Настоящее изобретение относится к новым фармацевтическим соединениям, способам их получения, фармацевтическим композициям, содержащим указанные соединения, и способам их использования.
Аденозинтрифосфат (АТФ) оказывает сильное фармакологическое действие на различные ткани. Активность АТФ и других внеклеточных адениновых нуклеотидов, таких, как аденозиндифосфат (АДФ) и аденозинмонофосфат (АМФ), опосредуется P2-пуриноцепторами. Однако действие АТФ в некоторых тканях, например, в мочевом пузыре, может быть снижено вследствие быстрого дефосфорилирования до АМФ паденозина под действием эктонуклеотидазы, присутствующей в этих тканях.
В последних работах, посвященных исследованию P2-пуриноцепторов, присутствующих в различных тканях, были использованы в качестве биологических зондов АТФ-аналоги; которые являются резистентными к дефосфорилированию.
В работе Cusack и др. (Br. J. Phatmacol., 1987, 90. 791 - 795), проведенной с использованием мочевого пузыря и tacnia coli морской свинки, описывается активность таких соединений, как моноангидрид 2-метилтио-5'-адениловой кислоты с метиленбифосфоновой кислотой, моноангидрид 2-метилтио-5'-адениловой кислоты с дихлорметиленбифосфоновой кислотой, и моноангидрид 2-метилтио-5'-адениловой кислоты с дифторометиленбисфосфоновой кислотой. В работе Stone и Cusack (Br. J. Phatm., 1989, 97, 631 - 635) описано использование inter alia моноангидрида 2-метилтио-5'-адениловой кислоты с дифторметиленбисфосфоновой кислотой в исследовании P2-пуриноцепторов в гиппокампе крысы. Maguire и Satchell в своей работе "Physiological and Regulatory Function of Adenosine and Adenine Nucleotides" (Ed. H.P. Baer & G.I. Drummond, Raven Press, New York, 1979, pp 33 - 43) раскрывают ингибирование taenia coli морской свинки соединением моноангидрида 2-хлоро-5'-адениловой кислоты с метиленбисфосфоновой кислотой.
В работе Cusack и Hourni (Nucleosides & Nucleosides, 1991, 10 (5), 1019 - 1028) также сообщается, что моноангидрид 2-метилтио-5'-адениловой кислоты с метиленбисфосфоновой кислотой ингибирует АДФ-α -S-индуцированную агрегацию тромбоцитов.
В Международной патентной заявке WO 92/17488 (Fisons pls) раскрывается ряд 2-замещенных АТФ-аналогов и их активность как ингибиторов агрегации тромбоцитов.
Авторами настоящей заявки была получена группа новых N-алкил-2-замещенных АТФ-аналогов, обладающих фармакологической активностью.
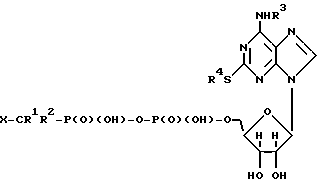
В соответствии со своим первым вариантом, настоящее изобретение относится к соединению формулы I:
где R1 и R2 незаисимо представляют собой водород или галоген;
R3 и R4 независимо представляют собой фенил или алкил, необязательно замещенный одним или несколькими заместителями, выбранными из OR5, C1-6-алкилтио, NR6R7, фенила, COOR3, и галогена;
R5, R6, R7 и R8 независимо представляют собой водород или C1-6-алкил; и
X представляет собой кислотную часть;
и к его фармацевтически приемлемым солям.
Соединения формулы I могут существовать в таутомерной, энантиомерной, и диастереомерной формах, которые также вхолят в объем настоящего изобретения.
Кроме того, настоящее изобретение относится к способу получения соединений формулы I и их солей, заключающемуся в том, что:
а) соединение формулы II или его соль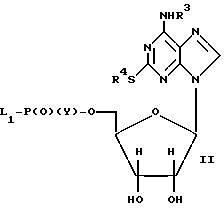
где R3 и R4 определены выше; L1 представляет собой уходящую группу; а Y представляет собой (i) OH, или (ii) уходящую группу L2, подвергают реакции с соединением формулы III или его солью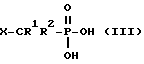
где R1, R2 и X определены выше, а затем в случае, когда у является L2, подвергают гидролизу;
b) удаляют защищенную группу из соответствующего защищенного соединения формулы I, в котором являются защищенными одна или несколько функциональных групп; а затем, если это необходимо, полученное соединение формулы I или его соль превращают в фармацевтически приемлемую соль, или наоборот.
В стадии (a) (i), где Y является OH, уходящими группами L1 могут быть амины, например, диалкиламины, либо насыщенные или ненасыщенные циклоамины; а предпочтительно, если такими уходящими группами являются морфолинил, имидазолил или триазолил. Указанную реакцию предпочтительно осуществляют в растворителе, предпочтительно, в полярном апротонном растворителе, таком, как пиридин, диметилформамид, ацетонитрил, гексаметилфосфоротриамид, N, N''-диметилпропиленмочевины, или 1-метил-2-пирролидинол. Эта реакция может быть проведена при температуре от -20oC до 100oC, например, при 10 - 30oC.
Соединения формулы II, в которых Y представляет собой OH, либо являются известными, либо они могут быть получены известными методами, например, методом, описанным в Международной патентной заявке WO 92/17499 (Jisons plc. ). Например, соединения формулы II, в которых L1 является морфолинилом, могут быть получены из соответствующих 5'-монофосфатов путем их обработки морфолином в присутствии конденсирующего агента, такого, как дициклогексилкарбодиимид, предпочтительно, в присутствии протонного растворителя или смеси растворителей, такой, как смесь т-бутанола и воды.
В стадии (a) (ii), когда Y является уходящей группой L2, уходящие группы L1 и L2 могут представлять собой галоген, например, хлор, L1 и L2 могут быть одинаковыми или различными, но предпочтительно, одинаковыми. Соединения формулы II, в которых Y представляет собой L2, могут быть получены из соответствующего нуклеозида посредством реакции этого нуклеозида с фосфорилирующим агентом, несущим три уходящие группы, т.е. POL1L2L3; а в частности, POCl3. Полученное соединение формулы II не должно быть обязательно выделено; например, оно может быть подвергнуто реакции in situ с соединением формулы III с последующим гидролизом, например, основным гидролизом с использованием Na2CO3.
Нуклеозиды и нуклеозид-5'-монофосфаты, используемые для получения соединений формулы II, либо являются известными соединениями, либо они могут быть получены известными методами из известных соединений (см. , например "Chemistry of Nucleosides and Nucleotides" Vol. 2, Ed. Leroy B. Town send, Plenum Press, 1991).
Соединения формулы III либо являются известными соединениями, либо они могут быть получены известными методами, описанными, например, в Международной патентной заявке WO92/17488 (Jisons plc.).
В рассматриваемой реакции может оказываться необходимым, чтобы функциональные группы, например, гидрокси- или аминогруппы, присутствующие в исходных соединениях, были защищенными, а поэтому, в стадии (b) могут быть удалены одна или несколько защитных групп.
Подходящими защитными группам и методами их удаления являются группы и методы, описанные, например, в "Protective Groups in Organic Synthesis", J. Green и P. G.M. Wutts, John Wiley & Sons Inc., 1991. Гидрокси-группы могут быть, например, защищены арилметильными группами, такими, как фенилметил, дифенилметил, или трифенилметил; ацильными группами, такими, как ацетил, трихлорацетил, или трифтороацетил; или тетрагидропираниловыми производными. Подходящими аминозащитными группами являются арилметильные группы, такие, как бензил, (R,S) α- фенилэтил, дифенилметил, или трифенилметил; и ацильные группы, такие, как ацетил, трихлорацетил, или трифтороацетил. Подходящими методами разблокирования являются гидрогенолиз, кислотный или основный гидролиз либо фотолиз. Арилметильные группы могут быть, например, удалены путем гидрогенолиза в присутствии металлического катализатора, например, палладированного угля. Тетрагидропиранильные группы могут быть отщеплены путем гидролиза в кислотных условиях. Ацильные группы могут быть удалены путем гидролиза с использованием основания, такого, как гидроксид натрия или карбонат калия; а такая группа, как трихлорацетил, может быть удалены путем восстановления, например, с использованием цинковой и уксусной кислоты.
Соединения формулы I или их соли могут быть выделены из их реакционных смесей с использованием стандартной техники.
Описания вышеупомянутых работ вводятся в настоящее описание в качестве ссылок.
Соли соединений формулы I могут быть получены реакцией свободной кислоты или ее соли, либо свободного основания, или его соли, или производного, с одним или несколькими эквивалентами соответствующего основания или кислоты. Эта реакция может быть осуществлена в растворителе или среде, в которой данная соль является нерастворимой; либо в растворителе, в котором данная соль является растворимой, например, таком, как этанол, тетрагидрофуран, или диэтиловый эфир, который может быть затем удален in vacuo, или путем осушки вымораживанием. Данная реакция может также представлять собой метатетический процесс, либо она может быть осуществлена на ионообменной смоле.
Фармацевтически приемлемыми солями соединений формулы I являются соли щелочных металлов, например, соли натрия и калия; соли щелочноземельных металлов, например, соли кальция и магния; соли элементов Группы III; например, алюминиевые и аммониевые соли. Подходящими солями органических оснований являются соли, образованные с гидроксиламином; низшими алкиламинами, например, метиламином или этиламином; замещенными низшими алкиламинами, например, гидроксизамещенными алкиламинами; либо с моноциклическими азотными гетероциклическими соединениями, например, пиперидином или морфолином; а также соли, образованные аминокислотами, например, такими, как аргинин, лизин, и т. п. или их N-алкиловыми производными; или аминосахарами, например, такими, как N-метил-D-глюкамин или глюкозамин. При этом предпочтительными являются нетоксичные физиологически приемлемые соли, хотя в некоторых случаях, например, для выделения или очистки продукта, могут быть также использованы и другие соли.
Соединения формулы I могут обладать таутомерией, например, имин-енаминовой таутомерией в 6-положении аденина. Указанные соединения могут также содержать одни или несколько асимметрических атомов углерода, а поэтому они могут существовать в виде оптических изомеров и/или диастереоизомеров. Диастереоизомеры могут быть разделены с использованием стандартной техники; например, хроматографии или фракционной кристаллизации. Различные оптические изомеры могут быть выделены с помощью стандартной методики, обычно используемой для разделения рацемических или других смесей данных соединений, например, такой, как фракционная кристаллизация или ВЭЖХ. Альтернативно, нужные оптические изомеры могут быть получены с помощью реакций соответствующих оптически активных исходных материалов в условиях, не благоприятствующих рацемизации.
Алкильными группами, представленными R3 - R8, могут быть прямые, разветвленные или циклические, насыщенные или ненасыщенные алкильные группы.
Галогенами, представленными R1 и R2, являются F, Cl, Br, или I. При этом предпочтительно, чтобы R1 и R2 были одинаковыми. Особенно предпочтительными являются соединения, в которых R1 и R2 представляют собой Cl.
Предпочтительными являются соединения, в которых R3 и R4 представляют собой C1-6-алкил, необязательно замещенный одним или несколькими заместителями, выбранными из OR5, C1-6-алкилтио, NR6R7, фенила, COOR8, и галогена.
Галогены, которые представлены R3 и R4, и которые могут быть замещены, являются Cl, Br и I, а предпочтительно F.
Особенно предпочтительными являются соединения, в которых R8 представляет собой C1-6-алкил, необязательно замещенный C1-6 -алкилтио-группой. Примерами конкретных алкильных групп, представленных R3, являются пропил, бутил, а особенно этил. Предпочтительными замещенными алкильными группами, представленными R3, является 2-(метилтио)этил.
Особенно предпочтительными являются соединения, в которых R4 представляет собой C1-6-алкил, необязательно замещенный одним или несколькими, например, тремя атомами галогена. Предпочтительными R4 - группами являются пропил и 3,3,3-трифторпропил.
Кислотными группами, представленными X, являются кислоты Бренстеда-Лоури, т. е. , группы, действующие как доноры протона. Указанные кислотные группы могут быть одноосновными или многоосновными. В качестве конкретных примеров таких кислотных групп могут служить -P(O)(OH)2, -SO3H, и -CO2H.
Прелпочтительными соединениями формулы I являются соединения, в которых представляет собой -P(O)(OH)2.
Соединения формулы I могут быть использованы для лечения млекопитающих, поскольку они обладают фармакологической активностью. В частности, они обладают активностью, способствующей предупреждению агрегации тромбоцитов.
Эффективность соединений формулы I как ингибиторов агрегации тромбоцитов может быть оценена исходя из их способности действовать как антагонисты по отношению к P2T-рецептору, см. Пример X.
Указанные соединения могут быть использованы в любых условиях, где имеет место агрегация тромбоцитов. Таким образом, эти соединения обладают антитромботическим действием и могут быть использованы для лечения или профилактики неустойчивой стенокардии, тромбоэмболического шока и заболеваний периферического кровообращения. Кроме того, указанные соединения могут быть использованы для лечения или профилактики остаточных явлений тромботических осложнений, возникающих в результате пластических операций на сосудах, тромболиза, эндартерэктомии, операций по пересадке на сердце и сосудах, гемодиализа, и искусственного кровообращения.
Другими показаниями к применению соединений настоящего изобретения является лечение или профилактика диссеминированного внутрисосудистого свертывания, тромбоза грубоких вен, преэклампсии/эклампсии, повреждения тканей после хирургических операций или травм, васкулитов, артериитов, тромбоцитемии, ишемии и мигрени.
В другом варианте своего осуществления настоящее изобретение относится к получению соединений формулы I, определенных выше, в виде фармацевтических средств.
Кроме того, настоящее изобретение относится к использованию соединений формулы I или их фармацевтисески приемлемых солей для изготовления фармацевтических композиций, предназначенных для лечения состояний, связанныъх с агрегацией тромбоцитов.
Дозы вводимых соединений могут широко варьироваться в зависимости от различных факторов, таких, как структура конкретного соединения формулы I, способ введения, физическое состояние пациента и тяжесть заболевания. Однако в основном дневная доза для человека составляет от 0,1 мг до 1000 мг, и может быть введена в виде разделенных доз до 6 раз в день. Если данное соединение вводится путем вливания, то типичная доза для человека составляет, например, 0,5 мкг/кг/мин.
Соединения настоящего изобретения вводятся в основном в виде фармацевтической композиции.
Так, например, в соответствии с первым вариантом настоящего изобретения фармацевтическая композиция, предпочтительно, включает в себя менее чем 80 мас. %, а более предпочтительно, менее чем 50 мас.%, например, от 0,1 до 20% соединения формулы I или его фармацевтически приемлемой соли, определенных выше, в сочетании с фармацевтически приемлемым разбавителем или носителем.
Настоящее изобретение также относится к способу получения указанной фармацевтической композиции путем смешивания ее ингредиентов.
Ниже приводятся примеры фармацевтических препаратов, которые могут быть использованы в целях настоящего изобретения, а также подходящих для этой цели разбавителей или носителей:
для внутривенных инъекций или вливаний - очищенная вода или физиологический раствор;
для ингаляций - необработанная лактоза;
для таблеток, капсул и драже - микрокристаллическая целлюлоза; фосфат кальция; диатомовая земля; сахар, такой, как лактоза, декстроза или маннит; тальк; стеариновая кислота; крахмал; бикарбонат натрия и/или желатин;
для суппозиториев - натуральные или отвержденные масла или воски.
Если данное соединение предназначено для использования в водном растворе, например, для вливаний, то эта композиция может включать в себя и другие добавки. Такими добавками могут быть, например, хелатообразующие агенты или секвестранты, антиоксиданты; добавки, корректирующие тоничность; pH-модифицирующие агенты и буферные добавки.
Если необходимо, то растворы, содержащие соединение формулы I, могут быть подвергнуты выпариванию, например, путем осушки вымораживанием или осушки распылением с получением твердой композиции, которая может быть затем переведена в другую форму непосредственно перед использованием.
Если данная композиция не является раствором, то предпочтительно, чтобы она была изготовлена в форме, имеющей средний диаметр от 0,01 до 10 мкм. Указанные композиции могут также содержать соответствующие консерванты, стабилизирующие и смачивающие агенты, солюбилизаторы, например, водорастворимый целлюлозный полимер, такой, как гидроксипропилметилцеллюлоза, или водорастворимый гликоль, такой, как пропиленгликоль; подслащивающие добавки, красители, и ароматизирующие добавки, Если необходимо, то композии настоящего изобретения могут быть изготовлены в виде препаратов с пролонгированным высвобождением.
В еще одном своем варианте настоящее изобретение относится к способу лечения заболеваний, связанных с агрегацией тромбоцитов, заключающемуся в том, что пациенту, страдающему указанным заболеванием, вводят терапевтически эффективное количество соединения формулы I, определенного выше.
Преимущество соединений настоящего изобретения заключается в том, что они продуцируют гораздо меньшее количество побочных эффектов, например, они обладают меньшей способнсотью к индуцированию гипотермии, как было установлено с помощью методики, описанной в Пример V, и, кроме того, они обладают большей продолжительностью действия, являются менее токсичными, более эффективными, более стабильными, более легко абсорбируются, более легко выводятся из организма, обнаруживают более широкий спектр активности или имеют другие преимущественные фармакологические свойства по сравнению с известными соединениями.
Ниже проиводятся примеры, иллюстрирующие, но не ограничивающие настоящее изобретение. В данный примерах, все температуры даны в градусах Цельсия, а названия соединений приводятся в соответствии со стандартной химической номенклатурой.
Пример 1
Моноангидрид N-этил-2-(пропилтио)-5'-адениловой кислоты с дихлорметиленбисфосфоновой кислотой, етранатриевая соль
а) N-Этил-2-(пропилтио)аденозин
9-(2,3,5-Три-O-ацетил -β- D-рибофуранозил)-6-хлоро-2-)-(пропилтио)пурин (1,3 г) и этиламин (1,6 мл) в диоксане (30 мл) и воде (30 мл) нагревали в герметично закрытом автоклаве в течение 20 часов при температуре 110oC. После охлаждения до комнатной температуры и выпаривания получили остаток, который перекристаллизовывали из этилацетата. В результате очистки с помощью хроматографии на двуокиси кремния (элюент: метанол/этилацетат, 1:15) было получено целевое соединение, а именно, N-этил-2-(пропилтио)аденозин (0,46 г).
MC (FAB): 370 (M+H, 100%), 238 (30%).
в) Моноаммониевая соль N-этил-2-(пропилтио)-5-адениловой кислоты
К перемешанному раствору продукта стадии а) (0,4 г) в триэтилфосфате (12 мл) при 0oC добавляли 0,66 г оксихлорида фосфора. Через 4,5 часа реакционную смесь выливали в смесь льда/воды (100 г), содержащую бикарбонат натрия (1,45 г). Через 45 минут раствор промывали эфиром (2 х 100 мл) и загружали на колонку с 50WX 8 (H+ форма). Эту колонку промывали водой до тех пор, пока pH элюата не становился равным 6, а затем элюировали 2 М гидроксидом аммония. После лиофилизации получали целевое соединение, а именно, моноаммониевую соль N-этил-2-(пропилтио)-5'-адениловой кислоты (0,32 г).
31P ЯМР δ (D2 O): 2,03 (с).
с) Моноангидрид N-этил-2-(пропилтио)-5'-адениловой кислоты с дихлорметиленбисфосфоновой кислотой, тетранатриевая соль
Целевой продукт стадии (b) (0,38 г) и трибутиламин (0,15 г) объединяли в небольшом объеме пиридина и полученный раствор выпаривали досуха. Затем этот раствор осушали путем азеотропной перегонки с пиридином (3 х 15 мл), а после этого с безводным N,N - диметилформамидом (ДМФ) (2 х 15 мл) и образовавшийся остаток растворяли в 10 мл безводного ДМФ. После добавления карбонилдиимидазола (0,66 г) реакционную смесь оставляли на 4 часа при комнатной температуре, а затем добавляли метанол (0,209 г). Через 30 минут добавляли 2,09 г моно(трибутиламмониевой) соли дихлорметиленбисфосфоновой кислоты в безводном ДМФ (30 мл) и полученную смесь перемешивали в течение 18 часов при комнатной температуре. После фильтрования и выпаривания получали остаток, который очищали с помощию хроматографии (DEAE-Сефадекс, элюент: O М 0,6 М бикарбонат триэтиламмония). В результате лиофилизации получали триэтиламмониевую соль, которую превращали в натриевую соль путем растворения в метаноле (2 мл) и добавления раствора иодида натрия (IM суспензия в ацетоне (30 мл)). Осадок собирали путем центрифугирования, промывали вышеуказанной суспензией в ацетоне (4 х 40 мл), и снова центрифугировали Образовавшееся твердое вещество растворяли в воде и лиофилизовали, в результате чего получали целевую соль в виде бесцветного порошка (0,25 г).
31 P-ЯМР δ (D2O): 9,00 (д, J = 18,6 Гц), 1,18 (дд, J = 18,6 Гц, J = 30,4 Гц), -9,35 (д, J = 18,6 Гц).
Пример 2
Моноангидрид N-бутил-2-(пропилтио)-5'-адениловой кислоты с дихлорметиленбисфосфоновой кислотой, тетранатриевая соль
а) N-Бутил-2-(пропилтио)аденозин
Целевое соединение, а именно, N-бутил-2-(пропилтио)-аденозин, получали способом, описанным в Примере (1а).
MC (FAB): 398 (M+H+: 100%).
b) Моноангидрид N-бутил-2-(пропилтио)-5'-адениловой кислоты с дихлорметиленбисфоновой кислотой, тетранатриевая соль
К раствору продукта стадии а) (1,8 г) в триэтилфосфате (50 мл), охлаждая льдом, по капле добавляли оксихлорид фосфора (1,39 г). Полученный раствор перемешивали в течение 4 часов при комнатной температуре.
К перемешанной суспензии моно(трибутиламмониевой) соли дихлорметиленбисфосфоной кислоты (5, 76 г) в триэтилфосфате (60 мл) добавляли трибутиламин (2,16 мл). После перемешивания в течение часа при комнатной температуре полученный раствор в течение 15 минут добавляли к раствору, описанному выше. Затем полученную смесь перемешивали в течение 4 часов, после чего реакционную смесь выливали в 5% водный раствор бикарбоната натрия (113 мл) и перемешивали в течение 18 часов. Полученный раствор промывали эфиром (4 х 50 мл), а затем осушали вымораживанием. В результате очистки (обращенно-фазовые С18-колонки с двуокисью кремния; элюент: 4% солевой раствор, а затем вода) получали целевую соль в виде бесцветного твердого вещества (0,69 г).
31P ЯМР δ (D2O): 9,70 (д, J = 18,4 Гц), 3,4 (дд, J = 18,4 Гц, J = 30,5 Гц), 9,19 (д, J = 30,5 Гц).
Пример 3
В соответствии со способом, описанным в Примере 2, были получены следующие соединения:
а) Моноангидрид N-пропил-2-(пропилтио)-5'-адениловой кислоты с дихлорометилденбисфосфоновой кислотой, тетранатриевая соль
I) N-Пропил-2-(пропилтио)аденозин
MC (FAB): 348 (M+H+, 100%).
II) Моноангидрид N-пропил-2-(пропилтио)-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой, тетранатриевая соль
31P ЯМР δ (D2O): 8,92 (д, J = 18,5 Гц), 1,07 (дд, J = 18,7 Гц, J = 29,0 Гц), -9,4 (д, J = 29,4 Гц).
в) Моноангидрид N-(1-метилэтил)-2-(пропилтио)-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой, диаммониевая соль
I) N-(1-Метилэтил)-2-(пропилтио)аденозин
MC (FAB): 384 (M+H+, 100%), 252.
II) Моноангидрид N-(1-метилэтил)-2-(пропилтио)-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой, диаммониевая соль
В результате очистки неочищенного продукта с помощью хроматографии (DEAE-Cефадекс; элюент 0 М - 0,6 М раствор бикарбоната аммония) получали целевую соль.
31P ЯМР δ (D2O): 8,71 (д, J = 18,6 Гц), 0,38 (дд, J = 19,1 Гц, J = 28,7 Гц), -9,49 (д, J = 29,0 Гц).
c) Моноангидрид N-(2-метоксиэтил)-2-(пропилтио)-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой, тетранатриевая соль
I) N-(2-Метоксиэтил)-2-(пропилтио)аденозин
MC (FAB): 400 (M+H+, 100%), 268.
II) Моноангидрид N-(2-метоксиэтил)-2-(пропилтио)-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой, тетранатриевая соль
31P-ЯМР δ (D2O): 9,05 (д, J = 18,7 Гц), 1,44 (дд, J = 18,8 Гц, J = 29,3 Гц), -9,40 (д, J = 29,5 Гц).
d) Моноангидрид N-циклопентил-2-(пропилтио)-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой, тетранатриевая соль
I) N-Циклопентил-2-(пропилтио)аденозин
MC (FAB): 410 (M+H+), 278 (100%).
II) Моноангидрид N-циклопентил-2-(пропилтио)-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой, тетранатриевая соль
1) Анализ для C19H26Cl2N5Na4O12P3S• 7H2O:
Вычислено: C 24,56; H 4,30; N 7,53; S 3,44 (%)
Найдено: C 24,43; H 4,43; N 7,52; S 3,76 (%)
e) Моноангидрид N-фенил-2-(пропилтио)-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой, тетранатриевая соль
I) N-Фенил-2-(пропилтио)аденозин
MC (FAB): 418 (M+H, 100%), 278.
II) Моноангидрид N-фенил-2-(пропилтио)-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой, тетранатриевая соль
31P-ЯМР δ (D2O): 8,98 (д, J = 18,3 Гц), 2,70 (дд, J = 18,3 Гц и J = 30,6 Гц), -9,89 (д, J = 30,6 Гц)
f) Моноангидрид N-(2,2,2-трифтороэтил)-2-(пропилтио)-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой, тетранатриевая соль
I) N-(2,2,2-Трифтороэтил)-2-(пропилтио)аденозин
MC (FAB): 424 (M+H+), 292 (100%).
II) Моноангидрид N-(2,2,2-трифторэтил)-2-(пропилтио)-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой, тетранатриевая соль
MC (FAB): 822, 820, 818 (M+H+), 115 (100%).
g) Моноангидрид N-(метоксикарбонилметил)-2-(пропилтио)-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой, триаммониевая соль
I) N-(Метоксикарбонилметил)-2-(пропилтио)аденозин
Анализ для C16H23N5O6S:
Вычислено: C 46,48, H 5,61, N 16,94, S 7,76 (%)
Найдено: C 46,44, H 5,43, N 16,80, S 7,67 (%)
II) Моноангидрид N-(метоксикарбонилметил)-2-(пропилтио)-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой, триаммониевая соль
Неочищенный продукт очищали с помощью хроматографии
(DEAE-Сефадекс; элюент: 0 М - 0,6 М раствор бикарбоната аммония), и получали целевую соль.
31P-ЯМР δ (D2O): 9,05 (д, J = 18,7 Гц), 1,44 (дд, J = 18,8 Гц, J = 29,3 Гц), -9,40 (д, J = 29,5 Гц).
h) Моноангидрид N-[2-(метилтио)этил]-2-(пропилтио)-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой, триаммониевая соль
I) N-[2-(Метилтио)этил]-2-(пропилтио)аденозин
MC (FAB): 416 (M+H+, 100%).
II) Моноангидрид N-[2-(метилтио)этил]-2-(пропилтио)-5'- адениловой кислоты с дихлорометиленбисфосфоновой кислотой, триаммониевая соль
После очистки выделенного сырого продукта с помощью хроматографии (ДЕАЕ-Сефадекс; элюент: O M-0,6 M раствор бикарбоната аммония) получали целевую соль.
31P-ЯМР δ (D2O): 8,68 (д, J=18,6 Гц), 0,33 (дд, J=18,9 Гц, J=29,0 Гц); -9,53 (д, J=29,0 Гц).
j) Моноангидрид N-[2-(N,N-диметиламино)этил]-2-(пропилтио)-5- адениловой кислоты с дихлорометиленбисфосфоновой кислотой, тринатриевая соль
I) N-[2-(N, N-Диметиламино)-этил] -2-(пропилтио)аденозин MC (FAB): 413 (M+H+, 100%).
II) Моноангидрид N-[2-(N,N-диметиламино)этил]-2-(пропилтио)-5'- адениловой кислоты с дихлорометиленбисфосфоновой кислотой, тринатриевая соль
MC (FAB): 789, 787, 785, (M+H+), 93 (100%).
Пример 4
Моноангидрид 2-(циклогексилтио)-N-5'-этил-5'- адениловой кислоты с дихлорометиленбисфосфоновой кислотой, тетранатриевая соль
a) 9-(2,3,5-Трис-O-ацетил -β- D-рибофуранозил)-6-хлоро-2- (циклогексилтио)пурин
К раствору 2-амино-9-(2,3,5-три-O-ацетил -β- D-рибофуранозил-6-хлоропурина (10,0 г) в ацетонитриле (200 мл) добавляли дициклогексилдисульфид (51,5 г) и изоамилнитрит (16,96 г). Полученный раствор дегазировали азотом, а затем нагревали при температуре 60oC в течение 16 часов. После концентрирования раствора остаток очищали (двуокись кремния; элюент:этилацетат/петролейный эфир, 1: 1) и получали целевое соединение, а именно, 9-(2,3,5-Три-O-ацетил -β- D-рибофуранозил)-6-хлоро-2-N-(циклогекситио)пурин (3,88 г) в виде оранжевой смолы. MC (EI): 528, 526 (M+ ), 43 (100%).
b) 2-(Циклогексилтио)-N-этил-аденозин
С использованием продукта стадии (a), было получено целевое соединение, а именно, 2-(циклогексилтио)-N-этил-аденозин, в соответствии с методикой, описанной в Примере 1(a). MC (FAB): 510 (M+H+, 100%).
c) Моноангидрид 2-(циклогексилтио)-N-этил-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой, тетранатриевая соль
Целевое соединение было получено способом, опсианным в Примере 2(b), за исключением того, что использовали продукт стадии (b).
31H-ЯМР δ (D2O): 9,85 (д, J=18,5 Гц), 3,85 (дд, J=18,5 Гц, J=30,4 Гц), -9,07 (д, J=30,4 Гц).
Пример 5
Моноангидрид N-этил-2-[(3,3,3-трифторопропил)тио]-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой, тринатриевая соль
a) 2-[(3,3,3-Трифторопропил)тио]аденозин
Суспензию, состоящую из гидрида натрия (60%, 1,453 г) и аденозин-2-тионмоногидрата (5,35 г) в ДМФ (80 мл) перемешивали в течение часа при комнатной температуре, а затем добавляли 3-хлоро-1,1,1-трифторпропан (6,6 мл). После перемешивания в течение 5 дней раствор концентрировали, а образовавшийся остаток распределяли между этилацетатом (250 мл) и водой (150 мл). Органическую фазу осушали, а затем концентрировали. В результате очистки (SiO2; элюент: дихлорметан/метанол, 9:1) получали целевое соединение, а именно 2-[(3,3,3-трифторопропил)тио] аденозин, в виде бесцветного твердого вещества (5,55 г).
MC (FAB): 396 (M+H+, 100%).
b) N-Ацетил-2-[(3,3,3-трифторопропил)тио]аденозин-2'-3',5'-триацетат
Полученный продукт стадии (a) (5,28 г) и безводный ацетат натрия (0,723 г) в уксусной ангидриде (42 мл) перемешивали в течение 6,5 часа при температуре 80oC. Этот раствор разводили водой (100 мл), перемешивали при комнатной температуре в течение 18 часов, а затем экстрагировали дихлорметаном (4х200 мл). Объединенные органические экстракты промывали насыщенным раствором бикарбоната натрия (200 мл), а затем выпаривали и образовавшийся остаток хроматографировали на двуокиси кремния (элюент:диэтиловый эфир:метанол, 97:3), в результате чего получали целевое соединение (N-ацетил-2-[(3,3,3-трифторопропил)тио]-аденозин-2',3',5'-триацетат) в виде бесцветной пены (5,35 г).
MC (FAB): 564 (M+H+), 139 (100%).
c) N-Ацетил-N-этил-2-[(3,3,3-трифторопровпил)тио] аденозин-2', 3',5'-триацетат
Полученный продукт стадии (b) (5,12 г) в ДМФ (100 мл) в течение 3 часов добавляли к суспензии гидрида натрия (60%, 0,443 г) в ДМФ (100 мл), содержащий этилиодид (2,2 мл). После перемешивания в течение 2 дней раствор выпаривали, а остаток растворяли в этилацетате (300 мл) и промывали водой (3х100 мл). Органическую фазу концентрировали, а затем остаток очищали (двуокись кремния; элюент: этилацетат:циклогексан, 1:1) и получали целевое соединение (N-ацетил-N-этил-2-[(3,3,3-трифторопропил)тио] аденозин-2', 3', 5'-триацетат) в виде желтой смолы (4,54 г).
MC (FAB): 592 (M+H+), 139 (100%).
d) N-Этил-2-[(3,3,3-трифторопропил(тио]аденозин
Полученный продукт стадии (c) (4,54 г) в растворе гидроксида натрия (0,1 М в метаноле, 155 мл) нагревали с обратным холодильником в течение 30 минут. После охлаждения до комнатной температуры добавляли 0,89 мл ледяной уксусной кислоты и полученный раствор концентрировали. В результате очистки (двуокись кремния; элюент:дихлорметан/метанол, 95:5) получали целевое соединение (N-этил-2-[(3,3,3-трифторопропил)тио]аденозин) в виде бесцветного твердого вещества (2,73 г).
MC (FAB): 424 (M+H+, 100%).
e) Моноангидрид N-этил-2-[(3,3,3-трифторопропил)тио]-5' -адениловой кислоты с дихлорометиленбисфосфоной кислотой, тринатриевая соль
Целевую соль получали способом, описанным в Примере 2b), используя продукт стадии (d).
31P-ЯМР δ (D2O): 8,89 (д, J=18,0 Гц), 2,34 (дд, J=18,0 Гц, J=30,3 Гц), -9,90 (д, J=30,0 Гц).
Пример 6
Нижеследующие соединения были получены способом, описанный в Примере 2:
a) Моноангидрид N-(2,2,2-трифтороэтил)-2-[(3,3,3- трифторопропил)тио]-5'-адениловой кислоты с дихлорометиленбиофосфоновой кислотой, триаммониевая соль
I. N-Ацетил-N-(2,2,2-трифтороэтил)-2-[(3,3,3-трифторопропил) тио]аденозин-2',3',5'-триацетат
MC (FAB): 646 (M+H+).
II. N-(2,2,2-Трифтороэтил)-2-[(3,3,3-трифторопропил)тио] -аденозин
MC (FAB): 478 (M+H+), 346 (100%).
III. Моноангидрид N-(2,2,2-трифтороэтил)-2-[(3,3,3- трифторопропил)тил] -5'-адениловой кислоты с дихлорометиленбисфосфоной кислотой, триаммониевая соль
31P-ЯМР δ (D2O: 8,82 (д, J=18,6 Гц) 0,63 (дд, J=18,9 Гц, J=28,9 гЦ), -9,43 (д, J=29,0 Гц).
b) Моноангидрид N-[2-(метиотио)этил]-2-[(3,3,3-трифторопропилтио]-5'-адениловой кислоты с дихлорометиленбисфоновой кислотой, триаммониевая соль
I. N-Ацетил-N-[2-(метилтио)этил] -2-[(3,3,3-трифторопропил) тио]аденозин-2', 3', 5'-триацетат
MC (FAB): 638 (M+H+), 139 (100%).
II. N-[2-(Метилтио)этил]-2-[(3,3,3-трифторопропил)тио]-аденозин
Анализ для C165H22F3N5O4S 2: (%)
Вычислено: C 40,90; H 4,72; N 14,92; S 13,70;
Найдено: C 40,70; H 4,82; N 14,79; S 13,60.
III. Моноангидрид N-[2-(метилтио)этил] -2-[(3,3,3-трифторпропил)тио]-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой, триаммониевая соль
Неочищенный продукт очищали с помощью хроматографии (DEAE-Сефадекс; элюент: O M-0,6 М раствора бикарбоната аммония), и получали целевую соль.
31P-ЯМР δ (D2O), 8,77 (д, J = 18,7 Гц), 0,38 (дд, J = 18,9 Гц, J = 27,4 Гц), -9,43 (д, J = 28,8 Гц).
Пример 7
Моногидрид N-(2-метоксиэтил)-2-[(3,3,3-трифторпропил)-тио] -5'- адениловый кислоты с дихлорметиленбифосфоновой кислотой, тетранатриевая соль
a) N-(2-Метоксиметил)-2-[(3,3,3-трифторпропил)тио]аденозил
Раствор, содержащий целевое соединение Примера (5b) (4,8 г), 2-бромэтилметиловый эфир (1,2 мл), и карбонат калия (1,77 г) в сухом ДМФ (190 мл), перемешивали при комнатной температуре в течение 3 дней. После добавления 2-бромэтилметилового эфира (1,2 мл) и карбоната калия (1,77 г) полученную смесь перемешивали в течение 24 часов при температуре 40oC. Реакционную смесь фильтровали, после чего фильтрат концентрировали с получением маслообразного вещества, которое распределяли между этилацетатом (200 мл) и водой (200 мл). Органическую фазу осушали, а затем концентрировали. Полученную смолу растворяли в 0,1 М растворе метоксида натрия в метаноле (180 мл), после нагревали с обратным холодильником в течение 45 минут. Эту смесь нейстрализовали с использованирем уксусной кислоты и концентрировали, а остаток очищали (SiO2, элюент:дихлорометан/метанол, 92:8) и получали целевое соединение (N-(2-метоксиэтил)-2-[(3,3,3-трифторпропил)тио]аденозин) в виде бесцветного твердого веществча (3,41 г).
MC (FAB): 454 (M+H+, 100%.
b) Моноангидрид N-(2-метокси этил)-2-[(3,3,3-трифторпропил)тио] -5'-адениловой кислоты с дихлорметиленбисфосфоновой кислотой, тетранатриевая соль
Целевую соль получали способом, описанным в Примере 2(b), используя соединение стадии (a).
31P-ЯМР δ (D2O): 9,88 (д, J = 19,0 Гц), 3,80 (дд, J = 19,0 Гц, J = 31,0 Гц); -9,12 (д, J = 31,0 Гц).
Пример X
Количественная оценка агонистической/антагонистической активности по отношению к P2T-рецептору с использованием промытых тромбоцитов человека
Венозную кровь человека (100 мл) разделяли на три равные части и помещали в 3 пробирки, каждая из которых содержала 3,2% тринатрийцитрата (4 мл) в качестве антикоагулянта. Затем эти пробирки центрифугировали в течение 15 минут при 240 х g, в результате чего получали обогащенную тромбоцитами плазму (PRP), к которой добавляли 300 нг.мл-1 простациклина (PG12, 3 мкл. мл-1 PRP c 1/10-разведением в физиологическом растворе, полученном от маточного раствора 1 мг.мл-1 в этаноле), для стабилизации тромбоцитов в процессе промывки. PRP-плазму, не содержащую эритроцитов, получали путем центрифугирования при 125 х g в течение 10 минут с последующим центрифугированием при 640 х g в течение 15 минут. Супернатант отбрасывали, а осадок тромбоцитов ресуспендировали в модифицированном, не содержащем кальция растворе Тироде (CFT) [(10 мл, состав: 137 мМ (8 г/л) NaCl; 11,9 мМ (1 г/л) NaHCO3; 0,38 мМ (0,06 г/л) NaH2PO4; 2,86 мМ (1 мл 20% раст./л) KCl; 1,05 мМ (1 мл 10% раств. /л) MgCl2; 5,55 мМ (1 г/л) дестрозы], который насыщали газом 95% O2/5% CO2, и поддерживали при 37oC. После добавления еще 300 нг/мл PG12 суспензию объединяли и центрифугировали при 640 х g в течение 15 минут. Затем супернатант отбрасывали и тромбоциты ресуспендировали в 10 мл CFT с последующим добавлением CFT до конечной концентрации тромбоцитов 2 • 105/мкл. Полученную суспензию хранили в 60-мл шприце при 3oC, с выпущенным воздухом.
Восстановление нормальной функции тромбоцитов после их PG12-ингибирования, в целях последования агрегации тромбоцитов, осуществляли не ранее чем через 2 часа после последнего ресуспендирования. Во всех исследованиях 430 мкл-аликвоты суспензии тромбоцитов добавляли в кремниевые кюветы для агрегации, содержащие CaCl2 - раствор (10 мкл 45 мМ раствора, конечная концентрация 1 мМ) и размешивали при 90 об/мин в агрегометре PAP4 (Biodata). Затем добавляли фибриноген человека (Sigma; F 4883) и 8-сульфофенилтеофилин (8-SPT, для блокирования любой P1 - агонистической активности соединений) до конечной концентрации 0,2 мг/мл (10 мкл 10 мг/мл раствора свертываемого белка в физиологическом растворе) и 3 • 10-4 М (10 мкл 5,6 мг/мл раствора в 6% глюкозе), соответственно. После этого начинали наблюдение за адгезией.
Протокол
а) Выбор субмаксимальной концентрации АДФ
Концентрацию АДФ, продуцирующую непосредственно субмаксимальный ответ, выбирали путем построения кривой концентрация/ответ в диапазоне 10 - 300 мкМ. Для этого соответствующий раствор АДФ добавляли в кювету в объеме 10 мкл через 20 минут после начала наблюдения за агрегацией. Ответную реакцию агрегации оценивали исходя из максимальной скорости изменения прозрачности, измеряемой с помощью счетчика со скошенным слоем. Субмаксимальную концентрацию АДФ, выбранную на этой стадии протокола, использовали для последующей оценки антагонистической активности соединений. Все измерения, проводимые для тромбоцитов от каждого донора, осуществляли дважды.
b) Оценка агонистической/антагонистической активности
Через 5 минут после начала наблюдения за агрегацией соответствующий раствор испытуемого соединения или физиологический раствор добавляли в кювету для агрегации в объеме 30 мкл для получения конечной концентрации 0,10 мкМ или 1000 мкМ. Агрегация на этой стадии указывала на агонистическую активность, и если она имела место, то ее оценку проводили путем сравнения с контрольными АДФ-ответами, полученными в стадии (a).
Если агрегация не наблюдалась, то через 15 минут после испытуемого соединения добавляли предварительно выбранную субмаксимальную концентрацию АДФ в объеме 10 мкл. Антагонистическую активность оценивали как % ингибирования контрольного ВДФ-ответа, в результате чего получали приблизительные значения IC50. Соединения, которые полностью ингибировали АДФ-ответ при начальных концентрациях, испытывали снова, но уже при более низком диапазоне концентраций. Соединения с IC50 < 10-8 М также подвергали повторному испытанию в отсутствии 8-SPT для подтверждения отсутствия какой-либо агонистической активности по отношению к P1, а также с 2-минутным (вместо 15-минутного) инкубированием для того, чтобы проверить зависит ли ингибирование от времени.
Результаты
Характерные результаты, полученные для соединений формулы I, представлены в Таблице 1 как отрицательные логарифмы антагоничтической активности (pIC50).
Пример V
Изменение гипотермической активности с использованием восприимчивых мышей
Для эксперимента использовали мышей CP/CD-1 (25-45 г). Этих мышей взвешивали и измеряли температуру ректально с использованием терморезисторного зонды. Затем каждое животное помещали в камеру, имеющую форму усеченного конуса, и хвостовую вену канюлировали с использованием иглы для подкожных инъекций (27G), соединенной с полиэтиленовой канюлепй и закрепленной клейкой лентой в нужном положении. Животных обрабатывали дозой используемого соединения, вводимой в виде 10-минутного внутривенного вливания со скоростью 0,5 мл/кг/мин. Ректальные измерения температуры проводили через 1/3; 1; 1,5; 2; 3; 4; 6 и 24 часа после завершения вливания.
Затем строили график зависимости среднего максимального снижения ректальной температуры от дозы вводимого соединения, и определяли дозу, требуемую для понижения температуры на 5o.

Описываются соединения формулы I, где R1 и R2 независимо представляют собой водород или галоген; R3 и R4 независимо представляют собой фенил или С12-6-алкил, необязательно замещенный одним или несколькими заместителями, выбранными из OR5, С1-6-алкилтио, NR6R7, COOR8 или галогена; R5, R6, R7, R8 независимо представляют собой С1-6-алкил; а Х является кислотным остатком, и его фармацевтически приемлемые соли. Кроме того, раскрываются способы получения указанных соединений. Соединения формулы I ингибируют агрегацию тромбоцитов. 2 c. и 7 з.п.ф-лы, 1 табл.
где R1 и R2 независимо представляют водород или галоген;
R3 и R4 независимо представляют фенил, С1-С6алкил, возможно замещенный заместителями OR5, С1-С6алкилтио, NR6R7, СООR8 или галогеном, где R5, R6, R7 и R8 независимо представляют водород или С1-С6алкил;
X - кислотный остаток,
или их фармацевтически приемлемые соли.
Моноангидрид N-пропил-2-(пропилтио)-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой;
Моноангидрид N-(1-метилэтил)-2-(пропилтио)-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой;
Моноангидрид N-(2-метоксиэтил)-2-(пропилтио)-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой;
Моноангидрид N-циклопентил-2-(пропилтио)-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой;
Моноангидрид N-фенил-2-(пропилтио)-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой;
Моноангидрид N-(2,2,2-трифторэтил)-2-(пропилтио)-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой;
Моноангидрид N-(метоксикарбонилметил)-2-(пропилтио)-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой;
Моноангидрид N-(2-метилтиоэтил)-2-пропилтио)-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой;
Моноангидрид N-[2-(N, N-диметиламино)этил] -2-(пропиотио)-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой;
Моноангидрид 2-(циклогексилтио)-N-этил-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой;
Моноангидрид N-(2,2,2-трифторэтил)-2-[3,3,3-трифторопропил)тио]-5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой;
Моноангидрид N-(2-метоксиэтил)-2-[3,3,3-трифторопропил)тио] -5'-адениловой кислоты с дихлорометиленбисфосфоновой кислотой; или любая из его фармацевтически приемлемых солей.
а) соединение общей формулы II: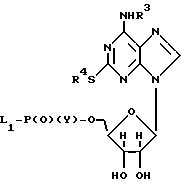
где R3 и R4 имеют вышеуказанные значения; L1 - уходящая аминогруппа или галоген; Y-OH или уходящая группа L2, где L2 - галоген, подвергают реакции с соединением общей формулы III или его солью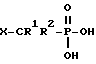
где R1, R2 и X имеют вышеуказанные значения,
и затем в случае, когда Y является L2, подвергают гидролизу,
в) удаляют защитные группы из соответствующего защищенного соединения формулы I, в котором защищены одна или несколько функциональных групп и затем при необходимости переводят полученное соединение формулы I в соль.
Приоритет по пунктам и признакам -10.02.93 - по пп.1 - 9, исключая значения R3 и R4.
| WO 9217488, A1, 1992 | |||
| British Journal of phamacology, 90, N 4, 1987 | |||
| Nucllosides and nucllotides, v.10, N 1 - 3, 1991 | |||
| WO 9011080 A, 1990 | |||
| Способ получения спин-меченого производного аденозинтрифосфата | 1977 |
|
SU764326A1 |

