Изобретение относится к переработке природных бутанов, более конкретно к способу совместного получения ди-н-бутена из природных алкил-трет-бутиловых эфиров.
Алкил-трет-бутиловые эфиры (в дальнейшем: АТБЭ) используют в качестве присадки к автомобильному бензину для повышения его октанового числа. Их получают путем присоединения алканолов к изобутену; данный процесс также называют этерификацией.
Изобутен может иметь четыре разных источкника: он может быть продуктом из установки крекинга с водяным паром, установки для получения пропиленоксида, нефтеперерабатывающих заводов (установок для каталитического крекинга в кипящем слое) и установок дегидрирования изобутана (см. R. A. Pogiiano и др. , Dehydrogenation-Based Ether Production -Adding Value to LPG and Gas Condensate, 1996 Petrochemical Review, издательство DeWitt & Company, г. Хьюстон, Тексас, США). В первых трех из перечисленных источников изобутен получается в качестве компонента фракции С4, то есть, в качестве непосредственно получаемого побочного продукта. При дегидрировании изобутана изобутен часто представляет собой косвенный побочный продукт, получаемый в таких установках, так как его исходное соединение, изобутан, также получается в качестве непосредственного побочного продукта в установках крекинга с водяным паром или на нефтеперерабатывающих заводах, или же путем изомеризации н-бутана, который со своей стороны представляет собой побочный продукт в установках крекинга с водяным паром и на нефтеперерабатывающих заводах. В настоящее время мировое производство АТБЭ составляет примерно 25 миллионов тонн в год, с тенденцией к повышению. Количество бутанов и бутенов, получаемых в качестве побочных продуктов в одной лишь установке крекинга или на одном лишь нефтеперерабатывающем заводе, является слишком небольшим для того, чтобы полностью пользоваться возможностью экономичного производства путем его расширения, в общем имеющейся в случае процесса получения АТБЭ. То есть, необходимо было бы собирать изобутен и/или изобутан (для дегидрирования) из установок крекинга или нефтеперерабатывающих заводов с тем, чтобы эксплуатировать установку для получения АТБЭ при оптимальной мощности. Альтернативно можно было бы собирать достаточное количество фракции C4 из установок указанного типа и на месте перерабатывать его с получением изобутена и изобутана. Однако проблема при обеих возможностях заключается в том, что перевозка жидких газов является дорогой, не в последнюю очередь из-за дорогостоящих мер безопасности.
Дибутен представляет собой смесь изомеров, которую получают при олигомеризации бутенов наряду с высшими бутеновыми олигомерами в результате димеризации и/или содимеризации бутенов, то есть, н-бутена и/или изобутена. Под ди-н-бутеном понимают продукт димеризации н-бутена, то есть, 1-бутена и/или 2-бутена. Важными компонентами ди-н-бутена являются 3-метил-2-гептен, 3,4-диметил-2-гексен и со второстепенным значением н-окгены. Диизобутен представляет собой ту смесь изомеров, которую получают в результате димеризации изобутена. Диизобутен является более разветвленным, чем дибутен, который, в свою очередь, является более разветвленным, чем ди-н-бутен.
Дибутен, ди-н-бутен и диизобутен являются исходными соединениями для получения изомерных нонанолов путем гидроформилирования и гидрирования получаемых таким образом альдегидов с 9 атомами углерода. Сложные эфиры данных нонанолов, в частности сложные эфиры фталевой кислоты, представляют собой пластификаторы, получаемые в крупном масштабе и используемые в первую очередь в поливинилхлориде. Нонанолы из ди-н-бутена являются в большей степени прямолинейными, чем нонанолы из дибутена, которые, однако, со своей стороны менее разветвлены, чем нонанолы из диизобутена. Сложные эфиры нонанолов из ди-н-бутена обладают техническими преимуществами по сравнению со сложными эфирами из других нонанолов и поэтому пользуются особым спросом.
Н-бутен для димеризации, так же как и изобутен, можно получать, например, из фракции С4 установки крекинга с водяным паром или установки для каталитического крекинга в кипящем слое. Фракции С4 перерабатывают, как правило, путем отделения 1,3-бутадиена с помощью избирательной промывки, например, с использованием N-метилпирролидона. Изобутен представляет собой желаемый и особенно ценный компонент фракции С4, потому его можно отдельно или в смеси с другими углеводородами с 4 атомами углерода подвергать химическому взаимодействию с получением продуктов, пользующихся большим спросом, например, с изобутаном до высокооктанового изооктана, или с метанолом до метил-трет-бутилового эфира, который представляет собой важнейший АТБЭ. После реакции изобутена в качестве остатка получают н-бутены, н-бутан и изобутан. Однако доля н-бутена среди продуктов расщепления из установки крекинга с водяным паром или нефтеперерабатывающего завода является сравнительно небольшой, а именно в установках крекинга с водяным паром она составляет порядка около 10 вес. % в пересчете на главный целевой продукт, то есть, этилен. Это означает, что с помощью установки крекинга с водяным паром, имеющей внушительную производительность 600 000 тонн этилена в год, получают лишь примерно 60 000 тонн н-бутена в год. Можно было бы увеличить количество получаемых н-бутенов (и изобутенов) путем дегидрирования н- и изобутана, получаемых вместе с н-бутенами в количестве примерно 15 000 тонн в год. Однако это не рекомендуется, потому что установки для дегидрирования требуют высоких капиталовложений и являются неэкономичными при столь небольшой мощности.
Как выше указывалось, изобутен представляет собой ценный продукт крекинга и поэтому, как правило, не имеется в распоряжении для изомеризации до н-бутена. Однако количество н-бутенов, непосредственно получаемых в установке крекинга с водяным паром или на нефтеперерабатывающем заводе, не хватает для получения достаточного количества ди-н-бутена для установки для получения нонанола, мощность которой достаточно высока с тем, чтобы она могла бы с точки зрения экономичности конкурировать с уже имеющимися большими установками для получения важных спиртов, служащих в качестве пластификаторов, например, 2-этилгексанола. Как выше указывалось, установки для получения пропиленоксида являются еще менее производительными, что касается получения н-бутенов. Это означает, что необходимо было бы собирать н-бутены из разных установок крекинга с водяным паром, нефтеперерабатывающих заводов или установок для получения пропиленоксида (или же обрабатывать фракцию С4 из разных источников с получением н-бутена) и совместно подвергать их олигомеризации с тем, чтобы достичь полной загрузки установки для получения нонанола, достаточно крупной для экономичной работы. Однако, как уже указывалось, перевозка жидких газов дорога.
Поэтому задачей изобретения является разработка способа, позволяющего получение н-бутена и изобутена в одном месте, т. е. без необходимости перевозки через большие расстояния, а именно в количестве, требуемом для совместного производства ди-н-бутена и АТБЭ на установке для получения ди-н-бутена, имеющей мощность, например, 200 000 - 800 000 т/г, и на такой же установке для получения АТБЭ, в частности, метил-трет-бутилового эфира, имеющей мощность, например, 300 000 - 800 000 т/г. Кроме того, была бы желательной разработка способа, позволяющего регулирование количественного соотношения н-бутена и изобутена в соответствии с желаемыми количествами ди-н-бутена и метил-трет-бутилового эфира.
Поставленная задача решается предлагаемым способом совместного получения ди-н-бутена и алкил-трет-бутиловых эфиров из природных бутанов за счет того, что
(а) природные бутаны разделяют на н-бутан и изобутан на стадии разделения,
(б) н-бутан подвергают дегидрированию на стадии дегидрирования до содержащей н-бутен смеси с последующей олигомеризацией полученного н-бутена на стадии олигомеризации до смеси олигомеров и выделением из последней ди-н-бутена,
(в) изобутан подвергают дегидрированию на стадии дегидрирования до смеси, содержащей изобутен, который на стадии этерификации подвергают взаимодействию с алканолом до алкил-трет-бутилового эфира.
Перед стадией разделения природные бутаны можно подвергать гидрированию на стадии гидрирования, при этом стадия разделения связана со стадией изомеризации, предназначенной для установления количественного соотношения н-бутана и изобутана в соответствии с желаемым количественным соотношением алкил-трет-бутилового эфира и ди-н-бутена.
Между стадией дегидрирования и стадией олигомеризацией в любой последовательности можно осуществлять стадию избирательного гидрирования и/или стадию очистки. При этом из смеси олигомеризации выделяют остаточные газы, которые, в случае необходимости после предварительной очистки, рециркулируют на стадию дегидрирования.
Между стадией дегидрирования и стадией этерификации можно осуществлять стадию избирательного гидрирования. Остаточные газы со стадии этерификации через стадию очистки можно рециркулировать на стадию дегидрирования. В качестве алканола предпочтительно используют этанол, изопропанол, изобу-танол или, в частности, метанол.
Предлагаемый способ отличается высокой гибкостью, так как в рамках мощности установки для получения ди-н-бутена и установки для получения АТБЭ количество ди-н-бутена и АТБЭ можно регулировать в зависимости от требований рынка.
Под природными бутанами понимают фракцию С4 "мокрых" компонентов природного газа и сопровождающих нефть газов, выделяемых из данных газов в жидкой форме путем охлаждения примерно до -30oС. Путем низкотемпературной перегонки из них получают бутаны, состав которых колебается в зависимости от месторождения. Однако в общем природные бутаны содержат примерно 30% изобутана и 65% н-бутана и, как правило, в качестве дальнейших компонентов примерно 2% углеводородов с 1 - 3 атомами углерода и примерно 3% углеводородов с числом атомов углерода выше четырех.
Природные бутаны можно без разделения использовать в качестве сырья в установках крекинга с водяным паром или в качестве присадки к автомобильному бензину. Их можно разделять на н-бутан и изобутан путем фракционной перегонки. Изобутан используют в крупном масштабе, например, для получения пропиленоксида путем соокисления пропилена и изобутана, а также в качестве агента алкилирования н-бутена соответственно изобутена до изооктана, который из-за его высокого октанового числа ценят в качестве присадки к автомобильному бензину. В отличие от этого имеются лишь менее важные области применения н-бутана. Последний служит, например, в качестве бутанового газа для отопительных целей или его используют в сравнительно небольших количествах для получения полимеров или сополимеров, или ангидрида малеиновой кислоты путем окисления с помощью воздуха.
Осуществление предлагаемого способа иллюстрируется с помощью технологической схемы, представленной на чертеже.
А) Получение ди-н-бутена
Сначала на стадии разделения 1 природный бутан, подаваемый по линии 2, разделяют на н-бутан, отводимый по линии 3, и изобутан, отводимый по линии 4. Разделение целесообразно осуществляют в высокоэффективной колонне, в которой н-бутан с помощью фракционной перегонки при низкой температуре или, предпочтительно, при повышенном давлении, целесообразно при 4 - 7 бар, отделают от изобутана, точка кипения которого в зависимости от давления примерно на 10 - 20oC ниже. При этом в качестве кубового продукта получают углеводороды с числом атомов выше четырех, н-бутан отводят в качестве бокового потока, а изобутан отводят в качестве головного продукта вместе с более легкими компонентами.
H-бутан подают на стадию дегидрирования 5, с которой по линии 6 отводят смесь, содержащую н-бутен. Дегидрирование осуществляют известными приемами. Целесообразно дегидрирование осуществляют в газовой фазе в присутствии твердого катализатора или катализатора, имеющегося в виде кипящего слоя, например, в присутствии окиси хрома (III) или, предпочтительно, платины на окиси алюминия, или цеолите в качестве носителя. В общем дегидрирование осуществляют при 400 - 800oС, предпочтительно 550 - 650oС. Обычно процесс осуществляют при атмосферном давлении или слегка повышенном до 3 бар давлении. Время пребывания в катализаторном слое в общем составляет в зависимости от катализатора, температуры и желаемой степени конверсии 1-60 мин. Таким образом, производительность обычно составляет 0,6 - 36 кг н-бутана на 1 м3 катализатора в 1 ч.
Целесообразно дегидрирование осуществляют лишь до остаточного содержания н-бутана в отводимой по линии 6 смеси, равного примерно 50%. При более высокой температуре можно достичь более высокой степени конверсии, однако при этом в растущей мере происходят крекинговые реакции, снижающие выход и вследствие образования отложений кокса срок службы катализатора дегидрирования. Оптимальные условия реакции, ведущие к достижению желательной степени конверсии, например вид катализатора, температура и время пребывания, можно легко определить путем предварительных опытов.
Как правило, после дегидрирования смесь содержит 90 - 95 % углеводородов с 4 атомами углерода и, кроме того, водород и ниже и выше летучие компоненты. Целесообразно смесь очищают перед олигомеризацией. На первой стадии очистки (не показана) путем конденсации отделяют фракцию С4 и вышекипящие компоненты. Конденсат подвергают перегонке под давлением, причем тоже конденсированные, растворенные углеводороды с 1 - 3 атомами углерода выходят из верхней части колонны. Из кубового продукта при дальнейшей перегонке в качестве главного продукта получают углеводороды с 4 атомами углерода, а в качестве остатка - сравнительно небольшое количество углеводородов с числом атомов углерода выше четырех.
Углеводороды с 4 атомами углерода в общем содержат небольшое количество, например 0,01 - 5 об. %, 1,3-бутадиена. Рекомендуется удалить этот компонент, потому что даже в гораздо меньшем количестве он может вредить катализатору олигомеризации. Пригодным методом для этого является избирательное гидрирование 7, которым, кроме того, повышается доля желательного н-бутена. Его можно проводить, например, в жидкой фазе с полностью растворенным водородом в стехиометрических количествах. В качестве катализаторов для избирательного гидрирования пригодны, например, никель и, в частности, палладий на носителе, например 0,3 вес. % палладия на активном угле или предпочтительно на окиси алюминия. Небольшое количество моноокиси углерода порядка частей на миллион поощряет избирательность гидрирования 1,3-бутадиена до моноолефина и тормозит образование полимеров (так называемого "зеленого масла"), дезактивирующих катализатор. В общем метод осуществляют при комнатной температуре или при повышенной температуре примерно до 60oС и при повышенном давлении, целесообразно составляющем до 20 бар. Таким образом, содержание 1,3-бутадиена во фракции С4 смеси дегидрирования снижается до величины менее чем 1 части на 1 миллион.
Кроме того, является целесообразным перед олигомеризацией на стадии 8 подавать полученную фракцию С4 смеси дегидрирования, уже почти полностью освобожденную от 1,3-бутадиена, на стадию очистки 9 с помощью молекулярного сита, причем удаляются дальнейшие вещества, вредящие катализатору олигомеризации, благодаря чему далее продлевается срок службы катализатора. К таким вредным веществам относятся соединения кислорода и серы. Целесообразно использовать молекулярное сито с размером пор 4-15 ангстрем, предпочтительно 7 - 13 ангстрем. В некоторых случаях по экономическим причинам является целесообразным пропускать смесь дегидрирования последовательно через молекулярные сита с разной величиной пор. Данный метод можно осуществлять в газовой, жидкой или смешанной газово-жидкой фазе. В зависимости от этого давление в общем составляет 1 - 200 бар. Целесообразно работают при комнатной температуре или при температуре до 200oС.
Химическая природа молекулярных сит играет меньшую роль, чем их физическая структура, то есть, в частности, размер пор. Это означает, что можно использовать самые разные молекулярные сита, кристаллические, естественные силикаты алюминия, например слоистые решетчатые силикаты, так же как и синтетические молекулярные сита, например, с цеолитной структурой. Имеются в торговле цеолиты типа А, Х и Y, среди других фирм Байер АГ, Дау Кемикальс Ко. , Юньон Карбайд Корпорейшн, Лапорт Индастриз Лтд. и Мобиль Ойль Ко. Для осуществления способа пригодны также синтетические молекулярные сита, которые наряду с алюминием и кремнием содержат и другие атомы, введенные путем катионного обмена, например, галлий, индий или лантан, а также никель, кобальт, медь, цинк или серебро. Кроме того, пригодны также синтетические цеолиты, в которых кроме алюминия и кремния путем соосаждения в решетку встроены еще дальнейшие атомы, например, бор или фосфор.
Как уже указывалось, стадия избирательного гидрирования 7 и стадия очистки 9 с помощью молекулярнго сита являются факультативными, предпочтительными мерами в рамках предлагаемого способа. Их последовательность в принципе не играет никакой роли, однако, предпочтительным является показанная на чертеже последовательность.
Полученную на стадии дегдрирования 5 смесь, в случае необходимости после описанной предварительной обработки, подают на стадию олигомеризации 8, которая представляет собой существенный прием предлагаемого способа. Олигомеризацию осуществляют известными методами. В общем работают в жидкой фазе, в присутствии, например, системы, состоящей из октоата никеля(11), хлорида этилалюминия и свободной жирной кислоты в качестве гомогенного катализатора, или, предпочтительно, одного из многочисленных известных, неподвижно установленных или суспендированных в смеси олигомеризации катализаторов на основе никеля и кремния, которые могут быть нанесены на носитель. Часто катализаторы дополнительно содержат алюминий. Другие пригодные катализаторы получают путем замены имеющихся на поверхности носителей частиц с положительным заряжением, например, протонов или натриевых ионов, ионами никеля. Это возможно в случае самых разных носителей, например, аморфного силиката алюминия, кристаллического силиката алюминия, цеолитов типа X, У и ZSM.
В зависимости от используемого катализатора олигомеризацию осуществляют целесообразно при 20 - 200oС и 1-100 бар. Время реакции (или контакта) в общем составляет 5-60 мин. Параметры способа, в частности вид катализатора, температуру и время контакта, согласуют друг с другом с тем, чтобы достигалась желаемая степень олигомеризации, то есть, главным образом димеризация. Само собой разумеется, что для этого нельзя добиваться полной конверсии при реакции, а целесообразно добиваются 30-70%-ной конверсии для каждого цикла реакции. Оптимальную комбинацию параметров способа можно без проблем устанавливать с помощью предварительных опытов.
Смесь олигомеризации 8 подают на стадию переработки 10, на которой выделяют остаточные газы, которые отводят по линии 11 и рециркулируют на стадию дегидрирования 5. В том случае, если на стадии олигомеризации 8 используют катализатор типа указанных жидких катализаторов, то желательно. чтобы перед рециркуляцией остаточные газы очищали с тем, чтобы обеспечить долговременную службу катализатора дегидрирования. Сперва смесь олигомеризации обрабатывают водой для экстракции катализатора. Затем выделенный остаточный газ сушат с использованием подходящего молекулярного сита, причем отделяются также другие побочные компоненты. После этого путем избирательного гидрирования, например в присутствии палладиевого катализатора, удаляют многократно ненасыщенные соединения, например, бутины, и в конце очищенный таким образом остаточный газ рециркулируют на стадию дегидрирования 5. Эти приемы очистки остаточного газа отпадают при использовании твердого катализатора для олигомеризации.
Из жидкой фазы смеси олигомеризации 8 на стаддии 10 путем фракционной перегонки выделяют ди-н-бутен, отводимый по линии 12, и тримерный н-бутен, то есть, изомерные додецены, отводимые по линии 13, причем ди-н-бутен в качестве главного продукта непосредственно пригоден для получения нонанолов. Додецены представляют собой желаемый побочный продукт. Их можно подвергать гидроформилированию, после чего продукты гидроформилирования подвергают гидрированию, и получаемые тридеканолы подвергают оксэти-лированию, в результате чего получают ценное сырье моющих средств.
Б) Получение АТБЭ
Изобутан со стадии разделения 1 подают на стадию дегидрирования 14, где его подвергают реакции до содержащей изобутен смеси, отводимой по линии 15. В отношении условий процесса данное дегидрирование не в значительной мере отличается от дегидрирования н-бутана на стадии 5. Изобутан подвергается дегидрированию легче, чем н-бутан, так что в рамках указанных для стадии 5 дегидрирования пределов эксплуатации можно выбрать в общем несколько более умеренные условия. И в случае данного процесса дегидрирования является целесообразным добиваться лишь конверсии примерно 50%.
Как выше описано для смеси, получаемой на стадии дегидрирования 5, смесь, получаемая на стадии дегидрирования 14, кроме углеводородов с 4 атомами углерода содержит водород и легколетучие компоненты (которые отчасти содержатся в природных бутанах и отчасти образуются при дегидрировании), а также компоненты с более высокой точкой кипения. Перед этерификацией получаемую на стадии дегидрирования 14 смесь целесообразно очищают на первой (не представленной на чертеже) стадии очистки, которая соответствует той стадии, которая описана выше в связи с очисткой смеси, получаемой на стадии дегидрирования 5.
Получаемую таким образом фракцию С4 в отводимой по линии 15 смеси целесообразно подают на стадию избирательного гидрирования 16, на которой диены, как пропадиен и 1,3-бутадиен, подвергаются избирательному гидрированию до моноолефинов. Диены образуются, например, из пропана, введенного в процесс с природными бутанами, из н-бутана, на стадии разделения 1, не полностью отделившегося от изобутана, или же они образуются в условиях дегидрирования путем изомеризации и/или крекинговых реакций. По меньшей мере при рециркуляции по линии 17 остаточных газов данные диены мешают реакции на стадии дегидрирования 14 и, хотя в меньшей степени, на стадии этерификации 18. Поэтому стадия избирательного гидрирования 16 может быть размещенной после стадии этерификации 18 в потоке отводимого по линии 17 остаточного газа перед стадией очистки 19 или после нее. Такая установка позволяет в случае необходимости уменьшить размеры реактора благодаря тому, что, естественно, объем потока остаточного газа меньше, чем объем смеси, отводимой по линии 15. Относительно условий процесса указывается на объяснения в связи со стадией избирательного гидрирования 7.
Получаемую на стадии дегидрирования 14 смесь, в случае необходимости после предварительного избирательного гидрирования, подают на стадию этерификации 18, где содержащийся в ней изобутен в общем известным методом подвергается взаимодействию с алканолом, подаваемым по линии 20, до АТБЭ, в частности до метил-трет-бутилового эфира. Кроме метанола в качестве алканола предпочитают еще этанол, изопропанол, изобутанол. Взаимодействие осуществляют в жидкой или в жидкогазовой фазе при 50 - 90oС и при давлении, устанавливающемся при соответствующей температуре. Целесообразно работают с небольшим избытком алканола, что приводит к повышению избирательности реакции изобутена и тормозит его димеризацию. В качестве катализатора используют, например, кислый бентонит или, предпочтительно, кислый ионит с большой величиной пор.
Из реакционной смеси стадии этерификации 18 путем перегонки выделяют остаточный газ, отводимый по линии 17, и возможно имеющийся избыток алканола отделяют от образовавшегося АТБЭ и отводят по линии 21. В случае метил-трет-бутилового эфира остаточный газ с метанолом образует азеотроп. Последний промывают водой и разделяют на водную фазу и остаточный газ, рециркулируемый на стадию дегидрирования 14, в случае необходимости через стадию избирательного гидрирования 16 (которая тогда соответствующим образом включена в установку) и/или стадию очистки 19, причем на последней целесообразно осуществляют обработку с использованием молекулярного сита, при которой удаляются, в частности, кислород- или серосодержащие примеси, мешающие катализатору дегидрирования. По меньшей мере часть остаточного газа можно также рециркулировать на стадию разделения 1 (не показано на схеме) с тем, чтобы избежать накопления н-бутана вследствие нечеткого разделения н-бутена и изобутана на стадии разделения 1. Из водной фазы, получаемой при промывке водой, выделяют метанол, который рециркулируют на стадию этерификации, и воду, которую вновь используют для промывки.
В) Регулирование количеств ди-н-бутена и АТБЭ
Целесообразно после стадии разделения 1 осуществляют стадию изомеризации 22, потому что это позволяет изменять количественное соотношение ди-н-бутена и АТБЭ. Возможности изменения ограничены лишь мощностью установок для получения ди-н-бутена и АТБЭ. С учетом необходимых капиталовложений редко обе установки будут строить с размером, позволяющим обработку всего потока полевого бутана лишь в одной из установок, причем другая установка не работает. Все же стадия изомеризации 22 дает возможность гибко реагировать на требования рынка.
В том случае, если природные бутаны 1 содержат ненасыщенные соединения, является целесообразным кроме стадии изомеризации 22 предусматривать еще стадию гидрирования 23, на которой данные ненасыщенные соединения подвергаются гидрированию, потому что они мешают изомеризации. Гидрирование осуществляют известным методом. Целесообразно работают в жидкой фазе и, в зависимости от катализатора, при комнатной температуре или же при повышенной температуре до 90oС и при давлении 4-20 бар, причем парциальное давление водорода составляет 1-15 бар. Используют обычные для гидрирования олефинов катализаторы, например 0,3% палладия на окиси алюминия.
Гидрированные природные бутаны подают на стадию разделения 1, где их разделяют вышеописанным методом на н-бутан, отводимый по линии 3, и изобутан, отводимый по линии 4. Если соотношение н-бутана и изобутана желают изменить в соответствии с требованиями обеих установок, то часть имеющегося в избытке изомера отводят и подают на стадию изомеризации 22. На схеме альтернативные возможности показаны штрихпунктирными линиями. На стадии изомеризации 22 отдельный изомер переводят в другой изомер максимум до равновесия, которое в зависимости от температуры составляет 40-55% н-бутана и 45-60 % изобутана. Изомеризация н-бутана и изобутана представляет собой известную реакцию. В общем работают в газовой фазе при 150-230oС и 14-30 бар, причем в качестве катализатора используют платину на окиси алюминия в качестве носителя, избирательность которого можно еще повысить путем добавления соединения хлора, например четыреххлористого углерода. Для предотвращения дегидрирования предпочтительно добавляют небольшое количество водорода. Избирательность изомеризации высока, крекинг до меньших кусков происходит лишь в незначительной мере (примерно 2%).
Получаемую на стадии изомеризации смесь 3 необходимо разделять на изомеры, для чего ее подают по линии 24 на и без того имеющуюся стадию разделения 1.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ БУТЕНОЛИГОМЕРОВ ИЗ ОЛЕФИНОВ ПО СИНТЕЗУ ФИШЕРА-ТРОПША | 1997 |
|
RU2189372C2 |
| СПОСОБ ПОЛУЧЕНИЯ БУТЕНОВЫХ ОЛИГОМЕРОВ ИЗ ПРИРОДНЫХ БУТАНОВ | 1997 |
|
RU2189373C2 |
| ТИОЭТЕРИФИКАЦИЯ МЕРКАПТАНОВ В СМЕСЯХ УГЛЕВОДОРОДОВ С | 2013 |
|
RU2628085C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВЫСШИХ ОКСО-СПИРТОВ | 1997 |
|
RU2183210C2 |
| СПОСОБ ПОЛУЧЕНИЯ 1-БУТЕНА И ПРОИЗВОДНОГО 1,3-БУТАДИЕНА | 2012 |
|
RU2585764C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛАРИЛСУЛЬФОНАТОВ | 2001 |
|
RU2312099C2 |
| СПОСОБ ПРЕДВАРИТЕЛЬНОЙ ОБРАБОТКИ В УСТАНОВКЕ МЕТАТЕЗИСА С ОБРАЗОВАНИЕМ ОКТЕНА | 2008 |
|
RU2460713C1 |
| АДСОРБЦИЯ СЕРНИСТЫХ СОЕДИНЕНИЙ ИЗ ОЛЕФИНОВЫХ СМЕСЕЙ ПРИ ПОМОЩИ ВОДОРОДА | 2016 |
|
RU2645159C2 |
| СПОСОБ ПЕРЕРАБОТКИ УГЛЕВОДОРОДНОЙ СМЕСИ | 2003 |
|
RU2252931C2 |
| СПОСОБ ЭТЕРИФИКАЦИИ-ГИДРИРОВАНИЯ | 1996 |
|
RU2165405C2 |
Использование: нефтехимия. Сущность: природные бутаны разделяют на н-бутан и изобутан, н-бутан подвергают дегидрированию с последующей олигомеризацией полученного н-бутена до смеси олигомеров и выделением из последней ди-н-бутена. Изобутан подвергают дегидрированию до смеси, содержащей изобутен, который на стадии этерификации подвергают взаимодействию с алканолом до алкил-трет-бутилового эфира. Технический результат - повышение экономичности процесса. 6 з. п. ф-лы, 1 ил.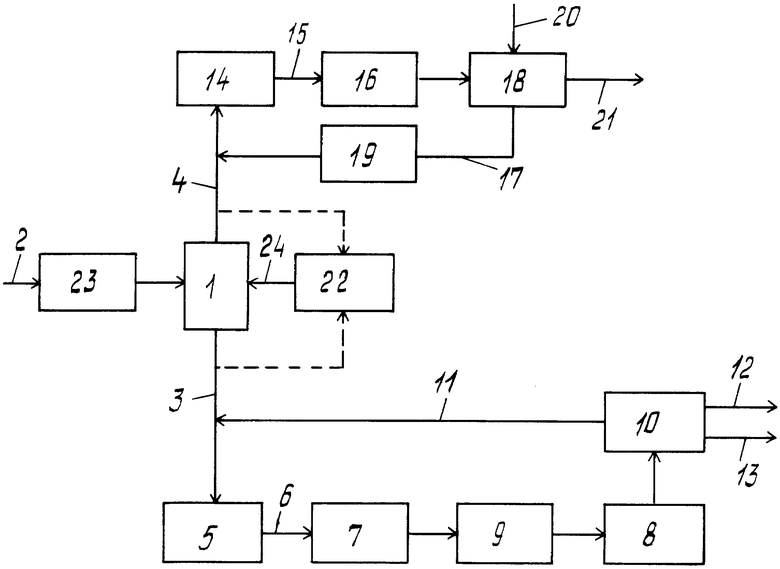
| СПОСОБ ПРОИЗВОДСТВА КУРИТЕЛЬНОЙ КОМПОЗИЦИИ ДЛЯ КАЛЬЯНА | 2015 |
|
RU2594139C1 |
| СПОСОБ ПРОИЗВОДСТВА КУРИТЕЛЬНОЙ КОМПОЗИЦИИ ДЛЯ КАЛЬЯНА | 2015 |
|
RU2594138C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЖЕЛЕЙНОГО МАРМЕЛАДА С ИСПОЛЬЗОВАНИЕМ КОНЦЕНТРИРОВАННОЙ ПАСТЫ ИЗ ТЫКВЫ | 2015 |
|
RU2603895C1 |
| US 5177282 А, 05.01.1993 | |||
| Способ получения олигомеров этилена | 1985 |
|
SU1351912A1 |

