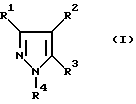
Изобретение относится к усовершенствованному способу получения пиразола и его производных формулы I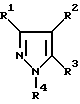
в которой радикалы R1-R4 имеют значения, указанные ниже,
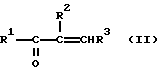
из α,β- ненасыщенных карбонильных соединений формулы II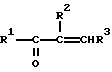
и гидразина либо производных гидразина формулы III
H2N-NHR4.
В EP-A 402722 описано получение пиразола и его производных из α,β- ненасыщенных карбонильных соединений и гидразина либо производных гидразина. Согласно описанному в этой публикации способу пиразолин, либо карбонильное соединение и гидразин или производное гидразина в смеси из серной кислоты и йода или соединения, высвобождающего йод либо йодистый водород, превращают in situ в требуемый пиразол. Выход согласно представленным примерам составляет в среднем приблизительно 78% по отношению к используемому производному гидразина.
Задачей настоящего изобретения является разработка способа, позволяющего получить простым путем пиразол и его производные с более высоким выходом с высокой степенью чистоты.
В соответствии с этой задачей предлагается способ получения пиразола и его производных общей формулы I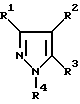
в которой радикалы R1-R4 обозначают водород или алкильную группу, линейную или разветвленную, предпочтительно C1-С10-алкильную группу, более предпочтительно С1-С8-алкильную группу и наиболее предпочтительно C1-С6-алкильную группу, причем эти группы могут быть разорваны гетероатомами, такими как азот, кислород и сера, и могут иметь заместители, выбранные из группы, включающей нитро, карбоксил, сульфонил, галоген, циклоалкил, бициклоалкил, арил и гетероарил; циклоалкильную группу или бициклоалкильную группу, предпочтительно С3-С8-циклоалкильную группу или С6-С10-бициклоалкильную группу, причем эти группы могут быть разорваны гетероатомами, такими как азот, кислород и сера, и могут иметь заместители, выбранные из группы, включающей нитро, карбоксил, сульфонил, галоген, алкил, циклоалкил, бициклоалкил, арил и гетероарил; арильную или гетероарильную группы, такие как фенил, нафтил и пиридил, причем эти группы могут иметь заместители, выбранные из группы, включающей нитро, карбоксил, сульфонил, галоген, алкил, циклоалкил, бициклоалкил, арил и гетероарил.
Способ заключается в том, что α,β- ненасыщенное карбонильное соединение формулы II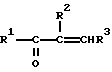
где R1-R3 имеют вышеуказанные значения,
подвергают взаимодействию с гидразином или его производным формулы III
H2N-NHR4,
где R4 имеет вышеуказанные значения,
в присутствии серной кислоты и йода или соединения, высвобождающего йод или йодистый водород. Отличием предлагаемого способа от известного является то, что сначала ведут взаимодействие α,β- ненасыщенного карбонильного соединения формулы II с гидразином или его производным формулы III без добавления растворителя и в отсутствии серной кислоты. Полученную при этом реакционную смесь на последующей стадии подвергают взаимодействию со смесью из серной кислоты и йода или соединения, высвобождающего йод или йодистый водород.
При этом под галогеном подразумеваются в первую очередь фтор, хлор и бром.
В качестве соответствующего углеводородного радикала следует назвать
алкильные группы, которые могут быть линейными либо разветвленными, например, С1-С10-алкил, предпочтительно С1-С8-алкил, прежде всего С1-С6-алкил, причем эти радикалы в свою очередь могут быть разорваны гетероатомами, такими как азот, кислород и сера, и могут иметь заместители, выбранные из следующей группы: нитро, карбоксил, сульфонил, галоген, циклоалкил, бициклоалкил, арил и гетероарил; циклоалкильные группы либо бициклоалкильные группы, например, С3-С8-циклоалкил либо С6-С10-бициклоалкил, причем эти радикалы в свою очередь могут быть разорваны гетероатомами, такими как азот, кислород и сера, и могут иметь заместители, выбранные из следующей группы: нитро, карбоксил, сульфонил, галоген, алкил, циклоалкил, бициклоалкил, арил и гетероарил; арильные группы либо гетероарильные группы, такие как фенил, нафтил и пиридил, причем эти радикалы в свою очередь могут иметь заместители из следующей группы: нитро, карбоксил, сульфонил, галоген, алкил, циклоалкил, бициклоалкил, арил и гетероарил.
Предлагаемый согласно изобретению способ пригоден в первую очередь для получения производных пиразола, в которых по крайней мере один из радикалов R1-R4 обозначает не водород.
Пиразол и его производные находят применение, например, в качестве побочных продуктов, используемых для получения обладающих фармакологическим действием соединений, действующих веществ в средствах защиты растений или же для изготовления красителей.
Более подробно способ согласно данному изобретению осуществляют следующим образом.
Вначале осуществляют взаимодействие α,β- ненасыщенного карбонильного соединения II с гидразином либо производным гидразина III путем их смешивания. Причем во время смешения температуру реакционной среды поддерживают, как правило, в диапазоне от 0 до 100oC, предпочтительно в пределах 10-70oC, прежде всего в пределах 20-50oC. Так как взаимодействие α,β- ненасыщенных карбонильных соединений формулы II с гидразином либо производным гидразина формулы III представляет собой экзотермическую реакцию, то при смешении указанных компонентов может потребоваться охлаждение реакционной смеси. Для приготовления смеси не имеет значения, какой из компонентов загружать в емкость первым или загружать ли исходные вещества одновременно либо раздельно. Для получения более однородной смеси после окончания загрузки перемешивание продолжают обычно еще в течение 10-60 мин, поддерживая температуру в указанных выше пределах. Согласно полученным аналитическим данным более длительное перемешивание не оказывает существенного влияния на улучшение реакции. Подвергаемые взаимодействию исходные вещества применяют, как правило, в практически стехиометрических количествах по отношению друг к другу, причем молярное соотношение карбонильного соединения II и производного гидразина III составляет обычно 1:0,65 - 1:1,25. Выбор другого соотношения компонентов не оказывает существенного влияния на процесс, однако по экономическим соображениям это делать нецелесообразно.
Гидразин, соответственно производное гидразина III, могут применяться в предлагаемом по изобретению способе как в виде гидратов или свободных оснований, так и в виде соответствующих гидразониевых солей. При использовании солей, не растворяющихся в реакционной среде, вследствие недостаточно тщательного перемешивания могут иметь место потери в выходе продукта. По этой причине предпочтительно использовать гидраты или свободные основания производных гидразина III.
Проведение процесса таким образом объясняется тем, что сначала при взаимодействии гидразинов с α,β- ненасыщенными карбонильными соединениями образуются соответствующие пиразолины и побочные продукты. Последующая переработка путем дистилляции, после удаления реакционной воды, позволяет получать желаемые продукты с весьма невысоким выходом, поскольку наряду с пиразолинами имеются также побочные продукты присоединения пиразолинов к исходным карбонильным соединениям, которые в свою очередь могут образовывать с гидразинами гидразоны и азины.
Полученную описанным выше путем реакционную смесь без последующей переработки подвергают затем взаимодействию со смесью из серной кислоты и йода либо соединения, высвобождающего йод или йодистый водород. При этом обработку проводят обычно аналогично тому, как это указано в европейской заявке EP-A 402722, а именно смесь из серной кислоты и йода, соответственно соединения, высвобождающего йод или йодистый водород, нагревают до 50-250oC, предпочтительно 70-200oC, преимущественно 100-180oC, и при этой температуре подвергают ее взаимодействию со смесью из первой стадии.
Однако, наряду с этим возможен и другой вариант, а именно полученную на первой стадии реакционную смесь можно вводить при более низких температурах, например, при 10-30oC, в смесь из серной кислоты и йода либо соединения, высвобождающего йод или йодистый водород. Однако по технологическим причинам целесообразно осуществлять это введение при более высокой температуре, так как в процессе смешения образуются соли и перемешивание обеих смесей может ухудшаться. Выбор температуры для введения одной смеси в другую согласно имеющимся данным не оказывает никакого влияния на выход продукта.
Реакция на этой второй стадии очевидно проходит аналогично тому, как проходит обменная реакция, описанная в EP-A 402722. На этой стадии, как правило, применяют серную кислоту в концентрации не менее чем 30 мас.%. Обычно применяют 40-99 мас. %-ную серную кислоту, предпочтительно 45-95 мас.%-ную серную кислоту.
Количество используемого в указанной реакции йода, или соответственно соединения, высвобождающего йод либо йодистый водород, составляет, как правило, 0,01-10 мол.%, предпочтительно 0,05-5 мол.%, прежде всего 0,1-2 мол.% по отношению к гидразину, соответственно к производному гидразина III.
Наряду с йодом и йодистым водородом в качестве соединений, высвобождающих йод либо йодистый водород, пригодны также, например, йодиды щелочных и щелочноземельных металлов, такие, как йодид лития, йодид натрия, йодид калия, йодид цезия, йодид магния и йодид кальция, а также йодиды других металлов; в принципе могут применяться все йодистые или йодистоводородные соединения, способные в соответствующих условиях реакции высвобождать йод либо йодистый водород. К ним относятся, например, такие другие неорганические йодистые соединения, как гипойодиды, йодиты, йодаты и перйодаты щелочных, щелочноземельных или других металлов, или органические йодистые соединения, например, алкилйодиды, как метилйодид.
Было найдено, что для дегидрирования, соответственно окисления йодида до йода оптимальная температура для проведения соответствующей реакции зависит от концентрации серной кислоты. Чем ниже концентрация серной кислоты, тем выше температура, необходимая для осуществления реакции. Поэтому для того, чтобы температуру реакции поддерживать в низком диапазоне, рекомендуется во время проведения реакции отгонять реакционную воду, равно как и воду, которая образуется при использовании гидратов.
Удаленная из смеси вода содержит большую часть введенного йодида в виде йода и йодистого водорода, который после восстановления, соответственно после нейтрализации, например, с помощью гидросульфита натрия, может быть возвращен.
После завершения реакции реакционной смеси дают остыть, при этом производное пиразола кристаллизуется в основном в виде соли серной кислоты.
Для выделения пиразола реакционную смесь нейтрализуют, после чего эту нейтральную смесь экстрагируют с помощью инертного органического, не смешиваемого с водой растворителя. Затем органическую фазу по обычной методике сушат и разделяют. Таким путем получают сырые пиразолы со степенью чистоты порядка 85-90%, которую однократной перегонкой можно довести до 99%.
Примеры.
1. Получение 3-метилпиразола.
1а. Взаимодействие кротонового альдегида и гидразингидрата.
К 115 г (2,3 моль) гидразингидрата добавляли 169,1 г (2,415 моль) кротонового альдегида, причем температуру охлаждением поддерживали на уровне 30oC. После окончания добавления смесь продолжали перемешивать еще в течение 30 мин при 25-30oC.
1б. Получение 3-метилпиразола.
Смесь из 720,8 г (5,06 моль) 68,8%-ной серной кислоты и 0,76 г (5,1 ммоль) йодида натрия нагревали до 155oC и при этой температуре смешивали со смесью, полученной в 1а. Во время добавления и затем еще в течение 30 мин после окончания добавления отгоняли воду. Выделенную таким путем воду после охлаждения реакционной смеси до 70oC повторно добавляли в смесь для разбавления последней. Разбавленную реакционную смесь с помощью 15%-ного едкого натра устанавливали на pH 8,5-9. В результате нейтрализации большую часть продукта получали в виде масла, которое было выделено путем декантирования. После экстрагирования водной фазы изобутанолом и последующего разделения соединенных органических фаз путем перегонки получали 172,5 г 3-метилпиразола (90,55% по отношению к количеству используемого гидразингидрата) со степенью чистоты 99% (по данным ЖХВД). Ткип: 88oC/10 мбар.
2. Получение 4-метилпиразола.
2а. Взаимодействие метакролеина и гидразингидрата.
К 115 г (2,3 моль) гидразингидрата добавляли 177,1 г (2,53 моль) метакролеина, причем температуру охлаждением поддерживали на уровне 30oC. После окончания добавления смесь продолжали перемешивать еще в течение 30 мин при 25-30oC.
2б. Получение 4-метилпиразола.
Смесь из 720,8 г (5,06 моль) 68,8%-ной серной кислоты и 1,00 г (6,7 ммоль) йодида натрия нагревали до 155oC и при этой температуре смешивали со смесью, полученной в 1а. Во время добавления и затем еще в течение 30 мин после окончания добавления отгоняли воду. Выделенную таким путем воду после охлаждения реакционной смеси до 50oC повторно добавляли в смесь для разбавления последней. Разбавленную реакционную смесь с помощью 15%-ного едкого натра устанавливали на pH 8,5. В результате нейтрализации большую часть продукта получали в виде масла, которое было выделено путем декантирования. После экстрагирования водной фазы изобутанолом и последующего разделения соединенных органических фаз путем перегонки получали 170,5 г 4-метилпиразола (89,52% по отношению к количеству используемого гидразингидрата) со степенью чистоты 99,2% (по данным ЖХВД). Ткип: 82oC/7 мбар.
3. Получение 3,4-диметилпиразола.
3а. Взаимодействие транс-2,3-диметилакролеина с гидразингидратом.
К 15,6 г (0,25 моль) 80%-ного гидразингидрата добавляли 22,1 г (0,2625 моль) транс-2,3-диметилакролеина, причем температуру охлаждением поддерживали на уровне 30oC. После окончания добавления смесь продолжали перемешивать еще в течение 30 мин при 25-30oC.
3б. Получение 3,4-диметилпиразола.
Смесь из 74,2 г (0,52 моль) 68,8%-ной серной кислоты и 0,5 г (3,3 ммоль) йодида натрия нагревали до 155oC и при этой температуре смешивали со смесью, полученной в 3а. Во время добавления и в течение последующих 30 мин после окончания добавления отгоняли воду. Выделенную таким путем воду после охлаждения реакционной смеси до 50oC повторно добавляли в смесь для разбавления последней. Разбавленную реакционную смесь с помощью 15%-ного едкого натра устанавливали на pH 8,5. При проведении этой реакции нейтрализации большую часть продукта получали в виде масла, которое можно выделить путем декантирования. После экстрагирования водной фазы изобутанолом и последующего разделения соединенных органических фаз путем перегонки получали 21,4 г 3,4-диметилпиразола (88,4% по отношению к количеству используемого гидразингидратом) со степенью чистоты 99,2%. Ткип: 96oC/10 мбар.
4. Получение 1,5-диметилпиразола,
4а. Взаимодействие кротонового альдегида с метилгидразином.
К 92 г (2 моль) метилгидразина добавляли 147 г (2,1 моль) кротонового альдегида, причем температуру охлаждением поддерживали на уровне 30oС. После окончания добавления смесь продолжали перемешивать еще в течение 30 мин при 25-30oC.
4б. Получение 1,5-диметилпиразола.
Смесь из 626,7 г (4,4 моль) 68,8%-ной серной кислоты и 0,66 г (4,4 ммоль) йодида натрия нагревали до 155oC и при этой температуре смешивали со смесью, полученной в 4а. Во время добавления и в течение последующих 30 мин после окончания добавления отгоняли воду. Выделенную таким путем воду после охлаждения реакционной смеси до 70oC повторно добавляли в смесь для разбавления последней. Разбавленную реакционную смесь с помощью 15%-ного едкого натра устанавливали на pH 8,5-9. В результате нейтрализации большую часть продукта получали в виде масла, которое можно выделить путем декантирования. После экстрагирования водной фазы изобутанолом и последующего разделения соединенных органических фаз путем перегонки получали 167,8 г 1,5-диметилпиразола (86,7% по отношению к количеству используемого метилгидразина) со степенью чистоты 99,2%. Ткип: 157oC/1013 мбар.
5. Получение 3-метилпиразола.
5а. Взаимодействие кротонового альдегида с гидразингидратом.
К 125 г (2,0 моль) 80%-ного гидразингидрата добавляли 147 г (2,1 моль) кретонового альдегида, причем температуру охлаждением поддерживали на уровне 30oC. После окончания добавления смесь продолжали перемешивать еще в течение 30 мин при 25-30oC.
5б. Получение 3-метилпиразола.
Смесь из 449,2 г (4,4 моль) 95%-ной серной кислоты и 0,66 г (4,4 ммоль) йодида натрия смешивали при 25oC со смесью, полученной в 5а, причем температуру охлаждением поддерживали на уровне 25oC. Затем температуру реакционной смеси доводили в течение 45 мин до 125oC и в течение 60 мин поддерживали на этом уровне. Во время нагрева и перемешивания воду отгоняли. Затем выделенную таким путем воду после охлаждения реакционной смеси до 70oC повторно добавляли в смесь для разбавления последней. Разбавленную реакционную смесь с помощью 10%-ного едкого натра устанавливали на pH 8,5-9. При нейтрализации большую часть продукта получали в виде масла, которое можно выделить путем декантирования. После экстрагирования водной фазы изобутанолом и последующего разделения соединенных органических фаз путем перегонки получали 143,4 г 3-метилпиразола (87% по отношению к количеству используемого гидразингидрата) со степенью чистоты 99,5%. Ткип: 88oC/1013 мбар.
6. Получение 3,5-дифенилпиразола.
6а. Взаимодействие бензилиденацетофенона и гидразингидрата.
К 6,25 г 80%-ного гидразингидрата (0,1 моль) добавляли 20,8 г (0,1 моль) бензилиденацетофенона. После окончания добавления смесь продолжали перемешивать еще в течение 4 ч при 70oC.
6б. Получение 3,5-дифенилпиразола.
Смесь 49 г 60%-ной серной кислоты (0,3 моль) и 0,2 г (1,33 ммоль) йодида натрия нагревали до 50oC и при этой температуре смешивали с полученным на предыдущей стадии реакционным продуктом. Смесь нагревали до 135oC, отгоняли 25 г воды и в течение еще 60 мин поддерживали при этой температуре. После добавления 100 г воды при 100oC и охлаждения до комнатной температуры добавляли 53,3 г (0,2 моль) 15%-ного едкого натра для установления pH реакционной смеси, равным 8. Продукт, полученный после фильтрования и сушки, подвергали перекристаллизации из этанола. В результате было получено 18,7 г бесцветных кристаллов указанного в заглавии продукта с Тпл 200-201oC, выход 85% от теоретического.
6в. Процесс проводили так, как описано выше, но вместо йодида натрия использовали 0,28 г (1,33 ммоль) йодата натрия. В результате было получено 18 г 3,5-дифенилпиразола, выход 81,8% от теоретического.
7. Получение 4-этилпиразола.
7а. Взаимодействие 2-этилакролеина и гидразингидрата.
К 12,5 г (0,2 моль) 80%-ного гидразингидрата добавляли 17,2 г (0,205 моль) 2-этилакролеина, причем температуру поддерживали на уровне 25oC путем охлаждения. Смесь продолжали перемешивать в течение 1 ч при 25oC и затем добавляли 0,2 г (1,33 ммоль) йодида натрия.
7б. Получение 4-этилпиразола.
Полученный на предыдущей стадии реакционный продукт вводили по каплям в течение 60 мин при 125oC в смесь 90,5 г (0,6 моль) 65%-ной серной кислоты и 0,1 г (0,67 ммоль) йодида натрия. Температуру реакционной смеси понижали до 110oC. Затем отгоняли 25 г воды, нагревали реакционную смесь до 125oC и выдерживали при этой температуре еще в течение 1 ч. После охлаждения к реакционной смеси добавляли 147,2 г (0,92 моль) 25%-ного едкого натра для нейтрализации и затем экстрагировали изобутанолом. Объединенные органические фазы подвергали вакуумной перегонке. В результате было получено 16 г 4-этилпиразола со степенью чистоты 99,6%. Выход продукта 83% от теоретического. Ткип: 90oC.
Сравнение способа по изобретению со способом, известным из ЕР-А-402722.
А. Получение 3-метилпиразола по способу согласно изобретению.
А1. Взаимодействие кротонового альдегида и гидразингидрата.
К 62,5 г (1,0 моль) 80%-ного гидразингидрата добавляли 73,5 г (1,05 моль) кротонового альдегида, причем температуру охлаждением поддерживали на уровне 30oC. После окончания добавления смесь продолжали перемешивать еще в течение 30 мин при 25-30oC.
А2. Получение 3-метилпиразола.
Смесь из 313,6 г (2,2 моль) 68,8%-ной серной кислоты и 0,33 г (2,2 ммоль) йодида натрия нагревали до 155oC и при этой температуре смешивали со смесью, полученной в 1а. Во время добавления и в течение 30 мин после окончания добавления воду отгоняли. Выделенную таким путем воду после охлаждения реакционной смеси повторно добавляли в эту смесь. Разбавленную реакционную смесь с помощью 15%-ного едкого натра устанавливали на pH 8,5. В результате нейтрализации большую часть продукта получали в виде масла, которое выделяли путем декантирования. После экстрагирования водной фазы изобутанолом и последующего разделения соединенных органических фаз путем перегонки получали 73,4 г 3-метилпиразола (89% по отношению к количеству используемого гидразингидрата со степенью чистоты 99,5% (по данным ЖХВД)). Ткип: 88oC/10 мбар.
Б. Получение 3-метилпиразола по способу, известному из ЕР-А-402722
Б1. Смесь из 313,6 г (2,2 моль) 68,8%-ной серной кислоты и 0,33 г (2,2 ммоль) йодида натрия нагревали до 155oC и при этой температуре одновременно смешивали с 62,5 г (1 моль) 80%-ного гидразингидрата и 73,6 г (1,05 моль) кротонового альдегида. Во время добавления и в течение 30 мин после окончания добавления воду отгоняли. Выделенную таким путем воду после охлаждения реакционной смеси до 50oC повторно добавляли в смесь для разбавления последней.
Б2. Разбавленную реакционную смесь с помощью 15%-ного едкого натра нейтрализовали и устанавливали pH 8,5. При проведении этой реакции нейтрализации большую часть продукта получали в виде масла, которое выделяли путем декантирования. После экстрагирования водной фазы изобутанолом и последующего разделения соединенных органических фаз путем перегонки получали 62,4 г 3-метилпиразола (75,7% по отношению к количеству используемого гидразингидрата) со степенью чистоты 99,5%. Ткип: 88oC/10 мбар.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПИРАЗОЛА | 1998 |
|
RU2192418C2 |
| СПОСОБЫ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ ДЛЯ ПОЛУЧЕНИЯ МЕТИЛАМИДОВ α-МЕТОКСИИМИНОКАРБОНОВЫХ КИСЛОТ | 1995 |
|
RU2146247C1 |
| КАРБАМАТЫ, ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ, ФУНГИЦИДНАЯ КОМПОЗИЦИЯ, СПОСОБ БОРЬБЫ С ГРИБАМИ | 1993 |
|
RU2129118C1 |
| СПОСОБ ПОЛУЧЕНИЯ АСИММЕТРИЧНО ЗАМЕЩЕННЫХ ТРИАЗИНОВ | 1994 |
|
RU2125995C1 |
| ФЕРМЕНТАТИВНО ОТЩЕПЛЯЕМЫЕ ЛИНКЕРЫ ДЛЯ ТВЕРДОФАЗНЫХ СИНТЕЗОВ | 1997 |
|
RU2198155C2 |
| ИМИНООКСИМЕТИЛЕНАНИЛИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), СРЕДСТВА ДЛЯ БОРЬБЫ С ВРЕДИТЕЛЯМИ, СПОСОБ БОРЬБЫ С ВРЕДИТЕЛЯМИ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1995 |
|
RU2143423C1 |
| ПРОИЗВОДНЫЕ ПЕПТИДА ИЛИ ИХ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2116312C1 |
| ПРОИЗВОДНЫЕ ФЕНИЛУКСУСНОЙ КИСЛОТЫ И СРЕДСТВО БОРЬБЫ ПРОТИВ НАСЕКОМЫХ И ПАУКООБРАЗНЫХ И ПРОТИВ ВРЕДОНОСНЫХ ГРИБОВ | 1995 |
|
RU2162075C2 |
| ПРОИЗВОДНЫЕ ФЕНИЛУКСУСНОЙ КИСЛОТЫ, ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ ДЛЯ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ СРЕДСТВА | 1995 |
|
RU2165411C2 |
| ХЛОРМЕТИЛДИАРИЛОКСИРАНЫ | 1991 |
|
RU2125997C1 |
В заявке описывается способ получения пиразола и его производных формулы I, в которой радикалы R1, R2, R3 и R4 обозначают водород или алкильную группу, линейную или разветвленную предпочтительно C1 - C10-алкильную группу и наиболее предпочтительно C1 - C8-алкильную группу и наиболее предпочтительно C1 - C6-алкильную группу, причем эти группы могут быть разорваны гетероатомами, такими как азот, кислород и сера, и могут иметь заместители, выбранные из группы, включающей нитро, карбоксил, сульфонил, галоген, циклоалкил, бициклоалкил, арил и гетероарил; циклоалкильную группу или бициклоалкильную группу, предпочтительно C3 - C8-циклоалкильную группу или C6 - C10-бициклоалкильную группу, причем эти группы могут быть разорваны гетероатомами, такими как азот, кислород и сера, и могут иметь заместители, выбранные из группы, включающей нитро, карбоксил, сульфонил, галоген, алкил, циклоалкил, бициклоалкил, арил и гетероарил; арильную или гетероарильную группы, такие как фенил, нафтил и пиридил, причем эти группы могут иметь заместители, выбранные из группы, включающей нитро, карбоксил, сульфонил, галоген, алкил, циклоалкил, бициклоалкил, арил и гетероарил; из α,β-ненасыщенного карбонильного соединения формулы II, где R1, R2 и R3 имеют указанные значения, и гидразина или его производных формулы III, где R4 имеет указанные значения, с использованием серной кислоты и йода или соединения, высвобождающего йод или йодистый водород, отличающийся тем, что сначала ведут взаимодействие α,β-ненасыщенного карбонильного соединения формулы II с гидразином или его производным формулы III без добавления растворителя и в отсутствии серной кислоты и полученную при этом реакционную смесь на последующей стадии подвергают взаимодействию со смесью из серной кислоты и йода, или соединения, высвобождающего йод или йодистый водород. Технический результат - получение простым путем пирадола и его производных с более высоким выходом и степенью чистоты. 5 з.п. ф-лы.
H2N - NHR4 (III)
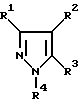
в которой радикалы R1 - R4 обозначают водород или алкильную группу, линейную или разветвленную, предпочтительно С1 - С10-алкильную группу, более предпочтительно С1 - С8-алкильную группу и наиболее предпочтительно С1 - С6-алкильную группу, причем эти группы могут быть разорваны гетероатомами, такими как азот, кислород и сера, и могут иметь заместители, выбранные из группы, включающей нитро, карбоксил, сульфонил, галоген, циклоалкил, бициклоалкил, арил и гетероарил; циклоалкильную группу или бициклоалкильную группу, предпочтительно С3 - С8-циклоалкильную группу или С6 - С10-бициклоалкильную группу, причем эти группы могут быть разорваны гетероатомами, такими как азот, кислород и сера, и могут иметь заместители, выбранные из группы, включающей нитро, карбоксил, сульфонил, галоген, алкил, циклоалкил, бициклоалкил, арил и гетероарил; арильную или гетероарильную группу, такую как фенил, нафтил и пиридил, причем эти группы могут иметь заместители, выбранные из группы, включающей нитро, карбоксил, сульфонил, галоген, алкил, циклоалкил, бициклоалкил, арил и гетероарил;
из α,β-ненасыщенного карбонильного соединения формулы II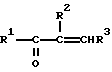
где R1 - R3 имеют указанные значения,
и гидразина или его производных формулы III
H2N-NHR4,
где R4 имеет указанные значения,
с использованием серной кислоты и йода или соединения, высвобождающего йод или йодистый водород, отличающийся тем, что сначала ведут взаимодействие α,β-ненасыщенного карбонильного соединения формулы II с гидразином или его производным формулы III без добавления растворителя и в отсутствие серной кислоты и полученную при этом реакционную смесь на последующей стадии подвергают взаимодействию со смесью из серной кислоты и йода или соединения, высвобождающего йод или йодистый водород.
| Способ получения 3-метилпиразола | 1979 |
|
SU833962A1 |
| Способ определения состава морской воды | 1988 |
|
SU1608511A1 |
| 0 |
|
SU402722A1 | |
| DE 3423930 A1, 1982 | |||
| Питательный насос | 1929 |
|
SU20964A1 |
| ТЕПЛОМАССООБМЕННИК | 0 |
|
SU366328A1 |
