Изобретение относится к способу получения бутеновых олигомеров, представляющих собой ценное сырье для получения спиртов в качестве пластификаторов, из природных бутанов. Поэтому изобретение относится к способу получения - кроме высших бутеновых олигомеров - исключительно ди-н-бутена в качестве дибутена.
Под ди-н-бутеном понимают продукт димеризации н-бутена, то есть 1-бутена и/или 2-бутена. Важными компонентами ди-н-бутена являются 3-метил-2-гептен, 3,4-диметил-2-гексен и со второстепенным значением н-октены. Ди-изо-бутен представляет собой ту смесь димеров, которую получают при димеризации изобутена. Ди-изо-бутен содержит более разветвленные молекулы, чем дибутен, который в свою очередь является более разветвленным, чем ди-н-бутен.
Дибутен, ди-н-бутен и ди-изо-бутен являются исходными соединениями для получения измерных нонанолов путем гидроформилирования и гидрирования получаемых таким образом альдегидов с 9 атомами углерода. Сложные эфиры данных нонанолов, в частности сложные эфиры фталевой кислоты, представляют собой пластификаторы, получаемые в крупном масштабе и используемые в первую очередь в поливинилхлориде. Нонанолы из ди-н-бутена являются в большей степени прямолинейными, чем нонанолы из дибутена, которые, однако, со своей стороны менее разветвлены, чем нонанолы из ди-изо-бутена.
Бутены можно использовать для димеризации, например, из фракции С4 установки крекинга с водяным паром или установки для каталитического крекинга в кипящем слое. Данную фракцию перерабатывают, как правило, путем отделения 1,3-бутадиена путем селективной промывки, например, с использованием N-метилпирролидона. Изо-бутен представляет собой желаемый и особенно ценный компонент фракции С4, потому его можно подвергать химическому взаимодействию с получением продуктов, пользующихся большим спросом, например, с изо-бутаном до высокооктанового изооктана, или с метанолом до метил-трет-бутилового эфира, который в качестве присадки к бензину улучшает его октановое число. После взаимодействия изо-бутена в качестве остатка получают н-бутены и н- и изо-бутен. Однако доля н-бутенов среди продуктов расщепления из установки крекинга с водяным паром или установки для каталитического крекинга в кипящем слое является сравнительно небольшой, а именно, она составляет порядка около 10 вес.% в пересчете на главный целевой продукт, то есть этилен. Это означает, что с помощью установки крекинга с водяным паром, имеющей максимальную производительность 600 000 тонн этилена в год, получают лишь примерно 60 000 тонн н-бутенов в год. Можно было бы увеличить количество получаемых н-бутенов (и изо-бутенов) путем дегидрирования н- и изо-бутана, получаемых вместе с н-бутенами в количестве примерно 15 000 тонн в год. Однако это не рекомендуется, потому что установки для дегидрирования требуют высоких капиталовложений и являются неэкономичными для столь небольшой производительности.
Известен способ получения бутеновых олигомеров из бутенов, в котором н- и изо-бутены, содержащиеся в бутанах, подвергают дегидрированию на стадии дегидрирования и смесь дегидрирования подвергают олигомеризации на стадии олигомеризации. Из смеси олигомеризации выделяют газ, который рециркулируют на стадию дегидрирования. Затем смесь подвергают разделению с получением целевых продуктов (FR 2594138, 14.08.87).
Однако количество н-бутенов, непосредственно получаемых в установке крекинга с водяным паром или установки для каталитического крекинга в кипящем слое, не хватает для получения достаточно большого количества дибутена для установки для получения нонанола, производительность которой достаточно высока с тем, чтобы она могла с экономической точки зрения соперничать с уже имеющимися большими установками для получения важных спиртов, служащих в качестве пластификаторов, например, 2-этилгексанола. Это означает, что необходимо было бы собирать н-бутены из разных установок крекинга с водяным паром или установок для каталитического крекинга в кипящем слое и совместно подвергать из олигомеризации с тем, чтобы достичь полной загрузки большой установки для получения нонанола. Этому препятствует высокая стоимость перевозки жидких газов, не в последнюю очередь из-за требуемых сложных мер безопасности.
Поэтому было бы желательным, если бы бутены имелись в распоряжении для олигомеризации в одном месте без перевозки через большие расстояния, а именно в количестве, требуемом для работы большой установки для получения нонанолов, имеющей производительность, например, 200 000 - 800 000 тонн в год. Кроме того, было бы желательно иметь способ получения бутеновых олигомеров, позволяющий выделять ценный ди-н-бутен из дибутена. И наконец, было бы желательно управлять способом с обеспечением получения, кроме высших бутеновых олигомеров, исключительно ди-н-бутена или ди-изо-бутена в качестве дибутена.
Таким образом, объект изобретения представляет собой способ получения бутеновых олигомеров, включающий дегидрирование бутана, каталитическую олигомеризацию продукта дегидрирования и выделение целевого продукта, отличающийся тем, что перед каталитической олигомеризацией осуществляют избирательное гидрирование и/или очистку молекулярным ситом в любой последовательности, при этом дегидрированию подвергают природный бутан.
В дальнейшем данный способ обозначается как вариант А.
Согласно предпочтительному варианту, далее обозначаемому как вариант Б, из продукта олигомеризации выделяют остаточный газ, который рециркулируют на дегидрирование, при необходимости, после предварительной чистки, а от олигомеров, остающихся после отделения остаточного газа, отделяют дибутен.
Согласно дальнейшему варианту способа, в дальнейшем обозначаемому как вариант В, дибутены четким фракционированием разделяют на ди-н-бутены и остальные дибутены.
Далее, согласно варианту Г получают исключительно ди-н-бутен, причем из природного бутана, прошедшего, при необходимости, предварительное гидрирование, путем фракционной перегонки выделяют н-бутан, подаваемый на дегидрирование, остаточный изо-бутан подвергают изомеризации до смеси н-бутана и изо-бутана, из которой путем фракционной перегонки выделяют н-бутан, который вместе с н-бутаном, непосредственно выделяемым из природного бутана, подают на дегидрирование, а остаточный изо-бутан рециркулируют на изомеризацию.
И наконец, согласно варианту Д получают исключительно ди-изо-бутен, причем из природного бутана, прошедшего при необходимости, предварительное гидрирование, путем фракционной перегонки выделяют изо-бутан, подаваемый на дегидрирование, остаточный н-бутан подвергают изомеризации до смеси н-бутана и изо-бутана, из которой путем фракционной перегонки выделяют изо-бутан, который вместе с изо-бутаном, непосредственно выделяемым из природного бутана, подают на дегидрирование, а остаточный н-бутан рециркулируют на изомеризацию.
Предлагаемый способ, то есть каждый из его вариантов А-Д, отличается высокой гибкостью. То есть в зависимости от требований рынка можно получать по желанию или исключительно ди-н-бутен, или дибутен, или одновременно ди-н-бутен и другие дибутены, или исключительно ди-изо-бутен, хотя последний редко будет желательным главным продуктом.
Под природными бутанами понимают фракцию С4 "мокрых" компонентов природного газа и сопровождающих нефть газов, выделяемых из данных газов в жидкой форме путем охлаждения примерно до -30oС. Путем низкотемпературной перегонки из них получают природные бутаны, состав которых колеблется в зависимости от месторождения. Однако в общем природные бутаны содержат примерно 30% изо-бутана и 65% н-бутана и, как правило, в качестве других компонентов примерно 2% углеводородов с 1-3 атомами углерода и примерно 3% углеводородов с числом атомов углерода выше четырех. Природные бутаны можно без разделения использовать в качестве сырья в установках крекинга с водяным паром или в качестве присадки к автомобильному бензину. Их можно разделять на н-бутан и изо-бутан путем фракционной перегонки. Изо-бутан используют в крупном масштабе, например, для получения пропиленоксида путем соокисления пропилена и изо-бутана, а также в качестве агента алкилирования н-бутена соответственно изо-бутена до изо-октана, который из-за его высокого октанового числа ценят в качестве присадки к автомобильному бензину. В отличие от этого имеются лишь менее важные области применения н-бутана. Последний служит, например, в качестве бутанового газа для отопительных целей, или его используют, в сравнительно небольших количествах, для получения полимеров, или сополимеров, или ангидрида малеиновой кислоты путем окисления с помощью воздуха. Раньше н-бутан также дегидрировали через н-бутен до 1,3-бутадиена, однако тем временем этот метод стал неэкономичным.
Так как изо-бутан представляет собой более желательный компонент природного бутана, н-бутан в крупном масштабе подвергают изомеризации до изо-бутана (см., например, R. A. Pogliano и др., Dehydrogenation-based Ether Production, 1996 г. , в Petrochemical Review, издательство DeWitt & Company, Хьюстон, Техас, США, Butamer®- Verfahren, стр. 6; и S. Т. Bakas, F. Nierlich и др. , Production of Ethers from Field Butanes and Refinery Streams, AlChE Summer Meeting, 1990 г., Сан Дьего. Калифорния, стр. 11). Поэтому варианты А, Б и В направлены именно на использование н-бутана, содержащегося в природном газе, из которого через ди-н-бутен в качестве промежуточного продукта получают предпочтительные нонанолы. Противоречит общей тенденции в технике, если согласно варианту Г даже обычно желательный изо-бутан подвергают изомеризации до н-бутана.
Предлагаемый способ поясняется ниже с помощью приложенной схемы (см. фиг. 1), в которой показаны ниже более подробно описанные варианты А, Б, В и Г и их обязательные и факультативные стадии. Природный бутан 1 в качестве потока 1а относится к вариантам А, Б и В, а альтернативный поток 1b относится к вариантам Г и Д.
Вариант А
Природные бутаны 1а сначала подвергают дегидрированию. Дегидрирование представляет собой со-дегидрирование. Следует отметить, что дегидрирование природного бутана, представляющего собой смесь компонентов с разным поведением при дегидрировании, осуществляется гладко. Условия процесса в основном соответствуют известным условиям, обычно устанавливаемым для н- и изо-бутана или других низких углеводородов. Так, например, S. Т. Bakas, F. Nierlich и др. , в указанном месте, стр. 12 и сл., описывают способ Oleflex®, который в общем пригоден для избирательного получения легких олефинов и с помощью которого изо-бутан можно дегидрировать до изо-бутена с избирательностью 91-93%. Другие источники в этой связи представляют собой G. С. Sturtevant и др. , Oleflex - Selective Production of Light Olefins, 1988 UOP Technology Conference, и европейская заявка ЕР 0 149 698. Целесообразно осуществлять дегидрирование в газовой фазе в присутствии твердого катализатора или катализатора, имеющегося в виде кипящего слоя, например, в присутствии окиси хрома (III) или предпочтительно платины на окиси алюминия или цеолите в качестве носителя. Как правило, дегидрирование осуществляют при температуре от 400 до 800oС, предпочтительно от 550 до 650oС. Обычно процесс осуществляют при атмосферном давлении или слегка повышенном до 3 бар давлении. Время пребывания в катализаторном слое в общем составляет, в зависимости от катализатора, температуры и желаемой степени конверсии, 1-60 минут. Таким образом, производительность обычно составляет 0,6-36 кг природного бутана на м3 в час.
Целесообразно проводить дегидрирование до того момента, чтобы в смеси 3 оставалось еще примерно 50% н- и изо-бутанов в неизмененном виде. При более высокой температуре можно достичь более высокой степени конверсии, однако при этом в растущей мере осуществляются крекинговые реакции, снижающие выход и вследствие образования отложений кокса срок службы катализатора дегидрирования. Оптимальные комбинации условий реакции, приведущие к достижению желательной степени конверсии, например, вид катализатора, температуру и время пребывания, можно легко определить путем предварительных опытов.
Как правило, после дегидрирования смесь 3 содержит 90-95% углеводородов с 4 атомами углерода и, кроме того, водород и низко- и высоколетучие компоненты, отчасти происходящие из природного бутана 1 и отчасти образовавшиеся на стадии 2 дегидрирования. Целесообразно очищать смесь перед олигомеризацией. На первой стадии очистки (не показанной на схеме) путем конденсации удаляют фракцию С4 и высоколетучие компоненты. Конденсат подвергают перегонке под давлением, причем тоже конденсированные, растворенные углеводороды с 1-3 атомами углерода выходят из верхней части колонны. Из кубового продукта путем дальнейшей перегонки в качестве главного продукта получают углеводороды с 4 атомами углерода, а в качестве остатка - сравнительно небольшое количество углеводородов с числом атомов углерода, превышающим четыре.
В зависимости от степени конверсии углеводороды с 4 атомами углерода в общем содержат небольшое количество, например, 0,01-5 об.% 1,3-бутадиена. Рекомендуется удалять этот компонент, потому что даже в гораздо меньшем количестве он может вредить катализатору олигомеризации. Пригодным методом для этого является избирательное гидрирование, которым, кроме того, повышается доля желательного н-бутена. Пригодный метод описан, например, в F. Nieriich и др. , в Erdöl und Kohle, Erdgas, Petrochemie, 1986 г., стр. 73 и сл. Согласно этому методу работают в жидкой фазе с полностью растворенным водородом со стехиометрическими количествами. В качестве катализаторов для избирательного гидрирования пригодны, например, никель и, в частности, палладий на носителе, например, 0,3 вес. % палладия на активном угле или, предпочтительно, на окиси алюминия. Небольшое количество моноокиси углерода порядка частей на миллион поощряет избирательность гидрирования 1,3-бутадиена до моноолефина и тормозит образование полимеров (так называемого "зеленого масла"), дезактивирующих катализатор. В общем метод осуществляют при комнатной температуре или при повышенной температуре примерно до 60oС и при повышенном давлении, целесообразно составляющем до 20 бар. Таким образом содержание 1,3-бутадиена во фракции С4 смеси дегидрирования снижается до величины менее чем 1 часть на миллион.
Кроме того, является целесообразным перед олигомеризацией подавать полученную фракцию С4 смеси дегидрирования 5, уже почти освобожденную от 1,3-бутадиена, на стадию 6 очистки, пропуская ее через молекулярное сито, причем удаляются вещества, вредные для катализатора олигомеризации, благодаря чему продлевается срок службы катализатора. К таким вредным веществам относятся соединения кислорода и серы. Данный метод описан F. Nierlich и др. в европейской заявке ЕР В1-0 395 857. Целесообразно использовать молекулярное сито с размером пор 4-15 ангстрем. В некоторых случаях по экономическим причинам является целесообразным пропускать смесь дегидрирования последовательно через молекулярные сита с разной величиной пор. Данный метод можно осуществлять в газовой, жидкой или смешанной газо-жидкостной фазе. В зависимости от этого давление в общем составляет 1-200 бар. Целесообразно работать при комнатной температуре или при температуре до 200oС.
Химическая природа молекулярных сит играет меньшую роль, чем их физическая структура, то есть, в частности, размер пор. Это означает, что можно использовать самые разные молекулярные сита, кристаллические, природные силикаты алюминия, например слоистые решетчатые силикаты, так же как и синтетические молекулярные сита, например, такие как с цеолитной структурой. Имеются в торговле цеолиты типа А, Х и Y, среди других, фирм Байер АГ, Дау Хемикальс Ко. , Юньон Карбайд Корпорейшн, Лапорт Индастриз Лтд. и Мобиль Ойль Ко. Для осуществления способа пригодны также такие синтетические молекулярные сита, которые наряду с алюминием и кремнием содержат и другие атомы, введенные путем катионного обмена, например, галлий, индий или лантан, а также никель, кобальт, медь, цинк или серебро. Кроме того, пригодны также синтетические цеолиты, в которых кроме алюминия и кремния путем со-осаждения в решетку встроены еще дополнительные атомы, например бор или фосфор.
Как уже указывалось, стадия 4 избирательного гидрирования и стадия 6 очистки с помощью молекулярного сита являются факультативными, предпочтительными мерами в рамках предлагаемого способа. Их последовательность в принципе не играет никакой роли, однако предпочтительной является показанная на чертеже последовательность.
Смесь дегидирования 7, в случае необходимости после описанной предварительной обработки, подают на стадию 8 олигомеризации, которая представляет собой существенный прием предлагаемого способа. Олигомеризация является со-олигомеризацией н-бутенов и изо-бутена, осуществляемой известным методом, описанным, например, F. Nierlich в Oligomerization for Better Gasoline, Hydrocarbon Processing, 1992 г. , стр. 45 и cл., или F. Nierlich и др., в европейской заявке ЕР-В1-0 395 857. В общем работают в жидкой фазе в присутствии, например, системы, состоящей из октоата никеля (II), хлорида этилалюминия и свободной жирной кислоты (см. патент DE 28 55 423), в качестве гомогенного катализатора, или, предпочтительно, один из многочисленных известных, неподвижно установленных или суспендированных в смеси олигомеризации катализаторов на основе никеля и кремния. Часто катализаторы дополнительно содержат алюминий. Так, например, в патенте DD 160 037 описано получение содержащего никель и алюминий катализатора осаждения на основе двуокиси кремния в качестве носителя. Другие пригодные катализаторы получают путем замены имеющихся на основе носителей частиц с положительным зарядом, например, протонов или ионов натрия, ионами никеля. Это возможно в случае самых разных носителей, например аморфного силиката алюминия (см. R. Espinoza и др., Appl. Kat, 31 (1987 г.), стр. 259-266), кристаллического силиката алюминия (см. патент DE 20 29 624), цеолитов типа ZSM (см. патент NL 8 500 459), цеолита типа Х (см. патент DE 23 47 235), цеолитов типов Х и Y (см. A. Barth и др., Z. Anorg. Allg. Chem., 521 (1985 год), стр. 207 - 214) и морденита (см. европейскую заявку ЕР-А-0 233 302).
Целесообразно осуществлять со-олигомеризацию в зависимости от используемого катализатора при температуре 20-200oС и давлении 1-100 бар. Время реакции (или контакта) в общем составляет 5-60 минут. Параметры способа, в частности вид катализатора, температуру и время контакта, согласуют друг с другом с тем, чтобы достигалась желаемая степень олигомеризации. Естественно, в том случае, если желаемым целевым продуктом являются нонанолы, олигомеризация представляет собой димеризацию. Само собой разумеется, что для этого нельзя добиться полной конверсии при реакции, а целесообразно добиваться 30-70%-ной конверсии для каждого цикла реакции. Оптимальную комбинацию параметров способа можно без проблем устанавливать с помощью предварительных опытов.
На стадии 10 разделения из смеси олигомеризации 9 выделяют остаточный газ 12, который рециркулируют на стадию 2 дегидрирования. В том случае, если на стадии 8 олигомеризации использовали катализатор типа указанных жидких катализаторов, то желательно, чтобы перед рециркуляцией остаточный газ 12 очищали с тем, чтобы щадить катализатор дегидрирования. Сперва смесь олигомеризации обрабатывают водой для экстракции катализатора. Затем отделенный остаточный газ 12 сушат с использованием подходящего молекулярного сита, причем отделяются также другие побочные компоненты. После этого путем избирательного гидрирования, например, в присутствии палладиевого катализатора удаляют многократно ненасыщенные соединения, например бутины, и в конце очищенный таким образом остаточный газ 12 рециркулируют на стадию дегидрирования 2. Эти меры очистки остаточного газа 12 отпадают при использовании твердого катализатора для олигомеризации.
Олигомеры 11, получаемые после отделения остаточного газа 12, пригодны в качестве присадки к автомобильному бензину для улучшения октанового числа благодаря их разветвленным компонентам.
Вариант Б
На стадии 13 перегонки олигомеры 11 разделяют на дибутены 14, тримеры 15, то есть изомерные додецены, и еще более высокие олигомеры, причем главная фракция состоит из желаемых дибутенов 14. Додецены можно подвергать гидроформилированию, продукты гидроформилирования можно подвергать гидрированию, и получаемые таким образом тридеканолы можно подвергать оксиэтилированию, в результате чего получают ценное сырье моющих средств. Дибутены 14 непосредственно пригодны в качестве сырья для получения нонанола.
Вариант В
В том случае, если важными являются специфические свойства нонанолов из ди-н-бутена, дибутены 14 на стадии 16 четкого фракционирования разделяют на ди-н-бутен 17 и остальные дибутены 18, которые как более разветвленные молекулы имеют более низкую точку кипения. Дибутены 18 также можно использовать для получения нонанолов или присадки к автомобильному бензину. Этот способ представляет собой выгодную альтернативу для варианта, согласно которому из смеси со-дегидрирования 7 путем перегонки выделяют н- и изо-бутены с последующей отдельной олигомеризацией данных изомеров. Этот вариант требовал бы две раздельные стадии олигомеризации, что привело бы к повышению требуемых капиталовложений и стоимости процесса по сравнению с одной единственной, хотя и большей стадией со-олигомеризации 8 в сочетании со стадией четкого фракционирования.
Вариант Г
Этот вариант осуществляют тогда, если в качестве дибутена желают получать исключительно ди-н-бутен. В том случае, если природный бутан 1b содержит олефиново ненасыщенные компоненты, то его целесообразно подавать сперва на стадию гидрирования 19, потому что данные компоненты могут мешать последующей изомеризации изо-бутана. Гидрирование осуществляют известным методом, описанным, например, К. Н. Walter и др. в The Hüls Process for Selective Hydrogenation of Butadien in Crude C4's, Development and Technical Application, совещание DGKM-Tagung Kassel, ноябрь 1993 г. То есть целесообразно работать в жидкой фазе, в зависимости от используемого катализатора при комнатной температуре или повышенной температуре до 90oС и при давлении 4-20 бар, причем парциальное давление водорода составляет 1-15 бар. Используют обычные для осуществления гидрирования олефинов катализаторы, например 0,3%-ный палладий на окиси алюминия.
Гидрированные природные бутаны 20 подают на стадию разделения 21, которая обычно осуществляется в высокоэффективной колонне, в которой н-бутан 22 и изо-бутан 23 разделяются путем фракционной перегонки. В колонне 21 работа осуществляется обычным образом, целесообразно при давлении 4-7 бар. Углеводороды с 1-3 атомами углерода получают в качестве кубового продукта, н-бутан 22 отводят по побочному потоку и вместе с остаточным газом 12 подают на стадию 2 дегидрирования, и изо-бутан 23, точка кипения которого на 10-20oС ниже, вместе с более легкими компонентами подают на стадию изомеризации 24, на которой изо-бутан превращается в н-бутан максимум до состава в зависимости от температуры примерно 40-55% н-бутана и 45-60% изо-бутана. Изомеризация н- и изобутана известна, хотя ее обычно осуществляют с целью получения изобутана (см. Н. W. Grote, Oil and Gas Journal, 56 (13, стр. 73 и сл., 1958 г. )). В общем работают в газовой фазе, целесообразно при температуре 150-230oС и при давлении 14-30 бар, используя в качестве катализатора платину на окиси алюминия в качестве носителя, избирательность которого можно еще повысить путем добавления к нему хлористого соединения, например, четыреххлористого углерода. Для предотвращения дегидрирования предпочтительно также добавляют небольшое количество водорода. Избирательность изомеризации до н-бутана высока, и расщепление на меньшие части происходит лишь в небольшом объеме (примерно 2%).
Смесь изомеризации 25 необходимо разделять на изомеры, для чего целесообразно использовать все же уже имеющуюся колонну 21, из которой н-бутан поступает на стадию 2 дегидрирования, не которой в отличие от вариантов А, Б и В не осуществляется совместное дегидрирование. В остальном вариант Г соответствует остальным вариантам. На стадии олигомеризации 8 осуществляется совместная олигомеризация, ведь н-бутен со стадии дегидрирования 2 является смесью 1-бутена и 2-бутена. Однако стадия четкого фракционирования 16 может отпадать, так как дибутен 14 уже представляет собой ди-н-бутен.
Вариант Д
Данный вариант выбирают в том случае, если желают получать исключительно ди-изо-бутен в качестве дибутена, хотя это будет происходить лишь в порядке исключения. В принципе данный вариант осуществляют аналогично варианту Г, причем, однако, из колонны 21 изобутан 22а подают на стадию 2 дегидрирования, на которой, также как согласно варианту Г, но иначе, чем в вариантах А, Б и В, не осуществляется совместное дегидрирование. Н-бутан 23а подают из колонны 21 на стадию 24 изомеризации, где его подвергают изомеризации до изобутана максимально до равновесия. Изобутан отделяют от н-бутана, для чего и в данном случае целесообразно использовать колонну 21, и также подают на стадию 2 дегидрирования, в то время как н-бутан рециркулируют на стадию 24 изомеризации. Таким образом н-бутан полностью превращают в изо-бутан. Смесь дегидрирования 3 целесообразно очищают описанным в связи с вариантом А образом. Олигомеризация на стадии 8 представляет собой гомоолигомеризацию, так как в ней участвует лишь изо-бутан, и на стадии 13 перегонки получают ди-изо-бутен. И в данном случае отпадает четкое фракционирование 16.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ СОВМЕСТНОГО ПОЛУЧЕНИЯ ДИ-Н-БУТЕНА И АЛКИЛ-ТРЕТ-БУТИЛОВЫХ ЭФИРОВ ИЗ ПРИРОДНЫХ БУТАНОВ | 1997 |
|
RU2178782C2 |
| СПОСОБ ПОЛУЧЕНИЯ БУТЕНОЛИГОМЕРОВ ИЗ ОЛЕФИНОВ ПО СИНТЕЗУ ФИШЕРА-ТРОПША | 1997 |
|
RU2189372C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВЫСШИХ ОКСО-СПИРТОВ | 1997 |
|
RU2183210C2 |
| СПОСОБ ПОЛУЧЕНИЯ 1-БУТЕНА И ПРОИЗВОДНОГО 1,3-БУТАДИЕНА | 2012 |
|
RU2585764C2 |
| ТИОЭТЕРИФИКАЦИЯ МЕРКАПТАНОВ В СМЕСЯХ УГЛЕВОДОРОДОВ С | 2013 |
|
RU2628085C2 |
| СПОСОБ ПРЕДВАРИТЕЛЬНОЙ ОБРАБОТКИ В УСТАНОВКЕ МЕТАТЕЗИСА С ОБРАЗОВАНИЕМ ОКТЕНА | 2008 |
|
RU2460713C1 |
| АДСОРБЦИЯ СЕРНИСТЫХ СОЕДИНЕНИЙ ИЗ ОЛЕФИНОВЫХ СМЕСЕЙ ПРИ ПОМОЩИ ВОДОРОДА | 2016 |
|
RU2645159C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛАРИЛСУЛЬФОНАТОВ | 2001 |
|
RU2312099C2 |
| СПОСОБ ПОЛУЧЕНИЯ МЕТИЛ-ТРЕТ.-БУТИЛОВОГО ЭФИРА И ПОЧТИ СВОБОДНОЙ ОТ ИЗОБУТЕНА СМЕСИ УГЛЕВОДОРОДОВ С4 | 2001 |
|
RU2250893C2 |
| СПОСОБ МНОГОСТАДИЙНОЙ КОНВЕРСИИ ЗАГРУЗКИ, СОДЕРЖАЩЕЙ ОЛЕФИНЫ С ЧЕТЫРЬМЯ, ПЯТЬЮ ИЛИ БОЛЕЕ АТОМАМИ УГЛЕРОДА, С ЦЕЛЬЮ ПОЛУЧЕНИЯ ПРОПИЛЕНА (ВАРИАНТЫ) | 2003 |
|
RU2299191C2 |
Использование: нефтехимия Н и изобутаны, содержащиеся в природных бутанах, подвергают дегидрированию на стадии, смесь дегидрирования подвергают олигомеризации. Путем дополнительного осуществления стадии изомеризации данным способом можно получать исключительно ди-н-бутен или ди-изо-бутен в качестве дибутена. Технический результат: упрощение технологии процесса. 5 з. п. ф-лы, 1 ил.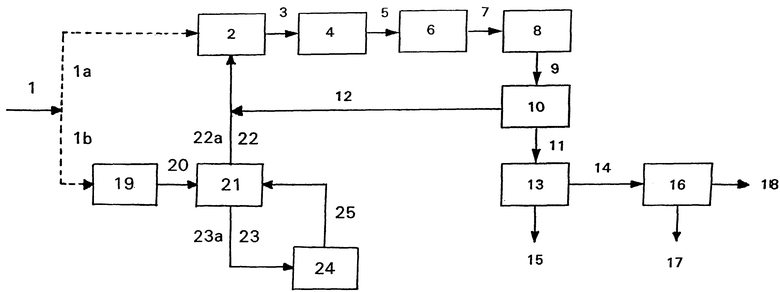
| СПОСОБ ПРОИЗВОДСТВА КУРИТЕЛЬНОЙ КОМПОЗИЦИИ ДЛЯ КАЛЬЯНА | 2015 |
|
RU2594138C1 |
| Способ получения олигомеров этилена | 1985 |
|
SU1351912A1 |
| СПОСОБ ПОЛУЧЕНИЯ ЖЕЛЕЙНОГО МАРМЕЛАДА С ИСПОЛЬЗОВАНИЕМ КОНЦЕНТРИРОВАННОЙ ПАСТЫ ИЗ ТЫКВЫ | 2015 |
|
RU2603895C1 |
| US 5177282 A, 05.01.1993. | |||

