Настоящее изобретение касается способа получения алкиларилсульфонатов, получаемых этим способом алкиларилсульфонатов и образующихся в качестве промежуточных продуктов алкилароматических соединений, применения алкиларилсульфонатов в качестве поверхностно-активных веществ, предпочтительно в составе моющих и чистящих средств, и применения содержащих алкиларилсульфонаты моющих и чистящих средств.
Алкилбензолсульфонаты (ABS) давно используют в составе моющих и чистящих средств в качестве поверхностно-активных веществ. Вначале использовали поверхностно-активные вещества на основе тетрапропилена, но поскольку они обладали неудовлетворительной способностью к биологическому расщеплению, впоследствии перешли к получению и использованию, по возможности, линейных алкилбензолсульфонатов (LAS). Однако линейные алкилбензолсульфонаты не обладают достаточно полным комплексом свойств, который позволял бы использовать их в любых сферах.
Так, например, следовало бы улучшить моющую способность сульфонатов в холодной воде или их поведение в жесткой воде. Наряду с этим желательно усовершенствовать определяемую их вязкостью и растворимостью пригодность к переработке, чтобы упростить приготовление соответствующих рецептур. Улучшения этих свойств достигают, благодаря ограниченному разветвлению соединений или смешиванию слаборазветвленных соединений с линейными соединениями, при этом необходимо соблюдать надлежащую степень разветвления и/или надлежащее соотношение линейных и разветвленных соединений. Чрезмерная степень разветвления уменьшает способность продукта к биологическому расщеплению. Слишком линейные продукты отрицательно влияют на вязкость и растворимость сульфонатов.
Кроме того, на свойства продуктов оказывает влияние содержание алканов с концевым (в положениях 2 и 3) и внутренним (в положениях 4, 5, 6 и так далее) замещением фенильными группами. С точки зрения качества продуктов (растворимости, вязкости, моющих свойств) предпочтительными могут быть продукты, содержащие около 30% 2-фенилалканов и около 50% 2- и 3-фенилалканов.
Существенным недостатком поверхностно-активных веществ с повышенным содержанием 2- и 3-фенилалканов может быть ухудшение их технологических свойств, обусловленное сильным повышением вязкости.
Вместе с тем, если содержание 2- и 3-фенилалканов отличается от оптимального, это может привести к ухудшению растворимости сульфонатов. Так, например, точка Крафта растворов линейных алкилбензолсульфонатов, обладающих слишком высоким или слишком низким содержанием 2- и 3-фенилалканов, на 10-20°С выше по сравнению с растворами алкилбензолсульфонатов с оптимальным содержанием 2- и 3-фенилалканов.
Существенное преимущество способа согласно изобретению состоит в том, что благодаря комбинированию реакций обмена и димеризации, получают уникальную смесь олефинов, используемую для алкилирования ароматического углеводорода, и после сульфирования и нейтрализации продуктов алкилирования получают поверхностно-активное вещество, обладающее сочетанием исключительно высоких технических характеристик (растворимости, вязкости, стабильности свойств в жесткой воде, моющей способности, способности к биологическому расщеплению). Что касается способности алкиарилсульфонатов к биологическому расщеплению, особенно предпочтительными являются продукты, которые не так сильно адсорбируются образующимся при осветлении шламом по сравнению с обычными линейными сульфонатами.
Итак, были разработаны алкилбензолсульфонаты с известной степенью разветвления.
Например, в патенте США US 3442964 описывается димеризация С5-С6-углеводородов, осуществляемая в присутствии содержащего переходный металл катализатора крекинга, в результате которой образуются олефины преимущественно с двумя или более разветвлениями. Алкилирование бензола этими олефинами приводит к образованию разветвленного алкилбензола. Например, смесь гексенов димеризуют в присутствии катализатора крекинга на основе диоксида кремния и оксида алюминия, и димеры используют для алкилирования, осуществляемого под влиянием плавиковой кислоты в качестве катализатора.
Международная заявка WO 88/07030 касается олефинов, алкилбензолов и алкилбензолсульфонатов, которые могут быть использованы в составе моющих и чистящих средств. Согласно этой заявке пропен димеризуют до гексена, который, в свою очередь, подвергают димеризации с образованием изомеров додецена, обладающих преимущественно линейной структурой. Последние используют для алкилирования бензола, осуществляемого в присутствии галогенидов алюминия и плавиковой кислоты.
В патенте США US 5026933 описана димеризация пропена или бутена до моноолефинов, по меньшей мере, 20% которых представляют собой C12-олефины со степенью разветвления, которой соответствует содержание от 0,8 до 2,0 метильных групп в расчете на одну алкильную цепь, причем разветвления образованы исключительно метильными группами. Ароматические углеводороды алкилируют в присутствии селективного катализатора, предпочтительно деалюминированного морденита.
Международная заявка WO 99/05241 касается чистящих средств, содержащих в качестве поверхностно-активного вещества алкиларилсульфонаты, получаемые димеризацией олефинов с образованием винилидинолефинов, алкилированием бензола в присутствии селективного катализатора, в частности морденита или бета (ВЕА), с последующим сульфированием.
Олефины, до последнего времени используемые для алкилирования, часто обладают слишком высокой или слишком низкой степенью разветвления, и образующиеся фенилалканы характеризуются неоптимальным соотношением концевых и внутренних фенильных групп. С другой стороны, такие олефины синтезируют из дорогих исходных соединений, например из пропена или α-олефинов, и содержание олефиновых фракций, представляющих интерес с точки зрения получения поверхностно-активных веществ, нередко не превышает 20%, что требует использования для их выделения дорогостоящих способов.
Задача настоящего изобретения состоит в создании способа получения алкиларилсульфонатов, которые, по меньшей мере, частично разветвлены и которые при использовании в составе моющих и чистящих средств обладают более предпочтительными свойствами по сравнению с известными соединениями. В частности, алкиларилсульфонаты согласно изобретению должны обладать пригодным для получения и использования комплексом свойств, включая способность к биологическому расщеплению, отсутствие чувствительности к ионам, придающим жесткость воде, растворимость и вязкость. Кроме того, способ получения алкиларилсульфонатов должен быть экономичным.
Согласно изобретению поставленная задача решается способом получения аликиарилсульфонатов, включающего следующие технологические стадии:
a) превращение смеси С4-олефинов в смесь олефинов, содержащую 2-пентен и/или 3-гексен, в присутствии катализатора реакции обмена и, при необходимости, выделение 2-пентена и/или 3-гексена,
b) димеризация полученного на стадии а) 2-пентена и/или 3-гексена в присутствии катализатора димеризации с образованием содержащей С10-С12-олефины смеси и, при необходимости, выделение С10-С12-олефинов,
c) взаимодействие полученной на стадии b) смеси С10-С12-олефинов с ароматическим углеводородом в присутствии катализатора алкилирования с образованием алкилароматических соединений, причем перед взаимодействием дополнительно могут быть добавлены линейные олефины,
d) сульфирование полученных на стадии с) алкилароматических соединений и нейтрализация продуктов сульфирования до алкиларилсульфонатов, причем перед сульфированием дополнительно могут быть добавлены линейные алкилбензолы,
e) при необходимости, смешивание полученных на стадии d) алкиларилсульфонатов с линейными алкиларилсульфонатами.
Благодаря комбинации реакции обмена С4-олефинов с последующей димеризацией и алкилированием ароматических углеводородов могут использоваться недорогие исходные соединения и способы получения, позволяющие получать желаемые продукты с высоким выходом.
Согласно изобретению было обнаружено, что путем реакции обмена C4-олефинов получают продукты, способные димеризоваться с образованием смесей слаборазветвленных С10-С12-олефинов. Последние предпочтительно могут использоваться для алкилирования ароматических углеводородов, в результате которого образуются продукты, которые после сульфирования и нейтрализации превращаются в поверхностно-активные вещества, обладающие ценным комплексом свойств, в частности низкой чувствительностью по отношению к ионам, придающим жесткость воде, оптимальными растворимостью и вязкостью и отличной моющей способностью. Кроме того, данный способ является чрезвычайно экономичным, поскольку производственный процесс может быть организован достаточно гибко и позволяет избежать образования побочных реакционных продуктов. Путем осуществляемой согласно изобретению реакции обмена исходных С4-олефинов получают линейные олефины с внутренними двойными связями, которые на последующей стадии димеризации превращают в разветвленные олефины.
Стадия а) способа согласно изобретению заключается в превращении смеси С4-олефинов в присутствии катализатора реакции обмена в содержащую 2-пентен и/или 3-гексен смесь олефинов и, при необходимости, выделении 2-пентена и/или 3-гексена. Реакция обмена может быть осуществлена, например, как описано в международной заявке WO 00/39085 или немецкой заявке на патент DE-A-10013253.
Наиболее простая форма осуществления реакции обмена олефинов (диспропорционирования) представляет собой обратимое, катализируемое металлами, сопровождающееся разрывом или формированием новых двойных связей переалкилиденирование олефинов, которое протекает в соответствии со следующим уравнением:

В особом случае в отличие от перекрестной реакции обмена или сореакции обмена ациклических олефинов, то есть взаимодействия двух разных олефинов (например, пропен+1-бутен → этен+2-пентен), может происходить самопроизвольная реакция обмена, в результате которой олефин превращается в смесь двух других олефинов разной молекулярной массы (например, пропен → этен+2-бутен). Если при перекрестной реакции обмена одним из исходных реагентов является этен, то в общем случае речь идет об этенолизе.
В качестве катализаторов реакции обмена в принципе пригодны гомогенные и гетерогенные соединения переходных металлов, в частности металлов из VI-VIII побочных групп периодической системы элементов, а также гомогенные и гетерогенные каталитические системы, содержащие такие соединения.
Согласно изобретению могут использоваться разные способы реакции обмена С4-олефинов.
Немецкая заявка на патент DE-A-199 32060 касается способа получения C5/С6-олефинов путем превращения исходной смеси, содержащей 1-бутен, 2-бутен и изобутен, в смесь С2-С6-олефинов, причем из бутенов, в частности, получают пропен. В качестве других продуктов реакции выделяют гексен и метилпентен. Этен в состав подвергаемых реакции обмена олефинов не входит. Образующийся при реакции обмена этен, при необходимости, возвращают в реактор.
Предпочтительный способ получения, при необходимости, пропена и гексена из исходного, содержащего олефиновые С4-углеводороды рафината II, отличается тем, что:
a) в присутствии катализатора метатезиса, содержащего, по меньшей мере, одно соединение металла из VIб, VIIб или VIII побочных групп периодической системы элементов, осуществляют реакцию метатезиса, то есть превращение содержащихся в исходной смеси бутенов и этена в смесь этена, пропена, бутенов, 2-пентена, 3-гексена и бутанов, причем мольное отношение этена к бутенам в исходной смеси составляет до 0,6: 1,
b) полученную на стадии а) реакционную смесь подвергают дистилляции, разделяя на две фракции, одна из которых, при необходимости, является легкокипящей, содержащей С2-С3-олефины фракцией А, а другая высококипящей, содержащей С4-С6-олефины и бутаны фракцией,
c) полученную, при необходимости, на стадии b) легкокипящую фракцию А подвергают дистилляции, разделяя на две фракции, одна из которых содержит этен, а другая пропен, причем фракцию, содержащую этен, возвращают на стадию а), а фракцию, содержащую пропен, выгружают в качестве целевого продукта реакции,
d) полученную на стадии b) высококипящую фракцию подвергают дистилляции, разделяя на три фракции, первая из которых является легкокипящей, содержащей бутены и бутаны фракцией В, вторая обладающей средней температурой кипения, содержащей 2-пентен фракцией С, а третья высококипящей, содержащей 3-гексен фракцией D,
e) причем фракцию В и, при необходимости, фракцию С полностью или частично возвращают на технологическую стадию а), а фракцию D и, при необходимости, фракцию С выгружают в качестве целевых продуктов.
Отдельные потоки и фракции могут содержать указанные выше соединения или состоять из этих соединений. В последнем случае не исключается возможность присутствия небольших количеств других углеводородов.
При этом фракцию С4-олефинов, предпочтительно состоящую из н-бутенов и бутанов, путем одноступенчатой реакции обмена, осуществляемой в присутствии гомогенных или предпочтительно гетерогенных катализаторов реакции обмена, при необходимости, с добавлением варьируемых количеств этена превращают в реакционную смесь, содержащую (инертные) бутаны, непревращенные 1-бутен и 2-бутен, а также этен, пропен, 2-пентен и 3-гексен в качестве продуктов реакции обмена. 2-Пентен и/или 3-гексен в качестве целевых продуктов выгружают, а прочие продукты и непревращенные соединения полностью или частично возвращают в реакцию обмена. Предпочтительным является возможно более полное их возвращение в реакцию обмена, причем выгрузке подлежит лишь небольшое количество углеводородов, предупреждающее переполнение. В идеальном случае дело до переполнения не доходит, и все соединения, за исключением 3-гексена, возвращают в реакцию обмена.
Согласно изобретению мольное отношение этена к бутенам, содержащимся в исходной смеси С4-углеводородов, составляет до 0,6:1, предпочтительно до 0,5: 1. Следовательно, по сравнению с уровнем техники используют лишь ограниченные количества этена.
Если дополнительный этен не подают, то в реакцию обмена возвращают лишь образующийся согласно способу этен, концентрация которого в реакционных продуктах не превышает 1,5% (смотри немецкую заявку на патент DE-A-19932060). Согласно изобретению возможно использование и больших количеств этена, причем они остаются гораздо меньшими по сравнению с известными способами получения пропена.
Согласно изобретению в реакцию обмена возвращают максимально возможные количества полученных в реакторе С4-, а, при необходимости, и C5-углеводородов. Речь, в частности, идет о возвращении непрореагировавших 1-бутена и 2-бутена, а также, при необходимости, образовавшегося н-пентена.
Если исходный поток С4-углеводородов содержит небольшие количества изобутена, могут образоваться также небольшие количества разветвленных углеводородов.
Количество дополнительно образующихся разветвленных С5 иС6-углеводородов в продуктах реакции обмена зависит от содержания изобутена в исходных С4-углеводородах, и это количество предпочтительно поддерживают на как можно более низком уровне (менее 3%).
Для более детального пояснения способа осуществления реакции обмена согласно изобретению происходящее в реакторе превращение разделяют на три следующие, наиболее важные реакции.
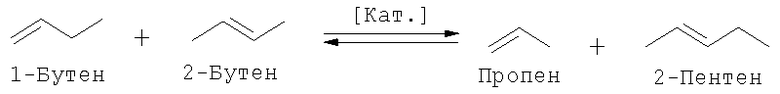
1. Перекрестная реакция обмена 1-бутена и 2-бутена
2. Самопроизвольная реакция обмена 1-бутена
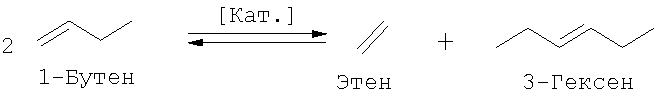
3. При необходимости, этенолиз 2-бутена
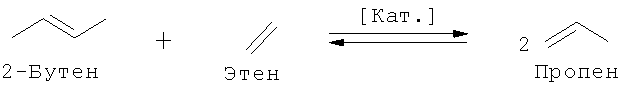
В зависимости от потребности в целевых продуктах: пропене и 3-гексене (под 3-гексеном подразумеваются и другие возможные изомеры гексена) или 2-пентене можно оказать целенаправленное воздействие на общий массовый баланс способа получения путем использования варьируемого количества этена и смещения равновесия за счет рециркуляции определенных потоков. Так, например, выход 3-гексена повышают, подавляя перекрестную реакцию обмена 1-бутена и 2-бутена за счет возвращения 2-пентена на стадию реакции обмена, вследствие чего 1-бутен не расходуется в этой реакции или расходуется в минимально возможном количестве. Тогда предпочтительно протекает самопроизвольная реакция обмена 1-бутена с образованием 3-гексена и дополнительного количества этена, последующее взаимодействие которого с 2-бутеном приводит к образованию пропена в качестве ценного продукта.
Смесь олефинов, содержащую 1-бутен, 2-бутен и, при необходимости, изобутен, получают в виде С4-фракции, в частности, реализуя различные процессы крекинга, например парофазный или жидкофазный каталитический крекинг. В качестве альтернативы возможно использование смеси бутенов, получаемых путем дегидрирования бутанов или димеризации этена. Входящие в состав С4-фракции бутаны ведут себя инертно. Диены, алкины или енины перед осуществляемой согласно изобретению реакции обмена удаляют обычными способами, в частности путем экстракции или селективного гидрирования.
Содержание бутенов (1-бутена, 2-бутена и изобутена) в составе используемой в способе С4-фракции составляет от 1 до 100 мас.%, предпочтительно от 60 до 90 мас.%.
Предпочтительным является использование С4-фракции, образующейся в результате парофазного или жидкофазного каталитического крекинга или дегидрирования бутана.
В качестве подвергаемой реакции обмена С4-фракции предпочтительно используют рафинат II, причем перед подачей в реакцию обмена его освобождают от осложняющих проведение этой реакции примесей, пропуская через адсорбционные слои, предпочтительно состоящие из оксидов алюминия с высокой удельной поверхностью или молекулярные сита.
Получаемую, при необходимости, на стадии b) легкокипящую, содержащую С2-С3-олефины фракцию А подвергают дистилляции, разделяя на фракцию, содержащую этен, и фракцию, содержащую пропен, первую из которых возвращают на стадию а), то есть подвергают реакции обмена, а вторую выгружают в качестве реакционного продукта.
Для осуществляемого на стадии d) разделения на легкокипящую фракцию В, фракцию со средней температурой кипения С и высококипящую фракцию D может использоваться, например, колонна с перегородками, причем фракцию В отбирают в головной части колонны, фракцию С в средней ее части, а фракцию D выгружают в качестве кубового остатка.
Однако для более удобного манипулирования различными количествами образующихся продуктов предусмотрен способ гибкого управления процессом, согласно которому предпочтительно осуществляют двухступенчатое разделение полученной на стадии b) высококипящей фракции, а именно предпочтительно сначала проводят ее дистилляцию, разделяя на легкокипящую, содержащую бутены и бутаны фракцию В, и высококипящую, содержащую 2-пентен и 3-гексен фракцию, которую затем путем дистилляции разделяют на фракции С и D.
Реакцию обмена предпочтительно осуществляют в присутствии гетерогенных катализаторов реакции обмена, не способных вызывать изомеризацию или обладающих ограниченной изомеризационной активностью, которые представляют собой соединения переходных металлов из VIб, VIIб или VIII побочных групп Периодической системы элементов на неорганических носителях.
Предпочтительным катализатором реакции обмена является оксид рения на носителе, предпочтительно на γ-оксиде алюминия или смешанном носителе, состоящем из оксида алюминия, оксида бора и диоксида кремния(Al2O3/В2O3/SiO2).
В частности, в качестве катализатора используют рениевый ангидрид на γ-оксиде алюминия (Re2О7/γ-Al2О3) с содержанием рениевого ангидрида от 1 до 20 мас.%, предпочтительно от 3 до 15 мас.%, особенно предпочтительно от 6 до 12 мас.%.
Реакцию обмена осуществляют в жидкой фазе предпочтительно при температуре от 0 до 150°С, особенно предпочтительно от 20 до 80°С и давлении от 2 до 200 бар, особенно предпочтительно от 5 до 30 бар.
Если реакцию обмена осуществляют в газовой фазе, то температура предпочтительно составляет от 20 до 300°С, особенно предпочтительно от 50 до 200°С, давление предпочтительно составляет от 1 до 20 бар, особенно предпочтительно от 1 до 5 бар.
Получение C5- и С6-олефинов и, при необходимости, пропена из синтезированной путем парофазного крекинга С4-фракции или рафинированной С4-фракции может состоять из следующих стадий.
(1) Выделение бутадиена и производных ацетилена, при необходимости, осуществляемое экстракцией бутадиена соответствующим селективным растворителем, с последующим селективным гидрированием /или селективное гидрирование бутадиена и ацетиленовых примесей, содержащихся в С4-фракции, с целью получения реакционного продукта, содержащего н-бутены и изобутен при практически полном отсутствии бутадиена и производных ацетилена.
(2) Выделение изобутена из полученного на предыдущей стадии реакционного продукта путем его взаимодействия со спиртом в присутствии кислого катализатора, приводящего к образованию соответствующего эфира, и выделение эфира и спирта, которое может быть осуществлено одновременно с этерификацией или после нее, с целью получения реакционного продукта, содержащего н-бутены и, при необходимости, кислородсодержащие примеси, причем образовавшийся эфир может быть выделен или подвергнут расщеплению для получения чистого изобутена, и после стадии этерификации может следовать стадия дистилляции для выделения изобутена, причем, при необходимости, одновременно с выделением эфира дистилляцией могут быть выделены также и содержащиеся в исходной С4-фракции С3-, изо-С4-, а также С5-углеводороды, или выделение изобутена из полученного на предыдущей стадии реакционного продукта путем олигомеризации или полимеризации, осуществляемой в присутствии кислого катализатора, сила кислоты которого пригодна для селективного выделения изобутена в виде олиго- или полиизобутена, с целью получения продукта, содержащего от 0 до 15% остаточного изобутена.
(3) Выделение кислородсодержащих примесей из полученных на предыдущей стадии продуктов путем поглощения специально подобранными адсорбентами.
(4) Реакция обмена полученного в результате осуществления стадий (1)-(3) рафината II указанным выше способом.
Стадию селективного гидрирования бутадиена и содержащихся в сырой С4-фракции ацетиленовых примесей предпочтительно осуществляют путем ее двухступенчатого жидкофазного контактирования с катализатором, представляющим собой, по меньшей мере, один металл, выбранный из группы никель, палладий и платина, предпочтительно палладий, на оксиде алюминия в качестве носителя, при температуре от 20 до 200°С, давлении от 1 до 50 бар, объемной подаче первичной С4-фракции от 0,5 до 30 м3 / м3 катализатора в час, отношении рецикла к первичному потоку от 0:1 до 30:1 и мольном отношении водорода к диолефинам от 0,5:1 до 50:1 с целью получения реакционных продуктов, наряду с изобутеном содержащих н-бутены (1-бутен и 2-бутен) в мольном соотношении от 2:1 до 1:10, предпочтительно от 2:1 до 1:3, и практически не содержащих диолефины и производные ацетилена. Для максимального выхода гексена предпочтительно использование избытка 1-бутена, для максимального выхода пропена предпочтительно использование избытка 2-бутена. Это означает, что в первом случае мольное отношение 1-бутена к 2-бутену может составлять от 2:1 до 1:1, во втором от 1:1 до 1:3.
Стадию экстракции бутадиена из сырой С4-фракции предпочтительно осуществляют, используя соответствующий селективный растворитель, выбранный из класса полярных апротонных соединений, в частности ацетон, фурфурол, ацетонитрил, диметилацетамид, диметилформамид и N-метилпирролидон, с целью получения продукта, подвергаемого последующему селективному гидрированию/изомеризации, после которого он содержит н-бутены (1-бутен и 2-бутен) в мольном соотношении от 2:1 до 1:10, предпочтительно от 2:1 до 1:3.
Стадию этерификации изобутена метанолом или изобутанолом, предпочтительно изобутанолом, осуществляют предпочтительно в присутствии кислого ионообменного вещества в каскаде из трех реакторов со стационарными слоями катализатора, пропуская реакционные продукты в направлении сверху вниз, причем температура на входе в реактор составляет от 0 до 60°С, предпочтительно от 10 до 50°С, на выходе от 25 до 85°С, предпочтительно от 35 до 75°С, давление от 2 до 50 бар, предпочтительно от 3 до 20 бар, отношение изобутанола к изобутену от 0,8:1 до 2,0:1, предпочтительно от 1,0:1 до 1,5:1, а общая степень превращения изобутена соответствует равновесному значению.
Стадию выделения изобутена путем его олигомеризации или полимеризации предпочтительно осуществляют, используя в качестве исходных продуктов составы, полученные в результате реализации описанных выше стадий экстракции бутадиена и/или селективного гидрирования, в присутствии катализатора, выбранного из класса гомогенных и гетерогенных кислот Брэнстеда или Льюиса (смотри немецкую заявку на патент DE-A-10013253).
Селективное гидрирование сырой С4-фракции
В связи с тем, что алкины, алкинены и алкадиены обладают склонностью к полимеризации или явно выраженной тенденцией к образованию комплексов с переходными металлами, их присутствие во многих процессах технического синтеза является нежелательным. Нередко эти ненасыщенные соединения оказывают чрезвычайно сильное отрицательное воздействие на используемые для технического синтеза катализаторы.
С4-фракция, образующаяся в результате парофазного крекинга, содержит большие количества соединений, обладающих несколькими ненасыщенными связями, в частности 1,3-бутадиен, 1-бутин (этилацетилен) и бутенин (винилацетилен). В зависимости от имеющегося в распоряжении способа переработки эти соединения либо экстрагируют (экстракция бутадиена) либо подвергают селективному гидрированию. В первом случае типичное остаточное содержание соединений с несколькими ненасыщенными связями составляет от 0,05 до 0,3 мас.%, во втором от 0,1 до 4,0 мас.%. Поскольку присутствие остаточных количеств указанных ненасыщенных соединений мешает дальнейшей переработке, необходимо дополнительно снизить их содержание до величины, составляющей менее 10 ч./млн. Для обеспечения как можно более высокого содержания бутенов в качестве ценных реакционных продуктов следует максимально ограничить их чрезмерное гидрирование, приводящее к образованию бутанов.
В качестве альтернативы: экстракция бутадиена из сырой С4-фракции
Предпочтительный способ выделения бутадиена основан на физическом принципе экстрактивной дистилляции. Благодаря введению селективного органического растворителя летучесть некоторых компонентов смеси, в рассматриваемом случае бутадиена, уменьшается. Поэтому бутадиен вместе с растворителем остается в кубе дистилляционной колонны, в то время как сопутствующие соединения, которые ранее не удалось выделить дистилляцией, могут быть удалены через верхнюю часть колонны. В качестве растворителей для экстракционной дистилляции используют главным образом ацетон, фурфурол, диметилацетамид, диметилформамид и N-метилпирролидон. Для переработки экстракционной дистилляцией особенно пригодны образующиеся в результате крекинга, обогащенные бутадиеном С4-фракции с относительно высоким содержанием алкинов, в частности метил-, этил и винилацетилена, а также метилалленов.
Упрощенный принцип экстракции бутадиена из сырой С4-фракции растворителем заключается в следующем. Полностью переведенную в газообразное состояние С4-фракцию вводят в нижнюю часть экстракционной колонны. Растворитель (диметилформамид, N-винилпирролидон) стекает навстречу поступающей из нижней части колонны газообразной смеси, поглощая обладающий наиболее высокой растворимостью бутадиен и небольшие количества бутенов. В нижнюю часть экстракционной колонны вводят некоторую часть выделенного чистого бутадиена с целью десорбции основной части поглощенных растворителем бутенов, которые покидают колонну через верхнюю часть. В другой, дегазационной колонне, бутадиен отделяют от растворителя отпариванием и подвергают заключительной дистилляции, получая чистый продукт.
После выделения бутадиена экстрактивной дистилляцией реакционные продукты обычно подают на вторую ступень очистки, заключающуюся в селективном гидрировании с целью уменьшения остаточного содержания бутадиена до величины, составляющей менее 10 ч./млн.
Основными компонентами С4-фракции после выделения бутадиена, называемой С4-рафинатом или рафинатом I, являются изобутен, 1-бутен, 2-бутены, а также н-бутан и изобутан.
Выделение изобутена из рафината I
Следующим, подлежащим выделению из С4-фракции соединением предпочтительно является изобутен, поскольку от прочих компонентов, входящих в состав этой фракции, он отличается разветвленностью и более высокой реакционной способностью. Наряду с возможностью селективного выделения изобутена посредством молекулярных сит, благодаря чему может быть получен продукт чистотой 99%, а адсорбированные в порах молекулярного сита н-бутены и бутан могут быть десорбированы высококипящим углеводородом, изобутен в первую очередь выделяют дистилляцией с использованием так называемого деизобутенизатора, благодаря которому изобутен вместе с 1-бутеном и изобутаном выводят через верхнюю часть колонны, а 2-бутен и н-бутан, содержащие остаточные количества изобутена и 1-бутена, остаются в кубе, или выделяют изобутен экстрактивным способом путем его взаимодействия со спиртом на кислых ионообменниках. В качестве спирта предпочтительно используют метанол (МТВЕ) или изобутанол (IBTBE), получая соответственно метил-трет-бутиловый и изобутил-трет-бутиловый эфиры.
Метил-трет-бутиловый эфир образуется в результате жидкофазного взаимодействия метанола и изобутена на кислых ионообменниках, осуществляемого при температуре от 30 до 100°С под небольшим избыточным давлением в двух реакторах или одном двухступенчатом шахтном реакторе до практически полного превращения изобутена в эфир (более 99%). В связи с зависящим от давления образованием азеотропных смесей метанола с метил-трет-бутиловым эфиром выделение чистого метил-трет-бутилового эфира требует осуществления многоступенчатой дистилляции, или в соответствии с более новой технологией чистый метил-трет-бутиловый эфир получают, выделяя метанол адсорбирующими смолами. Все остальные компоненты С4-фракции при этерификации изобутена сохраняются неизменными. Поскольку полимеризация незначительных количеств диолефинов и производных ацетилена может вызвать сокращение срока службы ионообменника, предпочтительно используют бифункциональные PD-содержащие ионообменники, на которых в присутствии небольших количеств водорода осуществляют селективное гидрирование диолефинов и производных ацетилена, не оказывающее влияния на протекание этерификации изобутена.
Метил-трет-бутиловый эфир (МТВЕ) используют главным образом в качестве добавки, повышающей октановое число автомобильного бензина. Как альтернатива метил-трет-бутиловый и изобутил-трет-бутиловый эфиры могут быть подвергнуты газофазному расщеплению на кислых оксидах при температуре от 150 до 300°С, приводящему к выделению чистого изобутена.
Дополнительная возможность выделения изобутена из рафината I состоит в непосредственной олигомеризации или полимеризации этого олефина. Используя кислые гомогенные и гетерогенные катализаторы, например триоксид вольфрама на диоксиде титана в качестве носителя, этим способом можно обеспечить степень превращения изобутена до 95%, получая реакционный продукт с остаточным содержанием этого олефина, не превышающим 5%.
Очистка потока рафината II адсорбентами
Для увеличения срока службы катализаторов реакции обмена необходима очистка подаваемого на стадию реакции обмена сырья, то есть выделение из него каталитических ядов, например воды, кислородсодержащих соединений, серы или серосодержащих соединений, а также органических галогенидов, которое осуществляют, используя защитный слой адсорбента.
Методы адсорбции и адсорбционной очистки описаны, например, в W.Kast, Adsorption aus der Gasphase, VCH, Weinheim (1988). Об использовании цеолитов в качестве адсорбентов сообщается в D.W.Breck, Zeolite Molecular Sieves, Wiley, New York (1974).
Удаление, особенно ацетальдегида из С3-С15-углеводородов, может быть осуществлено путем жидкофазной очистки в соответствии с европейской заявкой на патент ЕР-А-05832901.
Селективное гидрирование сырой С4-фракции
Примеси ненасыщенных соединений в полученной парофазным крекингом или рафинированием сырой С4-фракции подвергают двухступенчатому селективному гидрированию, причем прежде всего гидрируют бутадиен (1,2 и 1,3-бутадиен), а также алкины или алкенины. Согласно одному варианту осуществления изобретения полученная рафинированием С4-фракция может быть направлена непосредственно на вторую стадию селективного гидрирования.
Первую стадию гидрирования предпочтительно осуществляют на стационарном слое катализатора, содержащего от 0,1 до 0,5 мас.% палладия на оксиде алюминия в качестве носителя, в газожидкостной фазе (способ орошения) с рециркуляцией жидкости при температуре от 40 до 80°С, давлении от 10 до 30 бар, мольном отношении водорода к бутадиену от 10:1 до 50:1, объемной подаче первичной С4-фракции до 15 м3 / м3 катализатора в час и отношении рецикла к первичному потоку от 5:1 до 20:1.
Вторую стадию гидрирования предпочтительно осуществляют на стационарном слое катализатора, содержащего от 0,1 до 0,5 мас.% палладия на оксиде алюминия в качестве носителя, в газожидкостной фазе (способ орошения) с рециркуляцией жидкости при температуре от 50 до 90°С, давлении от 10 до 30 бар, мольном отношении водорода к бутадиену от 1:1 до 10:1, объемной подаче первичного продукта от 5 до 20 м3 / м3 катализатора в час и отношении рецикла к первичному потоку от 0:1 до 15:1.
Полученный в результате гидрирования реакционный продукт, называемый рафинатом I, наряду с изобутеном содержит 1-бутен и 2-бутен в мольном отношении от 2:1 до 1:10, предпочтительно от 2:1 до 1:3.
Альтернатива: выделение бутадиена из сырой С4-фракции экстракцией.
Для экстрагирования бутадиена из сырой С4-фракции используют N-метилпирролидон.
Согласно одному варианту осуществления изобретения реакционный продукт после экстракции направляют на вторую ступень описанного выше селективного гидрирования с целью удаления остаточных количеств бутадиена, причем в процессе гидрирования устанавливают желаемое соотношение между содержанием 1-бутена и 2-бутена.
Выделение изобутена этерификацией спиртами
На стадии этерификации осуществляют взаимодействие изобутена со спиртами, предпочтительно изобутанолом, на кислом катализаторе, предпочтительно кислом ионообменнике, получая соответствующий эфир, предпочтительно изобутил-трет-бутиловый эфир. В соответствии с одним вариантом осуществления изобретения этерификацию проводят в каскаде, состоящем из трех реакторов со стационарными слоями катализатора, пропуская через них реакционные продукты в направлении сверху вниз. Температура на входе в первый реактор составляет от 0 до 60°С, предпочтительно от 10 до 50°С, на выходе от 25 до 85°С, предпочтительно от 35 до 75°С. Давление в реакторе составляет от 2 до 50 бар, предпочтительно от 3 до 20 бар. Степень превращения изобутена при отношении изобутанола к изобутену от 0,8:1 до 2,0:1, предпочтительно от 1,0:1 до 1,5:1 составляет от 70 до 90%.
Температура на входе во второй реактор составляет от 0 до 60°С, предпочтительно от 10 до 50°С, на выходе от 25 до 85°С, предпочтительно от 35 до 75°С. Давление в реакторе составляет от 2 до 50 бар, предпочтительно от 3 до 20 бар. Степень превращения изобутена в результате осуществления второй ступени этерификации повышается, достигая 85-99%, предпочтительно 90-97%.
В третьем, обладающем наибольшими размерами реакторе, при одинаковой температуре реакционных продуктов на входе и выходе, составляющей от 0 до 60°С, предпочтительно от 10 до 50°С, достигают равновесной степени превращения изобутена. После этерификации изобутена и выделения образующегося эфира осуществляют его эндотермическое расщепление на кислых, предпочтительно гетерогенных катализаторах, например фосфорной кислоте на диоксиде кремния в качестве носителя, причем температура на входе составляет от 150 до 300°С, предпочтительно от 200 до 250°С, на выходе от 100 до 250°С, предпочтительно от 130 до 220°С.
При использовании в качестве исходного продукта С4-фракции, полученной путем жидкофазного каталитического крекинга, следует учитывать, что она содержит 1 мас.% пропана, от 30 до 40 мас.% изобутена и от 3 до 10 мас.% С5-углеводородов, что может оказать отрицательное воздействие на реализацию последующих технологических операций. Учитывая это, предусмотрена возможность путем дистилляции одновременно с эфиром выделять и указанные выше сопутствующие компоненты.
Остаточное содержание изобутена в полученном после осуществления этих технологических процессов продукте, называемом рафинатом II, составляет от 0,1 до 3 мас.%.
При повышенном содержании изобутена, например, если в качестве исходного продукта используют С4-фракцию, полученную путем жидкофазного каталитического крекинга, или при выделении изобутена путем его частичного превращения в полимер в присутствии соответствующего кислого катализатора, рафинат II согласно одному варианту осуществления изобретения может быть подвергнут дистилляции.
Очистка потока рафината II адсорбентами
Полученный в результате этерификации/полимеризации (и дистилляции) рафинат II подвергают очистке, используя для этого, по меньшей мере, один защитный слой, состоящий из оксидов алюминия с высокой удельной поверхностью, силикагеля, алюмосиликатов или молекулярных сит. Защитный слой служит для сушки С4-фракции, а также для удаления из нее соединений, способных вызвать отравление используемых при последующей реакции обмена катализаторов. Предпочтительными адсорбентами являются Selexsorb CD и CDO, а также молекулярные сита с размером пор 3Å и сита NaX (13X). Очистку производят в поглотительных колоннах, используя давление и температуру, при которых все компоненты находятся в жидком состоянии. При необходимости очистку подаваемых на стадию реакции обмена продуктов производят с целью их предварительного нагревания.
Получаемый в результате очистки рафинат II практически не содержит воду, кислородсодержащие примеси, органические хлориды и серосодержащие соединения.
При осуществлении этерификации посредством метанола (получении метил-трет-бутилового эфира) в связи с образованием диметилового эфира в качестве побочного продукта может возникнуть необходимость в комбинированном использовании нескольких последовательных ступеней очистки.
Предпочтительными катализаторами реакции обмена являются известные из литературы рениевые катализаторы, в частности, рениевый ангидрид (Re2O7) на γ-Al2О3 или смешанных носителях, например SiO2/Al2О3, В2O3/SiO2/Al2O3 или Fe2О3/Al2О3, с варьируемым содержанием металлов. Содержание рениевого ангидрида независимо от выбранного носителя составляет от 1 до 20%, предпочтительно от 3 до 10%.
Используют свежепрокаленные катализаторы, не требующие дополнительного активирования (например, посредством алкилирующего агента). Утратившие свою активность катализаторы могут быть многократно регенерированы выжиганием кокса при температуре выше 400°С в токе воздуха или атмосфере инертного газа.
Сравнение разных гетерогенных катализаторов показывает, что ангидрид рения на оксиде алюминия обладает активностью даже в весьма мягких условиях реакции обмена (при температуре от 20 до 80°С), в то время как катализаторы, представляющие собой МО3 на диоксиде кремния (М означает молибден (Мо) или вольфрам (W)), проявляют активность лишь при температурах, превышающих 100-150°С, в связи с чем в их присутствии может протекать побочная реакция изомеризации двойных связей.
Ниже приведены другие, пригодные для осуществления реакции обмена катализаторы:
- WO3/SiO2, полученный из (С5Н5)W(СО)зCl и SiO2 (J.Mol. Catal. 1995, 95, 75-83);
- трехкомпонентная система, состоящая из [Mo(NO)2(OR)2]n, SnEt4 и AlCl3 (J.Mol. Catal. 1991, 64, 171-178, и J.Mol. Catal. 1989, 57, 207-220);
- комплексы нитрида молибдена(VI), полученные из высокоактивных форкатализаторов (J.Organomet. Chem. 1982, 229, С19-С23);
- гетерогенные катализаторы МоО3 и WO3 на SiO2 в качестве носителя (J.Chem. Soc., Faraday Trans. / 1982, 78, 2583-2592);
- молибденсодержащие катализаторы на носителе (J.Chem. Soc., Faraday Trans. / 1981, 77, 1763-1777);
- активные вольфрамсодержащие катализаторы первой ступени (J.Am.Chem. Soc. 1980, 102(21), 6572-6574);
- ацетонитрил(пентакарбонил)вольфрам (J.Catal. 1975, 38, 482-484);
- трихлор(нитрозил)молибден(II) в качестве катализатора первой ступени (Z.Chem. 1974, 14, 284-285);
- W(CO)5PPH3/EtAlCl2 (J.Catal. 1974, 34, 196-202);
- WCI6/н-BuLi (J.Catal. 1973, 28, 300-303);
- WCI6/н-BuLi (J.Catal. 1972, 26, 455-458);
французский патент FR 2726563: О3ReO[Al(OR)(L)xО]nReO3 (R=углеводород с 1-40 атомами углерода, n=1-10, х=0 или 1 и L=растворитель); европейские заявки на патент ЕР-А-1910675 и ЕР-А-1290474, бельгийский патент BE 899897: каталитическая система, состоящая из вольфрама, двузамещенных фенольных остатков и четырех других лигандов, в том числе галогена, алкила и карбена;
французский патент FR 2499083: каталитическая система, состоящая из оксокомплекса переходного металла (вольфрама, молибдена или рения), образованного с кислотой Льюиса.
Патент США 4060468: каталитическая система, полученная из соли вольфрама, ароматического кислородсодержащего соединения, например 2,6-дихлорфенола и, при необходимости, молекулярного кислорода.
Бельгийский патент BE 776564: каталитическая система, полученная из соли переходного металла, металлоорганического соединения и амина.
Для увеличения срока службы катализаторов реакции обмена, прежде всего катализаторов на носителях, рекомендуется предварительная очистка подаваемых на соответствующую стадию продуктов посредством защитного адсорбционного слоя, что обеспечивает сушку С4-фракции, а также удаление из нее соединений, способных вызвать отравление катализаторов реакции обмена. Предпочтительными адсорбентами являются Selex-sorb CD и CDO, а также молекулярные сита с размером пор 3Å и сита NaX (13Х). Очистку производят в поглотительных колоннах, предпочтительно используя давление и температуру, при которых все компоненты находятся в жидком состоянии. При необходимости, очистку подаваемых на реакцию обмена продуктов производят с целью их предварительного нагревания. Может оказаться предпочтительным комбинирование нескольких последовательно реализуемых ступеней очистки.
Давление и температуру реакции обмена выбирают таким образом, чтобы все реагенты находились в жидком состоянии. Температура обычно составляет от 0 до 150°С, предпочтительно от 20 до 80°С, давление от 2 до 200 бар. Однако предпочтительным может оказаться осуществление реакции обмена в газовой фазе и/или использование катализатора, обладающего более низкой кислотностью, в частности, если реакции обмена подвергают продукты с повышенным содержанием изобутена.
Реакцию обмена, как правило, осуществляют в течение промежутка времени от 1 секунды до 1 часа, предпочтительно от 30 секунд до 30 минут, в непрерывном или периодическом режиме, используя в качестве реакторов, например, резервуары для сжатых газов, проточные трубы или устройства для дистилляции реактивов, причем предпочтительными являются проточные трубы.
Стадия b)
Синтезированные на стадии а) 2-пентен и/или 3-гексен димеризуют на стадии b) в присутствии катализатора димеризации, получая смеси, содержащие С10-С12-олефины, которые, при необходимости, выделяют.
В результате димеризации олефинов или смесей олефинов, полученных на стадии реакции обмена, образуются продукты димеризации, которые являются особенно благоприятными компонентами и особенно предпочтительными составами для последующего получения алкилароматических соединений, если
используют катализатор димеризации, содержащий, по меньшей мере, один элемент из VIII подгруппы Периодической системы элементов,
и состав катализатора и реакционные условия выбирают таким образом, чтобы получить смесь димеров, содержащую менее 10 мас.% соединений, обладающих структурным элементом формулы I (винилиденовая группа)
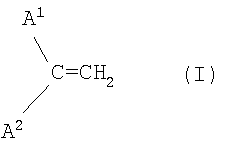
в которой А1 и А2 являются алифатическими углеводородными остатками.
Предпочтительно для димеризации используются внутренние, линейные пентен и гексен, входящие в состав продуктов реакции обмена. Особенно предпочтительно использование 3-гексена.
Димеризацию можно осуществлять, используя как гомогенные, так и гетерогенные катализаторы. Предпочтительным является гетерогенный катализ, поскольку в этом случае, с одной стороны, упрощается процесс выделения катализатора, что повышает экономичность способа, а, с другой стороны, отсутствует выделение причиняющих ущерб окружающей среде сточных вод, которые обычно образуются при выделении растворимых катализаторов, например, в результате их гидролиза. Другое преимущество гетерогенного способа состоит в том, что продукты димеризации не содержат галогенов, в частности хлора или фтора, в то время как в состав гомогенных растворимых катализаторов в общем случае входят галогенсодержащие лиганды или такие катализаторы используют в сочетании с галогенсодержащими сокатализаторами. Кроме того, галогены, содержащиеся в гомогенных каталитических системах, могут войти в состав продуктов димеризации, нанося существенный ущерб как качеству продуктов димеризации, так и их пригодности для дальнейшей переработки.
Для гетерогенного катализа целесообразно использование комбинаций оксидов металлов VIII подгруппы с оксидом алюминия на оксидах кремния и титана в качестве носителей, описанных, например, в немецкой заявке на патент DE-A-4339713. Гетерогенный катализатор может находиться в виде стационарного слоя, предпочтительно состоящего из грубодисперсных частиц размером от 1 до 1,5 мм, или суспензии с размером частиц от 0,05 до 0,5 мм. Димеризацию под влиянием гетерогенных катализаторов целесообразно осуществлять в герметичной системе при температуре от 80 до 200°С, предпочтительно от 100 до 180°С, и давлении, величина которого определяется температурой реакции, при необходимости, в атмосфере находящегося под избыточным давлением защитного газа. Для достижения оптимальной степени превращения реакционную смесь подвергают многократной рециркуляции с непрерывным отбором определенной части рециркулирующего продукта и добавлением исходной смеси.
В результате димеризации согласно изобретению получают смеси, состоящие из углеводородов с однократной ненасыщенностью, длина цепей которых преимущественно соответствует удвоенной длине исходных олефинов.
Катализаторы димеризации и реакционные условия в рамках приведенных выше параметров предпочтительно выбирают таким образом, чтобы, по меньшей мере, у 80% компонентов смеси димеров на 1/4-3/4, предпочтительно 1/3-2/3, длины главной цепи приходилось одно разветвление или два разветвления при соседних атомах углерода.
Характерной особенностью получаемых согласно изобретению смесей олефинов является очень высокое содержание разветвленных компонентов, как правило, превышающее 75%, в частности превышающее 80%, и ограниченное содержание неразветвленных компонентов, составляющее, как правило, менее 25%, в частности менее 20%. Другой их характерной особенностью является то, что к местам разветвления главной цепи димеров преимущественно присоединены группы с числом углеродных атомов, равным (у-4) и (у-5), причем «у» означает число атомов углерода в использованном для димеризации мономере. Если (у-5)=0, это означает полное отсутствие боковых групп.
Предпочтительными заместителями в точках разветвления главных цепей получаемых согласно изобретению смесей С12-олефинов являются метильные или этильные группы.
Еще одной характерной особенностью получаемых димеров является положение метильных и этильных групп вдоль главной цепи. Если речь идет о монозамещенных димерах, то метильная или этильная группа находится в точке Р=(n/2)-m главной цепи, причем n означает длину главной цепи, а m число атомов углерода в боковой группе. У двузамещенных продуктов один из заместителей находится в положении Р, а другой в положении Р+1, то есть замещает соседний атом углерода главной цепи. Характерному совокупному содержанию монозамещенных продуктов (с одним разветвлением) в получаемой согласно изобретению смеси олефинов соответствует интервал от 40 до 75 мас.%, а содержанию двузамещенных компонентов смеси соответствует интервал от 5 до 25 мас.%.
Получаемые вышеуказанным способом (смотри международную заявку на патент WO 00/39058) смеси олефинов представляют собой ценные промежуточные продукты, в частности, предназначенные для описанного ниже синтеза разветвленных алкилароматических соединений, из которых в дальнейшем получают поверхностно-активные вещества.
Стадия с)
На этой стадии осуществляют взаимодействие полученной на стадии b) смеси С10-С12-олефинов в присутствии катализатора алкилирования с ароматическими углеводородами, приводящее к образованию алкилароматических соединений.
Предпочтительным является использование катализатора алкилирования, обеспечивающего получение алкилароматических соединений с алкильным остатком, от одного до трех углеродных атомов которого обладают индексом Н/С, равным 1.
В принципе алкилирование может быть реализовано в присутствии любых, предназначенных для осуществления этой реакции катализаторов.
Хотя для алкилирования в принципе пригодны AlCl3 и HF, предпочтение отдают гетерогенным и селективным катализаторам. С точки зрения производственной безопасности и защиты окружающей среды предпочтительными сегодня являются твердые катализаторы алкилирования, к которым относятся, в частности, используемый в DETAL-процессе фторированный катализатор на основе кремния (Si) и алюминия (Al), ряд селективных катализаторов, металлоксидные катализаторы на носителях, а также слоистые силикаты и глины.
При выборе катализатора, несмотря на большое влияние используемого сырья, важно свести к минимуму происходящее в присутствии катализатора образование соединений, отличающихся тем, что в их алкильном остатке содержатся углеродные атомы с индексом Н/С, равным 0. Кроме того, катализатор должен обеспечивать образование соединений, в алкильном остатке которых в среднем от одного до трех углеродных атомов обладают индексом Н/С, равным 1. Этого можно достичь, в частности, подобрав соответствующие катализаторы, которые, с одной стороны, благодаря специфическим геометрическим параметрам подавляют образование нежелательных продуктов, а, с другой стороны, обеспечивают достаточно высокую скорость алкилирования.
Индекс Н/С означает число протонов, приходящееся на один содержащийся в алкильном остатке углеродный атом.
Наряду с этим при выборе катализаторов алкилирования особое внимание следует уделять их склонности к дезактивации. Недостатком одномерных систем пор в большинстве случаем является быстрое закупоривание последних продуктами деструкции и синтезируемыми веществами, в связи с чем предпочтение следует отдавать катализаторам с многомерными системами пор.
Для алкилирования могут использоваться катализаторы как природного, так и синтетического происхождения, некоторые свойства которых могут быть исследованы известными из литературы методами (например, путем ионного обмена, обработки острым паром, блокирования кислых центров, вымывания особых компонентов кристаллической решетки и так далее). Существенное значение для настоящего изобретения имеет то, чтобы катализаторы, хотя бы отчасти, обладали кислым характером.
В зависимости от способа использования катализаторы алкилирования могут находиться в виде порошка или в виде формованного изделия. Соединения, образующие матрицу формованных катализаторов, придают им достаточно высокую механическую стабильность, однако для обеспечения свободного доступа молекул к активным составным частям необходимо обеспечить достаточную пористость матрицы. Формованные катализаторы получают известными из литературы способами согласно уровню техники.
Ниже приводятся некоторые из катализаторов, которые могут использоваться для алкилирования.
AlCl3, AlCl3 на носителе (международный патент WO 96/26787), HF, H2SO4, ионные жидкости (например, международный патент WO 98/50153), перфторированные ионообменные смолы или NAFION/Silica (например, международный патент WO 99/06145), F-Si/Al (патент США US 5344997), цеолит типа Beta (например, международный патент WO 98/09929, патенты США US 5877370, US 4301316, US 4301317), фоязит (CN 1169889), слоистые силикаты, глины (европейский патент ЕР 711600), фторированный морденит (международная заявка WO 00/23405), морденит (европейский патент ЕР 466558), ZSM-12, ZSM-20, ZSM-38, маззит, цеолит L, канкринит, гмеллинит, оффретит, МСМ-22 и так далее. Предпочтительными являются 12-ядерные селективные цеолиты.
Предпочтительные условия реакции
Алкилирование осуществляют путем контактирования ароматических соединений (смеси ароматических соединений) и олефина (смеси олефинов) с катализатором в соответствующем реакторе, обработки реакционной смеси и выделения целевых продуктов.
Для алкилирования пригодны, например, трубчатые реакторы или реакторы с мешалками. Если катализатор находится в твердом состоянии, его можно использовать в виде суспензии, стационарного или псевдоожиженного слоя.
При использовании реактора со стационарным слоем катализатора возможно прямоточное или противоточное движение потоков реагентов. Возможно также использование способа каталитической перегонки.
Исходные реагенты могут находиться в твердом и/или жидком состояниях.
Реакционную температуру выбирают таким образом, чтобы можно было обеспечить возможно более полное превращение олефина при минимально возможном образовании побочных продуктов. Кроме того, при выборе температурного режима решающую роль играет вид используемого катализатора. Реакционной температуре соответствует интервал от 50 до 500°С, предпочтительно от 80 до 350°С, особенно предпочтительно от 80 до 250°С.
Давление зависит от способа осуществления алкилирования (типа реактора) и составляет от 0,1 до 100 бар, нагрузку на катализатор варьируют в интервале от 0,1 до 100.
При необходимости, исходные реагенты могут быть разбавлены инертными веществами, в качестве которых предпочтительно используют парафины.
Соотношение между ароматическим соединением и олефином обычно устанавливают в интервале от 1:1 до 100:1, предпочтительно от 2:1 до 20:1.
Используемые ароматические соединения
Могут использоваться любые ароматические соединения общей формулы Ar-R, в которой Ar означает моно- или бициклический ароматический углеводородный остаток, R означает водород, алкил с 1-5, предпочтительно с 1-3 атомами углерода, гидрокси или OR, предпочтительно водород или алкил с 1-3 атомами углерода. Предпочтительными ароматическими соединениями являются бензол и толуол.
Стадия d)
На стадии d) осуществляют сульфирование полученных на предыдущей стадии алкилароматических соединений и нейтрализацию продуктов сульфирования до алкиларилсульфонатов.
Алкиларильные соединения превращают в алкиларилсульфонаты путем
1) сульфирования, например, серным ангидрида (SO3), олеумом, хлорсульфоновой кислотой и другими агентами, предпочтителен серный ангидрид,
2) нейтрализации, например, натриевыми, калиевыми, аммонийными и магниевыми соединениями, предпочтительны натриевые соединения.
Сульфирование и нейтрализация подробно описаны в литературе, и их осуществляют согласно уровню техники. Сульфирование предпочтительно производят в реакторе с падающей пленкой, однако оно может быть реализовано и в реакторе с мешалкой. Более предпочтительным по сравнению с олеумом является использование для сульфирования серного ангидрида.
Смешивание
Полученные вышеописанным способом соединения (предпочтительно) подвергают дальнейшей переработке как таковые или предварительно смешивают их с другими алкиларильными соединениями, а затем направляют на дальнейшую переработку. Чтобы упростить этот процесс, может оказаться целесообразным непосредственное смешивание сырья, используемого для получения других вышеуказанных алкилароматических соединений с сырьем, используемым в настоящем способе, и последующее осуществление способа согласно изобретению. Так, например, целесообразным является смешивание слаборазветвленных олефинов из способа согласно изобретению с линейными олефинами. Могут использоваться также смеси алкиларилсульфокислот и алкиларилсульфонатов. Смешивание всегда осуществляют с учетом оптимального качества поверхностно-активных веществ, получаемых из алкиларильных соединений.
Сведения, касающиеся алкилирования, сульфирования и нейтрализации, приведены, например, в Surf. Sci. Ser. 56 (1996) Kapitel 2, Marcel Dekker, New York («Alkylarylsulfonates: History, Manufacture, Analysis and Environmental Properties») и цитируемой там литературе.
Стадия е)
На стадии е) алкиларилсульфонаты, полученные на стадии d), могут быть смешаны с линейными алкиарилсульфонатами.
Изобретение касается алкиларилсульфонатов, которые могут быть получены описанным выше способом.
Алкиларилсульфонаты согласно изобретению предпочтительно используют в качестве поверхностно-активных веществ, в частности, в составе моющих и чистящих средств. Изобретение касается также моющих и чистящих средств, содержащих, наряду с обычными ингредиентами, описанные выше алкиларилсульфонаты.
Ниже приведены примеры обычных ингредиентов моющих и чистящих средств согласно изобретению, не ограничивающие его объема.
Отбеливающие средства
Примерами отбеливающих средств являются пербораты щелочных металлов или пергидраты карбонатов щелочных металлов, в частности натриевые соли.
Примером пригодной органической пероксокислоты является надуксусная кислота, которую предпочтительно используют для промышленной стирки текстильных изделий или промышленной чистки.
Предпочтительно используемые составы для отбеливания или стирки текстильных изделий содержат перкарбоновые кислоты с 1-12 атомами углерода, диперкарбоновые кислоты с 8-16 атомами углерода, имидоперкарбоковые или арилдиперкарбоновые кислоты. Предпочтительными примерами пригодных перкарбоновых кислот являются надуксусная кислота, линейные или разветвленные октан-, нонан-, декан- или додеканмонопероксокислоты, декан- и додекандипероксокислоты, моно- и диперфталевые кислоты, моно- и диперизофталевые кислоты, моно- и дипертерефталевые кислоты, фталимидоперкапроновая и терефталоилдиперкарбоновая кислоты. Кроме того, могут использоваться полимерные пероксокислоты, например, на основе акриловой кислоты с пероксидной группой. Перкарбоновые кислоты могут использоваться в виде свободных кислот или соответствующих солей, предпочтительно солей щелочных и щелочноземельных металлов.
Активаторы отбеливания
Катализаторами отбеливания являются, например, кватернизированные имины и сульфонимины, в частности, описанные в патентах США US 5360568, US 5360569 и европейской заявке на патент ЕР-А-0453003, а также марганцевые комплексы, в частности, описанные в международной заявке WO-A 94/21777. Другие пригодные металлсодержащие катализаторы отбеливания описаны в европейских заявках на патент ЕР-А-0458397, ЕР-А-0458398 и ЕР-А-0549272.
Активаторами отбеливания являются, например, соединения из следующих классов веществ: полиацилированные сахара и производные сахаров, содержащие ацильные остатки с 1-10 атомами углерода, предпочтительно ацетильные, пропионильные, октаноильные, нонаноильные или бензоильные остатки, особенно предпочтительно ацетильные остатки. В качестве сахаров или производных сахаров пригодны моно- или дисахариды, а также их восстановленные или окисленные производные, предпочтительно глюкоза, манноза, фруктоза, сахароза, ксилоза или лактоза. Особенно пригодными активаторами отбеливания из данного класса соединений являются, в частности, пентаацетилглюкоза, тетраацетат ксилозы, 1-бензоил-2,3,4,6-тетраацетилглюкоза и 1-октаноил-2,3,4,6-тетраацетилглюкоза.
К другому пригодному классу веществ относятся ацилоксибензолсульфокислоты и соответствующие соли щелочных и щелочноземельных металлов, причем пригодны ацильные остатки с 1-14 атомами углерода. Предпочтительными являются ацетильные, пропионильные, октаноильные, нонаноильные и бензоильные, в частности ацетильные и нонаноильные остатки. Особенно предпочтительными активаторами отбеливания этого класса веществ являются ацетилоксибензолсульфокислоты, предпочтительно используемые в виде соответствующих натриевых солей.
Кроме того, пригодны эфиры O-ацилоксима, в частности O-ацетилацетоноксим, O-бензоилацетоноксим, бис-(пропилимино)карбонат, бис-(циклогексилимино)карбонат. Пригодные согласно изобретению ацилированные оксимы описаны, например, в европейской заявке на патент ЕР-А-0028432. Пригодные согласно изобретению эфиры оксимов описаны, например, в европейской заявке на патент ЕР-А-0267046.
Пригодными являются также N-ацилкапролактамы, в частности N-ацетилкапролактам, N-бензоилкапролактам, N-октаноилкапролактам и карбонил-бис-капролактам.
Другими пригодными активаторами отбеливания являются
- N-диацилированные и N,N'-тетраацилированные амины, например N,N,N',N'-тетраацетилметилендиамин и N,N,N',N'-тетраацетилэтилен-диамин, N,N-диацетиланилин, N,N-диацетил-п-толуидин или 1,3-диацилированные гидантоины, в частности 1,3-диацетил-5,5-диметилгидантоин;
- N-алкил-N-сульфонилкарбонамиды, например N-метил-N-мезилацетамид или N-метил-N-мезилбензамид;
- N-ацилированные циклические гидразиды, ацилированный триазол или уразол, например гидразид моноацетилмалеиновой кислоты;
- O,N,N-тризамещенные гидроксиламины, например O-бензоил-N,N-сукцинилгидроксиламин, О-ацетил-N,N-сукцинилгидроксиламин или O,N,N-триацетилгидроксиламин;
- N,N'-диметил-N,N'-диацетилсульфуриламид или N,N'-диэтил-N,N'-дипропионилсульфуриламид;
- трицилцианураты, например триацетилцианурат или трибензоилцианурат;
- ангидриды карбоновых кислот, например бензойный, м-хлорбензойный или фталевый ангидриды;
- 1,3-диацил-4,5-диацилоксиимидазолины, например 1,3-диацетил-4,5-диацетоксиимидазолин;
- тетраацетилгликолурил и тетрапропионилгликолурил;
- диацилированные 2,5-дикетопиперазины, например 1,4-диацетил-2,5-дикетопиперазин;
- продукты ацилирования пропилендимочевины и 2,2-диметилпропилендимочевины, например тетраацетилпропилендимочевина;
- α-ацилоксиполиацилмалонамиды, например α-ацетокси-N,N'-диацетилмалонамид;
- диацилдиоксогексагидро-1,3,5-триазины, например 1,5-диацетил-2,4-диоксогексагидро-1,3,5-триазин.
Кроме того, пригодны 1-алкил-(4Н)-3,1-бензоксазин-4-оны или 1-арил-(4Н)-3,1-бензоксазин-4-оны, например, описанные в европейских заявках на патент ЕР-В1-0332294 и ЕР-В 0502013.
В частности, пригодными являются 2-фенил-(4Н)-3,1-бензоксазин-4-он и 2-метил-(4Н)-3,1-бензоксазин-4-он.
Кроме того, пригодны катионные нитрилы, например, описанные в европейских патентах ЕР 303520 и ЕР 458391 А1. Примерами пригодных катионных нитрилов являются метосульфаты или тозилаты ацетонитрила триметиламмония, ацетонитрил N,N-диметил-N-октиламмония, пропионитрил 2-триметиламмония, 2-метилпропионитрил 2-триметиламмония, N,N'-диацетонитрил N-метилпиперазина и ацетонитрил N-метилморфолина.
Особенно пригодными кристаллическими активаторами отбеливания являются тетраацетилэтилидендиамин, NOBS, изоNOBS, карбонил-бис-капролактам, бис-(2-пропилимино)карбонат, бис-(циклогексилимино)карбонат, O-бензоилацетоноксим и 1-фенил-(4Н)-3,1-бензоксазин-4-он, антранил, фенилантранил, ацетонитрил N-метилморфолина, N-октаноилкапролактам, N,N'-диацетонитрил N-метилпиперазина, а также жидкие или плохо кристаллизующиеся активаторы отбеливания, выпускаемые в виде твердых продуктов.
Стабилизаторы отбеливания
Речь идет о добавках, способных адсорбировать, связывать следы тяжелых металлов или образовывать с ними комплексы. Примерами пригодных добавок согласно изобретению, которые обладают стабилизирующим отбеливание эффектом, являются полианионные соединения, в частности полифосфаты, поликарбоксилаты, полигидроксиполикарбоксилаты, растворимые силикаты в виде полностью или частично нейтрализованных солей щелочных или щелочноземельных металлов, в частности, нейтральных натриевых или магниевых солей, которые, однако, обладают относительно слабым стабилизирующим действием. Сильными, пригодными согласно изобретению стабилизаторами отбеливания являются, например, комплексообразующие добавки, в частности тетраацетат этилендиамина, нитрилотриуксусная кислота, метилглициндиуксусная кислота, β-аланиндиуксусная кислота, этилендиамин-N,N'-дисукцинат и фосфонаты, в частности этилендиаминтетраметиленфосфонат, диэтилентриаминпентаметиленфосфонат или гидроксиэтилиден-1,1-дифосфоновая кислота в виде кислот или частично или полностью нейтрализованных солей щелочных металлов, причем предпочтительными комплексообразователями являются натриевые соли указанных соединений.
Моющие средства согласно изобретению предпочтительно содержат, по меньшей мере, один стабилизатор отбеливания, особенно предпочтительно, по меньшей мере, один из приведенных выше сильных стабилизаторов.
Описываемые согласно одному варианту осуществления изобретения отбеливающие или моющие составы, предназначенные для стирки текстильных изделий, отбеливания и чистки в домашних условиях и промышленной сфере, могут содержать практически все компоненты, обычно содержащиеся в моющих, отбеливающих и чистящих средствах. В частности, могут быть созданы средства, специально предназначенные для обработки текстильных изделий при пониженной температуре, а также средства, пригодные для использования в нескольких температурных диапазонах, включая традиционную стирку белья с кипячением.
Основными компонентами средств для стирки текстильных изделий и чистящих средств, наряду с отбеливающими композициями, являются структурирующие вещества (модификаторы), то есть неорганические модификаторы и/или органические сомодификаторы, и поверхностно-активные вещества, в частности, анионные и/или неионные. Кроме того, если целесообразно, в состав указанных средств могут быть введены другие, обычно используемые вспомогательные добавки и сопутствующие вещества, в частности регуляторы, комплексообразователи, фосфонаты, красящие вещества, ингибиторы коррозии, ингибиторы потемнения окраски и/или полимеры, устраняющие загрязнения, ингибиторы передачи краски, катализаторы отбеливания, стабилизаторы пероксидов, электролиты, оптические отбеливатели, ферменты, парфюмерные масла, регуляторы пенообразования и активирующие вещества.
Неорганические модификаторы (структурирующие вещества)
В качестве неорганических модификаторов пригодны любые, обычно используемые модифицирующие добавки, в частности алюмосиликаты, силикаты, карбонаты и фосфаты.
Пригодными неорганическими модификаторами являются, например, обменивающие ионы алюмосиликаты, в частности цеолиты. Пригодны цеолиты различного типа, в частности цеолиты А, X, В, Р, MAP и HS в содержащем катионы натрия состоянии или в формах, в которых эти катионы частично заменены другими катионами, в частности катионами лития, калия, кальция, магния или аммония. Пригодные цеолиты описаны, например, в европейских заявках на патент ЕР-А 038591, ЕР-А 021491, ЕР-А 087035, патенте США US-A 4604224, патенте Великобритании GB-А 2013259, европейских заявках на патент ЕР-А 522726, ЕР-А 384070 и международной заявке на патент WO-A 94/24251.
Другими пригодными неорганическими модификаторами являются, например, аморфные или кристаллические силикаты, в частности аморфные дисиликаты, кристаллические дисиликаты, в частности слоистый силикат SKS-6 (изготовитель фирма Hoechst). Силикаты могут использоваться в виде соответствующих солей щелочных и щелочно-земельных металлов или аммония. Предпочтительно использование силикатов натрия, лития и магния.
Анионные поверхностно-активные вещества
Пригодными анионными поверхностно-активными веществами являются линейные и/или слаборазветвленные алкилбензолсульфонаты согласно изобретению.
Другими пригодными анионными поверхностно-активными веществами являются, например, сульфатированные жирные спирты с 8-22, предпочтительно 10-18 атомами углерода, например сульфатированные спирты с 9-11 атомами углерода, сульфатированные спирты с 12-13 атомами углерода, цетилсульфат, миристилсульфат, пальмитилсульфат, стеарилсульфат и сульфатированный жирный спирт на основе животного сала.
Другими пригодными анионными поверхностно-активными веществами являются сульфатированные этоксилированные спирты с 8-22 атомами углерода (алкилэфирсульфаты) и соответствующие растворимые соли. Соединения этого типа получают, например, путем алкоксилирования спирта с 8-22, предпочтительно с 10-18 атомами углерода, например жирного спирта, и последующего сульфатирования полученного продукта алкоксилирования. Для алкоксилирования предпочтительно используют этиленоксид, причем на 1 моль насыщенного спирта приходится от 2 до 50, предпочтительно от 3 до 20 молей этиленоксида. Алкоксилирование спиртов может быть осуществлено также посредством одного пропиленоксида или, при необходимости, с добавлением бутиленоксида. Кроме того, пригодными являются алкоксилированные спирты с 8-22 атомами углерода, содержащие этиленоксидные и пропиленоксидные или этиленоксидные и бутиленоксидные звенья. Алкоксилированные спирты с 8-22 атомами углерода могут содержать структурные единицы этиленоксида, пропиленоксида и бутиленоксида в виде блоков или статистически распределенных звеньев.
Другими пригодными анионными поверхностно-активными веществами являются N-ацилсаркозинаты, содержащие алифатические насыщенные или ненасыщенные ацильные остатки с 8-25 атомами углерода, предпочтительно с 10-20 атомами углерода, например N-олеилсаркозинат.
Анионные поверхностно-активные вещества вводят в состав моющих средств предпочтительно в виде солей. Пригодными являются соли щелочных металлов, в частности натрия, калия и лития, а также соли аммония, например соли гидроксиэтиламмония, ди(гидроксиэтил)аммония и три(гидроксиэтил)аммония.
Согласно изобретению моющие средства предпочтительно содержат линейные алкилбензолсульфонаты с 10-13 атомами углерода и/или слаборазветвленные алкилбензолсульфонаты.
Неионные поверхностно-активные вещества
В качестве неионных поверхностно-активных веществ пригодны алкоксилированные спирты с 8-22 атомами углерода, в частности алкоксилаты насыщенных спиртов или оксаспиртов. Алкоксилирование может быть осуществлено посредством этиленоксида, пропиленоксида и/или бутиленоксида. В качестве поверхностно-активных веществ могут использоваться любые алкоксилированные спирты, по меньшей мере, с двумя присоединенными молекулами упомянутых выше алкиленоксидов, причем они могут содержать этиленоксид, пропиленоксид и/или бутиленоксид в виде блоков или речь идет о продуктах присоединения со статистическим распределением звеньев алкиленоксидов. Для алкоксилирования одного моля спирта используют от 2 до 50, предпочтительно от 3 до 20 молей, по меньшей мере, одного алкиленоксида. Спирты предпочтительно содержат от 10 до 18 атомов углерода.
К другому классу пригодных неионных поверхностно-активных веществ относятся алкилфенолэтоксилаты с 6-14 атомами углерода в алкильных цепях, содержащие от 5 до 30 звеньев этиленоксида.
К другому классу пригодных неионных поверхностно-активных веществ относятся алкилполигликозиды с 8-22, предпочтительно 10-18 атомами углерода в алкильных цепях, чаще всего содержащие от 1 до 20, предпочтительно от 1,1 до 5 звеньев гликозида.
К другому классу неионогенных поверхностно-активных веществ относятся N-алкилглюкамиды общей структуры II или III
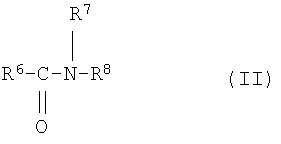
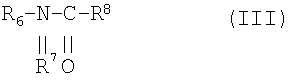
причем R6 означает алкил с 6-22 атомами углерода, R7 водород или алкил с 1-4 атомами углерода, R8 полигидроксиалкильный остаток с 5-12 атомами углерода и, по меньшей мере, тремя гидроксильными группами. R6 предпочтительно означает алкил с 10-18 атомами углерода, R7 метил и R8 полигидроксиалкильный остаток с 5 или 6 атомами углерода. Такие соединения получают, например, ацилированием продуктов восстановительного аминирования сахаров, причем ацилирование осуществляют посредством хлорангидридов карбоновых кислот с 10-18 атомами углерода.
Моющие средства согласно изобретению в качестве неионных поверхностно-активных веществ предпочтительно содержат спирты с 10-16 атомами углерода, этоксилированные посредством 3-12 молей этиленоксида, особенно предпочтительно этоксилированные жирные спирты.
Органические сомодификаторы
Пригодными органическими сомодификаторами являются, например, следующие низкомолекулярные поликарбоксилаты:
ди-, три- или тетракарбоновые кислоты с 4-20 атомами углерода, в частности янтарная, пропантрикарбоновая, бутантетракарбоновая, циклопентантетракарбоновая кислоты, а также алкилянтарная и алкенилянтарная кислоты с 2-16 атомами углерода в алкильных и алкенильных остатках;
гидроксикарбоновые кислоты с 4-20 атомами углерода, в частности яблочная, винная, глюконовая, глюкаровая, лимонная, лактобионовая кислоты, сахарозомоно-, ди и трикарбоновые кислоты;
аминополикарбоксилаты, в частности нитрилотриуксусная, метилглициндиуксусная, аланиндиуксусная, этилендиаминтетрауксусная и сериндиуксусная кислоты; соли фосфоновых кислот, в частности гидроксиэтандифосфоновая кислота, этилендиаминтетраметиленфосфонат и диэтилентриаминпентаметиленфосфонат.
Пригодными органическими сомодификаторами являются, например, следующие олиго- или поликарбоксилаты:
олигомалеиновые кислоты, в частности, описанные в европейских заявках на патент ЕР-А-451508 и ЕР-А-396303;
сополимеры и тройные сополимеры ненасыщенных дикарбоновых кислот с 4-8 атомами углерода и сомономеров, обладающих этиленовой ненасыщенностью, причем содержание звеньев этих сомономеров в сополимерах составляет
до 95 мас.% (сомономеры группы (i)),
до 60 мас.% (сомономеры группы (ii),
до 20 мас.% (сомономеры группы (iii).
В качестве ненасыщенных дикарбоновых кислот с 4-8 атомами углерода пригодны, например, малеиновая, фумаровая, итаконовая и цитраконовая кислоты, причем предпочтительной является малеиновая кислота.
В состав группы (i) входят обладающие этиленовой ненасыщенностью монокарбоновые кислоты с 3-8 атомами углерода, в частности акриловая, метакриловая, кротоновая и винилуксусная кислоты, причем предпочтительным является использование акриловой и метакриловой кислот.
В состав группы (ii) входят олефины с 2-22 атомами углерода, винилалкиловые эфиры с 1-8 атомами углерода в алкильных группах, стирол, виниловые эфиры карбоновых кислот с 1-8 атомами углерода, (мет)акриламид и винилпирролидон, причем предпочтительным является использование олефинов с 2-6 атомами углерода, винилалкиловых эфиров с 1-4 атомами углерода в алкильных группах, винилацетата и винилпропионата.
В состав группы (iii) входят (мет)акриловые эфиры спиртов с 1-8 атомами углерода, (мет)акрилонитрил, (мет)акриламиды аминов с 1-8 атомами углеродов, N-винилформамид и винилимидазол.
Если полимеры содержат мономерные звенья винилового эфира из группы (ii), последние могут являться частично или полностью гидролизованными до винилового спирта структурными единицами. Пригодные сополимеры и тройные сополимеры описаны, например, в патенте США US-A 3887806 и немецкой заявке на патент DE-A 4313909.
В качестве органических сомодификаторов предпочтительно пригодны следующие сополимеры дикарбоновых кислот: сополимеры малеиновой и акриловой кислот в массовом соотношении от 10:90 до 95:5, особенно предпочтительно от 30:70 до 90:10, обладающие молекулярной массой от 10000 до 150000;
тройные сополимеры малеиновой, акриловой кислот и винилового эфира карбоновой кислоты с 1-3 атомами углерода, причем массовое отношение малеиновой кислоты к сумме акриловой кислоты и винилового эфира составляет от 10:90 до 95:5, а массовое отношение акриловой кислоты к виниловому эфиру можно варьировать в интервале от 20:80 до 80:20 и
особенно предпочтительными органическими сомодификаторами являются следующие сополимеры дикарбоновых кислот:
тройные сополимеры малеиновой, акриловой кислот и винилацетата или винилпропионата, причем массовое отношение малеиновой кислоты к сумме акриловой кислоты и винилового эфира составляет от 20:80 до 90:10, а массовое отношение акриловой кислоты к виниловому эфиру можно варьировать в интервале от 30:70 до 70:30;
сополимеры малеиновой кислоты и олефинов с 2-8 атомами углерода в мольном соотношении от 40:60 до 80:20, причем особенно предпочтительными являются сополимеры малеиновой кислоты с этиленом, пропиленом или изобутеном в мольном соотношении 50:50.
Пригодными органическими сомодификаторами являются также сополимеры, получаемые путем прививки ненасыщенных карбоновых кислот к низкомолекулярным или гидрированным углеводам (смотри патент США US-A 5227446, немецкие заявки на патент DE-A-4415623 и DE-A-4313909).
Пригодными для прививки ненасыщенными карбоновыми кислотами являются малеиновая, фумаровая, итаконовая, цитраконовая, акриловая, метакриловая, кротоновая и винилуксусная кислоты, а также смеси акриловой и малеиновой кислот, количество которых по отношению к подлежащему прививке компоненту составляет от 40 до 95 мас.%.
С целью модифицирования могут быть дополнительно привиты и другие мономеры, обладающие этиленовой ненасыщенностью, количество которых по отношению к подлежащему прививке компоненту может достигать 30 мас.%. Пригодные для модифицирования мономеры могут быть выбраны из приведенных выше групп (ii) и (iii).
В качестве основы для прививки пригодны деструктированные полисахариды, в частности кислые или полученные путем ферментативной деструкции крахмал, инулин или целлюлоза, восстановленные гидрированием или гидрирующим аминированием и деструктированные полисахариды, в частности маннит, сорбит, аминосорбит и глюкамин, а также полиалкиленгликоли, обладающие среднемассовой молекулярной массой (Mw) до 5000, в частности полиэтиленгликоли, блоксополимеры этиленоксида с пропиленоксидом или бутиленоксидом, статистические сополимеры этиленоксида с пропиленоксидом или бутиленоксидом, алкоксилированные одно- или многоосновные спирты с 1-22 атомами углерода (смотри патент США US-A 4746456).
Из этой группы предпочтительно используют привитые деструктированные или деструктированные восстановленные крахмалы и привитые полиэтиленоксиды, причем к основе прививают от 20 до 80 мас.% мономера. Для прививки предпочтительно используют смесь малеиновой и акриловой кислот в массовом соотношении от 90:10 до 10: 90.
Полиглиоксиловые кислоты в качестве органических сомодификаторов описаны, например, в европейской заявке на патент ЕР-В-001004, патенте США US-A 5399286, немецкой заявке на патент DE-A-4106355 и европейской заявке на патент ЕР-А-656914. Концевые группы полиглиоксиловых кислот могут обладать различной структурой.
Полиамидокарбоновые кислоты и модифицированные полиамидокарбоновые кислоты в качестве сомодификаторов описаны, например, в европейских заявках на патент ЕР-А-454126, ЕР-В-511037, международной заявке WO-A 94/01486 и европейской заявке на патент ЕР-А-581452.
Кроме того, в качестве органических сомодификаторов предпочтительно используют полиаспарагиновую кислоту или продукты конденсации аспарагиновой кислоты с другими аминокислотами, моно- или дикарбоновыми кислотами с 4-25 атомами углерода и/или моно- или диаминами с 4-25 атомами углерода. Особенно предпочтительным является использование полиаспарагиновых кислот, полученных в среде фосфорсодержащих кислот и модифицированных посредством моно- или дикарбоновых кислот с 6-22 атомами углерода или моно- или диаминов с 6-22 атомами углерода.
Органические сомодификаторы на основе продуктов конденсации кротоновой кислоты с гидроксикарбоновыми кислотами или полигидроксильными соединениями описаны, например, в международных заявках WO-A 93/22362 и WO-A 92/16493. Такие продукты, содержащие карбоксильные группы, обычно обладают молекулярной массой до 10000, предпочтительно до 5000.
Ингибиторы потемнения окраски и полимеры, устраняющие загрязнения
Примерами пригодных, устраняющих загрязнения полимеров и ингибиторов потемнения окраски являются сложные полиэфиры, получаемые из полиэтиленоксидов, этиленгликоля и/или пропиленгликоля или ароматических и алифатических дикарбоновых кислот;
сложные полиэфиры, получаемые из полиэтиленоксидов с блокированными с одной стороны концевыми группами, двухосновных спиртов или спиртов с более высокой основностью и дикарбоновой кислоты.
Полиэфиры подобного рода описаны, например, в патенте США US-A 3557039, патенте Великобритании GB-A 1154730, европейских заявках на патент ЕР-А-185427, ЕР-А-241984, ЕР-А-241985, ЕР-А-272033 и патенте США US-A 5142020.
Другими пригодными полимерами для устранения загрязнений являются амфифильные полимеры, получаемые прививкой винилового и/или акрилового эфиров к полиалкиленоксиду, или соответствующие сополимеры (смотри патенты США US-A 4746456, US-A 4846995, немецкую заявку на патент DE-A-3711299, патенты США US-A 4904408, US-A 4846994 и US-A 4849126), а также модифицированные целлюлозы, в частности метилцеллюлоза, гидроксипропилцеллюлоза или карбоксиметилцеллюлоза.
Ингибиторы передачи краски
В качестве ингибиторов передачи краски используют, например, гомо- и сополимеры винилпирролидона, винилимидазола, винилоксазолидона и 4-винилпиридин-N-оксида с молекулярной массой от 15000 до 100000, а также сшитые тонкодисперсные полимеры на основе этих мономеров. О применении таких полимеров для указанной цели известно из немецкой заявки на патент DE-B-2232353, немецких заявок на патент DE-A 2814287, DE-A-2814329 и DE-A-4316023.
Ферменты
Пригодными ферментами являются, например, протеазы, амилазы, липазы и целлюлазы, в частности протеазы. Могут использоваться также сочетания нескольких ферментов.
Наряду с использованием композиций согласно изобретению в составе моющих и чистящих средств, предназначенных для домашней стирки текстильных изделий, они пригодны также для использования в сфере промышленной стирки и чистки. В этом случае в качестве отбеливающего компонента, как правило, используют надуксусную кислоту, добавляемую в виде водного моющего раствора.
Применение в средствах для стирки текстильных изделий
Типичный порошкообразный или гранулированный сверхмощный детергент согласно изобретению может обладать следующим составом:
- от 0,5 до 50 мас.%, предпочтительно от 5 до 30 мас.%, по меньшей мере, одного анионного и/или неионного поверхностно-активного вещества,
- от 0,5 до 60 мас.%, предпочтительно от 15 до 40 мас.%, по меньшей мере, одного неорганического модификатора,
- от 0 до 20 мас.%, предпочтительно от 0,5 до 8 мас.%, по меньшей мере, одного органического сомодификатора,
- от 2 до 35 мас.%, предпочтительно от 5 до 30 мас.% неорганического отбеливающего средства,
- от 0,1 до 20 мас.%, предпочтительно от 0,5 до 10 мас.% активатора отбеливания, при необходимости, смешанного с другими активаторами,
- от 0 до 1 мас.%, предпочтительно не более 0,5 мас.% катализатора отбеливания,
- от 0 до 5 мас.%, предпочтительно от 0 до 2,5 мас.% полимерного ингибитора передачи краски,
- от 0 до 1,5 мас.%, предпочтительно от 0,1 до 1,0 мас.% протеазы,
- от 0 до 1,5 мас.%, предпочтительно от 0,1 до 1,0 мас.% липазы,
- от 0 до 1,5 мас.%, предпочтительно от 0,2 до 1,0 мас.% удаляющего загрязнения полимера,
вспомогательные добавки и сопутствующие вещества (до 100%).
Предпочтительно используемыми в составе моющих средств неорганическими модификаторами являются карбонат натрия, гидрокарбонат натрия, цеолиты А и Р, а также аморфные и кристаллические силикаты натрия.
Предпочтительно используемыми в составе моющих средств органическими сомодифакторами являются сополимеры малеиновой и акриловой кислот, тройные сополимеры акриловой, малеиновой кислот и винилового эфира, а также лимонная кислота.
Предпочтительно используемыми в составе моющих средств неорганическими отбеливающими средствами являются перборат натрия и пергидрат карбоната натрия.
Предпочтительно используемыми в составе моющих средств анионными поверхностно-активными веществами являются линейные и слаборазветвленные алкилбензолсульфонаты согласно изобретению, сульфаты жирных спиртов и мыла.
Предпочтительно используемыми в составе моющих средств неионными поверхностно-активными веществами являются этоксилаты оксоспиртов с 11-17 атомами углеводорода, содержащие от 3 до 13 этиленоксидных звеньев, этоксилаты насыщенных спиртов с 10-16 атомами углеводорода, содержащие от 3 до 13 этиленоксидных звеньев, а также этоксилированные ненасыщенные и оксоспирты, дополнительно содержащие от 1 до 4 звеньев пропиленоксида или бутиленоксида.
Предпочтительно используемыми в составе моющих средств ферментами являются протеаза, липаза и целлюлаза. Из принятых в торговле ферментов к моющим средствам, как правило, добавляют от 0,05 до 2,0 мас.%, предпочтительно от 0,2 до 1,5 мас.% готовых ферментов. Пригодными протеазами являются, например, савиназа, десазим и эспераза, пригодной липазой, например, липолаза, пригодной целлюлазой, например, целлюзим (продукты фирмы Novo Nordisk).
Предпочтительно используемыми в составе моющих средств ингибиторами потемнения и полимерами, устраняющими загрязнения, являются привитые сополимеры с молекулярной массой от 2500 до 8000, получаемые прививкой винилацетата к полиэтиленоксиду (массовое соотношение от 1,2:1 до 3,0:1), полиэтилентерефталаты/оксиэтилентерефталаты, обладающие молекулярной массой от 3000 до 25000, получаемые из полиэтиленоксидов молекулярной массы от 750 до 5000, терефталевой кислоты и этиленоксида, причем мольное отношение полиэтилентерефталата к полиоксиэтилентерефталату составляет от 8:1 до 1:1, а также блокполиконденсаты в соответствии с немецкой заявкой на патент DE-A4403866.
Предпочтительно используемыми в составе моющих средств ингибиторами передачи краски являются растворимые сополимеры винилпирролидона и винилимидазола, обладающие молекулярной массой более 25000, а также тонкодисперсные сшитые полимеры на основе винилимидазола.
Порошкообразные и гранулированные моющие средства согласно изобретению могут содержать до 60 мас.% регулятора, в качестве которого обычно используют сульфат натрия. Однако предпочтительно моющие средства согласно изобретению обладают более низким содержанием регулятора, количество которого не превышает 20 мас.%, особенно предпочтительно не более 8 мас.%.
Моющие средства согласно изобретению могут обладать насыпной плотностью от 300 до 1200 г/л, в частности от 500 до 950 г/л. Современные компактные моющие средства, как правило, обладают высокой насыпной плотностью и являются гранулированными продуктами.
Изобретение поясняют следующие примеры.
Пример 1
Освобожденную от бутадиена С4-фракцию с общим содержанием бутенов 84,2 мас.% и мольным отношением 1-бутена к 2-бутенам от 1:1 до 1,06:1 непрерывно пропускают через трубчатый реактор, заполненный гетерогенным катализатором Re2О7/Al2О3, при температуре 40°С и давлении 10 бар. Нагрузка на катализатор составляет 4500 кг/м2·час. Выделенный дестилляцией реакционный продукт содержит следующие компоненты (в массовых процентах): этен 1,15%, пропен 18,9%, бутаны 15,8%, 2-бутены 19,7%, 1-бутен 13,3%, изобутен 1,0%, 2-пентен 19,4%, метилбутены 0,45%, 3-гексен 10,3%. Из этого продукта дистилляцией выделяют 2-пентен и 3-гексен со степенью чистоты, превышающей 99 мас.%.
Пример 2
Непрерывным способом осуществляют димеризацию 3-гексена, используя стационарный слой катализатора.
Катализатор: 50% NiO, 34% SiO2, 13% TiO2, 3% Al2О3 (в соответствии с немецким патентом DE 4339713) в виде частиц размером 1-1,5 мм (100 мл), кондиционированный в течение 24 часов при 160°С в токе азота.
Реактор: изотермический реактор диаметром 16 мм.
Нагрузка на катализатор: 0,25 кг/л·час.
Давление: от 20 до 25 бар.
Температура: от 100 до 160°С.
Дистиллят
Из собранного продукта дистилляцией выделена C12-фракция со степенью чистоты 99,9 мас.%.
Пример 3
2-Пентен, полученный в результате реакции обмена рафината II, подвергнут димеризации, осуществляемой непрерывным способом аналогично примеру 2 в присутствии никельсодержащего катализатора. В результате фракционированной дистилляции продуктов димеризации получена С10-фракция со степенью чистоты 99,5%.
Пример 4
Смесь 2-пентена и 3-гексена, полученная в результате реакции обмена рафината II, подвергнута димеризации, осуществляемой непрерывным способом аналогично примерам 2 и 3. В результате фракционированной дистилляции продуктов димеризации выделена фракция, содержащая децен, ундецен и додецен со степенью чистоты 99,5%.
Пример 5
Бензол алкилируют полученным согласно примеру 2 С12-олефином при его мольном отношении к бензолу 1:10, для чего реакционную смесь загружают в автоклав объемом 300 мл, снабженный мешалкой и корзиной, в которой находится цеолит типа морденита (25 мас.% по отношению к олефину). Автоклав закрывают, дважды пропускают через него азот и нагревают до температуры 180°С. Реакция продолжается в течение 12 часов, после чего автоклав охлаждают, отфильтровывают взвешенные частицы катализатора и полученную реакционную смесь анализируют, используя комбинированный метод газовой хроматографии/масс-спектрометрии. Отгоняют избыток бензола и легкокипящие продукты и полученную смесь алкилароматических соединений анализируют, используя комбинированный метод газовой хроматографии/масс-спектрометрии и С13-ядерный магнитный резонанс.
Пример 6
В четырехгорлую колбу объемом 2 литра (генератор серного ангидрида), снабженную магнитной мешалкой, термометром, капельной воронкой, пористым стеклянным фильтром для подводимого газа и устройством для выхода газа загружают 1900 г обедненного серным ангидридом олеума. Посредством выполненного из витона шланга, присоединенного к устройству для выхода газа, эта колба соединена с другой, трехгорлой колбой объемом 1 литр, снабженной плосколопастной мешалкой, термометром, пористым стеклянным фильтром для подводимого газа и устройством для выхода газа. В эту колбу загружают смесь полученных согласно примеру 5 алкилбензолов.
Обедненный серным ангидридом олеум в генераторе SO3 нагревают до температуры 120°С и в течение 30 минут посредством капельной воронки добавляют олеум концентрацией 65%. Уносимый током азота (80 л/час) газообразный серный ангидрид через патрубок диаметром 6 мм вводят в алкилбензол. Температура смеси алкилбензола с алкилбензолсульфокислотой медленно повышается до 40°С, и ее поддерживают на этом уровне, используя охлаждающую воду. Остаточный газ отсасывают водоструйным насосом. Мольное отношение серного ангидрида к алкилбензолу составляет 1,01:1.
Через 4 часа (период дозревания) образовавшуюся алкилбензолсульфокислоту стабилизируют, добавляя 0,4 мас.% воды, и нейтрализуют раствором едкого натра, превращая в алкилбензолсульфонат.
Пример 7
Бензол алкилируют полученной согласно примеру 4 смесью C10-/C11-/C12-олефинов при их мольном отношении к бензолу 1:10, для чего реакционную смесь загружают в автоклав объемом 300 мл, снабженный мешалкой и корзиной, в которой находится цеолит типа морденита (25 мас.% по отношению к смеси олефинов). Автоклав закрывают, дважды пропускают через него азот и нагревают до температуры 200°С. Реакция продолжается в течение 12 часов, после чего автоклав охлаждают, отфильтровывают взвешенные частицы катализатора и полученную реакционную смесь анализируют, используя комбинированный метод газовой хроматографии/масс-спектрометрии.
Отгоняют избыток бензола и легкокипящие продукты и полученную смесь алкилароматических соединений анализируют, используя комбинированный метод газовой хроматографии/масс-спектрометрии и С13-ядерный магнитный резонанс.
Пример 8
Полученную в примере 7 смесь алкилбензолов превращают в алкилбензолсульфонат аналогично примеру 6.
Пример 9
Бензол алкилируют полученной согласно примеру 4 смесью C10-/C11-/C12-олефинов при их мольном отношении к бензолу 1:2, для чего реакционную смесь загружают в автоклав объемом 300 мл, снабженный мешалкой и корзиной, в которой находится цеолит типа ZSM-12 (5 мас.% по отношению к смеси олефинов). Автоклав закрывают, дважды пропускают через него азот и нагревают до температуры 180°С. Реакция продолжается в течение 12 часов, после чего автоклав охлаждают, отфильтровывают взвешенные частицы катализатора и полученную реакционную смесь анализируют, используя комбинированный метод газовой хроматографии/масс-спектрометрии.
Отгоняют избыток бензола и легкокипящие продукты и полученную смесь алкилароматических соединений анализируют, используя комбинированный метод газовой хроматографии/масс-спектрометрии и С13-ядерный магнитный резонанс.
Пример 10
Полученную в примере 9 смесь алкилбензолов превращают в алкилбензолсульфонат аналогично примеру 6.
Пример 11
Бензол алкилируют С12-олефином, полученным согласно примеру 2, при его мольном отношении к бензолу 1:4, для чего реакционную смесь загружают в автоклав объемом 300 мл, снабженный мешалкой и корзиной, в которой находится цеолит типа Beta (10 мас.% по отношению к олефину). Автоклав закрывают, дважды пропускают через него азот и нагревают до температуры 180°С. Реакция продолжается в течение 12 часов, после чего автоклав охлаждают, отфильтровывают взвешенные частицы катализатора и полученную реакционную смесь анализируют, используя комбинированный метод газовой хроматографии/масс-спектрометрии. Отгоняют избыток бензола и легкокипящие продукты и полученную смесь алкилароматических соединений анализируют, используя комбинированный метод газовой хроматографии/масс-спектрометрии и С13-ядерный магнитный резонанс.
Пример 12
Полученную в примере 11 смесь алкилбензолов превращают в алкилбензолсульфонат аналогично примеру 6.
Пример 13
Бензол алкилируют полученной согласно примеру 4 смесью C10-/C11-/C12-олефинов при их мольном отношении к бензолу 1:4, для чего реакционную смесь загружают в автоклав объемом 300 мл, снабженный мешалкой и корзиной, в которой находится цеолит типа МСМ-22 (10 мас.% по отношению к смеси олефинов). Автоклав закрывают, дважды пропускают через него азот и нагревают до температуры 200°С. Реакция продолжается в течение 12 часов, после чего автоклав охлаждают, отфильтровывают взвешенные частицы катализатора и полученную реакционную смесь анализируют, используя комбинированный метод газовой хроматографии/масс-спектрометрии.
Отгоняют избыток бензола и легкокипящие продукты и полученную смесь алкилароматических соединений анализируют, используя комбинированный метод газовой хроматографии/масс-спектрометрии и С13-ядерный магнитный резонанс.
Пример 14
В нагретую до температуры 120°С четырехгорлую колбу объемом 10 литров насосом подают олеум (65%) в концентрированной серной кислоте со скоростью 1 л/час. Через серную кислоту со скоростью 130 л/час пропускают сухой воздух, который предварительно проходит через пористый стеклянный фильтр. Содержащий около 4% серного ангидрида воздушный поток поступает в реактор длиной 2 метра с падающей пленкой, в котором он при температуре около 40-50°С контактирует со смесью алкилбензолов из примера 13 (реактор снабжен двойной охлаждающей рубашкой, в которую подается вода с температурой 10-15°С). Мольное отношение серного ангидрида к алкилбензолу составляет 1,01:1, время реакции около 10 секунд. Полученную алкилбензолсульфокислоту перекачивают насосом в сосуд для дозревания, где ее выдерживают в течение 4-8 часов, после чего стабилизируют, добавляя 0,4 мас.% воды, и нейтрализуют раствором едкого натра, превращая в алкилбензолсульфонат.
Пример 15
Бензол алкилируют полученным согласно примеру 2 С12-олефином при его мольном отношении к бензолу 1:10 в четырехгорлой колбе объемом 2 литра, снабженной мешалкой, термометром, обратным холодильником с газоотводом и капельной воронкой. В колбу загружают бензол и AlCl3, повышают температуру до 80°С и медленно прибавляют олефин. Через 30 минут (период дозревания) колбу охлаждают, отфильтровывают взвешенные частицы катализатора и реакционную смесь нейтрализуют раствором едкого натра. Полученный продукт промывают водой и сушат. Отгоняют избыток бензола и легкокипящие продукты и полученную смесь алкилароматических соединений анализируют, используя комбинированный метод газовой хроматографии/масс-спектрометрии и С13-ядерный магнитный резонанс.
Пример 16
Полученную в примере 15 смесь алкилбензолов превращают в алкилбензолсульфонат аналогично примеру 6.
Пример 17
а) Непрерывном способом осуществляют димеризацию 3-гексена, полученного аналогично примеру 1, используя стационарный слой катализатора, аналогично приведенным в примере 2 образом. Полученная С12-фракция содержит следующие изомеры:
б) В трубчатый реактор, расположенный в печи с циркулирующим воздухом, подают 32 г катализатора (60% Н-морденита с соотношением SiO2:Al2O3=24,5 и 40% plural® SB фирмы Кондеа) в виде частиц размером 0,7-1,0 мм и активируют в течение 6 часов при температуре 500°С. Затем охлаждают и добавляют бензол и С12-фракцию, полученную на стадии а), в соотношении 10:1 моль, в количестве 0,32 г/г катализатора в час. Поддерживают рециркулирующий поток в количестве в 10 раз больше указанного подаваемого потока. Реакцию проводят при температуре 160°С и давлении 30 бар. Количество исходных веществ и полученную смесь C18-алкилароматических соединений после отгонки анализируют, используя комбинированный метод газовой хроматографии/масс-спектрометрии и С13-ядерный магнитный резонанс.
Свойства продукта:
степень разветвленности продукта составляет 1,1;
количество неразвлетвенных компонентов 16%;
степень разветвленности, исключая количество неразвлетвенных компонентов, 1,2;
доля структурных элементов в алкильной части, имеющих индекс H/С, равный 0, составляет максимально 10%.
Вышеприведенные данные, по мнению заявителя, дополнительно подтверждают возможность осуществления данного изобретения во всем испрашиваемом объеме охраны.
Кроме того, проводили опыт по способности к биологическому расщеплению (согласно Инструкциям ОЭСР №301 А - уменьшение растворимого органического углерода (DOC)- и ОЭСР №301 В - образование СО2- арилалкилсульфонатов, полученных предлагаемым способом, в частности с соединением примера 6, т.е. линейный алкилбензолсульфонат на основе додецена. При этом биологическое расщепление арилалкилсульфонатов в течение 17 дней составляет более 60%, а в течение 24 дней - более 70%. Таким образом, данные арилалкилсульфонаты обладают положительной способностью к биологическому расщеплению.
Далее проводили опыт по первичному моющему действию арилалкилсульфонатов по примеру 6. При этом использовали следующую композицию моющего средства (вес.%):
ПАВ (в виде смеси анионного и неионного ПАВ) 16% (10 вес.% в расчете на моющее средство арилалкилсульфоната по примеру 6 в качестве анионного ПАВ и 6 вес.% в расчете на моющее средство этоксилированного спирта с 13-15 атомами углерода, при алкосилировании которого использовали 7 моль этиленоксида на 1 моль спирта в качестве неионного ПАВ)
в количестве 4,5 г/л при температуре промывной воды 40°С, модуле ванны 1:12,5 и жесткости воды 3 ммоль/л, причем применяли 6 тканей, которые являются торговыми продуктами немецкой фирмы wfk Крефельд (ткани марки wfk 10D и wfk 20D) и швейцарской фирмы ЕМРА (ткани марки ЕМРА 101, ЕМРА 104, ЕМРА 141 и ЕМРА 143).
Первичное моющее действие измерялись следующим методом. Обработанные вышеуказанным образом ткани гладили. Затем измеряли отражение сглаженных тканей при длине волны 460 нм. В качестве контрольной системы или соответственно сравнительной системы использовали ткань, обработанную моющим средством, содержащим в качестве ПАВ 10 вес.% известного линейного арилалкилсульфоната в качестве анионного ПАВ и 6 вес.% этоксилированного спирта с 13-15 атомами углерода, при алкосилировании которого использовали 7 моль этиленоксида на 1 моль спирта, в качестве неионного ПАВ.
Приведенные в схемах данные представляют собой суммарную величину R по указанным 6 тканям.
Полученные данные по первичному моющему действию приведены в схемах, изображенных на фиг.1 и 2.
Из вышеприведенных данных однозначно вытекает, что арилалкилсульфонаты, полученные предлагаемым способом, обладают неожиданно улучшенными свойствами в отношении первичного моющего действия по сравнению с обычными арилалкилсульфонатами. Данный технический результат достигается и при комбинации арилалкилсульфонатов с дополнительным неионным ПАВ.

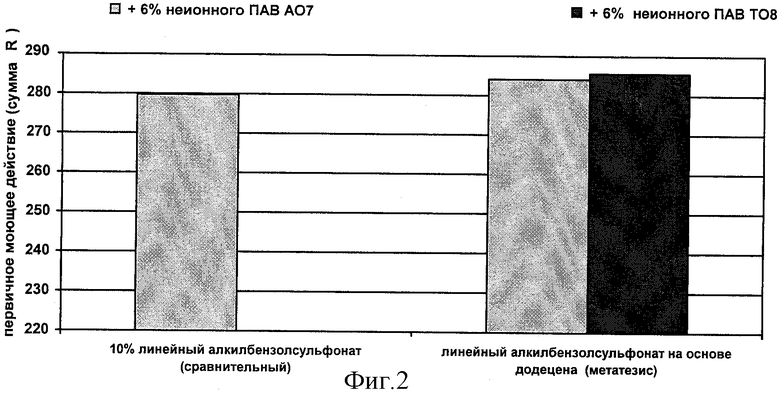
Изобретение касается способа получения алкиларилсульфонатов путем а) превращения смеси С4-олефинов в смесь олефинов, содержащую 2-пентен и/или 3-гексен, в присутствии катализатора реакции обмена и, при необходимости, выделения 2-пентена и/или 3-гексена, b) димеризации полученного на стадии а) 2-пентена и/или 3-гексена в присутствии катализатора димеризации с образованием содержащей С10-С12-олефины смеси и, при необходимости, выделения С10-С12-олефинов, с) взаимодействия полученной на стадии b) смеси С10-С12-олефинов с ароматическим углеводородом в присутствии катализатора алкилирования с образованием алкилароматических соединений, причем перед взаимодействием дополнительно могут быть добавлены линейные олефины, d) сульфирования полученных на стадии с) алкилароматических соединений и нейтрализации продуктов сульфирования до алкиларилсульфонатов, причем перед сульфированием дополнительно могут быть добавлены линейные алкилбензолы, и е) при необходимости, смешивания полученных на стадии d) алкиларилсульфонатов с линейными алкиларилсульфонатами. Описано также моющее средство на основе полученных согласно настоящему способу алкиларилсульфонатов. 2 н. и 5 з.п. ф-лы, 2 ил., 1 табл.
a) превращения смеси С4-олефинов в смесь олефинов, содержащую 2-пентен и/или 3-гексен, в присутствии катализатора реакции обмена и, при необходимости, выделения 2-пентена и/или 3-гексена,
b) димеризации полученного на стадии а) 2-пентена и/или 3-гексена в присутствии катализатора димеризации с образованием содержащей C10-C12-олефины смеси и, при необходимости, выделения C10-C12-олефинов,
c) взаимодействия полученной на стадии b) смеси C10-C12-олефинов с ароматическим углеводородом в присутствии катализатора алкилирования с образованием алкилароматических соединений,
d) сульфирования полученных на стадии с) алкилароматических соединений и нейтрализации продуктов сульфирования до алкиларилсульфонатов.
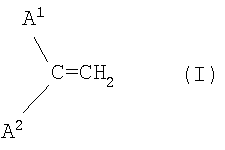
в которой А1 и А2 являются алифатическими углеводородными остатками.
| СПОСОБ ПОЛУЧЕНИЯ ВОДОРАСТВОРИМЫХ СУЛЬФИРОВАННЫХ ДИСПЕРГАТОРОВ | 1994 |
|
RU2117003C1 |
| US 3442964, 06.05.1969 | |||
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |

