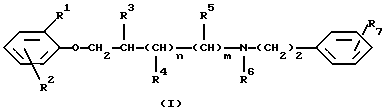
Данное изобретение относится к новым производным феноксиалкиламина, имеющим антиаритмическое действие, с двойным местом воздействия. Более конкретно, данное изобретение относится к новым производным феноксиалкиламина формулы (I)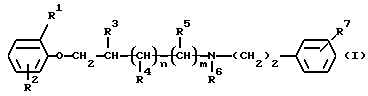
и их солям, а также к фармацевтическим композициям, содержащим данные соединения.
Известно, что сердечная аритмия представляет собой одно из наиболее серьезных сердечно-сосудистых заболеваний. Из этих заболеваний наиболее серьезным заболеванием является фибрилляция желудочков сердца, которая является наиболее частой непосредственной причиной внезапной смерти сердца и приводит к около 400000 смертям в год только в США. Фибрилляция предсердий также сопровождается серьезными осложнениями, которые наблюдаются почти у одного миллиона человек.
Наиболее широко используемым способом лечения аритмий является лекарственная терапия. Тем не менее, агенты, имеющиеся в настоящее время, не могут, в основном, считаться наилучшим решением данной проблемы: их использование может быть значительно ограничено их серьезными побочными эффектами. С этой точки зрения, отрицательные результаты, полученные из двух серий всеобъемлющих клинических исследований [CAST I и CAST II. Исследования CAST, N. Engl. J.Med.321, 406 (1989) и там же 327, 227 (1992)], прояснили, что некоторые так называемые антиаритмические агенты класса IC с медленной динамикой восстановления, которые ингибируют максимальную скорость реполяризации потенциала действия сердца, например, натриевый канал, уменьшают шансы к выживанию пациентов, восстанавливающихся после инфаркта миокарда [современная классификация антиаритмических агентов представлена, например, в А. О. Grant, Jr. и N.C.Durham: Am.Heart J.,123, 1130 (1992)]. Эти неблагоприятные результаты могут быть сродни проаритмическому (т.е. провоцирующему аритмию) эффекту и отрицательному инотропному [т.е. сердечная активность (минутный сердечный выброс)-ослабление] эффекту антиаритмических агентов класса IC. Далее, данные результаты оказались сходными с результатами, полученными для антиаритмических агентов класса III, привлекшими к себе повышенное внимание в начале 90-х [которые пролонгируют длительность потенциала действия и, следовательно, эффективный рефракторный период (сокращенно ЭРП)], которые имеют опасные побочные эффекты; клинические исследования d-соталола (химически, гидрохлорид d-N-[4-[l-гидрокси-2-(1-метилэтиламино)этил]фенил]метансульфонамида) в качестве прототипа агентов класса III были даже прерваны в группе пациентов из-за увеличения смертности, приписываемой проаритмическому эффекту [см., например, P.Matyus, A.Varro, J. Gy. Papp et al. : Med.Res.Rev. 17, 427 (1997)]. Существуют опасности или ограничения, соответственно, использования двух дополнительных классов антиаритмических агентов (II и IV), а также двух других подгрупп (IA и IB), блокирующих натриевый канал агентов класса I (особенно для классов IV и IA).
Исходя из вышесказанного, очевидно, что существует острая необходимость в новом, безопасном, но одновременно подходящем активном антиаритмическом агенте. Целью данного изобретения является разработка активного агента и композиции соответственно, отвечающих этим требованиям.
В литературе известно множество производных феноксиалкиламина. Однако как их химические структуры, так и биологическое действие отличны от соединений формулы (I) данного изобретения.
Ниже представлены ссылки, описывающие вещества со структурами, наиболее близкими к структурам данного изобретения.
В ЕР 245997 описаны бис(аралкил)амины и производные феноксиалкиламина с антиаритмическим действием, некоторые из которых также оказывают положительное инотропное действие. Согласно данному патенту эти соединения являются селективными антиаритмическими агентами класса III. Обнаружено, что производное формулы (VI)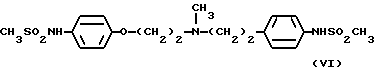
содержащее метансульфонамидогруппу в положениях 4 обоих фенильных колец, является наиболее активным.
В публикации заявки на патент Японии 06107614 описаны соединения, не попадающие в объем данного изобретения, которые могут быть охарактеризованы формулой (I), где R1, R2 и R3 являются водородом, оба n и m равны 0 или один из них равен 1; R6 является водородом, алкилом, формилом или алкилтиокарбамоилом и R7 является аминогруппой, замещенной п-хлорбензолсульфонилом, 2,4,6-триизопропилбензолсульфонилом, 8-хинолинсульфонилом, 1-нафтоилом или 2-пиридинкарбонилом. Согласно описанию данные соединения оказывают противоязвенное действие.
В патенте США 4044150 описаны 4'-[1-гидрокси-2-[(1-феноксиэтил)амино] этил]метансульфонанилид формулы (VII)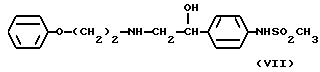
и его соли, которые имеют химическую структуру, приближенную к структуре части соединений данного изобретения. Согласно данному патенту эти соединения являются блокаторами β-адренергического рецептора и, как таковые, могут быть использованы в качестве гипотензивных средств.
Производные феноксиалкиламина, имеющие химические структуры, сходные со структурами соединений данного изобретения, описаны в патенте Великобритании 2088873, где феноксигруппа может быть моно- или дизамещенной водородом, галогеном, гидроксилом или алкокси, а фенильная группа(ы) с другой стороны алкиламиноалкильной цепи может быть ди- или тризамещенной пара-гидроксилом или аминогруппой, и/или мета-галогеном(ами), а также трифторметилом, циано или нитрогруппой(ами). Согласно указанному патенту эти соединения воздействуют на сердце и кровообращение, более конкретно, они имеют гипотензивное, антиаритмическое и/или кардиотоническое действия. Однако биологические данные, приведенные в описании, подтверждают только положительную инотропную эффективность соединений.
Наконец, интересно, что соединения, аналогичные соединениям формулы (I) данного изобретения, феноксигруппа которых замещена ациламиногруппой и фенильная группа на другом конце алкиламиноалкильной цепи замещена C1-4 алкилом или алкоксигруппой, и/или галогеном (который, следовательно, содержит заместитель, обратный соединениям данного изобретения) имеют противоопухолевый эффект (ЕР 494623).
Во время исследований настоящих авторов неожиданно было обнаружено, что соединения формулы (I) имеют сильное антиаритмическое действие и не демонстрируют вредных побочных эффектов. По способу действия эффекты агентов типа IB и III дают очень благоприятное антиаритмическое действие более широкого спектра, чем действие, характеризующее отдельные компоненты действия; оно является следствием комбинированного механизма. Одновременно, было установлено, что соединения формулы (I) не имеют серьезных побочных эффектов, которые являются характерными для соединений классов I и III.
Эти благоприятные фармакологические свойства соединений формулы (I) особенно удивительны в свете данных, представленных в литературе. А именно, относительно требований к химической структуре антиаритмических агентов классов IB и III имеется точка зрения, что антиаритмическое действие агентов класса IB является свойством ортодизамещенных феноксиалкиламино (или изостерических) систем, содержащих короткую алкильную цепь и аминозаместитель с небольшой пространственной способностью (например, токаинид, лидокаин); в то время как соединения, демонстрирующие сильный эффект агентов класса III, характеризуются структурой, содержащей две фенильные группы, связанные относительно длинной цепью, где обе фенильные группы обычно имеют парарасположенные, т.е. расположенные симметрично, притягивающие электроны заместители [для подробного анализа взаимоотношений структуры и действия см., например: P.Matyus, A.Varro, J.Gy. Papp et al.: Med. Res. Rev. 17, 427 (1997)].
Следовательно, неожиданностью даже для специалистов в данной области явилось то, что соединения формулы (I) данного изобретения, которые имеют структуру, коренным образом отличную от типичных представителей антиаритмических агентов обоих классов IB и III, обладают значительным антиаритмическим действием, ассоциированным с действием агентов обоих типов IB и III, при этом они не имеют проаритмического или отрицательного инотропного действия.
Из литературы хорошо известно, что соединения, имеющие двойное место воздействия, очень важны для фармацевтической химии [см., например, "The Practice of Medicinal Chemistry", стр.261-293, издание C.G. Wermuth, Academic Press, London (1996)]. В данном случае сочетание указанных выше двух антиаритмических механизмов действия даже более предпочтительно, чем обычно, так как оно дает качественно новые, крайне благоприятные свойства, а именно из-за одновременного присутствия обоих компонентов действия соединения формулы (I) не демонстрируют либо проаритмического, либо отрицательного инотропного эффекта. Другими словами, данное сочетание эффектов, объединенных в одной и той же молекуле, более предпочтительно с точки зрения фармакокинетики и безопасности лекарственного средства, по сравнению с сочетанием обеих отдельных молекул.
Взятые все вместе, при условии, что их токсичность низка, соединения формулы (I) могут считаться безопасными антиаритмическими лекарственными средствами нового типа с широким спектром действия.
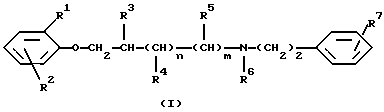
Таким образом, данное изобретение относится к соединениям формулы (I)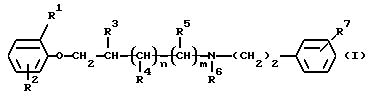
где R1 и R2 независимо друг от друга являются водородом, галогеном или C1-4 алкильной группой;
R3, R4 и R5 независимо друг от друга являются водородом или C1-4 алкильной группой;
R6 является водородом, C1-4 алкильной или бензильной группой;
R7 является нитрогруппой или аминогруппой, необязательно монозамещенной C1-4 алкильной, бензоильной,C1-4 алкилкарбонильной, C1-4 алкилсульфонильной, C1-4 алкилкарбамоильной или C1-4 алкилтиокарбамоильной группой;
n и m оба равны 0 или 1,
при условии, что R2 отличен от водорода, если R1 является водородом,
а также к их стереоизомерам или смесям стереоизомеров, их кислотно-аддитивным солям и гидратам, пролекарствам и метаболитам всех этих соединений.
В соединениях данного изобретения алкильные группы могут иметь прямую или разветвленную углеродную цепь; галоген может являться бромом, хлором или фтором.
Согласно вышесказанному, в формуле (I) R1 и R2 независимо друг от друга могут являться метилом, этилом, н-пропилом, изопропилом или бутилом, а также фтором, хлором или бромом. В случае, если один из R1 и R2 является водородом, другой должен быть отличен от водорода. Таким образом, замещенная феноксигруппа в формуле (I) может быть, например, 2,3-диметилфенокси, 2,4-диметилфенокси, 2,5-диметилфенокси, 2,6-диметилфенокси, 2,6-диэтилфенокси, 2-этил-6-метилфенокси, 2-хлор-6-метилфенокси, 2,6-дихлорфенокси, 2-бром-6-метилфенокси, 2,6-дибромфенокси, 2-бром-4-хлорфенокси, 2-метилфенокси, 2-этилфенокси, 2-изопропилфенокси, 2-хлорфенокси, 2-бромфенокси или 2-фторфенокси.
R3, R4, R5 и R6 независимо друг от друга могут быть водородом, метилом, этилом, н-пропилом, изопропилом или бутилом, кроме того, R6 также может быть бензилом; R7 может быть, например, нитро, амино, метиламино, этиламино, пропиламино, изопропиламино, бутиламино, ацетиламино, пропиониламино, изопропиониламино, бутаноиламино, бензоиламино, метансульфонамидо, этансульфонамидо, н-пропансульфонамидо, изопропилсульфонамидо, бутансульфонамидо, метилуреидо, этилуреидо, н-пропилуреидо, изопропилуреидо, метилтиоуреидо или этилтиоуреидо.
Предпочтительная группа соединений формулы (I) данного изобретения представлена соединениями, в которых R1 является алкилом, R2-R7 и n такие, как определено выше, и m равен 0.
Другая предпочтительная группа соединений данного изобретения включает соединения формулы (I), в которых R1 и R2 вместе являются 2,6-диалкильной группой, R3 и/или R6 является(ются) алкильной группой, R7 и n такие, как определено выше, и m равен 0.
Соединения формулы (I) данного изобретения могут быть получены с использованием нескольких известных способов.
а) Для получения соединений формулы (I), в которых R1 и R2 являются алкилом или галогеном или один из них является водородом, R3, R4, R5, R6, n и m такие, как определено выше, и R7 является нитро, например, соединение формулы (II)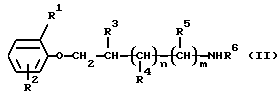
где R1, R2, R3, R4, R5, R6, n и m такие, как определено выше, подвергают взаимодействию с 4-нитрофенэтилбромидом. Эта реакция может проводиться при нагревании без какого-либо растворителя или в различных растворителях, например изопропаноле, бутаноле или ацетонитриле.
Часть аминосоединений формулы (II), используемых в качестве исходного материала, известны в литературе (например, патент BE 626725; патент США 3659019).
Соединения формулы (II), до настоящего времени не описанные в литературе, могут быть получены по известным способам. Получение новых соединений формулы (II), начиная с соединений формулы (V)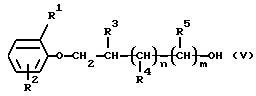
описано в примерах.
b) Для получения соединений формулы (I), в которых R1 и R2 являются алкилом, или один из них является водородом, R3, R4, R5, R6, n и m такие, как определено выше, и R7 является амино, например, соединение формулы (I), где R1, R2, R3, R4, R5, R6, n и m такие, как определено выше, и R7 является нитрогруппой, восстанавливают по известному способу. Данное восстановление может проводиться каталитическим гидрированием или каталитическим гидрированием с переносом, или другим способом, известным в литературе.
с) Получение соединений формулы (I), в которых R1 и R2 являются алкилом или один из них является водородом, R3, R4, R5, n и m такие, как определено выше, R6 является алкилом или бензилом и R7 является алкилсульфонамидо, бензамидо, ал-кил(тио)уреидо или алифатическим алкилкарбониламино (например, ацетиламино), может быть осуществлено, например, взаимодействием соединений формулы (I), где R1, R2, R3, R4, R5, R6, n и m такие, как определено выше, и R7 является аминогруппой, с реагентом, подходящим для введения сульфонильной группы, или с ацилирующими реагентами в различных растворителях.
Согласно предпочтительному варианту указанного выше способа с), соединения, где R7 является алкилсульфонамидо, бензамидо или алкилкарбониламино, подвергают взаимодействию в присутствии агента, связывающего кислоту, (например, триэтиламина). Эту реакцию проводят при комнатной температуре или, при необходимости, реакционную смесь охлаждают или нагревают.
Согласно другому предпочтительному варианту указанного выше способа с), соединения, в которых R7 является алкил(тио)уреидогруппой, подвергают взаимодействию в диоксане без какого-либо агента, связывающего кислоту, при комнатной температуре или при нагревании.
d) Для получения соединений формулы (I), в которых R1 и R2 являются алкилом, или один из них является водородом, R3, R4, R5, n и m такие, как определено выше, и R6 является алкилом и R7 является алкиламино, например, соединение формулы (I), где R1, R2, R3, R4, R5, R6, n и m такие, как определено выше, и R7 является алкилкарбониламиногруппой, может быть восстановлено по известному способу.
Согласно предпочтительному варианту способа d) данное восстановление проводят с использованием литийалюмогидрида при температуре кипения тетрагидрофурана.
e) Для получения соединений формулы (I), в которых R1 и R2 являются алкилом, галогеном или один из них является водородом, R3, R4, R5, n и m такие, как определено выше, и R6 является алкилом, водородом или бензилом и R7 является метансульфонамидогруппой, например, соединение формулы (III)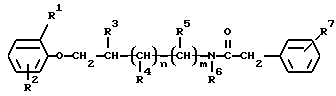
где R1, R2, R3, R4, R5, R6, R7, n и m такие, как определено выше, восстанавливают по известному способу.
Согласно предпочтительному варианту способа е) данное восстановление проводят с использованием литийалюмогидрида в тетрагидрофуране при его температуре кипения.
Соединения формулы (III), используемые в способе е), могут быть получены, например, взаимодействием аминов формулы (II) с (метансульфонамидо)фенилуксусной кислотой.
Согласно предпочтительному варианту указанного выше способа, используемого для получения соединений формулы (III), реакцию проводят в N,N-диметилформамиде в присутствии 4-метилморфолина, изобутилхлорформиата и триэтиламина при температуре -10oС и позже при 0oС или реакцию проводят в тетрагидрофуране в присутствии дициклогексилкарбодиимида.
f) Для получения соединений формулы (I), в которых R1 и R2 являются алкилом, галогеном или один из них является водородом, R3, R4, R5, R6, n и m такие, как определено выше, и R7 является метансульфонамидо, например, соединение формулы (II), где R1, R2, R3, R4, R5, R6, n и m такие, как определено выше, подвергают взаимодействию с N-[4-(2-бромэтил)фенил]метансульфонамидом.
Согласно предпочтительному варианту способа f) эту реакцию проводят без какого-либо растворителя при нагревании.
Соли соединений формулы (I) данного изобретения образуются непосредственно в реакции их получения или, если соединения формулы (I) выделены в их основной форме, соли могут быть получены из полученного основания растворением основания в подходящем растворителе, таком как метанол, этанол, изопропанол, этиловый эфир, этилацетат или в их смеси, и затем добавлением к раствору соответствующей кислоты, растворенной в подходящем растворителе. Соли могут быть выделены непосредственно фильтрацией или, необязательно, осаждением после добавления подходящего растворителя, или полным или частичным выпариванием растворителя.
Часто соли соединений формулы (I) данного изобретения содержат кислотный компонент в нестехиометрическом соотношении и/или они часто кристаллизуются в виде гидрата. Данное изобретение охватывает такие кристаллические формы тоже.
Соединения формулы (I) данного изобретения также могут необязательно содержать асимметрические атомы углерода, и поэтому они могут существовать в оптически активных и рацемических модификациях. Оптически чистые соединения могут быть получены из оптически чистых предшественников, используемых в качестве исходных материалов, или рацемический конечный продукт может быть растворен. В последнем случае оптические изомеры могут быть разделены, например, обработкой рацемического соединения, например, в метаноле, этаноле, этилацетате, ацетоне или другом растворителе, оптически активной кислотой, например, 0,5-2,0 молярным количеством [рассчитано для соединения формулы (I)], например D-винной кислоты, О,О-дибензоил-L-винной кислоты, N, N-диметилмоно-амида O, O-дибензоил-D-винной кислоты, L-тиазолидин-4-карбоновой кислоты или другой обычной оптически активной кислоты с получением диастереомерной соли одного оптического изомера, сразу же в чистом виде или необязательно после нескольких перекристаллизаций (в зависимости от используемой кислоты); другой изомер получают обработкой маточных жидкостей в основной форме или в форме их соли (что зависит от количества используемой кислоты) с последующей очисткой при необходимости.
Затем оптически активные основания могут быть выделены из их полученных таким образом солей и отделены, при желании, для получения из полученного основания кислотно-аддитивной соли с терапевтически приемлемой кислотой.
Как отмечено выше, соединения формулы (I) данного изобретения имеют полезное антиаритмическое действие.
Далее описаны результаты фармакологических исследований, которые доказывают электрофизиологическое действие на сердце IB и III класса соединений формулы (I) без отрицательного инотропного действия и действия, вызывающего аритмию, т.е. проаритмического действия, соответственно, которыми отличаются агенты классов IB и III. Наконец, антиаритмическое действие in vivo соединений формулы (I) будет показано на трех моделях аритмии.
Внеклеточные электрофизиологические измерения
Используемая методика по существу является методикой, описанной у Varro et al. [Arch. Int.Pnarmacodyn. Ther.292, 157-165 (1988)].
Правые желудочковые трабекулы получают из сердца дворняжек (вес от 5 до 10 кг каждая). Живые части сердца помещают в баню для органов (температура 37oС), содержащую модифицированную питательную среду Tyrode (Тайрода), и возбуждают точечным стимулированием с основной частотой 1 Гц. Внеклеточные биполярные платиновые электроды помещают на поверхность препаратов, с помощью которых регистрируют распространение поверхностных потенциалов; таким образом, становится возможным определение времени проводимости импульса или скорости проводимости импульса, соответственно, и эффективного рефракторного периода (ЭРП). Во время экспериментов частоту стимулирования временно изменяют от 0,5 Гц до 3 Гц. Тестируемые соединения непосредственно добавляют в баню для органов после разбавления маточного раствора до желаемой конечной концентрации. Действия соединений наблюдают по прошествии инкубационного периода 30-40 минут.
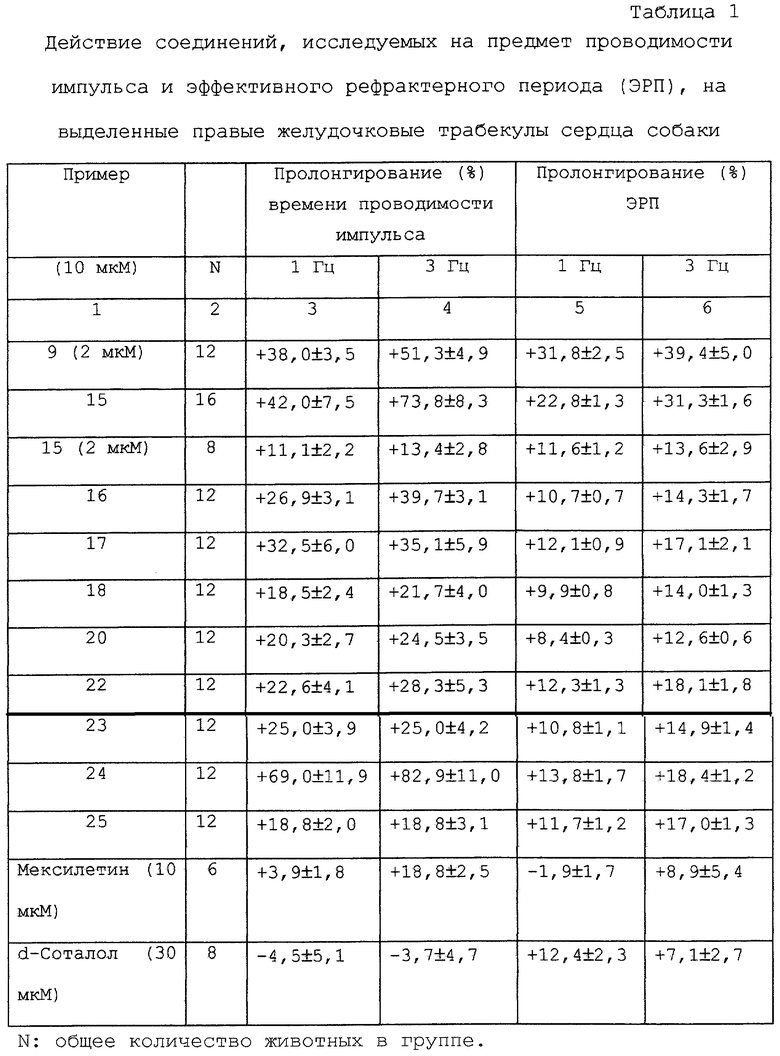
Результаты внеклеточных in vitro электрофизиологических измерений сердца показаны в таблице 1.
Среди исследуемых соединений соединения примеров 9, 15, 16 и 17 значительно пролонгируют время проводимости импульса в зависимости от частоты, т.е. они снижают скорость проводимости импульса вместе с одновременным пролонгированием эффективного рефракторного периода (ЭРП). Мексилетин (химически, 2-(2,6-диметилфенокси)-1-метилэтиламин гидрохлорид) используют в качестве ссылочного (эталонного) соединения, оказывающего действие в основном на проводимость импульса, в то время как d-Соталол демонстрирует значительное действие только на ЭРП.
Методики внутриклеточных микроэлектродов
Используемая методика по существу является методикой, описанной у Рарр et al. [J.Cardiovascular Pharmacol. Ther. 1, 287-296 (1996)].
Правые желудочковые папиллярные мышцы и волокна Пуркинье получают из сердец дворняжек (весом от 5 до 10 кг каждая). Препараты помещают в баню для органов (температура 37oС), содержащую модифицированную питательную среду Tyrode (Тайрода). Препараты возбуждают основной частотой 1 Гц. Во время эксперимента частоту стимулирования изменяют в широких пределах с помощью управляемого компьютером стимулятора. Внутриклеточные потенциалы действия регистрируют с помощью стеклянных капиллярных микроэлектродов, заполненных 3 н. раствором хлорида калия (стандартная методика внутриклеточных микроэлектродов). Полученный потенциал (ПП), амплитуду потенциала действия (АПД), а также продолжительность потенциала действия (ППД) измеряют с помощью IBM 386 - совместимого компьютера с программным обеспечением собственной разработки. Тестируемые соединения непосредственно добавляют в баню для органов после разбавления маточного раствора до желаемой конечной концентрации. Действие соединений наблюдают по прошествии инкубационного периода 30-40 минут.
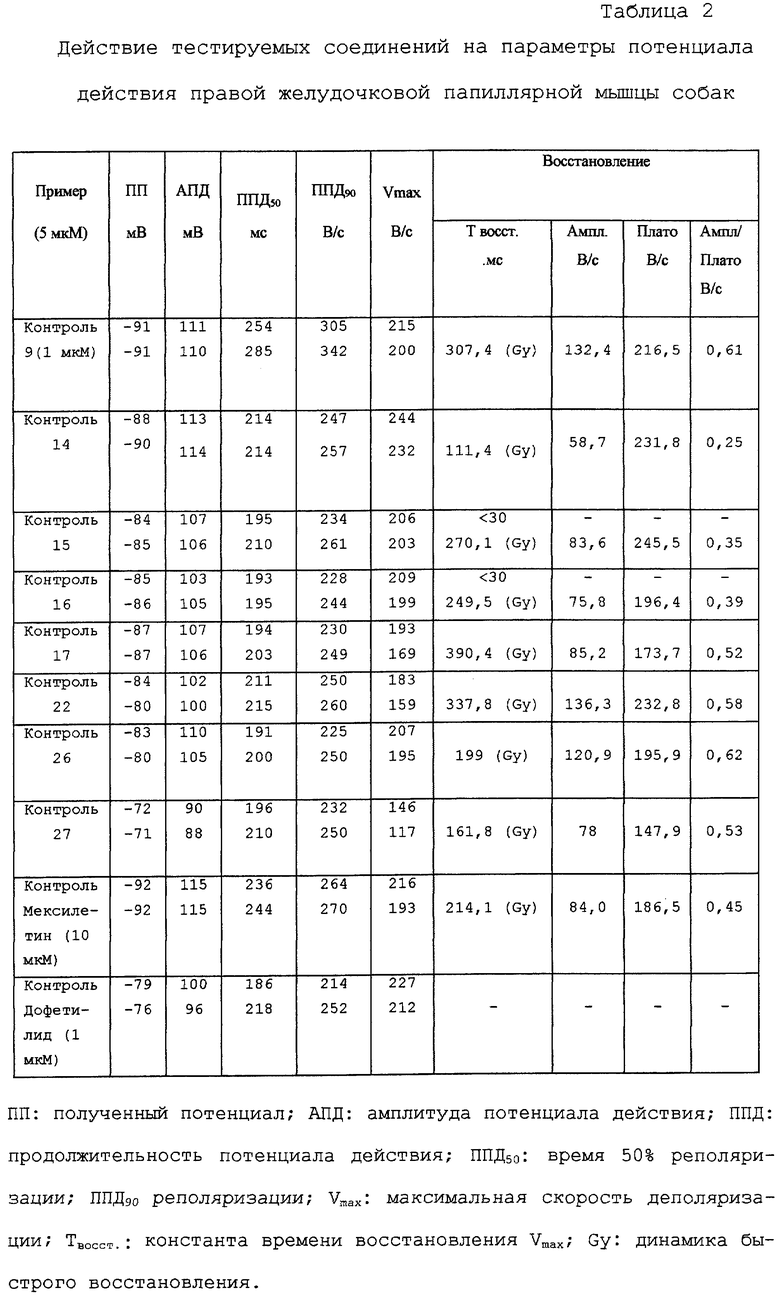
Результаты внутриклеточных электрофизиологических измерений сердца на папиллярных мышцах показаны в таблице 2. Среди тестируемых соединений соединения примеров 9, 14, 15, 16, 17, 22, 26 и 27 вызывают мексилетинподобное ингибирование Vmax, которое может быть охарактеризовано относительно значительной степенью динамики быстрого восстановления вместе с одновременным пролонгированием длительности потенциала действия (ППД) дофетилидподобное действие; химически дофетилид представляет собой N-[4-[2-[N-метил-N-[2-(4-метансульфонамидофенокси)этил] амино] этил] фенил] метансульфонамид). В данном эксперименте никаких существенных различий между действием соединения примера 15 и его энантиомеров (примеры 16 и 17) обнаружено не было.
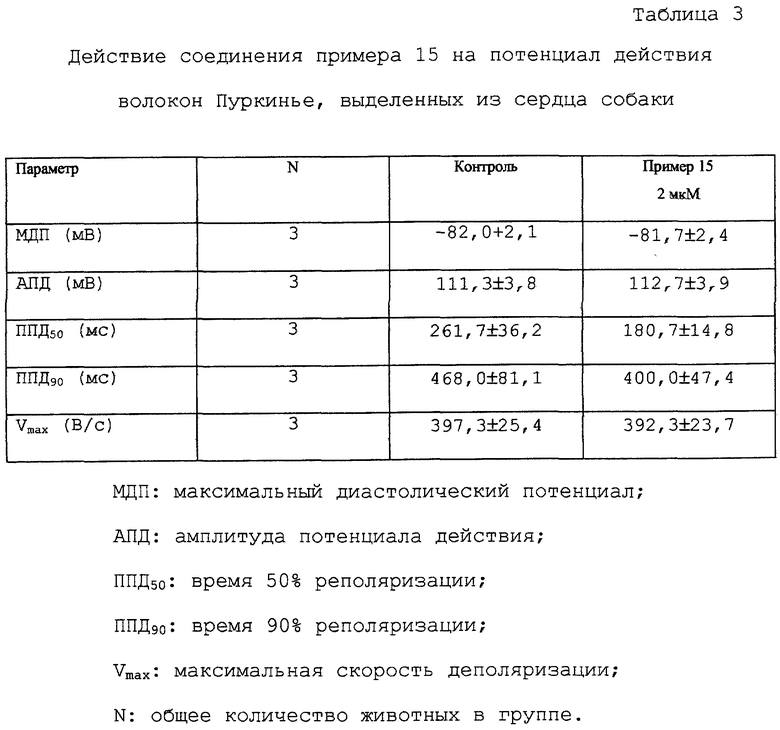
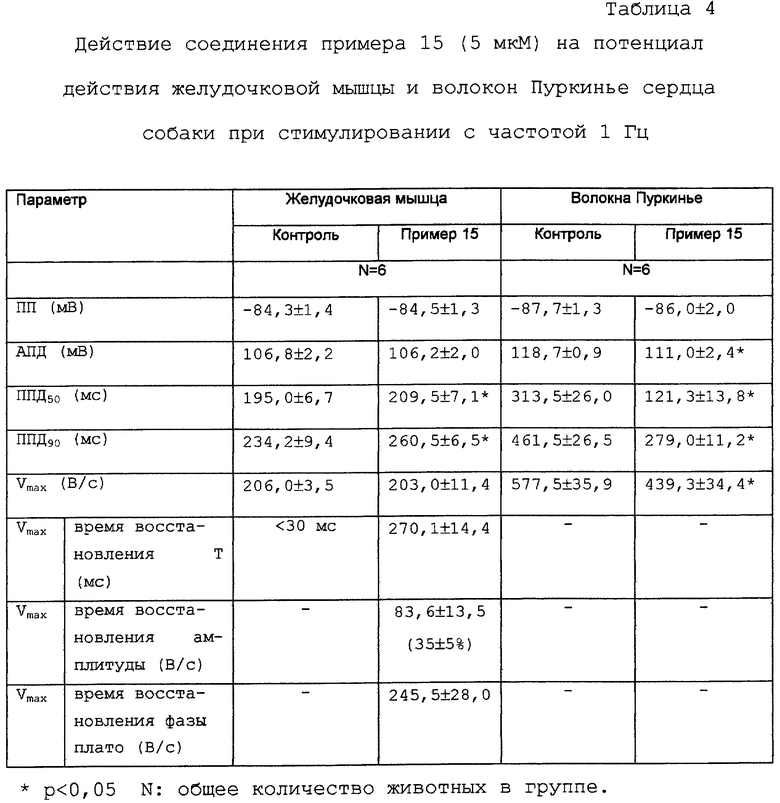
Действие тестируемых соединений данного изобретения на потенциал действия волокон Пуркинье, выделенных из сердца собаки, представлены для соединения примера 15 в таблицах 3 и 4.
Соединение примера 15 (5 мкМ) пролонгирует ППД желудочковой мышцы собаки и одновременно сокращает ППД волокон Пуркинье собаки в концентрациях 2 мкМ и 5 мкМ. Эти результаты являются крайне важными, так как можно ожидать, что соединение не будет повышать, а скорее понижать, неоднородность желудочковой реполяризации (которая является важным проаритмическим фактором).
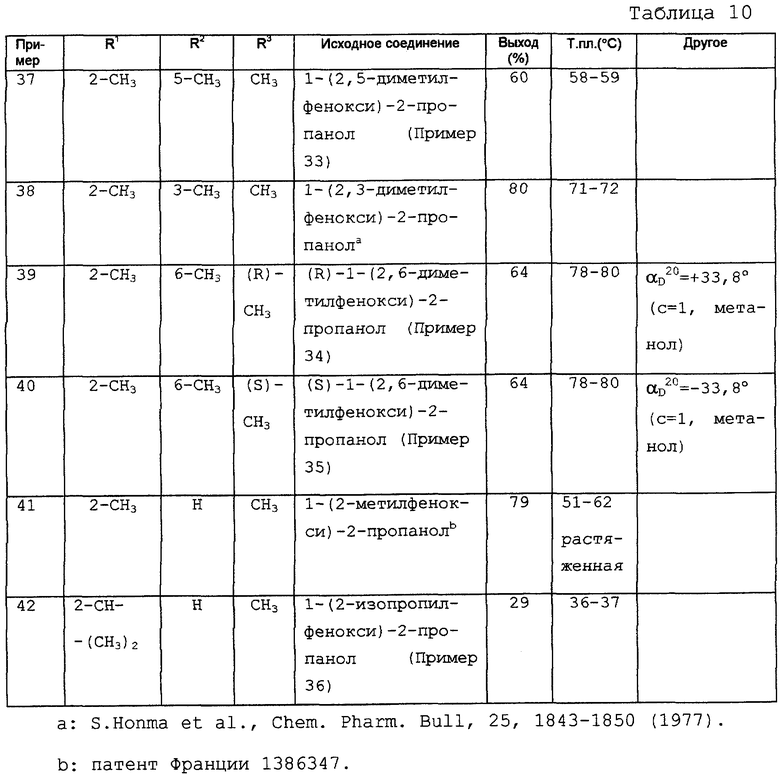
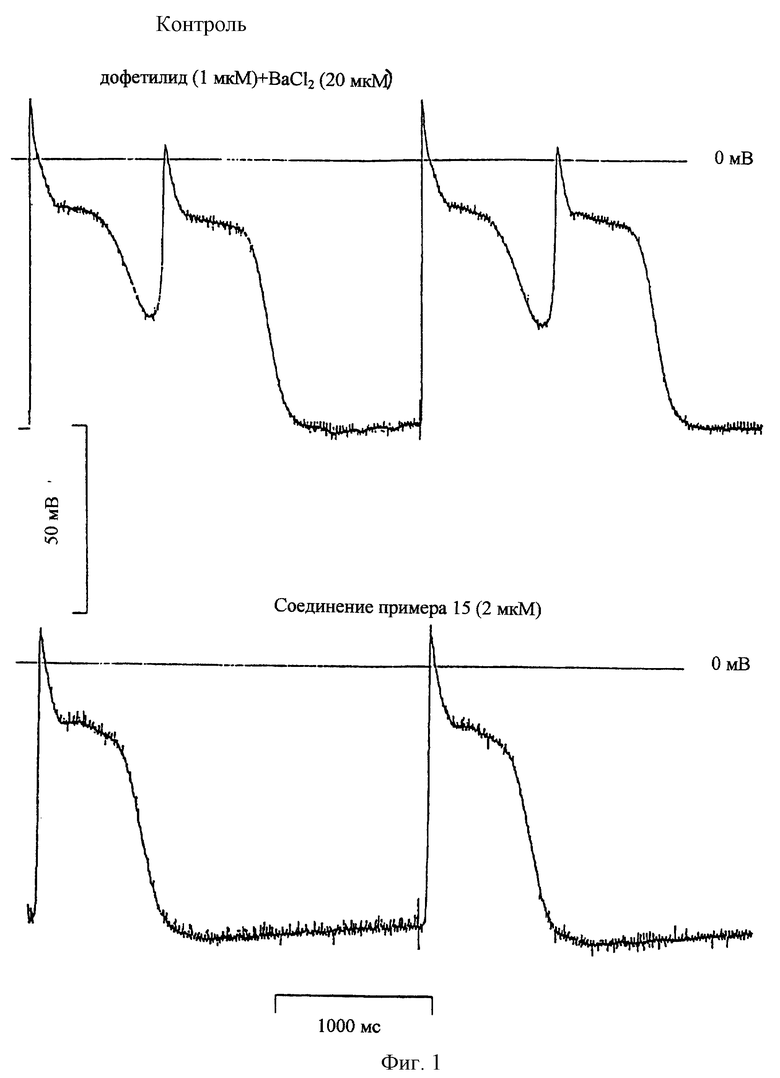
Исследование действия на раннюю постдеполяризацию (фиг. 1)
Раннюю постдеполяризацию (РПД) вызывают на волокнах Пуркинье собаки путем одновременного использования 1 мкМ дофетилида и 20 мкМ ВаСl2. Эти РПД отменяют добавлением соединения (2 мкМ) примера 15. Результат этих экспериментов показывает, что по сравнению с d-соталолом можно ожидать, что соединение примера 15 не будет вызывать тахикардию типа "трепетание-мерцание", т.е. проаритмическое осложнение, связанное с пролонгированием реполяризации.
Измерения сократительной способности in vitro
Используемая методика по существу является методикой, описанной у Virag et al. [Gen. Pharmac. 27, стр.551-556 (1996)].
Папиллярные мышцы сердец кроликов (вес 2-3 кг каждый) получают и помещают в баню для органов (температуре 37oС), содержащую модифицированную питательную среду Tyrode (Тайрода). Препараты стимулируют точечным стимулированием с основной частотой 1 Гц. Сократительную способность измеряют с использованием ауксотонической методики. Тестируемые соединения непосредственно добавляют в баню для органов после разбавления маточного раствора до желаемой конечной концентрации. Действие соединений наблюдают по прошествии инкубационного периода 30-40 минут.
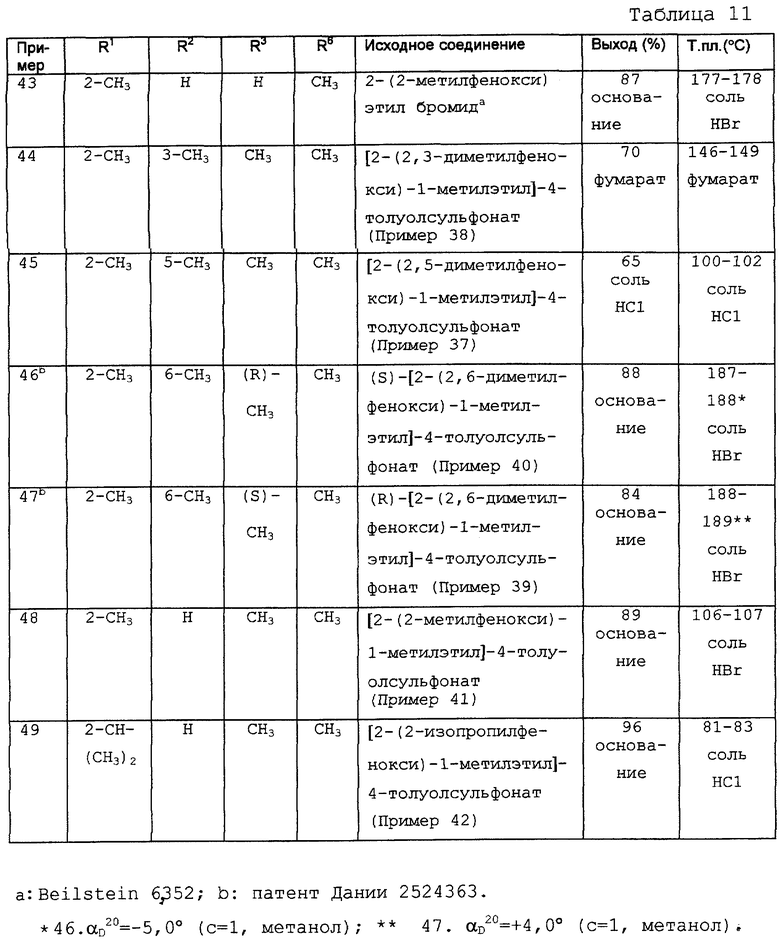
При использовании данной методики может быть получена полезная информация, касающаяся непосредственного действия на сердце исследуемых соединений. Действие соединения примера 15 на сократительную способность проиллюстрировано на правой желудочковой папиллярной мышце при частоте 1 Гц на фиг. 2. Результаты показали, что предполагаемая терапевтическая концентрация примера 15 не вызывает никакого отрицательного инотропного эффекта, потому что сократительная способность папиллярной мышцы не снижается при использовании концентрации ниже 30 мкМ.
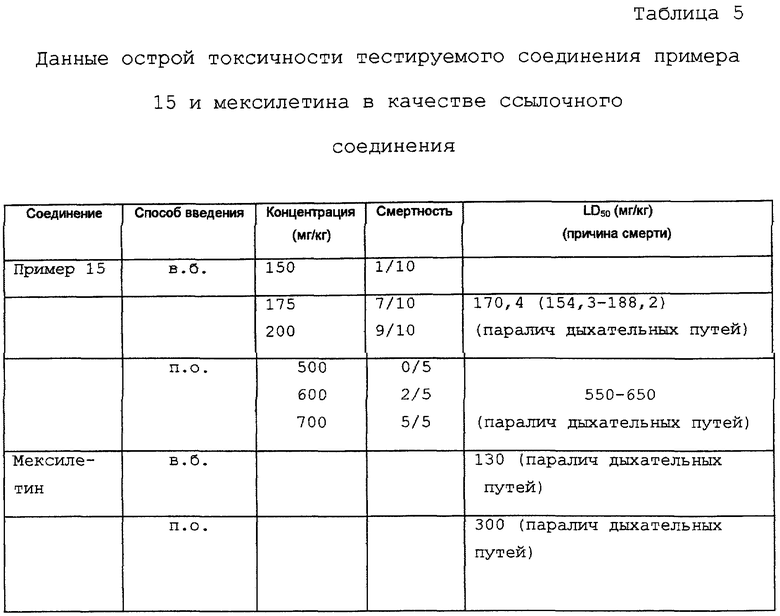
Острая токсичность
Определение значения LD50 на мышах
Животные: самцы мышей CDI (голодные), весящие 20-22 г каждый во время лечения.
Период наблюдения: 2 недели.
Носитель: 1-2% Tween-80 в дистиллированной воде или в физиологическом растворе соответственно.
Дозировка: 0,1 мл/10 г веса тела.
Результаты показаны в таблице 5.
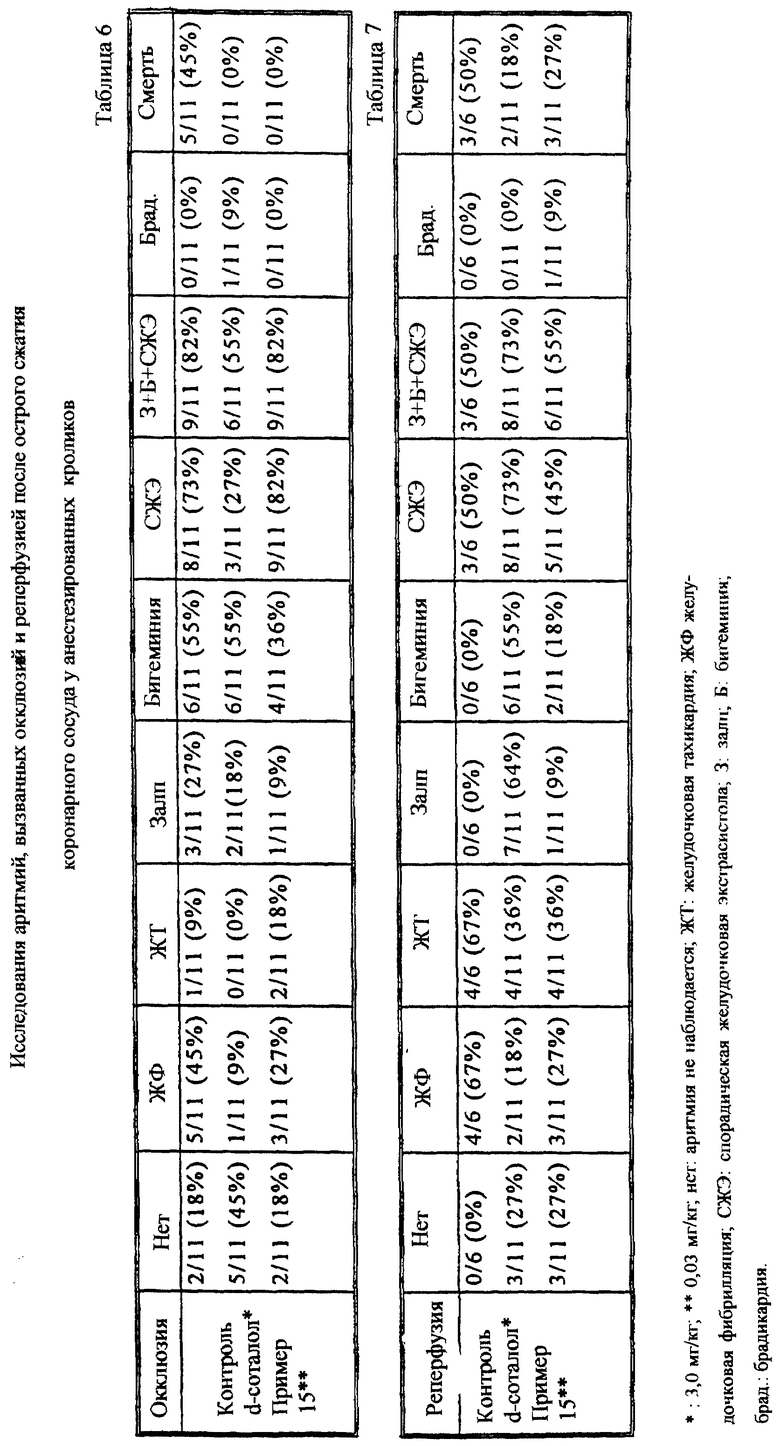
Исследование аритмий, вызванных окклюзией и реперфузией, после острого лигирования коронарной артерии у анестизированных кроликов
Используемая методика по существу является методикой, описанной у Thiemermann et al. [Br. J.Pharmacol.97, стр.401-408 (1989)].
Свободную петлю хирургической нити оборачивают вокруг левой огибающей коронарной артерии сердца после торакотомии анастезированных пентобарбиталом (30 мг/кг в. в.) самцов кроликов (вес 2-3 кг); оба конца нитяной петли выводят из грудной клетки через эластичную трубку. Регистрируют стандартную электрокардиограмму, используя игольчатые электроды под кожей. Кровяное давление животных постоянно измеряют с помощью катетера, расположенного в общей сонной артерии. После стабилизации кровяного давления и частоты сердечных сокращений свободную петлю затягивают с получением местной ишемии миокарда. Через 10 минут ишемии лигатуру высвобождают и вызывают реперфузию в течение 10 минут. Регистрируют степень выживания, частоту аритмий, время наступления аритмии и длину эпизодов. Животные получают физиологический раствор (контроль) внутривенно (2 мл/кг) или 0,03 мг/кг соединения примера 15 или 3 мг/кг дозы d-соталола за 5 минут до окклюзии.
С помощью данной методики может быть установлено, будет ли тестируемое соединение эффективным для защиты от аритмий после лигирования коронарной артерии. Проведенные эксперименты показали (см.таблицы 6 и 7), что соединение примера 15, вводимое в дозе 0,03 мг/кг, обладает сильным антиаритмическим действием на кроликах против реперфузионной аритмии, вызванной лигированием.
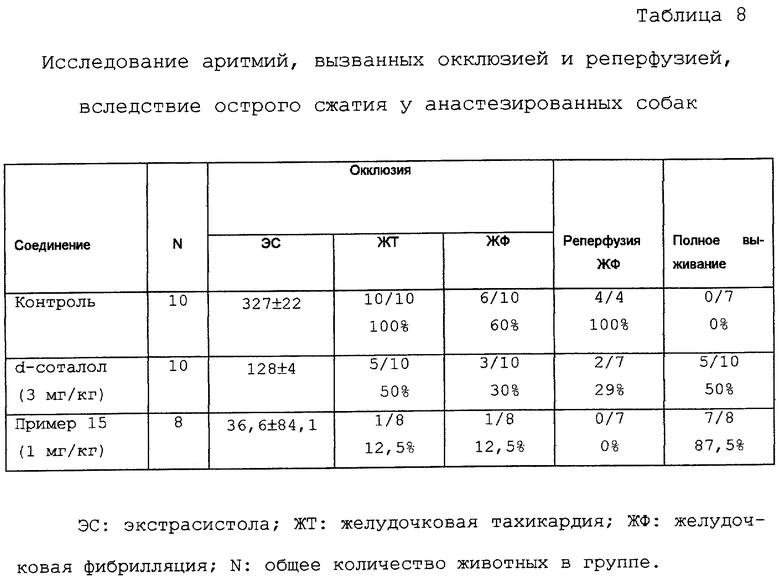
Исследование аритмий, вызванных окклюзией и реперфузией, вследствие острого коронарного лигирования у анестезированных собак
Используемая методика по существу является методикой, описанной у Vegh et al. [Basic Res. Cardiol. 82, 159-171 (1987)].
Дворняг обоих полов, весящих 10-20 кг каждая, анестезируют смесью хлоралозы (60 мг/кг) и уретана (200 мг/кг) и животных поддерживают на искусственном дыхании. После открытия грудной клетки нисходящее переднее ответвление (НПО) левой коронарной артерии сердца получают из первого основного коллатерального ответвления и вокруг него оборачивают свободную петлю из хирургической нити. Во время экспериментов давление крови животных измеряют с помощью канюли, введенной в левую бедренную артерию, и постоянно регистрируют параметры стандартной ЭКГ с помощью игольчатых электродов. Аритмии вызываются окклюзией в течение 25 минут затягивания петли вокруг НПО. Регистрируют степень выживания после летальной аритмии и частоту аритмий. 28 собак, используемых в эксперименте, делят на три группы. Соединение примера 15 вводят в дозе 1 мг/кг 8 животным; d-соталол дают в дозе 3 мг/кг 10 животным одной внутривенной инъекцией за 10 минут до сжатия коронарного сосуда. Контрольные собаки (10 животных) получают 5 мл физиологического раствора по той же методике, что и животные, подвергшиеся лечению.
С помощью данной методики может быть установлено будет ли тестируемое соединение эффективным для защиты от аритмии после лигирования коронарной артерии. Проведенные эксперименты показали (таблица 8), что соединение примера 15, вводимое в дозе 1 мг/кг, в более высокой степени защищает от аритмогенного действия лигирования и реперфузии коронарной артерии, чем d-соталол в дозе 3 мг/кг; наблюдения доказывают значительное антиаритмическое действие данного соединения.
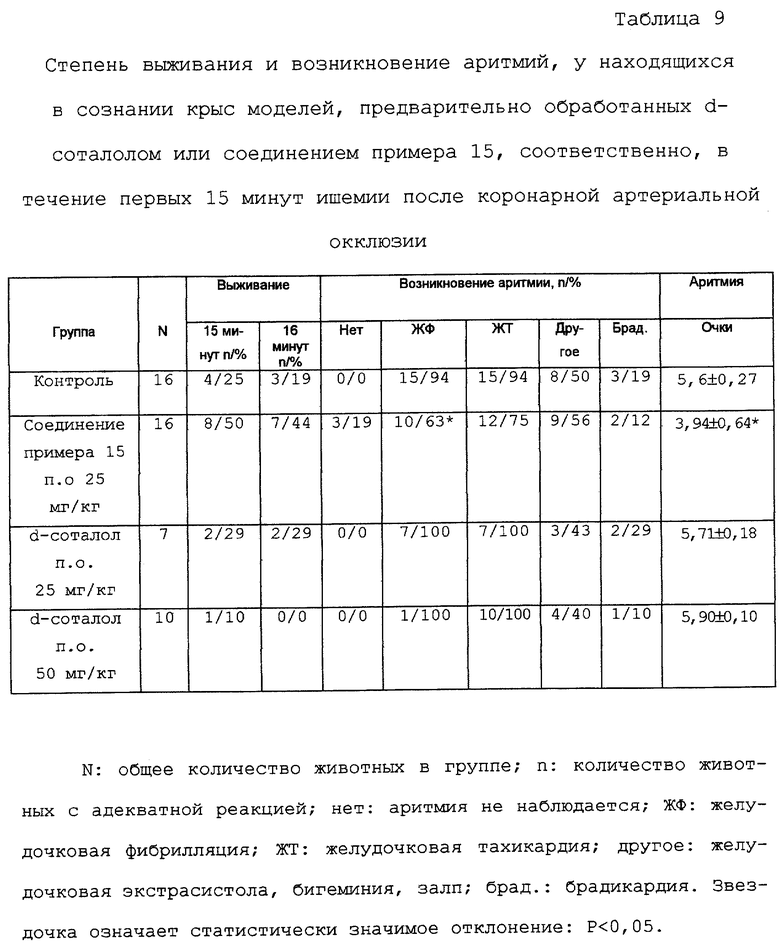
Аритмия, вызванная окклюзией, вследствие острого коронарного артериального лигирования у находящихся в сознании крыс
Используемая методика по существу является методикой, описанной у Lepran et al. [J.Pharmacol. Methods 9, стр.219-230 (1983)].
Во время предварительной операции самцам крыс, весящим 340-360 г каждый, устанавливают свободную петлю из хирургической нити вокруг левого нисходящего коронарного сосуда на примерно 2 мм от его начала. Концы петли выводят из грудной клетки под кожу, затем хирургический разрез закрывают. После полного выздоровления после этой предварительной операции (от 7 до 8 дней) острую ишемию миокарда вызывают сжатием лигатуры петли у находящихся в сознании, свободно передвигающихся животных. Постоянно регистрируют ЭКГ с помощью биполярных нагрудных электродов до лигирования коронарной артерии и в течение первых 15 минут ишемии. Определяют степень выживания, и развившиеся аритмии оценивают по методике Lambeth Convention [Walker et al.: Cardiovasc. Res. 22, 447-455 (1988)]. Для оценки аритмических эпизодов также используют систему очков, оценивая тяжесть эпизодов на основе системы очков от 0 до 6. Как d-соталол, так и соединение примера 15 соответственно суспендируют в 1% метилцеллюлозе и вводят перорально через желудочный зонд за 1 час до сжатия коронарного сосуда. Контрольные животные получают лечение в том же объеме, т.е. 5 мл/кг носителя.
У находящихся в сознании крыс, на модели аритмии после острой коронарной артериальной окклюзии, соединение примера 15 (имеющее смешанный механизм действия) показало антиаритмическое действие даже после перорального введения, в то время как d-соталол, пролонгируя только реполяризацию, доказал отсутствие активности в этих экспериментах (таблица 9). Другим предпочтительным эффектом использования соединения примера 15 является то, что при использовании дозы 25 мг/кг частота сердечных сокращений статистически не изменяется во время первых 15 минут инфаркта миокарда; по сравнению с частотой сердечных сокращений у контрольной группы без лечения, которая уменьшается на примерно 15%.
Соединения данного изобретения могут быть использованы у млекопитающих, включая человека, для ликвидации сердечных аритмий.
В терапевтических целях соединения данного изобретения и их терапевтически приемлемые соли могут быть использованы сами по себе или в виде фармацевтических композиций. Данное изобретение охватывает все такие композиции.
Эти фармацевтические композиции содержат количество соединения формулы (I) или его терапевтически приемлемой соли в качестве активного ингредиента, которое требуется для проявления действия, в смеси с носителями, наполнителями, разбавляющими агентами и/или другими фармацевтическими вспомогательными агентами, обычно используемыми при производстве лекарственных средств.
Подходящими носителями, разбавляющими агентами или наполнителями могут быть, например, вода, спирты, желатин, лактоза, сахароза, крахмал, пектин, стеарат магния, стеариновая кислота, тальк, различные масла животного или растительного происхождения, а также гликоли, например пропиленгликоль или полиэтиленгликоль. Фармацевтическими вспомогательными агентами являются, например, стабилизаторы, антиокислители, различные натуральные или синтетические эмульгаторы, диспергирующие агенты или увлажняющие агенты, красители, ароматизаторы, буферы, дезинтеграторы и другие вещества, способствующие биологической доступности активных агентов.
Фармацевтические композиции данного изобретения могут быть в обычных фармацевтических лекарственных формах. Такими обычными лекарственными формами являются, например, пероральные композиции (вводимые перорально), полученные с использованием фармацевтических вспомогательных агентов; они представляют собой твердые формы, такие как, например, таблетки, капсулы, порошки, пилюли, драже или гранулы, или жидкие композиции, такие как сиропы, эмульсии или суспензии; ректальные композиции, такие как суппозитории (которые могут вводиться ректально); и парентеральные композиции (вводимые вне желудочно-кишечной системы), такие как инъекции или вливания.
Хотя дозы соединений данного изобретения, требуемые для терапевтического действия, зависят от конкретного состояния и возраста пациента и определяются лечащим врачом, для лечения заболеваний, сопровождаемых аритмией, ежедневная пероральная или парентеральная, например внутривенная доза, соединения составляет от примерно 0,1 мг до 5,0 мг, предпочтительно примерно от 0,1 мг до 2,0 мг, рассчитанная на 1 кг веса тела.
Ниже проиллюстрированы соединения данного изобретения и способы их получения в примерах, не ограничивающих объем данного изобретения.
ПРИМЕР 1
N-[2-(2,6-Диметилфенокси)-1-метилэтил] -N-метил-2-(4-нитрофенил)этиламин гидрохлорид
К раствору, содержащему 12,0 г (62 ммоль) N-метил-2-(2,6-диметилфенокси)-1-метилэтиламина (патент BE 626725) в 40 мл изопропанола, добавляют 7,13 г (31 ммоль) 4-нитрофенэтилбромида и реакционную смесь кипятят с обратным холодильником в течение 5 часов. После выпаривания растворителя остаток растирают в порошок с 20 мл этилацетата. Осажденный гидробромид амина фильтруют и промывают этилацетатом. После выпаривания раствора полученный неочищенный продукт (основание) очищают колоночной хроматографией (элюент: дихлорметан/метанол= 9: 0,1). Общий выход (соль НС1) составляет 0,88 г (7,5%) белого кристаллического продукта, т.пл. 135-138oС.
1H-ЯMP (СDС13, очень разбавленный раствор): 1,15 (м, 3Н, С-СН3); 2,25 (с, 6Н, Аr-о-СН3); 2,90-4,20 (м, 10Н, O-CH2-CH-N, N-СН3, N-CH2-CH2-Ar); 6,85-7,10 (м, 3Н, Аr); 7,48 (д, 2Н) и 8,20 (д, 2Н) (Аr); 13,0 (ш. 1Н, NH+).
ПРИМЕР 2
N-[2-(2,6-Диметилфенокси)-N-метил-2-(4-нитрофенил)этиламин гидрохлорид
Целевое соединение получают по методике примера 1 за исключением того, что в качестве исходного материала используют N-метил-2-(2,6-диметилфенокси)этиламин (патент BE 626725), с получением целевого соединения с выходом 30% (соль НС1) белого кристаллического продукта, т.пл. 147-149oС.
1H ЯМР (ДМСО-d6): 2,22 (с, 6Н, Аr-СН3); 2,96 (д, 3Н, N-СН3); 3,20-3,80 (м, 6Н, СН2-СН2-N-СН2-СН2-Аr); 4,05-4,25 (м, 2Н, О-СН2-СН2); 6,80-7,00 (м, 3Н, Аr); 7,55 (д, 2Н) и 8,15 (д, 2Н) (п-нитрофенил); 11,54 (ш. 1Н, NH+).
ПРИМЕР 3
N-Метил-N-[2-(2-метилфенокси)этил-2-(4-нитрофенид)этиламин гидрохлорид
Целевое соединение получают по методике примера 1 за исключением того, что в качестве исходного материала используют N-метил-2-(2-метилфенокси)этиламин (пример 43), с получением 28% (соль НС1) белого кристаллического продукта, т.пл. 137-138oС.
1H ЯМР (ДМСО-d6): 2,12 (с,3Н, Аr-СН3); 2,95 (с,3Н, N-СН3); 3,28 (т, 2Н, CH2-CH2-Ar); 3,35-3,80 (м, 4Н, CH2-N-CH2); 4,42 (т, 2Н, O-CH2-CH2); 6,80-7,20 (м, 4Н, Аr); 7,60 (д, 2Н) и 8,22 (д, 2Н) (п-нитрофенил); 11,25 (ш. 1Н, NH4).
ПРИМЕР 4
(S)-N-[2-(2,6-Диметилфенокси)-1-метилэтил] -N-метил-2-(4-нитрофенил)этиламин
Целевое соединение получают по методике примера 1 за исключением того, что в качестве исходного материала используют (S) -N-метил-2- (2,6-диметилфенокси) -1-метилэтиламин (пример 47), с получением 17% (основание) маслянистого вещества, которое содержит 1% R-энантиомера на основании хирального исследования ВЭЖХ (Chiralcel OJ колонка); Rf=0,7 (этилацетат/метанол=9:1).
ПРИМЕР 5
(R)-N-[2-(2,6-Диметилфенокси)-1-метилэтил] -N-метил-2-(4-нитрофенил)этиламин
Целевое соединение получают по методике примера 1 за исключением того, что в качестве исходного материала используют (R)-N-метил-2-(2,6-диметилфенокси) -1-метилэтиламин (пример 46), с получением 17% (основание) масла, которое не содержит S-энантиомер на основании хирального исследования ВЭЖХ (Chiralcel OJ колонка); Rf=0,7 (этилацетат/метанол=9:1).
ПРИМЕР 6
N-Метил-N-[2-(2-метилфенокси)-1-метилэтил] -2-(4-нитрофенил) этиламин гидрохдорид
Целевое соединение получают по методике примера 1 за исключением того, что в качестве исходного материала используют N-метил-2-(2-метилфенокси)-1-метилэтиламин (пример 48), с получением 14% (соль НС1) белых кристаллов, т.пл. 140-142oС.
1H ЯМР (ДМСО-d6): 1,45 (м, 3Н, С-СН3); 2,10 (с) и 2,20 (с) (всего 3Н, Аr-СН3); 2,88 (м, 3Н, N-СН3); 3,20-3,60 (м, 4Н, N-СН2-СН2-Аr); 3,92 (м, 1Н, CH2-CH-N); 4,35 (м, 2Н, О-CH2-CH); 6,80-7,20 (м, 4Н, Аr); 7,60 (д, 2Н) и 8,22 (д, 2Н) (Аr); 11,20 (шс, 1Н, NH+).
ПРИМЕР 7
N-[2-(2,6-Диметилфенокси)этил]-N-метил-2-(4-аминофенил)этиламин
Раствор, содержащий 0,40 г (1,23 ммоль) N-[2-(2,6-диметилфенокси)этил] -N-метил-2-(4-нитрофенил)этиламина (пример 2) в 7 мл изопропанола добавляют к суспензии 0,1 г катализатора 10% палладия на углероде в 7 мл изопропанола. Суспензию гидрируют при атмосферном давлении. После завершения реакции катализатор отфильтровывают и промывают изопропанолом. Фильтрат выпаривают до постоянного веса при пониженном давлении с получением 0,37 г масляного основания (76%), Rf=0,35 (этилацетат/метанол=9:1).
ПРИМЕР 8
N-Метил-N-[2-(2-метилфенокси)этил]-2-(4-аминофенил)этиламин
Целевое соединение получают по методике примера 7 за исключением того, что в качестве исходного материала используют N-[2-(2-метилфенокси)этил]-N-метил-2-(4-нитрофенил)этиламин (пример 3), с получением 82% основания (масло), Rf=0,35 (этилацетат/метанол=9:1).
ПРИМЕР 9
N-[2-(2,6-Диметилфенокси)-1-метилэтил]-N-метил-2-(4-аминофенил)этиламин
Целевое соединение получают по методике примера 7 за исключением того, что в качестве исходного материала используют N-[2-(2,6-диметилфенокси)-1-метилэтил] -N-метил-2-(4-нитрофенил)этиламин (пример 1), с получением 90% масляного основания.
1H-ЯМР (СDСl3): 1,25 (д, 3Н, С-СН3); 2,32 (с, 6Н, Аr-СН3); 2,48 (с, 3Н, N-СН3); 2,65-2,85 (м, 4Н, N-CH2-CH2-Ar); 3,28 (м, 1Н, CH2-CH-N); 3,2-3,8 (шс, 2Н, NH2); 3,65 (дд, J=9,2 и 6,7 Гц, 1Н) и 3,92 (дд, J=9,2 и 5,7 Гц, 1Н) (O-СН2-СН); 6,66 (д, 2Н, Аr); 6,9-7,10 (м, 5Н, Ar).
ПРИМЕР 10
(S)-N-[2-(2,6-Диметилфенокси)-1-метилэтил] -N-метил-2-(4-аминофенил)этиламин
Целевое соединение получают по методике примера 7 за исключением того, что в качестве исходного материала используют (S)-N-[2-(2,6-диметилфенокси)-1-метилэтил] -N-метил-2-(4-нитрофенил)этиламин (пример 4), с получением 93% белого масла (основание), которое содержит от 1 до 2% R-энантиомера на основании хирального исследования ВЭЖХ (Chiralcel OJ колонка); Rf=0,35 (этилацетат/метанол=9:1).
ПРИМЕР 11
(R)-N-[2-(2,6-Диметилфенокси)-1-метилэтил] -N-метил-2-(4-аминофенил)этиламин
Целевое соединение получают по методике примера 7 за исключением того, что в качестве исходного материала используют (R)-N-[2-(2,6-диметилфенокси)-1-метилэтил] -N-метил-2-(4-нитрофенил)этиламин (пример 5), с получением 89% белого маслянистого основания, которое содержит 1% S-энантиомера на основании хирального исследования ВЭЖХ (Chiralcel OJ колонка); Rf=0,35 (этилацетат/метанол=9:1).
ПРИМЕР 12
N-Метил-N-[2-(2-метилфенокси)-1-метилэтил]-2-(4-аминофенил)этиламин
Целевое соединение получают по методике примера 7 за исключением того, что в качестве исходного материала используют N-метил-N-[2-(2-метилфенокси)-1-метилэтил]-2-(4-нитрофенил)этиламин (пример 6), с получением 89% маслянистого основания.
1Н-ЯМР (СDСl3): 1,16 (д, 3Н, С-СН3); 2,20 (с, 3Н, Аr-СН3); 2,40 (с, 3Н, N-СН3); 2,60-2,80 (м, 4Н, N-CH2-CH2-Ar); 3,30-3,70 (шс, 2Н, NH2); 3,70 (м, 1Н, CH2-CH2-N); 3,82 (дд, 1Н) и 4,02 (дд, 1Н) (O-CH2-CH); 6,60 (д, 2Н) и 7,00 (д, 2Н) (п-аминофенил); 6,72-6,90 (м, 2Н) и 7,08-7,20 (м, 2Н)(Аr).
ПРИМЕР 13
N-[4-[2-[N-Метил-N-[2-(2,6-диметилфенокси)этил] амино] этил]фенил]метансульфонамид гидрохлорид
К раствору, содержащему 1,44 г (4,82 ммоль) N-[2-(2,6-диметилфенокси)этил] -N-метил-2-(4-аминофенил)этиламина (пример 7) в 50 мл дихлорметана добавляют 0,50 г (4,95 ммоль) триэтиламина, затем к раствору, охлажденному до температуры 0oС, по каплям добавляют 0,63 г (5,5 ммоль) метансульфонилхлорида при той же температуре. После дальнейшего перемешивания реакционной смеси при температуре 0oС в течение 1 часа и промывания 30 мл насыщенного раствора бикарбоната натрия водную фазу экстрагируют дважды 20 мл дихлорметана каждый раз. Объединенные органические фазы дважды промывают 30 мл воды каждый раз, сушат и выпаривают с получением 0,44 г (94%) масляного основания. Выход соли НС1 составляет 0,244 г (47%) белых кристаллов, т.пл. 168-169oС.
1H ЯМР (ДМСО-d6): 2,25 (с, 6Н, Аr-СН3); 2,95 (с, 6Н, N-СН3 и S-СН3 наложение); 3,10 (т, 2Н, CH2-CH2-Ar); 3,25-3,75 (м, 4Н, СН2-N-СН2); 4,15 (т, 2Н, O-СН2-СН2); 6,85-7,05 (м, 3Н, Аr); 7,14 (д, 2Н) и 7,24 (д, 2Н) (4-метансульфонамидофенил); 9,75 (с, 1Н, Ar-NH-S); 11,20 (ш. 1Н, NH+).
ПРИМЕР 14
N-[4-[2-[N-Метил-N- [2- (2-метилфенокси) этил]амино] этил]фенил]метансульфонамид гидрохлорид
Целевое соединение получают по методике примера 13 за исключением того, что в качестве исходного материала используют N-метил-N-[2-(2-метилфенокси)этил] -2-(4-аминофенил)этиламин (пример 8), с получением 71% (НС1 соль) белых кристаллов, т.пл. 180-181oС.
1H ЯМР (ДМСО-d6): 2,15 (с,3Н, Аr-СН3); 2,90 (д, 3Н, N-СН3); 2,95 (с, 3Н, S-СН3); 3,10 (т, 2Н, СН2-СН2-Аr); 3,25-3,80 (м, 4Н, CH2-N-CH2); 4,44 (т, 2Н, O-СН2-СН2); 6,80-7,00 (м, 2Н, Аr) и 7,15-7,30 (м, 6Н) (Аr); 9,75 (с, 1Н, Ar-NH-S); 11,20 (ш. 1Н, NH+).
ПРИМЕР 15
N-[4-[2-[N-Метил-N-[2-(2,6-диметилфенокси)-1-метилэтил]амино]этил]фенил] метансульфонамид гидрохлорид
Целевое соединение получают по методике примера 13 за исключением того, что в качестве исходного материала используют N-[2-(2,6-диметилфенокси)-1-метилэтил] -N-метил-2-(4-аминофенил)этиламин (пример 9), с получением 56% (НС1 соль) белых кристаллов, т.пл. 224-227oС.
1H ЯМР (ДМСО-d6, при комнатной температуре): 1,45 (м, 3Н, С-СН3): 2,28 (с, 6Н, Аr-o-СН3); 2,90 (м,3Н, N-СН3); 2,96 (с, 3Н, 3-СН3); 3,00-3,50 (м, 4Н, N-CH2-CH2-Ar); 3,90 (м, 1Н, CH2-CH-N); 3,98-4,16 (м, 2Н, O-CH2-CH); 6,90-7,10 (м, 3Н, Аr); 7,14-7,32 (м, 4Н, Аr); 9,75 (с, 1Н, Ar-NH-S); 10,90 (шс, 1Н, NH+).
ПРИМЕР 16
(S)-N-[4-[2-[N-Метил-N-[2-(2,6-диметилфенокси)-1-метилэтил] амино] этил] фенил]метансульфонамид гидрохлорид
Целевое соединение получают по методике примера 13 за исключением того, что в качестве исходного материала используют (S)-N-[2-(2,6-диметилфенокси)-1-метилэтил] -N-метил-2-(4-аминофенил)этиламин (пример 10), с получением 63% (НС1 соль) белых кристаллов, т.п. 192-193oС.
На основе анализа хиральной ВЭЖХ (Chiralcel OJ колонка) данный продукт не содержит R-энантиомер.
1Н ЯМР (ДМСО-d6, при комнатной температуре): 1,45 (м, 3Н, С-СН3); 2,28 (с, 6Н, Аr-о-СН3); 2,90 (м, 3Н, N-СН3); 2,96 (с, 3Н, S-СН3); 3,00-3,50 (м, 4Н, N-CH2-CH2-Ar); 3,90 (м, 1Н, CH2-CH-N); 3,98-4,16 (м, 2Н, O-СН2-СН); 6,90-7,10 (м, 3Н, Аr); 7,14-7,32 (м, 4Н, Аr); 9,70 (с, 1Н, Ar-NH-S); 10,75 (шс, 1Н, NH+).
1H ЯМР (ДМСО-d6, 100oС): 1,45 (д,3Н, С-СНз); 2,28 (с, 6Н, Ar-o-СН3); 2,88 (с, 3Н, N-СН3); 2,96 (с, 3Н, S-СН3); 3,00-3,50 (м, 4Н, N-CH2-CH2-Ar); 3,85 (м, 1Н, CH2-CH-N); 4,04 (дд, J=10,6 и 5,1 Гц, 1Н) и 4,14 (дд, J=10,6 и 5,6 Гц, 1Н) (O-СН2-СН); 6,90-7,06 (м, 3Н, Аr); 7,16-7,30 (м, 4Н, Аr); 9,35 (с, 1Н, Ar-NH-S); 11,00 (шс, 1Н, NH+).
ПРИМЕР 17
(R)-N-[4-[2-[N-Метил-N-[2-(2,6-диметилфенокси)-1-метилэтил] амино] этил] фенил]метансульфонамид гидрохлорид
Целевое соединение получают по методике примера 13 за исключением того, что в качестве исходного материала используют (R)-N-[2-(2,6-диметилфенокси)-1-метилэтил] -N-метил-2-(4-аминофенил)этиламин (пример 11), с получением 55% (НС1 соль) белых кристаллов, т.пл. 190-191oС.
На основе анализа хиральной ВЭЖХ (Chiralcel OJ колонка) данный продукт не содержит S-энантиомер.
1H ЯМР (ДМСО-d6, при комнатной температуре): 1,45 (м, 3Н, С-СН3); 2,28 (с, 6Н, Аr-о-СН3); 2,90 (м, 3Н, N-СН3); 2,96 (с, 3Н, S-СН3); 3,00-3,50 (м, 4Н, N-CH2-CH2-Ar); 3,90 (м, 1Н, CH2-CH-N); 3,98-4,16 (м, 2Н, O-СН2-СН); 6,90-7,10 (м, 3Н, Аr); 7,14-7,32 (м, 4Н, Аr); 9,70 (с, 1Н, Ar-NH-S); 10,75 (шс, 1Н, NH+).
ПРИМЕР 18
N-[4-[2-[N-Метил-N-[2-(2-метилфенокси)-1-метилэтил] амино] этил] фенил] метансульфонамид гидрохлорид
Целевое соединение получают по методике примера 13 за исключением того, что в качестве исходного материала используют N-метил-N-[2-(2-метилфенокси)-1-метилэтил]-2-(4-аминофенил)этиламин (пример 12), с получением 60% (НС1 соль) белых кристаллов, т.пл. 165-167oС.
1H ЯМР (ДМСО-d6): 1,38-1,52 (м, 3Н, С-СН3); 2,10 (с) и 2,20 (с) (всего 3Н, Аr-СН3); 2,85 (м, 3Н, N-СН3); 2,96 (с, 3Н, S-СН3); 3,05-3,50 (м, 4Н, N-CH2-CH2-Ar); 3,94 (м, 1Н, CH2-CH-N); 4,20-4,40 (м, 2Н, O-CH2-CH); 6,80-7,00 (м, 2Н) и 7,10-7,35 (м, 6Н) (Аr); 9,72 (с, 1Н, Ar-NH-S); 10,70-11,00 (ш.1Н, NH+).
ПРИМЕР 19
N-[4-[2-[N-Метил-N-[2-(2,3-диметилфенокси)-1-метилэтил]амино]этил]фенил] метансульфонамид гидрохлорид
К раствору, содержащему 0,82 г (2 ммоль) N-[2-(2,3-диметилфенокси)-1-метилэтил]-N-метил-2-(4-метансульфонамидофенил)ацетамида (пример 50) в 20 мл абсолютного тетрагидрофурана добавляют 0,10 г литийалюмогидрида (ЛАГ) небольшими частями при перемешивании, затем смесь кипятят примерно в течение одного часа для завершения реакции. Развитие реакции подтверждают тонкослойной хроматографией (ТСХ). После завершения избыточный реагент расщепляют добавлением этилацетата, затем добавляют 10% раствор гидроксида натрия и смесь экстрагируют этилацетатом. Объединенные органические фазы промывают водой, затем экстрагируют 2 н. водной соляной кислотой. Кислую фазу подщелачивают до рН 9 добавлением 10% гидроксида натрия и затем экстрагируют этилацетатом. Экстракт этилацетата промывают водой, сушат и выпаривают при пониженном давлении с получением 0,56 г густого масла в виде остатка. Этот неочищенный продукт очищают, при желании, с помощью хроматографии на колонке с использованием смеси гексана и этилацетата в соотношении 2:1 или 1:1 соответственно. Соль НСl получают с выходом 0,38 г (45%) в виде аморфных белых хлопьев.
1H ЯМР (ДМСО-d6): 1,40-1,50 (м, 3Н, С-СН3); 2,00 (с), 2,08 (с) и 2,22 (с) (всего 6Н, Аr-СН3); 2,80-2,90 (м, 3Н, N-СН3); 2,95 (с, 3Н, 3-СН3); 3,00-3,50 (м, 4Н, N-CH2-CH2-Ar); 3,92 (м, 1Н, CH2-CH-N); 4,25-4,35 (м, 2Н, O-СН2-СН); 6,75-7,30 (м, 7Н, Аr); 9,75 (с, 1Н, Ar-NH-S); 10,8-11,1 (м, 1Н, NH+).
ПРИМЕР 20
N-[4-[2-[N-Метил-N-[2-(2,5-диметилфенокси)-1-метилэтил]амино]этил]фенил] метансульфонамид гидрохлорид
Целевое соединение получают по методике примера 19 за исключением того, что в качестве исходного материала используют N-[2-[2,5-диметилфенокси)-1-метилэтил] -N-метил-2-(4-метансульфонамидофенил)ацетамид (пример 51), с получением 40% НС1 соли в виде аморфного белого осадка.
1H ЯМР (ДМСО-d6): 1,40-1,50 (м, 3Н, С-СН3); 2,00 (с), 2,10 (с), 2,25 (с) и 2,26 (с) (всего 6Н, Аr-СН3); 2,80-2,90 (м, 3Н, N-СН3); 2,95 (с, 3Н, S-СН3); 3,00-3,50 (м, 4Н, N-CH2-CH2-Ar); 3,92 (м, 1Н, CH2-CH2-N); 4,20-4,35 (м, 2Н, O-CH2-СН); 6,60-7,30 (м, 7Н, Аr); 9,75 (с, 1Н, Ar-NH-S); 10,65-11,00 (м, 1Н, NH+).
ПРИМЕР 21
N-[4-[2-[N-Метил-N-[2-(2-хлорфенокси)-1-метилэтил] амино] этил]фенил]метансульфонамид гидрохлорид
Целевое соединение получают по методике примера 19 за исключением того, что в качестве исходного материала используют N-[2-(2-хлорфенокси)-1-метилэтил] -N-метил-2-(4-метансульфонамидофенил)ацетамид (пример 52), с получением 46% фумарата, т.пл. 152-153oС.
1H ЯМР (ДМСО-d6): 1,12 (м, 3Н, С-СН3); 2,40 (с, 3Н, N-СН3); 2,60-2,90 (м, 4Н, N-CH2-CH2-Ar); 2,92 (с, 3Н, S-СН3); 3,24 (м, 1Н, CH2-CH-N); 3,96 (дд, J=10,0 и 6,0 Гц, 1Н) и 4,10 (дд, J=10,0 и 5,6 Гц, 1Н), (O-CH2-CH); 6,60 (с, 2Н, фумаровая кислота); 6,90-7,45 (м, 7Н, Аr); 9,58 (с, 1Н, Ar-NH-S).
ПРИМЕР 22
N-[4-[2-[N-[2-(2,6-Диметилфенокси)-1-метилэтил]амино]этил]фенил]метансульфонамид гидрохлорид
Смесь, содержащую 0,58 г (2,08 ммоль) 2-(2,6-диметилфенокси)-1-метилэтиламина (патент США 3659019) и 0,3 г (1,08 ммоль) N-[4-(2-бромэтил)фенил] метансульфонамида (патент Великобритании 971041) плавят при температуре 120oС в течение 4 часов. Полученную смесь очищают хроматографией на колонке, элюируя смесью дихлорметана и метанола в соотношении 9:0,2. Полученные продукты растирают в порошок с гексаном и фильтруют с получением основания с выходом 64%, белые кристаллы, т.пл. 81-84oС.
1H-ЯМР (СDСl3): 1,14 (д, 3Н, С-СН3); 2,18 (с, 6Н, Аr-СН3); 2,86 (с, 3Н, S-СН3); 2,70-3,20 (м, 5Н, CH2-CH-N-CH2-CH2-Аr); 3,64 (д, 2Н, O-СН2-СН); 4,0-4,6 (ш., 2Н, NH); 6,80-7,00 (м, 3Н, Аr); 7,10-7,25 (м, 4Н, Ar).
Соль НСl получают с выходом 60%, белые кристаллы, т.пл. 210-211oС.
ПРИМЕР 23
N-[4-[2-[N-Метил-N-[3-(2,6-диметилфенокси)пропил] амино] этил] фенил]метансульфонамид гидрохлорид
Целевое соединение получают по методике примера 22 за исключением того, что в качестве исходного материала используют N-метил-3-(2,6-диметилфенокси)пропиламин (патент BE 626725), с получением 59% маслянистого основания.
1H-ЯМР (СDСl3, основание): 1,96 (м, 2Н, CH2-CH2-CH2); 2,26 (с, 6Н, Аr-СН3); 2,35 (с, 3Н, N-СН3); 2,58-2,82 (м, 6Н, СН2-СН2-N-СН2-СН2-Аr); 2,92 (с, 3Н, S-СН3); 3,75 (т, 2Н, O-CH2-CH2); 6,00-6,60 (1Н, NH); 6,82-7,02 (м, 3Н, 2,6-диметилфеноксигруппа); 7,16 (с, 4Н, 4-метансульфонамидофенильная группа).
Соль НСl получают с выходом 49%, белые кристаллы, т.пл. 115-117oС.
ПРИМЕР 24
N-[4-[2-[N-Метил-N-[4-(2,6-диметилфенокси)бутил] амино] этил]фенил]метансульфонамид гидрохлорид
Целевое соединение получают по методике примера 22 за исключением того, что в качестве исходного материала используют N-метил-4-(2,6-диметилфенокси)бутиламин (патент BE 626725), с получением 57% маслянистого основания.
1H-ЯМР (СDСl3, основание): 1,62-1,88 (м, 4Н, CH2-CH2-CH2-CH2); 2,26 (с, 6Н, Аr-СН3); 2,32 (с, 3Н, N-СН3); 2,45-2,85 (м, 6Н, CH2-CH2-N-CH2-CH2-Ar); 2,95 (с, 3Н, S-СН3); 3,75 (т, 2Н, O-CH2-CH2); 5,30-6,30 (1Н, NH); 6,82-7,02 (м, 3Н, 2,6-диметилфеноксигруппа); 7,16 (с, 4Н, 4-метансульфонамидофенильная группа).
Соль НСl получают с выходом 42,0%; только гигроскопические беловатые кристаллы.
ПРИМЕР 25
N-[4-[2-[N-Метил-N-[2-(2,6-диметилфенокси)-1-метилэтил]амино]этил]фенил] бензамид гидрохлорид
К раствору, содержащему 0,2 г (0,064 ммоль) N-[2-(2,6-диметилфенокси)-1-метилэтил] -N-метил-2-(4-аминофенил)этиламина (пример 9) в 1,7 мл ацетонитрила, добавляют 0,11 мл (0,768 ммоль) триэтиламина, затем добавляют 0,082 мл (7,04 ммоль) бензоилхлорида при перемешивании. Реакционную смесь перемешивают при комнатной температуре (охлаждают или нагревают, при необходимости). После завершения реакции реакционную смесь выпаривают при пониженном давлении, к остатку после выпаривания добавляют 10 мл воды и значение рН доводят до щелочного, при необходимости, добавлением раствора гидроксида натрия. Продукт экстрагируют три раза 20 мл этилацетата каждый раз. Органическую фазу промывают 50 мл воды, сушат и выпаривают с получением 0,24 г (90%) масла.
1H-ЯМР (СDСl3): 1,16 (д, 3Н, С-СН3); 2,25 (с, 6Н, Аr-СН3); 2,45 (с,3Н, N-СН3); 2,65-2,85 (м, 4Н, N-CH2-CH2-Ar); 3,26 (сх, 1Н, CH2-CH-N); 3,64 (дд, J= 9,5 и 6,1 Гц, 1Н) и 3,84 (дд, J=9,5 и 5,6 Гц, 1Н) (O-CH2-CH); 6,80-7,00 (м, 3Н, Аr); 7,10 (д, 2Н) и 7,80 (д, 2Н) (п-бензоиламинофенильная группа); 7,25-7,45 (м, 3Н) и 7,56 (д, 2Н) (бензоильная группа); 8,70 (с, 1Н, NH).
Соль НСl получают с выходом 0,19 г (66%), белые кристаллы, т.пл. 108-111oС.
ПРИМЕР 26
N-[4-[2-[N-Метил-N-[2-(2,6-диметилфенокси)-1-метилэтил]амино]этил]фенил] ацетамид полуторный гидрохлорид моногидрат
Целевое соединение получают по методике примера 25 за исключением того, что в качестве ацилирующего агента используют ацетилхлорид с получением 68% соли, белые кристаллы, т.пл. 104-106oС.
Анализ для C22H30N2O2•l,5 НСl•Н2O (молекулярный вес 427,20).
Рассчитано: С 61,85; Н 7,90; N 6,56; Cl 12,44; H2O 4,22%.
Найдено: С 61,79; Н 7,74; N 6,34; Cl 13,96; Н2O 4,70%.
1H ЯМР (ДМСО-d6): 1,40-1,50 (м, 3Н, С-СН3); 2,05 (с, 3Н, Ас); 2,15 (с, 6Н, Аr-СН3); 2,80-2,90 (м, 3Н, N-СН3);
2,90-3,60 (м, 4Н, N-CH2-CH2-Ar); 3,85 (м, CH2-CH-N); 4,05-4,15 (м, 2Н, O-CH2-CH); 6,80-7,05 (м, 3Н, Аr); 7,15-7,25 (м, 2Н) и 7,55-7,65 (м, 2Н) (п-ацетаминофенил); 10,15 (с, 1Н, Ar-NH-CO); 11,22 (ш. 1Н, NH+).
ПРИМЕР 27
1-Метил-3-[4-[2-[N-метил-[2-(2, 6-диметилфенокси] -1-метилэтил] амино] этил]фенил]тиомочевина полуторный гидрохлорид полугидрат
К раствору, содержащему 0,30 г (0,96 ммоль) N-[2-(2,6-диметилфенокси)-1-метилэтил]-N-метил-2-(4-аминофенил)этиламина (пример 9) в 2,5 мл диоксана, добавляют 0,077 г (1,05 ммоль) метилизотиоцианата. Реакционную смесь перемешивают при комнатной температуре (и, при необходимости, нагревают или добавляют еще метилизотиоцианат). После завершения реакции растворитель выпаривают при пониженном давлении. Остаток после выпаривания очищают хроматографией на колонке или растирают в порошок с небольшим количеством н-гексана, декантируют, к маслу добавляют 10 мл воды, значение рН доводят до щелочного добавлением раствора гидроксида натрия (при необходимости), затем продукт экстрагируют три раза 20 мл этилацетата каждый раз. Объединенные органические фазы промывают 50 мл воды, сушат и выпаривают с получением 0,18 г (48%) маслянистого основания.
1H-ЯМР (СDСl3): 1,16 (д, 3Н, С-СН3); 2,26 (с, 6Н, Аr-СН3); 2,42 (с, 3Н, N-СН3); 2,65-2,90 (м, 4Н, N-CH2-CH2-Ar); 3,10 (д, 3Н, NH-СН3); 3,25 (м, 1Н, CH2-CH-N); 3,62 (дд, J= 9,3 и 6,1 Гц, 1Н) и 3,85 (дд, J=9,3 и 5,9 Гц, 1Н) (O-CH2-CH); 6,04 (д, CS-NH-СН3); 6,85-7,00 (м, 3Н, Аr); 7,12 (д, 2Н) и 7,26 (д, 2Н) (п-тиоуреидофенильная группа); 8,00 (с, 1Н, Аr-NH-CS).
Соль получают с выходом 0,136 г (31%), гигроскопические белые кристаллы, т.пл. 120-122oС.
Анализ для C22H31N2OS•1,5 НСl•0,5Н2О (молекулярный вес 449,27).
Рассчитано: С 58,82; Н 7,52; N 9,35%.
Найдено: С 58,87; Н 7,61; N 8,66%.
ПРИМЕР 28
1-Этил-3-[4-[2-[N-метил-N-[2-(2,6-диметилфенокси)-1-метилэтил] амино] этил]фенил]мочевина
Целевое соединение получают по методике примера 27 за исключением того, что этилизоцианат используют в качестве реагента для получения маслянистого основания с выходом 59%.
1H-ЯМР (СDСl3): 1,05 (т, 3Н, СН2-СН3); 1,15 (д, 3Н, СН-СН3); 2,28 (с, 6Н, Аr-СН3); 2,40 (с, 3Н, N-СН3); 2,60-2,80 (м, 4Н, N-CH2-CH2-Ar); 3,08-3,28 (м, 3Н, N-CH2-СН3 и СН2-СН-N); 3,58 (дд, J=9,1 и 6,5 Гц, 1Н) и 3,86 (дд, J= 9,1 и 5,8 Гц, 1Н)(O-CH2-CH); 6,00 (т, 1Н, CO-NH-CH2); 6,82-7,02 (м, 3Н, о-диметилфеноксигруппа); 7,05 (д, 2Н) и 7,22 (д, 2Н) (п-этилуреидофенильная группа); 7,90 (с, 1Н, Ar-NH-CO).
ПРИМЕР 29
N- [2- (2, 6-диметилфенокси) -1-метилэтил] -N-метил-2- [4-(N'-этиламино)фенил]этиламин
Целевое соединение получают по методике примера 19 за исключением того, что в качестве исходного материала используют N-[4-[2-[N-метил-N-[2-(2,6-диметилфенокси)-1-метилэтил ] амино]этил]фенил]ацетамид (пример 26), с получением 60% маслянистого основания.
1H-ЯМР (CDCl3, основание): 1,17 (д, 3Н, СН-СН3); 1,21 (т, 3Н, СН2-СН3); 2,30 (с, 6Н, Аr-СН3); 2,42 (с, 3Н, N-СН3); 2,60-2,80 (м, 4Н, N-CH2-CH2-Ar); 3,11 (кв, 2H, N-CH2-СН3); 3,01 (м, 1Н, CH2-CH-N); 3,59 (дд, J=9,1 и 6,7 Гц, 1Н) и 3,86 (дд, J=9,1 и 5,6 Гц, 1H) (O-CH2-CH); 6,54 (д, 2H) и 7,01 (д, 2H) (п-этиламинофенильная группа); 6,86-7,06 (м, 3Н, о-диметилфеноксигруппа).
ПРИМЕР 30
N-[4-[2-[N-Пропил-N-[4-(2,6-диметилфенокси)этил] амино] этил] фенил] метансульфонамид
Целевое соединение получают по методике примера 22 за исключением того, что в качестве исходного материала используют N-[2-(2,6-диметилфенокси)этил] пропиламин (ЕР 152131), с получением 25% маслянистого основания.
1H-ЯМР (СDСl3, основание): 0,88 (т, 3Н, N-CH2-CH2-СН3); 1,52 (м, 2H, N-CH2-CH2-СН3); 2,27 (с, 6Н, Аr-СН3); 2,60 (т, 2H, N-CH2-CH2-СН3); 2,70-2,90 (м, 4Н, N-CH2-CH2-Ar); 2,95 (с, 3Н, S-СН3); 2,99 (т, 2H, O-CH2-CH2-N); 3,85 (т, 2H, O-CH2-CH2-N); 6,85-7,05 (м, 3Н, 2,6-диметилфеноксигруппа); 7,16 (с, 4Н, п-метансульфонамидофенильная группа).
ПРИМЕР 31
N-[4-[2-[N-Метил-N-[2-(2-изопропилфенокси)-1-метилэтил] этил] фенил] метансульфонамид гидрохлорид
Целевое соединение получают по методике примера 22 за исключением того, что в качестве исходного материала используют N-метил-2-(2-изопропилфенокси)-1-метилэтиламин (пример 49), с получением 52% маслянистого основания.
1H-ЯМР (СDСl3, основание): 1,12-1,22 (м, 9Н, СН-СН3 и Аr-СН-(СН3)2); 2,41 (с, 3Н, N-СН3); 2,70-2,80 (м, 4Н, N-CH2-СН2-Аr); 3,15-3,40 (м, 2Н, CH2-CH-N и Аr-СН-(СН3)2); 3,81 (дд, J=9,3 и 6,4 Гц, 1Н) и 4,00 (дд, J=9,3 и 5,4 Гц, 1Н)(O-СН2-СН); 6,77 (дд, J=8,0 и 1,0 Гц, 1Н); 6,92 (дд, J=7,5 и 1,0 Гц, 1Н); 7,10-7,25 (м, 6Н)(Аr).
Соль НСl получают с выходом 38%, белые кристаллы, т.пл. 86-89oС.
Получение исходных материалов
ПРИМЕР 32
4-(Метансульфонамидо)фенилуксусная кислота
В раствор, содержащий 5,44 г (53 ммоль) карбоната натрия в 36 мл воды, растворяют 3,2 г (20 ммоль) 4-аминофенилуксусной кислоты, добавляют 1,7 мл (2,48 г, 22 ммоль) метансульфонилхлорида одной порцией и смесь нагревают до температуры 85oС в течение 4 часов. После охлаждения и подкисления реакционной смеси до рН 3 концентрированной соляной кислотой смесь охлаждают в холодильнике в течение ночи, промывают водой и сушат с получением 2,7 г неочищенного продукта, который перекристаллизовывают из 6 мл горячей воды с получением 2,40 г целевого соединения в виде бежевых слоистых кристаллов, выход 53%, т.пл. 145-147oС.
1H ЯМР (ДМСО-d6): 2,95 (с, 3Н, S-СН3); 3,48 (с, 2Н, Аr-СН2-СО); 7,10 (д, 2Н) и 7,18 (д, 2Н) (Аr); 9,65 (ш. 1Н, NH); 12,25 (ш. 1Н, СООН).
Получение соединений формулы (V).
ПРИМЕР 33
1-(2,5-Диметилфенокси)-2-пропанол
К раствору, содержащему 3,67 г (30 ммоль) 2,5-диметилфенола в 16 мл 96% этанола, добавляют 2,61 г (45 ммоль) пропиленоксида и 0,44 г (3,2 ммоль) карбоната калия, затем суспензию кипятят при перемешивании в течение 6 часов. После фильтрации реакционной смеси и промывания ее этанолом растворитель выпаривают при пониженном давлении. Неочищенный продукт (полученный с выходом 100%) очищают дистилляцией при пониженном давлении, при необходимости. Выход составляет 85% бесцветной жидкости, температура кипения 104-107oС/0,26 кПа, Rf=0,1 (толуол).
ПРИМЕР 34
(R)-1-(2,6-диметилфенокси)-2-пропанол
Целевое соединение получают по методике примера 33 за исключением того, что в качестве исходных материалов используют 2,6-диметилфенол и R(+)-пропиленоксид с получением неочищенного продукта с выходом 100%. На основе анализа хиральной ВЭЖХ (Chiralcel OJ колонка) данный продукт имеет химическую чистоту 74,4% и не содержит S-энантиомера.
ПРИМЕР 35
(S)-1-(2,6-диметилфенокси)-2-пропанол
Целевое соединение получают по методике примера 33 за исключением того, что в качестве исходных материалов используют 2,6-диметилфенол и S-(-)-пропиленоксид с получением неочищенного продукта с выходом 100%. На основе анализа хиральной ВЭЖХ (Chiralcel OJ колонка) данный продукт имеет химическую чистоту 78% и не содержит R-энантиомера.
ПРИМЕР 36
1-(2-Изопропилфенокси)-2-пропанол
Целевое соединение получают по методике примера 33 за исключением того, что в качестве исходных материалов используют 2-изопропилфенол и пропиленоксид с получением неочищенного продукта с выходом 100%, Rf=0,l (толуол).
Получение соединений формулы (VI)
Общая методика
К раствору, содержащему 30 ммоль соединения формулы (V) в 10 мл пиридина, добавляют 7,63 г (40 ммоль) 4-толуол-сульфонилхлорида при комнатной температуре при перемешивании. После перемешивания в течение 3 часов реакционную смесь вливают в 55 мл воды. Продукт экстрагируют дважды 40 мл этилацетата каждый раз, органическую фазу промывают дважды 25 мл 2 н. соляной кислоты каждый раз, дважды 20 мл воды каждый раз, дважды 20 мл 1 н. раствором карбоната натрия каждый раз и, наконец, три раза 20 мл воды каждый раз, и после выпаривания растворителя, при необходимости, остаток кристаллизуют из смеси гексана и диизопропилового эфира.
По описанной выше методике получают следующие соединения формулы (IV)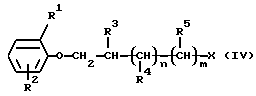
где R1, R2 и R3 такие, как определено в таблице 10, n и m равны 0, и Х является 4-толуолсульфонилокси группой.
Получение соединений формулы (II)
Общая методика
После взаимодействия 30 ммоль соответствующим образом замещенных соединений формулы (IV) (где Х является галогеном или 4-толуолсульфонилоксигруппой) с 60 мл 33% этанольного метиламина или замещенного амина при температуре 100oС в закрытой пробирке в течение 5 часов реакционную смесь выпаривают. Полученное масло помещают в 40 мл 2 н. соляной кислоты и промывают 10 мл этилацетата. Уровень рН водной фазы доводят до 10 добавлением 5 н. раствора гидроксида натрия, затем смесь экстрагируют три раза 50 мл дихлорметана каждый раз. Органическую фазу промывают дважды 40 мл воды каждый раз, сушат, фильтруют, затем раствор выпаривают. Полученное масло превращают в соль или очищают дистилляцией при пониженном давлении или используют далее в неочищенной форме.
По данной общей методике получают соединения формулы (III), где R1, R2, R3 и R6 такие, как определено в таблице 11, и n и m равны 0.
Получение соединений формулы (III)
ПРИМЕР 50
N-[2-(2,3-Диметилфенокси)-1-метилэтил] -N-метил-2-(4'- метансульфонамидофенил)ацетамид
К раствору, содержащему 0,80 г (3,5 ммоль) 4-метансульфонамидофенилуксусной кислоты (пример 32) в 20 мл N,N-диметилформамида (ДМФ), добавляют 0,46 мл (0,42 г, 4,2 ммоль) 4-метилморфолина, смесь охлаждают до температуры -10oС и добавляют 0,65 мл (0,70 г, 5 ммоль) изобутилхлорформиата. Через 10 минут добавляют раствор, содержащий 0,68 г (3,5 ммоль) N-метил-2-(2,3-диметилфенокси)-1-метилэтиламина (пример 44) в 3 мл ДМФ, предварительно охлажденный до температуры -10oС, затем значение рН смеси доводят до 8,5 добавлением триэтиламина. Реакционную смесь перемешивают при температуре -10oС в течение 1 часа, затем при температуре 0oС в течение 1 часа. Затем растворитель выпаривают при пониженном давлении и твердый остаток подщелачивают добавлением водного раствора аммиака и определяют между водой и этилацетатом. Водную фазу промывают дважды этилацетатом, объединенные органические фазы промывают 1 раз водой, сушат над сульфатом натрия и выпаривают. Таким образом, получают 1,13 г целевого соединения в виде аморфного вещества, которое подходит для следующей стадии без дальнейшей очистки. Выход 80%.
1H-ЯМР (СDСl3): 1,18-1,38 (м, 3Н, С-СН3); 2,10-2,20 (м, 3Н) и 2,25-2,35 (м, 3Н) (Аr-СН3); 2,85-3,05 (м, 6Н, N-СН3 и S-СН3); 3,72-4,05 (м, 4Н, O-СН2-СН и CO-CH2-Ar); 4,45 (м) и 5,15 (м) (всего 1Н, CH2-CH-N); 6,70 (дд, 1Н), 6,83 (т, 1Н) и 7,05 (дд, 1Н) (2,3-диметилфеноксигруппа); 7,10-7,22 (м, 4Н, 4-метансульфонамидофенильная группа); 7,50 (с, 1Н, Ar-NH-S).
ПРИМЕР 51
N-[2-(2,5-Диметилфенокси)-1-метилэтил] -N-метил-2-(4-метансульфонамидофенил)ацетамид
К раствору, содержащему 0,52 г (2,7 ммоль) N-метил-2-(2,5-диметилфенокси)-1-метилэтиламина (пример 45) в 15 мл тетрагидрофурана, добавляют 0,63 г (2,7 ммоль) 4-метансульфонамидофенилуксусной кислоты (пример 32) и 0,63 г (3,05 ммоль) дициклогексилкарбодиимида и реакционную смесь перемешивают при комнатной температуре в течение 16 часов. После фильтрации смеси фильтрат выпаривают при пониженном давлении и получают около 4,5 г бледно-желтоватой смолы, которую очищают хроматографией на колонке с использованием смеси этилацетата и гексана в качестве элюента. Таким образом, получают 0,76 г (70%) целевого соединения в виде аморфного материала.
1H-ЯМР (СDСl3): 1,18-1,36 (м, 3Н, С-СН3); 2,10-2,18 (м, 3Н) и 2,25-2,35 (м, 3Н)(Аr-СН3); 2,84-3,04 (м, 6Н, N-СН3 и S-СН3); 3,66 (с) и 3,80 (с) (всего 2Н, СО-СН2-Аr); 3,85-4,05 (м, 2Н, O-СН2-СН); 4,44 (м) и 5,10 (м) (всего 1Н, CH2-CH-N); 6,56-6,62 (м, 1Н), 6,62-6,74 (м, 1Н) и 6,96-7,04 (м, 1Н) (2,5-диметилфеноксигруппа); 7,05-7,20 (м, 4Н, 4-метансульфонамидофенильная группа); 7,50-7,80 (м, 1Н, Ar-NH-S).
ПРИМЕР 52
N-[2-(2-Хлорфенокси)-1-метилэтил] -N-метил-2-(4-метансульфонамидофенил)ацетамид
Целевое соединение получают по методике примера 50 за исключением того, что в качестве исходного соединения используют N-метил-2-(2-хлорфенокси)-1-метилэтиламин (Fr. M. 5912) с получением 68% выхода целевого соединения в виде аморфного материала.
1H-ЯМР (СDСl3): 1,25-1,40 (м, 3Н, С-СН3); 2,90-3,00 (м) и 3,12 (с) (всего 6Н, N-СН3 и S-СН3); 3,70 (с) и 3,85-4,20 (м) (всего 4Н, O-СН2-СН и СО-СН2-Аr); 4,52 (м) и 5,08 (м) (всего 1Н, СН2-CH-N); 6,85-7,40 (м, 8Н, Аr); 7,45-7,60 (м, 1Н, Ar-NH-S).
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ, СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2244709C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1997 |
|
RU2193029C2 |
| СОЕДИНЕНИЯ С ГИДРОКСИКАРБОНИЛЬНЫМИ-ГАЛОГЕНАЛКИЛЬНЫМИ БОКОВЫМИ ЦЕПЯМИ | 2000 |
|
RU2247106C2 |
| СИНТЕЗ НОВОГО КЛАССА ФТОРСОДЕРЖАЩИХ ЖИДКОКРИСТАЛЛИЧЕСКИХ СОЕДИНЕНИЙ С ИСПОЛЬЗОВАНИЕМ ХЛАДОНА 114В2 В КАЧЕСТВЕ ИСХОДНОГО СОЕДИНЕНИЯ | 2012 |
|
RU2505529C1 |
| 5-АМИНО-3-(2-АМИНОПРОПИЛ)-[1,2,4]ТИАДИАЗОЛЫ | 2011 |
|
RU2449997C1 |
| ПРОИЗВОДНЫЕ 1-(4-ИЗОКСАЗОЛ-5-ИЛ)-1Н-ПИРАЗОЛ-1-ИЛ)-2-МЕТИЛПРОПАН-2-ОЛА И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИРОРОВ ИЛ-17 И ИФН-ГАММА ДЛЯ ЛЕЧЕНИЯ АУТОИММУННЫХ ЗАБОЛЕВАНИЙ И ХРОНИЧЕСКОГО ВОСПАЛЕНИЯ | 2018 |
|
RU2785342C2 |
| ЗАМЕЩЕННЫЕ ГИДРОКСАМОВЫЕ КИСЛОТЫ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ И СПОСОБ СНИЖЕНИЯ УРОВНЕЙ TNFα | 1998 |
|
RU2199530C2 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА, СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2179554C2 |
| СПОСОБ ПОЛУЧЕНИЯ (1S,4R)-1-ИЗОПРОПИЛ-4-МЕТИЛ-10-АРИЛ-7,8,12,13-ТЕТРАОКСА-10-АЗАСПИРО[5.7]ТРИДЕКАНОВ | 2019 |
|
RU2726405C1 |
| ЛИГАНДЫ, ТРОПНЫЕ К ПРОСТАТИЧЕСКОМУ СПЕЦИФИЧЕСКОМУ МЕМБРАННОМУ АНТИГЕНУ И ИХ ПРИМЕНЕНИЕ ДЛЯ ПОЛУЧЕНИЯ ДВОЙНЫХ КОНЪЮГАТОВ С ТЕРАПЕВТИЧЕСКИМИ АГЕНТАМИ НА ИХ ОСНОВЕ ДЛЯ КОМБИНИРОВАННОЙ ТЕРАПИИ ПСМА ЭКСПРЕССИРУЮЩИХ ОПУХОЛЕЙ | 2023 |
|
RU2841078C1 |
В изобретении описываются соединения формулы (I), а также их стереоизомеры, кислотно-аддитивные соли и гидраты, где заместители имеют указанные в описании значения. Описывается также получение фармацевтической композиции, обладающей антиаритмической активностью. Далее приводится способ лечения сердечной аритмии у млекопитающих, включающий введение антиаритмического количества соединения формулы (I). 3 с. и 3 з.п. ф-лы, 11 табл., 2 ил.
где R1 и R2 независимо друг от друга являются водородом, галогеном или С1-4 алкильной группой;
R3, R4 и R5 независимо друг от друга являются водородом или С1-4 алкильной группой;
R6 является водородом, С1-4 алкильной группой;
R7 является нитрогруппой или аминогруппой, необязательно монозамещенной С1-4 алкильной, бензоильной, С1-4 алкилкарбонильной, С1-4 алкилсульфонильной, С1-4 алкилкарбамоильной или С1-4 алкилтиокарбамоильной группой;
n и m оба равны 0 или 1,
при условии, что R2 отличен от водорода, если R1 является водородом,
а также их стереоизомеры, кислотно-аддитивные соли и гидраты.
N-[4-[2-[N-метил-N-[2-(2,6-диметилфенокси)-1-метилэтил] амино] этил] фенил] метансульфонамида,
(S)-N-[4-[2-[N-метил-N-[2-(2,6-диметилфенокси)-1-метилэтил] амино] этил] фенил] метансульфонамида,
(R)-N-[4-[2-[N-метил-N-[2-(2,6-диметилфенокси)-1-метилэтил] амино] этил] фенил] метансульфонамида,
N-[4-[2-[N-метил-N-[2-(2-метилфенокси)этил] амино] этил] фенил] метансульфонамида,
N-[4-[2-[N-метил-N-[2-(2,6-диметилфенокси)-1-метилэтил] амино] этил] фенил] ацетамида,
N-[4-[2-[N-[2-(2,6-диметилфенокси)-1-метилэтил] амино] этил] фенил] метансульфонамида,
1-метил-3-[4-[2-[N-метил-N-[2-(2,6-диметилфенокси)-1-метилэтил] амино] этил] фенил] тиомочевины,
2-[4-аминофенил] -N-метил-N-[2-(2,6-диметилфенокси)-1-метилэтил] этиламина
и кислотно-аддитивных солей этих соединений.
| JP 63264466 А, 01.11.1988 | |||
| ПРОИЗВОДНЫЕ 1-АМИНО-2-ЗАМЕЩЕННОГО ФЕНИЛЭТАНОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 1992 |
|
RU2095344C1 |
| JP 05059011 А, 09.03.1993 | |||
| Drugs, Exp | |||
| Clin | |||
| Res., 1995, 21(4), 145-151 | |||
| Patol | |||
| Fiziol | |||
| Eksp., Ter., 1994, jul | |||
| - sep | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |

