Ссылка на рассматриваемые одновременно заявки
Ссылка делается на рассматриваемую одновременно заявку порядковый 08/973590, поданную 6 июня 1995 (дело поверенного РС9178), опубликованную как WO 96/39408 12 декабря 1996, которая раскрывает трициклические 5,6-дигидро-9Н-пиразоло[3,4-с] -1,2,4-триазоло[4,3-а] пиридины, имеющие биологическую активность как ингибиторы типа IV (PDE4) фосфодиэстеразы и производства фактора некроза опухоли (TNF), полезные для лечения астмы, бронхита, хронической обструктивной болезни легких, аллергического ринита, псориаза, дерматита, ревматоидного артрита и других воспалительных, аллергических и иммунологических болезней и состояний. Несколько способов для получения указанных трициклических соединений описываются в них, но ничто, что описывается, не указало бы обычному специалисту на улучшенный способ настоящего изобретения.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Класс соединений, полученных в соответствии с настоящим изобретением, был назван здесь как 8-циклопентил-6-этил-3-[замещенные]-5,8-дигидро-4Н-1,2,3а, 1,8-пентааза-ас-индацены, хотя этот класс соединений, являющихся трициклическими, был назван в данной области техники 5,6-дигидро-9Н-пиразоло[3,4-с]-1,2,4-триазоло[4,3-а]пиридинами. Каким бы предпочтительным образом указанный класс соединений не назывался, однако соединения, полученные в соответствии со способом настоящего изобретения, представляются следующей формулой (1.0.0):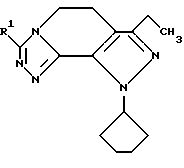
где R1 является членом, выбранным из группы, состоящей из водорода; (C1-C6)алкила; (C1-C4)алкокси; (C1-C4)алкокси(C1-C6)алкила; (C2-C8)алкенила; (C3-C7)циклоалкила и его 1'-метила; (C4-C7)циклоалкил(C1-C2)алкила; насыщенной или ненасыщенной (C4-C7)гетероциклической-(CH2)m-группы, где m равно 0, 1 или 2, включающей один или два гетероатома, выбранных из 0, S, S(=0)2, N, NR3, O и N или NR3, S или S(=O)2 и N или NR3, и N или NR3 и N или NR3, где R3 является водородом или (C1-C4)алкилом; и группы формулы (1.1.0):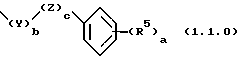
где а равняется 1-5, и b и с равны 0 или 1; R5 представляет водород, гидрокси, (C1-C4)алкил, (C2-C4)алкенил, (C1-C4)алкокси, (C3-C6) циклоалкокси, галоген, трифторметил, CO2R3a, CONR3aR3b, NR3aR3b, NO2, или SO2NR3aR3b; где R3a и R3b являются независимо водородом или (C1-C4)алкилом; Z является O, S, S(=O)2, С(=O), или NR3; и Y представляет - (C1-C4)алкилен- или - (С2-С4)алкенилен, любой из которых является необязательно монозамещенным гидрокси; где каждая вышеперечисленная алкильная, алкенильная, циклоалкильная, алкоксиалкильная или гетероциклическая группа является замещенной от 0 до 3 заместителями, выбранными из (C1-C2)алкила, трифторметила и галогена.
Вышеописанные пентааза-ас-индацены являются известными соединениями, имеющими биологическую активность как ингибиторы типа IV (PDE4) фосфодиэстеразы и производства фактора некроза опухоли (TNF). Эта биологическая активность делает указанные пентааза-ас-индацены полезными для лечения различных воспалительных, аллергических и иммунологических болезней и состояний, которые включают астму, бронхит, хроническую обструктивную болезнь легких, аллергический ринит, псориаз, дерматит и ревматоидный артрит. Вышеупомянутые терапевтические применения указанных пентааза-ас-индаценов хорошо установлены и приняты в данной области техники, как показано, например, в опубликованной заявке WO 96/39408, уже отмеченной выше. Применение ингибиторов PDE4 и TNF для лечения воспалительных, аллергических и иммунологических болезней и состояний также хорошо известно в данной области техники. См. например, WO 95/01980, опубликованную 19 января 1995 (дело поверенного РС8444А) и WO 96/12720, опубликованную 2 мая 1996 (дело поверенного РС8444С).
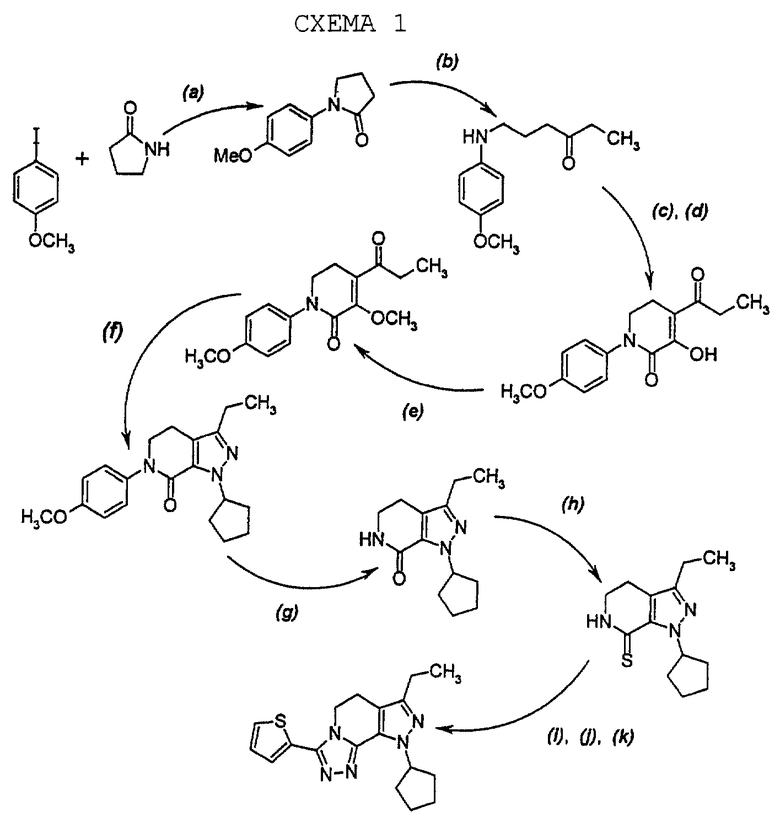
Способ получения 8-циклопентил-6-этил-3-[замещенных] -5,8-дигидро-4Н-1,2,3а, 7,8-пентааза-ас-индаценов, который известен в данной области техники и описывается в вышеупомянутой опубликованной заявке WO 96/39408, использует n-метоксифенильную N-защитную группу на начальных стадиях синтеза. Полный способ получения, изображенный для случая, где R1 является 2-тиенилом, представлен реакционной схемой 1, представленной в конце описания.
На стадии а полного синтеза 2-пирролидинон и 4-иоданизол нагреваются в присутствии порошка меди и карбоната калия, давая N-(4-метоксифенил)пирролидин-2-он, который на стадии b обрабатывается реактивом Гриньяра, этилмагнийбромидом, давая алифатический кетон после раскрытия кольца пирролидинона. Этот кетон выделяется и затем подвергается циклизации с образованием промежуточного соединения 3-гидрокси-1,2,5,6-тетрагидропиридин-2-она на стадиях с и d с использованием этилового эфира хлоангидрида щавелевой кислоты и гидрооксида натрия на стадии с и этилата натрия и этилового спирта на стадии d. Соответствующее 3-метоксильное промежуточное соединение получается на стадии е обработкой 3-метил-n-толилтриазином, после которой на стадии f получается промежуточный 4,5,6,7-тетрагидро-7-оксо-1H-пиразоло [3,4-с] пиридин циклизацией при использовании гидрохлорида циклопентилгидразина. 4-метоксифенильная N-защитная группа удаляется на стадии д обработкой азотнокислым, церием(1У)аммонием с получением лактамного промежуточного соединения, после которой на стадии h лактамное промежуточное соединение превращается в соответствующее тиолактамное промежуточное соединение обработкой пентасульфидом фосфора.
Трициклический конечный продукт получается на стадиях i, j и k обработкой безводным гидразином на стадии i, с последующей обработкой 2-тиофенкарбонилхлоридом на стадии j и кипячением в колбе с обратным холодильником на стадии k.
Однако, вышеописанный способ предшествующего уровня техники имеет ряд недостатков. Стадия а, например, является реакцией, проводимой без растворителей, в присутствии порошка меди и карбоната калия при температуре примерно 150oС. Когда реакция проводится в большем масштабе, чем применяемый для исследовательского синтеза, реакция стадии а становится экзотермической и после охлаждения может образоваться труднообрабатываемая твердая масса, если растворитель, например, этилацетат, не добавить немедленно к неочищенному расплаву, включающему реакционную смесь. Далее, на стадии е стоимость триазинового реагента, 3-метил-n-толилтриазина, является достаточно высокой, что создает проблему общей экономики способа по схеме 1, особенно, при рассмотрении в свете того факта, что выходы фактически всех стадий способа схемы 1 не являются оптимальными.
Кроме того, стадия b на которой получают алифатический кетон с помощью реактива Гриньяра, этилмагнийбромида, может быть проведена по существу без проблем в диэтиловом эфире, но в тетрагидрофуране, намного менее проблематичном растворителе, наблюдается тенденция к протеканию побочных реакций, ведущих к побочным продуктам и возможным проблемам стабильности. Аминокетон, защищенный n-метоксифенилом, полученный на стадии b, может быть недостаточно стабильным, чтобы он мог храниться. Другие проблемы могут возникать при синтезе и очистке циклопентилгидразинового реагента; и снятии n-метоксифениламидной защитной группы азотнокислым церийаммонием.
С дальнейшими проблемами можно сталкиваться при процедурах, связанных с использованием тиолактамной химии для введения триазольного компонента трициклического ядра конечных продуктов. Они включают использование безводного гидразина при введении триазольного кольца с помощью тиеноилхлорида. Безводный гидразин является опасным химическим веществом, дымящим на воздухе и способным взорваться во время перегонки, если присутствуют следы воздуха. Соответственно, в настоящее время в данной области техники необходим такой способ получения 8-циклопентил-6-этил-3-[замещенных]-5,8-дигидро-4Н-1,2,3а, 7,8-пентааза-ас-индаценов, который является менее проблематичным, более простым и имеет большую экономическую осуществимость. Отвечающий этой потребности способ получения настоящего изобретения представляется здесь в деталях.
ОПИСАНИЕ УРОВНЯ ТЕХНИКИ
Настоящее изобретение относится к области способов, применяемых для синтетического получения 8-циклопентил-6-этил-3-[замещенных]-5,8-дигидро-4Н-1,2,3а, 7,8-пентааза-ас-идаценов, которые являются известными соединениями, обладающими биологической активностью как селективные ингибиторы типа IV фосфодиэстеразы (PDE) и производства фактора некроза опухоли (TNF). Следовательно, способ настоящего изобретения выгодно обеспечивает уровень техники улучшенным способом получения соединений, которые, как известно, в свою очередь, применяются в лечении астмы, артрита, бронхита, хронической обструктивной болезни дыхательных путей, псориаза, аллергического ринита, дерматита и других воспалительных заболеваний, СПИДА, септического шока и других болезней у млекопитающих, особенно людей.
Со времени установления, что циклический аденозинфосфат (АМР) является внутриклеточным вторичным посредником, например, в E.W. Sutherland и Т. W. Rail, Pharmacol. Rev. 12, 265, (1960), ингибирование фосфодиэстераз было целью для модуляции и, соответственно, терапевтического вмешательства в область процессов болезни. Недавно отдельные классы PDE были установлены, например, в J. A. Beavo at al.,TiPS, 11, 150, (1990), и их селективное ингибирование привело к улучшенной лекарственной терапии. См., например, С. D. Nicholson, M. S, Hahid, TiPS, 12, 19, (1991). В частности, было признано, что ингибирование типа IV PDE может приводить к ингибированию высвобождения медиатора воспаления, например, в M.W. Verghese at al., J.Mol.Cell Car-diol., 12(Suppi. II), S 61, (1989) и релаксации гладкой мышцы дыхательных путей, например, в T. J. Torphy, "Direction for New Anti-Asthma Drugs", eds C.P. O'Donnell and C.G.A.Persson, 1988, 37 Birkhauser-Vertag.
Таким образом, соединения такие, как вышеупомянутые 8-циклопентил-6-этил-3-[замещенные] -5,8-дигидро-4Н-, 2,3а, 7,8-пентааза-ас-индацены, которые ингибируют тип IV PDE, но имеют слабую активность против других типов PDE, способны ингибировать высвобождение медиаторов воспаления и уменьшать напряжение гладкой мышцы дыхательных путей, не вызывая нежелательных сердечно-сосудистых или антитромботических эффектов. 8-циклопентил-6-этил-3-[замещенные] -5,8-дигидро-4Н-1,2,3а, 7,8-пентааза-ас-индацены также полезны как ингибиторы производства TNF, который, как установлено, вовлечен во многие инфекционные и аутоиммунные болезни. См., например, W.Friers, FEBS Letters, 285, 199, (1991). Кроме того, показано, что TNF является первичным медиатором ответа на воспаление, наблюдаемым при сепсисе и септическом шоке. См., например, С.Е. Spooner et al., Clinical Immunology and Immunopathology, 62, Sll, (1992).
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение касается улучшенного способа получения производного 8-циклопентил-6-этил-3-(замещенного)-5,8-дигидро-4Н-1,2,3а, 7,8-пентааза-ас-индацена формулы (1.0.0):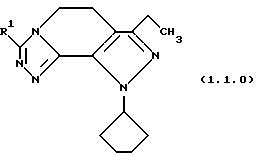
и его фармацевтически приемлемых солевых форм, в которой:
-R1 является членом, независимо выбранным из группы, состоящей из водорода; (C1-C6)алкила; (C1-C4)алкокси; (C1-C4)алкокси (C1-C4)алкила; (C2-C8)алкенила; (C3-C7) циклоалкила и его 1'-метила; (C3-C7) циклоалкил (C1-C2)алкила; насыщенной или ненасыщенной (С4-С7)гетероциклической-(СН2)n-группы, где n является целым числом, выбранным из 0, 1 или 2, включающей один или два гетероатома, независимо выбранных из О, S, S(=O)2, N, NR3, O вместе с N или NR3, S или 3(=О)2 вместе с N или NR3, и N или NR3 вместе с N или NR3; где:
-R3 является водородом или (C1-C4)алкилом; или
-R1 является группой формулы (1.1.0);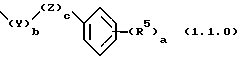
в которой:
- а является целым числом, выбранным из 1 до 5, включительно;
- b и с являются, каждый независимо, целым числом, выбранным из 0 и 1;
- R5 является членом, независимо выбранным из группы, состоящей из водорода, гидрокси, (C1-C4)алкила, (С2-С4)алкенила, (C1-C4)алкокси, (C3-C6)циклоалкокси, галогена, трифторметила, СО2R3a, CONR3aR3b, NR3aR3b, NO2 и SO2NR3aR3b; где
- R3a и R3b являются, каждый независимо, выбранным из группы, состоящей из водорода и (C1-C4)алкила;
- Z является O, S, S(=O)2, c(=O) или МR3; и
-Y представляет - (C1-C4)алкилен- или - (С2-С4)алкенилен-, любой из которых является необязательно монозамещенным гидрокси; где
- каждая вышеперечисленная алкильная, алкенильная, циклоалкильная, алкоксиалкильная или гетероциклическая группа является замещенной от 0 до 3 заместителями, включающими член, независимо выбранный из группы, состоящей из (C1-C2)алкила, трифторметила и галогена;
включающий:
(а) нагревание реакционной смеси γ-капролактона и п-метоксибензиламина без растворителя, посредством чего получается производное амида, N-защищенное п-метоксибензилом, формулы (2.0.0):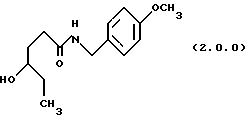
(в) восстановление указанного производного формулы (2.0.0), посредством чего получается производное аминоспирта, N-защищенное п-метоксибензилом, формулы (3.0.0.):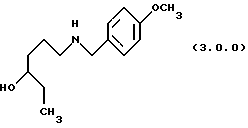
(с) ацилирование указанного производного аминоспирта формулы (3.0.0) этиловым эфиром хлорангидрида щавелевой кислоты, посредством чего получается производное этилового эфира оксаминовой кислоты, N-защищенное п-метоксибензилом, формулы (4.0.0):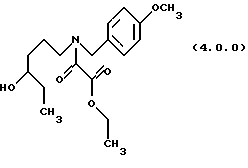
(d) окисление указанного производного этилового эфира формулы (4.0.0) оксаминовой кислоты, посредством чего получается производное оксаламидкетонна, N-защищенное п-метоксибензилом, формулы (5.0.0):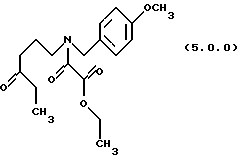
(е) циклизацию указанного производного оксаламидкетона формулы (5.0.0), посредством чего получается производное пиридинона, N-защищенное п-метоксибензилом, формулы (6.0.0):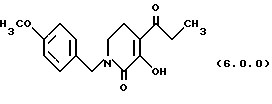
(f) O-метилирование указанного производного пиридинона формулы (6.0.0), посредством чего получается производное 3-метоксипиридинона, N-защищенное п-метоксибензилом, формулы (7.0.0):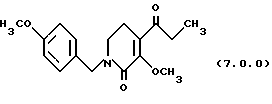
(q) обработку указанного производного 3-метоксипиридинона формулы (7.0.0) циклопентилгидразином, посредством чего получается производное пиразолопиридинона, N-защищенное п-метоксибензилом, формулы (8.0.0):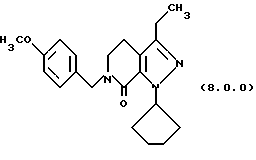
(h) снятие защитной группы с указаннного производного пиразолопиридинона формулы (8.0.0) удалением указанной п-метоксибензильной группы из него, посредством чего получается производное лактама формулы (9.0.0):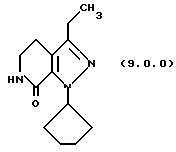
(i) этерификацию указанного производного лактама формулы (9.0.0), посредством чего получается соответствующее производное иминоэфира (имидат) формулы (10.0.0):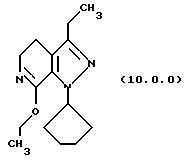
(j) обработку указанного производного иминоэфира (имидата) формулы (10.0.0) производных гидразида карбоновой кислоты формулы (11.0.0):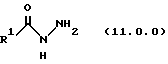
где R1 имеет то же самое значение, как изложено выше;
посредством чего получается указанное производное 8-циклопентил-6-этил-3-[замещенное] -5,8-дигидро-4Н-1,2,3а, 7,8-пентааза-ас-индацена формулы (1.0.0).
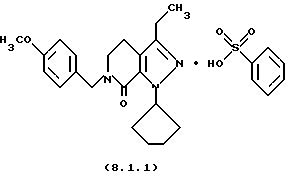
Настоящее изобретение также касается нескольких различных групп новых промежуточных соединений, которые применяются в вышеописанном способе получения производного 8-циклопентил-6-этил-3-[замещенного] -5,8-дигидро-4Н-1,2,3а, 7,8-пентааза-ас-индацена формулы (1.0.0). Одна группа таких новых промежуточных соединений включает тозилатные и безилатные соли производного пиразолопиридинона, N-защищенного п-метоксибензилом, формулы (8.1.0) и (8.1.1), соответственно: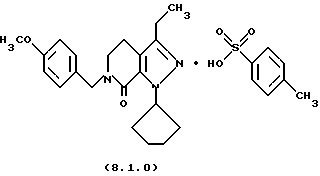
Другая группа новых промежуточных соединений настоящего изобретения включает производное иминоэфира (имидат) формулы (10.0.0):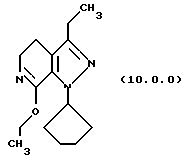
и его фармацевтически приемлемые солевые формы, включающие особенно его тозилатные и безилатные соли.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Улучшенный способ получения настоящего изобретения касается создания терапевтически полезных соединений формулы (1. 0. 0):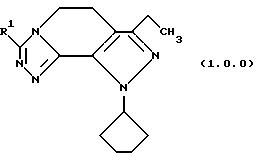
и их фармацевтически приемлемых солевых форм, где R1 является, между прочим, членом, независимо выбранным из группы, состоящей из водорода; (C1-C6)алкила; (C1-C4)алкокси; (C1-C4)алкокси (C1-C4)алкила; (C2-C8)алкенила; (C3-C7)циклоалкила и его 1'-метила; (C3-C7)циклоалкил(C1-C2)алкила; насыщенной или ненасыщенной (C4-C7)гетероциклической-(СН2)n-группы, где n является целым числом, выбранным из 0, 1 или 2, включающей один или два гетероатома, независимо выбранных из O, S, S(=O)2, N, NR3, O вместе с N или NR3, S или S(=O)2 вместе с N или NR3, и N или NR3 вместе с N или NR3;
где R3 является водородом или (C1-C4)алкилом.
Вышеописанные соединения формулы (1.0.0) называются вместе здесь как 8-циклопентил-6-этил-3-[замещенные] -5,8-дигидро-4Н-1,2,3а,7,8-пентааза-ас-индацены, и как уже обсуждалось, обладают биологической активностью как ингибиторы PDE4 и производства TNF. Улучшенный способ получения настоящего изобретения является подходящим для получения указанных соединений, где R1 фрагмент имеет значение (C1-C6)алкила; (C1-C4)алкокси(C1-C4)алкила; (C2-C8)алкенила; (C3-C7)циклоалкила и его 1'-метила; или (С3-С7)циклоалкил(C1-C2)алкила.
Выражение "и его 1'-метил", используемое в связи с определением R1 как (C3-C7)циклоалкил, означает, что возможная метильная группа присоединена к тому же атому углерода, которым указанная (C3-C7)циклоалкильная группа присоединена к трициклическому ядру соединений формулы (1.0.0). Необходимо отметить, что, такое определение R1 легко отличается от значения "(C3-C7)циклоалкил(C1-C2)алкил", в случае которого алкиленовый мостик, например, метиленовый, расположен между указанной (C3-C7)циклоалкильной группой и указанным трициклическим ядром. Соответственно, когда (C3-C7)циклоалкил имеет значение циклогексила, и присутствует 1'-метальная группа, R1 будет определяться как фрагмент формулы (1.2.0):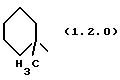
и будет называться как 3-метил-3-циклогексил.
В предпочтительных вариантах осуществления способ настоящего изобретения является особенно подходящим для получения соединений формулы (1.0.0), где R1 имеет значение метила, этила, н-пропила, изо-пропила, трет-бутила, циклопентила, циклогексила и 3-метил-3-циклогексила.
Улучшенный способ получения по настоящему изобретению является далее подходящим для получения соединений формулы (1.0.0), где R1 фрагмент имеет значение насыщенной или ненасыщенной (C4-C7) гетероциклической-(СН2)n-группы, где n является целым числом, выбранным из 0, 1 или 2, включающей один или два гетероатома, независимо выбранных из О, S, S(=O)2, N, NR3, O вместе с N или NR3, S или S(=O)2 вместе с N или NR3, и N или NR3 вместе с N или NR3; где R3 является водородом или (C1-C4)алкилом.
В предпочтительных вариантах осуществления способ настоящего изобретения является особенно подходящим для получения соединений формулы (1.0.0), где R1 имеет значение одной из следующих ненасыщенных (C5-C6)гетероциклических -(СН2)n-групп: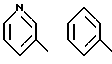
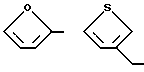
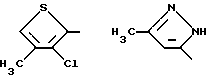
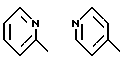
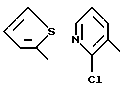
Улучшенный способ получения настоящего изобретения является далее подходящим для получения соединений формулы (1.0.0), где R1 фрагмент имеет значение группы формулы (1.1.0):
в которой а является целым числом, выбранным из 1 до 5, включительно, b и с являются каждый независимо целым числом, выбранным из 0 и 1; R5 является членом, независимо выбранным из группы, состоящей из водорода; гидрокси; (C1-C4)алкила; (C2-C4)алкенила; (C1-C4)алкокси; (C3-C6)циклоалкокси; галогена; трифторметила; CO2R3a; CONR3aR3b; NR3aR3b; NO2 и SO2NR3aR3b; где R3a и R3b являются, каждый независимо, выбранными из группы, состоящей из водорода и (C1-C4)алкила; Z представляет O, S, S(=O)2,С(=O) или NR3; и Y представляет - (C1-C4)алкилен- или - (C2-C4)алкенилен, любой из которых является необязательно монозамещенным гидрокси.
В предпочтительных вариантах осуществления способ настоящего изобретения является особенно подходящим для получения соединений формулы (1.0.0), где а равно 1 или 2; b равно 1; с равно 0; Y представляет -(C1-C2)алкилен-; и R5 представляет метил, метокси, гидроксил, хлор, иод или трифторметил. Соответственно, в более предпочтительных вариантах осуществления соединений, особенно подходящих для получения способом настоящего изобретения, R1 имеет значение одной из следующих групп:
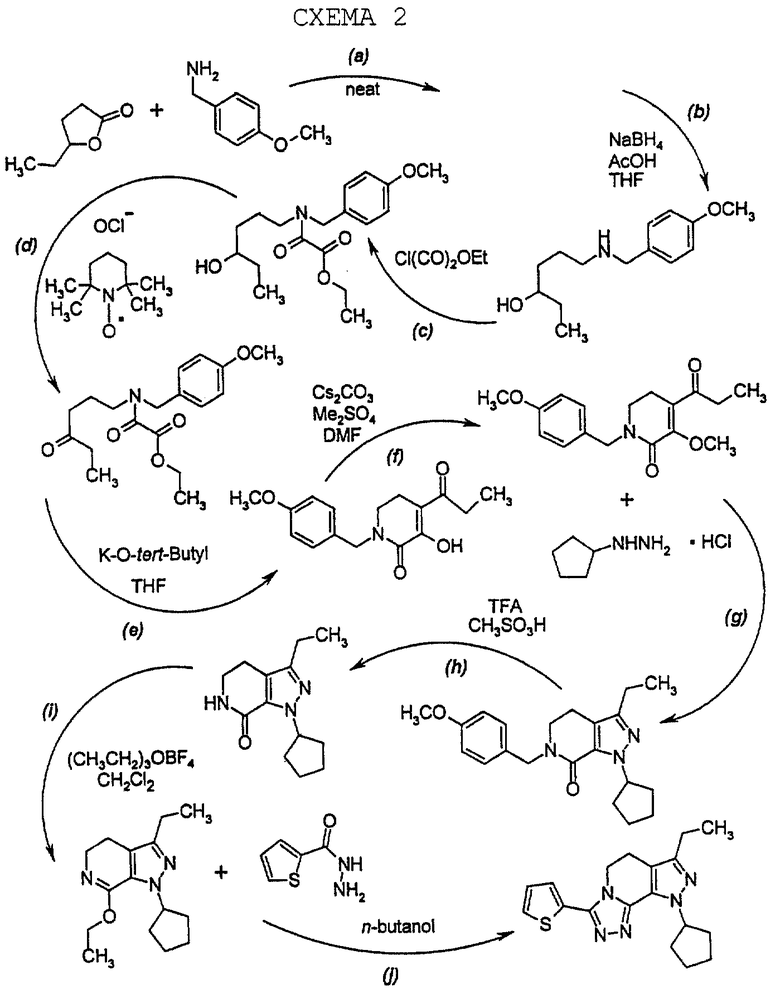
Улучшенный способ настоящего изобретения для получения соединения формулы (1.0.0) может быть проиллюстрирован реакционной схемой 2 (см. в конце описания), которая показывает получение представителей соединений формулы (1.0.0), где R1 представляет 2-тиенил.
На первой стадии, стадии (а) в вышепроиллюстрированной схеме 2, образуется реакционная смесь у-капролактона и п-метоксибензиламина, которая подвергается нагреванию для получения производного аминоспирта, N-защищенного п-метоксибензилом, формулы (2.0.0). Последовательность реакций этой стадии (а) может быть проиллюстрирована следующим образом: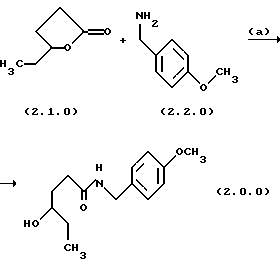
γ-капролактон формулы (2.1.0) реагирует с неразбавленным 4-метоксибензиламином формулы (2.2.0), т.е. без растворителя, и нагревается до температуры в интервале от 70o до 95o С, предпочтительно от 80o до 85o С, и выдерживается при этой температуре в течение от 12 до 24 часов, предпочтительно 16 часов. Амидный продукт формулы (2.0.0) получается при использовании общепринятых процедур разделения как кристаллическое твердое вещество. Эта стадия улучшается на основании процедур таких, как, например, восстановление γ-капролактона формулы (2.1.0) при использовании ди-изобутилалюминийгидрида (DiBAl-H) в хлористом метилене, с последующим восстановительным аминированием полученного лактола п-метоксибензиламином и натрийтриацетоксиборгидридом [NaHB(OAc)3] в смысле элиминирования восстановителя и растворителя, который иначе требовался бы на первой стадии.
Вторая стадия также приводит к получению более устойчивого аминоспиртового промежуточного продукта формулы (3.0.0). Следует отметить, что в качестве реагента лучше применять п-метоксибензиламин формулы (2.2.0), чем соответствующий п-метоксифениламин. Найдено, что, если такая п-метоксифенильная группа замещает n-метоксибензильную группу, присоединенную к атому азота аминоспиртового промежуточного соединения формулы (3.0.0), полученное соединение является нестабильным при ультрафиолетовом (UV) облучении. Стадия, включенная в эту реакцию, стадия (b) на схеме 2 выше, описывается в абзаце непосредственно ниже.
Амидный промежуточный продукт формулы (2.0.0), полученный в вышеописанной первой стадии способа настоящего изобретения, затем восстанавливается с образованием соответствующего аминоспирта формулы (3.0.0), который является N-защищенным п-метоксибензилом, как уже описано. Реакция стадии (b) может быть проиллюстрирована следующим образом: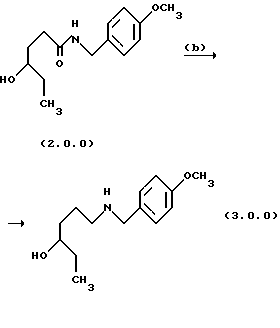
Вышепроиллюстрированное восстановление, выполненное на стадии (b), является восстановлением N-замещенного амида до соответствующего амина и осуществляется при использовании восстановителя для амидов. Такие восстанавливающие агенты являются известными специалисту в данной области техники и обычно состоят из восстановителя гидридного типа, например, комплекса борана с аммиаком, ВН3.NН3; комплекса борана с трет-бутиламином, (СН3)3СNН2 ВН3; комплекса борана с триметиламином, (СН3)3Н.ВН3; гидрата алюминия, АlН3; натрийбис(2-метоксиэтокси)алюминийгидрида, [(СН3ОСН2СН2О)2AlH2] Na или боргидрада натрия, NaBH4.
Предпочтительным восстановителем является боргидрид натрия, NaBH4, в то время как другие восстановители являются менее предпочтительными, например литийалюминийгидрид, LiAIH4, потому что он вызвал бы слишком энергичную реакцию. Восстановитель применяется совместно с источником протонов, который добавляется впоследствии и является предпочтительно слабой кислотой или THF раствором такой кислоты, например, уксусной кислоты. Восстановитель и источник протонов добавляются в подходящий растворитель такой, как метанол, этанол, диэтиловый эфир, муравьиная кислота, уксусная кислота, формамид и тетрагидрофуран THF. Предпочтительным растворителем является THF.
В предпочтительном способе выполнения стадии (b) натрийборгидридный восстановитель добавляется в THF растворитель, после чего 4-бензиламид 4-гидроксилгексановой кислоты формулы (2.0.0), полученный на стадии (а), добавляется в виде твердого вещества. Реакционная смесь после этого охлаждается, добавляется уксусная кислота в THF, и реакционная смесь нагревается до температуры слабого флегмообразования в интервале от 60o до 70oС в течение периода времени от 14 до 18 часов, предпочтительно, 16 часов. Водородный газ удаляется в течение реакции, и непрореагированный амид удаляется экстракцией этилацетатом после добавления 1н HCI для разложения избыточного реагента. После этого рН реакционной смеси повышается до 11, чтобы дать возможность аминоспиртовому промежуточному продукту формулы (3.0.0) быть извлеченным в этилацетат, и поддерживается для применения в последующей стадии (с).
Стадия (с) настоящего изобретения может быть проиллюстрирована следующей реакционной схемой: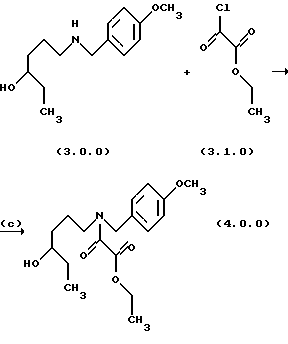
Вышепроиллюстрированное ацилирование, проведенное на стадии (с), является ацилированием амина хлоридом кислоты в водном щелочном растворе в соответствии с известными условиями "реакции Шоттена-Баумана". См. Schotten, Ber. 17, 2544 (1884): и Georg, Bioorg. Med. Chem. Letters.. 4, 335 (1994). Водная щелочь добавляется для связывания НС1, которая выделяется в течение реакции. В предпочтительном способе выполнения стадии (с) реакции ацилирования для этой цели применяется водный раствор гидрокарбоната натрия. Дополнительный растворитель, предпочтительно этилацетат, применяется для получения раствора этилового эфира хлорангидрида щавелевой кислоты, реагента формулы (3.1.0), так как реакционная смесь брала начало как этилацетатный раствор аминоспиртового промежуточного соединения формулы (3.0.0), полученного на стадии (b).
Ацилхлоридный реагент, применяемый на стадии (с) является этиловым эфиром хлорангидрида щавелевой кислоты формулы (3.1.0). Реакция является экзотермической; соответственно, этиловый эфир хлорангидрида щавелевой кислоты добавляется на протяжении времени, предпочтительно, от 20 до 30 минут, в то время как в то же самое время температура реакции предпочтительно поддерживается при 0o до 5oС. Реакция завершается в пределах короткого периода времени от 1 до 2 часов, но реакционная смесь необязательно перемешивается при комнатной температуре от 20o до 25oС в течение дополнительного периода времени от 14 до 18 часов, предпочтительно, 16 часов, чтобы дать возможность любому оставшемуся непрореагировавшему количеству этилового эфира хлорангидрида щавелевой кислоты быть удаленным разложением. Продукт формулы (4.0.0), масло, получается при использовании общепринятых методов разделения и является структурно этиловым эфиром оксаминовой кислоты, N-защищенным п-метоксибензильной группой. Это промежуточное соединение применяется как исходный материал в следующей стадии по существу без дополнительной очистки.
Стадия (d) способа настоящего изобретения может быть проиллюстрирована следующей реакционной схемой: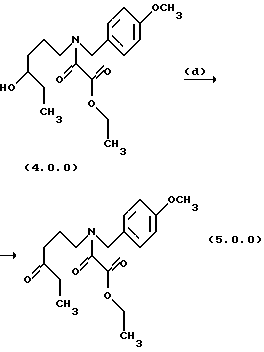
Вышепроиллюстрированное окисление, проводимое на стадии (d), является окислением фрагмента вторичного спирта в кето фрагмент, которое может быть выполнено при использовании сильных окислительных агентов при подходящих условиях окисления в соответствии со способами, о которых специалисты в данной области техники хорошо осведомлены. Например, подходящей является "реакция окисления Джонса", которая выполняется в присутствии хромовой кислоты, водной серной кислоты и ацетона, См.,например Bowden et. al., J. Chem. Soc., 39 (1946); или Ley and Madin, Comp. Org. Syn, 7, 253-256(1991). Способ является особенно полезным, так как он протекает быстро с высокими выходами и не затрагивает никакие из других присутствующих двойных связей. Способ также является очень прямым, так как он только требует, чтобы вторичный спирт формулы (4.0.0) растворялся в ацетоне и затем титровался "реактивом Джонса", состоящим из раствора хромовой кислотной и серной кислоты в воде.
Другой тип окислительного способа, подходящего для применения на стадии (d) настоящего изобретения, является окисление, включающее применение кислого хромата H2CrO4; и различных других каталитических композиций. окисления, включающих хром, например, оксида хрома, Cr2O3; гидроксида хрома, Сr(ОН)3•nН2O; ацетата хрома, СrСН3СОО)3. См. Cainelli; Cardillo Chromium Oxidations in Organic Chemistry', Springer: New York, 1984 для дальнейших деталей, касающихся хромовых катализаторов окисления и методов для их применения. Другим хорошо известным способом окисления вторичных спиртов в кетоны, который подходит для выполнения стадии (d), является "реакция окисления Саретта", применяющая СrО3-пиридиновый комплекс как катализатор окисления. См. , например, Poos et al., J.Am. Chem. Soc. 75, 422 (1953); или Hasan and Rocek, J. Am. Chem. Soc.. 97,1444, 3762 (1975).
Другие типы сильных катализаторов окисления и методы ддя их применения для превращения вторичного спирта, такого как спирт формулы (4.0.0), в соответствующий кетон такой, как кетон формулы (5.0.0), включают, но не ограничиваются ими, марганцовокислый калий, КМnО4; бром, Вг2; и тетраоксид рутения, RuO4.
Дальнейший пример подходящих катализаторов окисления и процедур для их применения для превращения вторичного спирта формулы (4.0.0) в соответствующий кетон формулы (5.0.0) и катализатора, который является предпочтительным для применения на стадии (d) способа настоящего изобретения, включает, но не ограничивается им, применение окислительного агента, гипохлорита натрия, в присутствии катализатора 2,2,6,6-тетраметил-1-пиперидинилокси, свободного радикала (TEMPO). Структура ТЕМПО катализатора может быть представлена следующей формулой (4.1.0):
В этом предпочтительном способе проведения окисления вторичного спирта формулы (4.0.0) для превращения его в кетон формулы (5.0.0) также является предпочтительным, чтобы раствор гипохлорита натрия был сделан свежим при выполнении стадии (d) растворением гипохлорита кальция и карбоната натрия в воде и регулированием рН итогового раствора от 9,0 до 10,0, предпочтительно 9,5, гидрокарбонатом натрия, с последующим фильтрованием указанного раствора для удаления оставшегося в растворе побочного продукта, карбоната кальция.
Далее в этом предпочтительном способе выполнения стадии (d) реакционная смесь включает вторичный спирт формулы (4.0.0), растворенный в хлористом метилене, СН2Сl2; и бромистый калий, КВr, растворенный в воде. TEMPO катализатор добавляется в реакционную смесь, которая затем охлаждается до температуры от 0o до 10oС, предпочтительно, от 0o до 5oС, после чего окислительный агент, гипохлорит натрия, медленно добавляется в реакционную смесь, которая поддерживается при
температуре от 10o до 20oС, предпочтительно от 10o до 15oС. Продукт представляет масло, который получается при использовании обычных методов разделения и применяется на следующей стадии способа без дополнительной очистки.
Еще более предпочтительный способ выполнения стадии (d), чем вышеописанный, включает применение полимера для поддержания окислительного агента, гипохлорита натрия, как активного иона ОСl- и/или TEMPO катализатора. См. McKillop; Young, Synthesis, 401-422 (1979).
Указанный еще более предпочтительный способ выполнения стадии (d) также включает применение межфазного катализа, так как реакция, имеющая место, является нуклеофильным замещением, в которой субстрат является относительно нерастворимым в воде и других полярных растворителях, в то время как нуклеофил является анионом, растворимым в воде, но не в субстрате или других органических растворителях. См. Dehmlow; Dehmlow Phase Transfer Catalysis, 2nd ed.; Verlag Chemie: Deerfield Beach. FL (1983).
Стадия (е) способа настоящего изобретения может быть проиллюстрирована следующей реакционной схемой: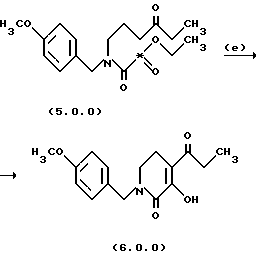
Вышепроиллюстрированная циклизация, проводимая на стадии (е), включает катализируемую основанием циклизацию сложных эфиров дикарбоновой кислоты с образованием β-кетоэфира. Звездочка ("*") в дикарбоновой кислоте формулы (5.0.0) указывает точку отделения одного из сложных эфиров с образованием этанольного побочного продукта, не показанного в вышеизложенной схеме реакции. Включенная циклизация является именной органической реакцией, называемой "реакция конденсации Дикмана". См. Dieckmann, Ber. 27, 102, 965 (1894); или Davis и Garrett, Comp. Org. Syn. 2, 806-829 (1991).
Реакция выполняется в присутствии относительно сильного основания такого, как этилат натрия или трет-бутилат калия, и в подходящем растворителе, например, сухом тетрагидрофуране, ди-изо-пропиловом эфире, метил-трет-бутиловом эфире и толуоле. Основание добавляется постепенно на протяжении периода от 15 до 45 минут, предпочтительно, 30 минут, в то время как температура реакционной смеси сохраняется ниже от 30o до 40oС, предпочтительно ниже 35oС. После этого реакция протекает до завершения в течение от 0,5 до 1,5 часов, обычно 1,0 часа, причем реакционная смесь находится при комнатной температуре, например, от 20o до 25oС. Продукт, твердое вещество, выделяется фильтрованием.
Стадия (f) способа настоящего изобретения может быть проиллюстрирована следующей реакционной схемой: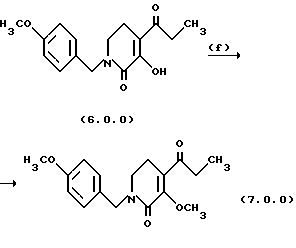
Вышепроиллюстрированная реакция включает O-метилирование производного пиридинона формулы (6.0.0), посредством чего получается производное 3-метокси-пиридинона N-защищенное n-метоксибензилом, формулы (7.0.0). Желательно провести селективное O-метилирование спиртовой группы без соответствующего С-метилирования; следовательно, некоторые реакции оказались неподходящими, например обработка йодистым метилом в ацетоне с карбонатом калия.
Одним успешным подходом, который представляет предпочтительный вариант осуществления способа настоящего изобретения, является алкилирование спиртовой группы неорганическим сложным эфиром, в частности, метилирование диметилсульфатом. В предпочтительном варианте осуществления эта реакция выполняется в диметилформамиде (DMF) в качестве растворителя в присутствии карбоната цезия, Cs2CO3, постепенным добавлением диметилсульфата на протяжении периода от 15 до 45 минут, предпочтительно, 30 минут, в то время как температура реакционной смеси сохраняется от 15o до 30oС, предпочтительно от 20o до 25oС. После этого реакционная смесь поддерживается при этой температуре и перемешивается в течение от 12 до 20 часов, обычно 16 часов. Продукт, масло, получается при применении общепринятых процедур разделения.
Стадия (q) способа настоящего изобретения может быть проиллюстрирована следующей реакционной схемой: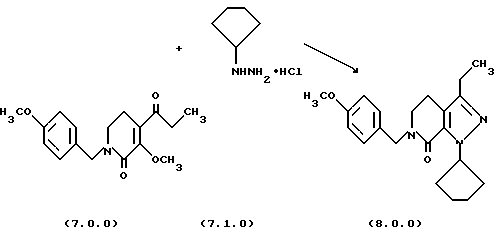
Вышепроиллюстрированная реакция включает получение пиразолсодержащего соединения формулы (8.0.0) обработкой производного 3-метоксипиридинона формулы (7.0.0) дигидрохлоридом циклопентилгидразина формулы (7.1.0). В предпочтительном варианте осуществления эта реакция выполняется в тетрагидрофурановом (THF) растворителе нагреванием реакционной смеси от 75o до 95oС, предпочтительно, 88oС, в течение от 8 до 16 часов, предпочтительно, 12 часов, в то время как реакционная смесь продувается азотом для удаления метанола, THF и HCl. Продуктом является густое, темное масло, которое может применяться в следующей стадии способа настоящего изобретения без дальнейшей обработки, или, альтернативно, может быть очищено как соль п-толуолсульфоновой кислоты или бензолсульфоновой кислоты при применении общепринятых процедур разделения.
Когда соединение формулы (8.0.0) должно быть очищено как соль п-толуолсульфоновой кислоты или бензолсульфоновой кислоты, в предпочтительном варианте осуществления оно растворяется в этилацетате и обрабатывается безводной n-толуолсульфоновой кислотой или безводной бензолсульфоновой кислотой, растворенной в этилацетате. Соответствующая соль кристаллизуется из реакционной смеси, которая затем охлаждается и фильтруется, обеспечивая чистую тозилатную или бензолсульфонатную соль.
Ключевым реагентом вышеописанной стадии (q) является дигидрохлорид циклопентилгидразина формулы (7.1.0), который может быть получен в соответствии с несколькими способами, известными в литературе. В предпочтительном варианте осуществления применяется способ, описанный в Syn. Comrn. 11, 43 (1981), в котором циклопентанол обрабатывается ди-трет-бутилазодикарбоксилатом и трифенилфосфином в соответствии со схемой реакции, которая может быть проиллюстрирована следующим образом: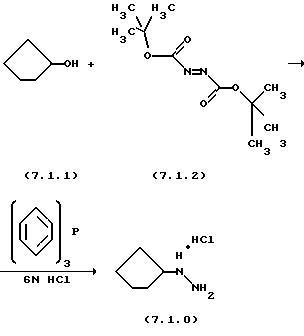
Вышеописанная реакция основывается на органической именной реакции, названной "реакция Митсунобу", которая включает конденсацию спиртов и кислых компонентов при обработке диалкилазодикарбоксилатов и триалкил- или триарилфосфинов, происходящую главным образом с инверсией конфигурации через промежуточную оксифосфониевую соль. См. Mitsunobu etal.. Bull. Chem. Soc. Japan 40, 935 (1967);
Brown etal.. Tetrahedron 50, 5469 (1994); Edwards etal., ibid. 5579; and Hughes, Org. React. 42, 335-656(1992).
В предпочтительном варианте осуществления для получения дигидрохлорида циклопентилгидразина формулы (7.1.0), циклопентанол формулы (7.1.1) и трифенилфосфин растворяются вместе в подходящем растворителе таком, как тетрагидрофуран (THF), и после этого реакционная смесь охлаждается до температуры от 2o до 8oС, предпочтительно, 5oС. Ди-трет-бутилазодикарбоксилат, растворенный в THF, затем добавляется в реакционную смесь на протяжении периода от 1 часа до 3 часов, предпочтительно, 2 часов, в то время как температура реакционной смеси сохраняется ниже 6oС. Реакционной смеси дается возможность нагреться до комнатной температуры, т.е. до 20o-25oС, и она перемешивается в течение от 4 часов до 6 часов, предпочтительно, 5 часов, после чего 6н HCI добавляется в реакционную смесь для удаления ВОС групп из продукта. Реакционная смесь затем перемешивается в течение дополнительного периода от 18 до 30 часов, предпочтительно 24 часов. Твердый продукт затем выделяется как дигидрохлоридная соль при применении общепринятых процедур разделения. Следует отметить, что основной продукт может быть или дигидрохлоридной солью, или моногидрохлоридной солью в зависимости от стехиометрии количества добавляемой 6н НСl. Любая соль действует хорошо в реакции вышеописанной стадии(q).
Стадия (h) способа настоящего изобретения, может быть проиллюстрирована следующей реакционной схемой: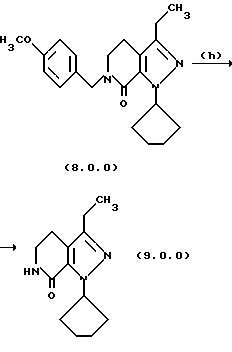
Вышепроиллюстрированная реакция включает снятие защитной группы с производного пиразолопиридинона формулы (8.0.0) удалением п-метоксибензильной группы, посредством чего образуется производное лактама формулы (9.0.0). Удаление п-метоксибензильной группы выполняется в соответствии с известными способами для снятия защитной группы с аминов, где защитная группа является п-метоксибензо льной группой. Далее отмечается, что реакция стадии (q), описанная подробно выше, и снятие защитной группы стадии (h) могут быть выполнены без выделения продукта стадии (q), то есть, обе реакции могут быть осуществлены совместно в том же самом реакционном сосуде.
В соответствии с предпочтительным вариантом осуществления способа настоящего изобретения стадия (h) выполняется при температуре от 50o до 60oС, предпочтительно, 55oС, что обычно требует охлаждения реакционной смеси после завершения стадии (q). После этого в реакционную смесь медленно добавляется трифторуксусная кислота (TFA), в то время как ее температура поддерживается при от 50o до 60oС, причем первоначальная загрузка TFA вызывает условия экзотермической реакции, которые требуют внешнего охлаждения. Метан-сульфоновая кислота, СН3SО3Н, затем добавляется в реакционную смесь, температура которой теперь поднимается до 65o-75oС, предпочтительно, 70oС, при которой реакционная смесь поддерживается в течение от 1 1/2 до 2 1/2 часов, предпочтительно, 2 часов. После этого реакционная смесь охлаждается до температуры 15o-30oС, предпочтительно, 20o-25oC, после чего твердый продукт лактам формулы (9.0.0) получается общепринятыми процедурами разделения.
Стадия (i) способа настоящего изобретения может быть проиллюстрирована следующей реакционной схемой: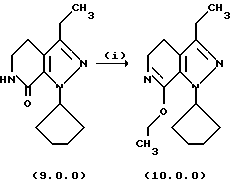
Вышепроиллюстрированная реакция включает этерификацию производного лактама формулы (9.0.0) в соответствующий иминоэфир, т.е. производное имидата формулы (10.0.0). Эта этерификация выполняется при применении тетрафторбората триэтилоксония, (СН3СН2)3ОВF4, агента, применяемого при получении ω-аминоэфиров из лактамов. См. Synth. Commun. 18, 1625 (1988).
В предпочтительном варианте осуществления способа настоящего изобретения для выполнения стадии (i) раствор тетрафторбората триэтилоксония, (СН3СН2)3ОВF4), в хлористом метилене медленно добавляется к суспензии производного лактама формулы (9.0.0) в хлористом метилене на протяжении периода от 30 до 50 минут, предпочтительно, 40 минут. После этого, реакционная смесь поддерживается при температуре от 15o до 25oС, предпочтительно от 18o до 22 oС, в течение периода от 18 до 24 часов, предпочтительно, 21 часа. Продукт, масло, получается при применении общепринятых процедур разделения.
Стадия (j) способа настоящего изобретения может быть проиллюстрирована следующей реакционной схемой: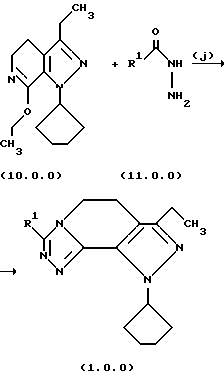
В предпочтительном варианте осуществления способа настоящего изобретения для выполнения стадии (j) раствор соединения формулы (10.0.0) в 1-бутаноле и гидразид 2-тиофенкарбоновой кислоты, или альтернативно, гидразид 2,2-диметилпропионовой кислоты, нагревается при температуре от 85o до 95oС, предпочтительно, 90oС на протяжении периода от 36 до 60 часов, предпочтительно, 48 часов. Продукт, белое твердое вещество и не совсем белое твердое вещество, соответственно, получается при применении общепринятых процедур разделения.
Выбор растворителя для растворения соединения формулы (9.0.0) и конкретного гидразида карбоновой кислоты, который должен быть применен для получения требуемого соединения формулы (1.0.0), зависит в значительной степени от способности кандидата на растворитель соответствующим образом растворять вышеупомянутые реагенты, также как иметь требуемую низкую точку кипения так, чтобы реакционная смесь могла нагреваться с обратным холодильником в течение длинных периодов времени без опасности разложения или реагентов, или конечного продукта. Растворитель должен быть доступен с высокой чистотой и по разумной стоимости. 1-бутанол является особенно подходящим в виде раствора 63% спирта и 37% воды, который образует азеотропную смесь, кипящую при 92oС. Другие подходящие растворители включают растворители, выбранные из группы, состоящей из н-амилового простого эфира, изо-амилацетата, изо-пентилового спирта и изо-пропилового спирта.
Следует отметить, что стадии (a)-(i) включительно способа настоящего изобретения, описанные подробно выше, все и каждая относятся к отдельным соединениям, превращаемым через каждую из реакций, изложенных в вышеупомянутых стадиях. Эти стадии, соответственно, не имеют никаких обобщенных смыслов. Ближайшая и вышепроиллюстрированная последняя стадия, стадия (j), с другой стороны, является пунктом в способе настоящего изобретения, где различные заместители, определяющие группу R1, вводятся в структуру конечного продукта определенного формулой (1.0.0). Таким образом, промежуточное соединение последней стадии и, следовательно, ключевое промежуточное соединение в способе настоящего изобретения включает производное иминоэфира (имидат) формулы (10.1.0):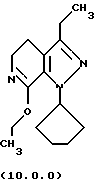
и его фармацевтически приемлемые солевые формы, включающие особенно его тозилатные и безилатные соли.
Ключевое промежуточное соединение формулы (10.0.0) последней стадии реагирует с гидразином соответствующей структуры для обеспечения требуемого значения R1 в конечных продуктах формулы (1.0.0). Реакция не только служит для введения требуемого заместителя R1 в соединение формулы (10.0.0), но она также служит для обеспечения дальнейшей циклизации с образованием "триазолильного" компонента три-циклического конечного продукта формулы (1.0.0). Как уже указывалось выше, конечные продукты формулы (1.0.0) назывались прежде 5,6-дигидро-9Н-пиразоло[3,4-с] -1,2,4-триазоло[4,3-а]пиридинами, хотя здесь предпочтительно называть указанные соединения формулы (1.0.0) 8-циклопентил-6-этил-3-[замещенными] -5,8-дигидро-4Н-1,2,3а, 7,8-пентааза-ас-индаценами.
Вышеупомянутый гидразин соответствующей структуры для обеспечения требуемого значения R1 представляет производное гидразида карбоновой кислоты формулы (11.0.0):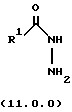
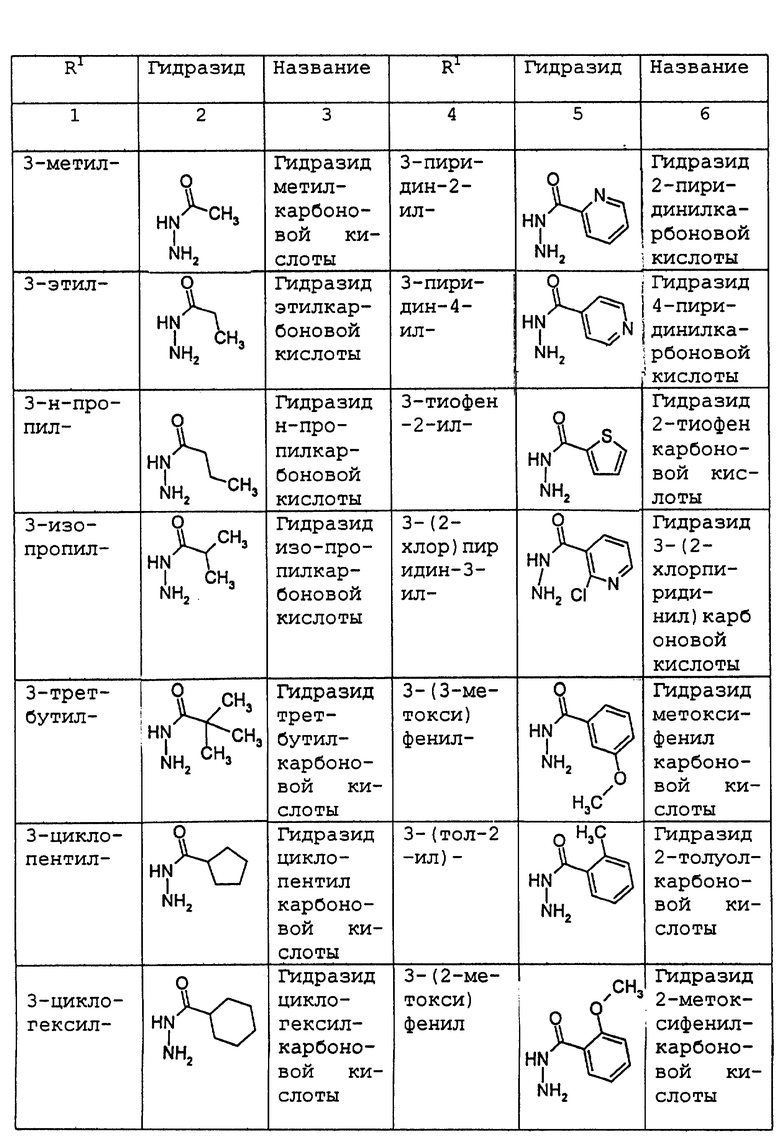
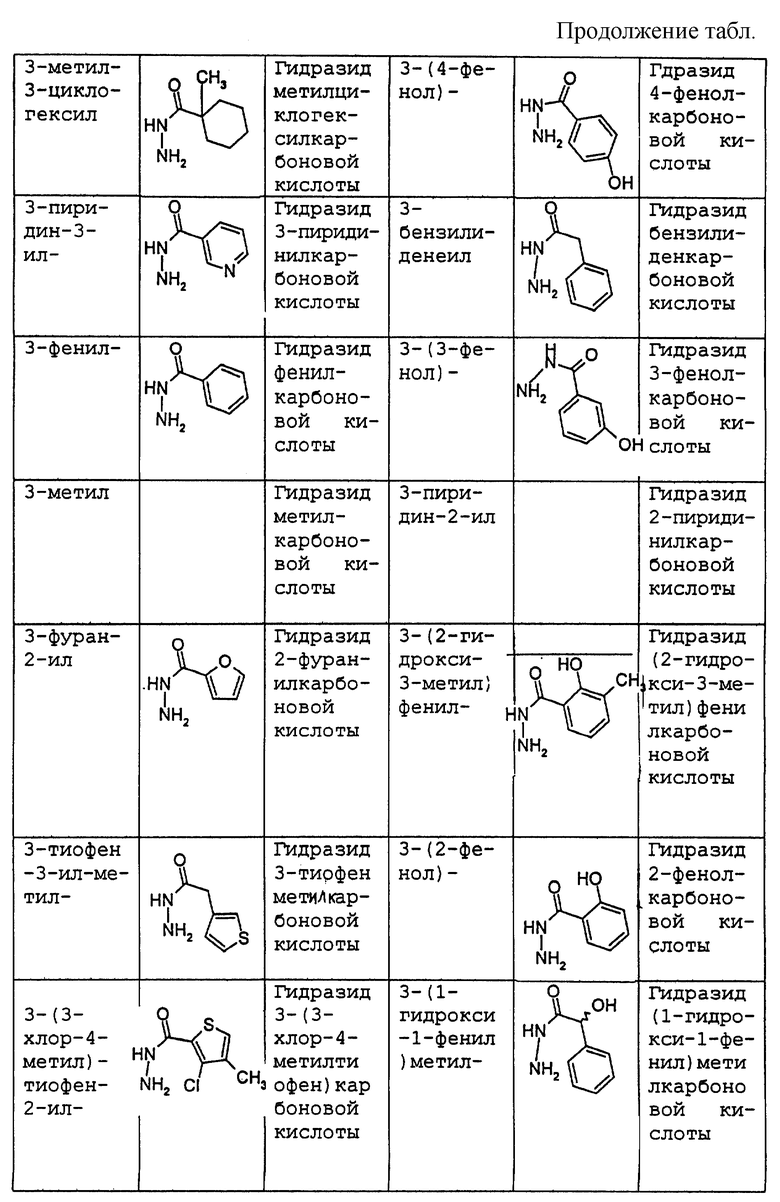
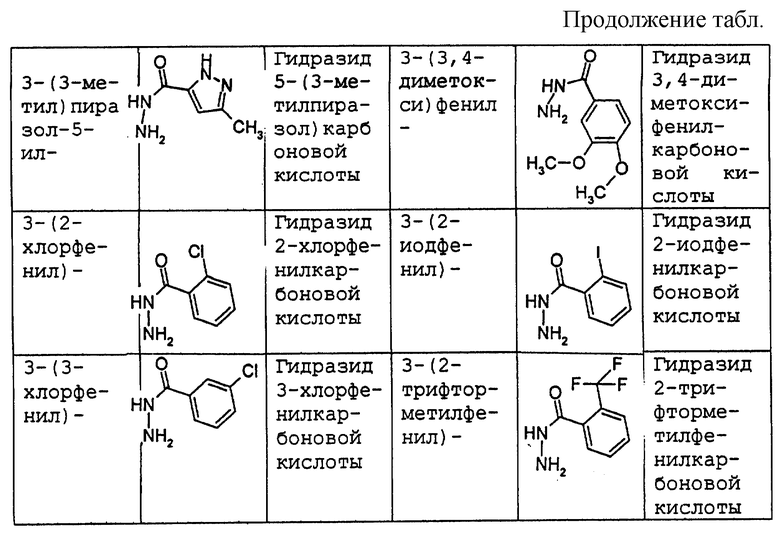
где R1 имеет то же самое значение, как изложено выше. В предпочтительных вариантах осуществления настоящего изобретения подходящее производное гидразида карбоновой кислоты формулы (11.0.0) представляет член, выбранный из группы, состоящей из таковых, изложенных следующим образом (см. таблицу).
Многие из вышеописанных реагентов, гидразидов карбоновой кислоты формулы (11.0.0), доступны коммерчески. Например, гидразид 2-тиофенкарбоновой кислоты доступен от Аldrich Chemical Company, St. Louis, МО 63178-9916 под номером в каталоге Т3261-1. Когда карбоновый гидразид коммерчески не доступен, например гидразид трет-бутилкарбоновой кислоты, он может быть получен при использовании способов, опубликованных в технической литературе и известных рядовым специалистам в области техники синтеза таких органических соединений. Такой способ разработан для получения гидразида трет-бутилкарбоновой кислоты, который более подходяще называть гидразидом 2,2-диметилпропанкарбоновой кислоты. Этот способ описывается ниже.
Способ, разработанный для получения гидразида 2,2-диметилпропанкарбоновой кислоты, является модификацией способа, описанного в опубликованной Европейской заявке ЕР 653419 (1995), переданной Shell Oil [Chem. Abs. 123: 32678b (1995)], который применяет триметилуксусную кислоту, гидрат гидразина и каталитическую TiO2. Реакция проводилась при применении н-пропанола как растворителя наряду с 1 мол% Ti(i-PrO)4, который гидролизуется немедленно при добавлении в реакционную смесь, давая аморфный активный катализатор TiO2. После того, как реакционная смесь нагревается с обратным холодильником в течение 24 часов, н-пропанольный растворитель отгоняется из реакциионного сосуда, азеотропно удаляя воду из реакционной смеси. После разбавления реакционной смеси свежим н-пропанолом твердый TiO2, активный катализатор, может быть отфильтрован из реакционной смеси. Остаток может упариваться и повторно суспендироваться в петролейном эфире, давая требуемый гидразид 2,2-диметил-пропанкарбоновой кислоты высокой чистоты и с 88% выходом.
Настоящее изобретение также относится к новым промежуточным соединениям, применяемым в вышеописанных стадиях способа для получения производного 8-циклопентил-6-этил-3-[замещенного] -5,8-дигидро-4Н-1,2,3а, 7,8-пентааза-ас-индацена формулы (1.0.0). Одна группа таких новых промежуточных соединений включает член, выбранный из группы, состоящей из тозилатных и безилатных солей производного пиразолопириди-нона N-замещенного n-метоксибензилом, формул (8.1.0) и (8.1.1), соответственно:
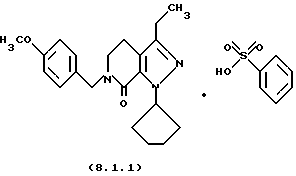
Вышеописанные промежуточные соли формулы (8.1.0) и формулы (8.1.1) применяются на стадии (h), как описано подробно выше.
Другая группа новых промежуточных соединений настоящего изобретения включает производное (имидиат) формулы (10.0.0):
и его фармацевтически приемлемые солевые формы, включающие особенно его тозилатные и безилатные соли. Тозилатные и безилатные соли могут быть представлены формулами (10.1.0) и (10.2.0) следующим образом:
Дальнейший предпочтительный вариант осуществления настоящего изобретения относится к способу получения соединений формулы (1.0.0), состоящему только из двух стадий, начинающихся исходными соединениями формулы (9.0.0), которые являются известными, как описано в схеме 1 подробно выше. Этот двухстадийный способ может быть представлен схемой 3 (см. в конце описания).
Соответственно, настоящее изобретение далее касается улучшенного способа получения производного 8-циклопентил-6-этил-3-[замещенного] -5,8-дигидро-4Н-1,2,3а,7,8,-пентааза-ас-индацена формулы (1.0.0):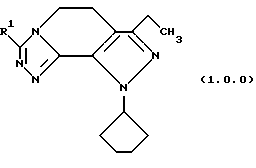
и его фармацевтически приемлемых солевых форм, в которой R1, как определено выше;
включающего:
(а) этерификацию производного лактама формулы (9.0.0):
посредством чего получается соответствующее производное иминоэфира (имидиат) формулы (10.0.0):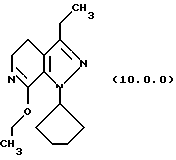
(b) обработку указанного производного иминоэфира (имидиата) формулы (10.0.0) производным гидразида карбоновой кислоты формулы (11.0.0):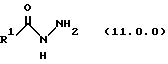
где R1 имеет то же самое значение, как изложено выше; посредством чего получается указанное производное 8-циклопентил-6-этил-3-[замещенное]-5,8-дигидро-4Н-1,2,3а,7,8-пентааза-ас-индацена формулы (1.0.0).
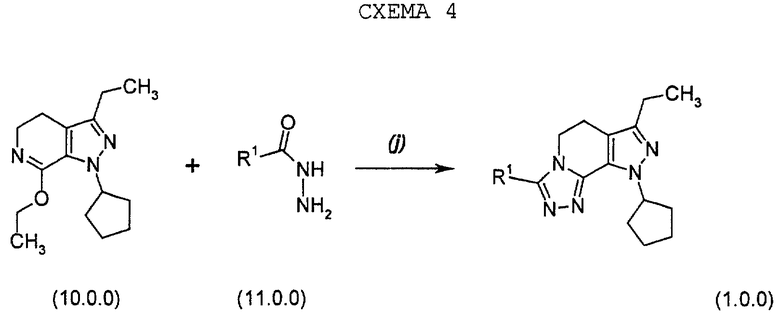
Следующий предпочтительный вариант осуществления настоящего изобретения относится к способу получения соединений формулы (1.0.0), состоящему из единственной стадии, начинающейся с нового промежуточного соединения формулы (10.0.0), которое может быть получено в соответствии со стадиями способа и процедурами, детализированными выше. Этот одностадийный способ может быть представлен схемой 4 (см. в конце описания).
Соответственно, настоящее изобретение далее еще касается улучшенного способа получения производного 8-циклопентил-6-этил-3-[замещенного]-5,8-дигидро-4Н-1,2,3а,7,8-пентааза-ас-индацена формулы (1.0.0).
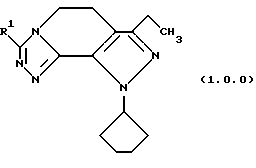
и его фармацевтически приемлемых солевых форм, в которой R1 является, как определено выше; включающего:
обработку производного имноэфира (имидата) формулы (10.0.0):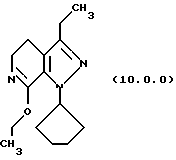
производным гидразида карбоновой кислоты формулы (11.0.0);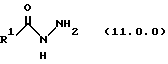
где R1 имеет то же самое значение, как изложено выше; посредством чего получается указанное производное 8-циклопентил-6-этил-3-[замещенное]-5,8-дигидро-4Н-1,2,3а,7,8-пентааза-ас-индацена формулы (1.0.0).
Предпочтительные варианты осуществления для выполнения различных стадий способа настоящего изобретения были описаны здесь. Соответственно, имеются предпочтительные варианты осуществления для выполнения полного способа настоящего изобретения. Один наиболее предпочтительный из таких предпочтительных вариантов осуществления описывается ниже.
Улучшенный способ получения производного 8-циклопентил-6-этил-3-[замещенного] -5,8-дигидро-4Н-1,2,3а, 7,8-пентааза-ас-индацена формулы (1.0.0).

и его фармацевтически приемлемых солевых форм, в которой R1 является таким, как определен выше; включающий:
(а) нагревание реакционной смеси γ-капролактона и п-метоксибензиламина без растворителя до температуры в интервале от 70o до 95oС, предпочтительно, от 80o до 85oС и выдержание при этой температуре в течение от 12 до 24 часов, предпочтительно, 16 часов, посредством чего получается производное амида, N-защищенное п-метоксибензилом, формулы (2.0.0):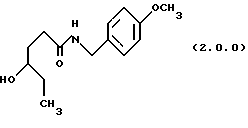
(b) восстановление указанного производного амидна формулы (2.0.0) при использовании восстановителей, выбранных из группы, состоящей из комплекса борана с аммиаком, ВН3. NН3; комплекса борана с трет-бутиламином, (СН3)3СNН2 ВН3; комплекса борана с триметиламином, (CH3)3NBH3; гидрида алюминия АlН3; натрийбис(2-метоксиэтокси)алюминийгидрида, [(СН3ОСН2СН2О)2AlH2] Na и боргидрида натрия NaBH4, предпочтительно, боргидрида натрия;
указанный восстановитель, применяемый совместно с источником протонов, включающим слабую кислоту или THF раствор такой кислоты, предпочтительно, уксусной кислоты; и причем указанный восстановитель и источник протонов добавляются в растворитель, выбранный из группы, состоящей из метанола, этанола, диэтилового эфира, муравьиной кислоты, уксусной кислоты, формамида и тетрагидрофурана, THF, предпочтительно, THF;
в котором после того, как указанный восстановитель добавляется к указанному растворителю, указанный амид формулы (2.0.0) добавляется в виде твердого вещества в указанную реакционную смесь, которая после этого охлаждается; указанный источник протонов в указанном растворителе добавляется в указанную реакционную смесь, которая затем нагревается до температуры слабого флегмообразования в интервале от 60o до 70oС в течение периода времени от 14 до 18 часов, предпочтительно 16 часов; причем водородный газ удаляется как побочный продукт, и непрореагировавший амид удаляется экстракцией этилацетатом после добавления 1н HCl, чтобы разложить избыточный реагент; и после этого поднимают рН указанной реакционной смеси от 10 до 12, предпочтительно, 11, чтобы дать возможность проэкстрагировать продукт формулы (3.0.0) в этилацетат и сохранить для применения на следующей стадии;
посредством чего получается производное аминоспирт N-защищенное п-метоксибензилом, формулы (3.0.0);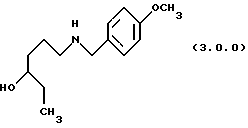
(с) ацилирование указанного производного аминоспирта формулы (3.0.0) в соответствии с условиями реакции Шоттена-Баумана для обработки амина хлорангидридом кислоты в водном щелочном растворе, предпочтительно, водном растворе гидрокарбоната натрия, в которой указанный хлорангидрид кислоты, предпочтительно, этиловый эфир хлорангидрида щавелевой кислоты, добавляется как раствор в растворителе, который является, предпочтительно, этилацетатом:
где реакция, которая имеет место, является экзотермической, вследствие чего указанный хлорангидрид кислоты, предпочтительно, этиловый эфир хлорангидрида щавелевой кислоты, добавляется на протяжении времени, предпочтительно, от 20 до 30 минут, и указанная температура реакции поддерживается от 0o до 5oС, пока указанная реакция не завершится за 1-2 часа; после чего указанная реакционная смесь необязательно перемешивается при температуре от 20o до 25oС в течение от 14 до 18 часов, предпочтительно 16 часов, чтобы дать возможность удалить непрореагировавший хлорангидрид кислоты, предпочтительно, этиловый эфир хлорангидрида щавелевой кислоты разложением;
посредством чего получается производное этилового эфира оксаминовой кислоты, N-защищенное п-метоксибензилом, формулы (4.0.0);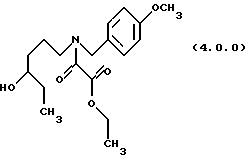
(d) окисление указанного производного этилового эфира оксаминовой кислоты формулы (4.0.0) с использованием сильных окислительных агентов при подходящих условиях окисления; при которых указанное окисление выполняется;
(i) при условиях реакции окисления Джонса, выполненное в присутствии хромовой кислоты, водной серной кислоты и ацетона; или
(ii) при использовании окислительного агента, гипохлорита натрия, в присутствии катализатора 2,2,6,6-тетраметил-1-пиперидинилокси, свободного радикала (TEMPO), в котором указанный раствор гипохлорита натрия делается свежим при выполнении указанного окисления, включающего: растворение гипохлорита кальция и карбоната натрия в воде и доведение рН полученного раствора от 9.0 до 10.0, предпочтительно, 9.5 гидрокарбонатом натрия, с последующим фильтрованием указанного раствора для удаления оставшегося побочного продукта, карбоната кальция, из указанного раствора, и далее
где реакционная смесь образуется в виде раствора указанного соединения формулы (4.0.0) в хлористом метилене, CH2Cl2; при добавлении к бромистому калию, КВr, растворенному в воде, к которому добавляется указанный TEMPO катализатор, и указанная реакционная смесь охлаждается до температуры от 0o до 10oС, предпочтительно, от 0o до 5oС; после чего указанный окислительный агент, гипохлорит натрия, медленно добавляется, в то время как указанная реакционная смесь поддерживается при температуре от 10o до 20oС, предпочтительно, от 10o до 15oС;
посредством чего получается производное оксаламидкетона N-защищенное n-метоксибензилом формулы (5.0.0):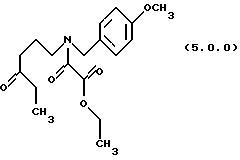
(е) циклизацию указанного производного оксаламидкетона формулы (5.0.0) при условиях реакции конденсации Дикмана, где реакция проводится в присутствии относительно сильного основания, выбранного из группы, состоящей из этилата натрия и трет-бутилата калия, в подходящем растворителе, включающем сухой тетрагидрофуран, ди-изо-пропиловый простой эфир, метил -трет-бутиловый простой эфир или толуол; где указанное основание добавляется постепенно на протяжении периода от 15 до 45 минут, предпочтительно 30 минут, в то время как температура указанной реакционной смеси сохраняется ниже 30o-40oС, предпочтительно, ниже 35oС, и указанная реакция протекает до завершения от 0,5 до 1,5 часа, обычно 1,0 часа, в то время как указанная реакциионная смесь находится при температуре 20oдо 25oС:
посредством чего получается производное пиридинона, N-защищенное n-метоксибензилом, формулы (6.0.0):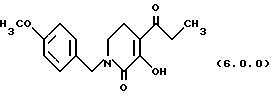
(f) O-метилирование указанного производного пиридинона формулы (6.0.0) метилированием диметилсульфатом; где реакционная смесь образуется диметилформамидным (DMF) растворителем в присутствии карбоната цезия, Сs2СО3, постепенным добавлением указанного диметилсульфата на протяжении периода от 15 до 45 минут, предпочтительно, 30 минут, в то время как температура указанной реакционной смеси сохраняется при температуре от 15o до 30oС, предпочтительно, от 20o до 25oС; и после этого указанная реакционная смесь поддерживается при указанной температуре и перемешивается в течение от 12 до 20 часов, обычно 16 часов;
посредством чего получается производное 3-метоксипиридинона, N-защищенное п-метоксибензилом, формулы (7.0.0):
(q) обработку указанного производного 3-метоксипиридинона формулы (7.0.0) дигидрохлоридом циклопентилгидразина; где реакционная смесь образуется тетрагидрофурановым (THF) растворителем и нагреванием указанной реакционной смеси до температуры 75o до 95oС, предпочтительно, 88oС, в течение от 8 до 16 часов, предпочтительно, 12 часов, в то время как указанная реакционная смесь продувается азотом для удаления метанола, THF и HCI;
посредством чего получается производное пиразолопиридинона, N-защищенное п-метоксибензилом, формулы (8.0.0):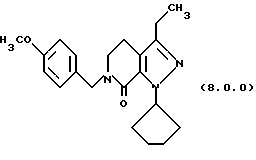
где указанное соединение формулы (8.0.0) может применяться в следующей стадии способа без дальнейшей обработки, или альтернативно, может быть очищено как соль п-толуолсульфоновой кислоты или бензолсульфоновой кислоты, растворением указанного соединения формулы (8.0.0) в этилацетате и после этого обработкой его безводной n-толуолсульфоновой кислотой, растворенной в этилацетате, или безводной бензолсульфоновой кислотой, растворенной в этилацетате; после чего соответствующая соль кристаллизуется из таким образом образованной реакционной смеси, которая затем охлаждается и фильтруется для получения чистой тозилатной или бензолсульфонатной соли;
(h) снятие защитной группы с производного указанного пиразолопиридинона формулы (8.0.0) удалением указанной п-метоксибензильной группы из него; где реакционная смесь сохраняется при температуре от 50o до 60oС, предпочтительно, 55oС; после чего трифторуксусная кислота (TFA) добавляется медленно, причем начальное добавление TFA вызывает условия экзотермической реакции, которые требуют внешнего охлаждения; после этого метансульфоновая кислота, СН3SО3Н, добавляется в указанную реакционную смесь, температура которой поднимается до от 65o до 75oС, предпочтительно, 70oС, при которой указанная реакционная смесь поддерживается в течение от 1 1/2 до 2 1/2 часов, предпочтительно, 2 часов, и после этого указанная реакционная смесь охлаждается до температуры от 15o до 30oС, предпочтительно от 20o до 25oС;
посредством чего получается производное лактама формулы (9,0.0):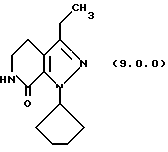
(i) этерификацию указанного производного лактама формулы (9.0.0) при использовании тетрафторбората триэтилоксония (СН3СH2)3ОВF4; где реакционная смесь создается медленным прибавлением раствора тетрафторбората триэтилоксония (СН3СН2)3ОВF4 в хлористом метилене к суспензии указанного производного формулы (9.0.0) в хлористом метилене на протяжении периода от 30 до 50 минут, предпочтительно, 40 минут; и после этого поддерживанием указанной реакционной смеси при температуре от 15o до 25oС, предпочтительно, от 18o до 22oС, в течение периода от 18 до 24 часов, предпочтительно 21 часа;
посредством чего получается соответствующее
производное иминоэфира (имидат) формулы (10.0.0)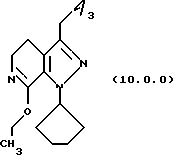
(j) обработку указанного производного иминоэфира (имидата) формулы (10.0.0) гидразидом карбоновой кислоты формулы (11.0.0);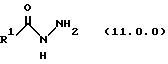
где R1 представляет 2-тиофен или трет-бутил; где реакционная смесь образуется раствором указанного соединения формулы (9.0.0) в 1-бутаноле и гидразиде 2-тиофенкарбоновой кислоты, или альтернативно, гидразиде 2,2-диметилпропанкарбоновой кислоты; и указанная реакционная смесь нагревается при температуре от 85o до 95oС, предпочтительно, 90oС на протяжении периода от 36 до 60 часов, предпочтительно, 48 часов;
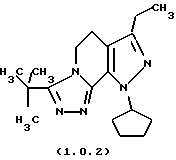
посредством чего получается 8-циклопентил-6-этил-3-тиофен-2-ил-5,8-дигидро-4Н-1,2,3а, 7,8-пентааза-ас-индацен формулы (1.0.1) и 8-циклопентил-6-этил-3-t-бутил-5,8-ди-гидро-4Н-1,2,3а, 7,8-пентааза-ас-индацен формулы (1.0.2):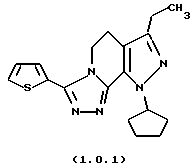
ИЛЛЮСТРИРУЕМЫЕ ПРИМЕРАМИ ПРЕДПОЧТИТЕЛЬНЫЕ ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Далее представлены получение и рабочие примеры предпочтительных вариантов осуществления настоящего изобретения с целью иллюстрации и для того, чтобы сделать даже более понятым специалисту технику выполнения способа получения настоящего изобретения. Однако примеры предназначаются только для цели демонстрации настоящего изобретения указанному специалисту и не должны быть восприняты каким-то образом ограничивающими объем и содержание настоящего изобретения, которое завершается формулой изобретения.
ПРИМЕР 1 4-метоксибензиламид4-гидроксигексановой кислоты (2.0.0)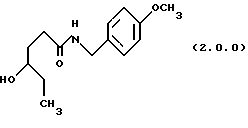
Гамма - капролактон (28,745 кг, 251,8 моля) и 4-метокси-бензиламин (38,0 кг, 277 молей) помещались в стеклянный 100 галлоный облицованный резервуар. Раствор нагревался до 80-850С и выдерживался при этой температуре в течение 16 часов. TLC тонкослойная хроматография на силикагелевых пластинках показала, что реакция завершилась. TLC система включала: этилацетат с детектированием при 254 нм. Этилацетат (18 галлонов, 68 л) медленно загружался в реакционный резервуар после охлаждения до 60oС. Гексан (общее количество 18 галлонов, 68 л) добавляли, пока помутнение не было достигнуто. После 1/2 часа, чтобы позволить начаться кристаллизации, добавлялся остаток гексана, суспензия охлаждалась до 25oС и гранулировалась в течение 3 часов. Твердое вещество собиралось фильтрованием и промывалось 1: 1 смесью этилацетата и гексана. Влажный осадок высушивался под вакуумом без дополнительного нагревания с получением 46,05 кг (72.8%) требуемого амида; т.пл. 81 -82oС.
'ПМР (СDСl3, 300 МГц) δ 7,18 (д, 2), 6,84 (д, 2), 6,27 (шир.с, 1), 4,32 (д, 2), 3,79 (с, 3), 3,50 (м., 1), 3,19 (шир. с, 1), 2,35 (т, 2), 1,85 (м., 1), 1,67 (м., 1), 1,49 (м., 2), 0,92 (т, 3).
Анал. Вычис. для C14H21MO3: С, 66,91; Н, 8,42; N, 5,57. Найдено: С, 67,26; Н, 8,71; N, 5,55.
ПРИМЕР 2 6-(4-метоксибензиламино)гексан-3-ол (3.0.0)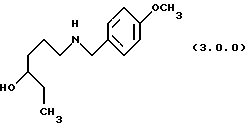
Тетрагидрофуран (121 галлон, 458 л) и боргидрид натрия (22,154 кг, 585,6 молей) загружались в заполненный чистым и сухим азотом 500 галлонный стеклянный облицованный резервуар. Суспензии позволялось перемешиваться в течение 30 минут при 20-25oС, затем 4-метоксибензиламид 4-гидроксигексановой кислоты (45,75 кг, 182 моля) добавлялся в виде твердого вещества. Через 30 минут реакция охлаждалась до 5-10oС и на протяжении 4 до 8 часового периода добавлялся раствор уксусной кислоты (9,1 галлонов, 34,4 л) в тетрагидрофуране (12 галлонов, 45,4 л) при сохранении температуры при 0 -10oС. Небольшое стравливание азота сохранялось в резервуаре, чтобы помочь удалить водород. Когда добавление завершалось, реакция нагревалась до 20-25oС и перемешивалась в течение часа. Температура реакции медленно увеличивалась до слабого флегмообразования (~ 66oС) и поддерживалась там в течение 16 часов. Реакция охлаждалась добавлением 1н HCI с сохранением температуры <25oС. Излишек тетрагидрофурана удалялся перегонкой при атмосферном давлении. Этилацетат добавлялся к полученному водному раствору для извлечения непрореагировавшего амида. Кислый водный раствор затем доводился до рН 11, чтобы дать возможность полученному амину проэкстрагироваться в этилацетат, и сохраняться для использования на следующей стадии. Аликвота этилацетатного раствора продукта отбиралась для предварительного определения конечного выхода и концентрации. Выход для этой крупномасштабной серии был 55.0%, который был меньше, чем выход, достигнутый при маломасштабных опытах (78.8%). При крупномасштабной процедуре было 12,8% невосстановленного исходного амида после охлаждения, что частично объясняло более низкий выход.
'ПМР (СDСl3,300 МГц) δ 7,21(д,2), 6,83 (д, 2), 3,78 (с, 3), 3,69 (с, 2), 3,41 (м. , 2), 2,78 (м., 1), 2,58 (м., 1), 1,71 (м., 2), 1,45 (м.,4), 0,95 (т,3). GC масс-спектр: m/e, 237 (М+).
ПРИМЕР 3 Этиловый эфир N-(4-гидроксигексил)-N-(4-метоксибензил) оксаминовой кислоты (4.0.0).
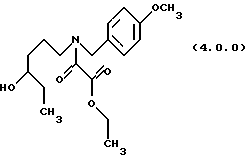
6-(4-Метоксибензиламино)гексан-3-ол (24 кг, 101,1 моля) в этилацетате (158 галлонов, 598 л) загружался в чистый и сухой заполненный азотом 500 галлонный резервуар. Этот раствор охлаждался до 0-5oС, затем раствор гидрокарбоната натрия (16,988 кг, 202,2 моля в 51 галлоне (193 л) воды) добавлялся при поддержании температуры от 0 до 5oС. Раствор этилового эфира хлорангидрида щавелевой кислоты (16,566 кг, 121,4 моля) в этилацетате (20 галлонов, 75,7 л) добавлялся при сохранении температуры от 0o до 5o С на протяжении периода времени примерно 25 минут. Реакции давали нагреться до температуры 20-25oС, при которой она завершалась согласно HPLC (жидкостная хроматография высокого разрешения). Реакция перемешивалась в течение дополнительных 16 часов, чтобы позволить разложиться любому оставшемуся этиловому эфиру хлорангидрида щавелевой кислоты. Нижний водный слой удалялся, и этилацетат промывался 49 галлонами (185,5 л) воды. Слои разделялись. Оставшийся этилацетат промывался раствором 2н НСl (5,6 галлонов (21,2 л) концентрированной HCI плюс 28,4 галлона (107,5 л) воды). Оставшийся этилацетат выпаривался под вакуумом с получением сырого результирующего амида в виде масла, 29,296 кг (85,9% теории).
'ПМР (СDСl3, 400 МГц) δ 7,18 (м., 2), 6,83 (м., 2), 4,41 (м., 1), 4,31 (м., 3), 3,76 (д, 3), 3,43 (м., 1), 3,25 (м., 1), 3,13 (т, 1), 2,00 (шир. с, 1), 1,80 - 1,26 (м. , 8), 0,87 (т, 3). ИК (без растворителя) 3456, 1739, 1654, 1513 cm-1. 13CMP (CDCl3, 100 мГц) δ 163,5, 162,1, 159,7, 159,0, 129,6, 129,2, 128,0, 127,1, 114,2, 114,1, 72,6, 72,5, 62,1, 55,3, 50,9, 47,0, 46,3, 43,6, 33,5, 33,4, 30,3, 30,2, 24,3, 22,9, 14,0, 9,9, GC масс-спектр: m/e, 337 (М+).
ПРИМЕР 4
Этиловый эфир N-(4-метоксибензил)-N-(4-оксогексил) оксаминовой кислоты (5.0.0)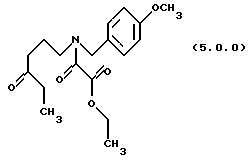
Бромистый калий (593 г, 5 молей) растворялся в воде (5 галлонов, 18.9 л) в 100 галлонном резервуаре. Добавлялся раствор оксаламидного спирта (33,62 кг, 99,6 молей) в хлористом метилене (34 галлона, 128,7 л). 2,2,6,6-тетраметил-1-пиперидинилокси (TEMPO), свободнорадикальный катализатор (150 г) добавлялся, и реакция охлаждалась до 0-5oС. Свежий раствор гипохлорита натрия (полученный из гипохлорита кальция (12,11 кг) и карбоната натрия (17,96 кг) в воде (100 галлонов, 378.5 л), доведенный до рН 9,5 гидрокарбонатом натрия (1,7 кг) и отфильтрованный для удаления карбоната кальция), добавлялся медленно с сохранением температуры при 10-15oС. После окончания реакции, слои разделялись, и водный слой экстрагировался 8 галлонами добавленного хлористого метилена. Объединенные органические слои промывались раствором, образованным концентрированной HCI (5,4 л) и йодистым калием (331 г) в воде (3,84 галлона, 14,5 л). Органический слой затем промывался раствором тиосульфата натрия (1197 г) в воде (5,3 галлона, 20 л). Хлористый метилен промывался 10 галлонами (37,85 л) воды и затем упаривался без вакуума до масла. Масло далее упаривалось после перенесения в 50 л реактор. Получался выход 33,407 кг продукта, но этот материал содержал 15 вес % хлористого метилена (по ЯМР). Исправленный выход был 28,396 кг (85.0% от теоретического).
'ПМР (СDСl3, 300 МГц) δ 7,18 (дд, 2), 6.82 (dd, 2), 4,49 (с, 1), 4,27 (м. , 3), 3,74 (д, 3), 3,22 (т, 1), 3,10 (т, 1), 2,34 (м., 4), 1,77 (м., 2), 1,29 (м. , 3), 0,98 (т, 3). 13СМР (СDСl3, 100 МГц) δ 163,2, 163,1, 162,3, 159,5, 159,2, 145,6, 129,7, 129,2, 127,96, 127,1, 114,2, 114,1, 112,1, 62,1, 55,2, 50,6, 46,1, 46,1, 42,7, 39,0, 38,1, 35,8, 21,5, 20,6, 13,9, 7,7. GC масс-спектр: m/e, 335(M+).
ПРИМЕР 5 3-гидрокси-1-(4-метоксибензил)-4-пропионил-5,6-дигидро-1H-пиридин-2-он (6.0.0)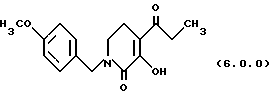
Оксаламидкетон (28,296 кг, 84,4 моля) растворялся в сухом тетрагидрофуране (28 галлонов, 106 л) в чистом и сухом 100 галлонном резервуаре. Этот раствор добавлялся к раствору трет-бутилата калия (10,392 кг) в тетрагидрофуране (42 галлона, 159 л) в 300 галлонном резервуаре на протяжении 30 минутного периода при сохранении температуры <35oС. После 1 часа при 20-25oС реакция завершалась по HPLC. Вода (98 галлонов, 371 л) добавлялась в реакцию, за которой следовало добавление изо-пропилового эфира (24 галлона, 90,8 л). Слои разделялись, и водный, содержащий продукт в виде его калиевой соли, промывался второй раз изо-пропиловым эфиром. Водный слой выпаривался частично в вакууме для удаления любого оставшегося THF и подкислялся до рН 2,1 добавлением 6н HCI (4 галлона, 15,1 л). Полученная суспензия фильтровалась, и твердые вещества промывались водой. Продукт высушивался на воздухе при 50oС, давая 17,9 кг продукта (73%); т. пл. 102-103oС.
'ПМР (CDCl3, 300 МГц) δ 7,20 (д, 2), 6,86 (д, 2), 4,60 (с, 2), 3,70 (с, 3), 3,33 (т, 2), 2,69 (к, 2), 2,56 (т, 2), 1,13 (т, 3).
ПРИМЕР 6
3-метокси-1-(4-метоксибензил)-4-пропионил-5,6-дигидро-lH-пиридин-2-он (7.0.0)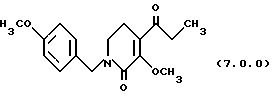
3-гидрокси-1-(4-метоксибензил)-4-пропионил-5,6-дигидро-1Н-пиридин-2-он (17,35 кг, 60 молей) и карбонат цезия (22,126 кг, 67.9 молей) добавлялись к сухому диметилформамиду (24 галлона, 90,8 л) в чистом, сухом 100 галлонном резервуаре. Суспензия перемешивалась полчаса для обеспечения диспергирования. Диметилсульфат (8,552 кг, 67,8 молей) добавлялся в чистом виде в течение 30 минут при сохранении температуры 20-25oС. Когда загрузка завершалась, воронка ополаскивалась в резервуар дополнительным DMF (500 мл). Реакцию перемешивали при 20-25o С в течение 16 часов. Реакцию разбавляли этилацетатом (108 галлонов, 408,8 л) и промывали водой (4х22 галлона (83,3 л)). Этилацетатный раствор промывался раствором, приготовленным из 6.94 литров 50% гидроксида натрия в 22 галлонах (83,3 л) воды, затем следовала промывка раствором, образованным 6,94 литрами концентрированной HCI в 22 галлонах (83,3л) воды. Органический раствор сушился промыванием рассолом (14 галлонов, 53 л). Этилацетат выпаривался под вакуумом до масла, которое было подходящим для применения в следующей стадии. Оцененный выход на основе ЯМР анализа остаточного растворителя был 89%. Небольшой образец выделялся для определения характеристик.
'ПНР (CDCl3, 300 МГц) δ 7,14 (д, 2), 6,78 (д, 2), 4,51 (с, 2), 3,88 (с, 3), 3,71 (с, 3), 3,2 (т, 2), 2,81 (к, 2), 2,42 (т, 2), 1,02 (т, 3). 13СМР (CDCl3, 100 мГц) 201,8, 159,1, 145,6, 129,3, 128,7, 126,5, 114,1, 60,2, 55,2, 49,6, 43,8, 37,0, 22,8, 8,1. GC масс-спектр: m/e, 303 (М+).
ПРИМЕР 7
Дигидрохлорид циклопентилгидразина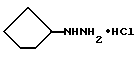
Циклопентанол (6,127 кг, 71.1 моля) и трифенилфосфин (18,667 кг, 71,25 моля) растворялись в тетрагидрофуране (40 галлонов) в чистом и сухом, заполненном азотом 100 галлонном резервуаре, и реакционная смесь охлаждалась до 5oС. Раствор ди-трет-бутилазодикарбоксилата (14,9 кг, 64,7 моля) в тетрагидрофуране (36 л) добавлялся на протяжении примерно 2 часов при сохранении температуры <6oС. Реакцию перемешивали в течение 5 часов, в то время как температуре давали медленно увеличиться до 20-25oС. 6н HCl (26,5 л) добавлялась к реакциионной смеси при 20oС. Реакцию перемешивали 24 часа при 20-25oС, при которой исходный материал реагировал. Вода (10 галлонов, 37,85 л) добавлялась, и тетрагидрофуран удалялся вакуумной перегонкой. Во время концентрирования оксид трифенилфосфина осаждался, и добавлялись дополнительные 20 галлонов (75,7 л) воды. Реакция охлаждалась, и хлористый метилен (30 галлонов, 113,6 л) добавлялся. Слои разделялись, и водный экстрагировался еще дважды хлористым метиленом (10 галлонов, 37,85 л). Водный слой перегонялся для удаления воды. По мере того как объем уменьшался, изопропиловый спирт (3х20 галлонов (75,7 л)) добавлялся для азеотропной отгонки остаточной воды. Полученная суспензия фильтровалась, и твердые вещества высушивались в вакуумном сушильном шкафу, давая 7,682 кг (68.6% от теоретического) на множественных партиях. Этот материал был охарактеризован, как являющийся дихлоридной солью; т.пл. 189-194o С.
'ПМР (ДМСО-d6, 300 МГц) δ 3,48 (м., 1), 1,79 (м., 2), 1,64 (м., 4), 1,49 (м., 2).
ПРИМЕР 8
1-циклопентил-3-этил-6-(4-метоксибензил)-1,4,5,6-тетрагидропиразоло[3,4-с]пиридин-7-он (8.0.0)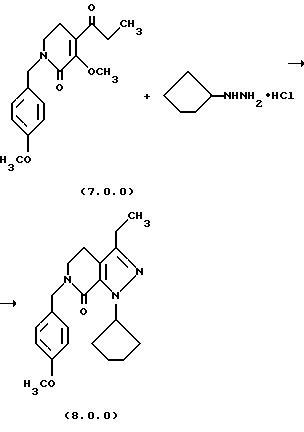
3-метокси-1-(4-метоксибензил)-4-пропионил-5,6-дигидро-1Н-пиридин-2-он (14,471 кг, 47,76 молей) растворялся в тетрагидрофуране (10,5 галлонов, 39,7 л) в чистом и сухом 100 галлонном резервуаре. Дигидрохлорид циклопентилгидразина (7,664 кг, 44,3 моля) добавлялся, и реакционная смесь нагревалась медленно до ~ 88oС, в то время как азот продувался через реакционную смесь для удаления метанола, THF и HCI. Реакция контролировалась HPLC, пока превращение не завершалось, которое требовало нагревания в течение ночи в большинстве случаев. Продукт реакции в резервуаре был густым темным маслом. Образец 1-циклопентил-3-этил-6-(4-метоксибензил)-1,4,5,6-тетрагидропиразоло[3,4-с]пиридин-7-она выделялся для определения характеристик.
'ПМР (СDСl3, 300 МГц) δ 7,23 (д, 2), 6,85 (д, 2), 5,72 (м., 1), 4,62 (с, 2), 3,77 (с, 3), 3,44 (т, 2). 2,62 (тик, 4), 2,06 (м., 4), 1,89 (м., 2), 1,67 (м. , 2), 1,17 (т. 3). 13СМР (СDСl3, 100 МГц) δ 159,5, 159,0, 148,0, 145,6, 129,6, 129,3, 118,5, 114,0, 112,9, 60,4, 55,2, 48,6, 47,2, 32,7, 24,4, 20,2, 19.9, 13,8. GC масс-спектр: m/e, 353 (M+).
Продукт этой стадии мог использоваться непосредственно в следующей стадии или очищаться как соль п-толуолсульфоновой кислоты или бензолсульфоновой кислоты, как описано.
ПРИМЕР 9
Получение п-толуолсульфоновой кислоты или бензолсульфоновой кислоты солей 1-циклопентил-3-этил-6-(4-метоксибензил)-1,4, 5,6-тетрагидропиразоло[3,4-с]пиридин-7-она
Сырой лактам (1 г, 2.83 ммоля) растворялся в этилацетате (5 мл) и обрабатывался раствором безводной п-толуолсульфоновой кислоты (0.487 г, 2,83 ммоля) в этилацетате (2 мл). Соль кристаллизовалась из смеси, которая затем охлаждалась и фильтровалась, давая 1,21 г чистой тозилатной соли как белого твердого вещества с 81% выходом; т.пл. 110-113.8oС.
Анализ. Bычислено для C28H35N3O5S: С, 63,98; Н, 6,71; N, 7,99; S, 6,10. Найдено: С, 63,83; Н, 6,69; N, 8,02; S, 6,14.
Соль бензолсульфоновой кислоты образовывалась тем же самым способом; т. пл. 126,6-131,4'С.
Анализ. Вычислено для C27H33N3O5S: С, 63,38; Н, 6,50; N, 8,21. Найдено: С, 63,09; Н, 6,48; N, 8,21.
Любая из этих кристаллических солей может быть применена в реакции снятия защитной группы с трифторуксусной кислотой и метансульфоновой кислотой, описанной в следующем примере.
ПРИМЕР 10
1-циклопентил-3-этил-1,4,5,6-тетрагидропиразоло[3,4-с] пиридин-7-он (9.0.0)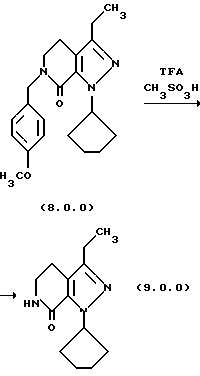
Реакционная смесь из предыдущего примера охлаждалась до 55oС и к ней медленно добавлялась трифторуксусная кислота (87,3 кг, 764 моля) при сохранении температуры между 50 - 60oС. Первая 1/3 загрузки была экзотермической и требовала внешнего охлаждения. Метансульфоновая кислота (6342 мл, 97,7 моля) добавлялась, и реакция нагревалась до ~70oС в течение двух часов. Реакция охлаждалась до 20-25oС, и хлористый метилен (17 галлонов, 64 л) добавлялся, с последующим медленным добавлением воды (17 галлонов, 64 л). Слои разделялись, и водный слой разбавлялся далее водой (6 галлонов, 22,7 л) и затем реэкстрагировался хлористым метиленом (6 галлонов, 22,7 л). Объединенные метиленхлоридные слои смешивались с водой (29 галлонов, 110 л) и затем доводились до рН~7,0 добавлением насыщенного гидрокарбоната натрия (приблизительно 45 галлонов, 170 л). Слои разделялись, и хлористый метилен отгонялся под атмосферным давлением до примерно 9 галлонов (35 л). Этилацетат (13 галлонов, 49 л) добавлялся, и реакционная смесь перегонялась до примерно 9 галлонов (35 л). Полученная суспензия охлаждалась н гранулировалась. Твердые вещества собирались фильтрованием, промывались этилацетатом и высушивались под вакуумом при 40o С под полным вакуумом. Выход был 7,91 кг, 71,2%; т.пл. 152-153oС.
'ПМР (СDСl3, 300 МГц) δ 5,61 (м., 2), 3,51 (дт, 2), 2,72 (т, 2), 2,62 (к, 2), 2,08 (м., 4), 1,90 (м., 2), 1,65 (м., 2), 1,40 (т, 3).
ПРИМЕР 11
1-циклопентил-7-этокси-3-этил-4,5-дигидро-1Н-пиразоло[3,4-с] пиридин (10.0.0)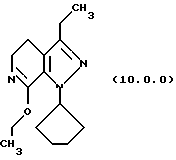
Раствор тетрафторбората триэтилоксония (3,371 кг, 17,74 молей) в хлористом метилене (10,8 л) медленно добавлялся к суспензии 1-циклопентил-3-этил-1,4,5,6-тетрагидропиразоло[3,4-с] пиридин-7-она (3,6 кг, 15,43 молей) в хлористом метилене (7,2 л) на протяжении периода примерно 40 минут. Раствору затем давали реагировать в течение примерно 21 часа при 18-22oС. После завершения реакции органический раствор промывался водным 10% карбонатом натрия (36 л) и выпаривался до масла, которое использовалось непосредственно в следующей стадии. Выход для этой стадии был 92.9%.
'ПМР (СDСl3, 300 МГц) δ 5,14 (квинтет, 1), 4,25 (к, 2), 3,62 (т, 2), 2,58 (м. , 4), 2,07 (м., 4), 1,88 (м., 2), 1,61 (м., 2), 1,35 (т, 3), 1,19 (т, 3). GC масс-спектр: m/e, 261 (М+).
ПРИМЕР 12
8-циклопентил-6-этил-3-тиофен-2-ил-5,8-дигидро-4Н-1,2,3а, 7,8-пентааза-ас-индацен
Раствор 1-циклопентил-7-этокси-3-этил-4,5-дигидро-1Н-пиразоло[3,4-с] пиридина (3,739 кг, 14,3 моля) и гидразида 2-тиофенкарбоновой кислоты (2,237 кг, 15,8 моля) нагревался в растворе 1-бутанола (37 л) до ~90oC в 50 галлонном резервуаре в течение 48 часов. При этой температуре некоторое количество 1-бутанола отгонялось для удаления воды азеотропно. Реакция концентрировалась до малого объема, и 4 галлона хлористого метилена (4 галлона, 15 л) добавлялись. Органические вещества промывались дважды 1н HCI (8 галлонов, 30,3 л) и концентрировались перегонкой до малого объема.
Иэопропиловый спирт (16 л) добавлялся к концентрату, и полученная суспензия охлаждалась и гранулировалась. Продукт собирался фильтрованием и высушивался в вакуумном сушильном шкафу при 40oC. Выход был 3,25 кг (67%) белого твердого вещества; т.пл. 126oC.
'ПМР (СDСl3, 300 МГц) δ 7,51 (м., 2), 7,28 (с, 1), 7,20 (дд, 1), 5,61 (м. , 1), 4,35 (т, 2), 3,00 (т, 2), 2,70 (к, 2), 2,18 (м., 4), 1,97 (м., 2), 1,62 (м., 2), 1,29 (т, 3).
Анализ. Вычислено для C18H21N5S: С, 63,69; Н, 6,24; N, 20,63. Найдено: С, 63,82; Н, 6,30; N, 20,77.
ПРИМЕР 13
3-трет-бутил-8-циклопентил-6-этил-5,8-дигидро-4Н-1,2,3а, 7,8-пентааза-ас-индацен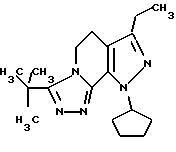
Раствор 1-циклопентил-7-этокси-3-этил-4,5-дигидро-1Н-пиразоло[3,4-с] пиридина (5 г, 19,4 ммоля) и гидразида 2,2-диметилпропанкарбоновой кислоты (2,48 г, 21,4 ммолл) нагревался в растворе 1-бутанола (30 мл) с обратным холодильником в течение 48 часов. Растворитель выпаривался при уменьшенном давлении, и остаточное масло растворялось в хлористом метилене. Органический раствор промывался 1н HCI (50 л) и высушивался над хлористым кальцием. Раствор фильтровался, выпаривался в вакууме и сырой продукт перекристаллизовывался из изопропилового спирта. Выход был 2,76 г (45%) не совсем белого твердого вещества; т.пл. 150-151oС.
'ПМР (СDСl3, 300 МГц) δ 5,50 (м.,1), 4,49 (т, 2), 3,15 (т, 2), 2,68 (к, 2), 2,13 (м., 4), 1,93 (м., 2), 1,70 (м., 2), 1,60 (с, 9), 1,24 (т, 3).
Анализ. Вычислено для C18H27N5: С, 68,97; Н, 8,68; N, 22,34. Найдено: С, 69,05; Н, 8,89; N, 22,46.

Описывается улучшенный способ получения (и его варианты) производных 8-циклопентил-6-этил-3-(замещенного)-5,8-дигидро-4Н-1,2,3а, 7,8-пентааза-ас-индацена формулы (1.0.0) и его фармацевтически приемлемых солей, R1 представляет (C1-С6) алкил или насыщенную или ненасыщенную (С4-С7) гетероциклилгруппу, содержащую один атом S, причем вышеуказанная алкильная и гетероциклическая группа необязательно является замещенной от 1 до 3 заместителями, независимо выбранными из (C1-C2) алкила, трифторметила и галогена, отличающийся тем, что исходя из γ-капролактона и n-метоксибензиламина многостадийным синтезом через новые промежуточные соединения формул (8.1.0), (8.1.1) и (10.0.0). Целевой продукт, полученный описываемым способом является ингибитором типа IV (PDE4) фосфодиэстеразы и производства фактора некроза опухоли (TNF) и полезен для лечения астмы, бронхита, аллергического ринита, псориаза, дерматита и других воспалительных, аллергических и иммунологических болезней. 5 с. и 2 з.п.ф-лы, 1 табл.
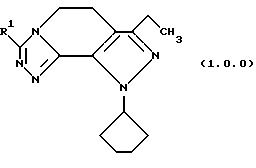
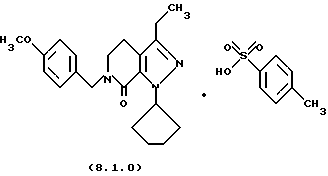
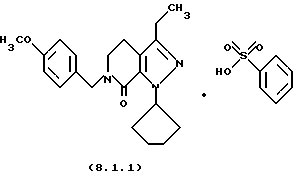
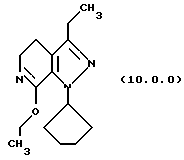
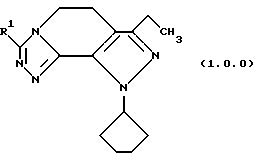
и его фармацевтически приемлемых солей, где R1 представляет (С1-С6)алкил или насыщенную или ненасыщенную (С4-С7)гетероциклилгруппу, содержащую один атом S, причем вышеуказанная алкильная и гетероциклическая группа необязательно является замещенной от 1 до 3 заместителями, независимо выбранными из (C1-C2)алкила, трифторметила и галогена, отличающийся тем, что (а) смесь γ-капролактона и п-метоксибензиламина без растворителя нагревают до температуры в интервале от 80 до 85oС в течение 16 ч с получением производного амида формулы (2.0.0)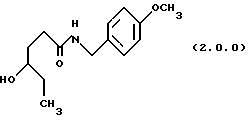
производное амида формулы (2.0.0) подвергают восстановлению с использованием боргидрида натрия в тетрагидрофуране в присутствии уксусной кислоты или ее раствора в тетрагидрофуране, причем боргидрид натрия добавляют к тетрагидрофурану, а амид формулы (2.0.0) добавляют в виде твердого вещества в полученную реакционную смесь, которую затем охлаждают и смешивают с уксусной кислотой или ее раствором, полученную смесь нагревают до температуры слабого флегмообразования в интервале 60-70oС в течение 16 ч; непрореагировавший амид удаляют экстракцией этилацетатом после добавления 1н. НСl, и после доведения рН указанной реакционной смеси до 11 экстрагируют аминоспирт формулы (3.0.0) этилацетатом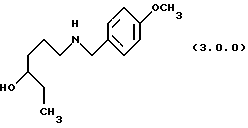
(с) полученный аминоспирт формулы (3.0.0) ацилируют путем постепенного добавления в течение 20-30 мин этилового эфира хлорангидрида щавелевой кислоты в этилацетате в соответствии с условиями реакции Шоттена-Баумана в присутствии водного раствора гидрокарбоната натрия при температуре от 0 до 5oС, с последующим выдерживанием реакционной смеси при указанной температуре в течение 1-2 ч; после чего указанную реакционную смесь перемешивают при температуре от 20 до 25oС в течение 16 ч, с получением этилового эфира оксаминовой кислоты формулы (4.0.0)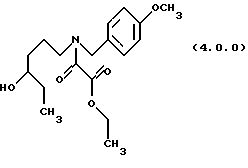
(d) полученный этиловый эфир оксаминовой кислоты формулы (4.0.0) подвергают окислению в хлористом метилене с использованием свежеприготовленного водного раствора гипохлорита натрия, в присутствии катализатора 2,2,6,6-тетраметил-1-пиперидинилокси, причем раствор гипохлорита натрия получают путем растворения гипохлорита кальция и карбоната натрия в воде и доведения рН полученного раствора до 9,5 гидрокарбонатом натрия, с последующим отделением побочного продукта карбоната кальция фильтрованием, а катализатор сначала добавляют к бромистому калию, растворенному в воде, полученную смесь объединяют с соединением формулы (4.0.0) в хлористом метилене, реакционную смесь охлаждают до температуры от 0 до 5oС, после чего медленно добавляют гипохлорит натрия, поддерживая температуру от 10 до 15oС, с получением оксаламидкетона формулы (5.0.0)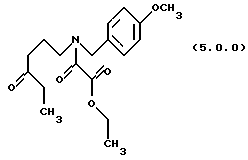
(е) оксаламидкетон формулы (5.0.0) подвергают циклизации в условиях реакции конденсации Дикмана, где реакция проводится в присутствии относительно сильного основания, состоящего из трет-бутилата калия в тетрагидрофуране, ди-изо-пропиловом простом эфире, метил-трет-бутиловом простом эфире или толуоле, причем основание добавляют постепенно на протяжении периода свыше 30 мин, поддерживая температуру реакционной смеси ниже 35oС, с последующим выдерживанием реакционной смеси при температуре 20-25oС в течение 1 ч и получением пиридинона формулы (6.0.0)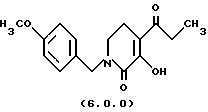
(f) пиридинон формулы (6.0.0) подвергают O-метилированию в среде диметилформамида в качестве растворителя в присутствии карбоната цезия, постепенным добавлением диметилсульфата на протяжении периода свыше 30 мин, при поддержании температуры реакционной смеси от 20 до 25oС, с последующим выдерживанием реакционной смеси при указанной температуре и перемешивании в течение 16 ч и получением 3-метоксипиридинона формулы (7.0.0)
(q) 3-метоксипиридинон формулы (7.0.0) подвергают обработке дигидрохлоридом циклопентилгидразина в среде тетрагидрофурана при нагревании реакционной смеси до 88oС в течение 12 ч и продувании ее азотом, с последующим получением пиразолопиридинона формулы (8.0.0)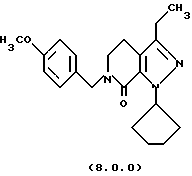
который, в случае необходимости, может быть очищен и выделен в виде соли п-толуолсульфоновой кислоты или бензолсульфоновой кислоты растворением указанного соединения формулы (8.0.0) в этилацетате и последующей обработкой его безводной n-толуолсульфоновой кислотой или безводной бензолсульфоновой кислотой, растворенными в этилацетате, с последующей кристаллизацией указанных солей из охлажденной реакционной смеси; (h) затем осуществляют снятие защитной n-метоксибензильной группы с пиразолопиридинона формулы (8.0.0) путем медленного добавления трифторуксусной кислоты при температуре 55oС, поддерживаемой за счет внешнего охлаждения, с последующим добавлением метансульфоновой кислоты в реакционную смесь при температуре 70oС, выдерживанием при этой температуре реакционной смеси в течение 2 ч и получением лактама формулы (9.0.0) при охлаждении реакционной смеси до температуры от 20 до 25oС: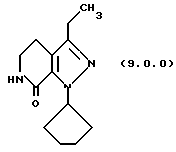
(i) суспензию лактама формулы (9.0.0) в хлористом метилене подвергают этерификации путем медленного прибавления раствора тетрафторбората триэтилоксония в хлористом метилене на протяжении периода свыше 40 мин, с последующим выдерживанием реакционной смеси при температуре от 18 до 22oС в течение 21 ч, и получением иминоэфира (имидата) формулы (10.0.0)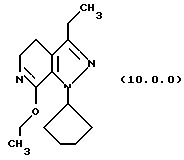
(j) раствор иминоэфира (имидата) формулы (10.0.0) обрабатывают в 1-бутаноле гидразидом карбоновой кислоты формулы (11.0.0)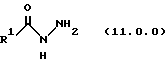
где R1 имеет вышеуказанные значения;
и реакционную смесь нагревают при температуре 90oС в течение периода свыше 48 ч, с последующим выделением целевого продукта.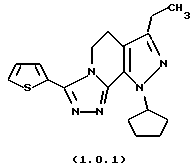
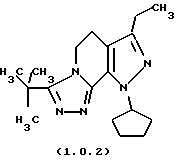
3. Способ получения производного 8-циклопентил-6-этил-3-(замещенного)-5,8-дигидро-4Н-1,2,3а, 7,8-пентааза-ас-индацена формулы (1.0.0)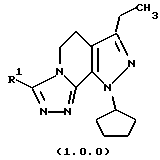
и его фармацевтически приемлемых солей, где R1 является таким, как определено в п. 1, отличающийся тем, что (а) пактам формулы (9.0.0)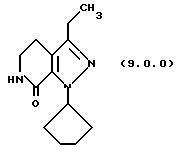
подвергают этерификации в условиях стадии (i) п. 1 с получением иминоэфира (имидата)формулы (10.0.0)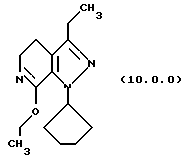
(b) иминоэфир (имидат) формулы (10.0.0) подвергают обработке гидразидом карбоновой кислоты формулы (11.0.0)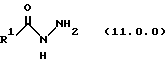
где R1 является таким, как определено в п. 1,
в условиях стадии (j) п. 1, с последующим выделением целевого продукта.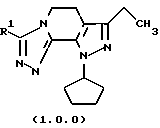
и его фармацевтически приемлемых солей, где R1 является таким, как определено в п. 1, отличающийся тем, что иминоэфир (имидат) формулы (10.0.0)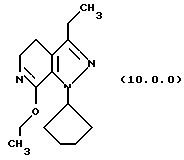
подвергают обработке гидразидом карбоновой кислоты формулы (11.0.0)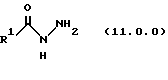
где R1 является таким, как определено в п. 1,
в условиях стадии (j) п. 1, с последующим выделением целевого продукта.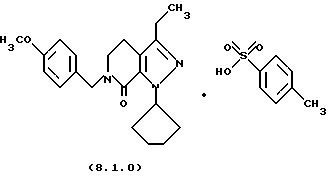
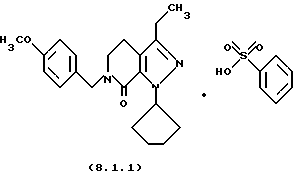
6. Иминоэфир (имидат) формулы (10.0.0)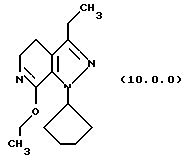
и его фармацевтически приемлемые соли.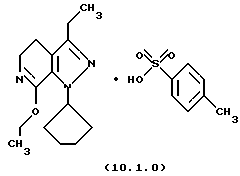
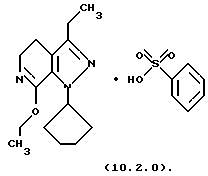
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| 0 |
|
SU343832A1 | |
| 3-Трифторметил-7-метилимидазо- @ 4,5-с @ -1,2,4-триазоло @ 4,3-а @ пиридин,обладающий спазмолитическим действием | 1982 |
|
SU1094303A1 |

