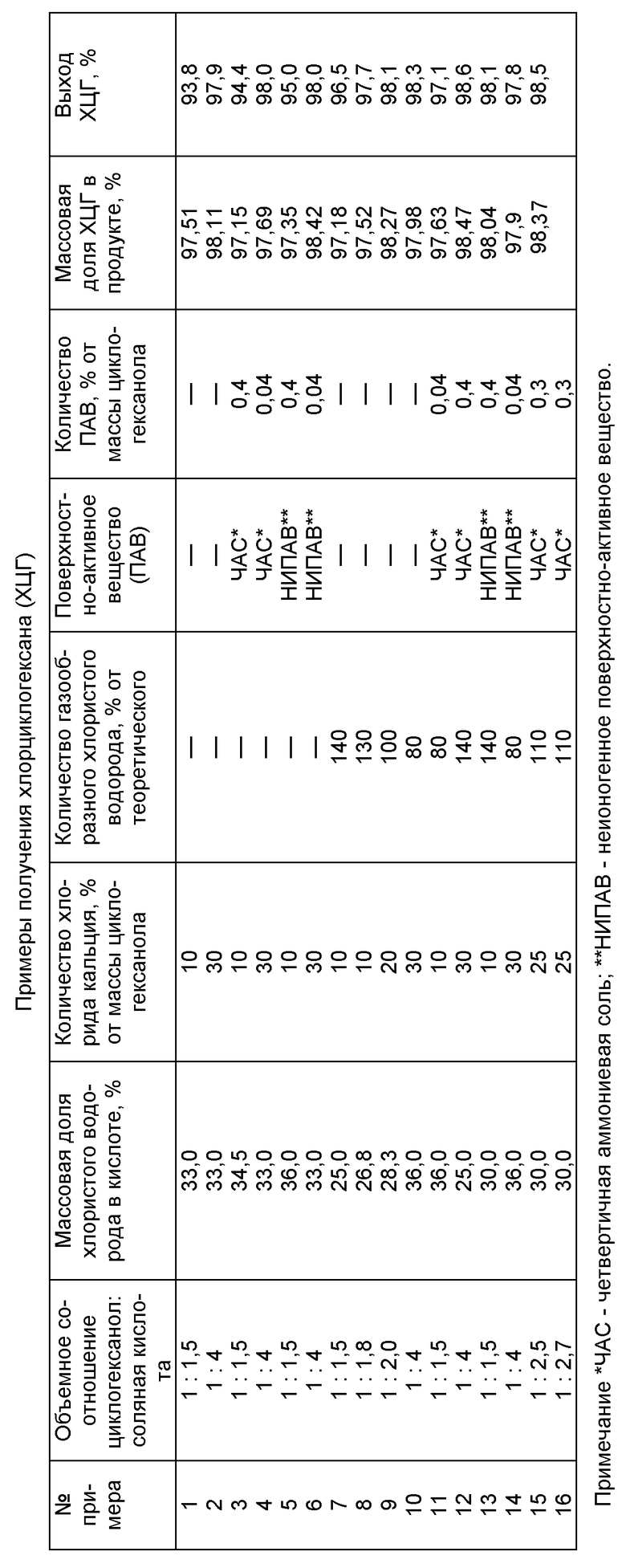
Предлагаемый способ иллюстрируется следующими примерами.
Пример 1 (типовая методика).
В реактор, снабженный термометром, механической мешалкой и обратным холодильником с газоотводной трубкой, помещают 50,92 г (52,9 см3, 0,4992 моля) циклогексанола с массовой долей (м. д.) основного вещества 98,2%, 79,4 см3 соляной кислоты с м.д. НСl 33.0% и 5,0 г хлорида кальция. Смесь перемешивают при температуре 40-77oС при атмосферном или избыточном давлении до 0,7 ати в течение 6-10 часов, затем охлаждают и отделяют органическую фазу от водной. Полученный сырец хлорциклогексана (ХЦГ) промывают водой и 5-10%-ным раствором хлорида натрия или 3-5%-ным раствором карбоната натрия, сушат над безводным хлоридом кальция и получают 56,96 г продукта с м. д. ХЦГ 97,51%. Выход ХЦГ составляет 93,8% от теоретического. Продукт с более высоким содержанием основного вещества получают дистилляцией при атмосферном давлении.
Пример 2.
Синтез проводят по методике примера 1, исходя из 50,92 г циклогексанола, 211,6 см3 соляной кислоты с м. д. НСl 33,0% и 15,0 г хлорида кальция. Продолжительность синтеза - 6 часов. Получают 59,08 г продукта с м. д. ХЦГ 98,11%. Выход ХЦГ составляет 97,9%.
Пример 3.
Синтез проводят по методике примера 1, исходя из 50,92 г циклогексанола, 79,4 см3 соляной кислоты с м. д. НСl 34,5%, 5,0 г хлорида кальция и 0,2 г ПАВ ионогенного типа, выбранного из группы четвертичных аммониевых солей: триметилбензиламмоний хлорид, триэтилбензиламмоний хлорид, трибутилбензиламмоний хлорид, тетрабутиламмоний бромид, триоктилбензиламмоний хлорид или их смеси. Продолжительность синтеза 6-8 часов. Органическую фазу отделяют и обрабатывают, как описано в примере 1. Получают 57,53 г продукта с м. д. ХЦГ - не менее 97,15%. Выход ХЦГ - 94,4%.
Пример 4.
Синтез проводят по методике примера 1, исходя из 50,92 г циклогексанола, 211,6 см3 соляной кислоты с м. д. НСl - 33,0%, 15,0 г хлорида кальция и 0,02 г ЧАС, выбранной из группы, приведенной в примере 3. Продолжительность взаимодействия - 6-7 часов. Получают 59,40 г продукта с м. д. ХЦГ - 97,69%. Выход ХЦГ - 98,0%.
Пример 5.
Синтез проводят по методике примера 1, исходя из 50,92 г циклогексанола, 79,4 см3 соляной кислоты с м. д. НСl - 36,0%, 5,0 г хлорида кальция и 0,2 г ПАВ неионогенного типа, выбранного из группы: полиэтиленгликоль, поливиниловый спирт, оксиэтилированные алкил- и диалкилфенолы, например, вспомогательные вещества ОП-7 или ОП-10. Продолжительность синтеза 6-7 часов. Получают 57,78 г продукта с м. д. ХЦГ - 97,35%. Выход ХЦГ - 95,0%.
Пример 6.
Синтез проводят по методике примера 1 при избыточном давлении до 0,7 ати, исходя из 50,92 г циклогексанола, 211,6 см3 соляной кислоты с м. д. НСl - 33,0%, 15,0 г хлорида кальция и 0,02 г ПАВ неионогенного типа, выбранного из группы, приведенной в примере 5. Продолжительность синтеза - 6-7 часов. Получают 58,96 г продукта с м. д. ХЦГ - 98,42%. Выход ХЦГ - 98,0%.
Пример 7 (типовая методика).
В реактор, снабженный термометром, обратным холодильником с газоотводной трубкой, механической мешалкой и газоподводящей трубкой с барботером, помещают 50,92 г циклогексанола, 79,4 см3 соляной кислоты с массовой долей НСl - 25,0% и 5,0 г хлорида кальция. При перемешивании и температуре в пределах 40-77oС при атмосферном или избыточном давлении до 0,7 ати в смесь через барботер подают 25,48 г (0,6988 моля или 140% от теоретически необходимого для полной конверсии циклогексанола) газообразного хлористого водорода в течение 6-8 часов. Затем смесь охлаждают, отделяют органическую фазу, обрабатывают ее, как описано в примере 1, и получают 58,8 г продукта с м. д. ХЦГ - 97,18%. Выход ХЦГ - 96,5%. Отделенный от органической фазы раствор хлорида кальция в соляной кислоте с м. д. НСl 26,8% в количестве 110,5 г используют непосредственно в следующей операции синтеза (пример 8).
Другие примеры, иллюстрирующие предлагаемый способ, приведены в таблице. Количество исходного циклогексанола с м. д. основного вещества 98,2% во всех примерах составляет 50,92 г. Примеры 8-15 проводят по методике примера 7 с использованием образующихся после деления фаз растворов хлорида кальция в соляной кислоте на стадии взаимодействия в последующих синтезах. Количество используемого в примерах 8-15 газообразного хлористого водорода составляет 14,56-25,48 г (0,3993-0,6988 моля) или 80-140% от теоретически необходимого для полной конверсии циклогексанола. В примерах 11, 12, 15 и 16 в качестве ЧАС используют соли, выбранные из группы, приведенной в примере 3. В примерах 13 и 14 в качестве неионогенного ПАВ используют соединение, выбранное из группы, приведенной в примере 5.
Таким образом, из представленных примеров следует, что предлагаемый способ позволяет увеличить выход хлорциклогексана на 3,8-8,6%, снизить количество используемого в процессе хлорида кальция в 1,7-5 раз и почти полностью утилизировать отработанный раствор хлорида кальция в соляной кислоте.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ДИЦИКЛОГЕКСИЛДИСУЛЬФИДА | 2001 |
|
RU2187501C1 |
| СПОСОБ ПОЛУЧЕНИЯ БЕНЗАЛЬДЕГИДА | 2000 |
|
RU2180329C2 |
| МОДИФИКАТОР ДЛЯ РЕЗИНОВЫХ СМЕСЕЙ (ВАРИАНТЫ) И СПОСОБ ЕГО ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 2001 |
|
RU2213108C2 |
| СПОСОБ ПОЛУЧЕНИЯ АНТРАНИЛОВОЙ КИСЛОТЫ | 2001 |
|
RU2187496C1 |
| СПОСОБ ПОЛУЧЕНИЯ ХЛОРИСТОГО БЕНЗИЛА | 1997 |
|
RU2125035C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИМЕРНОЙ СЕРЫ | 1998 |
|
RU2142406C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДИМЕРОВ АЛКИЛКЕТЕНОВ | 1999 |
|
RU2156761C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРА КИСЛОТНОЙ КОРРОЗИИ | 1998 |
|
RU2164551C2 |
| СПОСОБ ПОЛУЧЕНИЯ ХЛОРМЕТИЛА | 1993 |
|
RU2070188C1 |
| СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКИХ ХЛОРСОДЕРЖАЩИХ СОЕДИНЕНИЙ | 1998 |
|
RU2152381C1 |
Изобретение относится к способу получения хлорциклогексана, используемого для получения циклогексана, а также в качестве полупродукта в синтезе средств защиты растений и фармацевтических препаратов. Хлорциклогексан получают взаимодействием циклогексанола с концентрированной соляной кислотой при 40-77oС в присутствии хлорида кальция в количестве 10-30% от массы циклогексанола при атмосферном или избыточном давлении до 0,7 ати. Объемное соотношение циклогексанол : соляная кислота равно 1:1,5-1:4. Используют соляную кислоту с массовой долей хлористого водорода не менее 33,0% либо используют соляную кислоту с массовой долей хлористого водорода 25-36% и при этом процесс ведут в присутствии газообразного хлористого водорода, взятого в количестве 80-140% от теоретически необходимого количества, для полной конверсии циклогексанола, и/или в присутствии поверхностно-активных веществ ионогенного или неионогенного типа, взятых в количестве 0,04-0,4% от массы циклогексанола. В качестве поверхностно-активных веществ используют вещества, обладающие свойствами катализа межфазного переноса. Технический результат - повышение выхода конечного продукта, одновременная утилизация отходов процесса хлорирования. 5 з.п. ф-лы, 1 табл.
| СИДОРОВ Н.Г | |||
| Журнал общей химии, 1961, т.31, вып.7, с.2156 | |||
| VOGEL A.I | |||
| J | |||
| Chem | |||
| Societe, 1948, v.31, № 6, p.1809-1813 | |||
| DE 19604566 А1, 14.08.1997 | |||
| US 3689577 A, 05.09.1972. |

