Данное изобретение относится к хиназолин-производным и к способам их применения, особенно в качестве противораковых средств, для млекопитающих.
Большое количество способов лечения рака, используемых в настоящее время, основаны на применении соединений, которые ингибируют синтез ДНК. Такие соединения являются токсичными к клеткам вообще, но их токсическое действие, на быстро делящиеся опухолевые клетки, может быть полезным. Однако для повышения селективности действия по отношению к клеткам раковой опухоли разрабатываются альтернативные противораковые средства, механизмом действия которых не является прямое ингибирование синтеза ДНК.
Известно, что клетка может становиться клеткой раковой опухоли в результате трансформации части ее ДНК в онкоген (т.е. в ген, который при активации приводит к образованию клеток злокачественного образования). Большое количество онкоген-кодированных белков, которые являются аберрантными тирозинкиназами, обладают способностью вызывать такую трансформацию клеток. Кроме того, к пролиферативным заболеваниям, а в некоторых случаях к малигнантному фенотипу, также может приводить гиперэкспрессия нормальной прото-онкогенной тирозинкиназы.
Рецепторные тирозинкиназы представляют собой большие ферменты, которые соединяют клеточные мембраны, обладают внеклеточной областью, связывающей факторы роста, такие как эпидермальный фактор роста, трансмембранной областью и внутриклеточной частью, которая функционирует как киназа по отношению к фосфорилированным специфическим тирозиновым остаткам в белках и, следовательно, влияет на клеточную пролиферацию. Известно, что такие киназы часто являются аберрантно выраженными в обычных раковых заболеваниях человека, таких как рак молочной железы, рак желудочно-кишечного тракта, например толстой кишки, прямой кишки, желудка, лейкемия, рак яичников, бронхов, поджелудочной железы. Было показано также, что рецептор эпидермального фактора роста (EGFR), который обладает тирозинкиназной активностью, подвергается мутации и/или является гипервыраженным во многих раковых заболевания человека, таких как опухоли головного мозга, легких, клеток сквамозного эпителия, мочевого пузыря, желудка, молочной железы, головы и шеи, пищевода, женских половых органов и щитовидной железы.
Далее было установлено, что ингибиторы рецепторных тирозинкиназ полезны в качестве селективных ингибиторов роста раковых клеток млекопитающихся. Например, эрбстатин (erbstatin), ингибитор тирозинкиназы, у атимной голой мыши селективно ослабляет рост трансплантированной карциномы молочной железы, которую представляет тирозинкиназа рецептора эпидермального фактора роста (EGFR), но не оказывает воздействия на рост другой карциномы, которую не передает EGF рецептор.
Было показано, что другие соединения, такие как производные стирола, также обладают способностью ингибировать тирозинкиназу. Ранее, в трех опубликованных Европейских заявках: EP 0566226 A1, EP 0602851 A1 и EP 0520722 A1 - было показано, что некоторые хиназолин-производные обладают противораковой активностью, которая обусловлена их способностью ингибиторовать тирозинкиназы. В опубликованной Заявке PCT WO 92/20642 моно- и бициклические арил- и гетероарил-производные описывается как ингибиторы тирозинкиназы.
В патенте США 4012513 впервые описываются некоторые производные 1-(гетероциклил)-индол-3-илуксусной кислоты, которые обладают противовоспалительной, обезболивающей и жаропонижающей активностью.
Хотя описанные выше соединения, обладающие противораковой активностью, вносят значительный вклад в решение проблемы лечения раковых заболевания, исследования по усовершенствованию противораковых лекарственных средств продолжаются.
Краткое описание изобретения
Данное изобретение относится к хиназолин-производным, в частности 4-аминохиназолинам, которые полезны в качестве противораковых средств. Соединения данного изобретения имеют строение, представленное формулой I: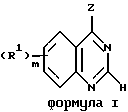
и их фармацевтически приемлемые соли и стереоизомеры имеют строение, где
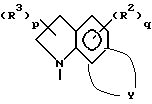
Z представляет собой A, B и C; при этом A представляет:
или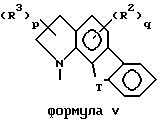
где X представляет собой метилен, тио-группу, - N(H)- или окси-группу;
Y - фрагмент, который вместе с частью бензольного кольца образует 5- или 6-членный ароматический или частично насыщенный цикл, который может содержать атом кислорода или серы;
T - метилен, -N(H)-, тио или окси;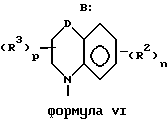
где D может представлять собой насыщенный углерод, окси или тио;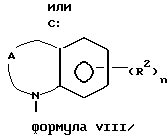
где A вместе с атомом азота и фрагментом бензольного кольца образует 7-9-членный моно-ненасыщенный моно-аза цикл;
где R1 в каждом случае независимо представляет собой
а. Трифторметил, галоген, нитро-, гидроксильную, амино-, циано-группу, (C1-C4)алкил, (C1-C4)алкокси, (C1-C4)алкоксикарбонил, (C1-C4)алканоилокси,
(C1-C4)алканоиламино, карбоксильную группу, фенокси, бензоилокси, карбамоил, моно- N или ди- N,N- ди-(C1-C4)алкилкарбамоил, моно- N или ди- N, N-(C1-C4)алкил-амино, моно- N или ди- N, N-(гидрокси(C2-C4)алкил)амино, моно- N- или ди- N,N-((C1-C4)алкокси(C2-C4)алкил)амино, анилино, пирролидин-1-ил, пиперидин-1-ил, морфолино, пиперазин-1-ил, 4-(C1-C4)алкилпиперазин-1-ил, (C1-C4) алкилтио, фенилтио или эти же группы, содержащие в качестве заместителя (C1-C4)алкил;
b. Гидрокси(C2 -C4)алкокси(C1-C4)алкил, (C1-C4) алкокси(C2-C4)алкокси-(C1-C4)алкил, гидрокси(C2-C4)алкилтио(C1-C4) алкил, (C1-C4)алкокси(C2-C4) алкилтио(C1-C4)алкил, гидроксиамино, бензоиламино, моно- N или ди- N,N-(C1-C4)алкилкарбамоилметиламино, карбамоилметиламино, (C1-C4)алкоксикарбониламино, (C1-C4) алканоиламино, карбоксиметиламино, (C1 -C4)алкоксикарбонилметиламино, (C1 -C4)алкоксиамино, (C2-C4)алканоилоксиамино, фенил (C1-C4)алкиламино, (C1-C4) алкилсульфониламино, бензолсульфонамидо, 3-фенилуреидо, 2-оксопирролидин-1-ил, 2,5-диоксопирролидин-1-ил, уреидо, (C1 -C4) алкокси (C1-C4)алкилкарбониламино, (C1-C4) алкилсульфинил, (C1 -C4)алкилсульфонил, (C1-C4) алкокси (C2 -C4) алкилтио, моно-, ди- или трифторметилокси, (C1 -C4)алкилендиокси, бензилокси, азидо, гуанидино, аминокарбонил, моно- N или ди- N,N-(C1-C4) алкиламинокарбонил, фенил (C1-C4) алкокси, карбонилметокси, (C1-C4)алкоксикарбонилметокси, карбамоилметокси, моно- N- или ди- N,N-(C1-C4) алкилкарбамоилметокси, моно- N или ди- N,N-(гидрокси(C2 -C4)алкил)карбоксамидо, моно- N- или ди- N,N-((C1 -C4)алкокси(C2-C4)алкил)карбоксамидо или бис (C1-C4)алкансульфонил)амидо; или
с. (C2 -C4) алкокси, (C2-C4) алкилтио, (C2 -C4)алканоилокси, (C2-C4)алкиламино, (C1 -C4)алкил(C1-C4)алкилендиокси или (C2 -C4) алканоиламино, каждая из перечисленных групп содержит в качестве заместителя амино-группу, галоген, гидроксильную группу, (C2-C4)алканоилокси, (C1-C4) алкокси-группу, моно- N- или ди-N,N-(C2-C4) алкиламино, моно- N или ди-N,N-(гидрокси(C1-C4)алкил)амино, моно- N- или ди-N,N-((C1 -C4)алкокси(C2-C4)алкил)амино, (C1 -C4)алканоиламино, фенокси, анилино, имидазол-1-ил, фенилтио, пиперидино, морфолино, пиперазин-1-ил, 4-(C1-C4)алкил-пиперазин-1-ил, карбокси, (C1 -C4)алкоксикарбонил, карбамоил, моно- N или ди-N,N -(C1-C4)алкилкарбамоил, карбоксамидо, моно- N- или ди- N, N-(C1-C4)алкилкарбоксамидо или моно- N- или ди-N, N-(гидрокси(C2-C4)алкил)карбоксамидо;
где любой фенил в R1-заместителе необязательно содержит один или два заместителя из группы, включающей галоген, нитро-группу, трифторметил, гидроксильную группу, (C1-C4)алкокси-группу или (C1-C4)алкил и указанная (C1-C4)алкилендиокси-группа обоими концами присоединяется к хиназолиновому фрагменту;
R2 в каждом случае независимо представляет собой моно, ди- или трифторметил, галоген, нитро, гидроксильную группу, амино, азидо, изотиоциано-группу, (C1-C4)алкил, фенил, тиенил, (C1-C4) алкокси, бензилокси, фенокси, (C2-C6)алкенил, (C2-C6)алкинил, (C1-C4)алкилендиокси, цианогруппу, бензоиламино, трифторметил-карбониламино-группу, (C1-C4) алканоиламино, (C1-C4)алканоил, N-моно- или N, N-ди(C1-C4)алкиламино, (C1 -C4)алкилсульфониламино, трифторметилсульфониламино, (C1-C4)алкилтио, (C1-C4)алкилсульфинил или (C1-C4)алкилсульфонил, пиррол-1-ил, пиперидин-1-ил или пирролидин-1-ил, где указанные фенил, бензоилокси, фенокси и бензоиламино-группы необязательно содержат один заместитель, выбранный из группы, включающей галоген, нитро-группу, трифторметил, гидроксильную группу или (C1-C4)алкил и указанная (C1-C4)алкилендиокси-группа присоединяется обоими концами к соседним атомам углерода на бензольном фрагменте;
R3 в каждом случае независимо представляет собой гидроксильную, амино-группу, N-моно- или N,N-ди(C1-C4)алкиламино, сульфо или (C1-C4)алкокси (при условии, что такие группы не соединены с атомом углерода цикла, который является соседним по отношению к атому кислорода, серы или -N-) или R3 в каждом случае независимо представляет собой карбокси, гидрокси(C1-C4) алкил, (C1 -C4)алкокси(C1-C4)алкил, амино(C1-C4)алкил, моно-N- или ди-N, N-(C1 -C4)алкиламино(C1-C4)алкил, морфолино(C1 -C4) алкил, 4-(C1-C4) алкилпиперазин-1-ил(C1-C4) алкил, карбокси (C1 -C4) алкил, (C1-C4)алкоксикарбонил, сульфо (C1-C4) алкил или (C1-C4) алкил;
где m принимает значения 0-3;
n принимает значения 0-4;
p принимает значения 0, 1 или 2; и
q принимает значения 0, 1 или 2;
при условии, что 4-(3,4-дигидро-2H-хинолин-1-ил)хиназолин не включен в указанный перечень.
Первую группу предпочтительных соединений формулы I составляют соединения, в которых
Z представляет собой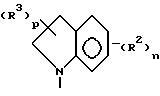
R1 в каждом случае независимо представляет собой гидроксильную группу, (C1-C4)алкокси, гидрокси(C2-C4)алкокси, амино(C2-C4)алкил, амино(C2-C4)алкокси, (C1-C4)алкокси(C2-C4)алкокси, (C1-C4)алкилендиокси, гидрокси(C1 -C4)алкил(C1-C4)алкилендиокси, (C1 -C4)алкокси(C1-C4)алкил(C1-C4) алкилендиокси, моно- N- или ди-N,N-(C1 -C4)алкиламино(C2-C4)алкокси, 3- или 4-(C1 -C4)алкокси-(2-гидрокси)-(C3-C4)алкокси, карбокси(C1-C4)алкокси, морфолино(C2-C4)алкокси, имидазол-1-ил(C2 -C4) алкокси, 4(C1-C4) алкилпиперазин-1-ил- (C2-C4)алкокси, (C1-C4)алкокси (C1-C4)алканоилокси, нитро, гидроксиламино, амино-группу, моно-N или ди-N, N-(C1-C4)алкиламино, (C1-C4)алканоиламино, гидрокси(C2 -C4)алкиламино, (C1-C4)алкокси(C2 -C4)алкиламино, (C1-C4)алкилсульфонамидо, морфолино, (C1-C4)алкилпиперазин-1-ил, бис(C1-C4)алкансульфонамидо, ди(C1-C4) алкиламино (C2-C4)алкиламино, (C1-C4) алкиламино(C2-C4)алкиламино, имидазол-1-ил, пиперидин-1-ил, пирролидин-1-ил, (C1-C4) алкокси (C1-C4)алкилкарбониламино, N-(C1-C4)алкил- N-(C1-C4)алканоиламино, карбоксильную группу, (C1-C4) алкоксикарбонил, (C1-C4) алкоксикарбонил(C1-C4)алкокси, амидо-группу, моно-N- или ди-N,N-(C1-C4)алкиламинокарбонил, моно- N или ди-N,N-(гидpoкcи(C2-C4)aлкил)aминoкapбoнил, (C1 -C4)алкил, гидрокси(C1-C4)алкил, моно- N- или ди-N,N-((C1-C4)алкокси (C1-C4)алкил)амино(C1-C4)алкил, моно- N или ди-N,N-(C1-C4)алкиламино(C1-C4)алкил, (C1-C4)алканоиламино(C1-C4)алкил,
(C1-C4)алкокси(C2-C4)алкокси(C1-C4) алкил, (C1-C4)алкилтио, (C1-C4)алкокси (C2-C4)алкилтио или гидрокси(C2-C4)алкилтио;
R2 в каждом случае независимо представляет собой нитро-группу, галоген, (C1-C4)алкил, пиррол-1-ил, гидроксильную, амино-группу, моно- N- или ди-N, N-(C1-C4) алкиламино, амино(C1-C4)алкил, азидо, этенил, этинил, (C1-C4)алкилендиокси, фенил или (C1-C4)алкилтио;
R3 в каждом случае независимо представляет собой гидрокси (C1-C4)алкил, (C1 -C4)алкил, амино(C1-C4)алкил, карбокси(C1-C4)алкил или моно-N- или ди- N,N-(C1-C4)алкиламино(C1-C4)алкил;
m принимает значения 0, 1 или 2;
p принимает значения 0 или 1; и
n принимает значения 0, 1, 2 или 3.
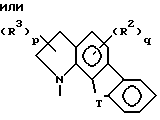
В пределах этой первой группы предпочтительных соединений формулы I первую группу особенно предпочтительных соединений составляют соединения, в которых
Z представляет собой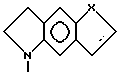
X представляет собой- N(H)-; и
R1 в каждом случае независимо замещен в положение 6 и/или 7.
Вторую группу особенно предпочтительных соединений в пределах указанной выше первой группы предпочтительных соединений формулы I составляют соединения, в которых
Z представляет собой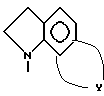
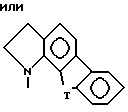
R1 в каждом случае независимо замещен в 6 или 7 положение.
Третью группу особенно предпочтительных соединений в пределах указанной выше группы предпочтительных соединений формулы I составляют соединения, в которых
Z представляет собой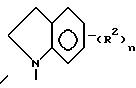
n принимает значения 1, 2 или 3;
m принимает значения 1 или 2;
R1 в каждом случае независимо замещен в положение 6 и/или 7 и представляет собой (C1-C4)алкил, (C1-C4)алкокси, гидрокси(C2-C4)алкокси, (C1-C4)алкокси(C2-C4)алкокси, карбокси(C1-C4)алкокси, (C1-C4)алкоксикарбонил(C1-C4)алкокси, имидозол-1-ил (C2-C4)алкокси, морфолино(C2-C4)алкокси, 4-(C1 -C4)алкилпиперазин-1-ил-(C1-C4)алкокси, (C1-C4)алкокси-2-гидрокси(C3-C4)алкокси, амино-группу, (C1-C4)алкиламино, ди-N,N-алкиламино, (C1-C4)алканоиламино, (C1-C4) алкилсульфониламидо, морфолино, (C1-C4)алкилпиперазин-1-ил, моно-N- или ди-N, -N (C1-C4)алкиламино(C2-C4)алкиламино; и
R2 в каждом случае независимо представляет собой 4-гидрокси, 4-амино, 5-фтор, 5-гидрокси, 5-амино, 6-галоген, 6-метил, 6-этенил, 6-этинил, 6-нитро или 7-метил.
Исключительно предпочтительными соединениями в пределах указанной выше группы особенно предпочтительных соединений являются соединения, в которых
R2 в каждом случае независимо представляет собой галоген, нитро-, гидроксильную группу или метил;
R1 представляет собой (C1-C4)алкокси или (C1-C4)алкил;
m равно 2; и
n принимает значения 1 или 2.
Другими исключительно предпочтительными соединениями в пределах указанной выше группы предпочтительных соединений являются соединения, в которых
а. m равно 2;
R1 представляет собой 6-метокси;
R1 представляет собой 7-метокси;
n равно 2;
R2 представляет собой 5-фтор; и
R2 представляет собой 6-бром;
b. m равно 2;
R1 представляет собой 6-(2-метоксиэтокси);
R1 представляет собой 7-(2-метоксиэтокси);
n равно 1; и
R2 представляет собой 6-хлор;
с. m равно 2;
R1 представляет собой 6-метокси;
R1 представляет собой 7-(2-гидроксиэтокси);
n равно 1; и
R2 представляет собой 6-хлор;
d. равно 1;
R1 представляет собой 6-амино;
n равно 1; и
R2 представляет собой 6-хлор;
е. m равно 2;
R1 представляет собой 6-метокси;
R1 представляет собой 7-(3-гидроксипропокси);
n равно 1; и
R2 представляет собой 6-хлор;
f. m равно 2;
R1 представляет собой 7-(2-имидазол-1-ил-этокси);
R1 представляет собой 6-метокси;
n равно 1; и
R2 представляет собой 6-хлор;
g. m равно 2;
R1 представляет собой 6-метокси;
R1 представляет собой 7-метокси;
n равно 1; и
R2 представляет собой 5-амино;
h. m равно 2;
R1 представляет собой 6-метокси;
R1 представляет собой 7-(2-метокси-этокси);
n равно 2;
R2 представляет собой 5-фтop и
R2 представляет собой 6-бром;
i. m равно 2;
R1 представляет собой 6-метокси;
R1 представляет собой 7-метокси;
n равно 2;
R2 представляет собой 5-амино;
R2 представляет собой 6-хлор; или
j. m равно 2;
R1 представляет собой 6-метокси;
R1 представляет собой 7-(2-гидрокси-3-метокси)пропокси;
n равно 1; и
R2 представляет собой 6-хлор.
Вторую группу предпочтительных соединений формулы I составляют соединения, в которых
Z представляет собой
B - шестичленный цикл с 0, 1 или 2 двойными связями, возможное расположение которых обозначено пунктирной линией;
n принимает значение 0-2;
R2 в каждом случае независимо представляет собой галоген, гидроксильную группу или (C1-C4)алкил;
m принимает значения 0, 1 или 2; и
R1 в каждом случае независимо замещен в 6 и/или 7 положение и представляет собой гидроксильную группу, (C1-C4)алкокси, гидрокси(C2-C4)алкокси, амино(C2-C4)алкил, амино(C2-C4)алкокси, (C1-C4)алкокси(C2-C4)алкокси,
(C1-C4)алкилендиокси, гидрокси (C1-C4)алкил (C1-C4)алкилендиокси, (C1-C4)алкокси (C1-C4)алкил(C1-C4)алкилендиокси, моно-N- или ди-N, N-(C1-C4)алкиламино(C2-C4)алкокси, 3- или 4-(C1-C4)алкокси-(2-гидрокси)(C3-C4) алкокси, карбокси(C1-C4)алкокси, морфолино(C2-C4)алкокси, имидазол-1-ил-(C2-C4)алкокси, 4(C1 -C4)алкилпиперазин-1-ил-(C2-C4)алкокси, (C1-C4)алкокси(C1-C4)алканоилокси, нитро-группу, гидроксиламино, амино, моно-N- или ди- N, N-(C1-C4)алкиламино, (C1-C4)алканоиламино, гидрокси (C2-C4)алкиламино, (C1-C4)алкокси (C2-C4)алкиламино, (C1-C4) алкилсульфонамидо, морфолино, (C1-C4)алкилпиперазин-1-ил, бис(C1-C4) алкансульфонамидо, ди-N,N-(C1-C4)алкиламино (C2-C4)алкиламино, (C1-C4)алкиламино (C2-C4)алкиламино, пиперидин-1-ил, имидазол-1-ил, пирролидин-1-ил, (C1-C4)алкокси(C1 -C4)алкилкарбониламино, N-(C1-C4)алкил-N-(C1-C4)алканоиламино, карбокси, (C1-C4)алкоксикарбонил, (C1-C4)алкоксикарбонил, (C1-C4) алкоксикарбонил(C1-C4)алкокси, амидо-группу, моно-N- или ди-N, N-(C1-C4)алкиламинокарбонил, моно-N- или ди- N, N-(гидрокси(C2-C4)алкил)амино-карбонил, (C1-C4)алкил, гидрокси(C1-C4)алкил, моно- N- или ди-N,N-((C1-C4)алкокси (C1-C4)алкил)амино(C1-C4)алкил, моно-N- или ди-N,N-(C1-C4)алкиламино(C1-C4)алкил, (C1-C4)алканоиламино(C1-C4)алкил,
(C1-C4)алкокси(C2-C4)алкокси (C1-C4)алкил, (C1-C4)алкилтио, (C1-C4)алкокси(C2-C4)алкилтио или гидрокси(C2-C4)алкилтио.
Третью группу предпочтительных соединений формулы I составляют соединения, в которых
Z представляет собой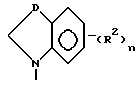
D - насыщенный углерод;
n принимает значения 0, 1 или 2;
R2 в каждом случае независимо представляет собой галоген, гидроксильную, амино, нитро-группу, трифторметил, этенил, этинил или (C1-C4)алкил;
m принимает значения 0, 1 или 2; и
R1 в каждом случае независимо замещен в 6 и/или 7 положении и представляет собой гидроксильную группу, (C1-C4)алкокси, гидрокси(C2-C4)алкокси, амино(C2-C4)алкил, амино(C2-C4)алкокси, (C1-C4)алкокси(C2-C4)алкокси,
(C1-C4)алкилендиокси, гидрокси(C1-C4)алкил (C1-C4)алкилендиокси, (C1-C4)алкокси(С1-C4)алкил(C1-C4)алкилендиокси, моно-N- или ди-N, N-(C1-C4)алкиламино(C2-C4)алкокси, 3- или 4-(C1-C4)алкокси-(2-гидрокси)-(C3-C4) алкокси, карбокси(C1-C4)алкокси, морфолино (C2-C4)алкокси, имидазол-1-ил(C2-C4) алкокси, 4-(C1-C4)алкил-пиперазин-1-ил (C2-C4)алкокси, (C1-C4)алкокси (C1-C4)алканоилокси, нитро-группу, гидроксиламино, амино-группу, моно-N-или ди-N,N-(C1-C4)алкиламино, (C1-C4)алканоиламино, гидрокси (C2 -C4)алкиламино, (C1-C4)алкокси (C2-C4)алкиламино, (C1-C4)алкилсульфонамидо, морфолино, (C1-C4)алкилпиперазин-1-ил, бис (C1-C4)алкансульфонамидо, ди-N, N- (C1-C4)алкиламино(C2-C4)алкиламино, (C1-C4)алкиламино (C2-C4)алкиламино, имидазол-1-ил, пиперидин-1-ил, пирролидин-1-ил, (C1-C4)алкокси(C1-C4) алкилкарбониламино, N-(C1 -C4)алкил-N-(C1-C4)алканоиламино, карбоксильную группу, (C1-C4)алкоксикарбонил, (C1-C4)алкоксикарбонил(C1-C4)алкокси, амидо, моно-N- или ди-N,N-(C1-C4)алкиламинокарбонил, моно-N или ди-N,N-(гидрокси(C2-C4)алкил) аминокарбонил, (C1-C4)алкил, гидрокси(C1 -C4)алкил, моно-N- и ди-N,N-((C1-C4)алкокси (C1-C4)алкил)амино(C1-C4)алкил, моно- N- или ди-N, N-(C1-C4)алкиламино(C1-C4) алкил, (C1-C4)алканоиламино(C1-C4)алкил,
(C1-C4)алкокси(C2-C4)алкокси (C1-C4)алкил, (C1-C4)алкилтио, (C1-C4)алкокси(C2-C4)алкилтио или гидрокси (C2-C4)алкилтио.
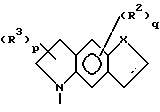
К третьей группе предпочтительных соединений формулы I относятся соединения, в которых
R2 в каждом случае независимо представляет собой галоген, нитро-, гидроксильную группу, (C1-C4)алкил или трифторметил;
R1 представляет собой (C1-C4)алкокси, (C1-C4)алкилендиокси или (C1-C4)алкил;
m равно 2; и
n принимает значения 1 или 2.
Еще одним предметом данного изобретения является группа предпочтительных соединений, соответствующих формуле I, которые имеют строение, представленное формулой IZ: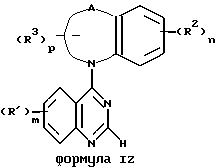
и фармацевтически приемлемые соли и гидраты, в которых
A вместе с фрагментом бензольного кольца при соединении с атомом азота образует конденсированный 5-9-членный моно-ненасыщенный, моно-аза-цикл, где указанный цикл, содержащий 6 или более членов может включать -О- или-S-;
каждый R1 независимо представляет собой водород, трифторметил, галоген, нитро-, гидроксильную, амино-группу, (C1-C4)алкил, карбоксильную группу, (C1 -C4)алкокси, (C1-C4)алкокси (C1-C4)алкилен, (C1-C4)алкокси(C2-C4)алкокси,
гидрокси(C2-C4)алкокси, трифторметилокси, (C1-C4)алкоксикарбонил, (C1-C4)алкилендиокси, (C1-C4)алкилсульфинил, (C1-C4)алкилсульфонил, пирролидин-1-ил, пиперидин-1-ил, морфолино, 4-(C1-C4)алкилпиперазин-1-ил, карбамоил, N, N-ди-(C1-C4)алкилкарбамоил, фенокси, бензилокси, гидроксиамино, (C1-C4)алкоксиамино, N,N-ди-(C1-C4)алкиламино, (C1-C4) алкоксикарбониламино, (C1-C4)алканоиламино, карбамоиламино или бензоиламино, указанные фенокси-, бензилокси- и бензоиламиногруппы необязательно содержат один заместитель, выбранный из группы, включающей галоген, нитро-группу, трифторметил, гидроксильную группу или (C1-C4)алкил, и указанная (C1-C4)алкилендиокси-группа соединена двумя концами с хиназолиновым фрагментом;
каждый R2 независимо представляет собой водород, трифторметил, галоген, нитро-, гидроксильную, амино-группу, (C1-C4)алкил, (C1-C4)алкокси, бензилокси, фенокси, (C1-C4)алкилендиокси, циано-, бензиламино-группу, (C1-C4)алканоил, N,N-ди-(C1-C4)алкиламино, (C1-C4)алкилтио, (C1-C4)алкилсульфинил или (C1-C4)алкилсульфонил, указанные бензилокси, фенокси и бензиламино группа необязательно содержит один заместитель, выбранный из группы, включающей галоген, нитро-группу, трифторметил, гидроксильную группу или (C1-C4)алкил и указанная (C1-C4)алкилендиокси-группа присоединена обоими концами к бензольному фрагменту;
каждый R3 независимо представляет собой гидроксильную группу, N,N-ди-(C1-C4)алкиламино, сульфо, (C1-C4)алкокси, при условии, что эти группы не присоединяются к атому углерода цикла, который является соседним к -О- или -S- или эти группы не находятся во втором положении, карбоксильная группа, гидрокси(C1-C3)алкилен, (C1-C4)алкокси(C1-C3)алкилен, N,N-ди- (C1-C4)алкиламино(C1-C3)алкилен, карбокси(C1-C3)алкилен, (C1-C4)алкоксикарбонил или сульфо(C1-C3)алкилен;
n принимает значения 1 или 2;
p принимает значения 0, 1 или 2; и
m принимает значения 1, 2 или 3.
Область данного изобретения включает также следующее соединения:
(6-Хлор-1-(6,7-метилендиоксихиназолин-4-ил)-2, 3-дигидро-1H-индол-5-ил)метиламин,
3-(6-Хлор-1-(6,7-бис- (2-гидроксиэтокси)-хиназолин-4-ил)-2,3-дигидро-1H-индол-3-ил) пропанол,
3-(4-(6-Фтор-7-метил-2, 3-дигидроиндол-1-ин)-7- (3-гидрокси-пропокси)хиназолин-6-илокси)пропан-1-ол,
6-Амино-7-гидроксиметил-4- (6-винил-2,3-дигидроиндол-1-ил)хиназолин,
4- (6-Этил-2,3-дигидроиндол-1-ил)-7-метокси-6-метилхиназолин,
1-(6, 7-Диметоксихиназолин-4-ил)-6-хлор-2,3-дигидро-1H-индол-4-ол,
(4- (6-Бром-3-(3-морфолин-4-илпропил)-2,3-дигидроиндол-1-ил)хиназолин- 6-ил)-метиламин,
(4-(6-Хлор-3-(3-диметиламинопропил)-2, 3-дигидроиндол-1-ил)-7-метоксихиназолин-6-ил)метанол,
3-(1-(6, 7-Бис-(2-метоксиэтокси)хиназолин-4-ил)-6-фтор-7-метил-2, 3-дигидро-1H-индол-3-ил)пропионовая кислота,
2-(4-(3-(3-Диметиламинопропил)-3,5,6,7-тетрагидро-2H-пирроло 2,3-f]индол-1-ил)-7-(2-гидроксиэтокси)хиналозил-6-илокси)этанол,
(2-(6-Хлор-1-(2,2-диметил-(1,3-) диоксоло[4,5-g]хиназолин-8-ил) -2,3-дигидро-1H-индол-3-ил)этил)диэтиламин,
N-(4- (6-Этинил-7-метил-2, 3-дигидроиндол-1-ил) -7-метоксихиназолин-6-илметил)ацетамид,
3-(4-(6-Бром-7-метил-2, 3-дигидроиндол-1-ил)-6-трифторметоксихиназолин-7-илокси) пропан-1-ол,
3-(6-Хлор-1-(6,7-бис-(2-метоксиэтокси) хиназолин-4-ил)-2,3- дигидро-1H-индол-3-ил) метанол,
6-Хлор-1- (7- (2-диметиламиноэтокси)хиназолин-4-ил)-2, 3-дигидро-1H-индол-4-ол и
3-(4-(6-Хлор-4-метиламино-2,3-дигидроиндол-1-ил)-6- (3-гидроксипропокси)хиназолин-7-илокси)пропан-1-ол.
В соответствии с еще одним аспектом данное изобретение относится к способу лечения гиперпролиферативного заболевания у млекопитающего посредством введения млекопитающему, страдающему гиперпролиферативным заболеванием, соединения формулы I в количестве, оказывающем лечебное действие на гиперпролиферативное заболевание.
Данное изобретение относится также к фармацевтическим композициям для лечения гиперпролиферативного заболевания у млекопитающих, которые включают соединение формулы I в количестве, оказывающем лечебное действие на гиперпролиферативное заболевание, и фармацевтически приемлемый носитель.
Термин "галоген" в данном описании означает хлор, бром, йод или фтор.
Термин "алкил" означает насыщенный углеводород с прямой или разветвленной углеродной цепочкой.
Термин "моно-ненасыщенный" означает, что в A цикле содержится только одна ненасыщенная связь, которая сопряжена с бензольным кольцом.
Те атомы углерода, у которых нет заместителей, соединены с водородом (т. е. углерод является нейтральным или обладает полным набором электронов - октетом).
В данном описании термин "инертный растворитель" относится к растворителю, который не взаимодействует с исходными веществами, реагентами, промежуточными продуктами таким образом, чтобы это оказывало влияние на выход конечного продукта.
Для специалиста понятно, что некоторые заместители, перечисленные в данном изобретении, будут химически несовместимыми друг с другом или с гетероатомами в соединениях и специалист будет избегать такого подбора заместителей при выборе соединений данного изобретения.
Другие отличительные черты и преимущества данного изобретения станут более понятными из описания и формулы изобретения, которые излагают суть данного изобретения.
Подробное описание изобретения
Схема 1 представлена в конце описания.
Соединения формулы I или их фармацевтически приемлемые соли могут быть получены любым известным способом, который подходит для получения соединений, химически родственных соединениям формулы I.
В общем случае хиназолины формулы I могут быть получены через промежуточное 4-амино-производное подходящего замещенного хиназолина с использованием соответственно замещенного амина.
Обычно соответственно замещенный 4-галогенхиназолин (или хиназолин, содержащий в четвертом положении подходящую уходящую группу, которая может быть замещена, такую как арилокси, алкилсульфонилокси, например трифторметансульфонилокси, арил- сульфонилокси, триалкилсилокси, циано, пиразоло, триазоло или тетрозоло), предпочтительно 4-хлорхиназолин, присоединяет подходящий амин в растворителе, таком как (C1-C6)спирт, диметилформамид, N-метилпирролидин-2-он, хлороформ, ацетонитрил, тетрагидрофуран (ТГФ), 1,4-диоксан, диметилсульфоксид или другой апротонный растворитель. Это присоединение может протекать в присутствии основания и предпочтительно протекает в присутствии карбоната или гидроксида щелочного или щелочноземельного металла или третичного аминного основания, такого как пиридин, 2,6-лутидин, коллидин, N-метилморфолин, триэтиламин, диизопропилэтиламин, 4-диметиламинопиридин или N,N-диметиланилин. Далее они называются подходящими основаниями. Смесь выдерживают при температуре в интервале от приблизительно комнатной температуры до температуры кипения с обратным холодильником, предпочтительно при температуре в интервале от приблизительно 35oC до приблизительно температуры кипения с обратным холодильником до тех пор, пока по существу можно будет зафиксировать отсутствие 4-галогенхиназолина, обычно в течение периода времени от приблизительно 2 часов до приблизительно 24 часов. Предпочтительно реакцию проводят под инертной атмосферой, такой как сухой газообразный азот.
В общем случае реагенты берут в стехиометрическом количестве при использовании аминного основания (или если не используют аминное основание, можно использовать избыток амина), однако для соединений, у которых используется соль амина (обычно соль соляной кислоты), предпочтительно применять избыток аминного основания, обычно используют дополнительный эквивалент аминного основания.
Для тех соединений, у которых стерически затруднено применение амина (таких как 2-алкилиндолин), или используется очень химически активный 4-галогенхиназолин, предпочтительно использовать в качестве растворителя трет-бутиловый спирт или полярный апротонный растворитель, такой как диметилформамид, диметилацетамид или N-метилпиролидин-2-он.
Далее описывается синтез ряда соединений формулы I с помощью подходящих реакций, приводящих к получению указанных выше соединений.
Для получения соединений формулы I, где R1 представляет собой амино- или гидроксиламино, используется восстановление соединения формулы I, в котором R1 представляет собой нитро-группу.
Восстановление можно удобно проводить любым из множества известных способов такого превращения. Восстановление можно проводить, например, гидрированием раствора нитро-соединения в инертном растворителе в присутствии подходящего металлического катализатора, такого как палладий или платина. Другими подходящими восстановителями являются, например, дитионит натрия в муравьиной кислоте или активированный металл, такой как активированное железо (полученное промыванием порошкообразного железа слабым раствором кислоты, такой как соляная кислота). Таким образом, восстановление можно проводить при нагревании смеси нитро-соединения и активированного металла в растворителе, таком как смесь воды и спирта, например метанола или этанола, до температуры в интервале, например, от 50 до 150oC, удобно при приблизительно 70oC.
Для получения соединений формулы I, в которых R2 представляет собой амино-группу, можно использовать восстановление соединения формулы I, в котором R2 представляет собой нитро-группу.
Для получения соединений формулы I, в которых R1 или R2 содержит фрагмент первичного или вторичного амина (отличный от амина, который использовался для взаимодействия с хиназолином), такой свободный амин предпочтительно защищают перед проведением описанной выше реакции с последующим снятием защиты, которое проводят после описанной выше реакции с 4-галоген-хиназолином.
Защитные группы описываются, например, в T.W.Greene and P.G.M.Wuts, "Protective Groups in Organic Synthesis", Second Ed., John Wiley and Sons, New York, 1991.
Можно использовать некоторые хорошо известные защитные группы атома азота. Такими группами являются (C1-C6)алкоксикарбонил, необязательно замещенный бензилкарбонил, арилкарбонил, тритил, винилоксикарбонил, о-нитрофенилсульфонил, дифенилфосфинил, п-толуолсульфонил и бензил. Помимо применения защитных групп для атома азота, можно проводить реакцию в хлорированном углеводородном растворителе, таком как хлористый метилен или 1,2-дихлорэтан, или в эфирном растворителе, таком как глим (glyme), диглим (diglyme) или тетрагидрофуран, в присутствии или в отсутствии третичного аминного основания, такого как тиэтиламин, диизопропилэтиламин или пиридин, предпочтительно триэтиламин, при температуре в интервале от приблизительно 0oC до приблизительно 50oC, предпочтительно при комнатной температуре. Защитные группы удобно также вводить в условиях реакции Шоттена-Баумана.
После описанной выше реакции присоединения амина защитная группа может быть удалена с помощью известных способов удаления защитных групп, которые хорошо известны квалифицированному специалисту, например для удаления третбутоксикарбонильной защитной группы используется обработка трифторуксусной кислотой в хлористом метилене.
Для получения соединений формулы I, в которых R1 представляет собой гидроксильную группу, предпочтительно расщепление соединения формулы I, в котором R1 представляет собой (C1-C4)алкокси.
Реакцию расщепления можно удобно проводить любым из множества известных методов такого превращения. Для такого О-деалкилирования можно применять обработку хиназолин-производного формулы I расплавленным пиридингидрохлоридом (20-30 экв.) при температуре от 150oC до 175oC. Такое превращение можно также осуществлять, например, обработкой хиназолин-производного (C1-C4)алкилсульфидом щелочного металла, таким как этанэтиолат натрия или, например, обработкой диарилфосфидом щелочного металла, таким как дифенилфосфид лития. Расщепление также можно удобно осуществить, например, при обработке хиназолин-производного тригалогенидом бора или алюминия, таким как трибромид бора. Такие реакции предпочтительно проводят в присутствии инертного растворителя при подходящей температуре.
Для получения соединений формулы I, в которых R1 или R2 является (C1-C4)алкилсульфинильной или (C1-C4)алкилсульфонильной группой, предпочтительно окисление соединения формулы I, в котором R1 или R2 представляет собой (C1-C4)алкилтио-группу.
Подходящими окислителями являются, например, вещества, хорошо известные как окислители тио-группы до сульфинил и/или сульфонил-группы, например перекись водорода, перкислота (такая как 3-хлорпербензойная, пермуравьиная или перуксусная кислота), пероксисульфат щелочного металла (такой как пероксимоносульфат натрия), триоксид хрома или газообразный кислород в присутствии платины. Окисление в общем случае проводят по возможности в мягких условиях с использованием стехиометрического количества окислителя для снижения риска окисления и повреждения других функциональных групп. В общем случае реакцию проводят в подходящем растворителе, таком как хлористый метилен, хлороформ, ацетон, тетрагидрофуран или трет-бутилметиловый эфир, при температуре в интервале, например, от -25oC до 50oC, удобно при комнатной температуре или при температуре, близкой к комнатной, например в интервале от 15 до 35oC. В том случае, когда целевым продуктом является соединение, содержащее сульфинильную группу, можно применять более мягкий окислитель, например метапериодат натрия или калия, удобно в полярном растворителе, таком как уксусная кислота или этанол. Далее будет показано, что когда целевым продуктом является соединение формулы I, содержащее (C1-C4)алкилсульфонил, оно может быть получено окислением соответствующего (C1-C4)алкилсульфинил-производного, а также соответствующего (C1-C4)алкилтио-производного.
Для получения соединений формулы I, в которых R1 представляет собой (C2-C4)алканоиламино-группу или замещенную (C2-C4)алканоиламино-группу, уреидо-, 3-фенилуреидо-, бензамидо- или сульфонамидо-группу, подходящим способом является ацилирование или сульфонилирование соединения формулы I, в котором R1 является амино-группой.
Подходящим ацилирующим агентом является, например, любой известный агент, применяемый для ацилирования амино-группы в ациламино-группу, например ацилгалогенид (например, (C2-C4)алканоилхлорид или бромид, бензоилхлорид или бензоилбромид), ангидрид алкановой кислоты или смешенный ангидрид (например, ангидрид (C2-C4)алкановой кислоты, такой как ангидрид уксусной кислоты или смешенный ангидрид, полученный в результате реакции алкановой кислоты с (C2-C4)алкоксикарбонилгалогенидом, например (C1-C4) алкоксикарбонилхлоридом, в присутствии подходящего основания). Для получения соединений формулы I, в которых R1 представляет собой уреидо- или 3-фенилуреидо-группу, подходящим ацилирующим агентом является, например, цианат, например цианат щелочного металла, такой как цианат натрия или, например, изоцианат, такой как фенилизоцианат. N-Сульфонилирование можно осуществлять, применяя подходящие сульфонилгалогениды или сульфонилангидриды, в присутствии третичного аминного основания. В общем случае ацилирование или сульфонилирование проводят в инертном растворителе при температуре в интервале, например, от -30 до 120oC, удобно при температуре, близкой к комнатной температуре.
Для получения соединений формулы I, в которых R1 представляет собой (C1-C4)алкокси- или замещенную (C1-C4)алкокси-группу или R1 представляет собой (C1-C4)алкиламино- или замещенную моно-N- или ди-N, N-(C1-C4)алкиламино-группу в качестве подходящего способа используют алкилирование, предпочтительно в присутствии подходящего основания, соединения формулы I, в котором R1 представляет собой гидроксильную или амино-группу.
Подходящим алкилирующим агентом является, например, любой известный алкилирующий агент, используемый для алкилирования гидроксильной группы в алкокси- или замещенную алкокси-группу, или для алкилирования амино-группы до алкиламино- или замещенной алкиламино-группы, например, алкил- или замещенный алкил-галогенид, например (C1-C4)алкилхлорид, -бромид или -йодид или замещенный (C1-C4)алкилхлорид, -бромид или -йодид, в присутствии подходящего основания в инертном растворителе при температуре в интервале, например, от 10 до 140oC удобно при комнатной или при температуре, близкой к комнатной.
Для получения соединений формулы I, в которых R1 представляет собой амино-, окси- или циано-замещенный (C1-C4)алкил-заместитель, можно применить реакцию, предпочтительно в присутствии подходящего основания, соединения формулы I, в котором R1 представляет собой (C1-C4)алкил-заместитель, замещаемую группу, с подходящим амино, спиртом или цианидом. Реакцию предпочтительно проводят в инертном растворителе или носителе при температуре в интервале, например, от 10 до 100oC, удобно проводить реакцию при комнатной или при температуре, близкой к комнатной.
Для получения соединений формулы I, в которых R1, R2 или R3 представляет собой карбокси-заместитель или заместитель, который содержит карбоксильную группу, необходимо провести гидролиз соединения формулы I, в котором R1, R2 или R3 представляет собой (C1-C4)алкоксикарбонил или заместитель, который содержит (C1-C4)алкоксикарбонильную группу.
Гидролиз можно удобно проводить, например, в щелочных условиях с использованием гидроксида щелочного металла, как показано в приведенных далее примерах.
Для получения соединений формулы I, в которых R1 представляет собой амино-группу, (C1 -C4)алкиламино, ди-[(C1-C4)aлкил]амино, пирролидин-1-ил, пиперидино, морфолино, пиперазин-1-ил, 4-(C1-C4)алкилпиперазин-1-ил или (C1-C4)алкилтио, предпочтительной может быть реакция соединения формулы I, где R1 представляет собой группу, которая может замещаться соответствующим амином или тиолом, реакцию удобно проводить в присутствии подходящего основания.
Реакцию обычно проводят предпочтительно в присутствии подходящего основания в инертном растворителе или наполнителе при температуре в интервале, например, от 10 до 180oC, удобно проводить реакцию при температуре в интервале от 100oC до 150oC.
Для получения соединений формулы I, в которых R1 представляет собой 2-оксопирролидин-1-ил или 2-оксопиперидин-1-ил, удобно применять циклизацию соединения формулы I, в котором R1представляет собой галоген (C2-C4)алканоиламино-группу, циклизацию проводят в присутствии подходящего основания.
Реакцию обычно проводят предпочтительно в присутствии подходящего основания в инертном растворителе или наполнителе при температуре в интервале, например, от 10 до 100oC, удобно проводить реакцию при комнатной температуре или при температуре, близкой к комнатной.
Для получения соединений формулы I, в которых R1 представляет собой карбамоил, замещенный карбамоил, алканоилокси- или замещенную алканоилокси-группу, удобно применять карбамоилирование или ацилирование соединения формулы I, в котором R1 представляет собой гидроксильную группу.
Подходящим ацилирующим агентом является, например, любой из известных агентов, применяемых для ацилирования гидрокси-арильных фрагментов с получением алканоилоксиарилов. Можно, например, применять (C2-C4)алканоилгалогениды, (C2-C4)алканоилангидриды или смешенные ангидриды и их подходящие замещенные производные, обычно в присутствии подходящего основания. Другим способом является соединение (C2-C4)алкановых кислот или их подходящих производных с соединением формулы I, в котором R1 представляет собой гидроксильную группу, с помощью конденсирующего агента, такого как карбодиимид. Для получения соединений формулы I, в которых R1 представляет собой карбамоил или замещенный карбамоил, подходящими карбамоилирующими агентами являются, например, цианат или алкил или арилизоцианат, обычно реакцию проводят в присутствии подходящего основания. Кроме того, подходящий промежуточный продукт такой, как хлорформиат, сукцинимидокарбонат или имидазолкарбонил-производное хиназолина формулы I, в котором R1 представляет собой гидроксильную группу, можно получать, например, обработкой указанного производного фосгеном (или фосгеновым эквивалентом), дисукциинимидокарбонатом или карбонилдиимидазолом. Образующееся производное затем может использоваться в реакции с соответствующим амином или замещенным амином для получения нужных карбамоил-производных.
Для получения хиназолин-производных формулы I, в которых R1 представляет собой аминокарбонил или замещенный аминокарбонил, предпочтительно применять аминолиз подходящего промежуточного производного хиназолина формулы I, в котором R1 является карбоксильной группой.
Активацию и присоединение соединения формулы I, в котором R1 представляет собой карбокси-группу, можно проводить различными способами, которые хорошо известны квалифицированному специалисту. Подходящими способами являются активирование карбоксила в виде галогенида кислоты, азида, симметричного или смешенного ангидрида или активного сложного эфира соответствующей реакционной способности для соединения с нужным амином. Примеры промежуточных продуктов такого типа и их получение описано в литературе, например в монографии: M. Bodonsky and A Bodansky, "The Practice of Peptide Synthesis". Springer,- Verlag, New York, 1984.
Образующиеся соединения формулы I можно, если это нужно, выделять и подвергать очистке стандартными способами, такими как отгонка растворителя и перекристаллизация или хроматография.
Исходные реагенты для представленных выше схем реакций (например, амины, хиназолины, защитные группы для аминогруппы) легко доступны или легко могут быть получены хорошо известными способами синтеза органических веществ. Например, получение 2,3-дигидро-1,4-бензоксазин-производных описано R.C. Elderfield, W.H. Todd, S.Gerber, Ch. 12 in "Heterocyclic Compounds", Vol. 6, R.C. Elderfield ed., John Wiley and Sons, Inc., N.Y., 1957.
Замещенные 2,3-дигидробензтиазинил-производные описываются в главе 13 книги R.C. Elderfield and E.E.Harris, in Vol. 6 of the Elderfield "Heterocyclic Comp. " Синтез 1,2,3, 4-тетрагидрохинолинов и их хинолиновых прекурсоров описывается, например, в "The chem. istry of Heterocyclic Compounds", vol. 32, Parts 1.2 and 3, G.Jones, ed., John Wiley and Sons, N.Y., 1977. Алкил и арил-замещенные 1,2,3,4-тетрагидрохинолины были получены традиционным способом - каталитическим гидрированием соответственно замещенных хинолинов с использованием Pt2/H2(газа) в метаноле (например, по методике, описанной M. Honel and F.W.Vierhopper, J.Chem. Soc. Perkin I 1980, 1933-1939). Синтез замещенных 2,3,4,5-тетрагидро-1H-бензо(b)азепинов описывается в G. R. Proctor, Ch.11, Vol.43, "The Chemistry of Heterocyclic Compounds", Part I, A.Rosowsky, ed., Wiley Interscience, N.Y. 1984. Некоторые замещенные 2,3,4,5-тетрагидро-1H-1-бензо[b] азепин-2-оны и 1,2,3,4,5,6-гексагидро-1-бензо [b] азоцин-2-оны, которые легко восстанавливаются в соответствующие 2,3,4,5-тетра-гидро-1H-бензо [b] азепины и l,2,3,5,6-гeкcaгидpo-1-бeнзo[b] азоцины соответственно описываются в публикациях E.C.Horning et al., J.Am. Chem. Soc. 74, 5153 (1952), and R. Huisgen et al., Liebigs Ann. Chem. 586, 30 (1954).
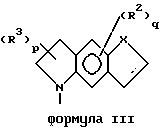
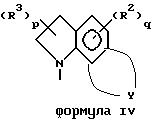
Кроме того, приведенное далее общее описание в сочетании со схемой 1 и способами получения (которые могут быть аналогичными) обеспечивают дополнительное пособие для получения индолин-основных аминов. Итак, в соответствии со схемой 1 целевые производные индола формул II, III, IV или V могут быть легко получены восстановлением соответствующих индол-производных формулы IX (обозначенные пунктирной линией кривые относятся к конденсированным моноциклическим и бициклическим фрагментам формул III, IV и V).
В общем случае соединения формулы IX, содержащие апротонные R2 или R3 заместители, обрабатывают ZnBH4 (получен из ZnCl2 и NaBH4 по методике, описанной в публикации W.J. Gensler et al., J. Am. Soc. 82, 6074-6081 (1960)) в эфирном растворителе, таком как диэтиловый эфир, при температуре в интервале от приблизительно 10oC до приблизительно 40oC, предпочтительно при комнатной температуре. Такая обработка описывается H.Korsuki et al., Heterocycles 26, 1771-1774 (1987). В соответствии с другим способом соединения формулы IX можно обрабатывать комплексом боран пиридин (или другим комплексом вида боран/тритичный амин) в отсутствие растворителя или в присутствии растворителя, такого как тетрагидрофуран, при температуре в интервале от приблизительно 10oC до приблизительно 30oC, предпочтительно при комнатной температуре, с последующей обработкой смеси кислотой, такой как соляная, трифторуксусная или уксусная кислота, с получением соединений формулы II, III, IV или V.
Целевые соединения формулы IX, в которой р равно 1 или 2, могут быть получены из соответствующего соединения формулы XIV посредством синтеза индола по Фишеру или при некотором его видоизменении ("The Fisher Indole Synthesis". B. Robinson, Wiley Interscience, N.Y. 1982; или U.Pindur, R. Adam, J.Het. Chem. 25, 1-8 (1988) и ссылки в этих публикациях).
Индолы формулы IX, в которых p равно 0, обычно могут быть получены из анилинов формулы XII двумя способами: либо через соединения формулы X, либо через соединения формулы XI. Однако получение через соединение формулы XI не является предпочтительным для нитро- или карбоалкокси-замещенных индолов. Таким образом, целевое соединение формулы IX можно получить из подходящего соединения формулы X (например, изотинов, окиндолов) восстановлением, или из подходящего соединения формулы XI посредством восстановительной циклизации. В общем случае соединение формулы X обрабатывают бораном в эфирном растворителе, таком как тетрагидрофуран, при температуре в интервале от приблизительно 0oC до приблизительно 30oC, предпочтительно при комнатной температуре. В общем случае соединение формулы XI обрабатывают боргидридом натрия в эфирном растворителе, таком как диоксан, при температуре в интервале от приблизительно 20oC до приблизительно 100oC, предпочтительно при температуре кипения.
Целевое изатин-производное формулы X можно получать из подходящего соединения формулы XII взаимодействием с хлоральгидратом и гидроксиламином с последующей каталитической циклизацией, такие процессы используют на основании описания в публикации Org. Syn., Coll. Vol. 1, 327-330.
Целевое соединение формулы XI может быть получено из подходящего соединения формулы XII с помощью орто-ацилирования в присутствии в качестве катализатора кислоты Льюиса. Обычный способ получения описан в T.Sugasawa et. al. , J.Org.Chem. 44 (4), 578-586 (1979). В общем случае соединение формулы ХII используют в реакции с 2-хлорацетонитрилом в присутствии трихлорида бора и вспомогательного кислотного катализатора, такого как хлорид алюминия, обычно в ароматическом растворителе, таком как ксилол, толуол или хлорбензол, при температуре в интервале от приблизительно 50oC до приблизительно температуры кипения с обратным холодильником.
Целевые аминоароматические соединения формулы XII могут быть получены из подходящих соответствующих нитроароматических соединений различными способами восстановления (такие способы восстановления описаны выше).
Некоторые целевые индолин-производные формулы II, III, IV или V могут быть получены из других описанных выше индолин-производных посредством дополнительных модификаций перед присоединением к хиназолиновым фрагментам формулы I. Например, подходящим образом замещенный целевой 5-гидроксииндол может быть получен из соответствующих индолинов гидрированием в соответствии с методикой, описанной в публикации H.J. Teuber and G.Staiger, Chem. Ber. 89, 489-508 (1956), с последующим восстановлением и получением соответствующего 5-гидроксиинодолина. В общем случае к подходящим образом замещенному индолину в ацетоне при нейтральной pH и при температуре в интервале от приблизительно 0oC до приблизительно 25oC добавляют нитрозидисульфонат калия в водном фосфатном буфере, в результате получают 5-гидроксииндол-производное, которое можно далее восстанавливать с помощью комплекса боран/пиридин/водная HCl для получения 5-гидроксииндолина.
Целевые броминдолин-производные, замещенные подходящим образом (например, содержащие заместители в четвертом или в шестом положении), можно получить бромированием соответствующих индолинов по методике, аналогично описанной в публикации Y. Miyake and Y.Kikugawa, J.Het. Chem. 20, 349-352 (1983). Эту методику можно применять для бромирования других больших циклов (например, 1,2,3,4-тетрагидрохинолинов, 2,3,4,5-тетрагидро-1H-бензо[b]азепинов и 1,2,3,4,5,6-гекса-гидро-бензо[b]азоцинов, особенно в 5/7, 6/8 и 7/9- положения). В общем случае, подходящий замещенный индолин взаимодействует с бромом в присутствии галогенофила, такого как сульфат серебра, в жестких кислотных условиях при температуре в интервале от 0oC до 25oC.
Кроме того, некоторые индолины, содержащие или не содержащие 3-алкил-заместители, можно удобно получать из подходящих 2-(2-галогенфенил)алкиламинов в соответствии с методикой, описанной в заявке Германии DE 3424900 A1.
Гидроксиалкилиндолины могут быть получены также восстановлением подходящих карбоновых кислот или их эфиров, например, в соответствии с методикой, описанной E.J.Corey et al., в статье, опубликованной в журнале J.Am.Soc. 92 (8), 2476-2488 (1970).
Целевые индолины, содержащие необходимые алкил-, алкенил-или аллил-заместители, могут быть получены из соответствующих триалкилсилил-защищенных 4-, 5- или 6-галогениндолинов с помощью добавления никель-фосфинового катализатора Гриньяра по методике, аналогичной приведенной в публикации K.Tamao et. al., Bull. Chem. Soc. Japan 49, 1958-1969 (1976). В общем случае защиту атома азота индолина осуществляют реакцией с трет-бутилдиметилсилилтрифлатом в галогенированном растворителе в присутствии третичного амина, N-силированный галоген-индол затем взаимодействует с подходящим алкил-, алкенил- или аллил-замещенным реактивом Гриньяра в эфирном растворителе в присутствии подходящего никель-фосфинового комплекса, обычно [Ni(dppe)Cl2]. В результате последующей обработки метанолом, содержащим следы кислоты, такой как трифторуксусная кислота, или анионом фтора в подходящем растворителе, таком как тетрагидрофуран, получают целевой индолин-продукт.
Кроме того, целевые замещенные алкенил- или алкинилиндолины могут быть получены в результате винилирования или алкилирования подходящего 4-, 5-, 6- или 7-галогениндолина в присутствии палладиевого катализатора, например, в соответствии с методиками, которые приведены в публикации V.N.Kalinin, Synthsis 1991, 413-432. Например, для алкинилиндолинов в общем случае соответствующий бром- или йодиндолин в диэтиламине обрабатывают алкил-, подходяще замещенный алкил- или триметилсилилацетиленом в присутствии каталитических количеств CuI или Pd(PPh3)4, при температуре кипения с обр./хол.
Дополнительно к приведенным ниже примерам, не ограничивающим область данного изобретения, получение различных индолинов, индолов, оксииндолов и изатинов, полезных в качестве промежуточных продуктов, описывается также в Heterocyclic Compounds with Indole and Carbazole Systems", W.C. Sumpter and F. M. Miller, in vol. 8 of "The Chemistry of Heterocyclic Compounds". Series, Interscience Publishers Inc., N.Y., 1954 и в публикациях, которые приведены в ссылках этих книг.
Некоторые хиназолины формулы I могут существовать как в сольватированной, так и в несольватированной формах, таких как, например, гидрированные формы. Следует учитывать то, что данное изобретение охватывает все как сольватированные, так и несольватированные формы, которые обладают активностью против гиперпролиферативных заболеваний.
Подходящей фармацевтически приемлемой солью хиназолин-производного данного изобретения является, например, кислотно-аддитивная соль хиназолин-производного данного изобретения, обладающего достаточной основностью, например кислотно-аддитивная соль, например, неорганической или органической кислоты, такой как соляная, бромистоводородная, серная, фосфорная, метансульфоновая, бензолсульфоновая, трифторуксусная, лимонная, молочная или малеиновая кислота. Кроме того, подходящей фармацевтически приемлемой основно-аддитивной солью хиналозин-производного данного изобретения, обладающего достаточной кислотностью, является соль щелочного металла, например лития, натрия или калия, соль щелочноземельного металла, например кальция или магния, аммониевая соль, или соль органического основания, которое дает физиологически приемлемый катион, например соль метиламина, диметиламина, триметиламина, пиперидина, морфолина или трис-(2-гидроксиэтил)амина. Все эти соли относятся к области данного изобретения и их можно получить удобными способами. Например, их можно получить просто контактированием кислотного и щелочного реагентов, обычно в стехиометрическом соотношении в водной, неводной или частично водной среде. Соли выделяют фильтрованием, высаживанием с помощью добавления среды, в которой соль не растворяется, предпочтительно эфирного или углеводородного растворителя, с последующим фильтрованием, упариванием растворителя или, в случае водных растворов, лиофилизацией.
Некоторые соединения формулы I содержат асимметричные атомы углерода. Диастериомерные смеси могут быть разделены на отдельные диастериомеры на основании различия в их физико-химических свойствах при помощи известных способов, например с помощью хроматографии и/или фракционной кристаллизации. Энантиомеры могут быть разделены с помощью превращения энантиомерных смесей в диастереомерную смесь в результате взаимодействия с соответствующим оптически активным соединением (например, спиртом или кислотой), разделением диастереомеров и конверсией (например, гидролизом) отдельных диастереомеров в соответствующий чистый энантиомер. Энантиомеры также могут быть разделены дифференциальной кристаллизацией в виде диастереомерных солей. Все такие изомеры, в том числе диастереомеры и энантиомеры, рассматриваются как часть данного изобретения.
Соединения данного изобретения являются сильными ингибиторами erbB-семейства онкогенных и протоонкогенных белковых тирозинкиназ, таких как рецептор эпидермального ростового фактора (EGFR-epidermal growth factor receptor), erbB2, HER3 или HER4, следовательно, могут применяться в терапии в качестве антипролиферативных средств (например, противораковых средств) у млекопитающих, особенно у людей. В частности, соединения данного изобретения представляют собой терапевтическое или профилактическое средство для лечения различных доброкачественных или злокачественных опухолей человека (опухолей почек, печени, мочевого пузыря, молочной железы, желудка, яичников, кишечника, простаты, поджелудочной железы, легких, вульвы, щитовидной железы, а также гепатокарциномы, саркомы, глиобластомы, различных опухолей головы и шеи) и других заболеваний, связанных с избыточным ростом ткани, таких как доброкачественная гиперплазия кожи (например, псориаз) или простаты (например ВРН). Такая активность в отношении заболеваний, связанных с доброкачественными опухолями может быть установлена стандартными методами, такими как описанные в журнале J.Invest. Dermatol. 98, 296-301 (1992). Кроме того, ожидается, что хиназолины данного изобретения могут обладать активностью против некоторых лейкозных и лимфоидных раковых заболеваний.
Соединения формулы I также усиливают ответы на общеизвестные способы химиотерапии и радиотерапию в зависимости от дозы и схемы применения, что основано на значительном синергизме, наблюдаемом между нейтрализующими антителами анти-EGFR и общепринятыми химиотерапевтическими средствами (J.Baselgo et al., J.Nat. Cancer Inst. 85, 1327-1333 (1993), Z.Fan et al., Cancer Res. 53, 4637-4642 (1993)).
Можно ожидать, что соединения формулы I полезны в лечении дополнительных заболеваний, в которых задействованы аберрантная экспрессия, взаимодействия вида лиганд/рецептор, активация или сигнализирующие события (Signalling events), связанные с различными белковыми тирозинкиназами, активность которых ингибируется соединениями формулы I.
Такие заболевания могут включать заболевания нейронной, астроцитальной, гипоталамной и другой гландулярной, макрофагиальной, эпителиальной, стомальной (относящейся к стоме) и бластосомной природы, в которые вовлечены аберрантная функция, экспрессия, активация или сигнализация erbB тирозинкиназ. Кроме того, соединения формулы I могут быть полезными в качестве терапевтического средства для применения в лечении воспалительных, ангиогенных (связанных с развитием кровеносных сосудов) и иммуногенных расстройств, в течение которых вовлечены как идентифицированные, так и еще не идентифицированные тирозинкиназы, которые ингибируются соединениями формулы I.
Активность этих соединений in vitro в ингибировании рецепторной тирозинкиназы (и последующего пролиферативного ответа, например, рака) может определяться по приведенной далее методике.
Активность соединений формулы I in vitro можно определить по количеству ингибирования фосфориляции экзогенного субстрата (например, Lys3-Gastrin или поли-Glu Tyr (4: 1) неупорядоченного сополимера (I.Posner et al., J. Biol. Chem. 267 (29), 20638-47 (1992) на тирозине через киназу рецептора эпидермального фактора роста испытываемым соединением по сравнению с контрольными условиями. Соответственно очищенный, растворимый рецептор EGF человека (96 нг) получают в соответствии с описанием, приведенным в статье: G.N.Gill, W. Weber, Methods in Enzymology 146, 82-88 (1987), из клеток вида A431 (American Type Culture Collection, Rockville, MD), предварительно инкубируют в микропробирке для центрофугирования (microfugal tube) с EGF (2 мкг мл) в смесевом растворе фосфорилирующего буфера и ванадата (PBV: 50 мМ HEPES, pH 7,4; 126 мМ NaCl; 24 мМ MgCl2; 100 мкл орто-ванадата) общим объемом 10 мкл - в течение 20-30 минут при комнатной температуре. Исследуемое соединение, растворенное в DMSO, разбавляют PBV, 10 мкл этого раствора смешивают со смесью EGF-рецептор/EGF и выдерживают при температуре 30oC в течение 10-30 минут. Реакцию фосфорилирования инициируют добавлением 20 мкл смеси 33PATP субстрат (120 мкМ LyS3-Gastrin (последовательность кода аминокислот: KKKGPWLEEEEEAYGWLDF), 50 мМ HERES pH 7,4, 40 мкМ ATP, 2 мкл Ci y-[33P]-ATP) к смеси EGF-рецептор/EGF и выдерживают в течение 20 минут при комнатной температуре. Реакцию останавливают добавлением 10 мкл соответствующего раствора (0,5 М ЭДТУК, pH 8; 2 мМ ATP) и 6 мкл 2N-ной HCl. Пробирки центрифугируют со скоростью 14000 об. мин при температуре 4oC в течение 10 минут. По 35 мкл супернатанта из каждой пробирки переносят с помощью пипетки на кружки из ватмана диаметром 2,5 см, эти кружки 4 раза обрабатывают 5%-ной уксусной кислотой (погружают в 1 л раствора в процессе каждой обработки), а затем сушат на воздухе. Это приводит к связыванию субстрата на бумаге и потерям несвязанного ATP при обработке. Введенный [33P] измеряют подсчетом с помощью жидкостной сцинтилляции. Введение при отсутствии субстрата (например, Lys3-gastrin) вычитают из общих величин как фоновое значение и вычисляют ингибирование в процентном отношении к контролю, где не использовалось испытываемое соединение (результаты испытания приведены в таблице 3).
Активность соединений формулы I in vivo может быть определена по количеству ингибирования роста опухоли испытываемым соединением относительно контроля. Ростингибирующее действие различных соединений в отношении опухолей измеряют в соответствии с методикой, приведенной в публикациях: Corbett. T. H. , et al., "Tumor Induction Relationships in Development of Transplantable Cancers of the Colon in Mice for Chemotherapy Assoys, with a Note on Carcinogen Structure", Canser Res, 35, 2434-2439 (1975); Corbett, T.H., et al., "A Mouse Colon-tumor Model for Experimental Therapy, Cancer Chemother. Rep (Part 2)", 5 169-186 (1975) и незначительно модифицированной. Опухоли вводят в левую часть живота с помощью инъекции шприцем выращенной культуры опухолевых клеток в количестве 1•106 ед. (клетки карциномы человека: MDA-MB-468 - карцинома молочной железы или HN5 - карцинома головы и шеи), суспендированных в 0,10 мл RPM1 1640. По прошествии достаточного промежутка времени, чтобы опухоли стали пальпируемыми (2-3 мм в диаметре), подопытных животных (атимную мышь) обрабатывают испытываемым веществом (обычно раствором в DMSO с концентрацией от 50 до 100 мг/мл с последующим разбавлением в соотношении 1: 9 1% Pluronic® P105 в 9%-ном растворе соли) посредством интраперитонеального (ip) или перорального (po) введения два раза в день (т.е. каждые 12 часов) в течение 5 дней. Для определения ингибирования роста опухоли измеряют два диаметра опухоли в миллиметрах Vermer-циркулем, размер опухоли (мг) вычисляют по формуле:
Вес опухоли (TuW) = (длина•[ширина]2)/2
в соответствии с методами, описанными в Geran, R.I., et al., Protocols for Screening Chemical Agents and Natural Products Against Animal Tumors and Other Biological Systems", Third Eddition. Cancer Chemother. Rep., 3, 1-104 (1972).
Ингибирование роста опухоли выражают в процентах по формуле:
Ингибирование (%) = (TuWкoнтpoль-TuWтecт)/TuWконтроль•100%
Место имплантации опухоли обеспечивает воспроизводимые эффекты доза/ответ различных химиотерапевтических агентов, а способ измерения (диаметр опухоли) является реализуемым способом оценки скоростей роста опухолей.
Введенное количество будет конечно зависеть от субъекта, которому необходимо лечение, степени поражения, способа введения и результатов предшествующего лечения. Однако эффективная доза заключается в области примерно 0,1-100 мг/кг, предпочтительно от 1 до 35 мг/кг в один прием или в несколько приемов. Для пациента весом 70 кг это составит количество от 0,05 до 7 г в день, предпочтительно от 0,2 до 2,5 г в день. Для местного введения (например, при псориазе) подходящая композиция должна включать от 0,01 до 5% соединения данного изобретения, предпочтительно от 0,05 до 0,5%. Предпочтительно при местном введении применять соединение непосредственно в месте поражения.
Фармацевтическая композиция данного изобретения может иметь форму, удобную для перорального введения, например изготавливаться в виде таблетки, капсулы, драже, порошка, композиции с длительным высвобождением действующего вещества, в виде раствора, суспензии, может иметь форму, удобную для парентеральной инъекции, например изготавливаться в виде стерильного раствора, суспензии или эмульсии, может иметь форму, удобную для местного введения, например изготавливаться в виде мази или крема, или может иметь форму, удобную для ректального введения, например, изготавливаться в виде суппозитория. Фармацевтическая композиция может изготавливаться в дозированных формах, подходящих для одного введения точно определенных доз. Фармацевтические композиции будут включать общепринятый фармацевтический носитель или наполнитель и в качестве активного ингредиента - соединение в соответствии с данным изобретением. Композиция может дополнительно включать другие лекарственные или фармацевтические средства, носители, адъюванты и т.д.
Фармацевтические композиции для неместного введения в соответствии с данным изобретением могут содержать от 0,1 до 95% соединения, предпочтительно 1%-70%. В любом случае композиция или лекарственный препарат, который подлежит введению, будет содержать количество соединения согласно данному изобретению, эффективное для смягчения или уменьшения симптомов заболевания у лечащегося субъекта, т.е. симптомов пролиферативных заболеваний, после завершения курса лечения.
Примерами форм, предназначенных для парентерального введения, являются растворы или суспензии соединения формулы I в соответствии с данным изобретением в стерильных водных растворах, например, применимы водный пропилен или коль или растворы декстрозы. Такие дозированные формы могут, при необходимости, содержать подходящий буфер.
Подходящими фармацевтическими носителями являются инертные разбавители или наполнители, вода и различные органические растворители. Фармацевтические композиции могут при необходимости содержать дополнительные ингредиенты, такие как вкусовые агенты, связующие агенты, наполнители и т.п. Так для перорального введения могут применяться таблетки, содержащие различные наполнители, такие как лимонная кислота, в сочетании с различными дезинтегрирующими агентами, такими как крахмал, альгиновая кислота и некоторые сложные силикаты, и связующими агентами, такими как сахароза, желатин и акация. Часто при получении таблеток полезно добавлять также смазывающие агенты, такие как стеарат магния, натрийлаурилсульфат и тальк. Твердые композиции аналогичного типа могут также применяться в мягких и твердых наполненных желатиновых капсулах. Предпочтительными материалами для таких капсул являются лактоза или молочный сахар и высокомолекулярные полиэтиленгликоли. Когда для перорального введения необходимы водные суспензии или эликсиры с большим количеством активного ингредиента, они наряду с обычными наполнителями, такими как вода, этанол, пропиленгликоль, глицерин или их смеси, могут содержать различные подслащивающие или вкусовые добавки, красители и, если это необходимо, эмульгирующие или суспендирующие вещества.
Способы получения различных фармацевтических композиций с дозированным количеством активного ингредиента хорошо известны и будут понятны специалисту из описания (см., например Remington's Pharmaceutical Science, Mack Publishing Company, Easter, Pa., 15th Eddition (1975)).
В качестве противоракового средства описанные выше соединения могут применяться без дополнительных лекарственных средств или их можно применять в сочетании с другими противоопухолевыми веществами или в сочетании с радиотерапией. Такое комплексное лечение может проводиться одновременным, последовательным циклическим или раздельным дозированием отдельных компонентов.
Следует учитывать, что изобретение не ограничивается конкретными воплощениями, приведенными в описании, но в пределах области данного изобретения допустимы различные изменения и модификации, которые могут осуществляться без выделения их области данного изобретения, которая определена в формуле изобретения.
Примеры
Анализ проводят с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (ВЭЖХ ОФ-18), образцы растворяют в водо-смешиваемом растворителе и вводят инъекцией в колонку (Perkin-Elmer RecosphereR),3x3C cartrige), 3 мм х 3 см, C18; доступна от Perkin Elmer Corp., Narwalk, CT 06959), которая соединяется с форколонкой Brownlee RP-8 Newgard (7 микрон, 3,2 мм х 15 мм, доступна от Applied Biosystems Inc. San Jose, CA 9511344), обе колонки предварительно уравновешивают в 200 мМ-ном буфере NH4OAc с pH 4,50. Образцы элюируют, используя линейный градиент (0-100%) системой MeCN/pH 4,50, 200 Мм NH4OAc спустя 10 минут со скоростью 3,0 мл/мин. Хроматограммы получают в области 240-400 нм, используется детектор Diode - структуры.
Газовую хроматографию и масс-спектрометрический анализ проводят на аппаратуре марки Hewlett Packard (5890 Series II) с колонкой 12 м HP-1 (200 М i. d. ). Градиент температуры составляет от 133oC (0-0,10 мин.) до 310oC при скорости нагрева 18oC/мин, в качестве носителя используется газообразный гелий, разделение компонентов осуществляют с использованием детектора пиков марки 5971 Mass Selective Detector.
Пример 1
4-(6-Хлор-2,3-дигидроиндол-1-ил)-7,8-дигидро-[1,4]диокси[2,3-g]хиназолин
К смеси 6-хлориндолина (52 мг, 0,330 ммоля) и пиридина (23,3 мг, 0,294 ммоля) в изопропаноле (3 мл) добавляют 4-хлор- 6,7-(этилендиокси)хиназолин (65 мг, 0,294 ммоля). Смесь нагревают до температуры дефлегмации под атмосферой сухого газообразного азота и выдерживают в этих условиях в течение 16 часов, после чего упаривают под вакуумом. Остаток экстрагируют смесью хлороформа и насыщенного водного раствора NaHCO3, органическую фазу промывают рассолом, сушат над сульфатом натрия и упаривают под вакуумом. Остаток подвергают быстрой хроматографии на кремнеземе, используя 30% раствор ацетона в гексане, в результате получают 84 мг 4-(6-хлор-2,3-дигидроиндол-1-ил)-7,8-дигидро-[1,4] диоксино[2,3- g]хиназолина в виде свободного основания (т.пл. 209-211oC; ГХ/МС: 339(M+); анал. ВЭЖХ с обр. фазой-С18 (ОФ-18 ВЭЖХ) - время удерживания: 5,02 мин.).
Пример 2
4-(6-Фтор-2, 3-дигидроиндол-1-ил)-6,7-диметоксихиназолин
К 6-фториндолину (274 мг, 2,0 ммоля) в сухом изопропаноле (10 мл) добавляют 4-хлор-6,7-диметоксихиназолин (225 мг, 1,0 ммоля). Смесь кипятят с обратным холодильником в течение 16 часов под атмосферой сухого газообразного азота, после чего растворитель отгоняют под вакуумом. Остаток парционно экстрагируют в хлороформе и 1М растворе гидроксида натрия, органическую фазу промывают рассолом, сушат над сульфатом натрия и упаривают под вакуумом. Остаток (535 мг) очищают быстрой хроматографией (кремнезем, элюент - 80% EtOAc/CH2Cl2), в результате получают 4-(6-фтор-2,3-дигидроиндол-1-ил)-6,7-диметоксихиназолин в виде свободного основания (239 мг) (ГХ/МС: 335(M+); ЖХ/МС: 326(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,37 мин.).
Для получения соли соляной кислоты этот продукт растворяют в минимальном количестве смеси CHCl3/Et2O и к полученной смеси при перемешивании добавляют по каплям 1М раствор соляной кислоты в диэтиловом эфире (1,0 мл). Образующуюся в виде выпавшего осадка соль соляной кислоты желтого цвета фильтруют, промывают сухим диэтиловым эфиром и петролейным эфиром и сушат под вакуумом до постоянной массы (310 мг, т.пл. 220oC (разл.)).
Пример 3
4-(6-Хлор-2,3-дигидроиндол-1-ил)-6,7-диметоксихиназолин
К смеси 6-хлориндолина гидрохлорида (100 мг, 0,526 ммоля) и пиридина (0,957 ммоля, 77 мкл) в сухом изопропаноле (8 мл) добавляют 4-хлор-6,7-диметоксихиназолин (107 мг, 0,478 ммоля). Смесь кипятят с обратным холодильником в течение 2 часов под атмосферой сухого газообразного азота, после чего растворитель отгоняют под вакуумом остаток парционно экстрагируют в хлороформе и 1М гидроксиде натрия, органическую фазу промывают рассолом, сушат над сульфатом натрия и упаривают под вакуумом. Остаток (171 мг) очищают быстрой хроматографией (кремнезем, элюент - 80% EtOAc/CH2Cl2), в результате получают 4-(6-хлор-2,3-дигидроиндол-1-ил)-6,7-диметоксихиназолин в виде свободного основания (148 мг) (Т.пл. 229-230oC; ЖХ/МС: 342(MH+); анал. ОФ-18 ВЭЖХ - время удерживания: 4,38 мин.).
Пример 4
4-(2, 3-Дигидроиндол-1-ил)-6,7-диметоксихиназолин
По методике, описанной в примере 2, из индолина (2 экв.) и 4-хлор-6,7-диметоксихиназолина (1,0 экв.) в изопропаноле получают указанный в заглавии продукт с выходом 94%. (Т.пл. 152-163oC; ЖХ/МС: 308(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4, 11 мин.).
Пример 5
6,7-диметокси-4(2-метил-2,3-дигидроиндол-1-ил)хиназолин
По методике, описанной в примере 1, из 2-метилиндолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв. ) в изопропаноле получают указанный в заглавии продукт с выходом 91%. (Т.пл. 154-156,5oC; ГХ/МС: 321(M+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,93 мин).
Пример 6
4-(4-Хлор-2, 3-дигидроиндол-1-ил)-6,7-диметоксихиназолин
По методике, описанной в примере 2, из 4-хлориндолина (2 экв.) и 4-хлор-6,7-диметоксихиназолина (1,0 экв.) в изопропаноле с выходом 92% получают указанный в заглавии продукт (т. пл. 172-179oC (разл. ); ЖХ/МС: 342(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,60 мин).
Пример 7
4-(3,4-Дигидро-2H-индол-1-ил)-6, 7-диметоксихиназолин
По методике, приведенной в примере 2, из 1,2,3,4-тетрагидрохинолина (2 экв. ) и 4-хлор-6,7-диметоксихиназолина (1,0 экв.) в изопропаноле с выходом 91% получают указанный в заглавии продукт (т.пл. 130-131oC; ЖХ/МС: 322(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,08 мин).
Пример 8
6,7-Диметокси-4-(6-метил-3, 4-дигидро-2H-хинолин-1-ил)хиназолин
По методике, приведенной в примере 2, из 6-метил-1,2,3,4-тетрагидрохинолина (2 экв.) и 4-хлор-6,7-диметоксихиназолина (1,0 экв.) в изопропаноле с выходом 94% получают указанный в заглавии продукт (т.пл. 147-148oC; ЖХ/МС: 336(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,51 мин).
Пример 9
6,7-Диметокси-4-(7-трифторметил- 3,4-дигидро-2H-хинолин-1-ил) хиназолин
По методике, приведенной в примере 2, из 6-трифторметил-1,2,3,4-тетрагидрохинолина (2 экв.) и 4-хлор-6,7-диметоксихиназолина (1,0 экв.) в изопропаноле с выходом 66% после перекристаллизации из смеси CHCl3/гексан получают указанный в заглавии продукт (т.пл. 184-185oC; ЖХ/МС: 390(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 5,10 мин).
Пример 10
4-(6-Хлор-2,3-дигидроиндол-1-ил)-6,7-диэтоксихиназолин
По методике, описанной в примере 1, из 6-хлориндолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв.) в изопропаноле с выходом 99% получают указанный в заглавии продукт. (Т.пл. 159-163oC; ГХ/МС: 369(M+); анал. ВЭЖХ ОФ-18 - время удерживания: 5,25 мин).
Пример 11
7-Бутокси-4-(6-хлор-2,3-дигидроиндол-1-ил)-6-метоксихиназолин
По методике, описанной в примере 1, из 6-хлориндолина (1,1 экв.) и 7-бутокси-4-хлор-6-метоксихиназолина (1,0 экв.) в изопропаноле с выходом 45% получают указанный в заглавии продукт (т.пл. 126-132oC; ГХ/МС: 383(M+); анал. ВЭЖХ ОФ-18 - время удерживания: 6,22 мин).
Пример 12
6,7-диметокси-4-(6-метил-2, 3-дигидро-индол-1-ил) хиназолин гидрохлорид
По методике, описанной в примере 1, из 6-метилиндолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв. ) в изопропаноле получают указанный в заглавии продукт с выходом 88%. Соль соляной кислоты получают из очищенного свободного основания по методике, приведенной в примере 2. (Т.пл. 231-232oC; ЖХ/МС: 322(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,32 мин).
Пример 13
6,7-Диметокси-4- (4-метил-2,3-дигидроиндол-1-ил)хиназолин
По методике, описанной в примере 1, из 4-метилиндолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв. ) в изопропаноле указанный в заглавии продукт получают с выходом 94%. (Т.пл. 174-175oC; ЖХ/МС: 322(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,85 мин).
Примеры 14 - 15
6,7-Диметокси-4- (цис-2, 3-диметил-2, 3-дигидроиндол-1-ил) хиназолин и 6,7-Диметокси-4-(4-транс-2,3-диметил-2,3- дигидроиндол-1-ил) хиназолин
По методике, описанной в примере 1, из коммерческой смеси цис/транс-2,3-диметилиндолина (1,1 экв.) и 4-хлор-6,7-диметок-сихиназолина (1 экв.) в изопропаноле указанные в заглавии продукты получают с выходом 65% и 15% соответственно, после хроматографирования рацематной смеси (кремнезем, элюент: 55-65% этилацетат/гексан). Для цис-изомера: Т.пл. 174-175oC; ЖХ/МС: 336(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 5,15 мин. Для транс-изомера: ЖХ/МС: 336(MH+); анал. ВЭЖХ ОФ-18 - время удерживания 4,83 мин.
Пример 16
4-(6-Йод-2,3-дигидроиндол-1-ил) -6,7-диметоксихиназолин гидрохлорид
По методике, описанной в примере 1, из 6-йодиндолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1экв.) в изопропаноле получают указанный в заглавии продукт в виде свободного основания с выходом 78%. Соль соляной кислоты получают из очищенного свободного основания по методике, приведенной в примере 2. (Т.пл. больше 230oC; ГХ/МС: 433(M+); анал. ВЭЖХ ОФ-18 - время удерживания: 5,20 мин).
Пример 17
4-(5-Фтор-2, 3-дигидроиндол-1-ил)-6,7-диметоксихиназолин
По методике, описанной в примере 1, из 5-фториндолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв. ) в изопропаноле получают указанный в заглавии продукт с выходом 81%. (Т.пл. 190-191oC; ЖХ/МС: 326(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,40 мин).
Пример 18 \ \\ 4-(6-хлор-2,3-дигидроиндол-1-ил)-6,7, 8-триметоксихиназолин
По методике, описанной в примере 1, из 5-хлориндолина (1,1 экв.) и 4-хлор-6,7,8-триметоксихиназолина (1 экв.) в изопропаноле получают указанный в заглавии продукт с выходом 20%. (Т.пл. 139-143oC; ГХ/МС: 371(M+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,70 мин).
Пример 19
4-(6-Бензилокси-2, 3-дигидроиндол-1-ил)-6, 7-диметоксихиназолин гидрохлорид
По методике, описанной в примере 1, из 6-бензилоксииндолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв.) в изопропаноле получают указанный в заглавии продукт с выходом 80%. Соль соляной кислоты получают из очищенного свободного основания по методике, приведенной в примере 2. (Т.пл. 249-250oC (разл.); ЖХ/МС: 414(M+); анал. ВЭЖХ ОФ-18 - время удерживания: 5,01 мин).
Пример 20
6,7-Диметокси-4- (6-метокси-2,3-дигидроиндол-1-ил)хиназолин гидрохлорид
По методике, описанной в примере 1, из 6-метоксииндолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв.) в изопропаноле получают указанный в заглавии продукт с выходом 91%. Соль соляной кислоты получают из очищенного свободного основания по методике, приведенной в примере 2. (Т.пл. 246-247oC (разл.); ЖХ/МС: 338(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,27 мин).
Пример 21
4-(6-Бром-2, 3-дигидроиндол-1-ил)-6,7-диметоксихиназолин гидрохлорид
По методике, описанной в примере 1, из 6-броминдолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв. ) в изопропаноле получают указанный в заглавии продукт с выходом 88%. Соль соляной кислоты получают из очищенного свободного основания по методике, приведенной в примере 2. (Т.пл. 245-248oC (разл. ); ЖХ/МС: 386, 388(MH+)-, анал. ВЭЖХ ОФ-18 - время удерживания: 4,95 мин).
Пример 22
4-(5-хлор-2,3-дигидроиндол-1-ил)-6,7-диметоксихиназолин
По методике, описанной в примере 1, из 5-хлориндолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв. ) в изопропаноле получают указанный в заглавии продукт с выходом 92%. (Т.пл. 190-191oC; ГХ/МС: 341(M+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,58 мин).
Пример 23
6,7-Диметокси-4-(5-метил-2,3-дигидроиндол-1-ил) хиназолин
По методике, описанной в примере 1, из 5-метилиндолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв. ) в изопропаноле указанный в заглавии продукт получают с выходом 94 %. (Т.пл. 180-181oC; ГХ/МС: 321(M+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,37 мин).
Пример 24
Метиловый эфир 1-(6,7-диметоксихиназолин-4-ил)-2,3-дигидро-1H-индол-2-карбоновой кислоты
К раствору 4-хлор-6,7-диметоксихиназолина (1,01 г, 4,50 ммоля) в ДМФА (10 мл) добавляют метиловый эфир DL-индолин-2- карбоновой кислоты (0,806 г, 4,55 ммоля) и пиридин (0,236 мл, 4,51 ммоля), смесь нагревают до температуры 80oC и выдерживают при этой температуре в течение 4,5 часов. Продукт выделяют с выходом 87% экстракцией и быстрой хроматографией по методике примера 1. (Т. пл. 186-189,5oC; ЖХ/МС: 366(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,28 мин).
Пример 25
4-(3,4-Дигидро-2Н-хинолин-1-ил)-6-метоксихиназолин
По методике, приведенной в примере 2, из 1,2,3,4-тетрагидрохинолина (2 экв. ) и 4-хлор-6-метоксихиназолина (1,0 экв.) в этаноле с выходом 59% получают указанный в заглавии продукт (Т.пл. 98oC; ЖХ/МС: 292(MH+).
Пример 26
6,7-Диметокси-4- (6-нитро-2,3-дигидроиндол-1-ил)хиназолин
По методике, описанной в примере 1, из 6-нитроиндолина (1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв. ) в изопропаноле получают указанный в заглавии продукт с выходом 91%. (Т.пл. 275oC (разл.); ГХ/МС: 352(M+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,30 мин).
Пример 27
4-(6-Бром-5-фтор-2,3- дигидроиндол-1-ил)- 6, 7-диметоксихиназолин гидрохлорид
По методике, описанной в примере 1, из 5-фтор-6-броминдолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв.) в изопропаноле получают указанный в заглавии продукт с выходом 68% (Т.пл. 258oC (разл.); ЖХ/МС: 404, 406(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 5,06 мин).
Пример 28
6,7-Диметокси-4-(7-метил-2,3-дигидроиндол-1-ил) хиназолин
По методике, описанной в примере 24, из 7-метилиндолина (1 экв.), 4-хлор-6,7-диметоксихиназолина (1 экв.) и пиридина (2 экв.) получают указанный в заглавии продукт с выходом 65%. (Т.пл. 146-147oC; ГХ/МС: 321(M+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,41 мин).
Пример 29
[1-(6,7-Диметоксихиназолин-4-ил)- 2,3-дигидро-1H-индол-2-ил] метанол
По методике, описанной в примере 1, из 2-гидроксиметилиндолина (1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв.) в изопропаноле получают указанный в заглавии продукт с выходом 57%. (Т.пл. 145,5-147oC; ЖХ/МС: 338(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,03 мин).
Пример 30
1-(6, 7-Диметоксихиназолин-4-ил)-2,3-дигидро-1H-индол-6-иламин
Раствор 6,7-Диметокси-4-(6-нитро-2,3-дигидро-индол-1-ил) хиназолина (0,50 г, 1,42 ммоля; из примера 26) в метаноле (10 мл) и соляной кислоты (1 мл) гидрируют в течение 18 часов водородом с давлением 45 пси в присутствии 10% палладия на углероде (105 мг). После этого смесь фильтруют через броунмереллит, фильтрат упаривают под вакуумом. Остаток переносят в 10%-ный раствор изопропанол/хлороформ, органическую фазу промывают насыщенным раствором бикарбоната натрия, 0,1 М раствором этилендиаминтетрауксусной кислоты (ЭДТУК) и рассолом. Органическую фазу сушат над сульфатом натрия, фильтруют и упаривают под вакуумом, в результате получают указанный в заглавии продукт в виде твердого вещества (423 мг; т.пл. 215-217,5oC; ЖХ/МС: 323(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 3,10 мин).
Пример 31
4-(6-изотиоцианато-2, 3-дигидроиндол-1-ил)-6,7-диметоксихиназолин
К раствору тиокарбонилдиимидазола (90 мг, 0,505 ммоля) в хлороформе (1 мл) в течение 2-х часов при температуре 5-10oC по каплям добавляют раствор 1-(6,7-диметоксихиназолин-4-ил)-2,3-дигидро-1H-индол-6-иламина (163 мг, 0,506 ммоля; из примера 30) в метилцианиде (8 мл). После перемешивания в течение 4 часов при температуре 5oC растворитель отгоняют под вакуумом и полученный остаток очищают методом быстрой хроматографии на кремнеземе (элюент: 1% раствор метанола в хлороформе), в результате получают 115 мг изотиоцианата, указанного в заглавии Т.пл. 215-217,5oC; ГХ/МС: 364(M+); анал. ВЭЖХ ОФ-18 - время удерживания: 5,60 мин).
Пример 32
6,7-Диметокси-4-(6-пиррол-1-ил-2,3-дигидроиндол-1-ил)хиназолин
К раствору 1-(6,7-Диметоксихиназолин-4-ил)-2,3-дигидро- 1H-индол-6-иламина (155 мг, 0,481 ммоля, из примера 30) в ледяной уксусной кислоте (4 мл) добавляют 1,4-диметокситетрагидрофуран (72,3 мкл, 0,558 ммоля). Смесь нагревают до температуры 90oC под атмосферой сухого газообразного азота, выдерживают при этой температуре в течение 4 часов, затем упаривают под вакуумом. Остаток растворяют в хлороформе, дважды промывают насыщенным водным раствором бикарбоната натрия и рассолом, органическую фазу сушат над сульфатом магния, фильтруют и упаривают под вакуумом, остаток хроматографируют на кремнеземе (50% смесь ацетон/гексан), в результате получают 71 мг 6-пирролил-производного. (Т.пл. 185-186oC; ГХ/МС: 372(M+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,60 мин).
Примеры 33-34
6,7-Диметокси-4-(транс-октагидроиндол-1-ил)хиназолин и 6, 7-Диметокси-4-(цис-октагидроиндол-1-ил)хиназолин
По методике, описанной в примере 24, указанные в заглавии продукты получают из цис/транс-пергидроиндола (1,1 экв.), триэтиламина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1,0 экв.) в ДМФА. Изомеры разделяют на кремнеземе, используя этил-ацетат в качестве элюента, в результате получают транс-изомер (т.пл. 130-131oC; ЖХ/МС: 314(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,05 мин) с выходом 16% и цис-изомер (т.пл. 123-124oC; ЖХ/МС; 314 (MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 3,89 мин) с выходом 46%.
Пример 35
4- (6-Бром-7-метил-2,3-дигидроиндол-1-ил)-6,7-диметоксихиназолин
По методике, описанной в примере 24, из 6-бром-7-метилиндолина (1,1 экв. ) и 4-хлор-6,7-диметоксихиназолина (1 экв. ) в ДМФА получают указанный в заглавии продукт с выходом 93%. (Т. пл.240-242oC; ЖХ/ МС: 400, 402 (MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 5,13 мин). Соль соляной кислоты получают по методике, описанной в примере 2: т.пл. 243-244oC.
Пример 36
4-(4-Бром-7-метил-2, 3-дигидроиндол-1-ил)-6,7-диметоксихиназолин
По методике, описанной в примере 24, из 4-бром-7-метилиндолина (1,1 экв. ) и 4-хлор-6,7-диметоксихиназолина (1 экв.) в ДМФА получают указанный в заглавии продукт с выходом 70%. (Т. пл.223-225oC; ЖХ/МС: 400, 402(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 5,23 мин).
Пример 37
4-(6-Бутил-2,3-дигидроиндол-1-ил)-6,7-диметоксихиназолин
По методике, описанной в примере 1, из 6-н-бутилаиндолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв.) в изопропаноле получают указанный в заглавии продукт с выходом 53%. (Т.пл.131-131oC; ГХ/ МС: 363(М+); анал. ВЭЖХ ОФ-18 - время удерживания: 5,41 мин).
Пример 38 6,7-Диметокси-4-(6-фенил-2,3-дигидроиндол-1-ил)хиназолин
По методике, описанной в примере 1, из 6-фенилиндолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв. ) в изопропаноле получают указанный в заглавии продукт с выходом 61%. (Т.пл. 175-177oC ГХ/ МС: 383(М+); анал. ВЭЖХ ОФ-18 - время удерживания: 6,03 мин).
Пример 39
5-(6,7-Диметоксихиназолин-4-ил)-6,7-дигидро-5H-[1,3] диоксоло[4,5-f]индол
По методике, описанной в примере 1, из 6,7-дигидро- 5H-[1,3]диоксоло[4,5-f]индола (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв.) в изопропаноле получают указанный в заглавии продукт с выходом 93%. (Т.пл. 225-228oC (разл. ); ЖХ/МС: 352(MH+); анал. ВЭЖХ ОФ-18 - время удерживания 3,91 мин).
Пример 40
4-(6-Изопропил-2,3-дигидроиндол-1-ил)-6, 7-диметоксихиназолин
По методике, описанной в примере 1, из 6-изопропилиндолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв.) в изопропаноле получают указанный в заглавии продукт с выходом 43%. (Т.пл.128-129oC; ЖХ/МС: 350(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 5,51 мин).
Пример 41
6,7-Диметокси-4-(6-пропил-2,3-дигидроиндол-1-ил)хиназолин
По методике, описанной в примере 1, из 6-пропилиндолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв.) в изопропаноле получают указанный в заглавии продукт с выходом 43%. (Т.пл. 140-141oC; ЖХ/МС: 350(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 5,73 мин).
Пример 42
4-(6-Азидо-2, 3-дигидроиндол-1-ил)-6,7-диметоксихиназолин
К раствору 1-(6,7-диметоксихиназолин-4-ил)-2,3-дигидро-1H-индол-6-иламина (104 мг, 0,323 ммоля; из примера 30) в 80%- ном растворе уксусной кислоты в воде (8 мл) при температуре 5oC добавляют раствор нитрита натрия (35 мг, 0,35 ммоля) в воде (200 мкл). После перемешивания в течение 10 минут при температуре 5oC к смеси добавляют раствор NaN3 (22 мг, 0,34 ммоля) в воде (200 мкл), смеси дают нагреться до 22oC и перемешивают ее в течение часа при этой температуре. После удаления растворителя лиофилизацией остаток растворяют в этилацетате, дважды промывают насыщенным водным раствором бикарбоната натрия, затем рассолом, органическую фазу сушат над сульфатом магния, фильтруют и упаривают под вакуумом. После хроматографирования остатка на кремнеземе (элюент: 2%-ный раствор метанола в хлороформе) получают 72 мг азид-замещенного продукта (т. пл. 184-186oC (разл.); ЖХ/МС: 349(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,70 мин).
Пример 43
[1-(6, 7-Диметоксихиназолин-4-ил)-2,3-дигидро-1H-индол-5-ил - (1-метилгептил)амин
По методике, описанной в примере 1, из [2,3-дигидро-1H-индол-5-ил], (1-метилгептил) амина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв.) в изопропаноле получают указанный в заглавии продукт с выходом 96%. (Т.пл. 61-63oC; ГХ/ МС: 434 (М+); анал. ВЭЖХ ОФ-18 - время удерживания: 6,91 мин).
Пример 44
Соль 6,7-Диметокси-4-(5-метокси-2, 3-дигидроиндол-1-ил)хиназолина и метансульфоновой кислоты
По методике, описанной в примере 1, из 5-метоксииндола (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв. ) в изопропаноле получают указанный в заглавии продукт с выходом 95%. Свободное основание подвергают очистке методом колоночной хроматографии (кремнезем, элюент: 45%-ный раствор ацетона в гексане), затем растворяют в минимальном количестве метиленхлорида и обрабатывают 1 экв. метансульфоновой кислоты в CH2Cl2, затем растворяют в нескольких объемах эфира для высаживания месилата, который фильтруют и сушат под вакуумом. (Т.пл. 285-292oC (разл.); ЖХ/МС: 338(М+); анал. ВЭЖХ ОФ-18 - время удерживания: 3,82 мин).
Пример 45
4-(5-Бензилокси-2,3-дигидроиндол-1-ил)-6,7-диметоксихиназолин
По методике, описанной в примере 1, из 5-бензилоксииндолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв.) в изопропаноле получают указанный в заглавии продукт с выходом 90%. (Т.пл.181-182oC; ЖХ/МС: 415(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,78 мин).
Пример 46
6,7-Диметокси-4- (6-пирролидин-1-ил-2,3-дигидроиндол-1- ил)хиназолин
К раствору 1-(6,7-диметоксихиназолин-4-ил)-2,3-дигидро-1H-индол-6-иламина (103 мг, 0,320 ммоля; из примера 30) в ДМФА (4 мл) добавляют 1,4-дибромбутан (42 мкл, 0,352 ммоля) и пиридин (34 мкл, 0,64 ммоля), смесь нагревают до 110oC и выдерживают при этой температуре под атмосферой газообразного азота в течение 36 часов. После этого смесь разбавляют насыщенным водным раствором бикарбоната натрия и экстрагируют смесью этилацетата и этанола (1: 1), органическую фазу затем промывают водой и рассолом, сушат над сульфатом натрия, фильтруют и упаривают под вакуумом. Хроматографирование (4% MeOH/CHCl3) остатка на кремнеземе приводит к получению 20 г указанного в заглавии продукта. (Т. пл. 198-205oC (разл); ГХ/МС: 376(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 5,05 мин).
Пример 47
Соль 4-(6-Хлор-5-фтор-2, 3-дигидроиндол-1-ил)-6,7-диметоксихиназолина и метансульфоновой кислоты
Смесь 6-хлор-5-фториндолина (157 мг, 0,9215 ммоля), пиридина (48 мкл) и 4-хлор-6,7-диметоксихиназолина (210 мг, 0,895 ммоля) нагревают на 120oC в N-метилпирролидин-2-оне (2 мл) под атмосферой газообразного азота и выдерживают в этих условиях в течение 36 часов. Затем смесь разделяют между этилацетатом и насыщенным водным раствором бикарбоната натрия, органическую фазу несколько раз промывают водой и рассолом, сушат над сульфатом магния, фильтруют и упаривают под вакуумом. После очистки остатка методом быстрой хроматографии (кремнезем, элюент: 40%-ный раствор ацетона в гексане) получают продукт (90 мг), который превращают в соль метансульфоновой кислоты по методике, описанной в примере 44. (Т.пл. 261-264oC; ЖХ/МС: 360 (MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,31 мин).
Пример 48
Соль 1-(6, 7-Диметоксихиназолин-4-ил)-2,3-дигидро-1H-индол-5-ола и метансульфоновой кислоты
По методике, описанной в примере 1 (с превращением в соль метансульфоновой кислоты по методике примера 44), из 5-гидроксииндолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв.) в изопропаноле получают указанный в заглавии продукт с выходом 90%. (Т.пл. 245-240oC (разл.); ЖХ/МС: 324(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 2,76 мин).
Пример 49
Соль 1-(6, 7-Диметоксихиназолин-4-ил)-6-метил-2,3-дигидро-1H-индол-5-ола и метансульфоновой кислоты
По методике, описанной в примере 1 (с преврашением в соль метансульфоновой кислоты по методике примера 44), из 5-гидрокси-6-метилиндолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв.) в изопропаноле получают указанный в заглавии продукт с выходом 45%. (Для свободного основания: Т.пл. 230oC; для соли: т.пл. 290oC (разл.) ЖХ/МС: 338 (MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 3,11 мин).
Пример 50
Соль 4-(6,7-Диметил-2,3-дигидроиндол-1-ил)-6,7-диметоксихиназолина и метансульфоновой кислоты
По методике, описанной в примере 47 (с превращением в соль метансульфоновой кислоты по методике примера 44), из 6,7-диметилиндолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв.) в N-метилпирролидин-2-оне получают указанный в заглавии продукт с выходом 92% (т.пл. 232-237oC (разл.); ГХ/МС: 335(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,44 мин).
Пример 51
Соль 1-(6, 7-Диметоксихиназолин-4-ил)-2,3,4,5-тетрагидро-1H-бензо[b] азепина и метансульфоновой кислоты
По методике, описанной в примере 47 (с превращением в соль метансульфоновой кислоты по методике примера 44), из 2,3,4,5-тетрагидро-1H-бензо[b]азепина (1,1 экв. ) и 4-хлор-6,7-диметоксихиназолина (1 экв.) в N-метилпирролидоне получают указанный в заглавии продукт с выходом 92 % (т.пл. 218,2-218,6oC; ЖХ/МС: 336(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,46 мин).
Пример 52
6,7-Диметокси-4-(5-нитро-2,3-дигидроиндол-1-ил)хиназолин
Используя методику, описанную в примере 47, в N-метилпирролидоне получают с выходом 42% аддукт 5-нитроиндолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1,0 экв. ) (т.пл. 245-249oC (разл.); ЖХ/МС: 353(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 3,95 мин).
Пример 53
4- 5-Азидо-2,3-дигидроиндол-1-ил)-6,7-диметоксихиназолин
Раствор 1-(6, 7-диметоксихиназолин-4-ил)-2,3-дигидро-1H-индол-5-иламина (155 мг, 0,481 ммоля; из примера 83 в виде свободного основания) в 80%-ном водном растворе уксусной кислоты (10 мл) обрабатывают по методике примера 42, в результате получают азид производной с выходом 47%. (Т.пл. 159-165oC. (разл.); ЖХ/МС: 349(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,93 мин).
Пример 54
Соль 6-хлор-1-(6, 7-диметоксихиназолин-4-ил)-2,3-дигидро-1H-индол-5-ола и метансульфоновой кислоты
По методике, описанной в примере 1 (с превращением в соль метансульфоновой кислоты по методике примера 44), из 6-хлор-5-гидроксииндолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв.) в изопропаноле получают указанный в заглавии продукт с выходом 49%. (Т.пл. 268-275oC; ЖХ/МС: 358 (MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 3,45 мин).
Пример 55
Соль 1- (6, 7-диметоксихиназолин-4-ил)-7-метил-2, 3-дигидро-1H-индол-5-ола и метансульфоновой кислоты
По методике, описанной в примере 1 (с превращением в соль метансульфоновой кислоты по методике примера 44), из 5-гидрокси-7-метилиндолина (1,1 экв. ) и 4-хлор-6,7-диметоксихиназолина (1 экв.) в изопропаноле получают указанный в заглавии продукт с выходом 73%. (Т.пл. 145-146oC (разл.); ЖХ/МС: 338(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 3,20 мин).
Пример 56
1-(6,7-диметоксихиназолин-4-ил) -6,7-диметил-2,3-дигидро-1H-индол-5-ол гидрохлорид
По методике, описанной в примере 47 (с превращением в соль соляной кислоты по методике примера 2), из 5-гидрокси-6,7-метилиндолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв.) в N-метилпирролидоне получают указанный в заглавии продукт с выходом 40%. (Т.пл. 207-209oC (разл.); ЖХ/МС: 352 (MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 3,48 мин).
Пример 57
Соль 4- (5, 7-дихлор-2, 3-дигидроиндол-1-ил)-6,7-диметоксихиназолина и уксусной кислоты
По методике, описанной в примере 47, из 5,7-дихлориндолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина (1 экв.) в N-метилпирролидиноне получают указанный в заглавии продукт с выходом 16%. Продукт выделяют в виде соли уксусной кислоты с помощью ВЭЖХ ОФ-18 (C18), используя градиентное элюирование (10-60% CH3CN /pH 4,5 50 мМ NH4OAc с последующей лиофилизацией. (Т.пл. 228-232,5oC; ЖХ/МС: 376(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 5,20 мин.)
Пример 58
4-(6-Хлор-5-нитро-2,3- дигидроиндол-1-ил)- 6,7-диметоксихиназолин
По методике, описанной в примере 47, из 6-хлор-5-нитроиндолина (1,1 экв. ) и 4-хлор-6,7-диметоксихиназолина (1 экв.) в N- метилпирролидоне получают указанный в заглавии продукт с выходом 24% (т. пл. 276-278oC; ЖХ/МС: 387(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,48 мин).
Пример 59
6-Хлор-1-(6,7-диметоксихиназолин-4-ил)-2,3-дигидро-1H-индол- 5-иламин гидрохлорид
К раствору 4-(6-Хлор-5-нитро-2, 3-дигидроиндол-1-ил)-6,7- диметоксихиназолина (150 мг, 0,386 ммоля; из примера 58) в тетрагидрофуране (5 мл) с 10%-ным палладием на углероде (21 мг) при температуре 40oС добавляют по каплям в течение 20 минут раствор NaH2PO2H2O (490 мг, 3,86 ммоля) в воде (0,5 мл) Смесь перемешивают в течение 5 часов при температуре 40 С. После этого раствор фильтруют через броунмиллерит, фильтрат упаривают под вакуумом, остаток растворяют в 10%-ном растворе изопропанола в хлороформе, промывают насыщенным водным раствором бикарбоната натрия, рассолом, сушат над сульфатом натрия, фильтруют и упаривают под вакуумом, в результате получают 121 мг (90%) продукта, который превращают в соль соляной кислоты по методике примера 2. (Т.пл. 223-228oC; ЖХ/МС: 357(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 3,68 мин).
Пример 60
4-(4-Бром-7-метил-2, 3-дигидроиндол-1-ил)-7-метоксихиназолин
По методике, описанной в примере 1, из 4-бром-7-метилиндолина (1,1 экв.) и 4-хлор-7-метоксихиназолина (1 экв.) в изопропаноле получают указанный в заглавии продукт с выходом 69%. (Т.пл. 134-135oC: ЖХ/МС: 370, 372(MH+); анал. ВЭЖХ ОФ-18 -время удерживания: 5,60 мин).
Пример 61 4-(6-Бром-7-метил-2,3-дигидроиндол-1-ил)-7-метоксихиназолин гидрохлорид
По методике, описанной в примере 1 (с получением соли соляной кислоты по методике примера 2), из 6-бром-7-метилиндолина (1,1 экв.) и 4-хлор-7-метоксихиназолина (1 экв.) в изопропаноле получают указанный в заглавии продукт с выходом 49%. (Т.пл. 200-205oC: ЖХ/МС: 370, 372(MH+); анал. ВЭЖХ ОФ-18 -время удерживания: 5,76 мин).
Пример 62
7-Метокси-4-(7-метил-2, 3-дигидроиндол-1-ил)хиназолин
По методике, описанной в примере 1, из 7-метилиндолина (1,1 экв.) и 4-хлор-7-метоксихиназолина (1 экв.) в изопропаноле получают указанный в заглавии продукт с выходом 79%. (Т.пл. 157-160oC; ЖХ/МС: 292(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,30 мин).
Пример 63
4-(6-Бром-5-фтор-2,3-дигидроиндол-1-ил)-7-метоксихиназолин гидрохлорид
По методике, описанной в примере 1 (с получением соли соляной кислоты по методике примера 2), из 6-бром-5-фториндо-лина (1,1 экв.) и 4-хлор-7-метоксихиназолина (1 экв.) в изо-пропаноле получают указанный в заглавии продукт с выходом 74%. (Т.пл. 252-252oC; ЖХ/МС: 374, 376(MH+); анал. ВЭЖХ ОФ-18 -время удерживания: 5,26 мин).
Пример 64
4-(6-Хлор-2,3-дигидроиндол-1-ил)-7-метоксихиназолин гидрохлорид По методике, описанной в примере 1 (с получением соли соляной кислоты по методике примера 2), из 6-хлориндолина (1,1 экв.) и 4-хлор-7-метоксихиназолина (1 экв. ) в изопропаноле получают указанный в заглавии продукт с выходом 82%. (Т.пл. для свободного основания: 140-141oC; для соли: т.пл.232-233oC; ЖХ/МС: 312(MH+); анал. ВЭЖХ ОФ-18 -время удерживания: 5,68 мин).
Пример 65
4-(6-Хлор-2,3-дигидроиндол-1-ил)-хиназолин-7-ол
К расплавленному пиридингидрохлориду (85 г) при температуре 170oC добавляют несколькими порциями 4-(6-хлор-2,3-дигидроиндол-1-ил)-7-метоксихиназолин (7,596 г, 24,4 ммоля; свободное основание из примера 64). Смесь нагревают в течение 75 минут, затем выливают в смесь лед/вода (примерно 600 мл). Выпавшее в осадок твердое вещество фильтруют, растворяют в 10%-ном растворе пропанол/хлороформ органическую фазу промывают насыщенным раствором бикарбоната натрия, затем рассолом, сушат над сульфатом натрия, фильтруют и упаривают под вакуумом, в результате получают 3,14 г продукта (т.пл. 270-280oC (разл. ); ЖХ/МС: 298(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,00 мин.).
Пример 66
4-(6-Хлор-2,3-дигидроиндол-1-ил)-7-(2-метоксиэтокси)хиназолин гидрохлорид
К раствору 4-(6-хлор-2, 3-дигидроиндол-1-ил)-хиназолин-7-ола (125 мл, 0,42 ммоля; из примера 65) в ДМФА (2 мл) добавляют воду (50 мкл), K2CO3 (тв. ) (116 мг, 0,84 ммоля), тетраметиламмонийгидрохлорид пентагидрат (15 мг, 0,08 ммоля) и 2-бромэтилметиловый эфир (43 мкл, 0,46 ммоля). Смесь перемешивают под атмосферой газообразного азота при температуре 50oC в течение 24 часов, после чего добавляют другую аликвоту (43 мкл) 2-бромэтилметилового эфира. Перемешивание продолжают при температуре 50oC в течение еще 36 часов, далее смесь экстрагируют и хроматографируют в соответствии с описанием, приведенным в примере 1, и затем полученный продукт превращают в соль соляной кислоты, как описано в примере 2, в результате получают 91 мг (61%) указанного в заглавии продукта (т.пл. 213-216oC (разл.); ЖХ/МС: 356(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,73 мин).
Пример 67
2-4-(6-Хлор-2,3-дигидроиндол-1-ил)хиназолин-7-илокси]этанол гидрохлорид
К раствору 4-(6-хлор-2, 3-дигидроиндол-1-ил)-хиназолин-7-ола (125 мл, 0,42 ммоля; из примера 65) в ДМФА (2 мл) добавляют воду (50 мкл), K2CO3 (тв. ) (116 мг, 0,84 ммоля), тетраметиламмонийгидрохлорид пентагидрат (15 мг, 0,0 ммоля) и 2-бромэтанол (39 мкл, 0,46 ммоля). Смесь перемешивают при температуре 50oC под атмосферой газообразного азота, после чего с интервалом 24 часа добавляют 3 другие аликвоты 2-бромэтанола (3 х 20 мкл). Далее экстракцию и хроматографирование выполняют в соответствии с методикой примера 1, а затем соль соляной кислоты получают по методике примера 2, в результате получают 76 мл (53%) указанного в заглавии продукта. (Т.пл. 215-217,5oC; ЖХ/МС: 342(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,09 мин).
Пример 68
3-[4-(6-Хлор-2, 3-дигидроиндол-1-ил)хиназолин-7-илокси] этанол гидрохлорид
К раствору 4-(6-хлор-2,3-дигидроиндол-1-ил)-хиназолин-7-ола (125 мл, 0,42 ммоля; из примера 65) в ДМФА (2 мл) добавляют воду (50 мкл), K2CO3 (тв. ) (116 мг, 0,84 ммоля), тетраметиламмонийгидрохлорид пентагидрат (15 мг, 0,08 ммоля), а затем и 3-бромпропанол (43 мкл, 0,46 ммоля). Смесь перемешивают под атмосферой газообразного азота при температуре 50oC в течение 24 часов перед добавлением другой аликвоты (11 мкл) 3-бромпропанола. Перемешивание продолжают при температуре 50oC в течение еще 24 часов, далее смесь экстрагируют и хроматографируют в соответствии с описанием, приведенным в примере 1, и затем полученный продукт превращают в соль соляной кислоты, как описано в примере 2, в результате получают 119 мг (70%) указанного в заглавии продукта (т.пл. 211-223oC (разл.); ЖХ/МС: 356 (МН+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,40 мин.; т.пл. свободного основания 137-138oC).
Примеры 69 - 70
4-(6-Хлор-2,3-дигидроиндол-1-ил)хиназолин-6,7-диол и 4-(6- Хлор-2, 3-дигидроиндол-1-ил)-6-метоксихиназолин-7-ол
4-(6-Хлор-2,3-дигидроиндол-1-ил)-6,7-диметоксихиназолин (1,00 г, 2,93 ммоля; из примера 3) добавляют к расплавленному гидрохлориду пиридина (10,14 г, 88 ммоля) при температуре 170oC. Смесь перемешивают при температуре 170oC в течение 0,5-1,0 часа (до тех пор, пока в реакционной массе исходный материал будет фиксироваться методом ВЭЖХ ОФ-18 в количестве менее 5%), а затем выливают в смесь вода/лед (110 мл). Выпавший осадок твердого вещества оранжевого цвета выделяют фильтрацией, сушат азеотропно с метилцианидом при температуре 40oC под вакуумом, затем растворяют в 15%-ном растворе изопропанола в хлороформе (75 мл), промывают насыщенным раствором бикарбоната натрия (х2). Органическую фазу сушат над сульфатом натрия, фильтруют и упаривают под вакуумом. Остаток (примерно 800 мг) обрабатывают хлороформом (60 мл) и фильтруют для выделения 6,7-диола (обычно 200 мл, 94%-ной чистоты, используют без дополнительной очистки) (т. пл. 200oC (разл. ); ЖХ/МС: 314(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,07 мин). Фильтрат подвергают быстрой хроматографии (кремнезем, градиентное элюирование 40-80% ацетон/метиленхлорид), получают примерно 550 мг чистого 6-метокси-хиназолин-7-ола (т. пл. 175oC (разл.); ЖХ/МС: 328(MH+); анал.ВЭЖХ ОФ-18 - время удерживания: 4,07 мин).
Пример 71
Соль 4-(6-Хлор-2, 3-дигидроиндол-1-ил)-6-метокси-7-(2-метокси-этокси)хиназолина и метансульфоновой кислоты
4-(6-Хлор-2, 3-дигидроиндол-1-ил)-6-метоксихиназолин-7-ол (100 мг, 0,305 ммоля; из примера 70) взаимодействует с 2-бром-этилметиловым эфиром по методике примера 66, последующую экстракцию и хроматографирование выполняют в соответствии с описанием, приведенным в примере 1, в результате получают 53 мг продукта, который превращают в соль метансульфоновой кислоты по методике примера 44. (Т.пл. 248oC (разл.); ЖХ/МС: 386 (MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,56 мин).
Пример 72
Соль 4-(6-хлор-2,3-дигидроиндол-1-ил)-6,7-бис (2-метоксиэтокси)хиназолина и метансульфоновой кислоты
К раствору 4-(6-хлор-2, 3-дигидроиндол-1-ил) хиназолин-6,7-диола (250 мг, 0,797 ммоля; из примера 69) в ДМФА (4 мл) добавляют воду (50 мкл), K2CO3 (тв.) (330 мг, 2,39 ммоля), тетраметиламмонийгидрохлорид пентагидрат (15 мг, 0,08 ммоля) и 2-бромэтилметиловый эфир (225 мкл, 2,39 ммоля). Смесь перемешивают под атмосферой газообразного азота при температуре 50oC под атмосферой газообразного азота в течение 5 часов, затем выделяют продукт в соответствии с описанием, приведенным в примере 1, в результате получают 131 мг продукта, который превращают в соль метансульфоновой кислоты по методике примера 44. (Т.пл. 185oC (разл.); ЖХ/МС: 430(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,68 мин).
Примеры 73-74
Этиловый эфир [4-(6-хлор-2,3-дигидроиндол-1-ил 6-метоксихиназолин-7-окси] уксусной кислоты и литиевая соль [4-(6-хлор-2,3-дигидроиндол-1-ил)-6-метоксихиназолин-7-окси] уксусной кислоты
К раствору 4-(6-хлор-2, 3-дигидроиндол-1-ил)-6-метоксихиназолин-7-ола (128 мг, 0,333 ммоля; из примера 70) в ДМФА (2 мл) добавляют трет-бутоксид калия (0,381 мл 1,0 М раствора в ТГФ), а затем в течение нескольких минут этиловый эфир 2-бромуксусной кислоты (47 мкл, 0,420 ммоля). Смесь перемешивают в течение 16 часов при температуре 20oC, а затем экстрагируют в смеси хлороформа и 5%-ного водного бикарбоната натрия. Органическую фазу промывают рассолом, сушат над Na2SO4, затем фильтруют и фильтрат упаривают под вакуумом. Остаток перекристаллизовывают из смеси хлороформ/гексан, в результате получают 30 мг эфира в виде бледно-желтого твердого вещества (т.пл. 175-176oC; ЖХ/МС: 413(MH+); анал.ВЭЖХ ОФ-18 - время удерживания: 5,34 мин).
Фильтрат, полученный в результате перекристаллизации, растворяют в метаноле (6 мл) и добавляют воду (3 мл) и гидроксид лития (30 мг, 0,666 ммоля). Смесь нагревают до кипения и кипятят в течение 1 часа, затем с помощью отгонки растворителя под вакуумом уменьшают объем примерно до 65% от первоначального, после чего промывают водную фазу диэтиловым эфиром. pH водной фазы доводят до 3,5 добавляя уксусную кислоту, смесь охлаждают до 4oC для осаждения свободной кислоты (73 мг), которую превращают в литиевую соль обработкой 1,00 экв. гидроксида лития в метаноле, и переводят в осадок, добавляя диэтиловый эфир (т.пл. 240-255oC (разл.); NEG-FAB: 390 (М-H); анал. ВЭЖХ ОФ-18 -время удерживания: 3,00 мин).
Пример 75
Соль 2-[4-(6-Хлор-2,3-дигидроиндол-1-ил) -6-метоксихиназолин-7-илокси] этанола и метансульфоновой кислоты
К раствору 4-(6-хлор-2,3-дигидроиндол-1-ил)-6-метоксихиназолин-7-ола (271 мг, 0,828 ммоля; из примера 70) в ДМФА (3 мл) добавляют тетраметиламмоний гидроксид пентагидрат (75 мг, 0,305 ммоля), K2CO3 (тв.) (171 мг, 1,24 ммоля), воду (50 мкл) и 2-бромэтанол (79 мкл, 1,11 ммоля). Смесь перемешивают при температуре 50oC, после чего ежедневно в течение четырех дней добавляют 2-бромэтанол (1,0 экв.). После этого смесь экстрагируют хлороформом и рассолом. Органическую фазу промывают насыщенным водным раствором бикарбоната натрия и рассолом, сушат над сульфатом натрия, фильтруют и упаривают под вакуумом. После быстрой хроматографии (кремнезем, 40% ацетон в метиленхлориде) остатка получают 269 мг (87%) свободного основания (т.пл. 205-206oC), из которого по методике примера 2 получают соль соляной кислоты (т. пл. 190-204oC (разл.)) или примера 44 получают соль метансульфоновой кислоты (т. пл. 233-237oC (разл.); ЖХ/МС: 372(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 3,90 мин).
Пример 76
4-(6-Хлор-2, 3-дигидроиндол-1-ил)-6-метокси-7 (2-морфолин-4-илэтокси) хиназолин-бис(метансульфонат) (соль)
К раствору 4-(6-хлор-2,3-дигидроиндол-1-ил)-6-метокси-хиназолин-7-ола (200 мг, 0,610 ммоля; из примера 70) и трифенилфосфина (160 мг, 0,610 ммоля) в сухом ТГФ (3 мл) под атмосферой азота по каплям в течение 10 минут при температуре 0oC добавляют диэтилазодикарбоксилат (106 мкл, 0,671 ммол), а затем раствор 4-(2-гидроксиэтил)морфолина (81 мкл, 0,671 ммоля) в сухом ТГФ (0,8 мл). Смеси дают нагреться до температуры 20oC, перемешивают 16 часов, а затем реакцию гасят добавлением воды (20 мкл) и полученную смесь упаривают под вакуумом. После быстрой хроматографии остатка (кремнезем, элюент: 45% ацетон хлороформ, 10% метанол/метиленхлорид) получают продукт в виде свободного основания (180 мг), который осаждают в виде его бис(метансульфонат) соли обработкой метансульфоновой кислотой (2,0 экв.) в растворе метиленхлорида. (Т.пл. 150-158oC (разл.); ЖХ/МС: 441(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 3,76 мин).
Пример 77
4-(6-Хлор-2, 3-дигидроиндол-1-ил)-7-(2-имидазол-1-ил-этокси)-6-метоксихиназолин бис (метансульфонат) (соль)
Указанный в заглавии продукт получают с выходом 50% из 4-(6-хлор-2,3-дигидроиндол-1-ил)-6-метоксихиназолин-7-ола и N-(2-гидроксиэтил)имидазола (1,1 экв. ) способом, аналогичным описанному в примере 76. (Т.пл. 162-168oC (разл.); ЖХ/МС: 422 (MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 3,94 мин).
Пример 78
Соль 1-[4-(6-хлор-2,3-дигидроиндол-1-ил)-6-метоксихиназолин- 7-илокси] -3-метоксипропан-2-ола и метансульфоновой кислоты
К раствору 4-(6-хлор-2, 3-дигидроиндол-1-ил)-6-метоксихиназолин-7-ола (125 мг, 0,381 ммоль; из примера 70) в ДМФА (2 мл) добавляют тетраметиламмоний гидроксид пентагидрат (15 мг, 0,076 ммоля), K2CO3 (тв.) (79 мг, 0,572 ммоля), воду (50 мкл) и метиловый эфир глицидной кислоты (37 мкл, 0,42 ммоля). Смесь перемешивают при температуре 50oC, после чего ежедневно в течение четырех дней добавляют метиловый эфир глицидной кислоты (0,5 экв.). После этого смесь экстрагируют хлороформом и рассолом. Органическую фазу промывают насыщенным водным раствором бикарбоната натрия и рассолом, сушат над сульфатом натрия, фильтруют и упаривают под вакуумом. После быстрой хроматографии (кремнезем, 25 - больше 40% ацетон/метиленхлорид) остатка с выходом 56% получают свободное основание (т.пл. 89-90oC), из которого по методике примера 44 получают соль метансульфоновой кислоты (т.пл. 180-187oC (разл.); ЖХ/МС: 416(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 3,81 мин).
Пример 79
Соль 2-[4-(6-хлор-2, 3-дигидроиндол-1-ил)-6-метоксихиназолин-7-илокси] -пропанола и метансульфоновой кислоты
К раствору 4-(6-хлор-2, 3-дигидроиндол-1-ил)-6-метоксихиназолин-7-ола (328 мг, 1,0 ммоль; из примера 70) в ДМФА (2 мл) добавляют тетраметиламмонийгидроксид пентагидрат (27 мг, 0,2 ммоля), K2CO3 (тв.) (276 мг, 2,0 ммоля), воду (50 мкл) и 3-бромпропанол (90 мкл, 1,0 ммоля). Смесь перемешивают при температуре 50oC в течение 16 часов. Последующую экстракцию, хроматографию (кремнезем, 55% ацетон/гексан) с получением свободного основания (т.пл. 152-153oC), а также дальнейшее получение соли проводят по методике примера 78, в результате получают продукт, указанный в заглавии с выходом 84%. (Т. пл. 195- 205oC (разл.); ЖХ/МС: 386(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 3,95 мин).
Пример 80
4-(6-Хлор-2,3-дигидроиндол-1-ил)-7,8-дигидро-[1,4] -ди- оксино[2,3-g] хиназолин-7-ил]метанол
К раствору 4-(6-Хлор-2,3-дигидроиндол-1-ил)хиназолин-6,7-диола (99 мг, 0,316 ммоля; из примера 69) в ДМФА (2 мл) добавляют воду (100 мкл), K2CO3 (тв. ) (65 мг, 0,473 ммоля) и тетраметиламмоний гидрохлорид пентагидрат (11 мг, 0,06 ммоля), после чего добавляют эпибромгидрин (28 мкл, 0,331 ммоля). Смесь перемешивают при температуре 50oC под атмосферой азота в течение 16 часов. Последующую экстракцию, хроматографию (кремнезем, 20% ацетон/метиленхлорид) проводят по методике примера 78, в результате получают указанный в заглавии продукт с выходом 11%. (Т.пл. 219-222oC (разл.); ЖХ/МС: 370(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 3,98 мин).
Пример 81
4- (6-Бром-5-фтор-2,3-дигидроиндол-1-ил)-6,7-бис(2-метокси-этокси)- хиназолин гидрохлорид
По методике, описанной в примере 1, из 6-бром-5-фториндолина (1,1 экв.) и 4-хлор-6,7-бис(2-метоксиэтокси)хиназолина (1 экв.) в изопропаноле получают указанный в заглавии продукт с выходом 78%. (Т.пл. свободного основания: 146-148oC). (Для соли: т.пл. 215-223oC (разл.); ЖХ/МС: 492, 494 (MH+); анал. ВЭЖХ ОФ-18- время удерживания: 4,64 мин).
Пример 82
4-(6-Бром-5-фтор-2,3-дигидроиндол-1-ил) -6-метокси-7-(2-метоксиэтокси) хиназолин гидрохлорид
По методике, описанной в примере 1, из 6-бром-5-фториндолина (1,1 экв.) и 4-хлор-6-метокси-7-(2-метоксиэтокси)хиназолина (1 экв. ) в изопропаноле получают указанный в заглавии продукт с выходом 58%. (Т.пл. свободного основания: 150-150,5oC). (Для соли: т.пл. 243-251oC (разл.): ЖХ/МС: 448, 450 (MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,79 мин).
Пример 83
1-(6, 7-диметоксихиназолин-4-ил)-2,3-дигидро-1H-индол-5-иламин гидрохлорид
(5-Нитроиндолин-1-ил)-6,7-диметоксихиназолин (412 мг, 1,17 ммоля), полученный в примере 52, гидрируют в течение 24 часов газообразным водородом с давлением 50 пси в среде, содержащей метанол (10 мл), уксусную кислоту (20 мл) с соляной кислотой (11,7 М раствор, 2 мл), в присутствии 10% палладия на углероде (200 мг). После выделения продукта, которое проводят по методике примера 30, и хроматографии (кремнезем, 5% метанол/хлороформ) получают продукт (91%, 372 мг), который превращают в соль соляной кислоты по методике примера 2. (Т. пл. 277-280oC (разл.); ЖХ/МС: 323(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 2,74 мин).
Пример 84.
Литиевая соль 1-(6,7-диметоксихиназолин-4-ил)-2, 3-дигидро-1H-индол-2-карбоновой кислоты
К раствору метилового эфира 1-(6,7-диметоксихиналозин-4-ил)-2,3-дигидро-1H-индол-2-карбоновой кислоты (500 мг, 1,37 ммоля; из примера 24) в смеси метанола (8 мл) с водой (4 мл) добавляют LiOH (56 мг, 1,37 ммоля). Раствор осторожно нагревают до температуры кипения (в течение приблизительно 5 минут) и охлаждают до 29oC, затем упаривают под вакуумом, в результате получают литиевую соль, указанную в заглавии (FAB-MC: 352 (MH+), 358 (M-H+Li)+; анал. ВЭЖХ ОФ-18 - время удерживания: 2,53 мин).
Пример 85
N-[1-(6,7-диметоксихиназолин-4-ил)-2,3-дигидро-1H-индол- -6-ил]ацетамид
К раствору 1-(6,7-диметоксихиназолин-4-ил)-2, 3-дигидро-1H-индол-6-иламина (99 мг, 0,307 ммоля; из примера 30) и пиридина (32 мкл, 0,620 ммоля) в хлороформе (5 мл) добавляют ацетилхлорид (33 мкл, 0,455 ммоля). Смесь кипятят в течение 2 часов, разбавляют хлороформом (15 мл), после этого органическую фазу промывают насыщенным водным раствором бикарбоната натрия, сушат над сульфатом натрия, фильтруют и упаривают под вакуумом. После хроматографии остатка (кремнезем, 5% метанол/хлороформ) получают 77 мг указанного в заглавии продукта. (Т. пл. 223-229oC.(разл); ГХ /МС: 364(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 3,42 мин).
Пример 86
N-[1-(6,7-диметоксихиназолин-4-ил)-2, 3-дигидро-1H-индол-6-ил]-2,2,2-трифторацетамид
К раствору 1-(6,7-диметоксихиназолин-4-ил)-2,3-дигидро-1H-индол-6-иламина (119 мг, 0,369 ммоля; из примера 30) и пиридина (42 мкл, 0,79 ммоля) в CH2Cl2 (5 мл) добавляют ангидрид трифторуксусной кислоты (112 мкл, 0,79 ммоля). Смесь перемешивают при температуре 20oC в течение 6 часов, гасят водой (5 мл), перемешивают в течение 30 минут и разбавляют хлороформом (15 мл). Органическую фазу отделяют и промывают насыщенным водным раствором бикарбоната натрия, сушат над сульфатом натрия, фильтруют и упаривают под вакуумом. После хроматографии остатка (кремнезем, 3% метанол хлороформ) получают 109 мг указанного в заглавии продукта. (Т.пл. 240oC (разл.); ГХ/МС: 418 (M+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,20 мин).
Пример 87
N-[1-(6,7-диметоксихиназолин-4-ил)-2,3-дигидро-1H-индол-5-ил ]ацетамид
К раствору 1-(6,7-диметоксихиназолин-4-ил)-2,3-дигидро-1H-индол-5-иламина (102 мг, 0,316 ммоля; свободное основание из примера 83) и 4-(N,N -диметиламино)пиридина (51,2 мг, 0,42 ммоля) в метиленхлориде (5 мл) добавляют ангидрид уксусной кислоты (88 мкл, 0,93 ммоля). Смесь перемешивают при температуре 20oC в течение 7,5 часов, гасят водой (5 мл), перемешивают 30 минут и разбавляют хлороформом (15 мл). Органическую фазу отделяют и промывают насыщенным раствором бикарбоната натрия, сушат над сульфатом натрия, фильтруют и упаривают под вакуумом. После хроматографии (кремнезем, элюент: 3,5% метанол/хлороформ) получают 53 мг указанного в заглавии продукта. (Т. пл. больше 250oC (разл.); ЖХ/МС: 338(MH +); анал. ВЭЖХ ОФ-18 -время удерживания: 2,79 мин).
Пример 88
N-[1-(6, 7-диметоксихиназолин-4-ил)-2,3-дигидро-1H-индол-5-ил]формамид
К ангидриду уксусной кислоты (88 мкл) при перемешивании при температуре 5oC добавляют муравьиную кислоту (42 мкл). Смесь перемешивают при температуре 20oC в течение часа, затем добавляют 1-(6,7-диметоксихиназолин-4-ил)-2,3-дигидро-1H-ин-дол-5-иламин (102 мг, 0,316 ммоля; свободное основание из примера 83). Смесь перемешивают при 20oC 1,5 часа, гасят водой (5 мл) перемешивают еще 30 минут, после чего разбавляют хлороформом (15 мл). Органическую фазу отделяют и промывают насыщенным водным раствором бикарбоната натрия, сушат над сульфатом натрия, фильтруют и упаривают под вакуумом. После хроматографии остатка (кремнезем, элюент: 4% метанол/метиленхлорид) получают 66 мг указанного в заглавии продукта. (Т.пл. 239-244oC (разл.); ЖХ/МС: 351(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 2,92 мин).
Пример 89
8-Бром-1-(6, 7-диметоксихиназолин-4-ил)-2,3,4,5-тетрагидро-1H-1H-бензо[b] азепин
По методике, описанной в примере 47, из 8-бром-2,3,4,5-тетрагидро-1H-бензо[b] азепина (1,1 экв. ) и 4-хлор-6,7-диметоксихиназолина (1,0 экв.) в N-метилпирролидоне получают указанный в заглавии продукт с выходом 58% (т. пл. 209-217oC; ЖХ/МС: 414, 416 (MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 5,47 мин).
Пример 90
1-(6,7-диметоксихиназолин-4-ил)- 2,3,4,5,6-гексагидро-1H-бензо[b]азоцин
По методике, описанной в примере 47, из 1,2,3,4,5,6-гексагидро-бензо[b] азоцина (1,1 экв. ) и 4-хлор-6,7-диметоксихиназолина (1,0 экв.) в N-метилпирролидоне получают указанный в заглавии продукт с выходом 67%. (Т.пл. 204-207oC; ЖХ/МС: 349 (M+); анал. ВЭЖХ ОФ-18 - время удерживания: 5,25 мин).
Пример 91
4-(6-Хлор-2,3-дигидроиндол-1-ил)-6-метоксихиназолин
Используя методику, приведенную в примере 1, указанный в заглавии продукт получают с выходом 77% из 6-хлориндолина и 4-хлор-6-метоксихиназолина. (Т.пл. 243oC; ЖХ/МС: 312(MH+)).
Пример 92
4-(3,4-Дигидро-2H-хинолин-1-ил)- 7-метокси-6- метилсульфанилхиназолин
Используя методику, приведенную в примере 1, указанный в заглавии продукт получают с выходом 35% из 1,2,3,4-тетрагидрохинолина и 4-хлор-7-метокси-6-метилсульфанилхиназолина. (Т.пл. 193-196oC; ЖХ/МС: 338(MH+)).
Пример 93
4-(6-хлор-2,3-дигидроиндол-1-ил) -6-метилихиназолин
Используя методику, приведенную в примере 24, указанный в заглавии продукт получают с выходом 15% из 6-хлориндолина и 4-хлор-6-метоксихиназолина. (Т.пл. 138-141oC; ЖХ /МС: 296 (MH+)).
Пример 94
4-(6-Хлор-2, 3-дигидро-индол-1-ил) -7-метокси-6-метилсульфанилхиназолин
Используя методику, приведенную в примере 1, указанный в заглавии продукт получают с выходом 18% из 6-хлориндолина и 4-хлор-7-метокси-6-метилсульфанилхиназолина. (Т.пл. 210oC; ЖХ/МС: 358(MH+)).
Пример 95
4-(6-Хлор-2,3-дигидроиндол-1-ил)-6,7-диметилихиназолин
Используя методику, приведенную в примере 24, указанный в заглавии продукт получают с выходом 16% из 6-хлориндолина и 4-хлор-6,7-диметилхиназолина. (Т.пл. 199oC; ЖХ/ МС: 310(MH+)).
Пример 96
7-Хлор-4-(6-хлор-2,3-дигидроиндол-1-ил) -6-(2-метоксиэтилсульфанил)хиназолин
Используя методику, приведенную в примере 1, указанный в заглавии продукт получают с выходом 38% из 6-хлориндолина и 4,7-дихлор-6-(2-метоксиэтилсульфанил)хиназолина. (Т.пл. 104-106oC; ЖХ/МС: 406(MH+)).
Пример 97
7-Хлор-4-(6-хлор-2,3- дигидроиндол-1-ил) хиназолин
Используя методику, приведенную в примере 24, указанный в заглавии продукт получают с выходом 50% из 6-хлориндолина и 4,7-дихлорхиназолина. (Т.пл. 189oC; ЖХ/МС: 316(MH+)).
Пример 98
8-Хлор-4-(6-хлор-2,3- дигидроиндол-1-ил) хиназолин
Используя методику, приведенную в примере 24, указанный в заглавии продукт получают с выходом 47% из 6-хлориндолина и 4,8-дихлорхиназолина. (Т.пл. 190oC; ЖХ/МС: 316(MH+)).
Пример 99
(6-Хлор-4-(6-хлор-2,3-дигидро-индол-1-ил) -7-метоксихиназолин
Используя методику, приведенную в примере 1, указанный в заглавии продукт получают с выходом 17% из 6-хлориндолина и 4,6-дихлор-7-метоксихиназолина. (Т.пл. 226oC; ЖХ/МС:346(MH+)).
Пример 100
7-Хлор-4-(6-хлор-2,3-дигидроиндол-1-ил) -6-метилсульфанилхиназолин
Используя методику, приведенную в примере 1, указанный в заглавии продукт получают с выходом 15% из 6-хлориндолина и 4,7-дихлор-6-метилсульфанилхиназолина. (Т.пл. 167-168oC; ЖХ/МС: 362 (MH+)).
Пример 101
(4-(6-Хлор-2, 3-дигидpoиндoл-1-ил)-7-метилихиназолин
Используя методику, приведенную в примере 24, указанный в заглавии продукт получают с выходом 14% из 6-хлориндолина и 4-хлор-7-метилхиназолина. (Т. пл. 265oC; ЖХ/МС: 296 (MH+)).
Пример 102
(4- (5-Бром-3,4-дигидро-2H-хинолин-1-ил)-6,7-диметоксихиназолин
Используя методику, приведенную в примере 1, указанный в заглавии продукт получают с выходом 4% из 5-бром-1,2,3,4-тетрагидрохинолина и 4-хлор-6,7-диметоксихиназолина. (Пленка; ЖХ/МС: 399 (MH+)).
Пример 103
4-(7-Бром-3,4-дигидро-2H-хинолин-1-ил)-6,7-диметоксихиназолин
Используя методику, приведенную в примере 1, указанный в заглавии продукт получают с выходом 11% из 7-бром-1,2,3,4-тетра-гидрохинолина и 4-хлор-6,7-диметоксихиназолина. (Пленка; ЖХ/МС: 399 (MH+)).
Пример 104
Этиловый эфир [4-(3,4-Дигидро-2H-хинолин-1-ил)-6-метокси-хиназолин- 7-илокси]уксусной кислоты
Используя методику, приведенную в примере 1, указанный в заглавии продукт получают с выходом 2% из 1,2,3,4-тетрагидрохинолина и этилового эфира (4-хлор-6-метоксихиназолин-7-илокси) уксусной кислоты. (Пленка, ЖХ /МС: 393 (MH+)).
Пример 105
Этиловый эфир [4-(3,4-Дигидро-2H-хинолин-1-ил)-7-этоксикарбонилметоксихинозолин -6-илокси]уксусной кислоты
Используя методику, приведенную в примере 1, указанный в заглавии продукт получают с выходом 19% из 1,2,3,4-тетрагидрохинолина и этилового эфира (4-хлор-7-этоксикарбонилметоксихиназолин-6-илокси)уксусной кислоты. (Пленка, ЖХ/МС: 465 (MH+)).
Пример 106
(4-(6-Хлор-2, 3-дигидроиндол-1-ил)-7-нитрохиназолин гидрохлорид
4-Хлор-7-нитрохиназолин (11,22 г, 53,5 ммоля) суспендируют в 35 мл изопропанола, обрабатывают 6-хлориндолином (8,25 г, 53,7 ммоля), кипятят с обратным холодильником в течение трех часов, затем медленно охлаждают до комнатной температуры. Продукт фильтруют и сушат на воздухе в течение ночи, в результате получают порошок ярко-желтого цвета; 13,18 г (68%): т.пл. 230oC (разл.); ЖХ/МС: 327 (MH+) 329 (M+2)H+.
Вычислено: C16H11CIN4O2: C 52,91; H 3,3; N 15,43; Cl 19,52.
Найдено: C 52,77; H 3,61; N 14,78; Cl 19,62.
Пример 107
4-(6-Хлор-2,3-дигидроиндол-1-ил)-хиназолин-7-ил амин гидрохлорид
4-(6-Хлор-2,3-дигидроиндол-1-ил)-7-нитрохинозолин гидрохлорид (2,16 г, 5,90 ммоля) суспендируют в 250 мл этанола, содержащих 0,21 г 10% палладия на углероде, и обрабатывают формиатом аммония (1,68 г, 26,6 ммоля). Спустя 16 часов смесь фильтруют через броунмиллерит и упаривают под вакуумом. Остаток растворяют в хлороформе, фильтруют и промывают раствором бикарбоната натрия и рассолом, затем сушат над сульфатом магния, фильтруют и упаривают под вакуумом, в результате получают технический продукт, который перекристаллизовывают из 50%-ного водного раствора этанола. Твердый продукт растворяют в метаноле, содержащем безводную соляную кислоту, и выливают в эфир, в результате после фильтрации и сушки под вакуумом получают указанное в заглавии вещество: 0,687 г (35%), т.пл. 282-283oC.
Пример 108
N-[4-(6-хлор-2, 3-дигидроиндол-1-ил)-хиназолин-7-ил]метансульфонамид
4-(6-Хлор-2,3-дигидроиндол-1-ил)-хиназолин-7-ил амин гидрохлорид (0,155 г, 0,465 ммоля) взаимодействует с метансульфонилхлоридом (0,0531, 0,465 ммоля) и триэтиламином (0,290 г, 2,87 ммоля) в 5 мл хлороформа в течение одного часа при температуре 0oC и в течение 16 часов при комнатной температуре, затем реакционную смесь выливают в 50 мл воды и экстрагируют этилацетатом (3х50 мл). Органические фракции после соединения промывают 50 мл рассола, сушат сульфатом магния, фильтруют и упаривают под вакуумом, в результате получают 0,121 г желтого остатка. После очистки с использованием прибора Chromatotron, снабженного селикагельной пластиной 2 мМ, и в качестве элюента 5% метанол в хлороформе. Чистый продукт выделяют выпариванием под вакуумом соответствующих фракций; 0,015 г (8,7%). Т.пл. 240-245oC (разл.).
Пример 109
N-[4-(6-Хлор-2, 3-дигидроиндол-1-ил)-хиназолин-7-ил]бисметан-сульфонамид
4-(6-Хлор-2,3-дигидроиндол-1-ил)-хиназолин-7-иламин гидрохлорид (0,1495 г, 0,449 ммоля) суспендируют в 5 мл метиленхлорида. В реакционную смесь добавляют триэтиламин (1,2 мл, 8,6 ммоля) и охлаждают до 0oC. После того, как реакционная смесь нагреется до комнатной температуры, добавляют метансульфонилхлорид (0,105 мл, 1,36 ммоля). Спустя 30 минут смесь выливают в 20 мл воды, экстрагируют метиленхлоридом (3х20 мл), промывают насыщенным водным раствором бикарбоната натрия и рассолом, сушат над сульфатом магния, фильтруют и после отгонки растворителя под вакуумом получают указанный в заглавии продукт в виде пенообразного вещества желтого цвета (0,177 г, 87%); ЖХ/МС: 453(MH+); 455 (M+2)H+).
Пример 110
N-[4-(6-Хлор-2, 3-дигидроиндол-1-ил)-хиназолин-7-ил] гидроксиламин
4-(6-Хлор-2, 3-дигидроиндол-1-ил)-7-нитрохиназолин гидрохлорид (2,90 г, 7,98 ммоля) растворяют в 700 мл метанола и гидрируют в течение 10 минут при 3 атм. в присутствии палладия на углероде. Реакционную смесь фильтруют через броунмиллерит и разбавляют 1,5 л диэтилового эфира. Продукт фильтруют и сушат под вакуумом, в результате получают твердый продукт ярко-желтого цвета, 2,23 г (80%): (Т.пл. 262-263oC (разл.); ГХ/МС: 313 (M+) 315 (M+2). Вычислено для С16H13ClN4O•HCl: С 55,03; H 4,04; N 16,04. Найдено: С 55,18; H 4,28; N 16,13.
Пример 111
[4-(6-Хлор-2,3-дигидроиндол-1-ил)-хиназолин-7-ил] метиламин гидрохлорид
4-(6-Хлор-2, 3-дигидроиндол-1-ил)-хиназолин-7-иламин гидрохлорид (0,104 г, 0,313 ммоля) растворяют в 3 мл ацетонитрила и обрабатывают 0,141 мл 37% водного формальдегида (1,74 ммоля). Цианоборгидрид натрия (0,036 г, 0,571 ммоля) медленно добавляют к реакционной смеси, а затем - 0,05 мл уксусной кислоты, после чего реакционную массу перемешивают при комнатной температуре в течение двух часов. Добавляют формальдегид (1,74 ммоля), спустя 30 минут цианоборгидрид натрия (0,571 ммоля), еще через 30 минут и затем через час в реакционную массу добавляют уксусную кислоту (по 0,05 мл). Через 2 часа реакционную смесь выливают в 50 мл диэтилового эфира, промывают 1N-ным раствором гидроксида натрия (3 х 50 мл), сушат над сульфатом магния, фильтруют и упаривают под вакуумом, в результате получают 0,913 г твердого вещества желтого цвета, которое содержит цианогидроборамин-производные моно- и диметилированного продуктов.
После хроматографирования (тонкослойная хроматография с автоматическим вращением (Chromatotron®, силикагельная пластина 2 мм, элюент: хлороформ, 10% метанол) получают 0,0156 г цианогидроборамин-производное монометилированного материала, которые растворяют в 1 мл этанола, обрабатывают 0,8 мл 1N HCl в метаноле и кипятят с об./х. в течение ночи. Образующуюся смесь выливают в 100 мл 1N раствора гидроксида натрия, экстрагируют хлороформом (4х50 мл), сушат над сульфатом магния, фильтруют и упаривают под вакуумом. Образующийся желтый остаток растворяют в минимальном количестве метанола, обрабатывают 1N HCl в метаноле, высаживают добавлением диэтилового эфира, фильтруют и сушат под вакуумом, в результате получают твердое вещество ярко-желтого цвета, 0,010 г (9%): (Т. пл. 270-274oC (разл. ); ЖХ/МС: 311 (MH+), 313 (M+2)H+).
Пример 112
[4-(6-Хлор-2,3-дигидроиндол-1-ил)-хиназолин-7-ил] -диметиламин гидрохлорид
Цианогидробармамин-производное продукта, выделенного в результате хроматографии в примере 111, обрабатывают аналогичным образом соляной кислотой в этаноле, в результате получают твердое вещество ярко-желтого цвета, 0,0145 г (13%): (Т.пл. 281-282oC (разл.): ЖХ/МС: 325 (MH+), 326(M+2)H+).
Пример 113
4- (6-Хлор-2, 3-дигидроиндол-1-ил)-6-нитрохиназолин
4-Хлор-6-нитрохиназолин (16,72 г, 79,8 ммоля) суспендируют в 250 мл изопропанола, обрабатывают 6-хлориндолином (12,25 г, 79,8 ммоля) кипятят с об. /х. в течение трех часов, затем медленно охлаждают до комнатной температуры. Продукт фильтруют и сушат на воздухе в течение ночи, в результате получают 12,30 г ярко-желтого порошка. Порошок растворяют в 100 г хлороформа, содержащего 16 мл триэтиламин, и кипятят с об.х. смесь 1 час. Охлажденную суспензию фильтруют, в результате получают очищенный указанный в заглавии продукт: 5,17 г (20%); (Т.пл. 260-261oC (разл.).
Пример 114
N-[4-(6-Хлор-2,3-дигидроиндол-1-ил)хиназолин-6-ил]формамид
4-(6-Хлор-2,3-дигидроиндол-1-ил)-7-нитрохиназолин (5, 17 г, 15,8 ммоля) растворяют в 60 мл 96% муравьиной кислоты. К полученному раствору медленно добавляют дитионит натрия (13,78 г, 79,2 ммоля), контролируя температуру с помощью ледяной бани. Спустя 30 минут приблизительно половину муравьиной кислоты отгоняют под вакуумом, а оставшуюся реакционную массу выливают в смесь воды и хлороформа, подщелачивают до pH 13 с помощью 6N-ного раствора гидроксида натрия, экстрагируют хлороформом (3х100 мл), промывают рассолом (100 мл), сушат над сульфатом магния, фильтруют и отгоняют растворитель под вакуумом, в результате получают пенообразное вещество ярко-желтого цвета, 2,71 г (53%), ЖХ/МС: 325(MH+), 327 (M+2)H+.
Пример 115
4-(6-Хлор-2,3-дигидроиндол-1-ил)хиназолин-6-иламин гидрохлорид
N-[4-(6-Хлор-2,3-дигидро-индол-1-ил)хиназолин-6-ил] -формамид (1,0055 г, 3,10 ммоля) суспендируют в 20 мл метанола, обрабатывают 20 мл 1N HCl в метаноле и перемешивают при комнатной температуре в течение 1 часа. В результате разбавления диэтиловым эфиром с доведением общего объема до 125 мл, получают твердое вещество желтого цвета, 0,8430 г (82%; дополнительно 0,089 г получают после охлаждения маточного раствора): (Т.пл. 289-290oC (разл.); ЖХ/МС: 297 (MH+), 299 (M+2)H+).
Пример 116
[4-(6-Хлор-2,3-дигидроиндол-1-ил) хиназолин-6-ил]метиламин
К суспензии литийалюминийгидрида (0,1726 г, 4,55 ммоля) в 5 мл тетрагидрофурана по каплям в течение 10 минут добавляют раствор N-[4-(6-хлор-2,3-дигидроиндол-1-ил) хиназолин-6-ил] -формамида (0,5010 г, 1,54 ммоля) в 6 мл тетрагидрофурана. Реакционную смесь перемешивают при комнатной температуре в течение 15 минут, затем реакцию гасят добавлением 0,2 мл воды, 0,2 мл 1N-ного водного раствора гидроксида натрия и 0,6 мл воды, смесь далее фильтруют и осадок на фильтре промывают водой и этилацетатом. Фильтрат экстрагируют этилацетатом (3х50 мл), промывают рассолом (100 мл), сушат над сульфатом магния, фильтруют и растворитель отгоняют под вакуумом. Остаток суспендируют в 10 мл метанола, обрабатывают 10 мл 1N HCl в метаноле и высаживают 20 мл диэтилового эфира. Осадок выделяют и сушат под вакуумом, в результате получают 0,312 г твердого вещества желтого цвета (64%): т.пл.259-261oC (разл. ), ЖХ/МС: 311(MH+), 313(M+2)H+); Вычислено для C17H15CIN4•HCl•5H2О С 54,55; H 5,12; N 14,97; найдено: С 54,27; H 5,00; N 14,99.
Пример 117
N-[4-(6-хлор-2,3-дигидроиндол-1-ил)хиназолин-6-ил]-N-метил- формамид
К соединению, полученному в примере 116 (0,22 г, 0,634 ммоля), добавляют 1 мл 50%-ного раствора ангидрида муравьиной кислоты в уксусной кислоте, спустя 1 час добавляют 10 мл этилового эфира, в результате чего образуется осадок. Осадок фильтруют, сушат под вакуумом, получают твердое вещество желтого цвета (0,14 г, 67%), т.пл. 178-179oC.
Пример 118
N-[4-(6-Хлор-2, 3-дигидроиндол-1-ил) хиназолин-6-ил]гуанидин гидрохлорид
4-(6-Хлор-2, 3-дигидроиндол-1-ил) хиназолин-6-иламин гидрохлорид (0,0994 г, 0,298 ммоля) суспендируют в 3 мл 1,2-дихлорэтана и обрабатывают триэтиламином (0,0415 мл, 0,298 ммоля). К суспензии добавляют уксусную кислоту (0,250 мл, 4,30 ммоля) и 3,5-диметилпиразол-1-карбоксамида (0,0880 г, 0,632 ммоля), смесь кипятят с об./х. в течение 18 ч. После охлаждения до комнатной температуры осадок выделяют фильтрацией, в результате получают твердое вещество рыжевато-коричневого цвета, 0,0387 г (35%): т.пл. 259-261oC. E1-MC: (высокоэффективное разделение) вычислено: 338,1047; найдено 338,1042.
Пример 119
N-[4-(6-Хлор-2, 3-дигидроиндол-1-ил) хиназолин-6-ил]-N',N''-ди- метилпропан-1,3-диамин
4-(6-Хлор-2, 3-дигидроиндол-1-ил) хиназолин-6-иламин гидрохлорид (0,525 г, 1,58 ммоля) соединяют с 2 мл свежеперегнанного 3-диметиламинопропилхлорида и нагревают до 120oC, после чего выдерживают при этой температуре в течение 1 часа. Жидкость желтого цвета отделяют, а оставшееся стеклообразное вещество красного цвета растворяют в воде, подщелачивают до pH 11 карбонатом калия, экстрагируют хлороформом (2х50 мл), промывают рассолом, сушат над сульфатом магния, фильтруют и сушат, в результате получают 0,272 г пенообразного вещества желтого цвета, содержащего исходный материал и моно- и диалкилированные продукты. Эту пенку растворяют в хлороформе, фильтруют через силикагель, затем продукт промывают на силикагеле 40% v/v раствором метанола в хлороформе, фильтрат упаривают под вакуумом, остаток растворяют в метаноле, обрабатывают 1N-ным раствором соляной кислоты в метаноле, разбавленным диэтиловым эфиром, фильтруют и сушат под вакуумом, в результате получают твердое вещество ярко-желтого цвета, 0,0995 г (15%): т.пл. 230oC (разл.); ЖХ/МС: 382(MH+), 384 (M+2)H+.
Пример 120
4-(6-Хлор-2, 3-дигидроиндол-1-ил)-6-морфолин-4-илхиназолин
4-Хлор-6-морфолинохиназолин (382 мг, 1,53 ммоля) и 6-хлориндолин (258 мг, 1,63 ммоля) нагревают до температуры кипения с об./х в 8 мл 1,2-дихлорэтана и пиридина (264 мг, 3,36 ммоля) и выдерживают при этой температуре в течение 16 часов. Реакционную смесь упаривают под вакуумом и парционируют между 100 мл этилацетата и 50 мл 5%-ного бикарбоната натрия. Органическую фракцию промывают дополнительным количеством 50 мл раствора бикарбоната натрия и 50 мл рассола сушат над сульфатом магния, фильтруют и упаривают под вакуумом до остатка. После хроматографирования остатка методом быстрой хроматографии (силикагель, 20% ацетан/метиленхлорид) полученный чистый продукт превращают в гидрохлорид с помощью растворения в метаноле, содержащем 1,1 эквивалент безводной соляной кислоты, и последующего высаживания эфиром, 396 мг (65%); т.пл. 277-279oC.
Пример 121
4- (6-Хлор-2,3-дигидроиндол-1-ил)-6-(4-метилпиперазин-1-ил)хиназолин
4-Хлор-6-(4-метилпиперазин-1-ил)хиназолин гидрохлорид (537 мг, 1,80 ммоля) и 6-хлориндолин (169 мг, 1,10 ммоля) кипятят с об./х. в 10 мл 1,2-дихлорэтана и в пиридине (350 мг, 4,40 ммоля) в течение 48 часов. Продукт выделяют в виде гидрохлорида по методике примера 120; 267 мг (58%); т.пл. 289-290oC.
Пример 122
4-(6-Фтор-7-метил-2, 3- дигидроиндол-1-ил)-6,7- диметоксихиназолин гидрохлорид
Используя методику, приведенную в примере 47 (с превращением свободного основания в соль соляной кислоты по методике примера 2), указанный в заглавии продукт получают с выходом 58% из 6-фтор-7-метилиндолина (1 1 экв.) и 4-хлор-6,7-диметоксихиназолина в N-метилпирролидиноне. (Т. пл. 220-225oC; ЖХ/МС: 340(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 4,63 мин).
Пример 123
1-(6,7-Диметоксихиназолин-4-ил) -2,3,6,7,8,9-гексагидpo-1H-бензо[g]индол гидрохлорид
Используя методику, приведенную в примере 47 (с превращением свободного основания в соль соляной кислоты по методике примера 2), указанный в заглавии продукт получают с выходом 58% из 2,3,6,7,8,9-гексагидро-1H-бензо[g] индола (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина в N-метилпирролидиноне. (Т. пл. 182-185oC; ЖХ/МС: 362(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 5,26 мин).
Пример 124
6,7-диметокси-4- (6-триметилсиланилэтинил-2,3-дигидроиндол-1-ил)-хиназолин
Используя методику, приведенную в примере 47, указанный в заглавии продукт получают с выходом 93% из 6-триметилсилинилэтинилиндолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина в N-метилпирролидиноне. (ЖХ/МС: 404(MH+); анал. ВЭЖХ ОФ-18 -время удерживания: 6,49 мин).
Пример 125
4-(6-Этинил-2,3-дигидроиндол-1-ил)-6,7-диметоксихиназолин
К раствору 6,7-диметокси-4-(6-триметилсиланилэтинил- 2,3-дигидроиндол-1-ил)хиназолина (1 экв.; 50 мг, 0,124 ммоля) в смеси метанола (2 мл) с тетрагидрофураном (2 мл) добавляют 1 М тетрабутиламмонийфторида (2 экв.; 0,248 мл) в тетрагидрофуране и смесь выдерживают при температуре 20oC в течение 16 часов. Растворители отгоняют под вакуумом, остаток растворяют в смеси этилацетат/диэтиловый эфир (1:1), промывают насыщенным водным раствором бикарбоната натрия, сушат над сульфатом натрия, фильтруют и упаривают под вакуумом. Продукт превращают в соль соляной кислоты по методике, приведенной в примере 2. (Т.пл. 241-243oC; ЖХ/МС: 332 (MH+); анал. ВЭЖХ ОФ-18 -время удерживания: 4,48 мин).
Пример 126
4-(6-Хлор-2,3-дигидроиндол-1-ил)-7-дифторметоксихиназолин
К раствору 4-(6-хлор-2,3-дигидроиндол-1-ил)хиназолин-7-ола (250 мг, 0,84 ммоля; из примера 65) в диметилформамиде (8 мл) добавляют KF (49 мг, 0,84 ммоля) и карбонат калия (120 мг). Через полученную смесь при перемешивании в течение 5-10 минут пропускают хлордифторметан (фреон 22) при температуре 0-5oC в реакторе под давлением. Реактор герметично запаивают и нагревают с перемешиванием до температуры 80oC, выдерживают в этих условиях в течение 4 часов, после чего методом ВЭЖХ ОФ устанавливают отсутствие исходных материалов. После экстракции и быстрой хроматографии на кремнеземе (элюент: 30% ацетон/гексан) по методике примера 78, получают 151 мг дифторметокси-производного, которое превращают в соль соляной кислоты по методике примера 2. (Т.пл. 250-256oC (разл.; ЖХ/МС: 348(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 5,64 мин).
Пример 127
1-(6, 7-диметоксихиназолин-4-ил)-1,2,3,5-тетрагидропирроло [2,3-f]индол
Используя методику, приведенную в примере 24 (с превращением свободного основания в соль соляной кислоты по методике примера 2), указанный в заглавии продукт получают с выходом 67% из 1,2,3,5-тетрагидропиррол [2,3-f]индола (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина в диметилформамиде. (Т.пл. 245-252oC; ЖХ/МС: 347(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 3,67 мин).
Пример 128
1-(6,7-диметоксихиназолин-4-ил)- 1,2,3,5,6,7-гексагидропиррол [2,3-f] индол
Используя методику, приведенную в примере 24, указанный в заглавии продукт получают с выходом 65% из 1,2,3,5,6,7-гексагидропиррол[2,3-f]индола (1,7 экв. ) и 4-хлор-6,7-диметоксихиназолина (1,0 экв.) в диметилформамиде (ЖХ/МС: 349 (MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 3,51 мин).
Пример 129
{ 3-[1-(6,7-диметоксихиназолин-4-ил)- 2,3-дигидро-1H-индол-3-ил]пропил}- диметиламин
Используя методику, приведенную в примере 24 (с превращением свободного основания в соль соляной кислоты по методике примера 2), указанный в заглавии продукт получают с выходом 52% из 3-(N,N-диметиламинопропил)индолина (1,1 экв.) и 4-хлор-6,7-диметоксихиназолина в диметилформамиде (1,0 экв.) в диметилформамиде. (Т.пл. 230-232oC; ЖХ/МС: 393(MH+); анал. ВЭЖХ ОФ-18 - время удерживания: 3,23 мин).
Способы получения индолинов из индолов
Восстановление индолов в индолины с помощью ZnBH4
Пример получения 1
6-метилиндолин
6-Метилиндол (2,785 г, 21,2 ммоля) в сухом диэтиловом эфире (30 мл) охлаждают до 0oC и к раствору добавляют раствор ZnBH4 в диэтиловом эфире (примерно 1,5 экв.; 215 мл 0,15 М). Смесь перемешивают в течение 3 дней при температуре 22oC в темноте, затем гасят добавлением 1М водного раствора соляной кислоты (до тех пор, пока не прекратится выделение водорода), затем подщелачивают до pH больше 10 2N-ным раствором гидроксида натрия. Эфирную фазу отделяют и промывают рассолом, сушат над сульфатом магния, фильтруют и упаривают под вакуумом, в результате получат 6-метилиндолин >90%-ной чистоты - в виде сиропа (2,82 г; ГХ/МС: время удерживания 0,63 минуты, M+ равно 133). Этот продукт очищают с помощью хроматографии (кремнезем, 25% этилацетат/гексан) или вакуумной перегонкой, но обычно используют без дополнительной очистки.
В табл. 1 представлены индолы, полученные аналогичным способом из соответственно замещенных индолов
Восстановление индолов с помощью комплекса боран пиридин
Пример получения 13
5-Метоксииндолин
К суспензии 5-метоксииндола (4,55 г, 31 ммоля) в тетрагидрофуране (10 мл) при температуре 0-5oC добавляют боран/пиридин-комплекс (15,5 мл, 8 М в BH3), а затем по каплям в течение 15 минут водную 6 N-ную соляную кислоту (50 мл). В процессе добавления соляной кислоты по каплям добавляют тетрагидрофуран для контроля пенообразования. Смесь перемешивают 45 минут при температуре 20oC, pH доводят примерно до 10 добавлением водного раствора гидроксида натрия и карбоната натрия и смесь экстрагируют хлороформом. Органические фракции промывают рассолом при pH 9-10, сушат над сульфатом магния, фильтруют и упаривают под вакуумом, в результате получают 5-метоксииндолин (96%; ГХ-МС: время удерживания: мин. M+ равно 149), который представляет собой указанный в заглавии продукт больше 97%-ной чистоты, которую устанавливают методом ПМР. Этот материал можно использовать без дальнейшей очистки или очищают высаживанием соли соляной кислоты посредством добавления 1 экв. 1N соляной кислоты в диэтиловом эфире в раствор свободного основания в этилацетате или эфире.
Приведенные далее индолины (см. табл. 2) получены аналогичным способом при обработке соответственно замещенных индолов 4-6 эквивалентами боран/пиридин-комплекса (5-гидроксииндол, используемый в качестве исходного материала получают по методике, описанной ниже).
Получение индолов из анилинов
Пример получения 25
6,7-Диметилиндол
К раствору трихлорида бора (200 мл 1M-ного раствора в ксилоле, 200 ммолей) при t 0-5oC по каплям в течение 20 минут добавляют 2,3-диметиланилин (22,2 мл, 182 ммоля) в сухом толуоле (110 мл), а затем по каплям в течение 10 минут 2-хлор-ацетонитрил (13,8 мл, 218 ммолей) и наконец одной порцией - трихлорид алюминия (27,0 г, 202 ммолей). Смесь нагревают до кипения с обрат. холод. в течение 1 часа, а затем выдерживают при температуре 80oC в течение 16 часов, затем охлаждают и добавляют 2N-ную соляную кислоту (190 мл). Смесь нагревают до 80oC и выдерживают 30 минут, затем охлаждают до 20oC, pH доводят до 3-4 с помощью 2 N-ного раствора гидроксида натрия. Полученную смесь экстрагируют метиленхлоридом, доводя pH до 3-4 после каждой экстракции. Органические экстракты соединяют, промывают рассолом, сушат над сульфатом натрия, фильтруют и упаривают под вакуумом. Остаток (28,9 г) перекристаллизовывают из смеси хлороформ/гексан, в результате получают 26,1 г чистого 2-амино-3,4-диметил-альфа-хлорацетофенона (ГХ-МС: 197 (M+)). Раствор этого продукта (5,02 г, 25,4 ммоля) в диоксане (125 мл) H2O (15 мл) обрабатывают NaBH4 (1,06 г, 28 ммолей), нагревают до кипения с обр. хол. в течение 4 часов. Смесь упаривают под вакуумом и экстрагируют в смеси H2O/метиленхлорид при pH 7. Органические экстракты соединяют, сушат над сульфатом магния, фильтруют и упаривают под вакуумом, в результате получают технический продукт (2,9 г), который может быть непосредственно восстановлен или его сублимируют в вакууме с получением 1,55 г указанного в заглавии чистого индола (ГХ-МС; 145 (M+)).
Пример получения 26
6,7,8,9-Тетрагидро-1H-бензо[g]индол
Этот продукт получают с выходом 76% из 5,6,7,8-тетрагидронафтил-1-амина через промежуточный альфа-хлорацетофенон по методике, описанной в примере получения 25 (ГХ-МС: 171 (M+)).
Пример получения 27
6-Фтор-7-метилиндол
Указанный в заглавии продукт получают с выходом 46% из 3-фтор-2-метиланилина через промежуточный альфа-хлорметилкетон по методике, описанной в примере получения 25 (ГХ-МС: 149 (M+)).
Получение замещенных изотинов из соответствующих анилинов и восстановление оксиндолов и изотинов с помощью комплекса BH3/тетрагидрофуран
Пример получения 28
1,2,3,5-Тетрагидропирроло[2,3-f]индол
К раствору хлоральгидрата (5,12 г, 31,0 ммоля) и сульфата натрия (73,8 г, 0,52 моля) в воде (68 мл) добавляют раствор 1-ацетил- 5-аминоиндолина (4,94 г, 28,03 ммоля) в концентрированной соляной кислоте (250 мл) и воде (17 мл). После растворения всех веществ к смеси добавляют гидроксиламингидрохлорид (6,27 г, 190 ммолей), раствор нагревают до кипения в течение 30 минут и выдерживают при температуре кипения в течение 30 минут. Осадок, образовавшийся после охлаждения смеси до 20oC, удаляют фильтрацией, промывают водой и сушат под вакуумом до постоянной массы, получают 6,68 г 1-ацетил-5-[2-(изонитрозо)ацетамидо]индола (ЖХ-МС: 248 (MH+)). Этот продукт добавляют небольшими порциями в течение 30 минут к концентрированной серной кислоте (20 мл) с температурой 50oC при перемешивании. После полного добавления смесь нагревают до 80oC, выдерживают в течение 10 минут при этой температуре, охлаждают до 20oC и выливают в смесь воды со льдом (300 мл). Образующийся осадок фильтруют, промывают водой и сушат под вакуумом, получают 6,02 г 1-ацетилизатин-производного (ЖХ-МС: 248 (M+NH4+). Раствор навески этого продукта (2,68 г, 11,4 ммоля) в смеси диоксан (23 мл)/6NHCl (25 мл) нагревают до 50oC и выдерживают при этой температуре 16 часов. В результате упаривания смеси под вакуумом при 40oC получают технический деацитилированный изатин (ЖХ-МС: 189(MH+)), который снова растворяют в тетрагидрофуране (50 мл) и восстанавливают непосредственно, добавляя 1М комплекса боран/тетрагидрофуран (114 мл, примерно 10 экв.). После перемешивания в течение 16 часов при 20oC смесь осторожно гасят водой (50 мл), разбавляют рассолом и доводят pH до 10-11, затем экстрагируют этилацетатом. Органические фракции соединяют, сушат под сульфатом натрия, фильтруют и упаривают под вакуумом, остаток перекристаллизовывают из смеси диэтиловый эфир гексан, получают чистый 1,2,3,5-тетрагидропирроло[2,3-f]индол (797 мг, ЖХ-МС: 159 (MH+).
Пример получения 29
6-Хлор-5-фториндол
К раствору 6-хлор-5-фтороксиндола (0,49 г, 2,64 ммоля) в тетрагидрофуране (5 мл) медленно в течение 30 минут добавляют 1М раствор BH3 в тетрагидрофуране (21,2 мл, 21,2 ммоля). Смесь перемешивают в течение 24 часов, затем медленно при осторожном перемешивании добавляют 1N-ный раствор гидроксида натрия. После перемешивания в течение 20 минут раствор экстрагируют смесью этилацетат/диэтиловый эфир (1:1), экстракты соединяют, промывают под вакуумом, получают 412 мг продукта в виде смеси 6-хлор-5-фториндола и 6-хлор-5-фториндолина (ГХ-МС: 169 и 171 (M+), соответственно), которую используют без дополнительной очистки.
Пример получения 30
5,6-Дихлориндол
Указанный в заглавии продукт получают с выходом 93% из 5,7-дихлоризотина (1 экв.) и BH3 в тетрагидрофуране (6 экв.) по методике, описанной в примере получения 6-хлор-5-фториндола (пример получения 29).
Пример получения 31
6-хлор-5-нитроиндол
Указанный в заглавии продукт получают с выходом 99% из 6-хлор-5-фториндола (1 экв. ) и BH3/тетрагидрофуран (6 экв.) по методике, описанной в примере получения 6-хлор-5-фториндола (пример получения 29) (продукт содержит в качестве примеси некоторое количество 6-хлор-5-нитроиндолина), и используют в последующем восстановлении без дополнительной очистки.
Получение 5-гидроксииндолов из соответственно замещенных индолинов
Пример получения 32
5-Гидрокси-6-метилиндол
К раствору 6-метилиндолина (266 мг, 2,0 ммоля) в ацетоне (25 мл) при температуре 20oC добавляют раствор нитрозодисульфоната калия (1,18 г, 4,4 ммоля, 2,2 экв.) в фосфатно-калиевом буфере (pH 7,0, 0,10 М, 80 мл). Смесь перемешивают 15 минут, затем четыре раза экстрагируют хлороформом (4х40 мл). Органические фракции промывают водой, сушат над сульфатом натрия и упаривают под вакуумом, в результате получают 290 мг технического продукта в виде твердого вещества темно-красного цвета (примерно 80%-ной чистоты, что определяют методом ПМР; ГХ-МС: 147 (M+)), который используют без дополнительной очистки.
Пример получения 33
6-Хлор-5-гидроксииндол
К раствору 6-хлориндолина (2,0 г, 13 ммолей) в ацетоне (110 мл) при температуре 0oC добавляют охлажденный раствор нитрозодисульфоната калия (7,69 г, 28,6 ммоля, 2,2 экв.) в фосфатно-калиевом буфере (pH 7,0, 0,13 М, 520 мл). Смесь перемешивают при температуре 0oC в течение 1 часа, экстрагируют хлороформом (250 мл, затем 2 х 75 мл). Органическую фракцию промывают рассолом, сушат над сульфатом натрия и упаривают под вакуумом, в результате получают 2,34 г твердого вещества темно-красного цвета. Смесь 6-хлориндолина, 6-хлориндола и 6- хлор-5-гидроксииндола распределяют между дегазированным раствором 1N водного гидроксида натрия (150 мл) и эфиром (3х25 мл). pH водной фазы доводят примерно до 4 с помощью уксусной кислоты, целевой 6-хлор-5-гидроксииндол (460 мл, ГХ-МС: 167 (M+)) выделяют экстракцией хлороформом и используют без дальнейшей очистки.
Примеры получения 34-35
5-Гидрокси-7-метилиндол и 5-гидрокси-6,7-диметилиндол
Эти промежуточные продукты получают из 7-метилиндолина и 6,7-диметилиндолина с выходами 73% и 68% соответственно по методике, приведенной для получения 5-гидрокси-6-метилиндола (пример получения 32).
Бромирование индолинов и высших аннилированных анилинов
Пример получения 36
8-Бром-2,3,4,5-тетрагидро- 1H-бензо[b]азепин
К раствору 2,3,5,5-тетрагидро-1H-бензо[b]азепина. (410 мг, 2,78 ммоля; из примера получения 41) в концентрированной серной кислоте (5 мл) добавляют сульфат серебра (438 мг, 1,41 ммоля). Смесь перемешивают до полного растворения сульфата серебра, затем в течение 15 минут при температуре 0-5oC добавляют жидкий бром (145 мкл, 2,80 ммоля). Смесь перемешивают в темноте и дают ей нагреться до комнатной температуры. Спустя 4 часа смесь переносят в смесь лед/вода (50 мл), доводят pH смеси до 12-14 осторожным добавлением 6N-ного раствора гидроксида натрия с охлаждением, затем фильтруют через броунмиллерит. Осадок промывают метилцианидом, хлороформом и диэтиловым эфиром. Водную фазу экстрагируют этилацетатом, органические экстракты, смывы соединяют, сушат над сульфатом магния, фильтруют и упаривают под вакуумом, получают 468 мг твердого вещества желтоватого цвета. 6-Бром-(48 мг) и 8-бром-изомеры (492 мг; ГХ-МС: 226,228 (M+)) разделяют быстрой хроматографией (кремнезем, 10% этилацетат/гексаны) и/или перекристаллизовывают (диэтиловый эфир гексаны).
Дополнительные способы получения индолинов
Способ получения 37
6-Йодиндолин
К суспензии 1-ацетил-6-аминоиндолина (17,6 г, 100 ммоля) в воде (100 мл) при перемешивании добавляют концентрированную серную кислоту (12,5 мл). После перемешивания в течение 10 минут раствор охлаждают до 0-5oC и добавляют по каплям в течение 1 часа раствор нитрата натрия (6,91 г, 100 ммоля) в воде. Свежеприготовленный раствор KI (20,04 г, 121 ммоля) в 1М серной кислоты (20 мл) по каплям добавляют к полученному темно-коричневому раствору при температуре 0-10oC. Смеси дают нагреться до температуры 22oC в течение примерно 30 минут, затем раствор нагревают до 55oC выдерживают при этой температуре до полного прекращения выделения газообразного азота. После охлаждения pH раствора доводят примерно до 11 и смесь экстрагируют хлороформом (3 х 150 мл). Органические экстракты соединяют, промывают рассолом, сушат над сульфатом магния, фильтруют и упаривают под вакуумом. Остаток, содержащий 1-ацетил-6-йодиндолин, обрабатывают КОН (14 мг, 250 ммолей) в кипящем метаноле (170 мл) и тетрагидрофураном (30 мл) под атмосферой азота в течение 72 часов. После охлаждения к смеси добавляют уксусную кислоту и растворители отгоняют под вакуумом. Остаток экстрагируют хлороформом (400 мл) и 5%-ным водным раствором бикарбоната натрия (250 мл), органическую фазу сушат над сульфатом магния, фильтруют и упаривают под вакуумом, остаток хроматографируют (кремнезем, 30% этилацетат/гексаны) получают чистый 6-йодиндолин (8,7 г; ГХ-МС: время удерживания 1,51 мин., M+ равно 245) в виде кристаллов рыжевато-коричневого цвета.
Пример получения 38
6-Триметилсиланилэтинилиндолин
К смеси CuI (77,5 мг, 0,407 молей), Pd (PPh3)4 (113 мг, 0,097 ммоля) и триметилсилилацетилена (0,432 мл, 3,О6 ммоля) в диэтиламине (без примесей газов) (10 мл) добавляют 6-йодиндолин (499 мг, 2,04 ммоля). Смесь перемешивают при кипячении с обратным холод. в атмосфере N2 в течение 30 минут. Растворитель отгоняют под вакуумом, остаток хроматографируют (кремнезем, 25% этилацетат/гексан), получают 364 мг чистого 6-триметилсиланилэтинилиндолина (ГХ-МС: 215 (M+)).
Пример получения 39
6-Хлориндолин
Этот продукт удобно получают в multigram scale циклизацией 2,4-дихлорфенэтиламина в присутствии карбоната натрия (1,2 мол. экв.) Cu2Cl2 (0,01 мол. экв. ) и 8-гидроксихинолина (0,012 мол. экв.) в изоамиловом спирте (1 объем) при температуре 130oC в течение 5 часов. После добавления гидразина (0,0055 объемов) и кипячения в течение 1 часа, смесь фильтруют, растворитель отгоняют под вакуумом (45oC @ примерно 10 мм рт.ст) и после отгонки под вакуумом (95-100oC при 2 мм рт.ст.) получают чистый 6-хлориндолин (65-95%).
Пример получения 40
Метиловый эфир DL-индолин-2-карбоновой кислоты
DL-Индолин-2-карбоновую кислоты (3,16 г, 19,4 ммоля) суспендируют в метилцианиде (50 мл) при перемешивании и обрабатывают эфирным раствором CH2N2 (примерно 0,5 М) до прекращения выделения газообразного азота. Раствор упаривают под вакуумом, остаток переносят в этилацетат (150 мл), промывают насыщенным водным раствором бикарбоната натрия (3 х 50 мл) и рассолом, сушат над сульфатом магния, фильтруют и упаривают под вакуумом до масла, содержащего метиловый эфир больше 95%-ной чистоты (3,36 г; ГХ/МС: 177 (M+)), который используют без дополнительной очистки.
Высшие системы аннилированных анилинов
Пример получения 41
2,3,4, 5-Тетрагидро-1H-бензо[b]азепин
К раствору 2,3,4,5-тетрагидро-1H-1-бензо[b]азепин-2-она (0,776 г, 4,81 ммоля) в тетрагидрофуране (10 мл) добавляют по каплям в течение 30 минут 1М BH3 в тетрагидрофуране (38,5 мл, 38,5 ммоля; 8 экв.). Раствор перемешивают в течение 24 часов при температуре 20oC и гасят добавлением по каплям дополнительного количества воды (40 мл). Смесь разбавляют рассолом и экстрагируют смесью этилацетат диэтиловый эфир (2:1). Органические экстракты промывают 1N-ным раствором гидроксида натрия, сушат над сульфатом магния, фильтруют и упаривают под вакуумом, получают 703 мг (примерно 99%) технического продукта (ГХ-МС: 147 (M+), который используют без дополнительной очистки.
Пример получения 42
1,2,3,4,5,6-Гексагидробензо[b]азоцин
Указанный в заглавии продукт получают восстановлением 1,2,3,4,5,6-гексагидро- 1-бензо[b] азоцин-2-она с помощью комплекса BH3 в тетрагидрофуране по методике примера получения 41.
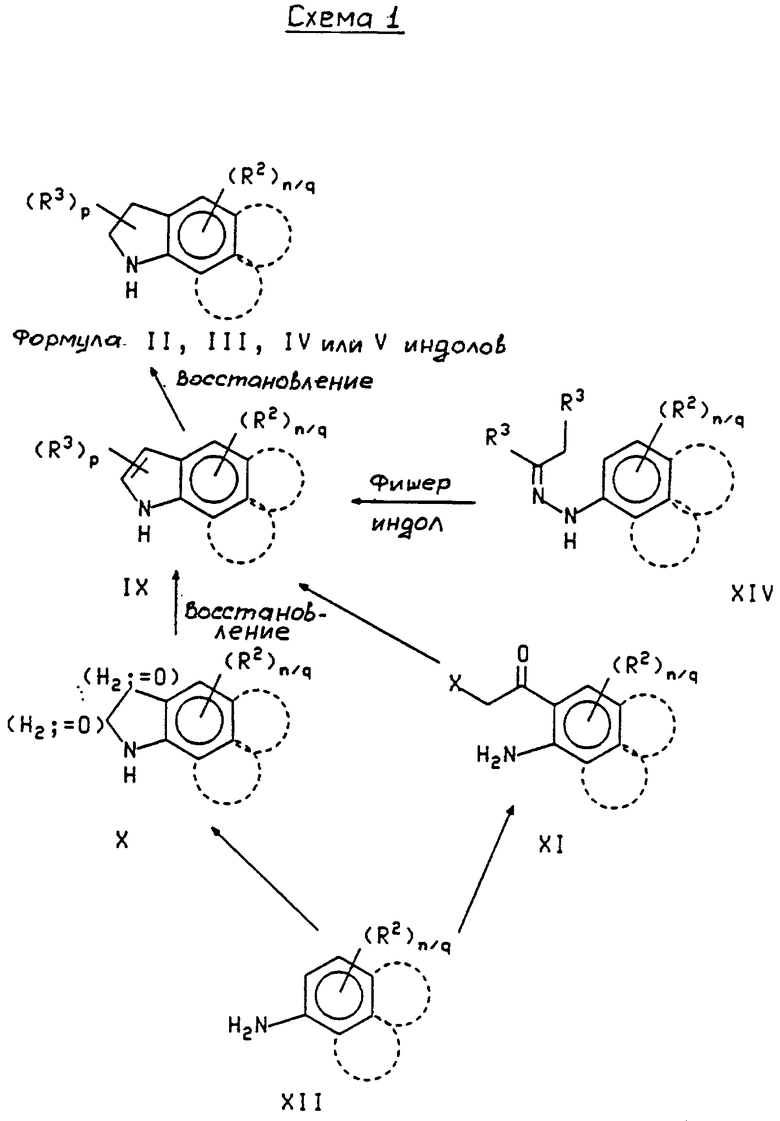
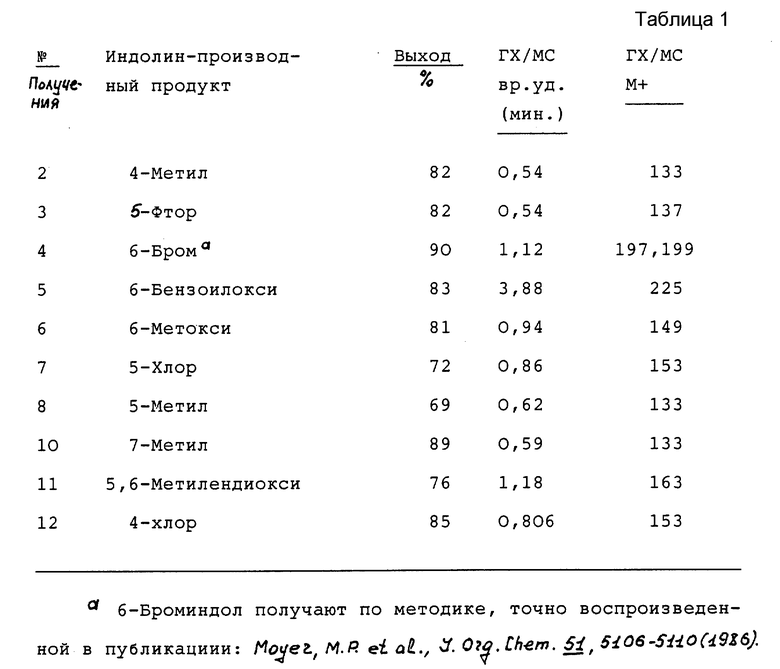

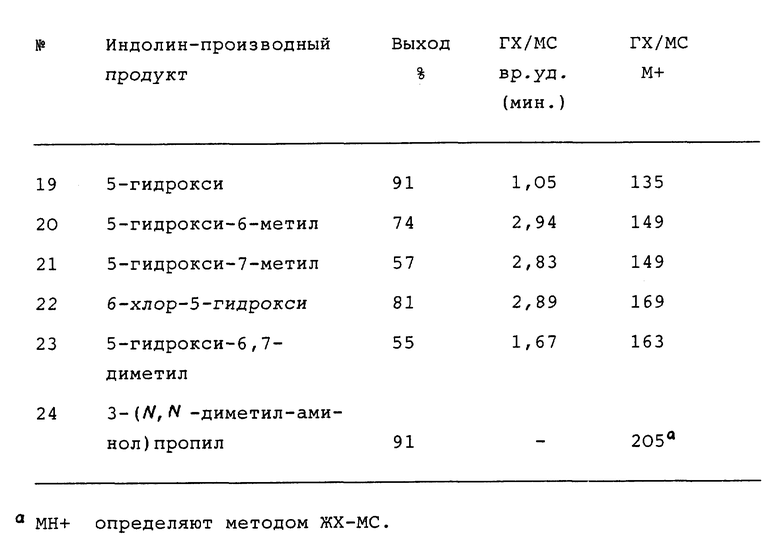
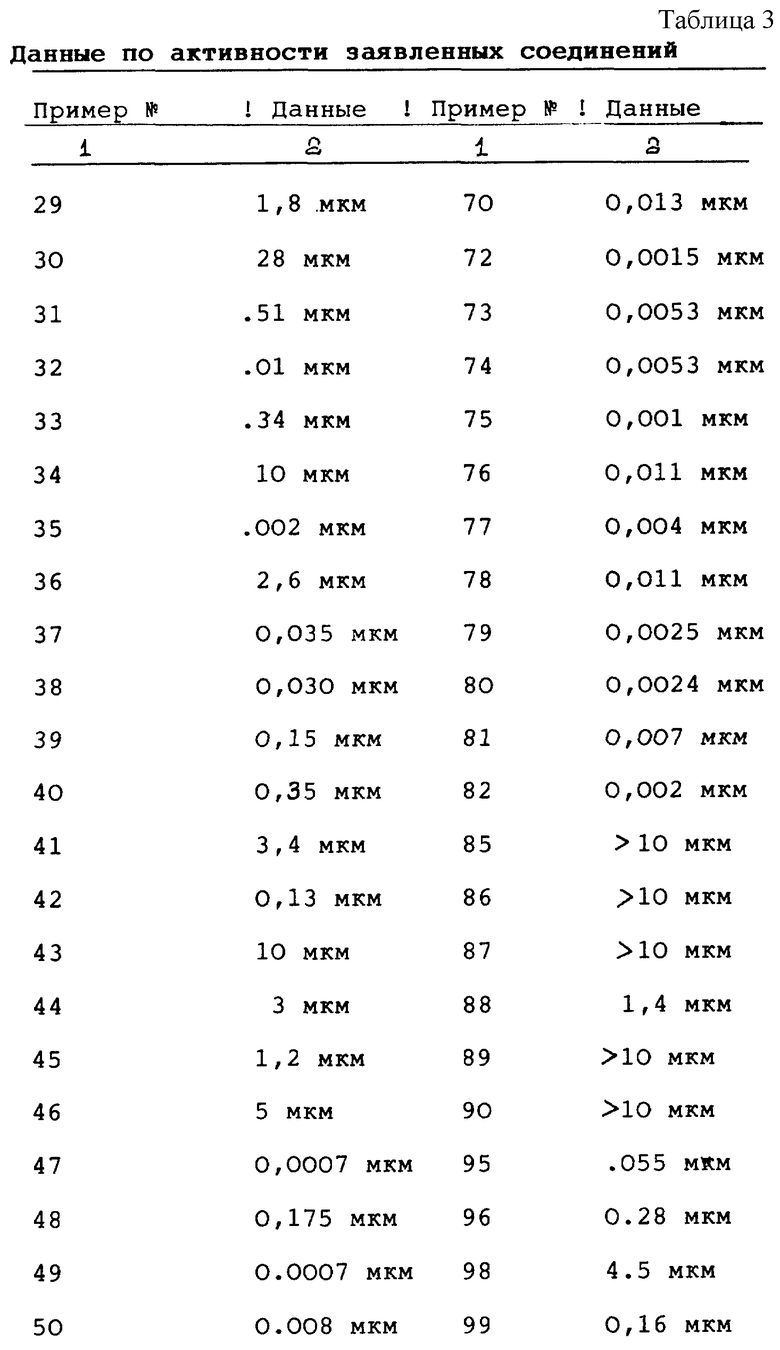
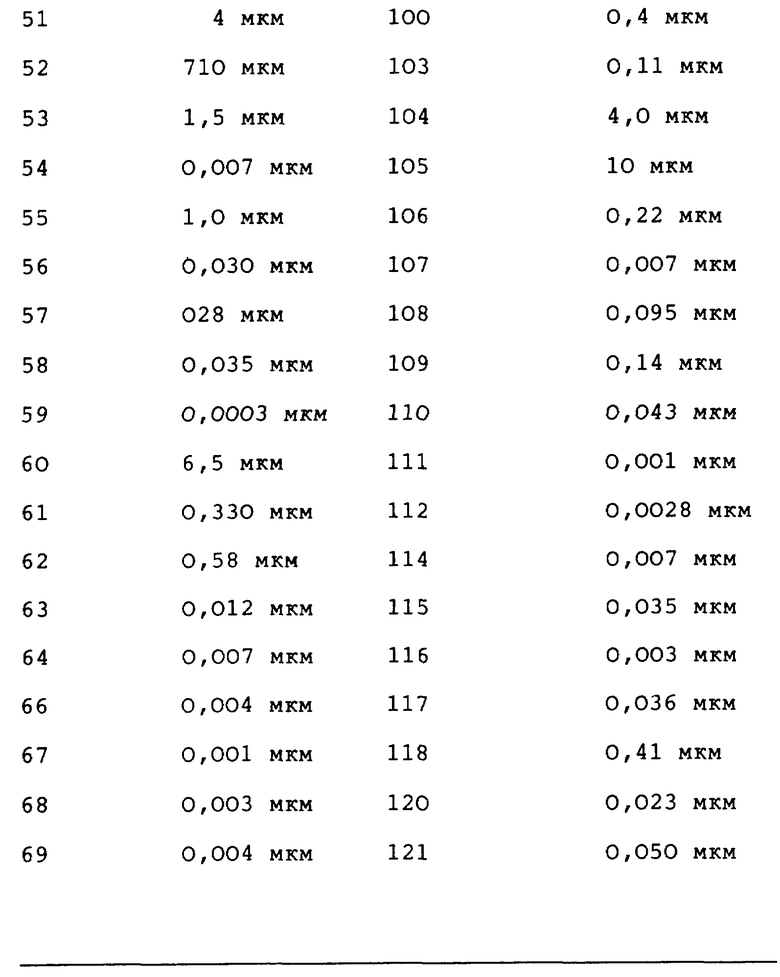
Описываются новые соединения общей формулы I
где значения R, m, Z указаны в 1 пункте формулы изобретения, проявляющие противораковую активность, а также фармацевтическая композиция на основе соединений формулы I. 2 с. и 6 з.п. ф-лы, 3 табл.
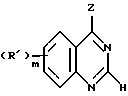
и их фармацевтически приемлемые соли и стереоизомеры, где Z представляет собой:
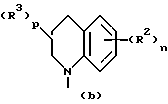
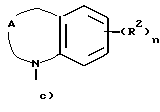
где А вместе с атомом азота и фрагментом бензольного кольца образует 7-9 членный мононенасыщенный моно-аза цикл,
R1 - в каждом случае, независимо, представляет собой: а) галоген-, нитро-, гидроксильную, амино-группу, (C1-C4)алкил, (C1-C4)алкокси-, моно -N- или ди-N,N,-(C1-C4)алкиламино, морфолино - или 4 - (C1-C4)алкил пиперазин-1-ил; в) (C1-C4)алкилсульфонил; трифторметилокси, алкоксикарбонилметокси; с) (C1-C4)алкоксигруппа, имидазол-1-ил;
m = 0, 1, 2, 3, n = 0, 1, p = 0 или 1;
R2 - в каждом случае, независимо, представляет собой галоген, нитро-, гидроксильную группу, амино-, азидо-, изотиоциано группу, (C1-C4) алкил, бензилокси, фенил, (C1-C4) алкоксигруппу, N-моно - (C1-C4) - алкиламино, пиррол-1-ил, пирролидин-1-ил, трифторметилкарбониламиногруппу;
R3 в каждом случае, независимо, представляет собой карбоксигруппу, (C1-C4)-алкоксикарбонил, при условии, что 4-/3,4-дигидро-2Н-хинолин-1-ил/ хиназолин исключается, обладающие противораковой активностью.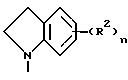
где n принимает значение 1,
m принимает значения 1 или 2;
R1 в каждом случае, независимо, расположен в положении 6 и/или 7 и представляет собой (C1-C4)алкил, (C1-C4)алкокси, моно-N- или ди-N,N-(C1-C4)-алкиламиногруппу, амино-группу;
R2 в каждом случае, независимо, представляет собой 4-гидрокси, 4-амино, 5-фтор, 5-гидрокси, 5-амино, 6-галоген, 6-метил, 6-нитро, 7-метил.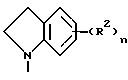
где n = 1, m = 1 или 2;
R1 в каждом случае, независимо, расположен в положении 6 и/или 7 и представляет собой (C1-C4)алкил, (C1-C4)алкокси, моно-N- или ди-N,N-(C1-C4)алкиламиногруппу, гидроксильную группу;
R2 в каждом случае независимо представляет собой галоген, гидроксильную, амино, нитрогруппу, (C1-C4) алкил.
