Область техники, к которой относится изобретение
Изобретение относится к новым галогенгидринам, сульфонам, триенам, которые являются полезными промежуточными соединениями для получения витамина А, и к способам получения промежуточных соединений и витамина А.
Краткое изложение сущности изобретения
Настоящее изобретение относится к
1. Соединению формулы [I]: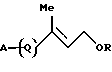
где R представляет атом водорода или защитную группу для гидроксильной группы; и
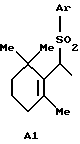
А представляет атом водорода, атом галогена или группу формулы А1: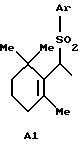
где Аr представляет арильную группу, которая может быть замещенной; и
когда А представляет A1, Q представляет Q1, Q2 или Q3: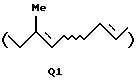

или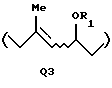
где R1 представляет атом водорода или защитную группу для гидроксильной группы; и
когда А представляет атом галогена, Q представляет Q3, определенную выше, или Q4: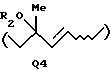
где R2 представляет атом водорода или защитную группу для гидроксильной группы; и когда А представляет атом водорода, Q представляет Q2;
2. Способу получения по крайней мере одного соединения, выбранного из группы, состоящей из соединения формулы [IIа]: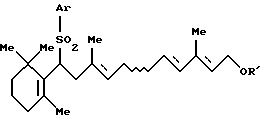
и соединения формулы [IIb]: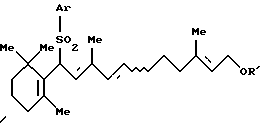
где Аr представляет арильную группу, которая может быть замещенной, и R' представляет защитную группу для гидроксильной группы, который включает дегидрирование соединения формулы [III]: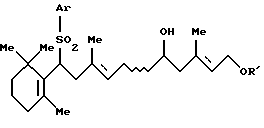
где Аr и R' - такие, как определены выше, в присутствии кислотного катализатора (далее способ называется "Способ А");
3. Способу получения соединения формулы [III]: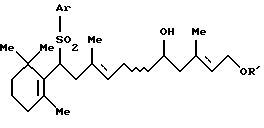
где Аr представляет арильную группу, которая может быть замещенной, и R' представляет защитную группу для гидроксильной группы, который включает взаимодействие соединения формулы [IV]: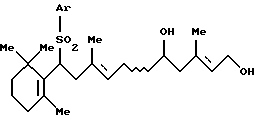
где Ar - такой, как определен выше, с защитным агентом в присутствии основания и катализатора фазового переноса (далее называется "Способ В");
4. Способу получения соединения формулы [V]: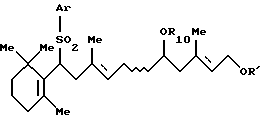
где Ar представляет арильную группу, которая может быть замещенной, R10 и R' одинаковые или разные и представляют защитную группу для гидроксильной группы, который включает взаимодействие сульфона формулы [VI]:
где Ar - такой, как определен выше, с галогенгидрином формулы [VII]: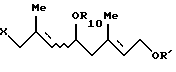
где R10 и R' - такие, как определены выше, и Х представляет атом галогена, в присутствии основания (далее этот способ называется "Способ С");
5. Способу получения галогенгидрина формулы [VII'];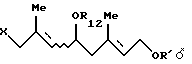
где R12 представляет ацильную группу, R' представляет защитную группу для гидроксильной группы и Х представляет атом галогена, который включает взаимодействие по крайней мере одного галогенгидрина, выбранного из группы, состоящей из соединения формулы [VII'а]: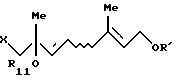
и соединения формулы [VII'b]:
где R11 представляет ацильную группу или атом водорода и R' представляет защитную группу для гидроксильной группы, с карбоновой кислотой формулы:
R12OH
где R12 - такой, как определен выше, в присутствии сильного кислотного катализатора (далее называется "Способ D");
6. Способу получения по крайней мере одного галогенгидрина, выбранного из группы, состоящей из соединения формулы [VII'a]: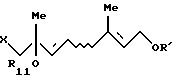
и соединения формулы [VII'b]: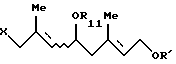
где R11 представляет ацильную группу или атом водорода и R' представляет защитную группу для гидроксильной группы, который включает взаимодействие триена формулы [VIII]: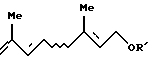
где R' - такой, как определен выше, с галогенирующим агентом и соединением формулы:
R11OH
где R11 - такой как определен выше (далее называется "Способ Е")
7. Способу получения триена формулы [VIII], определенного выше, который включает взаимодействие соединения формулы [IX]: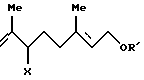
где Х представляет атом галогена и R' представляет защитную группу для гидроксильной группы, с основанием в присутствии палладиевого катализатора, фосфинового лиганда и межфазного катализатора (далее называется "Способ F"); и
8. Способу получения ретинола формулы [X]: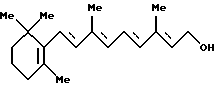
который включает взаимодействие соединения, выбранного из группы, состоящей из соединения формулы [IIa] и соединения формулы [IIb], таких, как определены выше, с основанием (далее называется "Способ G").
Описание предпочтительных вариантов изобретения
Указанное выше соединение формулы I включает:
Соединение [IIа]: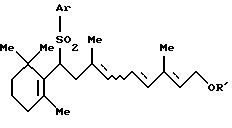
Соединение [IIb]: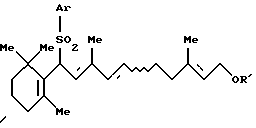
Соединение [V]: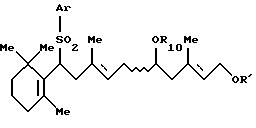
Соединение [VII]: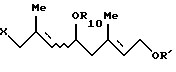
Соединение [VII'а]: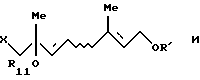
Соединение [VIII]:
В данном описании химическая связь, обозначенная как  , означает, что соединение, имеющее эту связь, включает Е-изомер или Z-изомер или оба изомера относительно двойной связи, связанной с указанной связью, и указанное выше соединение [I] имеет оптически активный изомер и его рацемат вследствие присутствия в соединении асимметрического углеродного атома, что может быть использовано в нижеследующих способах.
, означает, что соединение, имеющее эту связь, включает Е-изомер или Z-изомер или оба изомера относительно двойной связи, связанной с указанной связью, и указанное выше соединение [I] имеет оптически активный изомер и его рацемат вследствие присутствия в соединении асимметрического углеродного атома, что может быть использовано в нижеследующих способах.
Примеры группы Аr, которая может быть замещенной в вышеуказанных формулах, включают фенильную и нафтильную группы, которые могут быть замещенными.
Примеры заместителя включают по крайней мере один заместитель, выбранный из (C1-C5) алкильной группы (например, метила, этила, н-пропила, изопропила, н-бутила, трет-бутила, втор-бутила, н-пентила), (C1-C5)алкоксигруппы (например, метокси, этокси, н-пропокси, изопропокси, н-бутокси, трет-бутокси, втор-бутокси, н-пентокси), атома галогена (например, атома фтора, хлора, брома или йода) и нитрогруппы и подобных.
Конкретные примеры группы Аr, которая может быть замещенной, включают фенил, нафтил, о-толил, м-толил, п-толил, о-метоксифенил, м-метоксифенил, п-метоксифенил, о-хлорфенил, м-хлорфенил, п-хлорфенил, о-бромфенил, м-бромфенил, п-бромфенил, о-иодфенил, м-иодфенил, п-иодфенил, о-фторфенил, м-фторфенил, п-фторфенил, о-нитрофенил, м-нитрофенил, п-нитрофенил и подобные.
Примеры защитной группы для гидроксильной группы для R, R', R1, r2 и R10 включают:
ацильную группу (алифатическую или ароматическую ацильную группу, которая может быть замещенной), такую как ацетил, пивалоил, бензоил и п-нитробензоил;
силильную группу, такую как триметилсилил, трет-бутил-диметилсилил и трет-бутилдифенилсилил;
алкоксиметильную группу, такую как тетрагидрофуранил, тетрагидропиранил, метоксиметил, метоксиэтоксиметил и 1-этоксиэтил;
бензильную группу, которая может быть замещенной, такую как бензильная группа, п-метоксибензильная группа и тритильная группа;
(C1-С6)низшую алкильную группу, такую как трет-бутильная группа, метильная группа;
2,2,2-трихлорэтоксикарбонильную группу;
аллилоксикарбонильную группу и подобные.
Описанная выше ацильная группа может также включать такие группы, как определенные ниже для R11 и R12. Описанные выше силильная группа, алкоксиметильная группа и бензильная группа могут также включать группы, определенные ниже для R12.
Далее дано описание каждого из способов A-F получения указанного выше соединения [I] и способа G получения витамина А.
Способ А
По крайней мере одно соединение, выбранное из группы, состоящей из соединения формулы [IIа] и соединения формулы [IIb], можно получить способом, который включает дегидрирование определенного выше соединения формулы [III] в присутствии кислотного катализатора.
Примеры кислотного катализатора включают кислоту Льюиса, протонную кислоту, гетерополикислоту, ионообменную смолу и хлорангидрид кислоты.
Конкретные примеры кислоты Льюиса включают тетрахлорид титана, хлорид цинка, эфират трифторида бора, трифлат редкоземельного металла и подобные.
Протонная кислота может включать минеральную кислоту, такую как бромоводородная кислота, хлороводородная кислота и серная кислота, карбоновую кислоту, такую как бензойная кислота, уксусная кислота или трифторуксусная кислота, органическую сульфоновую кислоту, такую как метансульфоновая кислота или п-толуолсульфоновая кислота, или подобные.
Предпочтительным примером ионообменной смолы является смола сильно кислотного типа, которая имеет на конце сульфокислотную группу.
Примеры хлорангидрида кислоты включают тионилхлорид, оксихлорид фосфора и подобные.
Количество используемого кислотного катализатора составляет предпочтительно 0,1-1 моль на моль соединения [III] или 0,1-1 весовую часть на 1 весовую часть соединения [III].
В вышеуказанной реакции обычно используют органический растворитель. Примеры растворителя включают углеводородный растворитель, такой как н-гексан, циклогексан, н-пентан, толуол и ксилол;
эфирный растворитель, такой как диэтиловый эфир, тетрагидрофуран, диметоксиэтан и анизол;
галогенированный растворитель, такой как хлороформ, дихлорметан, 1,2-дихлорэтан, монохлорбензол и о-дихлорбензол; и
апротонный полярный растворитель, такой как N,N-диметилформамид, диметилсульфоксид, N,N-диметилацетамид, гексаметилфосфорный триамид.
Температура реакции обычно находится в пределах от -78oС до температуры кипения используемого растворителя и предпочтительно от -10oС до 50oС.
После завершения реакции соединение [IIа] или [IIb] может быть выделено с помощью традиционной последующей обработки. Полученное соединение может, при необходимости, быть очищено методом хроматографии на силикагеле или подобным методом.
Затем соединение, полученное путем вышеописанной реакции дегидрирования, можно освободить от защиты с получением спирта, проведя обычную реакцию удаления защиты так, как описано в Protective Groups in Organic Synthesis, Greene and Wuts, 2-ое издание, (1992), John Wiley & Sons, Inc., полное описание которого включено в данное описание в качестве ссылки.
Например, когда защитной группой является ацильная группа, удаление защиты можно обычно проводить взаимодействием соединения с основанием.
В качестве основания можно использовать алкоксид щелочного или щелочноземельного металла или подобные. Количество используемого основания обычно составляет 1 эквивалент или более по отношению к количеству соединения формулы [III].
В качестве растворителя для реакции можно использовать N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид, тетрагидрофуран, спирт, смешанный растворитель из спирта и воды, смешанный растворитель из тетрагидрофурана, воды и подобные.
Реакцию обычно проводят при температуре в пределах от 0oС до температуры кипения используемого в реакции растворителя.
Когда защитная группа представляет собой силильную группу или подобную, удаление защитной группы может быть проведено взаимодействием соединения с тетра-н-бутиламмоний-фторидом.
Когда защитной группой является 2,2,2-трихлорэтоксикарбонил, может быть проведено восстановительное удаление защиты с использованием цинковой пыли и уксусной кислоты.
Способ В
Соединение формулы [III] можно получить введением защитной группы в первичный спирт соединения [IV].
Введение защитной группы в первичную спиртовую группу соединения формулы [IV] обычно проводят реакцией соединения формулы [IV] с защитным агентом в присутствии основания и катализатора фазового переноса.
Защитный агент означает группу, состоящую из защитной группы и уходящей группы (например, активного атома галогена или ацилоксигруппы), и включает ацилгалогенид, бензилгалогенид, который может быть замещенным, алкоксиметилгалогенид, силилгалогенид и ангидрид кислоты.
Например, защитный агент включает соединение формулы R'Y, где R' представляет ацильную группу, алкоксиметильную группу, бензильную группу, которая может быть замещенной, или силильную группу, которая может быть замещенной тремя группами, выбранными из фенила и (C1-C6)низшей алкильной группы, и Y представляет атом галогена, такой как хлор, бром и йод, причем, когда R' представляет ацильную группу, то Y может представлять ацилоксигруппу, соответствующую ацильной группе, определенной выше для R'.
Ацильная группа может быть алифатической или ароматической ацильной группой, которая может быть замещенной, и может также включать те группы, которые определены ниже для R12. Конкретные ее примеры включают ацетил, пивалоил, бензоил, п-нитробензоил, 2,2,2-трихлорэтоксикарбонил и аллилоксикарбонил.
Примеры алкоксиметильной группы включают метоксиметил и метоксиэтоксиметил.
Примеры бензильной группы, которая может быть замещенной, включают бензильную группу, п-метоксибензильную группу и тритильную группу.
Примеры силильной группы включают триметилсилил, трет-бутилдиметилсилил, трет-бутилдифенилсилил и тому подобное.
Среди них предпочтительным является ацилгалогенид. А особенно предпочтительным является ацетилхлорид.
Примеры ангидрида кислоты включают уксусный ангидрид, пропионовый ангидрид, масляный ангидрид и подобные, причем предпочтительно используют уксусный ангидрид.
Защитный агент обычно используют в количестве примерно 0,1-1,1 моль на моль соединения формулы [IV].
В этой реакции в качестве основания используют органическое или неорганическое основание, предпочтительно неорганическое основание.
Примеры органического основания включают пиридин, 4-диметиламинопиридин, 3-этил-4-метилпиридин, 5-этил-2-метилпи-ридин, имидазол, 2-метилимидазол, 3-метилимидазол, 2-этил-4-метилимидазол, DBU, триметиламин, триэтиламин, диметилэтиламин, метилдиэтиламин, диизопропилэтиламин, трет-бутилдиметиламин и подобные.
Примеры неорганического основания включают гидроксид щелочного или щелочноземельного металла, карбонат щелочного или щелочноземельного металла, гидрокарбонат щелочного или щелочноземельного металла и подобные.
Конкретные примеры неорганического основания включают, например, гидроксид натрия, гидроксид калия, карбонат натрия, карбонат калия, гидрокарбонат натрия, гидрокарбонат калия, карбонат кальция, гидроксид кальция и подобные.
Твердое неорганическое основание предпочтительно используют в форме тонкоизмельченного порошка.
Количество используемого основания обычно находится в пределах 1-5 моль на моль соединения формулы [IV].
Обычно используют также катализатор фазового переноса. Примеры катализатора фазового переноса, используемого в реакции, включают соль четвертичного аммония, соль четвертичного фосфония, соль сульфония и подобные, причем предпочтительно используют соль четвертичного аммония.
В качестве соли четвертичного аммония используют соль, содержащую группы, необязательно выбранные из алкила и арила, имеющих 1-24 углеродных атома.
Примеры соли четвертичного аммония включают, например, тетраметиламмонийхлорид, тетраэтиламмонийхлорид, тетрапропиламмонийхлорид, тетрабутиламмонийхлорид, тетрапентиламмонийхлорид, тетрагексиламмонийхлорид, тетрагептиламмонийхлорид, тетраоктиламмонийхлорид, тетрагексадециламмонийхлорид, тетраоктадециламмонийхлорид, бензилтриметиламмонийхлорид, бензилтриэтиламмонийхлорид, бензилтрибутиламмонийхлорид, 1-метилпиридинийхлорид, 1-гексадецилпиридинийхлорид, 1,4-диметилпиридинийхлорид, тетраметил-2-бутиламмонийхлорид, триметилциклопропиламмонийхлорид, тетраметиламмонийбромид, тетраэтиламмонийбромид, тетрапропиламмонийбромид, тетрабутиламмонийбромид, тетрапентиламмонийбромид, тетрагексиламмонийбромид, тетрагептиламмонийбромид, тетраоктиламмонийбромид, тетрагексадециламмонийбромид, тетраоктадециламмонийбромид, бензилтриметиламмонийбромид, бензилтрибутиламмонийбромид, 1-метилпиридинийбромид, 1-гексадецилпиридинийбромид, 1,4-диметилпиридинийбромид, тетраметил-2-бутиламмонийбромид, триметилциклопропиламмонийбромид, бензилтриэтиламионийбромид, тетраметиламмонийиодид, тетрабутиламмонийиодид, тетраоктиламмонийиодид, третбутилэтилдиметиламмонийиодид, тетрадецилтриметиламмонийиодид, гексадецилтриметиламмонийиодид, октадецилтриметиламмонийиодид, бензилтриметиламмонийиодид, бензилтриэтиламмонийиодид, бензилтрибутиламмонийиодид и подобные.
Примеры соли четвертичного фосфония включают:
трибутилметилфосфонийхлорид,
триэтилметилфосфонийхлорид,
метилтрифеноксифосфонийхлорид,
бутилтрифенилфосфонийхлорид,
тетрабутилфосфонийхлорид,
бензилтрифенилфосфонийхлорид,
гексадецилтриметилфосфонийхлорид,
гексадецилтрибутилфосфонийхлорид,
гексадецилдиметилэтилфосфонийхлорид,
тетрафенилфосфонийхлорид,
трибутилметилфосфонийбромид,
триэтилметилфосфонийбромид,
метилтрифеноксифосфонийбромид,
бутилтрифенилфосфонийбромид,
тетрабутилфосфонийбромид,
бензилтрифенилфосфонийбромид,
гексадецилтриметилфосфонийбромид,
гексадецилтрибутилфосфонийбромид,
гексадецилдиметилэтилфосфонийбромид,
тетрафенилфосфонийбромид,
трибутилметилфосфонийиодид,
триэтилметилфосфонийиодид,
метилтрифеноксифосфонийиодид,
бутилтрифенилфосфонийиодид,
тетрабутилфосфонийиодид,
бензилтрифенилфосфонийиодид,
гексадецилтриметилфосфонийиодид.
Примеры соли сульфония включают: дибутилметилсульфонийхлорид, триметилсульфонийхлорид, триэтилсульфонийхлорид, дибутилметилсульфонийбромид, триметилсульфонийбромид, триэтилсульфонийбромид, дибутилметилсульфонийиодид, триметилсульфонийиодид и триэтилсульфонийиодид.
Количество используемого катализатора фазового переноса обычно находится в пределах примерно 0,01-0,2 моль, предпочтительно примерно 0,02-0,1 моль на моль соединения формулы [IV].
В этой реакции обычно используют органический растворитель. Примеры растворителя включают:
углеводородный растворитель, такой как н-гексан, циклогексан, н-пентан, толуол и ксилол;
эфирные растворители, такие как диэтиловый эфир, тетрагидрофуран, диметоксиэтан и анизол;
галогенированный растворитель, такой как хлороформ, дихлорметан, 1,2-дихлорэтан, монохлорбензол и о-дихлорбензол; и
апротонный полярный растворитель, такой как N,N-диметилформамид, диметилсульфоксид, N,N-диметилацетамид, гексаметилфасфорный триамид.
Реакцию обычно проводят при температуре от -78oС до температуры кипения используемого растворителя и предпочтительно в пределах от 0oС до 30oС.
Используемое выше соединение формулы [IV] может быть получено, например, удалением защитной группы в соответствии с традиционным методом, описанным выше в методике следующей за описанием способа А, для соединения формулы [V] , в котором защищены две гидроксильные группы.
Способ С
Соединение формулы [V] может быть получено способом, который включает взаимодействие сульфонового соединения формулы [VI] с галогенгидрином формулы [VII] в присутствии основания.
В галогенгидрине [VII] Х представляет атом галогена, такой как атом хлора, атом брома, атом йода или подобные.
R10 представляет такую же защитную группу, как определенная выше для R'.
Соединение формулы [VI] может быть получено способом, раскрытым в Chemistry Letters, 1985, 479.
Примеры основания включают алкиллитий, реактив Гриньяра, гидроксид щелочного металла, гидроксид щелочноземельного металла, гидрид щелочного металла, гидрид щелочноземельного металла, алкоксид щелочного металла и алкоксид щелочноземельного металла.
Конкретные его примеры включают н-бутиллитий, втор-бутиллитий, трет-бутиллитий, этилмагнийбромид, этилмагнийхлорид, гидроксид натрия, гидроксид калия, гидрид натрия, гидрид калия, метоксид натрия, метоксид калия, трет-бутоксид калия, трет-бутоксид натрия и подобные.
Количество основания обычно находится в пределах примерно 0,1-2 моль на моль сульфонового соединения формулы [VI].
В этой реакции можно, как описано выше, применять катализатор фазового переноса. Количество катализатора фазового переноса находится в пределах обычно примерно 0,01-0,2 моль, предпочтительно примерно 0,02-0,1 моль на моль сульфонового соединения формулы [VI].
В этой реакции обычно используют органический растворитель. Примеры растворителя включают эфирный растворитель, такой как диэтиловый эфир, тетрагидрофуран, диметоксиэтан и анизол, углеводородный растворитель, такой как н-гексан, циклогексан, н-пентан, толуол и ксилол, и апротонный полярный растворитель, такой как N,N-диметилформамид, диметилсульфоксид, N,N-диметилацетамид, гексаметилфосфорный триамид.
Реакцию обычно проводят при температуре в пределах от -78oС до температуры кипения используемого растворителя.
После завершения реакции сульфоновое соединение [V] может быть получено обычной последующей обработкой и дополнительно, при необходимости, может быть очищено хроматографией на силикагеле или подобным способом.
Способ D
Указанный выше галогенгидрин формулы [VII'] может быть получен способом, включающим взаимодействие соединения формулы R12OH, где R12 представляет ацильную группу с, по крайней мере, одним соединением, выбранным из галогенгидринов формул [VII'а] и [VII'b] в присутствии сильнокислотного катализатора.
Примеры ацильной группы для R11 в галогенгидрине формулы [VII'а] или [VII'b], используемом в этом способе, включают формил, ацетил, этоксиацетил, фторацетил, дифторацетил, трифторацетил, хлорацетил, дихлорацетил, трихлорацетил, бромацетил, дибромацетил, трибромацетил, цианацетил, пропионил, 2-хлорпропионил, 3-хлорпропионил, бутирил, 2-хлорбутирил, 3-хлорбутирил, 4-хлорбутирил, 2-метилбутирил, 2-этилбутирил, валерил, 2-метилвалерил, 4-метилвалерил, гексаноил, изобутирил, изовалерил, пивалоил, бензоил, о-хлорбензоил, м-хлорбензоил, п-хлорбензоил, о-ацетоксибензоил, о-метоксибензоил, м-метоксибензоил и п-метоксибензоил. Ацильная группа для R11 может также включать ацильные группы, перечисленные ниже для R12.
В галогенгидрине формулы [VII'а] или [VII'b] R' представляет защитную группу для гидроксильной группы.
Примеры атома галогена для Х включают атом хлора, атом брома и атом йода.
Примеры карбоновой кислоты формулы R12OH включают алифатическую карбоновую кислоту, имеющую 1-6 углеродных атомов, и ароматическую карбоновую кислоту, причем обе они могут быть замещенными атомом(ами) галогена, (С1-С3)алкоксигруппой(ами), цианогруппой(ами) или (C1-С3)ацилоксигруппой.
Конкретные примеры алифатической карбоновой кислоты включают муравьиную кислоту, уксусную кислоту, этоксиуксусную кислоту, фторуксусную кислоту, дифторуксусную кислоту, трифторуксусную кислоту, хлоруксусную кислоту, дихлоруксусную кислоту, трихлоуксусную кислоту, бромуксусную кислоту, дибромуксусную кислоту, трибромуксусную кислоту, циануксусную кислоту, пропионовую кислоту, 2-хлорпропионовую кислоту, 3-хлорпропионовую кислоту, н-масляную кислоту, 2-хлор-н-масляную кислоту, 3-хлор-н-масляную кислоту, 4-хлор-н-масляную кислоту, 2-метил-н-масляную кислоту, 2-этил-н-масляную кислоту, н-валериановую кислоту, 2-метил-н-валериановую кислоту, 4-метил-н-валериановую кислоту, гексановую кислоту, изомасляную кислоту, изовалериановую кислоту, пиваловую кислоту.
Примеры ароматической карбоновой кислоты включают бензойную кислоту, о-хлорбензойную кислоту, м-хлорбенэойную кислоту, п-хлорбензойную кислоту, ацетилсалициловую кислоту, о-анисовую кислоту, м-анисовую кислоту и п-анисовую кислоту. Количество ее особо не ограничено.
В качестве сильнокислотного катализатора используют органическую кислоту, такую как п-толуолсульфоновая кислота, бензолсульфоновая кислота, метансульфоновая кислота, трифторметансульфоновая кислота, камфорсульфоновая кислота и трифторуксусная кислота; сильнокислотные катионообменные смолы, такие как Nafion (торговый знак), Amberlyst (торговый знак) и Duolite (торговый знак); неорганические кислоты, такие как серная кислота, хлороводородная кислота и перхлорная кислота.
Количество сильнокислотного катализатора находится в пределах обычно примерно 0,01-0,5 моль, предпочтительно примерно 0,05-0,3 моль на моль галогенгидрина формулы [VII'а] или [VII'b].
Реакцию проводят при температуре обычно в пределах от -78oC до температуры кипения используемого растворителя, предпочтительно примерно 10-30oС.
После завершения реакции, например, в реакционную смесь добавляют воду и смесь экстрагируют, разделяют на фазы и концентрируют органический слой с получением галогенгидрина формулы [VII'], который далее, при необходимости, может быть очищен хроматографией на силикагеле.
Способ Е
По крайней мере одно соединение, выбранное из галогенгидринов формул [VII'а] и [VII'b] , можно получить взаимодействием триена формулы [VIII] с соединением R11ОН, где R11 представляет атом водорода или ацильную группу, и галогенирующим агентом.
Примеры галогенирующего агента включают хлорирующий агент, такой как хлор, гипохлористая кислота, трет-бутилгипохлорит, этилгипохлорит, гипохлорит натрия, гипохлорит калия, гипохлорит кальция, N-хлормочевина, N-хлорсукцинимид, хлорамин-Т и хлорамин-В; бромирующие агенты, такие как бром, гипобромистая кислота, гипобрамид кальция, N-бромацетамид и N-бромсукцинимид; и йодирующие агенты, такие как йод и N-иодсукцинимид.
Количество галогенирующего агента особо не ограничено, но обычно используют один моль галогенирующего агента на моль триена формулы [VII].
В качестве растворителя в реакции можно использовать простой эфир, такой как тетрагидрофуран, диоксан, диметоксиэтан и диэтиловый эфир; спирт, такой как трет-бутиловый спирт, трет-амиловый спирт и 2-пропанол; галогенированный углеводород, такой как дихлорметан, хлороформ и тетрахлорид углерода; кетон, такой как ацетон, метилизопропилкетон и метилизобутилкетон, апротонный полярный растворитель, такой как диметилсульфоксид, ацетонитрил, N,N-диметилформамид и N,N-диметилацетамид. Количество растворителя особо не ограничено.
Когда R11 представляет ацильную группу, она включает те ацильные группы, которые описаны выше для R12.
Количество карбоновой кислоты особо не ограничено, но обычно оно находится в пределах от примерно 1 до примерно 10 моль на моль триена [VIII].
Реакцию обычно проводят при температуре в пределах от примерно 15oС до 120oС.
В этом способе вместо карбоновой кислоты можно использовать воду, тогда R11 представляет атом водорода. Количество воды находится в пределах обычно примерно 1-100 моль, предпочтительно в пределах примерно 1-10 моль на моль триена формулы [VII]. Реакцию проводят при температуре в пределах обычно от -78oС до температуры кипения используемого растворителя, а предпочтительно 0-30oС.
После завершения реакции галогенгидрины [VII'а] и [VII'b] могут быть выделены традиционной последующей обработкой. Полученные региоизомеры могут быть разделены и очищены хроматографией на силикагеле.
Когда галогенгидрин [VII] имеет защитную группу, отличную от ацильной группы для R10, галогенгидрин может быть получен с помощью защиты либо гидроксильной группы галогенгидринов [VII'b], где R11 представляет атом водорода, либо группы, полученной путем удаления защиты ацильной группы R12 галогенгидрина [VII'].
Удаление защиты обычно проводят так, как описано выше в способе А.
Способ F
Триен формулы [VIII] может быть получен способом, включающим взаимодействие соединения формулы [IX] с основанием в присутствии палладиевого катализатора.
В этой реакции обычно используют гидроксид щелочного металла, такой как гидроксид натрия или гидроксид калия, в форме тонкодисперсного порошка. Обычно основание используют в количестве примерно 1-5 моль на моль соединения формулы [IX].
Примеры палладиевого катализатора включают димер аллилпалладийхлорида, ацетат палладия, оксид палладия, пропионат палладия, дихлорбис (трифенилфосфин) палладий, ди-μ-хлорбис(η-аллил)палладий, дихлор(η-1,5-циклооктандиен)палладий, дихлор(η-2, 5-норборнадиен)палладий, ди-хлорбис (ацетонитрил)палладий, дихлорбис(бензонитрил)палладий, дихлорбис(N,N-диметилформамид)палладий, бис(ацетилацетонато)палладий и подобные. Особо предпочтительным является димер аллилпалладий хлорида.
Количество палладиевого катализатора составляет обычно 0,05% по массе или более, предпочтительно 1% по массе или более на моль соединения [IX]. Верхний предел не ограничен, но по экономическим причинам он составляет предпочтительно 5% по массе или менее.
Для ускорения реакции обычно ее проводят в присутствии катализатора фазового переноса.
Примеры катализатора фазового переноса включают, как описано выше, соль четвертичного аммония, соль четвертичного фосфония и соль сульфония.
Количество катализатора фазового переноса составляет обычно 0,01-0,1 части по массе, предпочтительно 0,02-0,1 части по массе соединения [IX].
В этой реакции обычно используют безводный растворитель, примеры которого включают апротонный полярный растворитель, такой как N,N-диметилформамид, диметилсульфоксид и N,N-диметилацетамид; простой эфир, такой как диэтиловый эфир и тетрагидрофуран; ароматический углеводород, такой как толуол и ксилол; сложный эфир, такой как этилацетат или метилформиат; кетон, такой как ацетон; и спирт, такой как метанол, этанол, изопропиловый спирт или трет-бутиловый спирт. Реакцию обычно проводят при температуре в пределах от примерно 10oС до температуры кипения используемого растворителя.
После окончания реакции триен [VIII] может быть получен последующей традиционной обработкой и, при необходимости, может быть далее очищен хроматографией на силикагеле.
Соединение формулы [IX] можно легко синтезировать известным методом.
Способ G
Ретинол формулы [X] может быть преимущественно промышленно получен способом, включающим взаимодействие сульфона(ов) формул [IIa] и/или [IIb] с основанием.
Примеры основания, используемого в этой реакции, включают гидроксид щелочного металла, гидроксид щелочноземельного металла, гидрид щелочного металла, гидрид щелочноземельного металла, алкоксид щелочного металла и алкоксид щелочноземельного металла, а их конкретные примеры включают гидроксид натрия, гидроксид калия, гидрид натрия, гидрид калия, метоксид натрия, метоксид калия, трет-бутоксид калия, трет-бутоксид натрия и подобные.
Количество катализатора обычно составляет примерно 2-20 моль на моль сульфона(ов) формул [IIа] и/или [IIb].
В этой реакции обычно используют органический растворитель. Примеры растворителя включают углеводородный растворитель, такой как н-гексан, циклогексан, н-пентан, толуол и ксилол; простой эфир, такой как диэтиловый эфир, тетрагидрофуран, диметоксиэтан и анизол; и апротонный полярный растворитель, такой как N, N-диметилформамид, диметилсульфоксид, N,N-диметилацетамид или гексаметилфосфортриамид.
Температура реакции находится в пределах обычно от 0oС до температуры кипения используемого растворителя, предпочтительно от примерно -10oС до 50oС. По окончании реакции соединение формулы [X] может быть получено традиционной последующей обработкой и, при необходимости, может быть очищено хроматографией на силикагеле или подобным методом.
Далее настоящее изобретение проиллюстрировано в следующих примерах, которые не следует считать ограничивающими настоящее изобретение.
Пример 1
В сухую четырехгорлую колбу загружают в атмосфере азота 0,5 г (0,995 ммоль) 1-ацетокси-5-гидрокси-3,7-дииетил-9-(2,6,6-триметилциклогексен-1-ил)-9-(4-метилфенилсульфонил)-нона-2,6-диена (соединение II1-1) и 10 мл тетрагидрофурана. После растворения соединения [III-1] к раствору медленно добавляли при комнатной температуре 0,095 г (0,497 ммоль) тетрахлорида титана. После перемешивания при комнатной температуре в течение двенадцати часов исчезновение исходного материала подтверждали методом ТСХ (тонкослойная хроматография). Реакционную смесь прибавляли к 1% водному раствору гидроксида натрия и экстрагировали эфиром. Органический слой сушили над безводным сульфатом магния и затем концентрировали с получением неочищенного продукта. Полученный неочищенный продукт очищали хроматографией на силикагеле, получая соединение в виде бледно-желтого масла.
Пример 2
В сухую четырехгорлую колбу загружают под потоком азота 0,02 г соединения, полученного в примере 1, и 5 мл циклогексана. После растворения смеси к раствору добавляли 0,058 г (0,825 ммоль) метоксида калия и смесь перемешивали при 40oС в течение шести часов. После того как ТСХ подтвердила исчезновение исходных материалов, реакционную смесь добавляли к насыщенному водному раствору хлорида аммония и экстрагировали этилацетатом. Органический слой опять промывали насыщенным водным раствором хлорида аммония и сушили над безводным сульфатом магния. Из органического слоя удаляли растворитель, получив в результате неочищенный продукт в виде бледно-желтого масла. Анализ методом ЯМР подтвердил, что полученное соединение содержит в качестве основного компонента ретинол, имеющий только трансконфигурации.
В сухой четырехгорлой колбе растворяли в 5 мл толуола в атмосфере азота 0,01 г (0,035 ммоль) упомянутого выше неочищенного продукта. Затем, прибавив 0,003 г (0,035 ммоль) пиридина и 0,4 мг (0,004 ммоль) 4-диметиламинопиридина, к смеси медленно добавляли при комнатной температуре 0,004 г (0,035 ммоль) уксусного ангидрида, и полученную смесь перемешивали при указанной температуре в течение четырех часов. После того как ТСХ подтвердила исчезновение исходных материалов, к смеси добавляли 5% водный раствор хлористоводородной кислоты и толуола. Промыв смесь этим водным раствором, разделяли ее на два слоя. Полученный органический слой промывали насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия в указанном порядке и затем сушили над безводным сульфатом магния. Высушенный органический слой концентрировали с получением неочищенного продукта в виде желтого масла. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле. Сравнение результатов анализа методом ЯМР продукта с результатами аналогичного анализа стандартного образца транс-ретинолацетата полностью подтвердило, что в качестве основного компонента получили ретинолацетат, содержащий только трансконфигурации.
Пример 3
В сухой колбе растворяли в 20 мл гексана 60 мг (0,13 ммоль) 1,5-дигидрокси-З, 7-диметил-9-(2,6,6-триметилциклогексен-1-ил)-9-(4-метилфенилсульфонил)нона-2,6-диена (далее называемого как соединение [IV-I]) и к раствору прибавляли 3,4 мг (0,013 ммоль) н-додецилтриметиламмонийхлорида,
14 мг (0,13 ммоль) карбоната натрия и 13,3 мг (0,13 ммоль) уксусного ангидрида. После перемешивания при комнатной температуре в течение двадцати часов убеждались в исчезновении исходного материала и затем добавляли к реакционной массе воду. После экстракции эфиром органический слой промывали водным раствором хлорида аммония и насыщенным водным раствором хлорида натрия в указанном порядке. Органический слой сушили над безводным сульфатом магния и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали хроматографией на силикагеле, получая 1-ацетокси-5-гидрокси-3,7-диметил-9-(2,6,6-триметилциклогексен-1-ил)-9-(4-метилфенилсульфонил)нона-2,6-диен (далее называемый как соединение [III-I]) с выходом 92%.
Соединение [III-I] 1-НМР δ (СDСl3):
0,82 (6Н, с), 1,00 (6Н, с), 1,42 (3Н, с), 1,73 (3Н, с), 2,01 (3Н, с), 2,05 (3Н, с), 2,45 (3Н, с) 2,58-3,01 (2Н, м), 3,89 (1H, т, J = 1 Гц), 4,29-4,37 (1H, м), 4,58 (1H, д, J = 7 Гц), 5,14 (1H, д, J = 8 Гц), 5,23 (1H, д, J = 8 Гц), 5,41 (1H, т, J = 7 Гц), 7,31 (2Н, д, J = 8 Гц), 7,75 (2Н, д, J = 8 Гц).
Пример 4
Вводили 0,53 г (1,8 ммоль) β-циклогеранил п-толилсульфона (далее называемого как соединение [VI-I]) и растворяли в 20 мл тетрагидрофурана, после чего полученный раствор охлаждали до -60oС. При этой температуре в раствор по каплям добавляли 1,13 мл н-гексанового раствора, содержавшего 1,18 ммоль н-бутиллития, и полученную смесь держали при этой температуре. Затем в течение часа по каплям добавляли 5 мл тетрагидрофуранового раствора 0,3 г (0,9 ммоль) 8-бром-3,7-диметил-окта-2,6-диен-1,5-диацетата (далее называемого как соединение [VII-1] ). После перемешивания смеси при указанной температуре в течение трех часов убеждались с помощью ТСХ в исчезновении исходного материала. Реакционную массу добавляли к насыщенному водному раствору хлорида аммония и экстрагировали эфиром. Полученный органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. После удаления растворителя получили неочищенный продукт. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле, получая 1,5-диацетокси-3,7-диметил-9-(2,6, б-триметилциклогексен-1-ил)-9- 4-метилфенилсульфонил)-нона-2,6-диен (далее называемый как соединение [V-1]) в виде бледно-желтого масла с выходом 74%. (Значение Rf: 0,38, н-гексан-этилацетат = 3:1)
Соединение [V-1]
1-ЯМР δ (CDCl3):
0,76 (6Н, д, J = 14 Гц), 0,95 (6Н, д, J =14 Гц), 1,39 (3Н, с), 1,70 (3Н, с), 2,01 (3Н, с), 2,03 (3Н, с), 2,44 (3Н, с), 2,66-2,95 (2Н, м), 3,82-3,86 (1Н, м), 4,53 (2Н, д, J = 7 Гц), 5,10 (1Н, д, J = 9 Гц), 5,20 (1Н, д, 9 Гц), 5,34 (1Н, ш), 5,56 (1Н, ш), 7,33 (2Н, д, J = 8 Гц), 7,76 (2Н, д, J = 8 Гц). 13С-ЯМР δ (CDCl3):
15,1, 16,0, 16,1, 16,6, 18,8, 20,8, 20,9, 21,4, 28,2, 29,0, 35,5, 40,5, 40,8, 44,6, 60,8, 65,3, 65,5, 65,7, 68,3, 68,5, 68,8, 121,9, 127,1, 128,3, 129,4, 130,5, 130,6, 136,2, 137,1, 137,6, 137,7, 138,4, 144,0, 169,8, 170,0, 170,7.
Пример 5
К 0,53 г (1,8 ммоль) соединения [VI-1] добавляли 16 мл тетрагидрофурана и 4 мл гексаметилфосфорного триамида и соединение [VI-1] растворяли в растворителях. Затем к раствору добавляли при комнатной температуре 0,072 г (1,8 ммоль) гидроксида натрия и 0,058 г (0,18 ммоль) тетра-н-бутиламмонийбромида и смесь выдерживали при 40-45oС в течение трех часов. Охладив смесь до -60oС, добавляли по каплям в течение часа 5 мл тетрагидрофуранового раствора 0,3 г (0,9 ммоль) соединения [VII-1]. После перемешивания при указанной температуре в течение пяти часов смесь нагревали до 60oС и перемешивали при этой температуре в течение пяти часов. Убедившись с помощью ТСХ в израсходовании одного исходного соединения, реакционную массу загружали в насыщенный водный раствор хлорида аммония и экстрагировали эфиром. Полученный органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. После удаления растворителя получили неочищенный продукт. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле, получая соединение [V-1] в виде бледно-желтого масла с выходом 51%.
Пример 6
К 0,53 г (1,8 ммоль) соединения [VI-1] добавляли 20 мл N,N-диметилацетамида и соединение [VI-1] растворяли в растворителе. Затем к раствору прибавляли при 0oС 0,072 г (1,8 ммоль) гидроксида натрия и 0,058 г (0,18 ммоль) тетра-н-бутиламмонийбромида и добавляли по каплям в течение часа при 0oС 5 мл тетрагидрофуранового раствора 0,3 г (0,9 ммоль) соединения [VI1-1]. После перемешивания при указанной температуре в течение тридцати минут смесь нагревали до 50oС и перемешивали при этой температуре в течение пяти часов. Убедившись с помощью ТСХ в израсходовании одного исходного соединения, реакционную массу загружали в насыщенный водный раствор хлорида аммония и экстрагировали эфиром. Полученный органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. После удаления растворителя получили неочищенный продукт. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле, получая соединение [V-1] в виде бледно-желтого масла с выходом 59%.
Пример 7
В сухую колбу загружали 0,10 г (0,18 ммоль) соединения [V-1], 20 мл трет-бутилового спирта и 20 мл воды и к смеси добавляли при перемешивании 0,20 г (1,80 ммоль) трет-бутоксида калия. После перемешивания при 40oС в течение четырех часов к реакционной смеси, убедившись сначала с помощью ТСХ в исчезновении исходного соединения, добавляли насыщенный водный раствор хлорида аммония с последующим экстрагированием эфиром. Полученные органические слои объединяли и промывали водным раствором хлорида натрия. Объединенный органический слой сушили над безводным сульфатом магния и концентрировали с получением смеси, включающей три компонента. Полученную смесь разделяли колоночной хроматографией на силикагеле и в результате получили 1-ацетокси-5-гидрокси-3,7-диметил-9-(2,6,6-триметилциклогексен-1-ил)-9-(4-метилфенилсульфонил)-нона-2,6-диен [III-1] с выходом 31% и 5-ацетокси-1-гидрокси-3,7-диметил-9-(2,6,6-триметилциклогексен-1-ил)-9-(4-метилфенилсульфонил)-нона-2,6-диен [V-2] с выходом 37%.
Соединение [III-1] 1-ЯМР δ (СDСl3):
0,82 (6Н, с), 1,00 (6Н, с), 1,42 (3Н, с), 1,73 (3Н, с), 2,01 (3Н, с), 2,05 (3Н, с), 2,45 (3Н, с), 2,58-3,01 (2Н, м), 3,89 (1Н, т, J = 7 Гц), 4,29-4,37 (1Н, м), 4,58 (1Н, д, J = 7 Гц), 5,14 (1Н, д, J = 8 Гц), 5,23 (1Н, д, J = 8 Гц), 5,41 (1Н, т, J =7 Гц), 7,31 (2Н, д, J = 8 Гц), 7,75 (2Н, д, J = 8 Гц).
Соединение [V-2] 1H-ЯМР δ (СDСl3):
0,70 (6Н, д, J = 26 Гц), 0,88 (6Н, д, J = 26 Гц), 1,93 (3Н, с), 1,97 (3Н, с), 2,37 (3Н, с), 2,52-2,90 (2Н, м) 3,77-3,84 (1Н, м), 4,03 (2Н, т, J = 7 Гц), 5,05-5,14 (1Н, м), 5,33-5,36 (1H, 1м), 5,49, 5,51 (1Н, м), 7,24 (2Н, д, J = 8 Гц), 7,68 (2Н, д, J = 8 Гц).
Пример 8
К смешанному раствору 0,4 мл уксусного ангидрида и 1 мл уксусной кислоты, содержавшему 20 мг (0,10 ммоль) п-толуолсульфоновой кислоты, добавляли при перемешивании при 25oС 100 мг (0,34 ммоль) 8-бром-7-гидрокси-3,7-диметил-2,5-октадиенилацетата. После перемешивания смеси при 25oС в течение 25 минут добавляли 4 мл ионообменной воды и полученную смесь экстрагировали 1 мл н-гексана. Органический слой промывали 5%-ным водным раствором гидрокарбоната натрия, после чего сушили безводным сульфатом натрия. Полученный органический слой концентрировали при пониженном давлении с получением 58 мг бледно-желтого масла, содержащего 88% 5-ацетокси-8-бром-3,7-диметил-2,6-октадиенилацетата [VIII-1]. Чистый выход составлял 51%.
Пример 9
К смешанному раствору 40 мл уксусной кислоты и 40 мл тетрагидрофурана, содержащему 17,36 г (52,1 ммоль) смеси (1:1) 5-ацетокси-8-бром-3,7-диметил-2,6-октадиенилацетата [VII'b-1] и 7-ацетокси-8-бром-7-гидрокси-3,7-диметил-2,5-октадиенилацетата [VII'a-1] , добавляли при перемешивании при 0oС 260 мг (2,6 ммоль) серной кислоты. После перемешивания смеси при 0oС в течение четырех часов и при 25oС в течение двадцати часов добавляли к ней 120 мл ионообменной воды и 120 мл н-гексана. Разделив смесь на слои, водный слой дополнительно экстрагировали 80 мл н-гексана порциями по 40 мл. Объединенный органический слой промывали 40 мл 5%-ного водного раствора гидрокарбоната натрия и 40 мл насыщенного водного раствора хлорида натрия в указанном порядке и затем сушили над безводным сульфатом натрия. После концентрирования органического слоя при пониженном давлении получили 16,47 г (выход 73%) красновато-коричневого масла, содержавшего 77% 1,4-аддукта, и после хроматографии на силикагеле с использованием элюента, включающего н-гексан и этилацетат при отношении н-гексана к этилацетату, равном 10:1, получили 6,91 г 1,4-аддукт(5-ацетокси-8-бром-3,7-диметил-2,6-октадиенилацетата [VII'-l] в виде бледно-желтого масла с выходом 40%.
Соединение [VII'-1]
Rf: 0,49 (адсорбент: силикагель, элюент: н-гексан-этилацетат = 3:1).
1H-ЯМР δ (CDCl3):
1,74 (3Н, с), 1,85 (3Н, с), 2,02 (3H, с), 2,05 (3Н, с), 2,23 (1Н, дд, J = 13,5 Гц, 5,9 Гц), 2,38 (1Н, дд, J = 13,5 Гц, 7,3 Гц), 3,91 (2Н, с), 4,56 (2Н, д, J = 6,9 Гц), 5,38 (1Н, т, J = 6,9 Гц), 5,51 (1Н, д, J = 9,2 Гц), 5,59 (1Н, ддд, J = 9,2 Гц, 7,3 Гц, 5,9 Гц).
Пример 10
Смешанный раствор 40 мл диметилсульфоксида, содержащий 5,16 г (26,6 ммоль) 3,7-диметил-2,5,7-октатриенилацетата [VI11-1] и 0,48 г (26,6 ммоль) ионообменной воды, охлаждали до 10oС и прибавляли к смеси 4,78 г (26,6 ммоль) N-бромсукцинимида. Смесь перемешивали при комнатной температуре в течение сорока минут. Убедившись с помощью газовой хроматографии в израсходовании исходного соединения, прибавляли 40 мл ионообменной воды. Затем добавляли 40 мл этилацетата и отделяли органический слой. Водный слой экстрагировали 70 мл этилацетата порциями по 35 мл. Объединенный органический слой промывали 20 мл 5%-ного водного раствора гидрокарбоната натрия и 20 мл насыщенного водного раствора хлорида натрия в указанном порядке и затем сушили над безводным сульфатом натрия. Полученные путем концентрирования органического слоя при пониженном давлении 9,12 г желтого масла подвергли колоночной хроматографии на силикагеле с использованием элюента, включавшего н-гексан и этилацетат в отношении н-гексана к этилацетату, равном 5:1, а затем 3:1, и получили 4,84 г 8-бром-7-гидрокси-3,7-диметил-2,5-октадиенилацетата [VII'а-2] с выходом 63% и 1,28 г 8-бром-5-гидрокси-3,7-диметил-2,6-октадиенилацетата [VII'b-2] с выходом 17%.
Соединение [VII'а-2]
Rf: 0,30 (адсорбент: силикагель, элюент: н-гексан: этилацетат = 3:1)
1H-ЯМР δ (CDCl3):
1,43 (3Н, с), 1,70 (3Н, с), 2,06 (3Н, с) 2,34 (1Н, с), 2,78 (2Н, д, J = 6,9 Гц), 3,47 (2Н, с), 4,59 (2Н, д, J = 6,6 Гц), 5,38 (1Н, т, J = 6,6 Гц), 5,57 (1Н, д, J = 15,5 Гц), 5,74 (1Н, дт, J = 15,5 Гц, 6,9 Гц).
13С-ЯМР δ (CDCl3):
16,43, 20,94, 26,15, 41,98, 45,00, 61,17, 71,27, 119,43, 127,67, 135,38, 140,29, 170, 98.
Соединение [VII'b-2]
Rf: 0,14 (адсорбент: силикагель, элюент: н-гексан: этилацетат = 3:1)
1H ЯМР δ (СDСl3):
1,76 (с, 3Н), 1,82 (с, 3Н), 2,06 (с, 3Н), 2,20 (дд, J = 13,5 Гц, 5,6 Гц, 1Н), 2,30 (дд, J = 13,5 Гц, 7,9 Гц, 1Н), 2,67 (шс, 1Н), 3,95 (с, 2Н), 4,49 (ддд, J = 8,6 Гц, 7,9 Гц, 5,6 Гц, 1Н), 4,59 (д, J =6,9 Гц, 2Н), 5,42 (т, J = 6,9 Гц, 1Н), 5,58 (д, J = 8,6 Гц, 1Н).
13С-ЯМР δ (СDСl3):
14,94, 16,62, 20,76, 40,07, 46,81, 60,90, 66,09, 121,92, 132,65, 133,72, 137,48, 170, 89.
Пример 11
В колбу загружали 20,11 г (0,1 моль) 3,7-диметил-2,5,7-октатриенилацетата [VIII-1] и 100 мл уксусной кислоты и затем к смеси медленно добавляли 18,3 г (0,1 моль) N-бромсукцинимида. После перемешивания в течение 10-15 минут при комнатной температуре реакционная масса стала гомогенной. Через два часа, убедившись с помощью ТСХ в исчезновении исходных соединений, реакционную смесь вводили в воду и затем экстрагировали толуолом. Органический слой сушили над безводным сульфатом магния и затем концентрировали с получением смеси (примерно 1:1) соединений [VII'b-1] и [VII'a-1] с выходом 95%. Полученную смесь разделяли и очищали хроматографией на силикагеле, получая 8-бром-3,7-диметилокта-2,6-диен-1,5-диацетат [VII'b-1] в виде бледно-желтого масла с выходом 29% и соединение [VII'a-1] с выходом 30% и с выходом 31% в виде смеси.
Соединение [VII'b-1] 1H-ЯМР δ (СDСl3):
1,77 (3Н, с), 1,82 (3Н, с), 1,98 (3Н, с), 2,02 (3Н, с), 2,29 (2Н, ддд, J = 35 Гц, 8 Гц, 6 Гц), 3,89 (2Н, с), 4,55 (2Н, д, J = 7 Гц), 5,37 (1Н, т, J = 7 Гц), 5,48-5,62 (2Н, м).
Соединение [VII'a-1] 1H-ЯМР δ (СDСl3):
1,65 (3Н, с), 1,68 (3Н, с), 2,05 (3Н, с), 2,06 (3Н, с), 2,78 (2Н, д, J = 6 Гц), 3,75 (2Н, дд, J = 26 Гц, 11 Гц), 4,57 (2Н, д, J = 7 Гц), 5,35 (1Н, т, J = 7 Гц), 5,61-5,77 (2Н, м).
Пример 12
В сухую четырехгорлую колбу загружали 6,8 г (0,17 моль) измельченного в тонкий порошок гидроксида натрия, 2,2 г (8,5 ммоль) трифенилфосфина, 1,4 г (5,1 ммоль) тетра-н-бутиламмонийхлорида, 0,62 г (1,7 ммоль) димера аллилпалладийхлорида и 100 мл тетрагидрофурана. К смеси добавляли по каплям с перемешиванием при комнатной температуре в течение часа 150 мл тетрагидрофуранового раствора 40 г (0,17 моль) 6-хлор-3,7-диметилокта-2,7-диенилацетата [IX-1] . После перемешивания при комнатной температуре в течение трех дней, убедившись с помощью ТСХ в исчезновении исходных соединений, реакцию прекращали. Реакционную смесь выгружали в воду и экстрагировали эфиром. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Высушенный органический слой концентрировали с получением неочищенного продукта. Неочищенный продукт очищали хроматографией на силикагеле, получая 3,7-диметил-2,5,7-октатриенилацетат [VIII-1] в виде бледно-желтого масла с выходом 65%.
Соединение [VIII-1] 1H-ЯМР δ (СDСl3):
1,70 (3Н, с), 1,85 (3Н, с), 2,08 (3Н, с), 2,81 (2Н, д, J = 7 Гц), 4,58 (2Н, д, J = 7 Гц), 4,90 (2Н, с), 5,37 (1Н, т, J = 7 Гц), 5,61 (1Н, тд, J = 16 Гц, 7 Гц), 6,16 (1Н, д, J = 15 Гц).
Сравнительный пример 1
При проведении реакции и последующей обработки так же, как описано в примере 12, но без использования 2,2 г (8,5 ммоль) трифенилфосфина, 1,4 г (5,1 ммоль) тетра-н-бутиламмонийхлорида и 0,62 г (1,7 ммоль) димера аллилпалладийхлорида целевой продукт не получили, но извлекли непрореагировавший 6-хлор-З, 7-диметил-2,7-октадиен [IX-1] в виде бледно-желтого масла. Выход извлеченного продукта составлял 90%.
Ссылочный пример 1
В 100 мл н-гексана растворяли 40 г (20,4 ммоль) геранилацетата. После медленного добавления 17,1 г (70,0 ммоль) трихлоризоциануровой кислоты смесь выдерживали при температуре от -10oС до 0oС в течение шести часов. После завершения реакции оставшуюся трихлоризоциануровую кислоту и полученную в виде побочного продукта изоциануровую кислоту удаляли из системы путем фильтрования. Фильтрат промывали 5%-ным водным раствором гидрокарбоната натрия и ионообменной водой в указанном порядке и сушили над безводным сульфатом натрия. Удалив растворитель, получили неочищенный продукт. Неочищенный продукт подвергали колоночной хроматографии на силикагеле с получением целевого 6-хлор-3,7-диметил- 2,7-октатриенилацетата в виде бледно-желтого масла с выходом 86%.
Изобретение описывает новые родственные витамину А соединения формулы I, где R - Н или защитная группа для ОН, А - Н, галоген или группа формулы A1, где Ar - фенил, возможно замещенная С1-С5алкильной группой, и когда А = А1, Q = Q3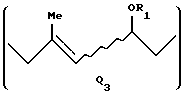
где R1 - Н или защитная группа для ОН, когда А - галоген, Q = Q3, или Q4
где R2 - Н или защитная группа для ОН, когда А - Н, Q = Q2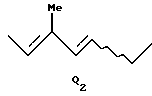 .
.
Описаны способа получения соединения I и витамина А.

6 с. и 15 з.п. ф-лы.
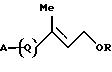
где R представляет атом водорода или защитную группу для гидроксильной группы;
А представляет атом водорода, атом галогена или группу формулы Аl
где Аr представляет фенильную группу, которая может быть замещена (C1-C5)алкильной группой;
когда А представляет A1, Q представляет Q3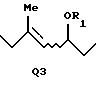
где R1 представляет атом водорода или защитную группу для гидроксильной группы; и
когда А представляет атом галогена, Q представляет Q3, определенный выше, или Q4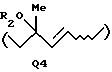
где R2 представляет атом водорода или защитную группу для гидроксильной группы;
когда А представляет атом водорода, Q представляет Q2
R представляет защитную группу для гидроксильной группы.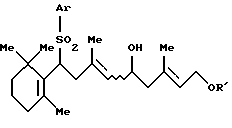
где Аr представляет фенильную группу, которая может быть замещена (C1-C5)алкильной группой,
R' представляет ацильную группу,
который включает взаимодействие соединения формулы (IV)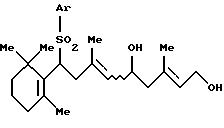
где Ar - такой, как определен выше,
с защитным агентом, который выбирают из ацилгалогенида или ангидрида карбоновой кислоты, в присутствии карбоната щелочного металла и катализатора фазового переноса.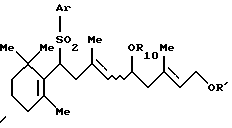
где Ar представляет фенильную группу, которая может быть замещена (С1-С5) алкильной группой;
R10 и R' одинаковые или различные и представляют защитную группу для гидроксильной группы,
который включает взаимодействие сульфона формулы (VI)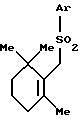
где Ar - такой, как определен выше,
с галогенгидрином формулы (VII)
где R10 и R' - такие, как определены выше;
Х представляет атом галогена,
в присутствии основания.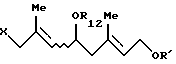
где R12 представляет (C1-C6)алифатическую ацильную группу;
R' представляет защитную группу для гидроксильной группы,
Х представляет атом галогена,
который включает взаимодействие, по крайней мере, одного галогенгидрина, выбранного из группы, включающей соединение формулы (VII'a)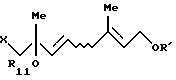
и соединение формулы (VII'b)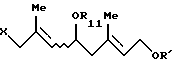
где R11 представляет (C1-C6)алифатическую ацильную группу или атом водорода;
R' представляет защитную группу для гидроксильной группы, с карбононой кислотой формулы
R12OH
где R12 - такой, как определен выше,
в присутствии сильнокислотного катализатора, выбранного из серной кислоты и п-толуолсульфоновой кислоты.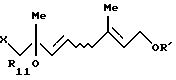
и соединение формулы (VII'b)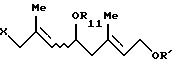
где R11 представляет (C1-C6)алифатическую ацильную группу или атом водорода;
R' представляет защитную группу для гидроксильной группы,
который включает взаимодействие триена формулы (VIII)
где R' - такой, как определен выше,
с галогенирующим агентом и соединением формулы
R11OH
где R11 - такой, как определен выше.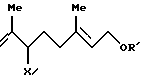
где Х представляет атом галогена;
R' представляет защитную группу для гидроксильной группы,
с основанием в присутствии палладиевого катализатора, фосфинового лиганда и катализатора фазового переноса.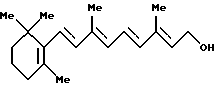
который включает стадии:
(а) обработку соединения формулы (III)
где Аr представляет фенильную группу, которая может быть замещена (С1-С5)алкильной группой;
R' представляет защитную группу для гидроксильной группы, тетрахлоридом титана, и
(b) взаимодействие соединения, полученного на стадии (а), с алкоксидом щелочного металла.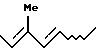
и R представляет защитную группу для гидроксильной группы.
Приоритет по пунктам
24.10.1997 по пп.2, 10, 11, 12;
25.08.1997 по пп.3, 4, 5, 6, 16, 20;
11.12.1997 по пп.13, 14;
27.01.1998 по п.18;
04.02.1998 по остальным пунктам.
| Способ получения производных витамина а | 1977 |
|
SU722483A3 |
| УСТРОЙСТВО ДЛЯ АВТОМАТИЧЕСКОЙ ДОЗИРОВАННОЙ СМАЗКИ ШТАМПОВ | 0 |
|
SU348813A1 |
| Helv | |||
| Chim | |||
| Acta, 59, 387, 1976. | |||

