Изобретение касается определенных инактиваторов фермента, которые применяются в медицине, преимущественно - в химиотерапии рака, особенно в сочетании с антиметаболическими антинеопластическими агентами, такими как 5-фторурацил (5-FU) (5-ФУ).
5-Фторурацил использовался в химиотерапии рака с 1957 г. К чувствительным к химиотерапии опухолям относятся: рак грудной железы, желудочно-кишечные злокачественные опухоли, рак головы и шеи; 5-фторурацил также применяют для повышения чувствительности к радиации. 5-Фторурацил быстро включается в метаболизм в печени (время полураспада - примерно от 8 до 20 минут) при участии фермента дигидропиримидин-дегидрогеназы (урацил-редуктазы). Сообщалось (Cancer Research, 46, 1094, 1986), что 5-/2-бромвинил/-урацил (БВУ) является ингибитором дигидротимидиндегидрогеназы, которая и тормозит метаболизм 5-фторурацила и увеличивает его антиопухолевую активность. Сообщалось также, что 5-/2-бpoмвинил/-2'-дезоксиуридин (который in vivo превращается в БВУ) увеличивает антиопухолевую активность 5-фторурацила и 5-дезокси-5-фторуридина - предшественника 5-фторурацила (Biochemical Pharmacology 38, 2885, 1989).
К сожалению, БВУ токсичен для людей.
Сейчас обнаружено, что ряд производных 5-замещенного урацила является инактиваторами урацил-редуктазы; они увеличивают уровень и время полураспада 5-фторурацила в плазме и повышают активность 5-фторурацила. Они также уменьшают обычно наблюдаемые колебания уровня 5-фторурацила в плазме разных пациентов.
Соответственно - первый аспект - в данной заявке предлагается инактиватор урацил-редуктазы, представляющий собой 5-замещенное или 5,6-дигидро-5-замещенное производное урацила, в котором 5-заместитель - это бром-, иод-, циан-, галоидозамещенный С1-4алкил, С2-6алкенил, I-гало С2-6алкенильная группа, С2-6алкенильная группа, галоидзамещенная С2-6алкинильная группа или их предшественники для применения в медицине, особенно в химиотерапии рака. Ингибитор урацил-редуктазы, вообще, будет применяться вместе с 5-фторурацилом и его пролекарством.
Под С2-6 алкинильной группой подразумевается алкинильная группа с прямой или разветвленной цепью, последняя включает алкинильную группу, замещенную циклоалкильной группой, содержащей, в целом, от 2 до 6 углеродных атомов.
В качестве галоидзаместителя в алкенильной или алкинильной группе предпочтительны бром, хлор или иод. Особенно предпочтительны галоидзамещенные винильная или этинильная группы. Обычно будет использоваться только один галоидзаместитель.
Следующим аспектом настоящего изобретения являются указанные выше производные урацила для производства лечебных препаратов, использующихся в химиотерапии рака. Лечебные препараты могут также применяться для защиты от токсичности 5-фторурацила, и, совместно с 5-фторурацилом или его предшественником, - для лечения псориаза, ревматоидного артрита или папилломной вирусной инфекции человека.
Следующий аспект - в данной заявке предлагается метод лечения или профилактики опухолей, включающий назначение эффективных количеств производных урацила, указанных выше, при лечении опухолей млекопитающих, в том числе и человека. Предпочтительно лечение в сочетании с 5-фторурацилом или его предшественником.
Еще один аспект - в настоящей заявке предлагается сочетание указанных выше производных урацила или его пролекарств с 5-фторурацилом или его пролекарством.
Лучшими являются производные урацила, в которых 5-заместитель - С2-6 алкинильная группа (может быть галоидзамещенной), пригодными - С2-4 алкинильные группы и предпочтительными - этинильные или пропинильные группы. В предпочтительных 1-галоалкенильных и алкинильных производных многократная связь находится в 1-й позиции. Особенно хорошими инактиваторами урацил-редуктазы для применения в соответствии с заявкой являются 5-этинилурацил и 5-пропинилурацил. Для такого применения используются также следующие инактиваторы:
5-цианоурацил
5-бромэтинилурацил
5-(1-хлорвинил)урацил
5-иодурацил
5-гекс-1-унилурацил
5-винилурацил
5-трифторметилурацил
5-бромурацил
Производные урацила, в которых 5-заместители - замещенные или незамещенные С3-6 алкильные группы, являются новыми соединениями, и это - следующий аспект данного изобретения.
Указанные выше пролекарства производных урацила - это соединения, из которых в процессе метаболизма in vivo получаются производные урацила. Эти предшественники могут иметь или не иметь собственную активность, но, обычно, они имеют низкую активность. Такие предшественники состоят из аналогов нуклеозидов, содержащих нуклеооснование, соответствующее указанным выше 5-замещенным соединениям урацила, например, нуклеозидные производные, содержащие рибозу, 2'-дезоксирибозу, 2',3'-дидезоксирибозун арабинозу или другой остаток сахара, который дополнительно может содержать 2'- или 3'-заместитель, такой как галоид - хлор или фтор; алкокси-, амино- или тиогруппы. Характерными примерами таких производных нуклеозидов являются 1-/в-D-арабинофуранозил/-5-проп-1-унилурацил и 2',3'-дидезокси-5-этинил-3'-фторуридин. Вообще, могут применяться и описанные далее соединения-аналоги пролекарств 5-ФУ. Под производными урацила (или инактиваторами урацилредуктазы) здесь подразумеваются и его предшественники.
Пролекарства 5-фторурацила (5-ФУ) - это соединения; которые в процессе метаболизма in vivo превращаются в 5-фторурацил, к ним относятся: 5-фторуридин, 5-фтор-2-дезоксиуридин, 5-фтор-2-дезоксицитидин, 5'-дезокси-4', 5-фторуридин, 5'-дезокси-5-фторуридин, 1-/2-тетрагидрофуранил/-5-фторурацил и 1-С1-8 алкилкарбомоил-5-фторурацил производные.
5-ФУ, его предшественник и описанные производные 5-урацила согласно данной заявке могут применяться в сочетании при введении комбинации компонентов либо совместно, например, в едином фармакологическом препарате, или же, лучше, отдельно, или последовательно в течение определенного периода времени, посредством чего и достигается желаемое терапевтическое действие комбинации. Лучше сначала вводить производное 5-урацила, а затем - 5-ФУ или его пролекарство, целесообразно вводить в период от 15 минут до 4-х дней, обычно - в период от 1 до 15 часов, главным образом - в период от 1 до 2-х часов со времени введения 1-го препарата.
5-ФУ или его пролекарство и производное 5-урацила в терапевтических целях могут вводиться любым соответствующим способом, включая введение через рот, прямую кишку, нос и местное (в том числе - защечное и подъязычное), введение через влагалище и парентеральное (включая подкожное, внутримышечное, внутривенное и внутрикожное). Большим преимуществом является то, что наилучший способ введения можно выбирать в зависимости от состояния и возраста пациента, природы инфекционного заболевания и других клинических факторов.
До настоящего времени было невозможно вводить 5-ФУ через рот, так как он разрушался урацил-редуктазой в желудочно-кишечном тракте. Однако, сейчас показано, что если 5-замещенное производное урацила (описанное ранее) вводится перед введением 5-ФУ (или его предшественника) через рот, в плазме наблюдается высокий и постоянный уровень 5-ФУ; это свидетельствует о том, что данное соединение не разрушается. Это является еще одним преимуществом настоящего изобретения. Предпочтительно вводить 5-ФУ в период от 15 минут до 4-х дней, обычно - в период от 1 до 15 часов, главным образом - в период от 1 до 2-х часов со времени введения 5-урацил производного.
Обычно в плазме пациентов наблюдается высокий уровень изменчивости концентрации 5-ФУ в результате приема определенной дозы 5-ФУ, этот уровень может быть обусловлен скоростями вывода 5-ФУ, различающимися у разных пациентов. Кроме того, у каждого пациента могут наблюдаться суточные колебания. Показано, что применение 5-замещенного производного урацила согласно данной заявке значительно снижает такую изменчивость у разных пациентов (см. Эксперимент 3).
Вообще, требуемые дозы 5-ФУ или его пролекарства находятся в пределах от 0,1 до 1000 мг на килограмм веса пациента в день, предпочтительно - от 0,1 до 200 мг на килограмм веса в день. Хотя при введении самого 5-ФУ предпочтительны дозы в пределах от 0,1 до 50 мг на килограмм веса пациента в день, предшественники 5-ФУ могут применяться в более высоких дозах. Дозы 5-ФУ или его предшественника могут вводиться в виде разовой дозы, например, содержащей от 5 до 3000 мг, лучше - от 20 до 1000 мг активного компонента на разовую дозу.
Проведенные с 5-ФУ эксперименты дают возможность полагать, что необходимо вводить такую дозу, при которой пик концентрации активного соединения в плазме составит примерно от 0,01 до 1,5 мкг/мл.
Производные 5-урацила можно вводить в дозах в пределах от 0,01 до 50 мг на килограмм веса пациента в день, лучше - от 0,01 до 10 мг/кг. В зависимости от применяемого производного предпочтительна доза в пределах от 0,01 до 0,4 мг на килограмм веса тела пациента в день. Иной предпочтительный режим введения - раз в неделю от 0,5 до 10 мг/кг.
Лучше, если необходимая доза изготовлена как одна, две или более меньших доз, вводимых через определенные интервалы в течение дня. Такие небольшие дозы могут вводиться в виде разовой дозы, например, содержащей от 1 до 200 мг, желательно от 2 до 100 мг, предпочтительно от 2 до 50 мг производного 5-урацила.
Инактиваторы урацил-редуктазы в 5-ФУ обычно применяются в соответствующем соотношении для того, чтобы значительно снизить имеющийся у пациента уровень урацил-редуктазы. Такое соотношение, основанное на отношении весов инактиватора урацил-редуктазы и 5-ФУ, обычно берется в пределах от 1:0,01 до 1-100, предпочтительно в пределах от 10:0,1 до 1:50, еще лучше -в пределах от 1:1 до 1:10.
5-ФУ или его пролекарства и производное 5-урацила желательно вводить в виде фармацевтических препаратов - либо в виде единичного фармацевтического препарата, содержащего оба компонента, либо - по отдельности, когда каждый препарат содержит один из компонентов данной комбинации. Производное 5-урацила усиливает действие 5-ФУ, что дает возможность применять низкие дозы 5-ФУ.
Таким образом, данное изобретение включает также фармацевтические препараты, в состав которых входит производное 5-урацила, описанное выше, в качестве необязательного - в сочетании с 5-ФУ или его производным, и, по крайней мере, один фармацевтически приемлемый носитель или инертный наполнитель.
Каждый носитель должен быть фармацевтически приемлемым в том смысле, что он должен быть совместим с другими компонентами препарата и не вредным для пациента. Препараты такого состава приспособлены для введения через рот, прямую кишку, нос и местного введения (включая защечное и подъязычное), введение через влагалище и парентеральное (в том числе - подкожное, внутримышечное, внутривенное и внутрикожное). Для удобства препараты могут изготавливаться в форме разовой дозы любым из хорошо известных в фармацевтике методом. Эти методы включают этап соединения активного компонента с носителем, который представлен одним или несколькими добавочными компонентами. В общем препараты изготовляют посредством равномерного и тщательного перемешивания активного компонента с жидкими носителями или тонкоизмельченными твердыми носителями, или и с теми, и с другими, а затем, в случае необходимости, приданием формы полученному продукту.
Препараты, описываемые в данной заявке, предназначенные для перорального введения, могут выпускаться в виде отдельных упаковок, таких как капсулы, облатки или таблетки, каждая из которых содержит определенное количество активного компонента; или могут выпускаться в виде порошков или гранул; в виде растворов или суспензий в воде или иной жидкости; в виде масляно-водных жидких эмульсий. Активный компонент может также быть изготовлен в виде большой пилюли, лекарственной кашки или пасты. Предпочтительно пероральное введение препарата.
Таблетка может быть изготовлена прессованием или формовкой, с одним или более добавочными компонентами. Прессованные таблетки можно получить в соответствующем аппарате прессованием активного компонента, находящегося в сыпучем состоянии, таком как порошок или гранулы, произвольно смешанным со связующим веществом (таким как повидон, желатин, оксипропилметилцеллюлоза), смазывающим агентом, инертным разбавителем: консервантом, разрыхлителем (например, натриевой солью гликолата крахмала, имеющим поперечные сшивки повидоном, имеющей поперечные сшивки натриевой солью карбоксиметилцеллюлозой), поверхностно-активным или диспергирующим агентом. Формованные таблетки могут быть изготовлены формованием в соответствующем аппарате смеси порошкообразного соединения, пропитанного инертным жидким разбавителем. Таблетки могут (но не обязательно) иметь покрытие или разделительную черту; и могут быть изготовлены так, чтобы обеспечить контролируемое высвобождение содержащегося в них активного компонента, например, с помощью оксипропилметилцеллюлозы, взятой в различных пропорциях, можно получить желаемый профиль высвобождения.
Препараты, предназначенные для местного введения через рот, включают лепешки, состоящие из активного компонента с вкусовыми добавками, обычно - сахарозой и аравийской камедью или трагакантом; пастилки, состоящие из активного компонента на инертной основе, такой как желатин и глицерин, или сахароза или аравийская камедь, и полоскания для рта, состоящие из активного компонента в соответствующем жидком носителе.
Препараты для введения через прямую кишку могут представлять собой свечи, изготовленные на соответствующей основе, включающей, например, масло какао или салицилат.
Препараты, предназначенные для введения через влагалище, могут представлять собой пессарии, тампоны, кремы, гели, пасты, пену или жидкость для опрыскивания, содержащую в дополнение к активному компоненту хорошо известные в фармацевтике соответствующие носители.
Препараты, предназначенные для парентерального введения, включают водные и неводные изотонические стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, антисептики и растворенные вещества, которые обеспечивают изотоничность вводимого препарата крови определенного пациента, водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и наполнители. Препараты можно выпускать в содержащих одну или несколько доз герметических сосудах, например, в ампулах, в флаконах; препараты можно хранить в высушенном при замораживании (лиофилизованном) виде, при этом непосредственно перед употреблением необходимо добавить только стерильный жидкий носитель, например, воду для инъекции. Самостоятельно можно приготовить растворы для инъекций и суспензии из стерильных порошков, гранул и таблеток типа описанных ранее.
Желательно, чтобы жидкие препараты с растворенным производным 5-урацила были забуферены - рН от 7 до 11, как правило, - от 9,5 до 10,5. Предпочтительны такие расфасованные дозы препарата, которые содержат дневную дозу или единичную дозу, часть дневной дозы (как описано выше) или соответствующую часть дозы активного компонента.
Упоминавшиеся ранее производные 5-урацила, применяемые согласно данной заявке в комбинации с 5-фторурацилом или его пролекарством, можно приготовить общепринятым способом. Например, описанные выше инактиваторы можно приготовить методами, описанными в: J. Meterocyl. Chem. 19/3/463-4 (1982) - для получения 5-этинилурацила; J. Chem. Soc. Perkin Trans 1/16/, 1655-70 (1981) для получения 5-(2-бромвинилурацила, 5-бромэтинилурацила и 5-/2-бром-1-хлорвинил/урацила; Nucleic Acids Chemistry т. 2, 927-30 (1978) для получения 5-цианурацила; Nucleic Acids Research. I/I/ 105-7 (1974) для получения 5-винилурацила; Z. Chem 17/II/ 415-16 (1977) для получения 5-трифторметилурацила; Nucleic Acids Research 3/10/, 2845 (1976) для получения 5-/1-хлорвинил/урацила.
Вышеупомянутые предшественники производных нуклеозида также можно получить общепринятыми способами, например, методами, описанными в Европейском патенте 356165 для получения 3'-фтор-2',3'-дидезокси-5-алкинилуридиновых соединений, таких как 2',3'-дидезокси-5-этинил-3'-фторуридин и Европейском патенте 272065 для получения 5-алкинилурациларабинозидов, таких как 1-/в-Д-арабинофуранозил/-5-проп-1-унилурацил.
Описанные выше новые соединения 5-С3-6 алкинилурацила, являющиеся предпочитаемыми производными 5-урацила для применения, соответственно описанному в заявке, могут быть получены одним из следующих способов:
а) обработкой 5-С3-6 алкинилуридинового соединения, превращающий его в желаемое соединение урацила; или
б) обработкой соединения урацила, замещенного в 5-м положении соответствующей отщепляемой группой, с С3-6 алкином для получения требуемого соединения урацила.
В описанном выше способе а) катализатором реакции может выступать фермент, например, при обработке соединения уридина - фермент тимидинфосфорилаза, преимущественно в забуференной среде при рН от 6 до 8.
В описанном выше способе б) соединение урацила, замещенное в 5-м положении подходящей отщепляемой группой, например иодом или бромом, обрабатывают С3-6 алкином в присутствии соответствующего палладиевого катализатора, такого как хлорид бис/трифенилфосфин/палладия (II), и иодида меди в аминовом растворителе, таком как триэтиламин.
Следующие примеры иллюстрируют данное изобретение.
ПРИМЕР 1. 5-Пропинилурацил
а) К размешиваемому раствору 2'-дезокси-5-пропинилуридина (Европейский патент 272065) (20 г, 75 мМ) в водном фосфатном буфере при рН 6,84 (1250 мл) добавляли очищенную тимидинфосфорилазу Е. coli (10.000 единиц) (Т.А. Кренитский и др., Biochemistry 20, 3615, 1981; Патент США 4.381.344) и щелочную фосфатазу (10.000 единиц) (Сигма тип YII-S из слизистой оболочки кишок быка) и смесь инкубировали при 37oС в течение 24 часов. Полученный белый осадок отфильтровывали, промывая водой (3 х 100 мл), этанолом (2 х 100 мл), простым эфиром (2 х 100 мл), высушивали под вакуумом над пятиокисью фосфора и получали указанное в названии соединение.
Т.пл. 275-280oС (разложение)
1Н ЯМР δ (d6ДMCO) 11,5-11,00 (bS, 2H, NH), 7,61 (1H, S, H-6), 1,95 ppm (3H, S, СН3)
При микроанализе вычислено для C7H6N2O2: С 56,00, Н 4,03, N 18,06. Найдено: С 55,92, Н 4,05, N 18,77.
б) 1-Арабинофуранозил-5-пропинилурацил (2,92 г, 20,4 мМ), 200 мл водного фосфата калия, рН 6,8, 4.000 единиц тимидинфосфорилазы (Креницкий Т.А. и др. , Biochemistry 20, 3615, 1981 и патент США 4.381.444), 4.000 единиц уридинфосфорилазы (Креницкий Т. А. и др. Biochemistry 20, 3615, 1981 и патент США 4.381.444) и 2.000 единиц щелочной фосфатазы (Бехрингер Маннхейм) размешивали при 40oС в течение пяти дней. Затем добавляли 8.000 единиц тимидин-фосфорилазы, 20.000 единиц уридин-фосфорилазы, 2.000 единиц щелочной фосфатазы и 30 единиц кислой фосфатазы (Бехрингер Маннхейм) и продолжали инкубировать еще в течение пяти дней. 5-Пропинилурацил, будучи менее растворимым, чем нуклеотид, осаждался из реакционной смеси.
Осадок и жидкость высушивали под вакуумом, затем 5-пропинилурацил дважды перекристаллизовывали из горячей воды и высушивали под вакуумом при комнатной температуре, получая 0,92 г (6,1 мМ) 5-пропинилурацила, выход 59%.
1Н ЯМР δ (d6ДMCO) 11,2 ppm уш.с. 2Н, 1Н и 3Н, 7,6 ppm (1Н, С, 6Н), 1.95 ррm (3Н, С, СН3).
CHN вычислено для C7H6N2O2: С 56,00, Н 4,03, N 18,66
Найдено: С 55,95, Н 4,03, N 18,60.
УФ-спектр: в 0,1М НСl максимум при 287 и 231 нм, в 50 мМ фосфате калия, рН 7,0 максимум при 287 и 231 нм, в 0,1М NaOH максимум при 306 и 240 нм.
При масс-спектрометрии наблюдается пик при молекулярном весе иона 151.
Пример 2. (5-Этинилурацил /ЭУ)
а) 5-(Триметилсилилэтинил)урацил
Раствор 5-иодоурацила (8 г, 30 мМ) в перегнанном триэтиламине (500 мл) и безводном ДМФ (10 мл) дегазировали в азоте, не содержащем кислород, в течение 15 минут. Затем добавляли хлорид бис(трифенилфосфин)палладия (11) (0,5 г), иодид меди (1) (0,5 г) и триметилсилилацетилен (10 г, 102 мМ), смесь нагревали при перемешивании при 50oС в течение 24 часов. Охлажденную реакционную смесь фильтровали, фильтрат выпаривали досуха и остаток растворяли в дихлорметане (500 мл). Органический раствор промывали 2% водным раствором динатриевой соли ЭДТА (3 х 250 мл), водой (3 х 200 мл), сушили (Nа2SО4) и выпаривали досуха. Остаток растирали с этанолом, получая первую порцию указанного в названии соединения. Было обнаружено, что твердый компонент, отфильтрованный из реакционной смеси, также содержал требуемый продукт, но в менее очищенном виде, поэтому его обрабатывали отдельно описанным выше способом, получая вторую порцию соединения.
1Н ЯМР δ (d6ДMCO) 11,75-10,85 (2Н, bS, 2Н), 7,75 (1H, S, H-6), 0,15 ррm (9Н, m, SiCH3).
б) 5-Этинилурацил
Раствор 5-триметилсилилэтинил/урацила (5,3 г, 24,4 мМ) в 0,2 М растворе метилата натрия в метаноле (400 мл) перемешивали при комнатной температуре в течение 3 часов и нейтрализовали до рН 7 разведенной соляной кислотой. Осадок отфильтровывали, промывали метанолом и высушивали, получая первую порцию указанного в названии соединения. Фильтраты и смывы объединяли, упаривали досуха и остаток кристаллизовали из метанола, получая вторую порцию продукта. Чистый продукт получали объединением двух полученных порций и дальнейшей перекристаллизацией из этанола.
Т.пл. 260oС (разложение).
1Н ЯМР δ (d6ДMCO) 11,6-10,8 (2Н, уш.с. 2Н), 7,8 (1Н, С, Н6), 4,03 ppm (1H, С, Н ацетилена).
Микроанализ: вычислено для C6H4N2O2: С 52,95, Н 2,96, N 20,58.
Найдено: С 52,04, Н 2,92, N 20,3.
ПРИМЕР 3. 5-Этинилурацил
а) 2,4-Диметокси-5-иод-пиримидин
В сухую круглодонную колбу объемом 1 л помещали 5-иодурацил (50 г, 0,21 М), хлорокись фосфора (300 мл) и N,N-диметиланилин (6 капель). Гетерогенную смесь прогревали при 120oС на масляной бане в атмосфере азота в течение 24 часов. Отгоняли хлорокись фосфора (при этом отгонялась и часть продукта). Затем реакционную смесь медленно и осторожно наливали поверх льда (1 л) с твердым бикарбонатом натрия, поддерживая внутреннюю температуру при или менее -20oС (это сопровождалось охлаждением в ацетоновой бане с сухим льдом). По окончании добавления рН реакционной смеси доводили до 7, добавляли твердый бикарбонат натрия. Смесь экстрагировали хлористым метиленом и органические фракции высушивали, пропуская через фазово-разделяющую бумагу. Неочищенный раствор 2,4-дихлор-5-иодпиримидина немедленно начинали добавлять по каплям к раствору, содержащему МеОН (400 мл) и метилат натрия (28,8 г, 0,533М). Это добавление занимало 1 час. Реакционную смесь затем перемешивали при комнатной температуре в течение ночи. Раствор нейтрализовали с помощью СО2 (газ), экстрагировали хлористым метиленом, высушивали над безводным Na2SO4, фильтровали и концентрировали. Неочищенный продукт адсорбировали на силикагеле (100 г) и наносили на испарительную хроматографическую колонку с 400 г силикагеля. Колонку элюировали смесью 90: 10 гексана:этилацетата (объем:объем). Соответствующие фракции объединяли и концентрировали, получая белое твердое вещество - соединение, указанное в названии.
Выход: 26,7 г (48%)
200 MHZ ЯМР CDCl3 δ = 3,97 (С, 3Н), 4,02 (С, 3Н), 8,43 (С, 1H).
б) 2,4-Диметокси-5-/b-триметилсилил/-этинилпиримидин
В сухую круглодонную колбу объемом 1 л в атмосфере азота помещали продукт, полученный на стадии а) (26,7 г, 0,10 М), безводный хлористый метилен (Олдрич, 150 мл), безводный Е3 (свежеперегнанный из гранулированного КОН, 250 мл). Несколько раз проводили откачивание и продувку через Firestone клапан. Шприцем добавляли триметилсилилацетилен (21,2 мл, 0,15М, Олдрич). Затем добавляли хлорид бис/трифенилфосфин/палладия (II) (Олдрич, 5,84 г, 8,32 мМ) и иодид меди (I) (Олдрич, 4,76 г, 25 мМ). Смесь прогревали при 60oС на масляной бане в течение 2 часов, охлаждали и фильтровали через целит. Фильтрат концентрировали под вакуумом. Остаток растворяли в толуоле (100 мл), затем толуол удаляли под вакуумом. Остаток переносили в хлористый метилен (200 мл), фильтровали и фильтрат экстрагировали 5% водным раствором динатриевой соли этилендиаминтетраацетата (3 х 100 мл Олдрич), Н2О (1 х 100 мл). Органический слой сушили, пропуская через фазово-разделяющую бумагу и концентрировали под вакуумом. Продукт чистили на Waters Prep 500, элюируя смесью 95:5 гексана:этилацетата (объем:объем). Неочищенный продукт адсорбировали на 100 г силикагеля и наносили на испарительную хроматографическую колонку с 400 г силикагеля. Колонку элюировали 97,5:2,5 гексана:этилацетата (объем: объем). Соответствующие фракции объединяли и концентрировали.
Выход: 16,94 г (73%).
Пробу (1,2 г) полученного соединения связывали на 6 г силикагеля и наносили на 50 г испарителей хроматографической колонны. Колонну элюировали гексан: этилацетатом 95: 5 (объем:объем). Соответствующие фракции объединяли, концентрировали, отгоняли с CH2Cl2 (2 х 30 мл) и высушивали под вакуумом, получая 1000 г указанного в названии соединения, температура плавления 72,5-73oС. Литературные данные: т. пл. 73-74oС Heterocyclic Chem, 19, 463 (1982).
в) 5-/В-триметилсилил/этинилурацил
В сухую 3-горлую круглодонную колбу в атмосфере азота помещали 2,4-диметокси-5-/b-триметилсилил/этинилпиримидин (6,5 г, 27,5 мМ), безводный ацетонитрил (120 мл Олдрич), иодид натрия (высушенный в печи под вакуумом при 80oС, 18 часов, 12,4 г, 82,7 мМ) и хлортриметилсилан (10,5 мл, 82,7 мМ, свежеперегнанный). Смесь прогревали с обратным холодильником в течение 3 часов и затем концентрировали под вакуумом. Остаток дигерировали с раствором, содержащим метанол (40 мл) и воду (20 мл), продукт отфильтровывали, получая 1,48 г (26%). Продукт растворяли в хлороформе, раствор адсорбировали на 7 г силикагеля, который затем наносили на испарительную хроматографическую колонну с 35 г силикагеля. Элюировали смесью хлороформ:метанол 95:5 (объем/объем), затем хлороформ:метанол 90:10 (объем:объем) и содержащие продукт фракции выпаривали, получая 1,23 г белого твердого вещества - соединения, указанного в названии.
г) 5-Этинилурацил
Раствор, содержащий 5-/b-триметилсилил/этинилурацил (3,85 г, 18,4 мМ) и метанол (370 мл) обрабатывали другим раствором, содержащим гидроокись натрия (2,3 г, 57,5 мМ) и воду (18 мл). Смесь перемешивали при комнатной температуре в течение 2 часов и затем концентрировали под вакуумом. Остаток суспендировали в воде (35 мл) и рН доводили до 5, используя 0,1N HCl. Твердые вещества растворялись, затем при рН 5 образовывался второй осадок. Продукт отфильтровывали, промывали водой и затем высушивали под вакуумом, получая 2,3 г (92%) 5-этинилурацила в виде светло-бежевого порошка.
При микроанализе вычислено для С6Н4N2O2: С 52,95, Н 2,96, N 20,58. Найдено: С 52,79, Н 3,02, N 20,44.
ПРИМЕР 4. 5-Этинилуридин
а) 2',3'-5'-Три-О-ацетил-5-иодуридин
В сухую 250 мл круглодонную колбу помещали 5-иодуридин (10 г, 27 мМ Олдрич), безводный пиридин (30 мл) и ангидрид уксусной кислоты (30 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут в атмосфере азота и растворитель удаляли под вакуумом. Соединение разбавляли толуолом (2 х 50 мл) и толуол удаляли под вакуумом. Продукт чистили на 75 г испарительной хроматографической колонне, элюцию проводили 90:10 (объем: объем) СНСl3: МеОН. Соответствующие фракции объединяли и концентрировали, получая указанное в названии соединение в виде белой пены. Полученный продукт использовали на следующей стадии.
б) 2',3',5'-Три-O-ацетил-5-[2-(триметилсилил)этинил]уридин
В сухую круглодонную колбу, снабженную обратным холодильником, (в атмосфере азота) помещали продукт, полученный на стадии а) (27 мМ), безводный хлористый метилен (260 мл Олдрич) и безводный триэтиламин (260 мл, свежеперегнанный из гранулированного NaOH). Несколько раз проводили откачивание и продувку системы азотом и затем оставляли в атмосфере азота. Следующим добавляли (триметилсилил)ацетилен (11,65 мл, 82 мМ, Олдрич), затем - иодид меди (1) (Олдрич, 1,57 г, 8,2 Мм) и хлорид бис/трифенилфосфин/палладия 11 (Олдрич, 1,85 г, 2,6 мМ). Смесь прогревали при 60oС на масляной бане в течение 30 минут, охлаждали и фильтровали. Фильтрат концентрировали под вакуумом. Остаток переносили в СН2Сl2 (300 мл), фильтровали, промывали 5% водным раствором динатриевой соли этилендиаминтетраацетата (2 х 75 мл), водой (100 мл), сушили над Na2SO4, фильтровали и концентрировали под вакуумом.
Полученное соединение связывали с 50 г силикагеля и наносили на испарительную хроматографическую колонку с 400 г силикагеля, элюцию проводили СНСl3. Фракции, содержащие продукт, объединяли и концентрировали, получая указанный в названии продукт в виде светло-желтой пены.
Выход: 13 г.
300 MHZ ЯМР СDСl3 δ 8,2 (уш.с, 2Н, 1H), 7,77 (С, 1H, Н6), 6,11 (g, H1', 1H), 2,22 (С, 3Н, ОАс), 2,11 (С, 3Н, ОАс), 0,22 (С, 9Н, SiMe3).
в) 5-Этинилуридин
Продукт, полученный на стадии б) (9,5 г, 24 мМ), растворяли в метаноле (200 мл) и разводили раствором, содержащим натрий (0,8 г) и метанол (100 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем нейтрализовали, используя смолу Дауэкс 50W-X8 (Н+форма). Смолу отделяли фильтрованием и промывали метанолом. Фильтрат концентрировали под вакуумом, получая 4,85 г твердого вещества бежевого цвета. Соединение чистили на Waters Prep 500 на колонке C18 с реверсионной фазой, получая 1,2 г указанного в названии продукта (белое твердое вещество). Неочищенные фракции хроматографировали повторно. Дополнительно было получено 1,94 г продукта.
Выход: 49%.
Вычислено: С 49,25%, Н 4,47%, N 10,44%
Найдено: С 49,07%, Н 4,53%, N 10,32%
200 MHZ ЯМР (ДМСО d6) δ 11,60 (уш.с., NH, 1H), 8,36 (С, Н6, 1H), 5,72 (g, J=4,3 H' H1, 1H), 4,01(C, 1H, C=C-Н).
Следующие примеры иллюстрируют фармакологические препараты, в которых "Активный компонент" - это 5-пропинилурацил, 5-этинилурацил или другой инактиватор урацил-редуктазы, описанный выше, или смесь инактиватора с 5-фторурацилом.
ПРИМЕР 5. Препараты в таблетках
Препараты 5А, 5В и 5С изготовлены мокрым гранулированием компонентов (за исключением стеарата магния) с раствором повидона и последующим высушиванием гранул, добавлением стеарата магния и прессованием (табл.1).
Следующий препарат 5Д изготовлен непосредственно прессованием смешанных компонентов. Использовалась лактоза прямо в прессованном виде.
Препарат 5Д - мг/таблетку
Активный компонент - 5
Лактоза - 155
Авицель РН 101 - 100 - 260
Следующий препарат 5Е представляет собой таблетку с контролируемым высвобождением препарата, изготовлен мокрым гранулированием компонентов (за исключением стеарата магния) с раствором повидона, последующим высушиванием гранул, добавлением стеарата магния и прессованием.
Препарат 5Е - мг/таблетку
Активный компонент - 5
Оксипропилметиоцеллюлоза (Methocel КЧМ Premium) - 110
Лактоза, В.Р. - 50
Повидон, В.Р. - 28
Стеарат магния - 7 - 200
ПРИМЕР 6. Препараты в капсулах
Следующие препараты 6А и 6В изготовляли смешиванием непрессованных компонентов, препараты помещали в плотные капсулы, состоящие из 2-х частей.
Препарат 6А - мг/капсулу
Активный компонент - 10
Лактоза, В.Р. - 250
Натриевая соль гликолата крахмала - 25
Стеарат магния - 5 - 290
Препарат 6В - мг/капсулу
Активный компонент - 5
Предварительно желатинированный крахмал NF-15 - 245 - 250
Препарат 6С - мг/капсулу
Активный компонент - 10
Macrogol 4000, В.Р. - 340 - 350
Macrogol 4000, В.Р. плавили и диспергировали в нем активный компонент. Расплав тщательно перемешивали и затем им заполняли состоящую из 2-х частей плотную желатиновую капсулу.
ПРИМЕР 7. Препараты для инъекций
Активный компонент - 10 мг
Стерильный, очищенный от пирогеновых примесей, пирофосфатный буфер (рН 10), q.s. до - 10 мл
Активный компонент растворяли в большей части фосфатного буфера (35-40oС), затем доводили объем и фильтровали через микропоровый фильтр в 10-мл капсулу из желтого стекла (тип 1), закрывали стерильной пробкой и герметизировали.
ПРИМЕР 8. Свечи - мг/свечу
Активный компонент, 63 мкм* - 10
Твердый жир, В.Р. (Витепзоль Н15-нейтральный Dynamit) - 1790 - 1800
* Активный компонент использовали в виде порошка, в котором, по крайней мере, 90% частиц имели размеры 63 мкм и менее.
Одну пятую часть Витепзоля Н15 растворяли в сосуде с паровой рубашкой при температуре не выше 45oС. Активный компонент просеивали через 200-мкм сито и добавляли к расплавленному основанию при перемешивании, используя мешалку, снабженную режущей головкой, до получения хорошо перемешанного дисперсного раствора. Поддерживая температуру смеси при 45oС, к суспензии добавляли остальной Витепзоль Н15 и перемешивали до получения гомогенной смеси. Суспензию пропускали через 250-мкм сито из нержавеющей стали и охлаждали при постоянном перемешивании примерно до 40oС. При температуре от 38 до 40oС 1,8 г смеси фильтровали в подходящую пластиковую форму. Свечи охлаждали до комнатной температуры.
Для проверки эффективности 5-замещенных соединений урацила, описанных в заявке, был проведен ряд экспериментов.
ЭКСПЕРИМЕНТ 1. Определение инактивации урацил-редуктазы
Урацил-редуктазу (1 микромоль) (дигидропиримидиндегидрогеназа, ЕС 1.3.1.2), очищенную из печени быка, инкубировали со 100 микромолями инактиватора и 5 мМ дитиотрейтола (восстановитель фермента) при 37oС в течение 30 минут в 0,05 М трис-НСl при рН 8,0. Фермент и инактиватор разводили в 100 раз в буфере для пробы, содержавшем 200 микромолей НАДФ.Н, 200 микромолей тимина и 1 мМ дитиотрейтола в трис-НСl при рН 8.0. Активность фермента определяли спектрофотометрически. Полученные активности корректировали с учетом скорости окисления НАДФ.Н оксидазой, которая составляла менее 10% скорости тиминзависимого окисления НАДФ.Н. Процент инактивации фермента равнялся 100% минус процент остаточной ферментативной активности. Значения, данные в скобках, - кажущиеся контанты скорости реакции инактивации фермента (реакция первого порядка), полученные в подобных экспериментах, где активность фракций определяли как функцию времени инкубации 50 микромолей инактиватора с ферментом.
Результаты представлены ниже:
Соединение - % инактивации
5-Этинилурацил - 100 (100)
5-Цианурацилa - 100 (14)
5-Пропинилурацил - 100 (8)
5-Бромэтинилурацил - 100 (8)
5-/1-Хлорвинил/урацил - 100 (5)
5-Иодурацил - 100 (4)
5-Гекс-1-нилурацилa - 90
5-Винилурацил, a,в - 86
5-Трифторметилурацил - 75
5-Бромурацил - 75
а Ингибирование было обратимым, так как фермент, обработанный этим производным, медленно восстанавливал активность после 100-кратного разведения в смеси для пробы.
в Эти нуклеооснования были получены in situ обработкой соответствующего нуклеозида тимидин-фосфорилазой (40 единиц/мл) в 35 мМ калий-фосфате в течение 20 минут перед добавлением к урацил-редуктазе. Исходные нуклеозиды не являлись инактиваторами.
Изучали эффективность 5-этинилурацила (ЭУ), как указано в следующих экспериментах со 2 по 4 и представлено на чертежах, где:
на фиг.1 показано увеличение уровней урацила и тимина через 24 часа после введения крысам через рот различных доз ЭУ;
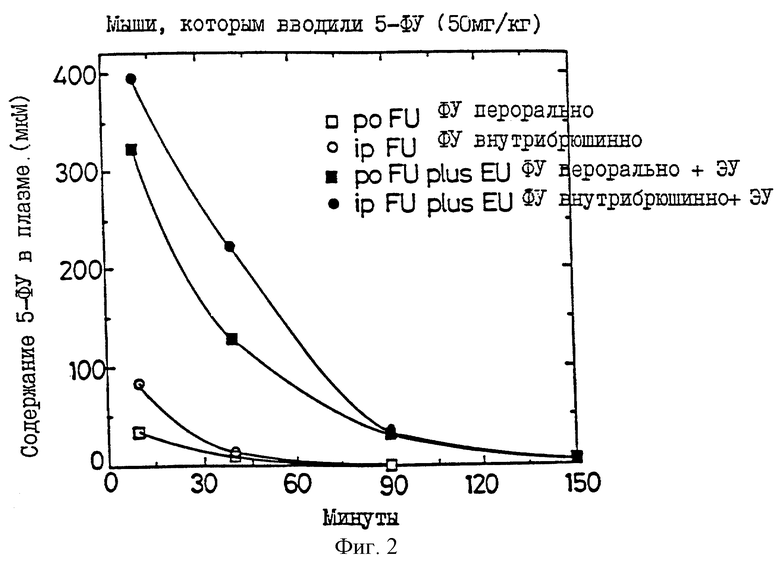
фиг. 2 показано, что ЭУ повышает уровни 5-фторурацила (5-ФУ) в плазме. Мышам вводили дозы либо через рот (перорально), либо внутрибрюшинно (введение 5-ФУ). 5-Этинилурацил в дозах 2 мг/кг вводили внутрибрюшинно за 90 минут до введения 5-ФУ.
ЭКСПЕРИМЕНТ 2. Инактивация урацил-редуктазы (in vivo). У мышей, крыс, собак и обезьян, которым вводили малые количества 5-этинилурацила (ЭУ), наблюдалось быстрое и значительное увеличение уровней урацила и тимина в плазме. Максимальный эффект достигался при введении крысам перорально примерно 0,1 мг/кг, мышам - подкожно - от 0,5 до 1 мг/кг и собакам - внутривенно - приблизительно 1 мг/кг; вероятно, при этом происходит полная инактивация урацил-редуктазы. У мышей, собак и крыс такие дозы увеличивают уровни урацила в плазме от, приблизительно, 3 мМ примерно до 50-60 мкМ. Уровень урацила в плазме снижается до нормального через 24 часа (время полужизни = 10 часам). На фиг.1 показано повышение уровней урацила и тимина в плазме у крыс через 4 часа после введения через рот различных доз 5-этинилурацила, обусловленное инактивацией урацил-редуктазы. ЕД50 составляет 0,01 мг/кг.
ЭКСПЕРИМЕНТ 3. Влияние на уровень ФУ в плазме
У мышей и крыс, которым предварительно вводили 5-этинилурацил (ЭУ), а затем давали ФУ, поддерживались более высокие уровни ФУ в плазме, чем у тех мышей, которым предварительно не вводили ЭУ. К тому же обычная изменчивость ГУ в плазме у крыс, которым вводили ФУ через рот в дозах 50 мг/мл, ликвидировалась при предварительном введении ЭУ. АУС кривых зависимости концентраций ФУ в плазме от времени составляли 41,126,68 (среднее = 78±55%) против 417, 446 и 426 (среднее = 430±3%) для крыс, которым предварительно не вводился ЭУ, и для крыс, которым предварительно вводили ЭУ, соответственно.
ЭКСПЕРИМЕНТ 4. Усиление антиопухолевой активности 5-фторурацила с помощью 5-этинилурацила (ЭУ) у мышей
Мышам имплантировали опухоль прямой кишки 38 в "нулевой" день. Мышам (по 8 в группе) 5-ФУ в дни с 1 по 9 включительно в дозах, указанных в табл.2, ЭУ вводили внутрибрюшинно в дозах 2 мг/кг за 30 минут до введения указанных доз 5-ФУ.



Предложен новый инактиватор урацил-редуктазы. Это 5-этинилурацил. Он усиливает действие 5-фторурацила и применяется при лечении рака. Предложены также фармацевтическая композиция на основе 5-этинилурацила и способ ее получения. Предложены фармацевтическая комбинация для усиления действия или снижения токсичности 5-фторурацила. Комбинация содержит отдельные или смешанные компоненты 5-этилурацил и 5-фторурацил или его пролекарство для одновременного, раздельного или последовательного введения. Изобретение расширяет арсенал инактиваторов урацил-редуктазы. 4 с. и 12 з.п. ф-лы, 2 ил., 2 табл.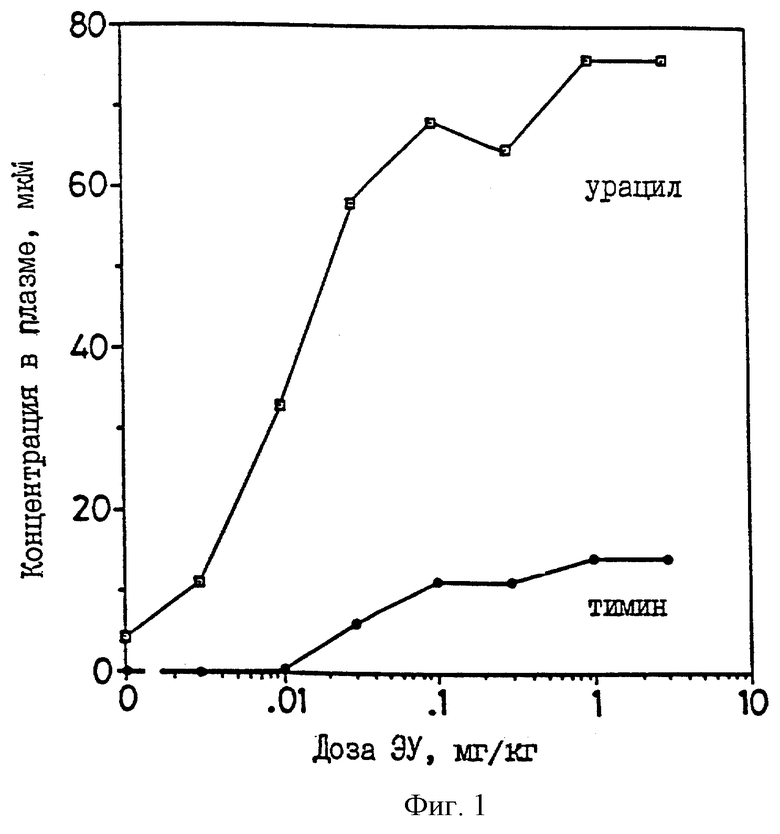
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| P.J | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Part IV | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| NOCLEIC ACIDS RESEARCH, vol | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Tatsumi, Kunihiko et al.: "Inhibitory effects of pyrinidine, barbituric acid and pyridine/derivatives on 5-fluorouracil degradation in rat liver extracts", Jpn | |||
| J | |||
| Cancer Res | |||
| Кузнечная нефтяная печь с форсункой | 1917 |
|
SU1987A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| ЕР, 0272065, А2, 22 June 1988 | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Claude Desgranges et al, "Effect of (E)-5-(2-Bromovinyl) uracil on the Catabolism and Antitumor Activity of 5-fluorouracill in Rats and Leukemic Mice", Cancer research, vol 46, 1986, see page 1094 - page 1101. | |||

