Изобретение относится к способу получения бутенолигомеров из олефинов по синтезу Фишера-Тропша. Бутенолигомеры представляют собой ценные исходные вещества для получения спиртов. Предпочтительными бутенолигомерами являются изомерные октены, которые представляют собой димерные бутены и которые, следовательно, называют также дибутеном. Пользующимся большим спросом дибутеном является ди-н-бутен. Поэтому изобретение относится также к способу, в котором ди-н-бутен отделяется от дибутена. Кроме того, изобретение относится к способам, в результате проведения которых получают, кроме высших бутенолигомеров, в качестве дибутена исключительно ди-н-бутен, при этом в качестве побочных продуктов получают ценные алкил-трет-бутиловые эфиры.
Дибутен представляет собой смесь изомеров, которая образуется наряду с высшими бутенолигомерами в результате димеризации и/или со-димеризации бутенов, то есть н-бутена и/или изобутена при олигомеризации бутенов. Ди-н-бутеном называют продукт димеризации н-бутена, то есть 1 -бутена и/или 2-бутена. Существенными компонентами ди-н-бутена являются 3-метил-2-гептен, 3,4-диметил-2-гексен и, в меньшей степени, н-октены. Ди-изобутен представляет собой смесь изомеров, образующуюся в результате димеризации изобутена. Ди-изобутен содержит более разветвленные молекулы, чем дибутен, который, со своей стороны, более разветвлен, чем ди-н-бутен.
Дибутен, ди-н-бутен и ди-изобутен являются исходными веществами для получения изомерных нонанолов путем гидроформилирования и гидрирования образующихся альдегидов с 9 атомами углерода. Сложные эфиры этих нонанолов, в частности сложные эфиры фталевой кислоты, представляют собой пластификаторы, которые производят в значительном количестве и употребляют прежде всего для получения поливинилхлорида. Нонанолы из ди-н-бутена являются менее разветвленными, чем нонанолы из дибутена, которые со своей стороны менее разветвлены, чем нонанолы из ди-изобутена. Сложные эфиры нонанолов из ди-н-бутена именно из-за своей более линейной структуры имеют технологические преимущества по сравнению со сложными эфирами из нонанолов на основе дибутена и ди-изобутена и пользуются большим спросом.
Бутены для димеризации можно получать, например, из фракции С4, полученной в результате крекинга с водяным паром, или каталитического крекинга в псевдоожиженном слое. Фракцию перерабатывают, как правило, путем отделения 1,3-бутадиена селективной промывкой, например, N-метилпирролидоном. Изобутен является желаемым и особо ценным компонентом фракции С4, так как он подвергается химической реакции с получением пользующихся большим спросом продуктов. Он подвергается, например, взаимодействию с изобутаном с получением высокооктанового изооктана или же с алканолом, таким, как метанол, с получением метил-трет-бутилового эфира, который в качестве присадки к автомобильному бензину улучшает его октановое число. После реакции изобутена остаются н-бутен, н- и изобутан. Однако доля н-бутена в продуктах расщепления крекинга с водяным паром или каталитического крекинга в псевдоожиженном слое относительно низка. Она составляет менее 10% от веса главного целевого продукта этилена. На установке для крекинга с водяным паром с производительностью 600000 т этилена в год получают лишь около 60000 т н-бутена в год. Количество н-бутена (а также изобутена), правда, можно повышать путем дегидрирования 15000 т н- и изобутана в год, которые получают наряду с н-бутеном. Это, однако, не рекомендуется, так как установки для дегидрирования требуют высоких капиталовложений и являются нерентабельными в случае такой незначительной производительности.
Как уже упомянуто, изобутен представляет собой пользующийся большим спросом крекинг-продукт и поэтому, как правило, не может быть использован для олигомеризации. Однако количество н-бутенов, непосредственно полученных на установке для крекинга с водяным паром или установке каталитического крекинга в псевдоожиженном слое, не хватает для производства достаточного количества дибутена для установки получения нонанолов, производительность которой является такой большой, что она может экономически конкурировать с имеющимися крупными установками получения значительных спиртов для получения пластификаторов, таких, как 2-этилгексанол. Это означает, что необходимо собирать полученный на разных крекинг-установках н-бутен и подвергать его олигомеризации для покрытия потребности крупной установки производства нонанолов в дибутене. В качестве альтернативы можно собирать еще неразделенные фракции С4 из разных крекинг-установок и перерабатывать на месте до н-бутена. Этому противостоит, однако, тот факт, что транспорт сжиженных газов стоит дорого, не в последнюю очередь из-за проведения требуемых дорогостоящих мер безопасности.
Поэтому желательно предоставлять бутены для олигомеризации на одном лишь месте без перевозки через большие расстояния в количествах, требуемых для эксплуатации большой установки для получения нонанолов, имеющей, например, производительность 200000-800000 т/год. Также желательно использовать при этом бутены, которые неизбежно получают в результате проведения способа, направленного на получение других олефинов, и таким образом улучшить создание стоимости этого способа. Кроме того желательно иметь способ получения бутенолигомеров, в котором ценный ди-н-бутен можно отделять от дибутена. В дальнейшем желательно такое управление способом, чтобы кроме высших бутенолигомеров в качестве дибутена образовался исключительно ди-н-бутен, а в качестве желаемых побочных продуктов получались простые алкил-трет-бутиловые эфиры. Также желательно иметь возможность регулировать количественное соотношение этих веществ.
Способ согласно изобретению поясняется блок-схемами на приложенных фигурах 1 и 2, в которых изложены нижеописанные варианты способа с обязательными и факультативными стадиями способа.
Изобретение представляет собой способ получения бутенолигомеров из олефинов по синтезу Фишера-Тропша, в котором содержащиеся в подаваемой по линии 1 фракции С4 олефинов по синтезу Фишера-Тропша бутены подвергают олигомеризации на стадии 2 олигомеризации, а из отводимой по линии 3 смеси олигомеризации получают отводимый по линии 4 дибутен.
Этот способ с его разными факультативными стадиями, которые в нижеследующем поясняются с помощью фигуры 1, называют вариантом А.
Вариантом Б, которому соответствует фигура 2, называют способ, в котором сначала на стадии 5 этерификации содержащийся в подаваемой по линии 1 фракции С4 изобутен подвергают взаимодействию с подаваемым по линии 25 алканолом с получением отводимого по линии 26 алкил-трет-бутилового эфира и олигомеризуют лишь оставшийся н-бутен с получением в качестве дибутена исключительно ди-н-бутена, отводимого по линии 6. В особой форме выполнения варианта Б имеется стадия 27 изомеризации, на которой непрореагировавшийся на стадии 2 олигомеризации н-бутен изомеризуют до изобутена, который возвращают на стадию 5 этерификации.
В частности, последняя форма выполнения варианта Б отличается высокой гибкостью. В зависимости от требований рынка можно получать изменяющиеся количества ди-н-бутена и алкил-трет-бутилового эфира.
Фракции С4 в качестве исходного вещества способа
Исходным веществом предлагаемого способа является подаваемая по линии 1 фракция С4, полученная из способа Фишера-Тропша, в котором, как известно, из синтез-газа (окиси углерода и водорода) производят углеводороды или же кислородсодержащие продукты. Синтез-газ можно получать из самых различных энергоносителей, таких, как, например, природный газ, мазут, остаточное масло, бурый уголь и каменный уголь, практически в любых количествах.
Синтез Фишера первоначально был разработан для получения бензина, однако после Второй мировой войны интерес концентрировался прежде всего на разработке способов получения богатых альфа-олефинами фракций углеводородов с низким числом атомов углерода. Здесь подобно как на установках крекинга с водяным паром на первом плане стоит оптимизация выхода этилена, а также пропилена, так как эти олефины являются основными компонентами многих важных химических продуктов, таких, как, например, полиэтилен, полипропилен, поливинилхлорид, окись этилена и окись пропилена.
Получение главным образом короткоцепных углеводородов с 2-6 атомами углерода, преобладающим образом представляющихся олефины и, в частности, альфа-олефины, обеспечивает, например, способ согласно заявке ЕР 0 216 972. Его существенный признак - выбор определенного катализатора, а именно однофазной, карбидсодержащей и восстановленной шпинели без носителя, образующейся из окисного соединения эмпирической формулы FexCоy04, в которой х и у означают целые или десятичные числа, при условии, что сумма х + у = 3 и соотношение х/у составляет по меньшей мере 7. Поверхность катализатора составляет по меньшей мере около 0,1 до 5 м2/г. В качестве промотора катализатор может содержать соединение металла группы IA или IIА периодической системы элементов.
Катализатор является пирофорным. Поэтому его дезактивируют с помощью небольших количеств кислорода в инертном газе для улучшения обращения с ним. Его можно применять в виде суспензии в инертной органической жидкости, такой, как высококипящие парафины, ароматические углеводороды или простые эфиры, третичные амины или смеси таких веществ, в количествах от 10 до 50 г сухого катализатора на 500 г органической жидкости. До подачи синтез-газа катализатор кондиционируют, то есть вновь активируют, путем промывки азотом и обработки водородом при повышенной температуре.
Соотношение окиси углерода и водорода в синтез-газе может колебаться в широких пределах и преимущественно составляет 1:1 до 2:1. Оптимальная температура для получения желаемых короткоцепных альфа-олефинов с 2-6 атомами углерода равняется 230-270oС, в частности 240-260oС. Эта температура является критической, так как при повышенных температурах образуется больше метана, а низкие температуры способствуют образованию воскообразных продуктов. Способ осуществляют, как правило, при давлении в диапазоне от 350 до 2200 кПа.
В подобных условиях и в способе с неподвижным слоем катализатора предпочтительно производят углеводороды с 2-6 атомами углерода, большинство которых представляет собой олефины, и предпочтительно альфа-олефины. Фракцию С4 стандартным образом выделяют из углеводородной смеси, преимущественно путем фракционной перегонки при низкой температуре и/или повышенном давлении.
Необходимо подчеркнуть, что в качестве источника фракции С4 пригодна не только эта специальная форма выполнения способа Фишера-Тропша. Можно скорее применять любую фракцию С4, полученную из установок, работающих по методу Фишера-Тропша. В Южноафриканской Республике по оптимированным способам Фишера-Тропша в промышленном масштабе производят больше 400000 т этилена и 500000 т пропилена в год. Недостаток этих способов заключается в том, что неизбежно образуется примерно такое же количество высших углеводородов и, в частности, углеводородов с 4 атомами углерода, для которых практически не существует подходящего применения, кроме их применения в качестве присадки к карбюраторным топливам. Но даже для топлива фракция С4 скорее не пригодна из-за ее высокой доли олефинов, которые склонены к осмолению, и из-за высокого давления пара, являющегося экологической нагрузкой. Таким образом способ согласно изобретению обеспечивает полезное применение неизбежно образующихся веществ и улучшает создание стоимости способа Фишера-Тропша.
Вариант А
Из подаваемого по линии 7 энергоносителя на стадии 8 синтез-газа производят синтез-газ соответствующего его назначению состава, который по линии 9 подают на стадию 10 синтеза Фишера-Тропша, где производят, например, по способу согласно заявке ЕР 0 216 972, богатую олефинами смесь углеводородов с большой долей углеводородов с 2-6 атомами углерода. Смесь по линии 11 подают на стадию 12 разделения, где из смеси стандартным образом получают отводимую по линии 1 фракцию С4, например путем сжижения газообразных компонентов при температуре -30oС и фракционной перегонки. Полученная фракция С4 имеет, как правило, следующий состав:
1-Бутен - 70±10%
Изобутен - 10±5%
2-Бутен - 10±5%
н-/Изобутен - 12±3%
1,3-Бутадиен - Следы
Можно повышать выход отводимой по линии 1 фракции С4 путем димеризации поступающегося на стадии 12 разделения этилена на стадии 12а димеризации. Для этого пригоден, например, способ получения торгового продукта димерсола, описанный автором Y. Chauvin и др. в источнике "Erdol, Erdgas, Kohle" 106, 7/8 (1990 г. ), стр. 309 и cл. Работают в жидкой фазе с катализатором типа катализаторов Циглера на основе соединения никеля, которое активируют с помощью металлоорганического соединения. "Дегенерированная полимеризация" додимера протекает в умеренных условиях при температуре около 20-80oС. Конверсия по циклу составляет 50-90%. Смесь димеризации возвращают на стадию 12 разделения.
Подаваемая по линии 1 фракция С4 содержит следы 1,3-бутадиена. Рекомендуется удалить эти диены, так как даже ничтожные количества диенов могут повреждать катализатору олигомеризации. Подходящим способом является селективное гидрирование 13, которое, кроме того, повышает долю желаемого н-бутена. Подходящий способ описан, например, автором F. Nierlich и др. в источнике "Erdol & Kohle, Erdgas, Petrochemie, 1986 г., стр. 73 и ел. В жидкой фазе способа работают с полностью растворенным водородом в стехиометрических количествах. В качестве селективных катализаторов гидрирования пригодны, например, никель и, в частности, палладий на носителе, например 0,3 вес. % палладия на активном угле или предпочтительно на окиси алюминия. Небольшое количество окиси углерода в диапазоне ч/милл. способствует селективности гидрирования 1,3-бутадиена до моноолефина и противодействует образованию полимеров, так называемому зеленому маслу, инактивирующих катализатор. Способ осуществляют при комнатной температуре или слегка повышенной температуре до 60oС и при повышенном давлении, целесообразно составляющих до 20 бар. Содержание 1,3-бутадиена в подаваемой по линии 1 фракции С4 таким образом снижается до величин ниже 1 ч/милл., которые не мешают при олигомеризации.
Кроме того целесообразно перед поступлением на стадию 2 олигомеризации максимально освобожденную от 1,3-бутадиена фракцию С4 подавать на стадию 14 очистки, представляющую собой молекулярное сито, благодаря чему удаляются другие вредные для катализатора олигомеризации вещества и повышается срок его службы. К вредным веществам относятся соединения кислорода и серы. Очистка с помощью молекулярных сит описана автором F. Nierlich и др. в патенте ЕР 0 395 857. Целесообразно используют молекулярное сито с диаметром пор, равным 4-15  преимущественно 7-13
преимущественно 7-13  В некоторых случаях по экономическим причинам целесообразно смесь дегидрирования последовательно подавать через молекулярные сита с разными размерами пор. Способ можно осуществлять в газовой фазе, жидкой фазе или газожидкой фазе. Давление соответственно составляет, как правило, 1 - 200 бар. Целесообразно работают при комнатной температуре или повышенных температурах до 200oС.
В некоторых случаях по экономическим причинам целесообразно смесь дегидрирования последовательно подавать через молекулярные сита с разными размерами пор. Способ можно осуществлять в газовой фазе, жидкой фазе или газожидкой фазе. Давление соответственно составляет, как правило, 1 - 200 бар. Целесообразно работают при комнатной температуре или повышенных температурах до 200oС.
Химические свойства молекулярных сит менее важны, чем их физическая характеристика, то есть, в частности размер, пор. Следовательно, можно применять самые различные молекулярные сита, например кристаллические, естественные силикаты алюминия, например силикаты со слоистой решеткой, а также синтетические молекулярные сита, например сита с цеолитной структурой. Цеолиты типа А, Х и Y можно получать в качестве торговых продуктов фирм Байер АГ, Доу Кемикл Ко., Юнион Карбайд Корпорейшн, Лапорте Индустриз Лтд. и Мобил Ойл Ко. Пригодными являются также такие синтетические молекулярные сита, которые наряду с алюминием и кремнием содержат еще другие, введенные путем катионообмена атомы, например галлий, индий или лантан, а также никель, кобальт, медь, цинк или серебро. Кроме того пригодны синтетические цеолиты, у которых наряду с алюминием и кремнием в решетку введены путем смешанного осаждения еще другие атомы, например бор или фосфор.
Как уже упомянуто, стадия 13 селективного гидрирования и стадия 14 очистки с молекулярным ситом являются факультативными, преимущественными мерами для способа согласно изобретению. Их последовательность в принципе может быть любой, однако предпочитается указанная на фиг.1 последовательность.
Подаваемую по линии 1 фракцию С4, в случае необходимости предварительно обработанную описанным образом, подают на стадию 2 олигомеризации, представляющую собой существенную часть предлагаемого способа. Олигомеризация является со-олигомеризацией н-бутена и изобутена, осуществляемой общеизвестным образом, описанным, например, автором F. Nierlich в источнике "Oligomerization for Better Gasoline", Hydrocarbon Processing, 1992 г., стр. 45 и сл., или же F. Nierlich и др. в уже названном патенте ЕР 0 395 857. Как правило, работают в жидкой фазе и в качестве гомогенного катализатора применяют, например, систему, состоящую из октоата никеля (II), хлорида этилалюминия и свободной кислоты жирного ряда (см. патент DE 28 55 423), или же предпочтительно применяют один из многочисленных известных катализаторов в неподвижном слое или суспендированных в смеси олигомеризации катализаторов на основе никеля и кремния. Катализаторы часто дополнительно содержат алюминий. Так в патенте ГДР 160 037 описано получение содержащего никель и алюминий катализатора осаждения на двуокиси кремния в качестве носителя. Другие пригодные катализаторы получают путем замены находящихся на поверхности носителей положительно заряженных частиц, таких, как протоны или ионы натрия, ионами никеля. Это удается в случае самых различных носителей, таких, как аморфный силикат алюминия (см. R. Espinoza и др. в источнике "Appl. Kat", 31, 1987 г., стр. 259-266), кристаллический силикат алюминия (см. патент DE 20 29 624), цеолиты типа ZSM (см. патент NL 8 500 459), цеолиты типа Х (см. патент DE 23 47 235), цеолиты типа Х и Y (см. A. Barth и др. в источнике "Z. Anorg. Allg. Chem." 521, 1985 г., стр. 207-214) и морденит (см. заявку ЕР 0 233 302).
Со-олигомеризацию целесообразно осуществляют в зависимости от катализатора при температуре 20-200oС и давлении 1-100 бар. Время реакции (или же время контактирования) составляет, как правило, 5-60 мин. Параметры способа, в частности вид катализатора, температура и время контактирования, согласуют так, чтобы желаемая степень олигомеризации была достигнута. В случае нонанолов в качестве желаемого целевого продукта речь прежде всего идет о димеризации. Само собой разумеется, что для этого реакцию нельзя осуществлять с целью полной конверсии. Целесообразно намеревается достижение степени конверсии, составляющей 30-70% по циклу. Оптимальные комбинации параметров способа можно определять без проблем путем проведения предварительных испытаний.
Из подаваемой по линии 3 смеси олигомеризации на стадии 15 разделения фракции С4 отделяют отводимый по линии 16 остаточный газ, который в качестве обратного газа по линии 17 частично возвращают на стадию 2 олигомеризации и частично выводят из цикла по линии 18 в качестве отработанного газа. Возвращение части отводимого по линии 16 остаточного газа требуется, потому что, как уже упомянуто, конверсия на стадии 2 олигомеризации является неполной. Отводимый по линии 18 отработанный газ необходимо вывести из цикла, чтобы отводить небольшие количества (12±3%) н-/изобутана, обычно имеющиеся во фракции С4. Конечно, отводимый по линии 18 отработанный газ содержит также бутены, главным образом н-бутен, так как предпочтительно олигомеризуется изобутен, уже имеющийся во фракции С4 в небольшом количестве. Отводимый по линии 16 остаточный газ разделяют на отводимый по линии 17 обратный газ и отводимый по линии 18 отработанный газ в таком соотношении, чтобы содержание н- и изобутана в отводимом по линии 16 остаточном газе повысилось не слишком сильно. В стационарном состоянии его держат преимущественно ниже 70 об. %.
Остающиеся после отделения остаточного газа олигомеры по линии 19 подают на стадию 20 разделения олигомеров, где их разделяют путем перегонки на отводимый по линии 4 дибутен, отводимый по линии 21 трибутен (или додецен) и отводимый по линии 22 остаток. Отводимый по линии 21 трибутен представляет собой желаемый побочный продукт. Его можно подвергать гидроформи-лированию, продукты гидроформилирования можно гидрировать и полученные таким образом тридеканолы можно этоксилировать, в результате чего получают ценный компонент для моющих средств. Отводимый по линии 4 дибутен непосредственно пригоден как исходное вещество для получения нонанолов, тем более что он является относительно линейным из-за высокой доли н-бутена во фракции С4. Если важны особые свойства нонанолов из ди-н-бутена, то отводимый по линии 4 дибутен можно разделять на стадии 23 четкой ректификации на отводимый по линии 6 ди-н-бутен и на отводимые по линии 24 ди-изобутены, которые в качестве более разветвленных молекул кипят при более низких температурах. Последние можно также применять для получения спиртов для получения пластификаторов или в случае необходимости после гидрирования в качестве присадки к автомобильному бензину.
Вариант Б
В варианте Б из подаваемой по линии 1 фракции С4 на стадии 5 этерификации общеизвестным образом отделяют изобутен в результате его взаимодействия с подаваемым по линии 25 алканолом с получением отводимого по линии 26 алкил-трет-бутилового эфира. Реакция является практически селективной, так как оба изомера н-бутена значительно меньше реакционноспособны, чем изобутен. На стадии 2 олигомеризации олигомеризуют исключительно н-бутен, так что в качестве дибутена образуется исключительно ди-н-бутен, отводимый по линии 6.
Подходящими алканолами, подаваемыми по линии 25, являются прежде всего алканолы с 1 - 6 атомами углерода, например метанол, этанол или изобутанол. Их взаимодействие с изобутеном описано, например, в источнике "Methyl-Tert-Butyl Ether", Ullmanns Encyclopedia of Industrial Chemistry, том А 16, стр. 543 и сл. Реакция осуществляется в жидкой фазе или в газожидкой фазе при температуре 50-90oС при давлении, устанавливающемся при соответствующей температуре. Целесообразно работают с небольшим избытком алканола, за счет чего повышается селективность конверсии изобутена и уменьшается его димеризация. В качестве катализатора используют, например, кислый бентонит или кислый ионообменник с крупными порами. Из жидкой смеси этерификации в результате перегонки получают отводимый по линии 26 алкил-трет-бутиловый эфир и в случае необходимости избыточный алканол, который можно возвращать в реакцию.
Отделение изобутена путем его взаимодействия с алканолом рекомендуется, в частности, тогда, если имеется возможность применения алкил-трет-бутилового эфира, в частности метил-трет-бутилового эфира в качестве повышающей октановое число присадки к автомобильному бензину.
В случае особой формы выполнения варианта Б имеется стадия 27 изомеризации, на которой часть н-бутена превращают до изобутена. Дополнительный изобутен возвращают на стадию 5 этерификации. Таким образом можно варьировать количественное соотношение ди-н-бутена и алкил-трет-бутилового эфира и согласовать его с потребностями рынка.
Изомеризацию олефинов также называют скелетной изомеризацией. Стадию 27 изомеризации целесообразно подключают к стадии 15 разделения фракции С4. Отводимый по линии 16 остаточный газ, содержащий до 70 об. % н- и изобутана, затем разделяют на три парциальных потока, а именно на отводимый по линии 17 обратный газ, отводимый по линии 18 отработанный газ и отводимый по линии 28 газ изомеризации. Изомеризация н-бутена до изобутена была разработана в недавнем прошлом. Обзор разных способов приведен автором F. Nierlich в источнике "Recent Developments in Olefin Processing for Cleaner Gasoline", Oil Gas European Magazine, 1992 г. , 4, стр. 31 и cл. Общая черта всех способов заключается в том, что н-бутен смешивают с водяным паром и пропускают его через кислый катализатор, например кислый цеолит, при температуре 500-600oС и атмосферном давлении. При этом н-бутен в самом благоприятном случае изомеризуют до изобутена до достижения равновесия, составляющего в зависимости от температуры 35-50% изобутена и 65-50% н-бутена. Из подаваемой по линии 29 смеси изомеризации под давлением отделяют высококипящие компоненты с помощью промывки 30 циркулирующей водой. Не конденсированные в этих условиях части подаваемой по линии 29 смеси изомеризации, выходящие из верха колонны в качестве азеотропа, состоящего (в основном) из углеводородов с 4 атомами углерода и воды, подают на разделительную стадию 31 - в колонну. Из верха колонны выходит азеотроп, состоящий из низкокипящих компонентов и воды, а в нижней части колонны получают углеводороды с 4 атомами углерода и небольшие количества высококипящих компонентов, которые не осаждались на стадии 30 промывки циркулирующей водой и которые на последующей разделительной стадии 32, также представляющей собой колонну, в качестве кубового продукта отделяют от выходящих из головной части углеводородов с 4 атомами углерода, которые возвращаются на стадию 5 этерификации.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ СОВМЕСТНОГО ПОЛУЧЕНИЯ ДИ-Н-БУТЕНА И АЛКИЛ-ТРЕТ-БУТИЛОВЫХ ЭФИРОВ ИЗ ПРИРОДНЫХ БУТАНОВ | 1997 |
|
RU2178782C2 |
| СПОСОБ ПОЛУЧЕНИЯ БУТЕНОВЫХ ОЛИГОМЕРОВ ИЗ ПРИРОДНЫХ БУТАНОВ | 1997 |
|
RU2189373C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВЫСШИХ ОКСО-СПИРТОВ | 1997 |
|
RU2183210C2 |
| ТИОЭТЕРИФИКАЦИЯ МЕРКАПТАНОВ В СМЕСЯХ УГЛЕВОДОРОДОВ С | 2013 |
|
RU2628085C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛАРИЛСУЛЬФОНАТОВ | 2001 |
|
RU2312099C2 |
| СПОСОБ ПРЕДВАРИТЕЛЬНОЙ ОБРАБОТКИ В УСТАНОВКЕ МЕТАТЕЗИСА С ОБРАЗОВАНИЕМ ОКТЕНА | 2008 |
|
RU2460713C1 |
| СПОСОБ КОНВЕРСИИ УГЛЕВОДОРОДНОЙ ЗАГРУЗКИ | 2003 |
|
RU2294916C2 |
| СПОСОБ МНОГОСТАДИЙНОЙ КОНВЕРСИИ ЗАГРУЗКИ, СОДЕРЖАЩЕЙ ОЛЕФИНЫ С ЧЕТЫРЬМЯ, ПЯТЬЮ ИЛИ БОЛЕЕ АТОМАМИ УГЛЕРОДА, С ЦЕЛЬЮ ПОЛУЧЕНИЯ ПРОПИЛЕНА (ВАРИАНТЫ) | 2003 |
|
RU2299191C2 |
| ПОЛУЧЕНИЕ ПРОМЫСЛОВЫХ УГЛЕВОДОРОДОВ | 2015 |
|
RU2692491C2 |
| СПОСОБ ПОЛУЧЕНИЯ 1-БУТЕНА И ПРОИЗВОДНОГО 1,3-БУТАДИЕНА | 2012 |
|
RU2585764C2 |

Использование: нефтехимия. Сущность: содержащиеся во фракции С4 углеводороды по синтезу Фишера-Тропша бутены подвергают олигомеризации на стадии олигомеризации и из смеси олигомеризации получают дибутен, который на стадии четкой ректификации разделяют на ди-н-бутен и ди-изобутен. В одной форме выполнения способа содержащийся в олефинах по синтезу Фишера-Тропша этилен димеризуют, и смесь димеризации возвращают на стадию 12 разделения фракции С4. В другой форме выполнения способа между стадией 12 разделения фракции С4 и стадией 13 селективного гидрирования предусматривают стадию 5 этерификации, на которой содержащийся в подаваемой по линии 1 фракции С4 изобутен подвергают взаимодействию с подаваемым по линии 25 спиртом с получением отводимого по линии 26 алкил-трет-бутилового эфира и лишь оставшийся н-бутен олигомеризуют на стадии 2 олигомеризации, так что в качестве отводимого по линии 4 дибутена образуется исключительно ди-н-бутен, отводимый по линии 6. Кроме того, может иметься стадия 27 изомеризации, на которой непрореагировавшийся на стадии 2 олигомеризации н-бутен изомеризуют до изобутена, который подают на стадию 5 этерификации. Технический результат - повышение эффективности способа. 6 з.п.ф-лы, 2 ил.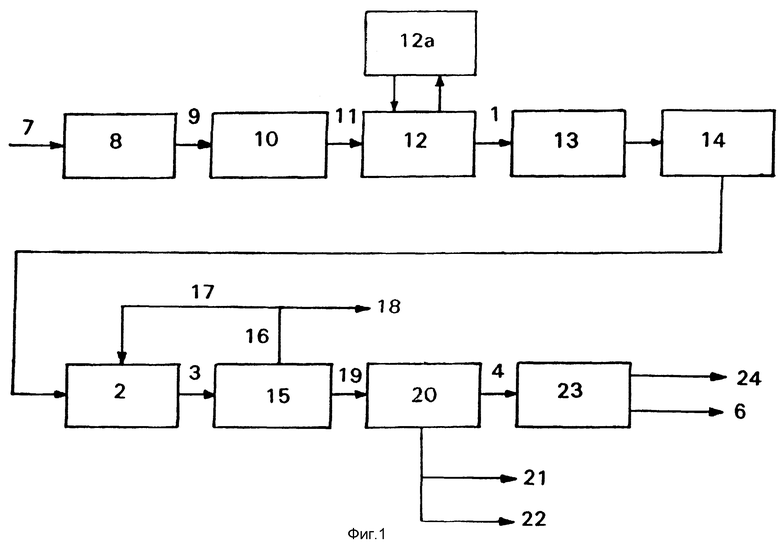
| СПОСОБ ПРОИЗВОДСТВА КУРИТЕЛЬНОЙ КОМПОЗИЦИИ ДЛЯ КАЛЬЯНА | 2015 |
|
RU2594138C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЖЕЛЕЙНОГО МАРМЕЛАДА С ИСПОЛЬЗОВАНИЕМ КОНЦЕНТРИРОВАННОЙ ПАСТЫ ИЗ ТЫКВЫ | 2015 |
|
RU2603895C1 |
| SU 1432970 А1, 27.08.1999 | |||
| US 5177282 А, 05.01.1993. | |||

