Изобретение относится к лекарственным средствам, содержащим в качестве активного начала, по меньшей мере, одно соединение общей формулы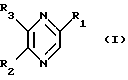
в виде стереоизомерных форм и их солей с минеральными или органическими кислотами, и к новым соединениям формулы I, их солям с минеральными или органическими кислотами и способу их получения.
В общей Формуле I:
R1 обозначает стереоизомерные формы цепи
-(СНОН)3-СН2ОН (II)
и либо R2 обозначает атом водорода, а R3 обозначает стереоизомерные формы цепи
-СН2-(СНОН)2-СН2ОН (III),
либо R2 обозначает стереоизомерные формы цепи
(СНОН)3-СН2ОН (II)
или
-СН2-(СНОН)2-CH2OH (III),
a R3 обозначает атом водорода,
за исключением
- фруктозазина формулы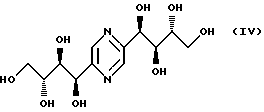
- деоксифруктозазина формулы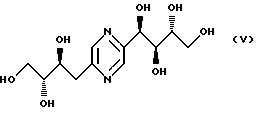
- и соединения формулы
Лекарственные средства по изобретению содержат, таким образом, по меньшей мере, один стереоизомер следующих соединений:
или соль одного из этих соединений с органической или минеральной кислотой за исключением фруктозазина, деоксифруктозазина и соединения формулы VI.
Предпочтительными лекарственными средствами согласно изобретению являются те, которые содержат в качестве активного начала, по меньшей мере, одно соединение формулы I, выбранное из следующих соединений:
1-[5-(1R, 2R, 3R, 4-тетрагидроксибутил)пиразин-2-ил] бутан-1R,2R,3R, 4-тетраол,
1-[5-(1R, 2R, 3S,4-тетрагидроксибутил)пиразин-2-ил]бутан-1R,2R,3S,4-тетраол,
1-[5-(1R, 2S, 3S,4-тетрагидроксибутил)пиразин-2-ил]бутан-1R,2S,3S,4-тетраол,
1-[5-(1S, 2R, 3R,4-тетрагидроксибутил)пиразин-2-ил]бутан-1S,2R,3R,4-тетраол,
1-[5-(1S, 2R, 3S,4-тетрагидроксибутил)пиразин-2-ил]бутан-1S,2R,3S,4-тетраол,
1-[5-(1S, 2S, 3R,4-тетрагидроксибутил)пиразин-2-ил]бутан-1S,2S,3R,4-тетраол,
1-[5-(1S, 2S, 3S,4-тетрагидроксибутил)пиразин-2-ил]бутан-1S,2S,3S,4-тетраол,
1-[5-(2R,3R,4-тригидроксибутил)пиразин-2-ил]бутан-1R,2R,3R,4-тетраол,
1-[5-(2R,3S,4-тригидроксибутил)пиразин-2-ил]бутан-1R,2R,3S,4-тетраол,
1-[5-(2S,3S,4-тригидроксибутил)пиразин-2-ил]бутан-1R,2S,3S,4-тетраол,
1-[5-(2R,3R,4-тригидроксибутил)пиразин-2-ил]бутан-1S,2R,3R,4-тетраол,
1-[5-(2R,3S,4-тригидроксибутил)пиразин-2-ил]бутан-1S,2R,3S,4-тетраол,
1-[5-(2S,3R,4-тригидроксибутил)пиразин-2-ил]бутан-1S,2S,3R,4-тетраол,
1-[5-(2S,3S,4-тригидроксибутил)пиразин-2-ил]бутан-1S,2S,3S,4-тетраол,
1-[6-(2R,3R,4-тригидроксибутил)пиразин-2-ил]бутан-1R,2R,3R,4-тетраол,
1-[6-(2R,3S,4-тригидроксибутил)пиразин-2-ил]бутан-1R,2R,3S,4-тетраол,
1-[6-(2R,3R,4-тригидроксибутил)пиразин-2-ил]бутан-1S,2R,3R,4-тетраол,
1-[6-(2R,3S,4-тригидроксибутил)пиразин-2-ил]бутан-1S,2R,3S,4-тетраол,
1-[6-(2S,3R,4-тригидроксибутил)пиразин-2-ил]бутан-1S,2S,3R,4-тетраол,
1-[6-(2S,3S,4-тригидроксибутил)пиразин-2-ил]бутан-1S,2S,3S,4-тетраол,
или соли этих соединений с органическими или минеральными кислотами,
и еще более предпочтительными лекарственными средствами являются те, которые содержат в качестве активного начала, по меньшей мере, одно соединение формулы I, выбранное из следующих соединений:
1-[5-(1R, 2R, 3R,4-тетрагидроксибутил)пиразин-2-ил]бутан-1R,2R,3R,4-тетраол,
1-[5-(2R,3R,4-тригидроксибутил)пиразин-2-ил]бутан-1R,2R,3R,4-тетраол,
1-[5-(2S,3R,4-тригидроксибутил)пиразин-2-ил]бутан-1S,2S,3R,4-тетраол,
1-[6-(2R,3S,4-тригидроксибутил)пиразин-2-ил]бутан-1S,2R,3S,4-тетраол,
1-[6-(2R,3R,4-тригидроксибутил)пиразин-2-ил]бутан-1R,2R,3R,4-тетраол,
или соли этих соединений с органическими или минеральными кислотами.
Известными являются следующие соединения:
- фруктозазин, деоксифруктозазин и соединение формулы VI, описанные в патенте JP 43-13469; Ann., 644, 122-127 (1961); Agr. Biol. Chem., 39 (5), 1143-1148 (1975);
- стереоизомеры приведенных ниже общих формул VIa, VIb, VIc и VId: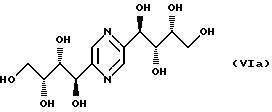
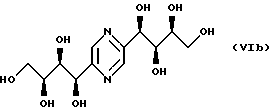


описанные в патенте JP 43-13469; Carbohyd. Res., 26 (2), 377-384 (1973); J. Anal. Appl. Pyrolysis, 13, 191-198 (1988);
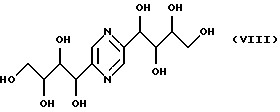
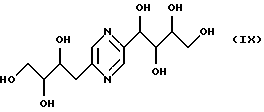
соединения общих формул VII, VIII и IX, являющиеся производными глюкозы, фруктозы, маннозы, галактозы, описанные в японском патенте JP 53-90401.
Однако о применении этих соединений в качестве лекарственных средств не сообщалось, и такое их применение является предметом настоящего изобретения.
Соединения формулы I или их соли с минеральными или органическими кислотами, за исключением следующих соединений:
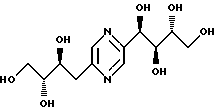




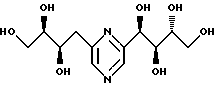
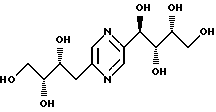
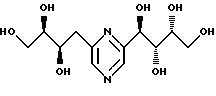
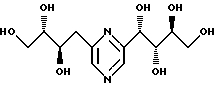
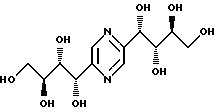

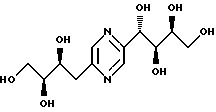
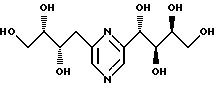
являются новыми соединениями и, как таковые, являются частью изобретения.
Предпочтительными соединениями формулы I являются следующие:
1-[5-(1R,2R,3S,4-тетрагидроксибутил) пиразин-2-ил]бутан-1R,2R,3S,4-тетраол, 1-[5-(1S,2R,3R,4-тетрагидроксибутил)пиразин-2-ил]бутан-1S,2R,3R,4-тетраол, 1-[5-(1S,2S,3R,4-тетрагидроксибутил)пиразин-2-ил]бутан-1S,2S,3R,4-тетраол, 1-[5-(1S,2S,3S,4-тетрагидроксибутил)пиразин-2-ил]бутан-1S,2S,3S,4-тетраол, 1-[5-(2R, 3S,4-тригидроксибутил)пиразин-2-ил]бутан-1R,2R,3S,4-тетраол, 1-[5-(2S, 3S, 4-тригидроксибутил)пиразин-2-ил] бутан-1R,2S,3S,4-тетраол, 1-[5-(2R, 3R, 4-тригидроксибутил)пиразин-2-ил]бутан-1S,2R,3R,4-тетраол, 1-[5-(2S, 3R, 4-тригидроксибутил)пиразин-2-ил]бутан-1S,2S,3R,4-тетраол, 1-[6-(2R, 3S, 4-тригидроксибутил)пиразин-2-ил} бутан-1R, 2R,3S,4-тетраол, 1-[6-(2R,3R, 4-тригидроксибутил)пиразин-2-ил] бутан-1S, 2R, 3R,4-тетраол, 1-[6-(2S,3R,4-тригидроксибутил)пиразин-2-ил] бутан-1S, 2S, 3R,4-тетраол и соли этих соединений с минеральными или органическими кислотами.
Из приведенных выше соединений предпочтительны:
1-[5-(2S, 3S, 4-тригидроксибутил)пиразин-2-ил] бутан-1R,2S,3S,4-тетраол, 1-[5-(2R, 3R, 4-тригидроксибутил)пиразин-2-ил] бутан-1S,2R,3R,4-тетраол, 1-[5-(2S, 3R, 4-тригидроксибутил)пиразин-2-ил] бутан-1S,2S,3R,4-тетраол и соли этих соединений с минеральными или органическими кислотами, наиболее предпочтительным соединением является 1-[5-(2S,3R,4-тригидроксибутил)пиразин-2-ил] бутан-lS,2S,3R,4-тетраол или соль этого соединения с минеральной или органической кислотой.
Стереоизомерные формы соединений общей формулы I получают из стереоизомерных форм приведенных ниже соединений, используемых в способе получения согласно изобретению.
Стереоизомеры соединений формулы I, в которых R1 обозначает стереоизомерные формы цепи -(СНОН)3-СН2ОН (II), R2 обозначает атом водорода и R3 обозначает стереоизомерные формы цепи -СН2-(СНОН)2-СН2ОН (III), т.е. соединения, обозначаемые формулой VII, могут быть получены действием формиата аммония на альдозу или на смесь двух альдоз из правовращающего или левовращающего ряда общей формулы
СНО-СНОН-R1 (X)
в которой R1 имеет то же значение, что и в формуле I.
Эта реакция может быть преимущественно проведена при температуре от 15 до 100oС и предпочтительно в водной среде.
Альдозы могут быть приобретены в готовом виде или получены из:
а) коммерчески доступных альдоз:
- реакциями эпимеризации с применением или адаптацией методов, описанных в Adv. Carbonydr. Chem., 13, 63 (1958), в частности, в щелочной среде с использованием разбавленного водного раствора натриевой щелочи (0,03-0,05%) при температуре от 20 до 40oС;
- реакциями удлинения цепи путем применения или адаптации методов, описанных в "The Carbohydrates", издатели W. Pigman и D. Horton, Academic Press, New-York, Volume IA, 133 (1972), и в частности, путем образования циангидрина исходной альдозы (например, действием цианида натрия в водном растворе при температуре от 10 до 30oС в присутствии натриевой щелочи при рН, близком к 9) с последующим гидролизом образовавшейся при этом нитрильной функции в соответствующую кислотную функцию с применением или адаптированном методов, описанных в Organic Synthesis, том I, стр.436 и том III, стр.85 (например, с помощью хлористоводородной или концентрированной серной кислоты в водном растворе при температуре от 20oС до температуры кипения реакционной среды с обратным холодильником) и восстановлением карбоксильной функции в соответствующую альдегидную с применением или адаптированием методов, описанных в J. Amer. Chem. Soc. 71, 122 (1949), в частности, с помощью борогидрида щелочного металла (например, борогидрида натрия) в водном растворе при температуре от 20oС до температуры кипения реакционной среды;
- реакциями укорочения цепи с применением или адаптацией методов, описанных в "The Carbohydrates", издатели W. Pigman и D. Horton, Academic Press, New-York, Volume IB, 1980, стр.929, или в Сhеm. Ber., 83, 559 (1950), и в частности путем превращения альдегидной функции альдозы в соответствующий гидроксиламин с применением или адаптированием методов, описанных в Organic Synthesis, том II, стр.314 (например, с помощью гидроксиламингидрохлорида в водном растворе в присутствии основания, такого как карбонат натрия, при температуре от 20 до 50oС) с последующим действием 3,4-динитрофторбензола в присутствии диоксида углерода, основания, такого как гидрокарбонат натрия, в водном растворе и алифатического спирта (например, изопропилового спирта) при температуре от 50 до 80oС;
b) соответствующих аллиловых спиртов путем применения или адаптации методов, описанных в Science 220, 949 (1983), и в частности с помощью трет-бутилгидропероксида в присутствии комплекса титана (IV), такого как изопропилат титана (IV), и оптически чистого диалкилтартрата (например, диэтилтартрата) с последующим последовательным действием тиофенолятом натрия, п-хлорпербензойной кислотой в уксусном ангидриде и гидридом диизопропилалюминия.
Стереоизомеры сахара формулы Х могут быть стереоизомерами альдоз с 6 атомами углерода, из которых предпочтительно используются D-гулоза, D-галактоза, D-аллоза, D-альтроза, D-идоза, D-талоэа, L-глюксза, L-манноза, L-галактоза, L-аллоза, L-альтроза, L-идоза, L-талоза, L-гулоза.
Стереоизомеры соединений формулы I, в которых R1 обозначает стереоизомерные формы цепи -(СНОН)3-СН2ОН (II), R2 обозначает стереоизомерные формы цепи -(СНОН)3-СН2OН (II) и R3 является атомом водорода, т.е. соединения, обозначаемые формулой VIII, могут быть получены обработкой в щелочной среде аминоальдозы или смеси двух аминоальдоз общей формулы
CHO-CH(NH2)-R1 (XI)
возможно, в форме солевого аддукта, такого как гидрохлорид, где R1 имеет то же значение, что и в общей формуле I.
Процесс ведут преимущественно при температуре, близкой к 20oС, используя предпочтительно аммиачный, предпочтительно 28%-ный раствор.
Аминоальдозы формулы XI могут быть приобретены в готовом виде или получены с применением или адаптацией методов, описанных, например, в:
a) Methods Carbohyd. Chem., 7, 29 (1976), состоящих в превращении альдегидной функции соответствующей альдозы в нитроэтиленовую группу с помощью нитрометана в щелочной среде (например, этилат натрия) с последующей обработкой полученного продукта последовательно насыщенным аммиачным раствором при температуре от 20 до 30oС, водным раствором Ba(OH)2 при 20-30oС и, наконец, разбавленной (10-15%) серной кислотой при 20-30oС,
b) "The Amino Sugar", издатель R.W. Jeanioz, Academic Press, New-York, 1969, стр. 1, или "The Carbohydrates", издатели W. Pigman и D. Horton, Academic Press, New-York, Volume IB, 1980, стр.664, состоящих в превращении альдегидной функции соответствующей альдозы в иминогруппу, исходя из первичного ароматического амина (например, анилина), с последующей реакцией с синильной кислотой при температуре от 0 до 20oС и затем с водородом в присутствии палладия в растворителе, таком как простой эфир (например, тетрагидрофуран) или алифатический спирт (например, этанол или метанол), при температуре от 20 до 50oС.
Стереоизомеры аминоальдозы формулы XI могут быть аминоальдозами с 6 атомами углерода, причем преимущественно используется D-галактозамин, который может быть также использован в виде солевого аддукта, например гидрохлорида.
Стереоизомеры соединений формулы I, в которых R1 обозначает стереоизомерные формы цепи -(СНОН)3-СН2ОН (II), R2 обозначает стереоизомерные формы цепи -СН2-(СНОН)2-СН2OН (III) и R3 является атомом водорода, т.е. соединения, обозначаемые формулой IX, могут быть получены
либо из аминоальдозы или смеси двух аминоальдоз общей формулы
CHO-CH(NH2)-R1 (XI)
в которой R1 имеет то же значение, что и в общей формуле I, в кислой среде, преимущественно в среде уксусной кислоты, и предпочтительно при температуре от 15 до 100oС,
либо из кетозы или смеси двух кетоз общей формулы
HOCH2-CO-R1 (XII)
в которой R1 имеет то же значение, что и в общей формуле I, действуя формиатом аммония и проводя реакцию преимущественно при температуре от 15 до 100oС и предпочтительно в водной среде.
Кетозы формулы XII могут быть приобретены в готовом виде или получены с применением или адаптацией методов, описанных в:
a) Adv. Carbohyd. Chem., 13, 63 (1958), состоящих в реакции соответствующей альдозы либо с основанием, таким как гидроксид кальция, гидроксид натрия, пиридин или хинолин, либо с кислотой, такой как серная кислота, в водном растворе или чистой фазе при температуре от 20 до 50oС;
b) Tetrahedron Asymmetry, 7 (8), 2185 (1966), J. Amer. Chem. Sоc., 118 (33), 7653 (1966), J. Org. Chem., 60 (13), 4294 (1995), Tetrahedron Lett., 33 (36), 5157 (1992), J. Amer. Chem. Soc., 113 (17), 6678 (1991), Angew. Chem., 100 (5), 737 (1988), J. Org. Chem., 57, 5899 (1992), состоящих, например, в конденсации либо гидроксипирувальдегида, 1,3-дигидроксиацетона, 1,3-дигидроксиацетона-монофосфата или гидрокспировиноградной кислоты с замещенным в положении 2 2-гидроксиацетальдегидом (включая оптически чистый), возможно, в присутствии фермента, например транскетолазы. Эту реакцию проводят преимущественно в водном растворе при температуре от 20 до 50oС, возможно, в присутствии основания (например, натриевой щелочи), хлорида бария, хлорида магния или хлорида цинка. Производные, содержащие 2-гидроксиацетальдегидную группу, могут быть приобретены в готовом виде или получены из альдоз с применением или адаптацией методов, описанных в Р. Collins, R. Ferrier, Monosaccharides. Their Chemistry and their roles in Natural Products, издатель J. Wiley (1995), и М. Bols, Carbohydrate Building Blocks, издатель J. Wiley (1996).
Преимущественно используемым стереоизомером аминоальдозы формулы XI является D-галактозамин.
Стереомизомеры соединений формулы XII могут быть стереоизомерами кетоз с 6 атомами углерода, из которых предпочтительно используемыми являются D-псикоза, D-сорбоза, D-тагатоза, L-псикоза, L-фруктоза, L-сорбоза и L-тагатоза.
Реакционные смеси, получаемые этими различными описанными выше способами, обрабатывают с использованием традиционных физических (например, упаривание, экстракция, перегонка, хроматография, кристаллизация) или химических (например, получение солей) методов.
Соединения формулы I могут быть превращены в солевые аддукты с минеральными или органическими кислотами действием этими кислотами в органических растворителях, таких как спирты, кетоны, простые эфиры и хлорсодержащие растворители. Эти соли также составляют часть изобретения.
В качестве примеров фармацевтически приемлемых солей можно назвать следующие солевые аддукты с минеральными и органическими кислотами: ацетат, пропионат, сукцинат, бензоат, фумарат, малеат, оксалат, метансульфонат, изотионат, теофиллинацетат, салицилат, метилен-бис-b-оксинафтоат, гидрохлорид, сульфат, нитрат и фосфат.
Следующие примеры более детально иллюстрируют применяемый в соответствии с изобретением способ получения продуктов.
ПРИМЕР 1.
Раствор 1,0 г D-сорбозы и 3,5 г формиата аммония в 4 см3 воды кипятят в течение 0,5 часа с обратным холодильником и затем оставляют охлаждаться до комнатной температуры. Смесь далее концентрируют при пониженном давлении (2,7 кПа) при температуре, близкой к 40oС. Бурый остаток забирают последовательно этиловым эфиром и толуолом и досуха упаривают. Новый остаток забирают этанолом и фильтруют. Фильтрат упаривают, получая красновато-коричневое масло. Операцию повторяют несколько раз до тех пор, пока не прекратится появление осадка. Полученный в результате этого остаток очищают хроматографией на колонке с силикагелем (0,063-0,200 мм), элюируя смесью этанол/н-бутанол/28%-ный водный аммиак/вода в объемном отношении 8/2/2/1. Фракции, содержащие целевой продукт, объединяют и концентрируют при пониженном давлении (2,7 кПа) при температуре, близкой к 40oС. Полученный желтый клейкий твердый продукт забирают смесью этанол/метанол в количестве, достаточном для получения раствора, к которому затем добавляют этиловый эфир до начала появления осадка, который отфильтровывают. После кристаллизации продукта получают 0,15 г 1-[5-(2R,3R,4-тригидроксибутил)пиразин-2-ил]бутан-1S, 2R,3R,4-тетраола в виде бежевого твердого вещества с т.пл. 90oС.
Спектр ЯМР 1H (400 МГц, (СD3)2SО-d6, δ в м.д.): 2,85 и 2,93 (2дд соответственно, J = 13 и 9 Гц и J = 13 и 4 Гц, 2Н: СН2 5α); от 3,25 до 3,55 (мт, 6Н: СН 2γ - СН2 2δ - СН 5γ и СН2 5δ); 3,76 (мт, 1Н: СН 2β) ; 3,91 (мт, 1Н: СН 5β); от 4,35 до 4,65 (мф, 6Н: ОН в 2β - ОН в 2γ - ОН в 2δ - ОН в 5β - ОН в 5γ и ОН в 5δ); 4,78 (т, J = 4,5 Гц, 1Н: СН 2α) ; 5,39 (д, J = 4,5 Гц, 1Н: ОН в 2α); 8,43 (с, 1Н: =СН в 6); 8, 61 (с, 1Н: = СН в 3).
α
ПРИМЕР 2.
Суспензию, содержащую 1,0 г D-галактозамингидрохлорида и 0,73 см3 диэтиламина, перемешивают 1 час и фильтруют. Фильтрат упаривают, остаток растворяют в 10 см3 28%-ного аммиака и перемешивают при комнатной температуре в течение 3 недель. Смесь после этого концентрируют при пониженном давлении (2,7 кПа) при температуре, близкой к 40oС, получая желтое масло, которое забирают метанолом и фильтруют. Фильтрат упаривают с образованием оранжевого масла, которое очищают хроматографией на колонке с силикагелем (0,04-0,063 мм), элюируя смесью этанол/н-бутанол/28%-ный аммиак/вода в объемном отношении 8/2/2/1. Фракции, содержащие целевой продукт, объединяют и концентрируют при пониженном давлении (2,7 кПа) при температуре, близкой к 40oС, получая желто-оранжевый клейкий твердый продукт. Последний кристаллизуют из метанола, отфильтровывая твердый продукт с получением 0,12 г 1-[5-(1R,2R,3R,4-тетрагидроксибутил)пиразин-2-ил] бутан-1R,2R,3R,4-тетраола в виде бежевого порошка с т.пл. 109oС.
Спектр ЯМР 1H (400 МГц, (CD3)2SO-d6, δ в м.д.): от 3,35 до 3,50 (м, 4Н: СН2О 2δ и СН2О 5δ); от 3,70 до 3,85 (м, 4Н: СН 2β - СН 2γ - СН 5β и СН 5γ) ; 4,24 (д, J = 8 Гц, 2Н: ОН в 2β и ОН в 5β); 4,41 (д, J = 6,5 Гц, 2Н: ОН в 2γ и ОН в 5γ) ; 4,50 (т уширенный, J = 6 Гц, 2Н: ОН в 2δ и ОН в 5δ); 4,64 (2дд, J = 7 и 6 Гц, 2Н: СН 2α и СН 5α); 5,48 (д, J = 6 Гц, 2Н: ОН в 2α и ОН в 5α) ; 8,56 (с, 2Н: = СН в 3 и =СН в 6).
ПРИМЕР 3.
Раствор 1,0 г D-галактозы и 3,5 г формиата аммония в 4 см3 воды кипятят в течение 0,5 часа с обратным холодильником и затем оставляют охлаждаться до комнатной температуры. Смесь фильтруют и остаток концентрируют при пониженном давлении (2,7 кПа) при температуре, близкой к 40oС, получая красно-коричневый остаток, который последовательно забирают этанолом и этиловым эфиром и затем досуха упаривают. Остаток растирают в этиловом эфире, отфильтровывают и растворяют образовавшееся бурое твердое веществo в этаноле. К раствору добавляют гидроксид натрия до рН 12 и перемешивают в течение 40 час, наблюдая образование осадка. Реакционную смесь фильтруют и концентрируют фильтрат при пониженном давлении (2,7 кПа) при температуре, близкой к 40oС, получая желтое твердое вещество, которое забирают смесью метанол/этиловый эфир и фильтруют. Фильтрат упаривают, остаток растворяют в метаноле и добавляют спиртовой раствор НСl до рН 2. Образовавшийся при этом осадок отфильтровывают и фильтрат концентрируют. Остаток очищают хроматографией на колонке с силикагелем (0,040-0,063 мм), элюируя смесью этанол/н-бутанол/водный аммиак/вода в объемном отношении 8/2/1/1. Фракции, содержащие целевой продукт, объединяют и концентрируют при пониженном давлении (2,7 кПа) при температуре, близкой к 40oС. Полученный белый твердый продукт перекристаллизуют из метанола, получая 90 мг 1-[5-(2R, 3R,4-тригидроксибутил)пиразин-2-ил]бутан-1R,2R,3R,4-тетраол в виде
белых твердых кристаллов с т.пл. 146oС.
Спектр ЯМР 1Н (400 МГц, (СD3)2SО-d6, δ м.д.): 2,86 (2дд, J = 14 и 9 Гц, 1Н: 1Н из СН2 5α; 2,9,2 (дд, J = 14 и 3,5 Гц, 1Н: другой Н из СН2 5α ; от 3,30 до 3,60 (м, 5Н: СН2О 2δ - СН2O 5δ и СН 5γ); от 3,70 до 3,85 (м, 2Н: СН 2γ и СН 2β); 3,90 (м, 1Н: СН 5β); 4,22 (д, J = 7 Гц, 1Н: ОН в 2β); 4,38 (д, J = 6,5 Гц, 1Н: ОН в 2γ); 4,43 (д, J = 7 Гц, 1Н: ОН в 5β); от 4,40 до 4,55 (м, 2Н: ОН в 2δ и ОН в 5δ); от 4,55 до 4,70 (м, 2Н: СН 2α и ОН в 5γ); 5,44 (д, J = 6 Гц, 1Н: ОН в 2α); 8,43 (с, 1Н: = СН в 6); 8,54 (с, 1Н: = СН в 3).
α
ПРИМЕР 4.
Раствор 10,0 г L-сорбозы и 7,0 г формиата аммония в 28 см3 воды кипятят в течение 2,5 часа с обратным холодильником и оставляют охлаждаться до комнатной температуры. Смесь далее концентрируют при пониженном давлении (2,7 кПа) при температуре, близкой к 50oС. Красно-коричневый пастообразный остаток очищают хроматографией на колонке с силикагелем (0,020-0,045 мм), элюируя смесью этанол/н-бутанол/водный аммиак/вода в объемном отношении 8/2/2/1. Фракции, содержащие целевой продукт, объединяют и концентрируют при пониженном давлении (2,7 кПа) при температуре, близкой к 50oС. Полученное красно-коричневое масло (9,1 г) забирают смесью 100 см3 этанола и 10 см3 воды. Смесь доводят до кипения, обрабатывают 0,9 г животного угля и фильтруют через бумажный фильтр. Фильтрат концентрируют при пониженном давлении (2,7 кПа) при температуре, близкой к 50oС, получая красно-коричневое масло (6,2 г). Последнее забирают смесью 50 см3 этанола и 1,5 см3 воды и перекристаллизуют. Полученные кристаллы отфильтровывают, удаляют влагу и промывают той же смесью. После сушки до постоянного веса получают 0,86 г 1-[5-(2S,3S,4-тригидроксибутил)пиразин-2-ил]бутан-1R,2S,3S,4-тетраола в виде бежевого кристаллического вещества с т.пл. 116oС.
Спектр ЯМР 1Н (400 МГц, (CD3)2SO-d6, δ в м.д.): 2,87 (АВ limit, 2Н: СН2 5α); от 3,30 до 3,60 (м, 6Н: СН 2γ - СН2О 2δ - СН 5γ и СН2О 5δ); 3,76 (м, 1Н: СН 2β); 3,90 (м, 1Н: СН 5β); 4,77 (д, J = 5,5 Гц, 1Н: СН 2α) ; 8,43 (с уширенный, 1Н: = СН в 6); 8,61 (с уширенный, 1Н: = СН в 3).
α
ПРИМЕР 5.
Раствор 5,0 г L-глюкозы и 5,2 г формиата аммония в 20 см3 воды кипятят в течение 2 час с обратным холодильником и оставляют охлаждаться до комнатной температуры. Смесь далее концентрируют при пониженном давлении (2,7 кПа) при температуре, близкой к 50oС. Черный пастообразный остаток забирают метанолом, растирают, отфильтровывают нерастворимую фракцию и промывают ее метанолом. Фильтрат концентрируют при пониженном давлении (2,7 кПа) при температуре, близкой к 50oС, получая 7,5 г красно-коричневого масла, которое очищают хроматографией на колонке с силикагелем (0,020-0,045 мм), элюируя смесью этанол/н-бутанол/водный аммиак/вода в объемном отношении 8/2/2/1. Фракции, содержащие целевой продукт, объединяют и концентрируют при пониженном давлении (2,7 кПа) при температуре, близкой к 50oС. Остаток последовательно забирают зтанолом и эфиром, после чего вновь концентрируют. Полученное при этом масло (0,6 г) забирают 5 см3 воды и лиофилизуют, получая таким образом 0,47 г 1-[6-(2S,3S,4-тригидроксибутил)пиразин-2-ил]бутан-1R,2S,3S,4-тетраола в виде бурого лиофилизата.
Спектр ЯМР 1H (400 МГц, (СD3)2SO-d6, температура 383 К, δ в м.д.): 2,94 и 3,03 (2дд соответственно, J = 14 и 9 Гц и J = 14 и 4 Гц, 1Н каждый: СН2 6α); от 3,40 до 3,70 (м, 6Н: СН 2γ - СН2О 2δ - СН 6γ и СН2О 6δ); 3,88 (т, J = 4 Гц, 1Н: СН 2β); 4,01 (м, 1Н: СН 6β); 4,84 (д, J = 4 Гц, 1Н: СН 2α); 8,42 (с, 1Н: = СН в 5); 8,57 (с, 1Н: = СН в 3).
α
ПРИМЕР 6.
Раствор 5,0 г L-глюкозы и 8,8 г формиата аммония в 14 см3 воды кипятят в течение 3 час с обратным холодильником и оставляют охлаждаться до комнатной температуры. Смесь далее концентрируют при пониженном давлении (2,7 кПа) при температуре, близкой к 65oС. Красно-коричневый пастообразный остаток забирают метанолом, растирают, отфильтровывают нерастворимую фракцию и промывают ее метанолом. Фильтрат концентрируют при пониженном давлении (2,1 кПа) при температуре, близкой к 50oС. Повторяют эту операцию с этанолом, получая 6,2 г красно-коричневого масла. Последнее очищают хроматографией на колонке с силикагелем (0,020-0,045 мм), элюируя смесью этанол/н-бутанол/водный аммиак в объемном отношении 8/2/1. Фракции, содержащие целевой продукт, объединяют и концентрируют при пониженном давлении (2,7 кПа) при температуре, близкой к 60oС. Полученное при этом масло (0,5 г) забирают 14 см3 этанола, фильтруют в горячем виде и кристаллизуют. После сушки до постоянного веса при температуре, близкой к 40oС, получают 0,35 г 1-[6-(2R,3S,4-тригидроксибутил)пиразин-2-ил]бутан-1S,2R,3S,4-тетраола в виде бежевых кристаллов с т.пл. 114oС.
Rf = 0,3 при хроматографировании в тонком слое силикагеля с элюированием смесью этанол/н-бутанол/водный аммиак/вода в объемном отношении 8/2/2/1.
ПРИМЕР 7.
Раствор 2,0 г D-псикозы и 3,2 г формиата аммония в 3,4 см3 воды кипятят в течение 2 час с обратным холодильником и оставляют охлаждаться до комнатной температуры. Смесь далее разбавляют 25 см3 этилацетата и декантируют. Водную фазу промывают 25 см3 этилацетата и концентрируют при пониженном давлении (2,7 кПа) при температуре, близкой к 70oС. Красно-коричневый маслянистый остаток забирают 100 см3 этанола, растирают, отфильтровывают нерастворимую фракцию и промывают ее этанолом. Фильтрат концентрируют при пониженном давлении (2,7 кПа) при температуре, близкой к 45oC, получая красно-коричневое пастообразное вещество (1,6 г). Последнее очищают хроматографией на колонке с силикагелем (0,020-0,045 мм), элюируя смесью этанол/вода в объемном отношении 199/1, и затем на колонке с силикагелем (0,020-0,045 мм), элюируя смесью этилацетат/уксусная кислота/вода в объемном отношении 30/12/10, и, наконец, на колонке с силикагелем (0,020-0,045 мм) под давлением приблизительно 1,5xl05 Па, элюируя смесью этанол/н-бутанол/водный аммиак в объемном отношении 8/2/1. Фракции, содержащие целевой продукт, объединяют и концентрируют при пониженном давлении (2,7 кПа) при температуре, близкой к 50oС. Полученное твердое вещество янтарного цвета (0,22 г) забирают смесью 5 см3 этанола и 0,25 см3 воды, фильтруют в горячем состоянии и затем перекристаллизуют. Образовавшиеся кристаллы отфильтровывают, промывают этанолом и удаляют влагу. После сушки до постоянного веса получают 65,5 мг 1-[5-(2S,3R,4-тригидроксибутил)пиразин-2-ил]бутан-1S,2S,3R,4-тетраола в виде кристаллического порошка охристого цвета с т.пл. 141oС.
Спектр ЯМР 1Н (400 МГц, (CD3)2SO-d6, δ в м.д.): 2,75 и 3,08 (2дд соответственно, J = 14 и 10 Гц и J = 14 и 2,5 Гц, 1Н каждый: CH2 5α; от 3,30 до 3,50 (м, 4Н: СН 2γ - СН 5γ - 1Н из СН2О 2δ и 1Н из CH2O 5δ); 3,60 (м, 2Н: другой Н из СН2O 2δ и другой Н из СН2О 5δ); 3,79 (м, 2Н: СН 2β и СН 5β); 4,36 и 4,45 (2т, J = 5,5 Гц, 1Н каждый: ОН в 2δ и ОН в 5δ); 4,58 - 4,64 - 4,71 и 4,78 (4д соответственно, J = 4,5 Гц - J = 6,5 Гц - J = 5 Гц и J = 5,5 Гц, 4Н: ОН); 4,82 (т, J = 5,5 Гц, 1Н: СН 2α); 5,53 (д, J = 5,5 Гц, 1Н: ОН в 2α); 8,41 (с уширенный, 1Н: = СН в 6); 8,60 (с уширенный, 1Н: = СН в 3).
ПРИМЕР 8.
Раствор 5,0 г D-галактозы и 8,8 г формиата аммония в 14 см3 воды кипятят в течение 45 мин с обратным холодильником и оставляют охлаждаться до комнатной температуры. Смесь далее разбавляют 50 см3 этилацетата и декантируют. Водную фазу дважды промывают 50 см3 этилацетата и концентрируют при пониженном давлении (2,7 кПа) при температуре, близкой к 65oС. Красно-коричневый пастообразный остаток забирают 100 см3 этанола и растирают. Супернатант концентрируют при пониженном давлении (2,7 кПа) при температуре, близкой к 45oС (операцию один раз повторяют). Красно-коричневый твердый остаток последовательно забирают метанолом, этанолом и затем диэтиловым эфиром, после чего досуха упаривают при пониженном давлении (2,7 кПа) при температуре, близкой к 45oС. Остаток очищают хроматографией на колонке с силикагелем (0,020-0,045 мм) под давлением 1,5х105 Па, элюируя смесью этанол/н-бутанол/водный аммиак в объемном отношении 8/2/1. Фракции, содержащие целевой продукт, объединяют и концентрируют при пониженном давлении (2,7 кПа) при температуре, близкой к 40oС. Полученное желтое твердое вещество (0,26 г) забирают смесью 3 см3 этанола и 0,25 см3 воды, фильтруют в горячем состоянии и затем перекристаллизуют. Полученный твердый продукт отфильтровывают и удаляют влагу. После сушки при пониженном давлении (2,7 кПа) при температуре, близкой к 25oС, получают 119 кг 1-[6-(2R,3R,4-тригидроксибутил)пиразин-2-ил] бутан-1R, 2R, 3R,4-тетраола в виде пастообразного продукта янтарного цвета, плавящегося при 90-130oС (паста).
Спектр ЯМР 1Н (400 МГц, (СD3)2SО-d6, δ в м.д.): 2,89 (АВ limit, 2Н: СН2 6α; от 3,30 до 3,55 (м, 5Н: СН2О 2δ - СН2O 6δ и СН 6γ); от 3,70 до 3,85 (м, 2Н: СН 2γ и СН 2β); 3,92 (м, 1Н: СН 6β); 4,64 (д, J = 8,5 Гц, 1Н: СН 2α); 8,38 (с, 1Н: = СН в 5); 8,45 (с, 1Н: = СН в 3).
Соединения формулы I обладают полезными фармакологическими свойствами, проявляя гипогликемическую активность.
Гипогликемическую активность соединений формулы I определяют по гипергликемической реакции, возникающей при пероральном введении глюкозы мышам с нормальным уровнем глюкозы по следующей методике.
Мышей Swiss альбиносов весом от 22 до 26 г выдерживают без пищи в течение 2 час. В конце этого периода измеряют уровень глюкозы и непосредственно после этого мышам вводят перорально глюкозу в дозе 2 г/кг. Спустя 30 мин вновь измеряют уровень глюкозы. Отбирают мышей с уровнем глюкозы выше 170 мг/дл для определения на них гипогликемической активности соединений по изобретению.
Отобранных таким образом мышей разделяют на группы, состоящие из, по меньшей мере, 10 животных каждая. В разных группах животные получают дозы от 3 до 50 мг/кг продукта в носителе, таком как вода или смесь метилцеллюлоза/твин, с водой один раз в день с помощью желудочной трубки. Лечение продолжают в течение 4 суток. На четвертые сутки, после последнего введения препарата, животные получают дозу глюкозы (2 г/кг) и через 20-40 мин измеряется уровень глюкозы. Степень (в процентах) ингибирования гипергликемической реакции на прием глюкозы рассчитывают из сравнения с гипергликемической реакцией, измеряемой в группе, получающей только носитель.
В этом испытании соединения по изобретению демонстрируют ингибирование гипергликемии на 10% или более.
Соединения общей формулы I согласно изобретению обладают низкой токсичностью. Их летальная доза DL50 при оральном введении у мышей выше 2000 мг/кг.
В терапии человека эти соединения представляют интерес для профилактики и лечения диабета, в частности диабета типа II (диабет NID), диабета тучных, диабета пятидесятилетних, метаплеторического диабета (при полнокровии), диабета пожилых и легкого диабета. Соединения могут использоваться как дополнение к инсулинотерапии при инсулинозависимом диабете, и в этом случае они позволяют постепенно снижать дозу инсулина; при неустойчивом диабете; при инсулинонезависимом диабете, дополняя сульфамиды-гипогликемианты, когда последние не обеспечивают достаточного понижения уровня сахара. Соединения могут быть также использованы при осложнениях диабета, таких как гиперлипемии, расстройства липидного обмена, дислипемии, ожирение. Они могут быть также использованы для профилактики и лечения атеросклеротических расстройств и их осложнений (коронопатии, инфаркт миокарда, кардиомиопатии, переход этих трех осложнений в недостаточность левого желудочка сердца, различные артериопатии, артерииты нижних конечностей с хромотой и переходом в язвы и гангрену, цереброваскулярная недостаточность и ее осложнения, половое бессилие, обусловленное сосудистыми нарушениями), диабетической ретинопатии и всех ее проявлений (увеличение капиллярной проницаемости, расширение и тромбоз капилляров, микроаневризмы, артериовенозный шунт, расширение вен, пятнистое и точечное кровотечения, эксудаты, пятнистые отеки, проявления пролиферантной ретинопатии: новообразование сосудов, рубцы пролиферантного ретинита, кровотечения стекловидного тела, отслойка сетчатки), диабетической катаракты, различных форм диабетической нейропатии (периферические полиневропатии и их проявления, такие как парестезии, гиперстезии и боли, мононевропатии, радикулопатии, автономные невропатии, диабетические амиотрофии), проявлений диабетической стопы (язвы нижних конечностей и стопы), диабетической нефропатии в ее диффузной и узелковой формах, атероматоза (повышение уровня липопротеинов высокой плотности (HDL), способствующее удалению холестерина из атеромных бляшек, понижения уровня липопротеинов низкой плотности (LDL), понижение отношения LDL/HDL, ингибирование окисления липопротеинов низкой плотности, уменьшение липкости бляшек), гиперлипемий и дислипемий (перхолестеринемии, гиперглицеридемии, нормализация уровня жирных кислот, нормализация урикемии, нормализация уровня апопротеинов А и В), катаракты, повышения артериального давления и его последствий.
Лекарственные средства по изобретению состоят из соединения по изобретению или комбинации таких соединений в чистом виде или в виде композиции, в которой также содержится фармацевтически совместимый продукт, который может быть как инертным, так и физиологически активным. Лекарственные средства по изобретению могут применяться перорально, парентерально, ректально или наружно.
В качестве твердых композиций для перорального введения могут быть использованы таблетки, пилюли, порошки (желатиновые капсулы, облатки) или гранулы. В этих композициях активное начало согласно изобретению смешивают в токе аргона с одним иди несколькими инертными разбавителями, такими как крахмал, целлюлоза, сахароза, лактоза или кремнезем. Эти композиции могут также содержать и другие не являющиеся разбавителями вещества, например один или несколько смазывающих агентов, таких как стеарат магния или тальк, краситель, оболочка (драже) или лак.
В качестве жидких композиций для перорального введения могут быть использованы фармацевтически приемлемые растворы, суспензии, эмульсии, сиропы и эликсиры, содержащие инертные разбавители, такие как вода, этанол, глицерин, растительные масла или парафиновое масло. Эти композиции могут также содержать и другие, не являющиеся разбавителями вещества, например увлажняющие агенты, подсластители, загустители, ароматизаторы и стабилизаторы.
Стерильные композиции для парентерального введения являются преимущественно водными или неводными растворами, суспензиями или эмульсиями. В качестве растворителя или носителя могут быть использованы вода, пропиленгликоль, полиэтиленгликоль, растительные масла (в особенности, оливковое масло), инъецируемые эфиры органических кислот, например этилолеат, или другие подходящие органические растворители. Эти композиции могут также содержать добавки, в частности увлажнители, изотонизирующие агенты, амульгаторы, диспергенты и стабилизаторы. Стерилизация композиций может быть произведена несколькими способами, например асептической фильтрацией, введением стерилизующих агентов, облучением или нагревом. Композиции могут быть также приготовлены в стерильной твердой форме для растворения непосредственно перед применением в стерильной воде или любой другой пригодной для инъекций стерильной среде.
Композиции для ректального введения представляют собой суппозитории или ректальные капсулы, содержащие, кроме активного вещества, эксципиенты, такие как масло какао, полусинтетические глицериды или полиэтиленгликоли.
Композиции для наружного применения могут представлять собой, например, кремы, лосьоны, глазные капли (мази), жидкости для полоскания, капли для носа или аэрозоли.
Дозы зависят от желаемого эффекта, длительности лечения и пути введения. Обычно они составляют от 150 до 600 мг в сутки при пероральном введении для взрослых при разовых дозах в пределах от 50 до 200 мг в расчете на активное вещество.
Обычно врач определяет необходимую дозу в зависимости от возраста, веса и остальных факторов, относящихся к пациенту.
Композиции по изобретению иллюстрируют следующие примеры.
ПРИМЕР А.
Приготовляют обычным способом желатиновые капсулы, содержащие 50 мг активного вещества и характеризующиеся следующим составом, мг:
Активное вещество - 50
Целлюлоза - 18
Лактоза - 55
Коллоидальный оксид кремния - 1
Натриевый карбоксиметилкрахмал - 10
Тальк - 10
Стеарат магния - 1
ПРИМЕР В.
Приготовляют обычным способом таблетки, содержащие 50 мг активного вещества, и характеризующиеся следующим составом, мг:
Активное вещество - 50
Лактоза - 104
Целлюлоза - 40
Поливидон - 10
Натриевый карбоксиметилкрахмал - 22
Тальк - 10
Стеарат магния - 2
Коллоидальный оксид кремния - 2
Смесь гидроксиметилцеллюлозы, глицерина, оксида титана (72/3,5/24,5) в количестве, достаточном для приготовления 1 таблетки массой 245 мг
ПРИМЕР С.
Приготовляют обычным способом раствор для инъекций, содержащий 50 мг активного вещества и характеризующийся следующим составом:
Активное вещество, мг - 50
Бензойная кислота, мг - 80
Бензиловый спирт, мл - 0,06
Бензоат натрия, мг - 80
95%-ный этанол, мл - 0,4
Гидроксид натрия, мг - 24
Пропиленгликоль, мл - 1,6
Вода в достаточном количестве, мл - До 4
Изобретение относится также к применению соединений общей формулы I для получения фармацевтических композиций, пригодных для лечения и профилактики диабета и осложнений диабета.

Изобретение относится к производным полигидроксиалкилпиразина формулы I, используемым в качестве активного начала в лекарственном средстве, в которой R1 обозначает стереоизомерные формы цепи (II): -(СНОН)3-СН2ОН, R2 обозначает атом водорода и R3 обозначает стереоизомерные формы цепи (III): -СН2-(СНОН)2-СН2ОН или R2 обозначает стереоизомерные формы цепей (II): -(СНОН)3-СН2ОН или (III): СН2-(СНОН)2-СН2ОН и R3 обозначает атом водорода. Описаны также способы получения соединений формулы I на их основе. Соединения проявляют гипогликемическую активность и в терапии представляют интерес для профилактики и лечения диабета. 5 с. и 7 з.п.ф-лы.

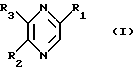
в которой R1 обозначает стереоизомерные формы цепи
-(СНОН)3-СН2ОН (II)
и либо R2 обозначает атом водорода, а R3 обозначает стереоизомерные формы цепи
-СН2-(СНОН)2-СН2ОН (III)
либо R2 обозначает стереоизомерные формы цепи
-(СНОН)3-СН2ОН (II)
или
-СН2-(СНОН)2-CH2OH (III)
a R3 обозначает атом водорода, за исключением
фруктозазина формулы
деоксифруктозазина формулы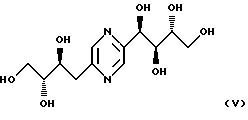
и соединения формулы
или соль названных выше соединений с органическими или минеральными кислотами.
1-[5-(1R, 2R, 3R, 4-тетрагидроксибутил)пиразин-2-ил] бутан-1R, 2R, 3R, 4-тетраол,
1-[5-(1R, 2R, 3S, 4-тетрагидроксибутил)пиразин-2-ил] бутан-1R, 2R, 3S, 4-тeтраол,
1-[5-(1R, 2S, 3S, 4-тетрагидроксибутил)пиразин-2-ил] бутан-1R, 2S, 3S, 4-тетраол,
1-[5-(1S, 2R, 3R, 4-тетрагидроксибутил)пиразин-2-ил] бутан-1S, 2R, 3R, 4-тетраол,
1-[5-(1S, 2R, 3S, 4-тетрагидроксибутил)пиразин-2-ил] бутан-1S, 2R, 3S, 4-тетраол,
1-[5-(1S, 2S, 3R, 4-тетрагидроксибутил)пиразин-2-ил] бутан-1S, 2S, 3R, 4-тетраол,
1-[5-(1S, 2S, 3S, 4-тетрагидроксибутил)пиразин-2-ил] бутан-1S, 2S, 3S, 4-тетраол,
1-[5-(2R, 3R, 4-тригидроксибутил)пиразин-2-ил] бутан-1R, 2R, 3R, 4-тетраол,
1-[5-(2R, 3S, 4-тригидроксибутил)пиразин-2-ил] бутан-1R, 2R, 3S, 4-тетраол,
1-[5-(2S, 3S, 4-тригидроксибутил)пиразин-2-ил] бутан-1R, 2S, 3S, 4-тетраол,
1-[5-(2R, 3R, 4-тригидроксибутил)пиразин-2-ил] бутан-1S, 2R, 3R, 4-тетраол,
1-[5-(2R, 3S, 4-тригидроксибутил)пиразин-2-ил] бутан-1S, 2R, 3S, 4-тетраол,
1-[5-(2S, 3R, 4-тригидроксибутил)пиразин-2-ил] бутан-1S, 2S, 3R, 4-тетраол,
1-[5-(2S, 3S, 4-тригидроксибутил)пиразин-2-ил] бутан-1S, 2S, 3S, 4-тетраол,
1-[6-(2R, 3R, 4-тригидроксибутил)пиразин-2-ил] бутан-1R, 2R, 3R, 4-тетраол,
1-[6-(2R, 3S, 4-тригидроксибутил)пиразин-2-ил] бутан-1R, 2R, 3S, 4-тетраол,
1-[6-(2R, 3R, 4-тригидроксибутил)пиразин-2-ил] бутан-1S, 2R, 3R, 4-тетраол,
1-[6-(2R, 3S, 4-тригидроксибутил)пиразин-2-ил] бутан-1S, 2R, 3S, 4-тетраол,
1-[6-(2S, 3R, 4-тригидроксибутил)пиразин-2-ил] бутан-1S, 2S, 3R, 4-тетраол,
1-[6-(2S, 3S, 4-тригидроксибутил)пиразин-2-ил] бутан-1S, 2S, 3S, 4-тетраол,
или солей этих соединений с органическими или минеральными кислотами.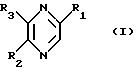
в которой R1 обозначает стереоизомерные формы цепи
-(СНОН)3-СН2ОН (II)
либо R2 обозначает атом водорода, а R3 обозначает стереоизомерные формы цепи
-СН2-(СНОН)2-СН2ОН (III)
либо R2 обозначает стереоизомерные формы цепи
- (СНОН)3-СН2ОН (II)
или
-СН2 - (СНОН)2-CH2OH (III)
R3 обозначает атом водорода,
или их соли с минеральными или органическими кислотами, за исключением соединений формул (см. графическую часть).
1-[5-(1R, 2R, 3S, 4-тетрагидроксибутил)пиразин-2-ил] бутан-1R, 2R, 3S, 4-тетраол,
1-[5-(1S, 2R, 3R, 4-тетрагидроксибутил)пиразин-2-ил] бутан-1S, 2R, 3R, 4-тетраол,
1-[5-(1S, 2S, 3R, 4-тетрагидроксибутил)пиразин-2-ил] бутан-1S, 2S, 3R, 4-тетраол,
1-[5-(1S, 2S, 3S, 4-тетрагидроксибутил)пиразин-2-ил] бутан-1S, 2S, 3S, 4-тетраол,
1-[5-(2R, 3S, 4-тригидроксибутил)пиразин-2-ил] бутан-1R, 2R, 3S, 4-тетраол,
1-[5-(2S, 3S, 4-тригидроксибутил)пиразин-2-ил] бутан-1R, 2S, 3S, 4-тетраол,
1-[5-(2R, 3R, 4-тригидроксибутил)пиразин-2-ил] бутан-1S, 2R, 3R, 4-тетраол,
1-[5-(2S, 3R, 4-тригидроксибутил)пиразин-2-ил] бутан-1S, 2S, 3R, 4-тетраол,
1-[6-(2R, 3S, 4-тригидроксибутил)пиразин-2-ил] бутан-1R, 2R, 3S, 4-тетраол,
1-[6-(2R, 3R, 4-тригидроксибутил)пиразин-2-ил] бутан-1S, 2R, 3R, 4-тетраол,
1-[6-(2S, 3R, 4-тригидроксибутил)пиразин-2-ил] бутан-1S, 2S, 3R, 4-тетраол
и соли этих соединений с минеральными или органическими кислотами.
1-[5-(2S, 3S, 4-тригидроксибутил)пиразин-2-ил] бутан-1R, 2S, 3S, 4-тетраол,
1-[5-(2R, 3R, 4-тригидроксибутил)пиразин-2-ил] бутан-1S, 2R, 3R, 4-тетраол,
1-[5-(2S, 3R, 4-тригидроксибутил)пиразин-2-ил] бутан-1S, 2S, 3R, 4-тетраол
и соли этих соединений с минеральными или органическими кислотами.
CHO-CHOH-R1 (X)
в которой R1 имеет то же значение, что и в п. 3,
выделяют продукт и превращают его при желании в соль.
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| СПОСОБ РАСЧЕТА ОПТИЧЕСКОЙ СИЛЫ ИОЛ У ДЕТЕЙ РАННЕГО ВОЗРАСТА С МОНОКУЛЯРНОЙ ВРОЖДЕННОЙ КАТАРАКТОЙ | 2012 |
|
RU2495615C1 |
| Способ получения -3,5, 3,5-тетраоксо-1,2 дипиперазинопропана | 1976 |
|
SU615077A1 |

