Изобретение относится к новым сложным кетоэфирам и кетоамидам, которые представляют собой ингибиторы ферментов, в частности цистеин-протеаз, таких как, калпаин (зависящее от кальция протеазы цистеина) и его изоферменты и катепсины, например, катепсины В и L.
Калпаины представляют собой внутриклеточные, протеолитические ферменты из группы так называемых цистеин-протеаз и имеются во многих клетках. Фермент калпаин активируется повышенной концентрацией кальция, причем различают между калпаином I или мк-калпаином, который активируется мк-молярными концентрациями ионов кальция, и калпаином II или м-калпаином, который активируется ммолярными концентрациями ионов кальция (см. публикацию П. Джонсон, Int. J. Biochem. 1990, 22(8), 811-22). В настоящее время имеются также и другие изоферменты калпаина (см. К. Зузуки и др., Biol. Chem. Hoppe-Seyler, 1995, 376(9), стр. 523-9.
Предполагают, что калпаины играют важную роль в различных физиологических процессах. К этому относятся расщепление регуляторных протеинов, таких как, например, протеин-киназа С, цитоскелетные протеины, такие как MAP 2 и спектрин, мышечные протеины, расщепление протеинов при ревматоидном артрите, нейропептидный метаболизм, протеины при активации тромбоцитов, протеины в митозе и другие калпаины, которые приведены в публикациях М.Л.Баррет и др. Life Sci. 1991, 48, стр. 1659-69 и К.К.Ванг и др., Trends in Pharmacol. Sci. 1994, стр. 15, 412-9.
В различных патофизиологических процессах наблюдались повышенные уровни калпаина, например, в случае ишемии сердца (например, инфаркта сердца) почки или центральной нервной системы (например, инсульта), воспалений, мышечных дистрофий, катаракта глаз, повреждения центральной нервной системы (например, травма) или болезни Альцгеймера (см. вышеупомянутый источник К.К. Ванг и др.). Поэтому предполагается связь этих болезней с повышенным внутриклеточным уровнем кальция. Вследствие этого зависящие от кальция процессы чрезмерно активируются и больше не поддаются физиологической регуляции. В соответствие с этим чрезмерная активность калпаинов может также вызвать патофизиологические процессы.
Поэтому имеются утверждения, что ингибиторы калпаиновых ферментов могут быть полезными для лечения этих болезней. Различные исследования это подтверждают. Так, например, Сеунг-Чиул Хонг и др., Stroke 1994, 25(3), стр. 663-9 и Р.Т.Бартус и др., Neurological Res. 1995, 17, стр. 249-58 показали нейрозащитное действие ингибиторов калпаинов при острых нейродегенеративных нарушениях, имеющихся, например, после инсульта. Также и после экспериментальных мозговых повреждений ингибиторы калпаина способствовали смягчению дефицитов функции памяти и нейродвигательные нарушения (см. К.Э.Саатман и др. Proc. Natl. Acad. Sci. США, 1996, 93, стр. 3428-3433). Авторы К.Л. Эдельштайн и др. Proc. Natl. Acad. Sci. CША, 1995, 92, стр. 7662-2, обнаружили защитное действие ингибиторов калпаина на поврежденные гипоксией почки. Иошида, Кен Иши и др., Jap. Circ. J. 1995, 59 (1), стр. 40-8 смогли показать благоприятные эффекты ингибиторов калпаина после сердечных повреждений, которые были вызваны ишемией или реперфузией. В связи с тем, что ингибиторы калпаина тормозят выделение β-АР4-протеина, было предложено их потенциальное применение при терапии болезни Альцгеймера (см, И.Хигаки и др. Neuron, 1995, 14, стр. 651-59). Выделение интерлейкина-1α также подавляется ингибиторами калпаина (см. Н. Вантанабе и др., Cytokine 1994, 6(6), стр. 597-601. Далее было найдено, что ингибиторы калпаина оказывают цитотоксическое действие на опухолевые клетки (см. Э. Шиба и др. 20th Meeting Int. Ass. Breast Cancer Res. , Сендай Jp, 1994, 25.-28-го сентября, Int. J. Oncol. 5 (Suppl.), 1994, стр. 381).
Другие возможные области применения ингибиторов калпаина приведены в публикации Trends in Pharmacol. Sci., 1994, 15, стр. 412-8 (автор: К.К.Ванг).
Ингибиторы калпаина уже были описаны в литературе. Однако в подавляющем большинстве это или необратимые или пептидные ингибиторы. Необратимые ингибиторы являются, как правило, алкилирующими средствами и имеют тот недостаток, что они реагируют в организме неселективно или нестабильны. Так, например, эти ингибиторы часто проявляют нежелательные побочные явления, такие как токсичность, и поэтому они ограничены в их применении или вообще неприменимы. К необратимым ингибиторам причисляются, например, эпоксиды Е 64 (см. Biochem. Biophys. Res. Commun. 1989, 158, стр. 432-5), α-галоген-кетоны (см. X.Англикер и др. J. Med. Chem. 1992, 35, стр. 216-20) и дисульфиды (см. Р.Матсуэда и др. Chem. Lett. 1990, стр. 191 -194).
Многие известные обратимые ингибиторы цистеин-протеаз, такие как калпаин, представляют собой пептидные альдегиды, в частности дипептидные и трипептидные альдегиды, как, например, Z-Val-Phe-H (MDL 28170) см. С.Меди, Trends in Biol. Sci. 1991, 16, стр. 150-3) и соединения, описанные в европейском патенте 520336. В физиологических условиях пептидные альдегиды имеют часто тот недостаток, что вследствие имеющейся реакционноспособности они нестабильны, могут быстро метаболизироваться, склонны к неспецифичным реакциям, которые могут быть причиной токсических эффектов (см. Й.А.Ферентц и Б. Кастро, Synthesis 1983, стр. 676-78). Применение пептидных альдегидов при лечении болезней является таким образом ограниченным или не имеющим смысла. Поэтому нет ничего удивительного в том, что только некоторые альдегиды применяются в качестве активного начала, а именно прежде всего тогда, когда альдегидная группа стабилизируется, например, посредством образования полуацеталя.
Прогрессом является открытие того, что определенные пептидные производные кетона представляют собой также ингибиторы цистеин-протеаз, в частности калпаина. Так, например, производные кетона известны в качестве ингибиторов при серин-протеазах, причем кетогруппа активируется электронопритягивающей группой, например, трифторметилом. При цистеин-протеазах производные кетона, у которых кетогруппа активирована трифторметильной группой или подобными группами, являются мало или совсем не эффективными (см. М.Р.Ангеластро и др. J. Med. Сhem. 1990, 33, стр. 11-13). Неожиданно до сих пор в качестве эффективных ингибиторов калпаина могли быть найдены только производные кетона, при которых, с одной стороны, находящиеся в α-положении уходящие группы вызывают необратимое ингибирование и, с другой стороны, производное карбоновой кислоты активирует кетогруппу (см. М.Р.Ангеластро и др. в вышеупомянутом источнике, а также заявки WO 92/11850; WO 92/12140; WO 94/00095 и WO 95/00535). Однако из этих кетоамидов и сложных кетоэфиров до сих пор только пептидные производные были описаны как эффективные (см. Жаожао Ли и др., J. Med. Chem. 1993, 36, стр. 3472-80; С.Л.Харбенсон и др., J. Chem. 1994, 37, стр. 2918-29 и М.Р.Ангеластро в вышеупомянутом источнике).
Задачей настоящего изобретения является разработка непептидных ингибиторов, которые производятся от более стабильных кетонов и которые не имеют общих проблем пептидов (метаболическая стабильность, плохой проход через оболочки клеток и т.д.).
Объектом настоящего изобретения являются производные пипередин-кетокарбоновой кислоты формулы (I)
где R1-CO-R4, -SO2-R4, -CONH-R4, COOR4, -C(=N)-R4, -C(=O)-NHR4 и -C(= S)-NHR4, где R4 означает разветвленный или неразветвленный C1-C6-алкил, причем цепь из двух или более атомов углерода может иметь одну двойную или тройную связь и может быть замещена одним или двумя кольцами, такими как фенильное, нафталиновое, хиноксалиновое, хинолиновое, изохинолиновое, пиримидиновое, тиофеновое, бензотиофеновое, бензофурановое, пиримидиновое, тиазольное, изотиазольное, триазольное, имидазольное, циклогексильное, циклопентильное, флуореновое, индольное, бензимидазольное, оксазольное, изооксазольное и фурановое кольцо, причем каждое из колец может иметь еще максимально два радикала R5, означающего разветвленный или неразветвленный С1-С4-алкил, -O-С1-С4-алкил, ОН, Cl, F, Br, J, СF3, NO2, NH2, CN СООН, СОО-С1-C4-алкил, -NНСО-С1-C4-алкил, NHCOPh, NHSO2-C1-C4-алкил, NHSO2Ph,
-SO2-С1-С4-алкил и -SO2Ph, где Ph означает фенил,
R2-разветвленный или неразветвленный C1-C6-алкил, который может иметь еще фенильное, пиридиновое или нафтильное кольцо, которое со своей стороны может быть замещено максимально двумя радикалами R5, имеющими вышеуказанное значение,
R3-группы -OR6 и -NHR6, где R6 означает водород, фенильное кольцо, которое может иметь один или два радикала R5, имеющих вышеуказанное значение, разветвленный или неразветвленный C1-C6-алкил, который может содержать одну двойную или тройную связь и может иметь кольцо, как, например, фенильное, нафталиновое, пиридиновое, пиримидиновое, пиперидиновое, пирролидиновое, морфолиновое, тиофеновое, хинолиновое и изохинолиновое кольцо, причем ароматические кольца могут еще иметь максимально два радикала -NR7R8, где R7 и R8 независимы друг от друга и означают водород или разветвленный или неразветвленный C1-C6-алкил, или максимально два радикала R5, имеющих вышеуказанное значение, и их таутомерные и изомерные формы, а также их возможные, физиологически переносимые соли
Предпочтительными являются производные пиперидин-кетокарбоновой кислоты общей формулы (I), в которой
R1 означает группы -C(=O)R4, -SO2R4,
R2 разветвленный или неразветвленный C1-C6-алкил, бензил и -СН2-пиридил,
R3 группы -OR6 и -NHR6, а
R4, R5 и R6 имеют вышеприведенные значения.
Особенно предпочтительными являются производные пиперидин-кетокарбоновой кислоты общей формулы (I), в которой
R1 означает группы -C(=O)R4, -SO2R4,
R2 - С1-С4-алкил и бензил,
R3 - группа -NHR6,
R4 - группа -CH=CH-R9, причем R9 может быть фенилом, нафталином или хинолином, и
R6 - водород, С1-С4-алкил, который может быть замещен фенилом, пиридином или морфолином.
Соединения формулы (I) могут применяться как рацематы или как чистые энантиомерные соединения или как диастереомеры. Если желательны чистые энантиомерные соединения, то их можно получать, например, за счет того, что с помощью подходящих оптически активных оснований или кислот проводят классическое расщепление рацемата соединений формулы (I) или их промежуточных продуктов.
Объектом изобретения также являются соединения формулы (I) в их мезомерной или таутомерной форме, например, такие, при которых кетогруппа в формуле (I) имеется в виде енольного таутомера.
Объектом настоящего изобретения являются также физиологически приемлемые соли соединений (I), которые можно получать посредством взаимодействия соединений (I) с подходящими кислотами или основаниями.
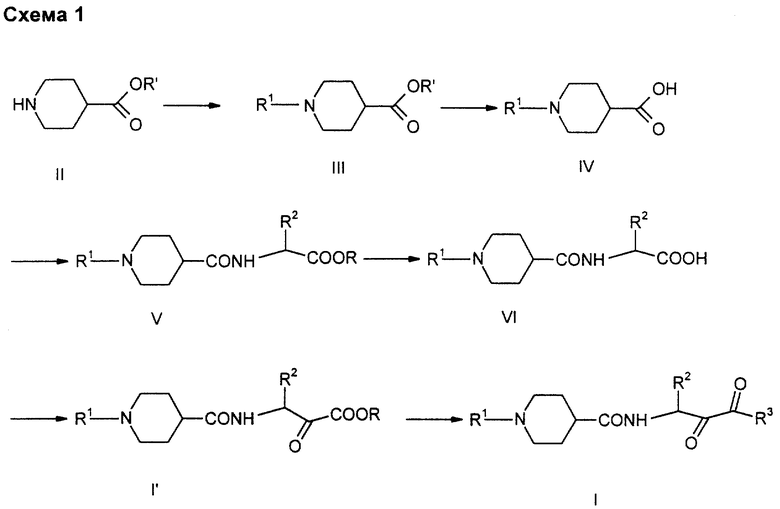
Получение производных пиперидин-карбоновой кислоты (I) может производиться различными методами, показанными на схемах 1 и 2.
Исходя из пиперидин-карбоновой кислоты II получают производное III взаимодействием при обычных условиях с "активированными" производными кислоты R1-L-, где L представляет собой уходящую группу, такую как хлор, имидазол и N-гидроксибензотриазол. Эта реакция происходит в безводном, инертном растворителе, таком как метиленхлорид, тетрагидрофуран и диметилформамид, при температуре от -20 до +25oС и как правило при обычных условиях, изложенных в Хоубен-Вейль, Methoden der organischen Chemie, 4-ое изд. том 5, раздел V.
Сложные эфиры пиперидин-карбоновой кислоты III преобразуются в кислоты IV взаимодействием с кислотами или основаниями, такими как гидроокись лития, гидроокись натрия или гидроокись калия в водной среде или в смесях из воды и органических растворителей, таких, как спирты или тетрагидрофуран, при комнатной или повышенной температуре, как, например, при 25-100oС.
Эти кислоты IV подвергают взаимодействию с производным α-аминокислоты, причем используют обычные условия, которые описаны выше и которые приведены в Хоубен-Вейль.
Производные V, которые представляют собой, как правило, сложные эфиры, превращаются аналогично вышеописанному гидролизу в кетокарбоновые кислоты VI. Аналогичной реакцией получают сложные кетоэфиры VII, причем работают по методу, описанному Жаожао Ли, J. Med. Chem., 1993, 36, стр. 3472-80. Согласно этому методу карбоновую кислоту, такую как по формуле (VI), при повышенной температуре (50-100oС) подвергают взаимодействию с хлорангидридом сложного моноэфира щавелевой кислоты в таком растворителе, как тетрагидрофуран, и после этого полученный таким образом продукт подвергают взаимодействию с такими основаниями, как метанолят натрия, в этаноле при температуре 25-80oC с получением сложного кетоэфира I' согласно изобретению. Сложные кетоэфиры I' могут быть, например, гидролизированы описанным выше образом в кетокарбоновую кислоту согласно изобретению.
Превращение в кетоамиды I' производится также аналогично методу по Жаожао Ли и др. в присутствии кислоты Льюиса, как, например, бортрифторидэтерата, в инертных растворителях, таких как метиленхлорид, при комнатной температуре, причем получается дитиан. Эти производные подвергают взаимодействию с аминами R3-Н в полярных растворителях, таких как спирты, при температуре 0-80oС. При этом получают кетоамиды I'.
Альтернативный метод представлен на схеме 2. Пиперидин-карбоновые кислоты IV подвергают взаимодействию с производными аминогидроксикарбоновой кислоты VII (см. С.Л.Харбенсон и др. J. Med. Chem. 1994, 37, стр. 2918-29) при обычных условиях образования пептидной связи (см. вышеупомянутую публикацию Хоубен-Вейль) с получением амидов VIII. Эти производные VIII могут окисляться до производных кетокарбоновой кислоты I согласно изобретению. Для этого можно использовать различные обычные реакции окисления по Сверну или аналогичное Сверну окисление (см. К.Р. Ларок, Comprenhensive Organic Transformations, изд. VCH Publisher, 1989, стр. 604 и сл.), как, например, окисление по Сверну или аналогичное Сверну окисление (см. Т.Т.Тидвелл, Synthesis 1990, стр. 857-70) или гипохлоритом натрия/ТЕМРО (С.Л.Харбенсон и др., см. выше).
Если соединения VIII представляют собой сложный α-гидроксиэфир (X=О-алкил), то они могут быть гидролизированы до карбоновой кислоты IX, причем работают аналогично вышеуказанным методам, однако предпочтительно с гидроксидом лития в смесях воды с тетрагидрофураном при комнатной температуре. Получение других сложных эфиров или амидов Х производится путем взаимодействия со спиртами или аминами при вышеописанных условиях. Производное спирта Х может снова подвергаться окислению с получением производных кетокарбоновой кислоты I' согласно изобретению.
Получаемые в рамках настоящего изобретения производные кетона I представляют собой ингибиторы цистеин-протеаз, в частности таких цистеин-протеаз, как калпаин I и II и катепсин В, соответственно L.
Ингибиторное действие производных кетона I определяли посредством известных в литературе испытаний ферментов, причем в качестве масштаба активности определяли концентрацию ингибитора, при которой ингибируется 50% активности фермента (=IС50). Производные кетона I измерялись таким образом на их ингибиторное действие в отношении калпаина I, калпаина II и катепсина В.
Испытание по определению ингибирования катепсина В
Ингибиторное действие на катепсин В определяли аналогично методу по С. Хаснайн и др., J. Biol. Chem. 1993, 268, стр. 235-40. К 88 мкл катепсина В (катепсин В из печени человека, продукт фирмы Калбиохем), разбавленного до 5 един. в 500 мкмоль буферного раствора, добавляли 2 мкл раствора ингибитора в диметилсульфоксиде (конечная концентрация: 100 мкмоль до 0,01 мкмоль). Эту смесь на 60 мин при комнатной температуре (25oС) предварительно инкубировали и потом инициировали реакцию посредством добавки 10 мкл, 10 ммоль Z-Arg-Arg-pNA (в буфере с 10% диметилсульфоксида). За ходом реакции наблюдали 30 мин при 405 нм с помощью считывающего устройства микротитровых пластинок. Из максимальных подъемов определяли значения IC50.
Испытание с применением калпаина I и II
Испытание ингибиторных свойств ингибитора калпаина производили в буфере с 50 ммоль трис-HCl, рН 7,5; 0,1 моль NaCl; 1 ммоль дитиотреитола; 0,11 ммоль CaCl; 1 ммоль дитиотреитола, 0,11 ммоль CaCl2, причем применяли флуорогенный субстрат калпаина Suc-Leu-Tyr-AMC (25 ммоль растворено в ДМСО, продукт фирмы Бахем, Швейцария) (см. Сасаки и др. J. Biol. Chem. 1984, том 259, стр. 12489-12494). Выделяли человеческий мк-калпаин из эритроцитов, следуя методам по Кроалл и ДеМартино (ВВА 1984, том 788, стр. 348-355) и Грейбилл и др. (Bioorg. & Med. Lett. 1995, том 5, стр. 387-392). После нескольких хроматографических приемов (содержащая диэтиламиноэтиловые группы сефароза, фенил-сефароза, супердекс 200 и Blue-сефароза) получали фермент с чистотой >95%, определенной электрофорезом на полиакриламидном геле с применением додецилсульфата натрия, вестерн-блоттингом и N-концевым секвенированием. Флуоресценцию продукта расщеплением, 7-амино-4-метилкумарина (АМС), определяли с помощью флуориметра Spex-Fluorolog при λэкс=460 нм. За 60-минутное измерение расщепление субстрата является линейным и автокаталитическая активность калпаина малой, если испытания проводились при температуре 12oС (см. геттерджи и др. 1996, Bioorg. & Med. Chem. Lett., том 6, стр. 1619-1622). Ингибиторы и субстрат калпаина подавали в испытательную среду в качестве растворов в диметилсульфоксиде (ДМСО), причем конечная концентрация ДМСО не должна превышать 2%.
Типичная испытательная смесь сводилась к тому, что в кювету емкостью 1 мл, содержащую буфер, подавали 10 мкл субстрата (конечная концентрация 250 мкм) и затем 10 мкм мк-калпаина (конечная концентрация: 2 мкг/мл, т.е. 18 нмоль). Вызванное калпаином расщепление субстрата измеряли в течение 15-20 мин. После этого производили подачу 10 мкл ингибитора (50-100 мкмоль растворено в (ДМСО) и измерение ингибирования расщепления определяли еще 40 мин. Значение Ki определяли по обычному уравнению для обратимого ингибирования, т. е. Ki: =I(V0V1)-1; причем I= концентрация ингибитора, V0= начальная скорость подачи ингибитора; V1= скорость реакции в равновесии.
Испытание с помощью тромбоцитов для определения клеточной активности ингибиторов калпаина.
Вызванное калпаином расщепление протеинов в тромбоцитах было проведено как описано Жаожао Ли и др. в J. Med. Chem., 1993, 36, стр. 3472-3480. Человеческие тромобциты выделяли из свежей донорской крови, смешанной с цитратом натрия, и устанавливали до 107 клеток/мл в буфере из 5 ммоль HEPES, 140 ммоль NaCl и 1 мг/мл альбумина сыворотки крупного рогатого скота, рН 7,3.
Тромбоциты (0,1 мл) предварительно инкубировали 5 минут посредством 1 мкл различных концентраций раствора ингибитора в ДМСО. После этого производили подачу ионофора кальция А23187 (1 мкмоль в тесте) и кальция (5 ммоль в тесте) и инкубировали еще 5 минут при температуре 37oС. После центрифугирования тромбоциты помещали в буфер, содержащий полиакриламидный гель, обработанный додецилсульфонатом натрия, кипятили 5 минут при 95oС и протеины разделяли на 8%-ном геле. Расщепление обоих протеинов, связывающего актин протеина и талин, прослеживали с помощью количественной денситометрии, так как после подачи кальция и ионофора эти протеины исчезли и в области молекулярной массы 200 КД образовалась новая полоса. Отсюда определяли полумаксимальную активность фермента.
Индуцированная глутаматом гибель клеток кортикальных нейронов
Тест проводили аналогично тесту, описанному Д.В.Чой, М.А.Маулучи-Гедде и А. Р.Кригштайн (1987) "Glutamate neurotoxicity in cortical cell culture", J. Neurosci. 7, стр. 357-368.
Из 15-дневних эмбрионов мышей препарировали половины коры головного мозга и ферментативно (трипсин) получали отдельные клетки. Эти клетки (глия и кортикальные нейроны) высеивали в микротитровые пластинки с 24 углублениями. Через три дня (при покрытых ламинином пластинках) или через семь дней (при покрытых орнитином пластинках) с помощью 5-фтор-2-дезоксиуридина проводили митотическое деление клеток (митотическую обработку). 15 дней после препарирования клеток посредством добавки глутамата (15 мин) вызывали гибель клеток. После удаления глутамата подавали ингибиторы калпаина. Через 24 часа определяли повреждение клеток посредством выявления лактатдегидрогеназы в насадочной жидкости культур клеток.
Имеется утверждение, что калпаин играет роль также и при апоптотической гибели клеток (см. публикацию М.К.Т.Сквие и др. J.Cell. Physiol. 1994, 159, стр. 229-237; Т. Пател и др., Faseb Journal 1996, 590, 587-597). Поэтому в другом тесте вызывали гибель клеток в человеческой клеточной линии кальцием в присутствии кальциевого ионофора. Ингибиторы калпаина должны попасть в клетки и там ингибировать активность калпаина, чтобы предотвратить вызванную гибель клеток.
В человеческой клеточной линии NT2 можно вызвать гибель клеток посредством кальция в присутствии ионофора А23187. 105 клеток/на углубление подавали в микротитровые пластинки за 20 часов до испытания. Затем клетки инкубировали с различной концентрацией ингибиторов в присутствии 2,5 мкмоль ионофора и 5 ммоль кальция. К реакционной смеси прибавляли через 5 часов 0,05 мл ХТТ (Cell Proliferation Kit II, фирмы Берингер, Маннгейм, DE). Оптическую плотность определяли приблизительно через 17 часов в соответствии с указаниями изготовителя с помощью считывающего устройства Easy Reader EAR 400 фирмы SLT. Оптическая плотность, при которой погибла половина клеток, высчитывается из обоих контрольных значений с клетками без ингибиторов, которые инкубировали в отсутствии и в присутствии ионофора.
Производные кетона I представляют собой ингибиторы цистеин-протеаз, таких как калпаин I и II, а также катепсин В и L, и могут применяться при борьбе с заболеваниями, связанными с повышенной активностью ферментов калпаина и/или катепсина. Вследствие этого данные производные кетона I могут применяться для лечения нейродегенеративных заболеваний, которые возникают после ишемии, травм, массивных кровоизоляций и нейродегенеративных заболеваний, таких как деменция в результате многократного инфаркта, болезнь Альцгеймера, болезнь Хантингтона (хорея) и, кроме того, для лечения повреждений сердца после сердечной ишемии, повреждений почек после почечной ишемии, повреждений скелетных мышц, мышечной дистрофии, повреждений, вызванных пролиферацией гладких мышечных клеток, коронарных спазм сосудов, церебральных спазм сосудов, катаракты глаз и/или рестеноза кровеносного русла после ангиопластии.
К тому же производные кетона I могут быть полезными при хемотерапии опухолей и их метастазов и для лечения заболеваний, при которых имеется высокий уровень интерлейкина-1, например, при воспалениях и/или ревматических заболеваниях.
Лекарственные формы согласно изобретению содержат наряду с обычными вспомогательными веществам терапевтически эффективное количество соединений I.
Для локального наружного применения, например, как пудра, мазь или спрей, активное начало может содержаться в формах в обычной концентрации. Как правило, активное начало содержится в количестве от 0,0001 до 1 вес.%, предпочтительно, от 0,001 до 0,1 вес.%.
При внутреннем применении лекарственные формы дают в отдельных дозах. В отдельной дозе дают на кг веса 0,1-100 мг. Лекарственные формы могут даваться ежедневно в одну или несколько дозировок в зависимости от вида и серьезности заболевания.
В соответствии с желаемым видом применения лекарственные формы согласно изобретению содержат наряду с активным началом обычные в галенике наполнители и растворители. Для локального наружного применения могут использоваться фармацеватически-технические вспомогательные вещества, такие, как этанол, изопропанол, оксиэтилированное касторовое масло, оксиэтилированное гидрированное касторовое масло, полиакриловая кислота, полиэтиленгликоль, полиэтиленгликостеарат, этоксилированные спирты жирного ряда, парафиновое масло, вазелин и ланолин. Для внутреннего применения пригодны, например, молочный сахар, пропиленгликоль, этанол, крахмал, тальк и поливинилпирролидон.
Кроме того, лекарственные формы могут содержать антиокислители, такие как токоферол и бутилированный оксианизол и бутилированный окситолуол, а также улучшающие вкус вещества, стабилизаторы, эмульгаторы и смазочные вещества.
Содержащиеся наряду с активным началом вещества, а также вещества, применяемые при изготовлении фармацевтических лекарственных форм, должны быть токсикологически приемлемыми и совместными с соответствующим активным началом. Изготовление лекарственных форм происходит обычным образом, например, посредством смешения активного начала с другими обычными носителями и разбавителями.
Получение лекарственных форм осуществляют посредством известных специалистам способов (см. например, X.Сукер и др. Pharmazeutische Technologie, изд. Thieme Verlag, Штутгарт, 1991).
Лекарственные препараты могут даваться различным образом, например, перорально, парентерально, внутривенно посредством вливаний, подкожно, внутрибрюшинно и локально. Возможны такие лекарственные формы, как таблетки, эмульсии, растворы для вливания и инъекций, пасты, мази, гели, кремы, лосьоны, пудры и спрей.
Примеры
Пример 1
Сложный этиловый эфир 4-метил-2-оксо-3-(1-(Е-3-фенил-1-акрилоил)пиперидин-4-ил)амидовалерьяновой кислоты
а) 1-(Е-фенил-1-акрилоил)пиперидинил-4-карбоновая кислота
32,0 г (0,248 моль) пиперидин-4-карбоновой кислоты растворяют в 500 мл пиримидина и после этого порциями смешивают с 43,3 г (0,26 моль) хлорангидрида коричной кислоты. Получаемую смесь перемешивают в течение 16 часов при комнатной температуре. Потом реакционную смесь концентрируют под вакуумом и остаток распределяют между 2 М соляной кислотой и этиловым эфиром уксусной кислоты. Органическую фазу отделяют, сгущают и концентрируют под вакуумом. Получают 47,0 г (76%) продукта. Т.пл.: 178-179oС.
б) Сложный метиловый эфир 4-метил-2(1-(Е-3-фенил-1-акрилоил)-пиперидин-4-ил)амидомасляной кислоты
20,0 г (77,1 ммоль) продукта стадии а) и 12,5 г (77,1 ммоль) гидрохлорида сложного метилового эфира L-валина подают в 350 мл метиленхлорида и при охлаждении льдом смешивают по каплям с 25,6 мл (185,1 ммоль) триэтиламина. Смесь перемешивают 1 час и потом прибавляют 3,1 г (23,1 ммоль) 1-гидрокси-1Н-бензотриазола. Реакционную смесь охлаждают до 0oС и после этого порциями смешивают с 14,8 г (77,1 ммоль) N'-(3-диметиламинопропил)-N-этилкарбодиимида. Полученную смесь перемешивают в течение 16 часов при комнатной температуре. После этого органическую фазу промывают водой, водным раствором гидрокарбоната натрия, 5%-ным раствором лимонной кислоты и снова водой, сгущают и концентрируют под вакуумом. Получают 27,3 г (96%) продукта.
1H-ЯМР (CDCl3): δ= 0,9(6Н), 1,6-2,0(3Н), 2,2(1Н), 2,5(1Н), 2,8(1Н), 3,2(1Н), 3,8(3Н), 4,2(1Н), 4,6(1Н), 4,7(1Н), 6,0(1Н), 6,9(1Н), 7,3-7,6(5Н) и 7,6(1Н) млн.дол.
в) 4-метил-3-1-(Е-3-фенил-1-акрилоил) пиперидин-4-ил)амидомасляная кислота
27,0 г (72,5 ммоль) продукта стадии б) растворяют в 200 мл тетрагидрофурана и смешивают с 3,5 г (145 моль) гидроокиси лития, растворенной в 250 мл воды. Получаемую смесь перемешивают в течение 1 часа при комнатной температуре. После этого удаляют тетрагидрофуран под вакуумом и полученный водный раствор экстрагируют этилацетатом. Потом его нейтрализуют 1 М соляной кислотой и снова экстрагируют. Органическую фазу сушат и концентрируют под вакуумом, при этом получают 26 г (100%) продукта.
1H-ЯМР (CDCl3): δ= 1,0(6Н), 1,6-2,2(1 Н), 2,9(1Н), 3,2(1Н), 4,6(2Н), 6,4(2Н), 6,4(1Н), 6,9(1Н), 7,3-7,6(5Н) и 7,7(1Н) млн.дол.
г) Сложный этиловый эфир 4-метил-2-оксо-3-(1-(Е-3-фенил-1-акрилоил)пиперидин-4-ил)-амидовалерьяновой кислоты
26,0 г (72,5 ммоль) продукта стадии в) 0,9 г (7,25 ммоль) 4-диметиламинопиридина и 23,4 мл (0,29 моль) пиридина растворяют в 150 мл безводного тетрагидрофурана. После этого по каплям добавляют 16,2 мл (0,15 моль) хлорангидрида моноэтилового эфира щавелевой кислоты с таким расчетом, что температура поднимается до приблизительно 50oС. На протяжении следующих трех часов кипятят с обратным холодильником. Реакционную смесь перемешивают в течение 16 часов при комнатной температуре. Потом прибавляют осторожно 100 мл воды и перемешивают снова приблизительно 30 минут. Смесь распределяют между водой и этилацетатом. Органическую фазу промывают несколько раз водой, сушат и концентрируют под вакуумом.
Полученный таким образом сложный енольный эфир растворяют в 200 мл этанола, смешивают с 0,47 г (5,6 ммоль) этанолята калия и перемешивают 16 часов при комнатной температуре. После этого смесь концентрируют под вакуумом и остаток подвергают хроматографической очистке (растворитель: метиленхлорид/метанол =20/1), при этом получают 10,8 г (36%) продукта.
МС (бомбардировка быстрыми атомами): m/е=414 (М+).
Пример 2
Амид 4-метил-2-оксо-3-(1-(Е-3-фенил-1-акрилоил)пиперидин-4-ил)амидовалерьяновой кислоты
а) Сложный этиловый эфир 2,2-этилендимеркапто-4-метил-Е-3-(1-(3-фенил)-1-акрилоил)-пиперидин-4-ил)амидовалерьяновой кислоты
6,0 г (14,6 ммоль) продукта примера 1 г) и 1,5 мл (17,5 ммоль) 1,2-этандитиола растворяют в 20 мл безводного метиленхлорида и смешивают с 4 мл ботрифторидэтерата. Смесь перемешивают в течение 16 часов при комнатной температуре. После этого разбавляют 10 мл метиленхлорида и 3 раза промывают насыщенным раствором хлорида натрия. Органическую фазу сушат и концентрируют в вакууме, при этом получают 7,3 г сырого продукта, который без очистки подают на стадию б).
б) Амид 4-метил-2-оксо-3-(Е-1-(3-фенил)-1-акрилоил)пиперидин-4-ил)-амидовалерьяновой кислоты
1,7 г (3,6 ммоль) продукта стадии а) подают в 20 мл 2 М этанольного раствора аммиака и перемешивают в течение 16 часов при комнатной температуре. После этого смесь концентрируют под вакуумом и подвергают хроматографической очистке (растворитель: метиленхлорид/метанол =40/3), при этом получают 0,22 г продукта.
МС (бомбардировка быстрыми атомами): m/е=385 (М+).
Пример 3
Амид N-этил-4-метил-2-оксо-3-(1-(Е-3-фенил-1-акрилоил)пиперидин-4-ил) амидовалерьяновой кислоты
1,7 г (3,6 ммоль) продукта примера 2а) подвергают взаимодействию с этанольным раствором этиламина в условиях примера 2б. При этом получают 0,15 г продукта.
МС (бомбардировка быстрыми атомами): m/е=413 (М+).
Пример 4
Амид 4-метил-N-(3-(морфолино-1-ил)-пропил)-2-оксо-3-(1-(Е-3-фенил-1-акрилоил)пиперидин-4-ил)-амидовалерьяновой кислоты
1,3 г (3,6 ммоль) продукта примера 2а) подвергают взаимодействию с 0,8 г (5,4 ммоль) 3-(морфолино-1-ил)-пропиламина в условиях примера 2б). Получают 1,1 г продукта.
МС (бомбардировка быстрыми атомами): m/е=512 (М+).
Пример 5
Амид 4-метил-2-оксо-3-(1-(Е-3-фенил-1-акрилоил)пиперидин-3-ил)-амидо-N-2-(пиримид-2-ил)этилвалерьяновой кислоты.

1,3 г (2,8 ммоль) продукта примера 2а) подвергают взаимодействию с 0,7 г (5,5 ммоль) 2-(2-аминоэтил)пиридина аналогично примеру 2б). При этом получают 0,85 г продукта.
МС (бомбардировка быстрыми атомами): m/е=490 (М+).
Пример 6
Сложный этиловый эфир 2-оксо-4-фенил-3-(1-(Е-3-фенил-1-акрилоил)-пиперидин-4-ил)-амидомасляной кислоты
а) Сложный метиловый эфир 3-фенил-3-(1-(Е-3-фенил-1-акрилоил)-пиперидин-4-ил)-амидопропионовой кислоты
Продукт получают аналогично примеру 1б) из промежуточного продукта примера 1а) и сложного метилового эфира фенилаланина.
1H-ЯМР (CDCl3): δ=1,6-2,0(3Н), 2,35(1Н), 2,9(1Н), 3,0-3,3(4Н), 3,7(3Н), 4,1(1H), 4,6(1Н), 4,9(1H), 6,9(1Н), 7,1(2Н) и 7,2-7,7(9Н) млн.дол.
б) 3-фенил-3-(1-(Е-3-фенил-1-акрилоил)-пиперидин-4-ил)-амидопропионовая кислота
Продукт получают аналогично примеру 1в) из промежуточного продукта примера 6а).
1H-ЯМР (CDCl3): δ=1,4-2,0(4Н), 2,3(1H), 2,8(1Н), 3,0-3,4(3Н), 4,0(1Н), 4,6(1Н), 4,9(1H), 4,9(1H), 6,2(1Н), 6,8(1Н), 7,0-7,8(11Н) и ок. 8,2(шир.) млн.дол.
в) Сложный этиловый эфир 2-оксо-4-фенил-3-(1-(Е-3-фенил-1-акрилоил)-пиперидин-4-ил)-амидомасляной кислоты
Продукт получают аналогично примеру 1г) из промежуточного продукта примера 6б).
МС (бомбардировка быстрыми атомами): m/е=462 (M+).
Пример 7
Амид N-(3-морфолин-1-ил)пропил)-2-оксо-4-фенил-3-(1-(Е-3-фенил-1-акрилоил)-пиперидин-4-ил)-амидомасляной кислоты
Продукт получают аналогично примеру 2б) из продукта примера 6 и 1-(3-аминопропил)-морфолина.
1H-ЯМР (CDCl3): δ= 1,4-1,9(6Н), 2,3-2,6(6Н), 2,8(1Н), 3,3(2Н), 3,3-3,5(3Н), 3,6-3,8(4Н), 4,1(2Н), 4,6(1Н), 5,5(1Н), 6,1(1Н), 6,9(1Н), 7,1(1Н), 7,2-7,7(10Н) и 8,9(1Н) млн.дол.
Пример 8
Амид 2-оксо-4-фенил-3-(1-(Е-3-фенил-1-акрилоил)-пиперидин-4-ил)-амидомасляной кислоты.

Продукт получают аналогично примеру 2б) из продукта примера 6 и этанольного раствора аммиака.
1H-ЯМР (D6-ДМСО): δ= 1,2-1,9(4Н), 2,4(1Н), 2,7-2,9(2Н), 3,0-3,2(2Н), 4,1-4,3(3Н), 5,1(1Н) и 7,0-8,2(14Н) млн.дол.
Пример 9
Амид 4-метил-N-(2-(морфолин-1-ил)этил)-2-оксо-3-(1-(Е-3-фенил-1-акрилоил)-пиперидин-4-ил)амидовалерьяновой кислоты
Продукт получают аналогично примеру 2б) из промежуточного продукта примера 2а) и 1-(2-аминоэтил)-морфолина.
МС: m/е=498 (M+).
Пример 10
Сложный этиловый эфир 2-оксо-3-(1-(Е-3-фенил-1-акрилоил)-пиперидин-4-ил)амидовалерьяновой кислоты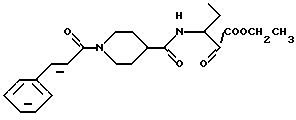
а) Сложный этиловый эфир 3-(1-(Е-3-фенил-1-акрилоил)-пиперидин-4-ил)-амидомасляной кислоты
Продукт получают аналогично примеру 1б) из промежуточного продукта примера 1а) и сложного этилового эфира 2-аминомасляной кислоты.
1H-ЯМР (CDCl3): δ= 0,9(3Н), 1,6-2,0(6Н), 2,5(1Н), 2,9(1Н), 3,2(1Н), 3,8(3Н), 4,2(1Н), 4,5-4,7(2H), 6,3(1H), 6,9(1H), 7,4(3Н), 7,6(2Н) и 7,7(1Н) млн.дол.
б) 3-(1-(Е-3-фенил-1-акрилоил)-пиперидин-4-ил)-амидомасляная кислота
Продукт получают аналогично примеру 1в) из промежуточного продукта примера 10а).
1H-ЯМР (D6-ДМСО): δ= 0,9(3Н), 1,3-1,9(6Н), 2,6(1Н), 2,7(1Н), 3,1 (1Н), 4,1(1Н), 4,3(1Н), 4,5(1Н), 7,2-7,6(5Н), 7,7(2Н), 8,0(1Н) и 12,5 (шир.) млн. дол.
в) Сложный этиловый эфир 2-оксо-3-(1-(Е-3-фенил-1-акрилоил)-пиперидин-4-ил)-амидовалерьяновой кислоты
Продукт получают аналогично примеру 1г) из промежуточного продукта примера 10б).
1H-ЯМР (CDCl3): δ= 0,9(3Н), 1,4(3Н), 1,8-2,2(6Н), 2,5(1Н), 2,8(1Н), 3,2(1Н), 4,2(1Н), 4,4(2Н), 4,6(1Н), 5,1(1Н), 6,7(1Н), 6,9(1Н), 7,4(3Н), 7,5(2Н) и 7,7(1Н) млн.дол.
Пример 11
Амид 2-оксо-3-(1-(Е-3-фенил-1-акрилоил)-пиперидин-4-ил)-амидовалерьяновой кислоты
Продукт получают аналогично примеру 2а) и 2б) из продукта примера 10 и этанольного раствора аммиака.
MC: m/e=371 (M+).
Пример 12
Сложный этиловый эфир 3-(1-(2-нафтилсульфонил)-пиперидин-4-ил)-амидо-2-оксо-4-фенилмасляной кислоты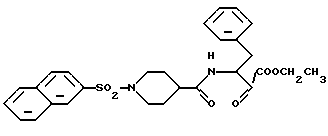
а) 1-(2-нафтилсульфонил)-пиперидин-4-карбоновая кислота
26,0 г (0,2 моль) пиперидин-4-карбоновой кислоты растворяют в 250 мл пиридина и при комнатной температуре порциями смешивают с 47,6 г (0,2 моль) хлорангидрида 2-нафтилсульфоновой кислоты. Получаемую смесь перемешивают около 5 часов при комнатной температуре. Реакционную смесь концентрируют в вакууме и остаток распределяют между этилацетатом и 2 М соляной кислотой. Органическую фазу сушат и концентрируют в вакууме. Получают 48,5 г (75%) продукта.
б) Сложный этиловый эфир 3-(1-(2-нафтилсульфонил)-пиперидин-4-ил)-амидо-2-оксо-4-фенилпропионовой кислоты
Продукт получают аналогично примеру 1б) из промежуточного продукта примера 12а).
1H-ЯМР (D6-ДМСО): δ= 1,1(3Н), 1,4-1,8(5Н), 2,3-2,6(2Н), 2,7-3,2(Н), 3,5-3,8(2Н), 4,0(2Н), 4,5(1Н), 7,2(4Н), 7,7(3Н), 8,1-8,3(3Н) и 8,5(1Н) млн. дол.
в) 3-(1-(2-нафтилсульфонил)-пиперидин-4-ил)-амидо-2-оксо-4-фенилпропионовая кислота
Продукт получают аналогично примеру 1в) из промежуточного продукта примера 12б).
1Н-ЯМР (D6-ДМСО): δ=1,3-1,8(5H), 2,3-2,6(3H), 2,8-3,2(2H), 3,4-3,8(2H), 4,4(1Н), 7,2(4Н), 7,7(3Н), 8,0-8,3(4Н) и 8,4(1Н) млн.дол.
г) Сложный этиловый эфир 3-(1-(2-нафтилсульфонил)-пиперидин-4-ил)-амидо-2-оксо-4-фенилмасляной кислоты
Продукт получают аналогично примеру 1г из промежуточного продукта примера 12в).
1H-ЯМР (D6-ДМСО): δ=1,2(3Н), 1,3-1,9(4Н), 2,2(1Н), 2,3-2,5(2Н), 2,8(1Н), 3,1(1Н), 3,6(2Н), 4,2(2Н), 4,4(1Н), 7,0-7,3(5Н), 7,7(3Н), 8,0-8,3(3Н) и 8,4(2Н) млн.дол.
Пример 13
Амид 3-(1-(2-нафтилсульфонил)-пиперидин-4-ил)-амино-2-оксо-4-фенилмасляной кислоты
Продукт получают аналогично примеру 2а) и 2б) из продукта примера 12.
МС (бомбардировка быстрыми атомами): m/е=493 (М+).
Пример 14
Амид N-(3-(морфолин-1-ил)прол-1-ил)-3-(1-(2-нафтилсульфонил)-пиперидин-4-ил)-амидо-2-оксо-4-фенилмасляной кислоты
Продукт получают аналогично примеру 2а) и 2б) из продукта примера 12 и 1-(3-амино-прол-1-ил)-морфолина.
МС (бомбардировка быстрыми атомами): m/е=620 (M+).
Пример 15
Амид 3-(1-(2-нафтилсульфонил)-пиперидин-4-ил)-амидо-2-оксо-4-фенил-N-(2-(2-пиридил)-этил)масляной кислоты
Продукт получают аналогично примеру 2а) и 2б) из продукта примера 12 и 2-(2-аминоэтил)-пиридина.
МС (бомбардировка быстрыми атомами): m/е=598 (М+).
Пример 16
Амид 3-(S)-(1-(2-нафтоил)-пиперидин-4-ил)-амидо-2-оксо-4-фенилмасляной кислоты
а) Амид 3-(S)-(N-трет. -бутоксикарбонил-амино)-2-(R, S)-гидрокси-4-фенилмасляной кислоты
17,7 г (60 ммоль) 3-(S)-N-трет.-бутоксикарбонил-амино)-2-(R, S)-гидрокси-4-фенилмасляной кислоты (см. публикацию С.Л.Харенсон., J. Med. Chem. 1994 и др. 37, стр. 2918-29) и 8,1 г (60 ммоль) 1-гидроксибензотриазола растворяют в 150 мл безводного диметилформамида. При -5oС последовательно добавляют 12,6 г (66 ммоль) гидрохлорида (N'-(3-диметиламинопропил)-N-этилкарбодиимида и 48 мл (приблизительно 2 молярного) этанольного раствора аммиака и перемешивают около часа при этой температуре. После этого смесь перемешивают еще 16 часов при комнатной температуре. Потом добавляют 500 мл воды и экстрагируют этилацетатом. Органическую фазу промывают разбавленным раствором едкого натрия и водой, сушат и концентрируют в вакууме. Остаток обрабатывают еще н-гептаном и осадок отсасывают. Получают 13,5 г (76%) продукта.
1H-ЯМР (D6-ДМСО): δ= 1,3(9H), 2,6-2,9(2H), 3,7(1H), 5,7(1H), 6,2(1H) и 7,3(5H) млн.дол.
б) Амид 3-(S)-амино-2-(R, S)-гидрокси-4-фенилмасляной кислоты
13,4 г (46 ммоль) продукта стадии а) растворяют в 300 мл метиленхлорида и смешивают с 100 мл трифторуксусной кислоты. Получаемую смесь перемешивают 1 час при комнатной температуре и потом концентрируют в вакууме. Остаток распределяют между водой и простым диэтиловым эфиром и после этого водную фазу концентрируют в вакууме. Получают 12,3 г (88%) продукта в виде трифторацетата.
в) Амид 2-(R, S)-гидрокси-3-(S)-(1-(2-нафтоил)-пиперидин-4-ил)-амидо-4-фенилмасляной кислоты
Аналогично стадии б) 1,1 г (3,6 ммоль) продукта стадии б) подвергают взаимодействию с 1,0 г (3,6 ммоль) 1-(2-нафтоил)-пиперидин-4-карбоновой кислоты. Получают 1,0 г (61%) продукта.
1H-ЯМР (D6-ДМСО): δ= 1,2-1,9(6Н), 2,6-3,2(4Н), 3,6(1H), 3,6(1H), 3,7-4,0(1H), 4,0(1H), 4,2-4,6(2H) и 7-8,2(14Н) млн.дол.
г) Амид 3-(S)-(1-(2-нафтоил)-пиперидин-4-ил)-амидо-2-оксо-4-фенилмасляной кислоты
0,46 г (1 ммоль) продукта стадии в) и 0,4 г (4 ммоль) триэтиламида растворяют в 10 мл диметилсульфоксида и при комнатной температуре смешивают с 0,48 г (3 ммоль) комплекса пиридина и трехокиси серы, растворенного в 5 мл диметилсульфоксида. Получаемую смесь перемешивают 16 часов. После этого добавляют 150 мл воды и отсасывают осадок. Получают 0,33 г (72%) продукта.
МС: m/е=457 (М+).
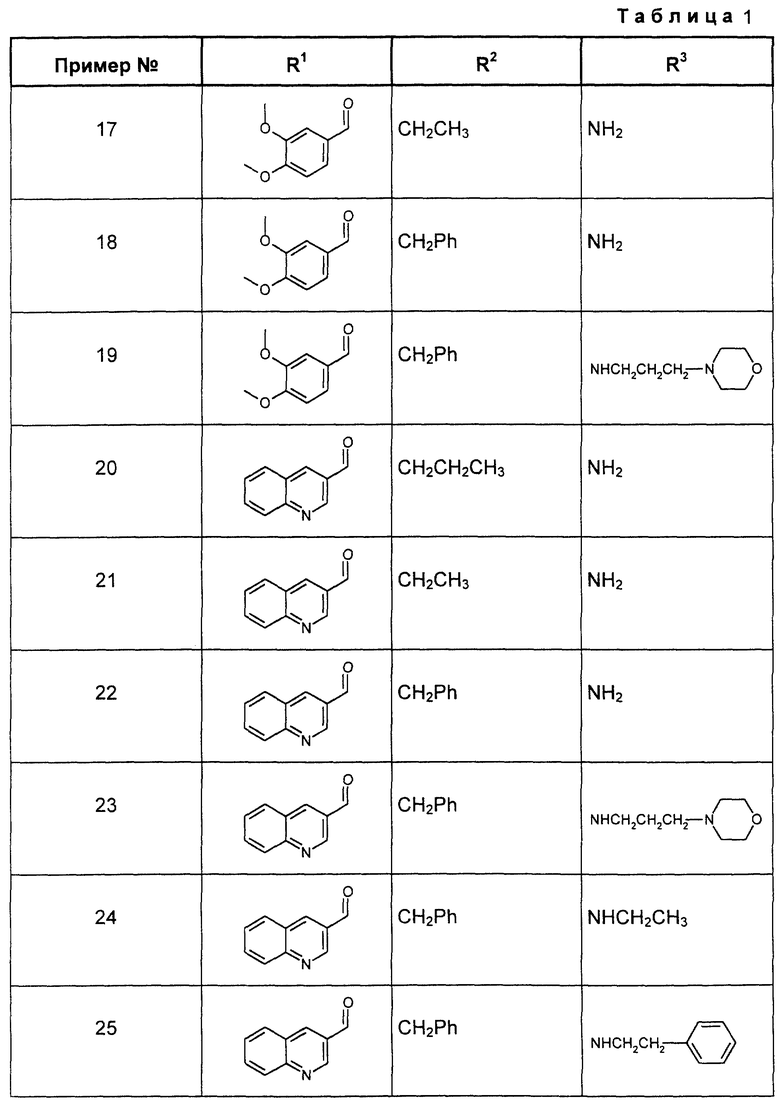
Аналогично примеру 16 можно получать еще соединения общей формулы (I), сведенные в таблице 1, где условное сокращение Рh означает фенил.
Заявитель представляет для подверждения совокупности признаков следующие данные по активности производных пипернидин-кетокарбоновой кислоты формулы I в таблице 2. Указанные данные получают испытанием калпаина I.
Кроме того, заявитель сообщает, что производные пиперидин-кетокарбоновой кислоты формулы (I) согласно изобретению относят к категории малотоксичных веществ.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ КЕТОБЕНЗАМИДЫ | 1997 |
|
RU2190599C2 |
| НОВЫЕ БЕНЗАМИДОАЛЬДЕГИДЫ | 1997 |
|
RU2189973C2 |
| СПОСОБ ПОЛУЧЕНИЯ АСИММЕТРИЧНО ЗАМЕЩЕННЫХ ТРИАЗИНОВ | 1994 |
|
RU2125995C1 |
| ПРОИЗВОДНЫЕ ФЕНИЛУКСУСНОЙ КИСЛОТЫ И СРЕДСТВО БОРЬБЫ ПРОТИВ НАСЕКОМЫХ И ПАУКООБРАЗНЫХ И ПРОТИВ ВРЕДОНОСНЫХ ГРИБОВ | 1995 |
|
RU2162075C2 |
| СПОСОБЫ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ ДЛЯ ПОЛУЧЕНИЯ МЕТИЛАМИДОВ α-МЕТОКСИИМИНОКАРБОНОВЫХ КИСЛОТ | 1995 |
|
RU2146247C1 |
| ПРОИЗВОДНЫЕ БЕНЗАМИДОКСИМОВ, ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ, ФУНГИЦИДНЫЕ СРЕДСТВА, СПОСОБ БОРЬБЫ С ФИТОПАТОГЕННЫМИ ГРИБАМИ | 1998 |
|
RU2192412C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ АЦЕТАМИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ И ИНГИБИТОРЫ ПРОТЕАЗ НА ИХ ОСНОВЕ | 1997 |
|
RU2181360C2 |
| АМИДЫ КАРБАМОИЛКАРБОНОВОЙ КИСЛОТЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, СРЕДСТВО И СПОСОБЫ ДЛЯ БОРЬБЫ С ВРЕДОНОСНЫМИ ГРИБАМИ | 1995 |
|
RU2145956C1 |
| ПРОИЗВОДНЫЕ 2-[(2-АЛКОКСИ-6-ТРИФТОРМЕТИЛПИРИМИДИН-4-ИЛ)ОКСИМЕТИЛЕН]ФЕНИЛУКСУСНОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СРЕДСТВО ДЛЯ БОРЬБЫ С ВРЕДИТЕЛЯМИ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ | 1995 |
|
RU2166500C2 |
| ПРОИЗВОДНЫЕ ФЕНИЛУКСУСНОЙ КИСЛОТЫ, ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ ДЛЯ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ СРЕДСТВА | 1995 |
|
RU2165411C2 |




Изобретение относится к новым производным пиперидин-кетокарбоновой кислоты формулы (I), где R1 - COR4 или SO2R4, R4 означает алкенил, замещенный фенилом или пиридином, нафтил, хиноксалинил, хинолинил, бензотиофенил, дигидроксифенил или пиридил, замещенный ациламиногруппой, R2 - C1-С6-алкил, который может быть замещен фенилом или пиридилом, R3 - группы -OR6 или -NHR6, где R6 означает водород, C1-С6-алкил, который может быть фенилом, пиридином или морфолинилом, их таутомерным и изомерным формам и солям. Соединения формулы (1) проявляют ингибирующие свойства по отношению к ферментам, в частности цистеин-протеаз, таких, как калпаин, и могут найти применение в медицине. 2 з.п. ф-лы, 2 табл.

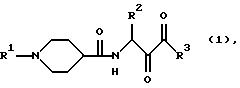
где R1-CO-R4 или -SO2-R4, где R4 означает алкенил, замещенный фенилом или пиридином, нафтил, хиноксалинил, хинолинил, бензотиофенил, дигидроксифенил или пиридил, замещенный ациламиногруппой;
R2 - разветвленный или неразветвленный C1-C6-алкил, который может быть замещен фенилом или пиридилом;
R3 - группы -OR6 или -NHR6, где R6 означает водород, разветвленный или неразветвленный C1-С6-алкил, который может быть замещен фенилом, пиридином или морфолинилом,
и их таутомерные и изомерные формы, а также их возможные физиологически переносимые соли.
| Способ получения 1,4-диметил-2,5-диизопропилбензола | 1974 |
|
SU520336A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Экономайзер | 0 |
|
SU94A1 |
| ПРОИЗВОДНОЕ N-ЗАМЕЩЕННОГО 1-ГЕКСИЛ-4-ФЕНИЛ-4-ПИПЕРИДИНКАРБОКСАМИДА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1992 |
|
RU2039043C1 |
