Данное изобретение относится к усовершенствованному способу получения известных соединений аминов, которые можно использовать в качестве лекарственных средств.
В Европейском патенте ЕР 279937 описывается группа соединений, которые показаны в качестве противосудорожных средств. Соединение примера 1, гидрохлорид 2-амино-N-(1,2-дифенил-1-метилэтил)ацетамида (которое имеет название гидрохлорид ремацемида INN), находится на клинических испытаниях.
Известные способы получения 2-aмино-N-(1,2-дифенил-1-метилэтил)ацетамида и его аналогов имеют недостаток, выражающийся в низких выходах. В примере 1 Европейского патента ЕР 279937 описывается получение гидрохлорида 2-амино-N-(1,2-дифенил-1-метилэтил)ацетамида с выходом только 32% относительно исходного материала 1,2-дифенил-2-пропиламина. Данный способ включает сочетание Cbz-глицина с 1,2-дифенил-2-пропиламином в присутствии DCC с последующим удалением Cbz-группы гидрогенолизом.
Кроме того, продукты таких известных способов требуют значительной очистки перед тем, как они могут быть использованы в фармацевтических препаративных формах. В настоящее время неожиданно было обнаружено, что другой способ имеет в качестве преимущества значительно повышенный выход и, кроме того, обеспечивает получение целевого продукта с хорошей чистотой.
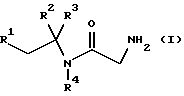
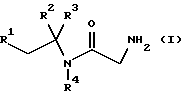
В первом аспекте данное изобретение, следовательно, предлагает способ получения соединения формулы (I) или его фармацевтически приемлемой соли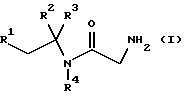
где R1 и R2 независимо представляют фенил или 4-фторфенил;
R3 представляет водород, алкил C1-4 или метоксикарбонил;
R4 представляет водород или метил;
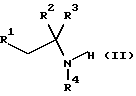
который включает реакцию соединения формулы II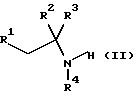
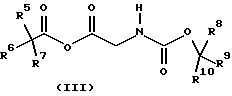
где R1, R2, R3 и R4 имеют значения, определенные в формуле (I), с соединением формулы (III)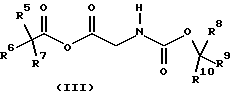
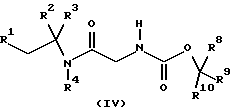
где R5, R6, R7, R8, R9 и R10 независимо представляют C1-6-алкил с получением соединения формулы (IV)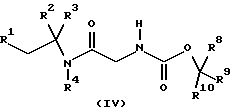
где R1, R2, R3, R4, R8, R9 и R10 имеют значения, определенные в формуле (I), с последующим удалением защитной группы и затем, необязательно, образованием фармацевтически приемлемой соли.
Фармацевтически приемлемые соли соединений формулы (I) включают кислотно-аддитивные соли, в частности гидрохлоридные соли. Такие соли получают с использованием стандартных методик, известных в данной области.
Подходяще R1 и R2 независимо представляют фенил или 4-фторфенил, предпочтительно, R1 и R2 оба представляют фенил.
Подходяще R3 представляет водород, C1-4-алкил или метоксикарбонил, предпочтительно R3 представляет C1-4-алкил, в частности метил.
Подходяще R4 представляет водород или метил, предпочтительно, R4 представляет водород.
Более предпочтительно использование вышеуказанного способа для получения соединения формулы (I), которое представляет собой 2-амино-N-(1,2-дифенил-1-метилэтил)ацетамид или его фармацевтически приемлемую соль. Подходящие соли включают кислотно-аддитивные соли, такие как гидрогалогенидные соли, предпочтительно, гидрохлоридную соль.
Подходяще R5, R6, R7, R8, R9 и R10 независимо представляют C1-6-алкил, предпочтительно, R5, R6, R7, R8, R9 и R10 представляют все метил, так что R8, R9 и R10 образуют часть Вос-защитной группы.
Смешанные ангидриды формулы (III) получают реакцией соединения формулы (V)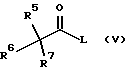
где R5, R6 и R7 имеют значения, определенные в формуле (III), и L представляет уходящую группу, с соединением формулы (VI)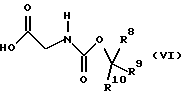
где R8, R9 и R10 имеют значения, определенные в формуле (III). Подходяще L представляет уходящую группу, в частности галоген, и, предпочтительно, хлор. Образование смешанных ангидридов формулы (III) и их реакцию с соединениями формулы (II) проводят, предпочтительно, в температурном интервале от примерно -30 до примерно 10oС, предпочтительно, от примерно -10oС до примерно 10oС, более предпочтительно, при примерно -5oС. Предпочтительно, смешанные ангидриды формулы (III) не выделяют, а подвергают взаимодействию с соединениями формулы (II) по методике в одном резервуаре.
Получение смешанного ангидрида из соединений (V) и (VI), предпочтительно, проводят в присутствии органического основания, такого как третичный органический амин, например, диизопропиламин, N-метилморфолин, и триалкиламины, такие как триметиламин и триэтиламин. Предпочтительные основания включают триэтиламин. Предпочтительно, соединение формулы (V) смешивают с соединением формулы (VI) с последующим добавлением амина. Было обнаружено, что эта методика снижает потребность в избыточных реагентах для ускорения завершения реакции соединений формулы (V) и (VI).
Соединения формулы (V) и (VI) коммерчески доступны или могут быть получены с использованием стандартных методик. Например, предпочтительное соединение формулы (V), где R5, R6 и R7 представляют метил и L представляет хлор, является коммерчески доступным пивалоилхлоридом. Предпочтительное соединение формулы (VI), где R8, R9 и R10 представляют метил, является коммерчески доступным Вос-глицином.
Реакцию смешанного ангидрида можно проводить в любом подходящем растворителе. Примеры подходящих растворителей включают диметоксиэтан, трет-бутилметиловый простой эфир, ТГФ, хлороформ, ксилол, толуол и дихлорметан. Реакцию смешанного ангидрида проводят, предпочтительно, с использованием толуола или дихлорметана в качестве растворителя и, более предпочтительно, дихлорметана.
Реакцию смешанного ангидрида проводят, предпочтительно, в атмосфере инертного газа, предпочтительно, в атмосфере азота.
После образования соединений формулы (IV), предпочтительно, следует стадия удаления защитной группы, в которой защитную группу удаляют любыми стандартными методами. Когда защитная группа представляет группу Вос, ее, предпочтительно, удаляют кислотным гидролизом. Его можно проводить в подходящем растворителе, таком как изопропанол. В таком случае продукты данного способа имеют такую чистоту, что их можно использовать в фармацевтических препаративных формах без дополнительной очистки. Альтернативно, продукт можно очистить общепринятыми способами, например, перекристаллизацией из подходящей системы растворителей, такой как метанол/IPA (изопропанол). Следовательно, в следующем аспекте изобретение предлагает способ, как описано выше, включающий перекристаллизацию соединения формулы (I) или его соли.
Новые промежуточные соединения также образуют аспект изобретения. Следовательно, изобретение предлагает соединение формулы (III), как определено выше.
Соединения формулы (II) можно получить способами, описанными в Европейском патенте ЕР 279937. Соединение формулы (III) можно получить способами, хорошо известными специалистам в данной области, и как иллюстрируется ниже.
Следующие общепринятые аббревиатуры, используемые в данном описании, хорошо известны специалистам в данной области:
Cbz - бензилоксикарбонил
Вос - трет-бутилоксикарбонил
DCC - дициклогексилкарбодиимид
Изобретение иллюстрируется следующими примерами.
Пример 1
Получение гидрохлорида 2-амино N-(1,2-дифенил-1-метилэтил)ацетамида
(а) 2-(трет-Бутилоксикарбониламино)-N-(1,2-дифенил-1-метилэтил)ацетамид
В реакционный сосуд загружают дихлорметан (360 л) и Вос-глицин (32,6 кг, 186,3 моль). Осторожно добавляют триэтиламин (18,8 кг, 186,1 моль), поддерживая температуру ниже 25oС. Смесь охлаждают до температуры ниже -5oС и пивалоилхлорид (триметилацетилхлорид, 22,4 кг, 185,9 моль) в дихлорметане (40 л) добавляют с такой скоростью, чтобы поддерживать температуру ниже -5oС. Смесь промывают дихлорметаном (2 л). Смесь затем перемешивают при температуре ниже -5oС в течение 2-2,5 час и затем добавляют в виде твердого вещества гидрохлорид 1,2-дифенил-2-пропиламина (40 кг, 161,1 моль, сухая масса, действительная масса, включающая влагу, 42,94 кг), от начала до конца поддерживая температуру ниже -5oС. Затем добавляют триэтиламин (28,4 кг, 281,2 моль), поддерживая температуру ниже -5oС. Смесь промывают дихлорметаном (2 л). Смесь перемешивают при температуре ниже -5oС в течение 2,75 - 3,25 час, добавляют воду (400 л) и смесь перемешивают в течение по меньшей мере 15 мин. Органический слой отделяют и промывают разбавленной хлористоводородной кислотой, приготовленной из концентрированной хлористоводородной кислоты (40 л) и воды (400 л). Органический слой снова отделяют и затем примерно 80% (360 л) дихлорметана удаляют перегонкой. Затем загружают изопропиловый спирт (140 л) и перегонку продолжают до тех пор, пока температура головной части не достигнет 80oС. Раствор затем охлаждают и добавляют изопропиловый спирт до получения общей массы 200 кг. Этот раствор затем делят на две части по массе и каждую половину используют непосредственно в следующей стадии.
(b) Гидрохлорид 2-амино-N-(1,2-дифенил-1-метилэтил)ацетамида
В реакционный сосуд загружают половину продукта со стадии (а) в растворе пропан-2-ола из вышеуказанной реакции (общая масса 100 кг), дополнительный пропан-2-ол (179,4 кг) и метанол (61,2 л). Сосуд продувают азотом и затем к раствору добавляют концентрированную хлористоводородную кислоту (15,8 кг). Раствор нагревают до температуры образования флегмы в течение 2-3 час и затем фильтруют через находящийся на одной линии фильтр во второй сосуд. Первый реакционный сосуд и фильтр промывают при помощи метанола (8 кг) во второй сосуд. Этот сосуд затем нагревают и растворитель перегоняют для удаления избыточного метанола. Перегонку продолжают до тех пор, пока не будет удалено 125 кг дистиллята (смесь метанола и пропан-2-ола). Сосуд охлаждают до -5oС в течение примерно 2 час, конечный продукт отделяют фильтрованием и промывают холодным (-5oС) пропан-2-олом (25 л). Продукт сушат на поддоне в вакууме, получая конечный продукт в виде не совсем белого твердого вещества (21,4 кг, общий выход 87% из гидрохлорида 1,2-дифенил-2-пропиламина). Продукт был достаточно чистым для использования в фармацевтической препаративной форме без дополнительной очистки.
Пример 2
Получение гидрохлорида 2-амино-N-(1,2-дифенил-1-метилэтил)ацетамида (гидрохлорид ремацемида)
(а) Смесь гидрохлорида 1,2-дифенил-2-пропиламина (40 г, 0,1507 моль) и толуола (100 мл) перемешивают при комнатной температуре в атмосфере азота. В виде одной порции добавляют триэтиламин (46,1 мл, 0,3315 моль) и продолжают перемешивание получаемой смеси.
(b) Смесь ВОС-глицина (30,3 г, 0,1733 моль) и толуола (486 мл) перемешивают в атмосфере азота. В виде одной порции добавляют пивалоилхлорид (21,3 мл, 0,1733 моль). Сразу после добавления содержимое реактора охлаждают до -5oС. В течение одного часа добавляют триэтиламин (17,5 г, 0,1733 моль), поддерживая содержимое сосуда при -5oС. После завершения добавления получаемую смесь перемешивают в течение 2 часов. Суспензию гидрохлорида 1,2-дифенил-2-пропиламина, полученную в (А), добавляют в течение 30 минут, поддерживая температуру сосуда -5oС. После завершения этого добавления реакционную смесь перемешивают в течение дополнительных 3 часов и затем добавляют воду (250 мл) и внутренней температуре дают повыситься до 20oС. Смесь перемешивают в течение 45 минут и затем слой толуола отделяют. Анализ ВЭЖХ показывает 96,4% превращение в целевой ВОС-ремацемид. Этот раствор используют непосредственно в стадии удаления защитной группы.
(c) Раствор в толуоле ВОС-ремацемида (содержащий по расчету 0,3013 моль ВОС-ремацемида) перемешивают при 65oС. В течение 10 минут добавляют хлористоводородную кислоту (52 мл) и затем смесь перемешивают при 65oС в течение 1,5 часа. В течение этого времени происходит осаждение твердого вещества.
Смесь нагревают до температуры образования флегмы и растворитель собирают путем перегонки. Собирают 310 мл растворителя и температура головной части достигает 98oС. Смесь охлаждают до -5oС, затем фильтруют и реакционный сосуд промывают толуолом (2 х 30 мл). Влажная масса составляет 127,7 г. Отфильтрованный остаток сушат в вакууме при 80oС, получая 88,2 г сырого гидрохлорида ремацемида, что составляет выход 96,1% относительно гидрохлорида 1,2-дифенил-2-пропиламина.
Пример 3
Кристаллизация гидрохлорида ремацемида
Смесь гидрохлорида ремацемида (50 г) и метанола (200 мл) перемешивают и нагревают до температуры образования флегмы с образованием раствора. Это был минимальный объем метанола, требуемый для образования раствора. Раствор фильтруют через 0,2m и затем промывают при помощи метанола (12 мл).
Раствор повторно нагревают до температуры образования флегмы и растворитель удаляют путем перегонки, причем собирают 50 мл дистиллята. Затем добавляют изопропанол (400 мл), который инициирует осаждение. Перегонку продолжают, причем собирают еще 410 мл дистиллята. Смесь охлаждают до -5oС, разбавляют изопропанолом (100 мл) и очищенный гидрохлорид ремацемида собирают фильтрованием, затем сушат в вакууме при 65oС. Масса высушенного очищенного материала составляет 46,05 г (выход 92%).
Настоящее изобретение относится к способу получения соединения формулы (I), где R1 и R2 представляют собой фенил или 4-фторфенил, R3 представляет собой водород, алкил или метоксикарбонил, R4 представляет собой водород или метил, взаимодействием соединения формулы (II) с соединением формулы (III), где R5, R6, R7, R8, R9, R10 представляют собой алкил, с получением соединения формулы (IV) и с последующим удалением защитной группы, затем необязательно образованием фармацевтически приемлемой соли. Предложенный способ имеет в качестве преимущества значительно повышенный выход и обеспечивает получение целевого продукта с хорошей чистотой. 15 з.п.ф-лы.
где R1 и R2 независимо представляют фенил или 4-фторфенил;
R3 представляет водород, С1-4 алкил или метоксикарбонил;
R4 представляет водород или метил;
включающий реакцию соединения формулы II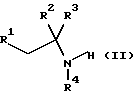
где R1, R2, R3 и R4 имеют значения, определенные в формуле (I),
с соединением формулы (III)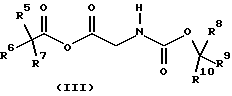
где R5, R6, R7, R8, R9 и R10 независимо представляют С1-6-алкил,
с получением соединения формулы (IV)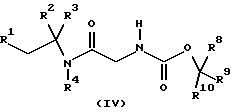
где R1, R2, R3, R4, R8, R9 и R10 имеют значения, определенные в формуле (I),
с последующим удалением защитной группы и затем необязательно образованием фармацевтически приемлемой соли.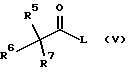
где R5, R6 и R7 имеют значения, определенные в формуле (III);
L представляет уходящую группу,
с соединением формулы (VI)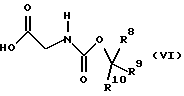
где: R8, R9 и R10 имеют значения, определенные в формуле (III), с последующим добавлением органического амина.
| УСТАНОВКА ДЛЯ СВАРКИ ТЕРМОПЛАСТОВ | 0 |
|
SU279937A1 |
| Tetrahedron Letters, 1995, vol.36, n.2, р.211, tab.1 | |||
| Способ запирания вентилей инвертора | 1977 |
|
SU955508A1 |
| Способ получения -замещенныхАМидОВ -АМиНОКАРбОНОВыХ КиС-лОТ | 1977 |
|
SU795456A3 |

